DOI:
10.1039/D2RA01918C
(Paper)
RSC Adv., 2022,
12, 13381-13392
Enhanced photocatalytic hydrogen evolution and ammonia sensitivity of double-heterojunction g-C3N4/TiO2/CuO†
Received
24th March 2022
, Accepted 27th April 2022
First published on 4th May 2022
Abstract
The performance of semiconductor photocatalysts has been limited by rapid electron–hole recombination. One strategy to overcome this problem is to construct a heterojunction structure to improve the survival rate of electrons. In this context, a novel g-C3N4/TiO2/CuO double-heterojunction photocatalyst was developed and characterized. Its photocatalytic activity for hydrogen production from water–methanol photocatalytic reforming was explored. Methanol is always used to eliminate semiconductor holes. The g-C3N4/TiO2/CuO double-heterojunction photocatalyst with a narrow bandgap of ∼1.38 eV presented excellent photocatalytic activity for hydrogen evolution (97.48 μmol (g h)−1) under visible light irradiation. Compared with g-C3N4/TiO2 and CuO/TiO2, the photocatalytic activity of g-C3N4/TiO2/CuO for hydrogen production was increased approximately 7.6 times and 1.8 times, respectively. Below 240 °C, the sensitivity of g-C3N4/TiO2/CuO to ammonia was approximately 90% and 46% higher than that of g-C3N4/TiO2 and CuO/TiO2, respectively. The enhancement of the photocatalytic activity and gas sensing properties of the g-C3N4/TiO2/CuO composite resulted from the close interface contact established by the double heterostructure. The trajectory of electrons in the double heterojunction conformed to the S-scheme. UV-vis, PL, and transient photocurrent characterization showed that the double heterostructure effectively inhibited the recombination of e−/h+ pairs and enhanced the migration of photogenerated electrons.
1. Introduction
With rapid economic development, the demand for clean energy is constantly increasing.1–3 Therefore, the exploitation of renewable clean energy is a focus of attention globally. Hydrogen has been widely studied because it is nontoxic, pollution-free and renewable and has a high calorific value.4,5 To date, many encouraging methods have been obtained, such as chemical, physical-chemical or electrocatalytic hydrogen evolution.6,7 Therefore, photocatalytic technology for hydrogen production has received more attention because it is clean, inexpensive, and environmentally friendly.8 Since the initial report of water splitting for hydrogen evolution based on photo-assisted electrochemical technology in 1972, photocatalysis has been accepted as a prospective method for hydrogen production.9 Although prominent progress has been made in photocatalytic techniques, how to effectively inhibit the rapid recombination of photogenerated electrons and holes is still a problem to be solved.10
One strategy to reduce the electron–hole recombination of semiconductors is to add a sacrificial agent (hole scavenger) to the reaction solution. The efficiency of photocatalysts for hydrogen production in pure water is not ideal. Methanol is the most commonly used sacrificial agent and can eliminate the holes that are generated after electron transition. More surviving electrons are used in the reduction reaction to produce hydrogen. The hydrogen atoms result partly from water cracking and partly from the dehydrogenation of hole scavengers. Methanol is considered a possible candidate fuel with several advantages: a simple molecular structure, a high hydrogen carbon ratio and the ability to extract it from biomass energy.
It is also one of the common methods to enhance the density and survival rate of photogenerated electrons by constructing heterojunctions.11,12 To make better use of sunlight, many researchers are looking for photocatalytic semiconductors with low band gap energy.13 Graphitic carbon nitride (g-C3N4) is a burgeoning photocatalyst and has a narrow band gap, physical-chemical stability, and low cost characteristics. Nevertheless, the practical application of bare g-C3N4 is still hindered by the problems of a high electron–hole recombination rate and low quantum efficiency.14 In general, the construction of a heterojunction between g-C3N4 and other semiconductors can enhance g-C3N4 photocatalytic performance. Heterojunctions encourage light excitation of e−/h+ pairs and prolong their life for separation. However, although titanium dioxide (TiO2,∼3.2 eV) has a wide band gap and low visible light utilization, its catalytic performance is still high.15 As shown in preceding records, the band gap of TiO2 was opportunely matched with the band gap of g-C3N4.16 Combining TiO2 with g-C3N4 to produce a new composition can increase the flat plate potential and reduce the band gap of the composite. This is conducive to the electronic transition between heterogeneous interfaces. In addition, the photoelectrons (e−) in the conduction band (CB) of g-C3N4 can be diverted to the CB of TiO2. The process can control the rapid recombination of electrons and holes. In addition, metal oxides have great cocatalytic properties due to their excellent conductivity, such as CeO2/g-C3N4,17 NiO/TiO2,18 ZnO/In2O3,19 CuO/ZnO,20 CuO/WO3,21 etc. Among these metallic oxides, copper oxide (CuO) has become a research hotspot in the preparation of heterojunction photocatalysts attributable to the low toxicity of raw materials and absorption ability in the visible light spectrum. As a p-type semiconductor with a narrow band gap, CuO has shown excellent photocatalytic performance. Studies have shown that putting CuO into the structure of TiO2 can result in an increase in photocatalytic activity owing to the internal electric field.22 In recent years, the S-scheme (step-scheme)23,24 photocatalytic system was considered an interpretation to explain the result that presented to decrease the recombination rate, energy separation of generated charges and retaining excellent redox ability. Vairamuthu Raj et al.25 researched g-C3N4/TiO2/CuO S-scheme composites as effective photocatalysts in the field of photocatalytic degradation. However, the g-C3N4/TiO2/CuO catalysts H2 evolution of the photocatalytic performance has not been verified.
On this basis, we designed and constructed g-C3N4/TiO2/CuO heterojunction structures. In this structure, TiO2 and g-C3N4 are n-type semiconductors, and CuO is a p-type semiconductor. Therefore, this special structure contains double p–n heterostructures at the same time. The double p–n heterobonding electron transition provides sufficient space, which can effectively reduce the recombination rate of electron holes, prolong the electron lifetime and improve the performance of H2 production. The cross section of the p–n heterojunction also has a certain synergistic effect. Double heterojunctions can independently control e− and h+ injection/extraction while maintaining a high photogenerated carrier migration rate.26 Therefore, this work provides unique insight into the design of a multicomponent photocatalyst heterojunction mechanism.
2. Experiment section
2.1 Materials
Tetrabutyl titanate (TBOT) and melamine (MA) were purchased from Shanghai Ron Chemical Technology Co., Ltd. Titanium dioxide (P25, 20 nm) was purchased from Macklin Co., Ltd. Anhydrous ethanol, concentrated hydrochloric acid (HCl, 32 wt%) and methanol were obtained from Sinopharm Chemical Reagent Co., Ltd. Copper acetate (Cu(CH3COO)2) was procured from Tianjin BASF Chemical Co. Ltd. All chemicals were of analytical purity without further purification.
2.2 Synthesis
2.2.1 Synthesis of g-C3N4, TiO2 and CuO. MA powder (5 g) was placed and heated at 500 °C for 2 h in a muffle furnace. When cooled, the g-C3N4 sheet was obtained and ground into powder in a mortar. TiO2 powder was prepared in triplicate by a sol–gel strategy. First, TBOT (17 mL) was added into a 200 mL beaker. Then, anhydrous ethanol (40 mL) was added dropwise to the beaker through a peristaltic pump (2 rpm), and the resulting solution I was continuously stirred for 30 min. Ethanol (60 mL) and deionized water (2.5 mL) were added to the second 200 mL beaker. When the pH of the resulting mixing solution II was adjusted to 1–2 with HCl (32 wt%), solution II was added to solution I slowly by a peristaltic pump (2 rpm) to form solution III. After stirring for 2 h, solution III was allowed to stand for 24 h to obtain the colloid and then dried overnight to acquire the TiO2 precursor. Next, the precursor was calcined for 2 h to prepare powder. Cu(CH3COO)2 powder (2.51 g) was added to deionized water (50 mL) and stirred for 15 min to form a homogeneous solution, which was heated at 100 °C and continuously stirred until evaporation. The obtained solids were dried overnight and ground to powder in a mortar. Then, the powder was calcined at 500 °C in air for 2 h to acquire CuO powder.
2.2.2 Synthesis of g-C3N4/TiO2 and CuO/TiO2. The previously prepared g-C3N4 powder (1 g) and TiO2 powder (1 g) were added to deionized water (50 mL) and stirred evenly. The mixing solution was heated at 100 °C and stirred successively to evaporation. Afterward, the obtained sample was dried overnight in an oven and ground to powder. g-C3N4/TiO2 was acquired by calcining the powder at 500 °C in air for 2 h. The g-C3N4 powder was replaced with the previously prepared CuO powder, and the above steps were repeated to obtain the CuO/TiO2 catalyst.
2.2.3 Synthesis of g-C3N4/TiO2/CuO. g-C3N4/TiO2 powder (1 g), Cu(CH3COO)2 powder (2.51 g) and deionized water (50 mL) were blended together. The mixed solution was heated to evaporation at 100 °C. The mixture was dried in an oven overnight and calcined in air at 500 °C for 2 h to obtain g-C3N4/TiO2/CuO powder.
2.3 Catalyst characterization
The phases and crystal structure characteristics of the catalysts were examined by X-ray diffraction (XRD, D8-advance, brukeraxs). The chemical functional groups of the samples were explored by Fourier transform infrared spectroscopy (FTIR, Nicolet 5700, Thermo Nicolet Corporation) in the range of 4000 to 500 cm−1 with a PerkinElmer spectrophotometer. The micromorphology and elemental mapping were observed by scanning electron microscopy (SEM, FEI Sirion 200). Transmission electron microscopy (TEM, FEI Tecnai G2F 20) and selected area electron diffraction (SAED) were used to characterize the morphology and the crystal structure of the fabricated photocatalyst. X-ray photoelectron spectroscopy (XPS, AXIS Supra) was carried out to analyze the chemical bonds of substances and valence state. The Brunauer–Emmett–Teller (BET) specific surface area and pore structure properties of the photocatalysts were obtained employing a quantachrome instrument nitrogen adsorption apparatus at 75 K. The optical absorption characteristics and band gap calculation were measured by ultraviolet visible diffuse reflectance spectroscopy (UV-vis, Shimadzu, UV3600). At an excitation wavelength of 320 nm, room-temperature photoluminescence emission spectra (PL, Edinburgh, FLS9800) were measured to study the suppression of the e−/h+ recombination rate. The instantaneous photocurrent response was tested by an electrochemical workstation (Edinburgh, FLS9800) via the standard three-electrode configuration, including the Ag/AgCl electrode with K2S2O8 (0.05 M) and Na2SO4 (0.5 M) as the electrolyte.
2.4 Photocatalytic performance
A closed gas circulation/evacuation system was used to collect the amount of H2 generated by the water–methanol solution in the presence of the above photocatalysts. In a standard photocatalytic process, photocatalyst (0.05 g), methanol (25 mL) and deionized water (25 mL) were added to a 0.2 L hermetic quartz container and blended together. The resulting solution was stirred under dark conditions for at least 30 minutes to achieve desorption and adsorption equilibrium. Afterward, the air was completely expelled from the airtight quartz container by N2 in 40 min. The reactant solution was kept at ∼6 °C and irradiated under a 300 W Xe lamp during the whole photocatalytic process. The gaseous products were measured by gas chromatography (Beijing Zhongjiao Jinyuan Technology Co., Ltd, GC-7920) with a TDX-01 column and thermal conductivity detector (TCD). Gaseous products were taken every 30 min for detection, and the experimental period was not less than 6 h.
2.5 Gas sensitivity test
First, a certain amount of catalyst was scattered into absolute ethanol to form a ropy solution. Afterward, an appropriate amount of catalyst-containing solution was applied to the ceramic tube. The ceramic tubes were fixed on the welding table by spot welding, and electrifying resistances were formed through a resistance wire. The coated electrodes were put in a dehumidifier for 24 h. Then, each sensor board was installed in a homemade poison gas chamber with a volume of 18 L. The sensor plate was attached to the gas sensing test system (WS-30, Winse) to obtain response signals of the sensor in terms of resistance. Ammonia (100 ppm) was injected into the gas chamber and converted to steam on a high-temperature platform. There was a small fan in the air chamber, which could diffuse the steam evenly. The response value of the sample to ammonia was detected at 260 °C and calculated using (1)27where Ra represents the sensor resistance in bare air and Rg represents the sensor resistance in measurement gases.
3. Results and discussion
3.1 Crystal phase and functional group analysis
The XRD spectrum analysis patterns of the prepared catalysts are shown in Fig. 1. In the pattern of pure CuO, diffraction peaks for CuO (JCPDS Card No. 00-089-5895) at 35.44°, 38.58°, 48.72°, 53.56°, 57.99°, 61.49°, 65.91° and 67.60° could be observed, corresponding to the (002), (111), (112), (020), (202), (113), (310), and (220) monoclinic crystalline planes, respectively.28 The all diffraction peaks of CuO could be evidently observed in g-C3N4/TiO2/CuO and CuO/TiO2, which proves that CuO is successfully introduced into the g-C3N4/TiO2/CuO and CuO/TiO2 catalysts. This is also confirmed in the following FTIR spectra. The diffraction peaks at 25.50°, 37.76°, 48.04°, 55.06° and 62.76° could be ascribed to the TiO2 (101), (004), (200), (211) and (204) planes, respectively.29 This confirms that the crystal form of TiO2 is anatase. And the diffraction peaks of TiO2 are disappeared in the spectra of g-C3N4/TiO2/CuO and CuO/TiO2, except the (101) diffraction peak. We speculate the peaks of TiO2 are covered by the diffraction peaks of CuO. The two diffraction peaks at 13.6° and 27.7° in the XRD spectrum of g-C3N4 correspond to the (100) and (002) peaks, respectively. The characteristic peak of (100) is related to the stacking of periodic array motifs of tris-triazine in the plane, while the (002) peak is due to the long-range planar stacking of aromatic rings.30 Simultaneously, there are other miscellaneous peaks caused by incomplete condensation of melamine. No characteristic peaks of g-C3N4 are found in the XRD images of g-C3N4/TiO2 and g-C3N4/TiO2/CuO. There are several possibilities for this phenomenon. One reason is that the characteristic peaks of g-C3N4 are covered by the strong peaks of TiO2. Another probable reason might be that g-C3N4 is decomposed by TiO2.30 The third is that it may be due to the high dispersion of g-C3N4 in the composites.17 However, the uniform loading of C and N elements in the EDS spectrum confirms the existence of g-C3N4. The crystallite sizes are 22.86, 11.17, 13.23, 20.52, 5.33 and 19.39 nm for CuO, g-C3N4, g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO, respectively. And they are calculated from X-ray line broadening (ω) according to the Scherrer formula (2):12| |
D = 0.89 λ/ω![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ
| (2) |
where λ is the X-ray wavelength and λ = 0.15406 nm. D presents the crystallite size, θ is the peak position and ω is FWHM (radians).
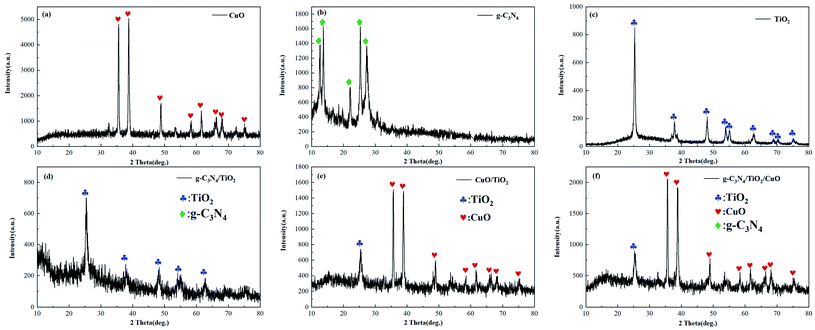 |
| | Fig. 1 XRD images of CuO, g-C3N4, TiO2, g-C3N4/TiO2, CuO/TiO2, g-C3N4/TiO2/CuO. | |
The FTIR spectra of the CuO, g-C3N4, g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO catalysts are displayed in Fig. 2. For g-C3N4, the bands related to the breathing of triazine units and stretching vibration of N–H are perceived at 802 and 3117 cm−1.31 Interestingly, these two peaks at the corresponding positions almost disappear in the g-C3N4/TiO2/CuO catalyst, which may be due to another calcination at 500 °C in the preparation of the catalyst. At 1200–1650 cm−1, four other distinct peaks are observed at 1094, 1332, 1464, and 1611 cm−1, which are derived from the representative stretching modes of the C–N heterocycles.32 Similarly, compared with g-C3N4, the strength of the four typical peaks of g-C3N4 becomes weaker in the g-C3N4/TiO2/CuO catalyst. The band at 532 cm−1 is ascribed to vibrations of Cu–O bonds.33 Moreover, no other bands are found in the range of 605 to 660 cm−1. The existence possibility of the Cu2O phase is completely excluded.34 The prime characteristic peak of g-C3N4 almost appears in the g-C3N4/TiO2 complex, which confirms its successful synthesis. Similarly, the successful synthesis of CuO/TiO2 and g-C3N4/TiO2/CuO is indicated through the retaining characteristic peaks of CuO and g-C3N4.
 |
| | Fig. 2 FTIR images of CuO, g-C3N4, TiO2, g-C3N4/TiO2, CuO/TiO2, g-C3N4/TiO2/CuO. | |
3.2 Morphology and element distribution of g-C3N4/TiO2/CuO
SEM and EDS were conducted to investigate the morphology and element distribution of the g-C3N4/TiO2/CuO catalyst. From Fig. S1(b–f),† it can be unambiguously observed that the dispersion of C and N elements is completely uniform, which confirms that g-C3N4 is equally supported on the composite catalyst. At the same time, Ti seems to have agglomerated, while Cu is evenly dispersed on the surface of TiO2 under the influence of selective deposition. CuO and g-C3N4 clusters are tightly wrapped around TiO2 because of the selective deposition method and direct contact with the TiO2 surface, resulting in the establishment of a double heterojunction due to the synergistic effect between the three components and the balance of the Fermi level.
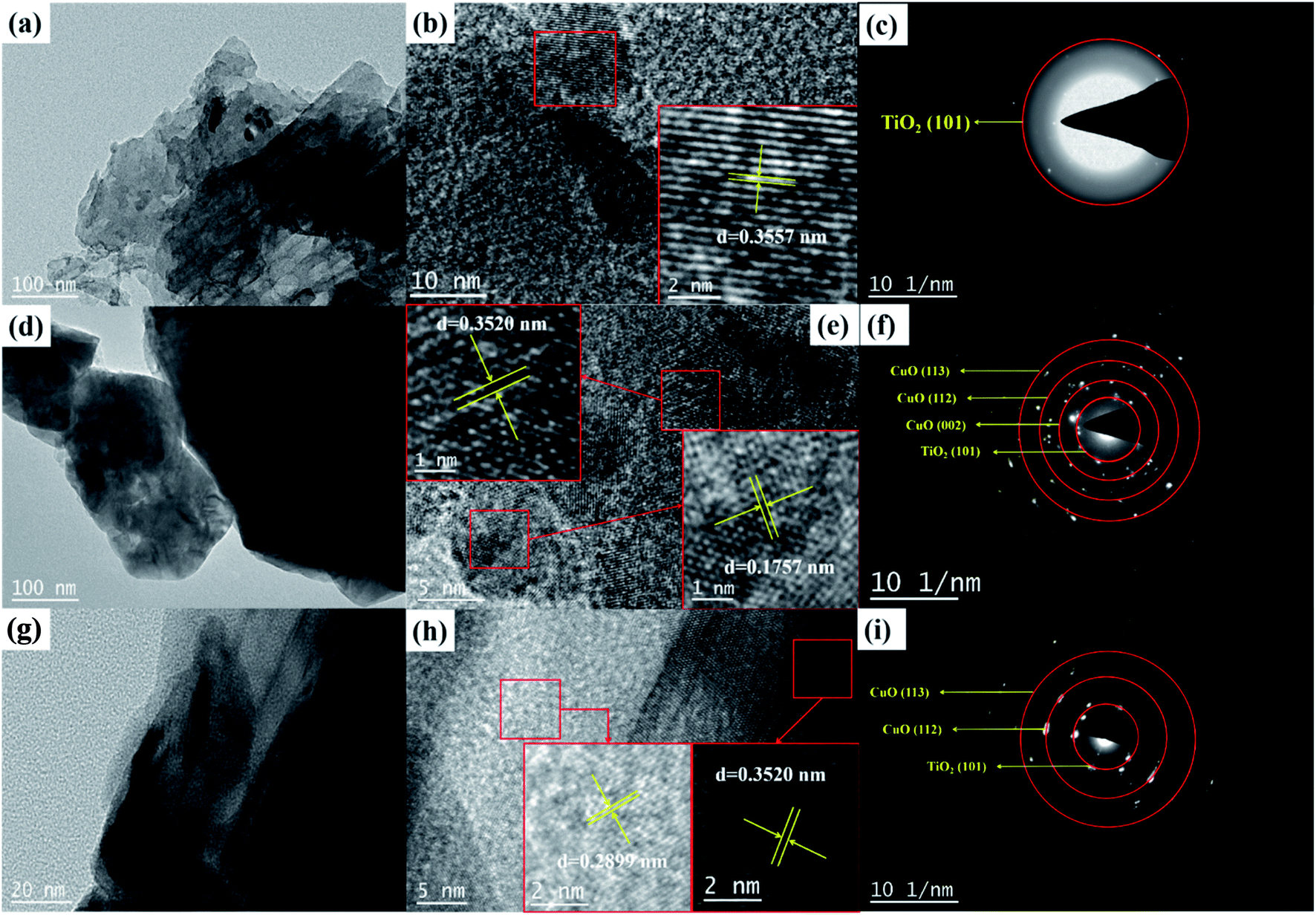
The successful preparation of g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO heterojunction photocatalysts are further verified by TEM and SAED images. Fig. 3(a and b) are the TEM image of g-C3N4/TiO2 catalyst. In Fig. 3(b), the lattice stripe with d = 0.3557 nm can be obtained by measuring the grid spacing, which corresponds to the (101) crystal plane of TiO2 (JCPDS PDF#21-1227).35 In Fig. 3(d), it can be seen that CuO is directly contact with TiO2 in CuO/TiO2 photocatalyst. CuO particles are smaller on the left and TiO2 particles are larger on the right. In Fig. 3(e), the lattice stripes with d = 0.3520 nm and d = 0.1757 nm are measured, which correspond to the (101) crystal plane of TiO2 and the (112) crystal plane of CuO (JCPDS Card No. 00-089-5895), respectively.28 The TEM of g-C3N4/TiO2/CuO is exhibited in Fig. 3(g–h), which indicate that the g-C3N4/TiO2/CuO material is a heterojunction nanocomposite with combined geometry. In Fig. 3(h), the lattice stripes with d = 0.3520 nm and d = 0.2899 nm are obtained by measuring the grid spacing, which correspond to the (101) crystal plane of TiO2 and the (−110) crystal plane of CuO. Heterojunction structure of g-C3N4/TiO2/CuO can enlarge the active sites and enhance the dynamic sites of reactant molecular adsorption. This is conducive to the separation of electron–hole, resulting in significant photocatalytic activity.20 Fig. 3(c) shows the SAED diagram of g-C3N4/TiO2 heterojunction, indicating the formation of single crystal heterojunction composites. Fig. 3(f and i) show the SAED diagram of CuO/TiO2 and g-C3N4/TiO2/CuO heterojunction, which exhibit the formation of polycrystalline heterojunction composites. The existence of g-C3N4 was not detected in the TEM images of g-C3N4/TiO2 and g-C3N4/TiO2/CuO, which are consistent with the results of XRD.
 |
| | Fig. 3 (a and b) Representative TEM micrographs and (c) SAED image of g-C3N4/TiO2, (d and e) TEM micrographs and (f) SAED image of CuO/TiO2, (g and h) TEM micrographs and (i) SAED image of g-C3N4/TiO2/CuO. | |
3.3 Valence and chemical bond analysis of g-C3N4/TiO2/CuO
The valence state and chemical bond of the g-C3N4/TiO2/CuO catalyst were analyzed by XPS characterization analysis. As shown in Fig. 4(a), the presence of Cu, C, N, Ti and O elements can be clearly distinguished in the full spectrum diagram. Fig. 4(b) displays the spectrum of Cu 2p. The bands at 933.2 and 953.5 eV are attributed to Cu 2p3/2 and Cu 2p1/2, respectively, and the other three shake-up satellite peaks appear at 941.6, 943.5 and 962.3 eV, which indicate that Cu2+ is formed.36 Meanwhile, there is no characteristic peak of Cu+. To verify the chemical bond of g-C3N4 in g-C3N4/TiO2/CuO, high-resolution spectra of C 2s and N 1s are obtained, as shown in Fig. 4(c) and (d). Three peaks are observed at 284.7, 285.3, and 288.5 eV in the XPS spectrum of C 1s (Fig. 4(c)). The first two peaks belong to polluted C, while the latter corresponds to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N binding mode. The three peaks at 398.9, 400.0, and 401.1 eV in the XPS spectral profile of N 1s (Fig. 4(d)) are explained to NC–N,N–(C)3, and N–H chemical bond, respectively.37 The Ti 2p spectrum (Fig. 4(e)) shows two prime peaks at 458.5 and 464.2 eV corresponding to the respective contributions of Ti 2p3/2 and Ti 2p1/2, which imply that Ti exists by Ti4+ formation.38 As seen from Fig. 4(f), the peaks of O 1s at 529.8 and 531.5 eV are derived from the contribution of the –OH and Ti–O–Ti chemical bonds, respectively. Since g-C3N4/TiO2/CuO is calcined in an air atmosphere, more oxygen vacancies are produced, leading to a clear peak shift of O 1s.39
C–N binding mode. The three peaks at 398.9, 400.0, and 401.1 eV in the XPS spectral profile of N 1s (Fig. 4(d)) are explained to NC–N,N–(C)3, and N–H chemical bond, respectively.37 The Ti 2p spectrum (Fig. 4(e)) shows two prime peaks at 458.5 and 464.2 eV corresponding to the respective contributions of Ti 2p3/2 and Ti 2p1/2, which imply that Ti exists by Ti4+ formation.38 As seen from Fig. 4(f), the peaks of O 1s at 529.8 and 531.5 eV are derived from the contribution of the –OH and Ti–O–Ti chemical bonds, respectively. Since g-C3N4/TiO2/CuO is calcined in an air atmosphere, more oxygen vacancies are produced, leading to a clear peak shift of O 1s.39
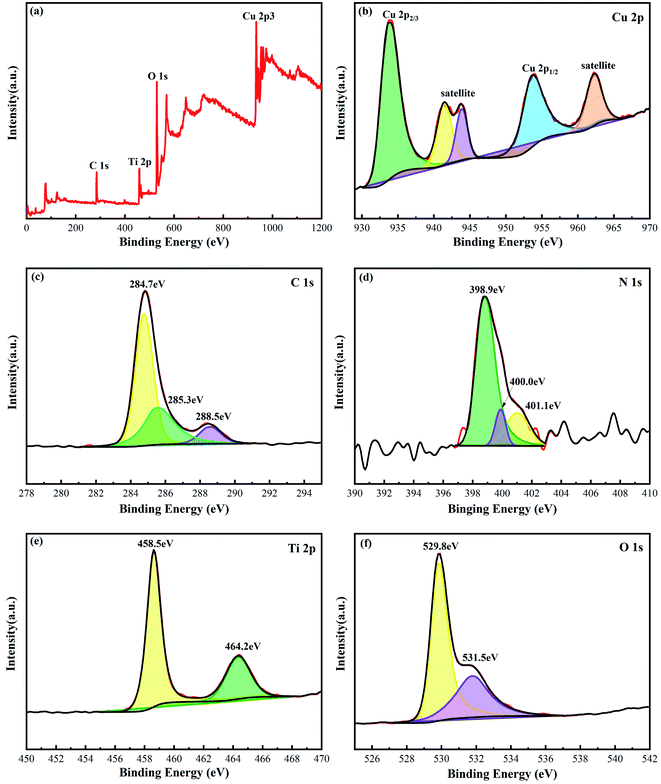 |
| | Fig. 4 XPS spectra of g-C3N4/TiO2/CuO sample: (a) survey XPS spectrum, (b) Cu 2p, (c) C 1s, (d) N 1s (e) Ti 2p, (f) O 1s. | |
3.4 Textural properties
The N2 adsorption–desorption isotherm and pore size distribution characteristics for the as-prepared catalysts are exhibited in Fig. 5. The specific surface areas of g-C3N4/TiO2 and CuO/TiO2 are only 28.51 and 6.34 m2 g−1, respectively, while that of the g-C3N4/TiO2/CuO ternary catalyst is 3.38 m2 g−1 and lower than those of the other two catalysts, as shown in Table 2, which is caused by the combination of CuO deposition and g-C3N4 pore coverage.16 As expected, the g-C3N4/TiO2/CuO catalyst also gives the lowest pore volume of 0.009 cm3 g−1, corresponding to a pore diameter of 10.71 nm, which is higher than those of the other two catalysts. A large aperture can increase the refractive index of incident light to improve the utilization of light. This is consistent with the photocatalytic capacity of H2 production below. On the other hand, g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO catalysts all display type IV isotherm while presenting a discrete hysteresis loop (H3 type) under the relative pressure (P/P0, 0.40–1.0) range. The phenomenon are caused by the slit-like pores and capillary condensation exist in the mesoporous.40
 |
| | Fig. 5 The N2 adsorption–desorption isotherm (inset: Barrett–Joyner–Halenda (BJH) mesoporous size distribution) of the (a) g-C3N4/TiO2, (b) CuO/TiO2, and (c) g-C3N4/TiO2/CuO, respectively. | |
Table 1 Comparison photocatalytic H2 production performance of relevant g-C3N4 and CuO-based heterojunction photocatalysts
| Photocatalysts |
Cocatalysts |
Sacrificial agents |
Light source (wavelength/nm) |
Activity (μmol h−1 g−1) |
Ref. |
| SiO2/g-C3N4/CdS QDs |
None |
Na2S and Na2SO3 |
Visible light |
225.1 |
41 |
| TiO2/g-C3N4 |
Pt |
Methanol |
450 W Xe lamp |
74.7 |
42 |
| g-C3N4/CdS |
None |
Lactic acid |
300 W Xe lamp (λ > 400 nm) |
392.84 |
43 |
| g-C3N4/CeO2 |
Pt |
Lactic acid |
300 W Xe lamp (λ > 420 nm) |
73 |
44 |
| CuO/MoO3 |
None |
Methanol |
Visible light |
98.5 |
45 |
| CuO/CdS |
None |
Lactic acid |
300 W Xe lamp (λ > 420 nm) |
3317.5 |
46 |
| CuO/ZnO |
None |
Na2SO4 |
Three tungsten lamps (200 W, 410–800 nm) |
340 |
47 |
| CuO/WO3/CdS |
None |
MB |
300 W Xe lamp |
44.5 |
48 |
| CuO/Ag–TiO2 |
Ag |
Methanol |
450 W Hg lamp (200–800 nm) |
180 |
49 |
| g-C3N4/TiO2/CuO |
None |
Methanol |
300 W Xe lamp |
97.48 |
This work |
Table 2 The pore characteristics of g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO
| Catalyst |
Surface area (m2 g−1) |
Vtotal (cm3 g−1) |
Pore size (nm) |
| g-C3N4/TiO2 |
28.51 |
0.065 |
9.11 |
| CuO/TiO2 |
6.34 |
0.012 |
7.35 |
| g-C3N4/TiO2/CuO |
3.38 |
0.009 |
10.71 |
3.5 H2 production performance and gas sensitivity of different catalysts
The photocatalytic performances of g-C3N4, CuO, g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO for H2 production are revealed in Fig. 6. The CuO and g-C3N4 catalysts exhibit low hydrogen evolution rates of 13.33 and 6.01 μmol (g h)−1, respectively. This is affected by the properties of CuO and g-C3N4. The combination of CuO or g-C3N4 with TiO2 further improves the H2 production rate to 53.56 μmol (g h)−1 and 12.87 μmol (g h)−1, respectively. The H2 generation rate of the CuO/TiO2 heterojunction catalyst is 4.0 times that of CuO, while g-C3N4/TiO2 gives a hydrogen evolution rate of 2.1 times that of g-C3N4. The heterojunction would be a dominating factor for the promotion of H2 production capacity. The photocatalytic performance of g-C3N4/TiO2/CuO was further explored to inspect whether the double heterojunction can promote the H2 generation capacity. As seen in Fig. 6, in the case of double-heterostructure construction, the H2 evolution rate exhibits a palpable enhancement and obtains a first-rank value at g-C3N4/TiO2/CuO (97.48 μmol (g h)−1). Compared with g-C3N4/TiO2 and CuO/TiO2, the photocatalytic activity of g-C3N4/TiO2/CuO for H2 production is increased by 7.6 times and 1.8 times, respectively. In order to verify the commercial prospects of g-C3N4/TiO2/CuO double-heterostructure photocatalyst, the hydrogen production performance of g-C3N4/TiO2/CuO was compared with that of commercial P25 photocatalyst. The result show that the H2 generation rate of the g-C3N4/TiO2/CuO catalyst is 3.8 times that of P25. And the comparison photocatalytic H2 production performance of relevant g-C3N4 and CuO-based heterojunction photocatalysts in recent years are exhibited in Table 1.
 |
| | Fig. 6 (a) Photocatalytic H2 production rate and (b) H2 production histograms of CuO, g-C3N4, P25, g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO. | |
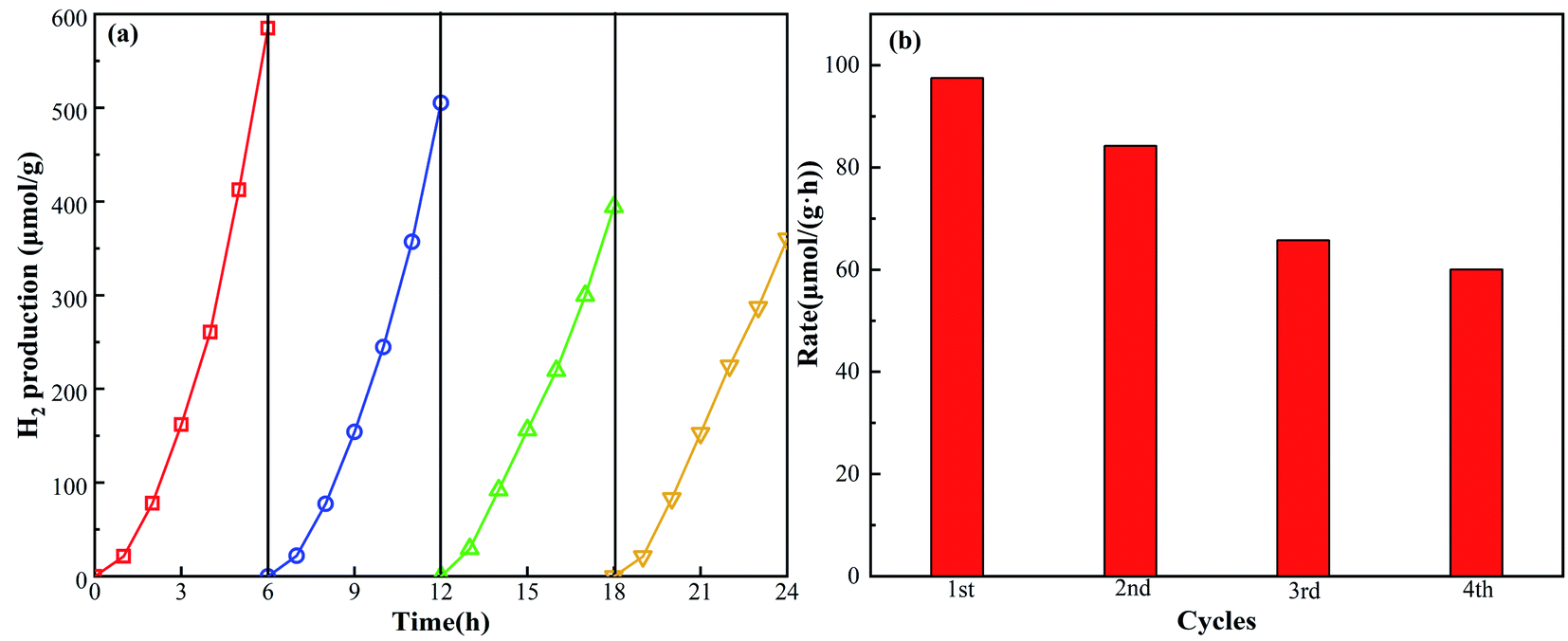
Because of the excellent H2 production ability of the g-C3N4/TiO2/CuO catalyst, it is important to explore its cycle stability. As shown in Fig. 7, in the 24 h (4 continuous cycles) H2 evolution process, the stability of the double-heterojunction g-C3N4/TiO2/CuO photocatalyst decreases with the passage of time and the increase in cycle times. The hydrogen evolution rate decreases from 97.48 μmol (g h)−1 in the first cycle to 60.05 μmol (g h)−1 in the fourth cycle. After that, the H2 generation rate of the g-C3N4/TiO2/CuO catalyst basically stabilizes. We speculate that the reason for this outcome is the carbon deposition of the catalyst owing to strong light. Furthermore, the loss due to the catalyst being stirred to the inner wall of the reactor cannot be ignored. The above results indicate that the successful construction of double heterojunctions can effectively improve the photocatalytic functioning of H2 production. Thus, promoting the stability of the g-C3N4/TiO2/CuO catalyst and exploring the mechanism of the double heterojunction in the photocatalytic process of H2 production are important issues.
 |
| | Fig. 7 (a) The cycle curve of the g-C3N4/TiO2/CuO catalyst and (b) the histogram of the g-C3N4/TiO2/CuO catalyst. | |
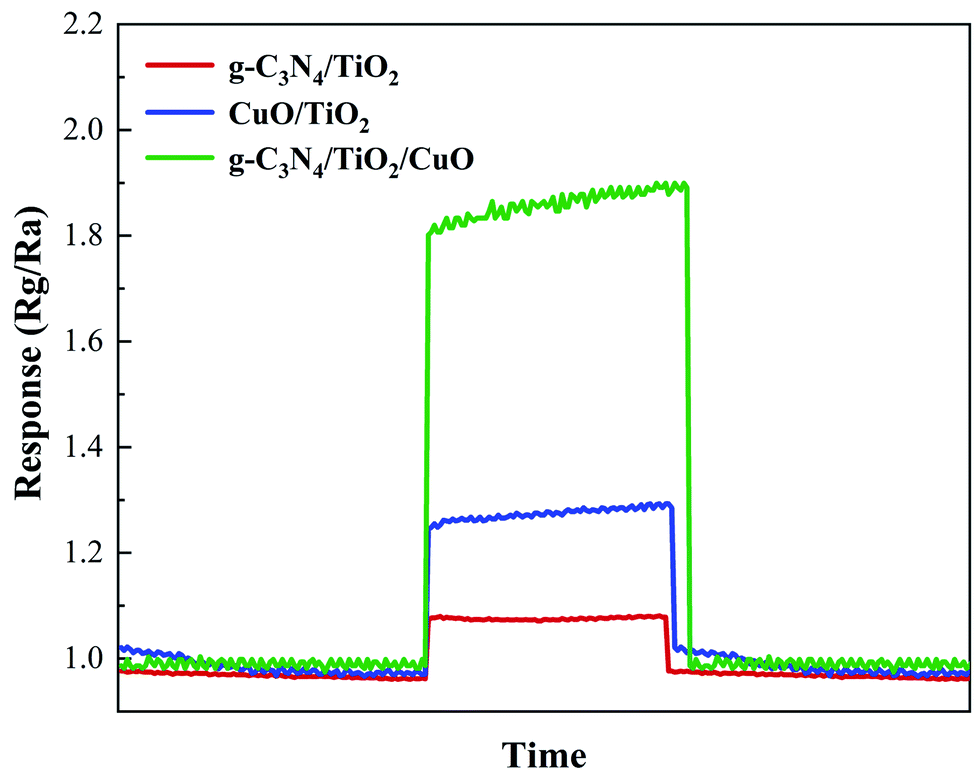
The double-heterojunction g-C3N4/TiO2/CuO semiconductor was tested by ammonia sensitivity performance. As shown in Fig. 8, g-C3N4/TiO2 has a very weak response to ammonia. Meanwhile, the response value of CuO/TiO2 is also weak. The establishment of a double heterojunction significantly improves the response value of the g-C3N4/TiO2/CuO semiconductor to ammonia. The maximum response value of g-C3N4/TiO2/CuO to ammonia is approximately 1.9. The reasons for the improvement of ammonia sensitivity are summarized as electrical effects, synergistic effects and structural effects. The conductivity of the material is affected by the interface barrier formed by the heterojunction.50 Heterojunctions are constructed for direct contact of each component to form synergistic effects.51
 |
| | Fig. 8 Ammonia sensitivity performance test images of g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO. | |
3.6 Analysis of electron hole recombination rate
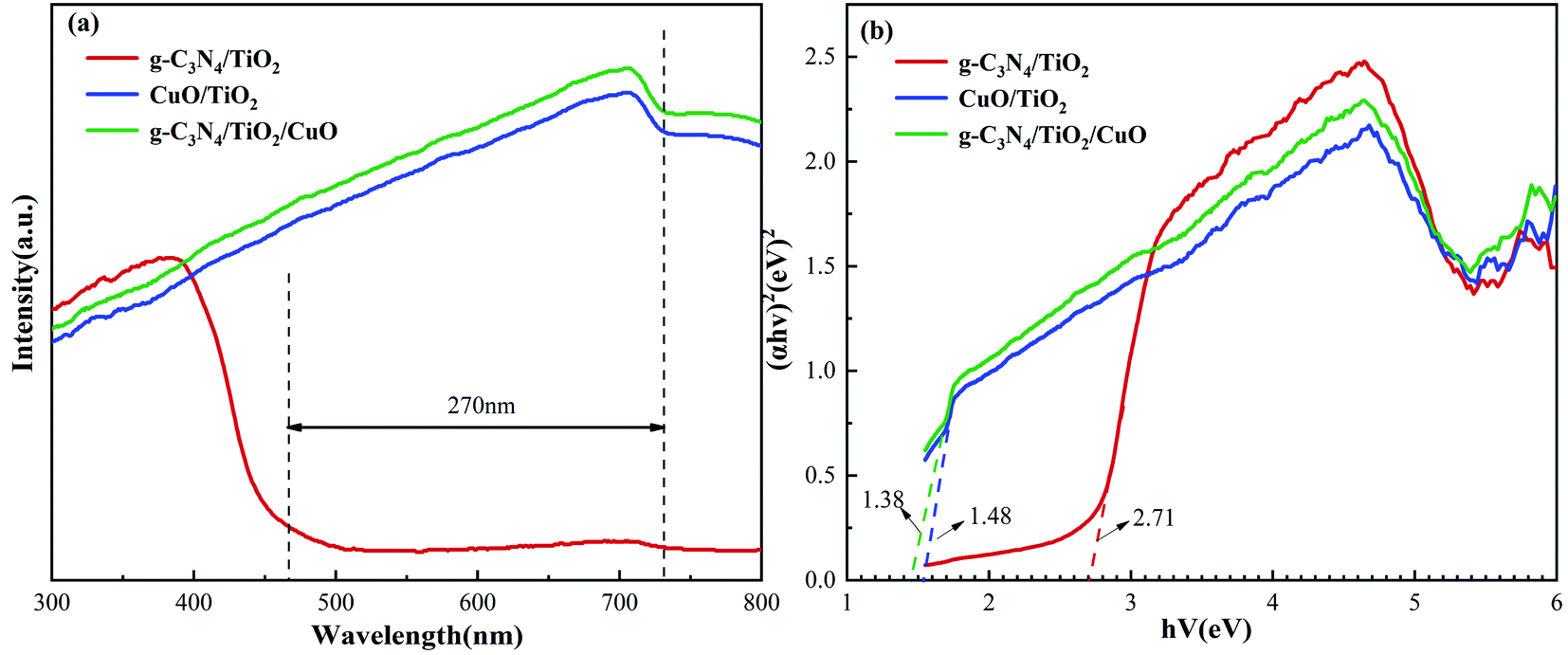
Fig. 9(a) shows the UV-vis opical spectra of g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO. As a result of introducing g-C3N4 with a narrow band gap, the absorption edge of g-C3N4/TiO2 reaches nearly 460 nm. Due to the loading of CuO, the light absorption ranges of the CuO/TiO2 and g-C3N4/TiO2/CuO heterojunction catalysts are obviously widened, whose absorption edges are up to nearly 730 nm. The extension of the absorption edge in the visible range can be explained by surface plasmon resonance.52 Because of the d to d trajectory transition of Cu2+ in CuO NPs, it is more likely to prolong visible light absorption.53 Moreover, compared with the CuO/TiO2 catalyst, the g-C3N4/TiO2/CuO catalyst further promotes the absorption of visible light. This also provides evidence for the inference that double heterojunctions are conducive to the improvement of catalyst performance. In addition, the band gap width of these synthetic photocatalysts can be calculated from the Tauc diagram.54 In Fig. 9(b), the light absorption ranges of g-C3N4/TiO2, TiO2/CuO, and g-C3N4/TiO2/CuO are 2.71, 1.50, and 1.38 eV, respectively. The introduction of a double heterojunction is conducive to modifying the band gap and broadening the optical absorption range. Obviously, the improvement of light absorption capacity is an important factor to improve H2 evolution capacity.
 |
| | Fig. 9 (a) UV-vis absorption spectra and (b) transformed Kubelka–Munk functions of g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO. | |
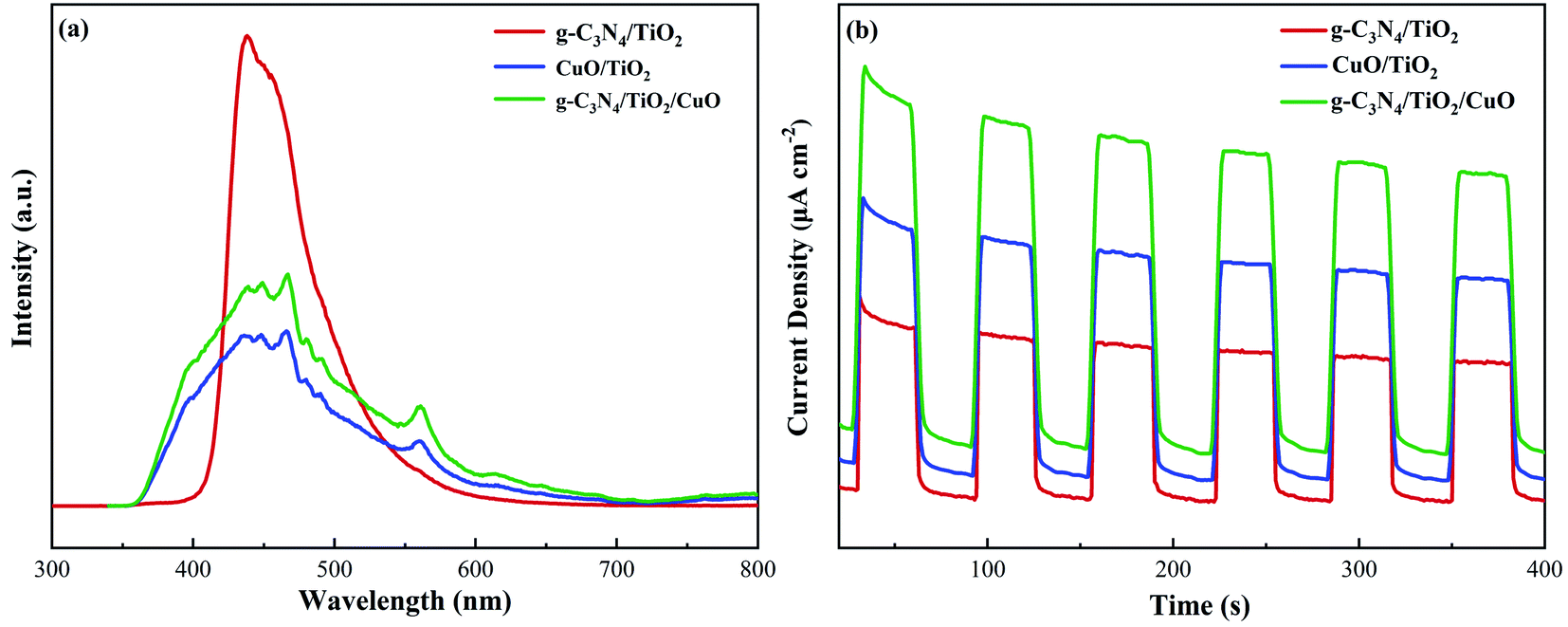
The separation of photogenerated electron holes and the transfer of electrons are significant factors to evaluate photocatalytic activity. Fig. 10(a) shows the PL spectra of the g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO samples. The separation efficiency of photogenerated electron holes can be verified from the figure. The PL intensity of g-C3N4/TiO2/CuO is clearly lower than that of g-C3N4/TiO2 and CuO/TiO2, which means a lower electron–hole recombination of g-C3N4/TiO2/CuO. This result further confirms that the g-C3N4/TiO2/CuO double heterojunction is successfully constructed. As shown in Fig. 10(b), during the instantaneous photocurrent test process, when the light source is turned off, it immediately returns to the initial state. The process can be repeated, which means the excellent structural stability of the catalyst.55 Moreover, the transient photocurrent density of g-C3N4/TiO2/CuO is 2.0 times higher than that of g-C3N4/TiO2 and 1.3 times higher than that of CuO/TiO2. This is mainly because the ternary heterojunction structure effectively promotes electron charge separation and improves light absorption.54 The density and transfer velocity of photogenerated electrons are increased. Therefore, the increase in photocurrent can be mainly due to effective photogeneration, separation, and transfer. Which is conducive to photocatalysis process.
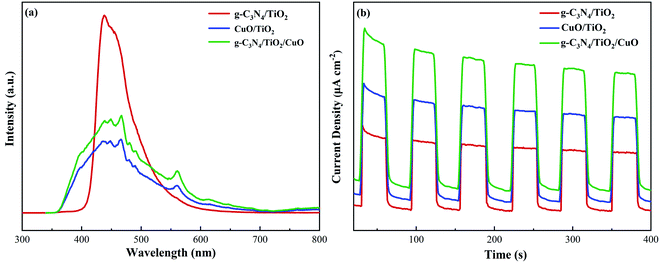 |
| | Fig. 10 (a) PL emission spectra and (b) transient photocurrent spectra of g-C3N4/TiO2, CuO/TiO2, and g-C3N4/TiO2/CuO. | |
3.7 Explanation of electron trajectory by S-scheme
Fig. 11 exhibits the H2 production mechanism of the g-C3N4/TiO2/CuO photocatalyst in water–methanol. The double p–n heterostructure of g-C3N4/TiO2/CuO was constructed to ensure high electron mobility. The recombination rate of electron hole pairs is reduced, and the H2 evolution rate is increased. The visible light is first absorbed by the surface layer of the g-C3N4 material. Then, the g-C3N4 layer is penetrated by visible light to compel electrons that jump from the VB to CB. When the incident light becomes scattered light, it will refract between the g-C3N4 and TiO2 layers owing to the existence of a protective layer. Meanwhile, the CuO material will produce an electron resonance swing effect owing to light excitation and then generate electron hole pairs via Landau damping attenuation. As shown in Fig. 11, we can explain the trajectory of electron–hole pairs between semiconductors by the S-scheme. The band edge locations of the g-C3N4, TiO2 and CuO catalysts are determined by the empirical formula ECB = χ − Ee − 0.5. Where ECB is the edge energy of CB, Ee is the free electron potential (∼4.5 eV),56 and χ indicates the Mulliken absolute electronegativity of atomic components. For instance, g-C3N4 = χ ∼4.72 eV, CuO = χ ∼5.81 eV, and TiO2 = χ ∼5.81 eV.57 Eventually, the ECB values of g-C3N4, TiO2 and CuO are −1.13, −0.29 and 0.65 eV, respectively. Likewise, EVB is the edge potential of the VB, and the calculation formula is EVB = ECB + Eg.56 Eg is the bandgap energy of the as-defined semiconductor (g-C3N4 = 2.7 eV, Eg of TiO2 = 3.2 eV, and Eg of CuO = 1.32 eV), so the ECB values of g-C3N4, TiO2 and CuO are 1.57, 2.91, and 1.97 eV, respectively.
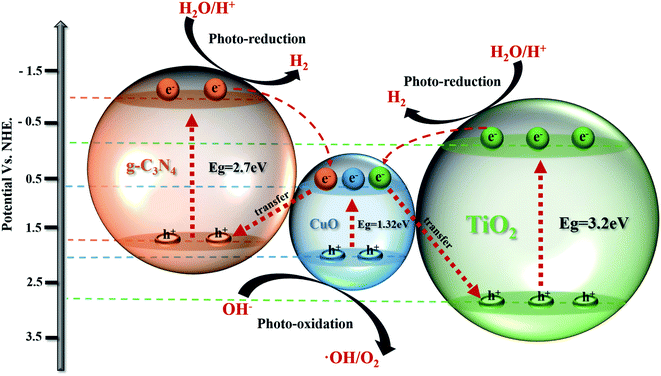 |
| | Fig. 11 Illustration of the photocatalytic H2 production mechanism of g-C3N4/TiO2/CuO. | |
Based on the above discussions, the CB of CuO is higher than that of g-C3N4 and TiO2, which means that some electrons may migrate from the CB of g-C3N4 and TiO2 to the CB of CuO. Then, the electrons of CuO are drivably transferred to the VB of TiO2 and g-C3N4 based on the S-scheme mechanism.25 The S-scheme heterojunction consists of reduction photocatalysts (RP, high CB and VB positions) and oxidation photocatalysts (OP, low CB and VB positions) with staggered band structures. RP has a smaller work function than OP, which leads to the spontaneous migration of electrons in RP to OP by van der Waals forces. An electron depletion layer is formed near the RP interface, resulting in its positive charge. Similarly, an electron stacking layer is formed near the OP interface, resulting in its negative charge. Meanwhile, RP and OP generate an internal electric field that accelerates the transfer of photon-generated carrier. In the S-scheme, the electrons stored in the CB of RP are reduced, and the holes in the VB of OP are oxidized. The S-scheme retains the strong redox ability of the photocatalysts, which is attributed to partly meaningless electron–hole recombination.58 The Fermi energy of OP and RP should correspond to the same energy level to achieve the balance of the Fermi energy level. The photogenerated electrons in the CB of OP are shifted to combine with holes in the VB of RP by band bending and Coulomb force. In the g-C3N4/TiO2/CuO ternary catalyst, TiO2 and g-C3N4 are used as RP and CuO as OP. Here, CuO acts as an electron acceptor and plays a bridging role. CuO not only functions as a structural defect to capture electrons but also plays an electronic medium role to promote electron–hole pair separation. These electrons will remain in the VB of g-C3N4 and TiO2 and combine with their holes. This facilitates the separation of original electron hole pairs on g-C3N4 and TiO2, thus improving the H2 production rate of the material.
4. Conclusion
A novel ternary heterostructure g-C3N4/TiO2/CuO photocatalyst was successfully developed and used in water–methanol for hydrogen evolution under visible light irradiation. Benefiting from the synergistic effect of the S-scheme and the double p–n heterostructures, g-C3N4/TiO2/CuO photocatalyst presented considerable H2 generation performance. Based on the S-scheme, the electrons in the CB of CuO and the holes in the VB of g-C3N4 and TiO2 are recombined. Useless electrons and holes are recombined. Band bending, internal electric field, and Coulomb attraction are the main factors that trigger this phenomenon. This study further explained the mechanism of the S-scheme, and the ternary catalyst provides an idea for further design and synthesis with a low electron–hole recombination rate and strong redox properties. We believe that more efforts should be made to improve the stability of photocatalysts in order to obtain better performance of hydrogen production. At the same time, the preparation of small particle size, high specific area and excellent morphology of the photocatalysts are the next step forward direction.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully thank the support for this research from National Natural Science Foundation of China (No. 51676116), Special Project Fund of "Taishan Scholar" of Shandong Province (No. tsqn202103066), Youth Innovation Support Program of Shandong Colleges and Universities (2019KJD013), Zibo Key R & D Project (2019ZBXC300) and Enterprise Commissioned Project (9101/2212221).
References
- C. Liao, J. T. Erbaugh, A. C. Kelly and A. Agrawal, Renew. Sustain. Energy Rev., 2021, 145, 111063 CrossRef.
- A. M. Mitrašinović, Energy, 2021, 237, 121510 CrossRef.
- S. J. Phang, V.-L. Wong, L.-L. Tan and S.-P. Chai, Appl. Mater. Today, 2020, 20, 100741 CrossRef.
- C.-W. Huang, B.-S. Nguyen, J. C. S. Wu and V.-H. Nguyen, Int. J. Hydrogen Energy, 2020, 45, 18144–18159 CrossRef CAS.
- C. Tarhan and M. A. Çil, J. Energy Storage, 2021, 40, 102676 CrossRef.
- M. Zhou, Q. Weng, Z. I. Popov, Y. Yang, L. Y. Antipina, P. B. Sorokin, X. Wang, Y. Bando and D. Golberg, ACS Nano, 2018, 12, 4148–4155 CrossRef CAS PubMed.
- S. Qu, W. Chen, J. Yu, G. Chen, R. Zhang, S. Chu, J. Huang, X. Wang, C. Li and K. Ostrikov, J. Power Sources, 2018, 390, 224–233 CrossRef CAS.
- Y. Liu, S. Ding, Y. Shi, X. Liu, Z. Wu, Q. Jiang, T. Zhou, N. Liu and J. Hu, Appl. Catal., B, 2018, 234, 109–116 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- C. Lops, A. Ancona, K. Di Cesare, B. Dumontel, N. Garino, G. Canavese, S. Hérnandez and V. Cauda, Appl. Catal., B, 2019, 243, 629–640 CrossRef CAS PubMed.
- L. Zhang, P. Ma, L. Dai, S. Li, W. Yu and J. Guan, Catal. Sci. Technol., 2021, 11, 3834–3844 RSC.
- L. Zhang, Q. Zhang, H. Xie, J. Guo, H. Lyu, Y. Li, Z. Sun, H. Wang and Z. Guo, Appl. Catal., B, 2017, 201, 470–478 CrossRef CAS.
- C. Xu, L. Cao, G. Su, W. Liu, H. Liu, Y. Yu and X. Qu, J. Hazard. Mater., 2010, 176, 807–813 CrossRef CAS PubMed.
- R. Malik, V. K. Tomer, N. Joshi, T. Dankwort, L. Lin and L. Kienle, ACS Appl. Mater. Interfaces, 2018, 10, 34087–34097 CrossRef CAS PubMed.
- R. Ma, J. Sun, D. H. Li and J. J. Wei, J. Catal., 2020, 392, 165–174 CrossRef CAS.
- B. Chen, J. Yu, R. Wang, X. Zhang, B. He, J. Jin, H. Wang and Y. Gong, Sci. China Mater., 2022, 65, 139–146 CrossRef CAS.
- W. Zhao, T. She, J. Zhang, G. Wang, S. Zhang, W. Wei, G. Yang, L. Zhang, D. Xia, Z. Cheng, H. Huang and D. Y. C. Leung, J. Mater. Sci. Technol., 2021, 85, 18–29 CrossRef.
- C. Yu, M. Li, D. Yang, K. Pan, F. Yang, Y. Xu, L. Yuan, Y. Qu and W. Zhou, Appl. Surf. Sci., 2021, 568, 150981 CrossRef CAS.
- A. Das, M. Patra, M. Kumar P, M. Bhagavathiachari and R. G. Nair, Mater. Chem. Phys., 2021, 263, 124431 CrossRef CAS.
- K. P. Sapkota, I. Lee, S. Shrestha, A. Islam, A. Hanif, J. Akter and J. R. Hahn, J. Environ. Chem. Eng., 2021, 9, 106497 CrossRef CAS.
- S. Dursun, S. N. Koyuncu, İ. C. Kaya, G. G. Kaya, V. Kalem and H. Akyildiz, J. Water Proc. Eng., 2020, 36, 101390 CrossRef.
- M. Edelmannová, K.-Y. Lin, J. C. S. Wu, I. Troppová, L. Čapek and K. Kočí, Appl. Surf. Sci., 2018, 454, 313–318 CrossRef.
- S. Wageh, A. A. Al-Ghamdi, R. Jafer, X. Li and P. Zhang, Chin. J. Catal., 2021, 42, 667–669 CrossRef CAS.
- J. Chen, T. Liu, H. Zhang, B. Wang, W. Zheng, X. Wang, J. Li and J. Zhong, Appl. Surf. Sci., 2020, 527, 146788 CrossRef CAS.
- R. Rajendran, S. Vignesh, S. Suganthi, V. Raj, G. Kavitha, B. Palanivel, M. Shkir and H. Algarni, J. Phys. Chem. Solids, 2022, 161, 110391 CrossRef CAS.
- N. Oh, S. Nam, Y. Zhai, K. Deshpande, P. Trefonas and M. Shim, Nat. Commun., 2014, 5, 3642 CrossRef PubMed.
- D. Li, Y. Tang, D. Ao, X. Xiang, S. Wang and X. Zu, Int. J. Hydrogen Energy, 2019, 44, 3985–3992 CrossRef CAS.
- R. Koutavarapu, K. Syed, S. Pagidi, M. Y. Jeon, M. C. Rao, D.-Y. Lee and J. Shim, Chemosphere, 2022, 287, 132015 CrossRef CAS PubMed.
- J. Ma, J. Dai, Y. Duan, J. Zhang, L. Qiang and J. Xue, Renewable Energy, 2020, 156, 1008–1018 CrossRef CAS.
- J. Wang, J. Huang, H. Xie and A. Qu, Int. J. Hydrogen Energy, 2014, 39, 6354–6363 CrossRef CAS.
- M. Hu, Z. Xing, Y. Cao, Z. Li, X. Yan, Z. Xiu, T. Zhao, S. Yang and W. Zhou, Appl. Catal., B, 2018, 226, 499–508 CrossRef CAS.
- J. Pan, Z. Dong, B. Wang, Z. Jiang, C. Zhao, J. Wang, C. Song, Y. Zheng and C. Li, Appl. Catal., B, 2019, 242, 92–99 CrossRef CAS.
- A. George, D. M. A. Raj, A. D. Raj, B.-S. Nguyen, T.-P. Phan, T. Pazhanivel, K. Sivashanmugan, R. L. Josephine, A. A. Irudayaraj, J. Arumugam and V.-H. Nguyen, Mater. Lett., 2020, 281, 128603 CrossRef CAS.
- H. B. Uma, S. Ananda and M. S. V. Kumar, Chem. Data Collect., 2021, 32, 100658 CrossRef CAS.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Balkus, ACS Catal., 2012, 2, 949–956 CrossRef CAS.
- M. W. Kadi, R. M. Mohamed and A. A. Ismail, Ceram. Int., 2020, 46, 8819–8826 CrossRef CAS.
- A. Kumar, M. Khan, J. He and I. M. C. Lo, Appl. Catal., B, 2020, 270, 118898 CrossRef CAS.
- M. Logar, I. Bračko, A. Potočnik and B. Jančar, Langmuir, 2014, 30, 4852–4862 CrossRef CAS PubMed.
- C. Byrne, L. Moran, D. Hermosilla, N. Merayo, Á. Blanco, S. Rhatigan, S. Hinder, P. Ganguly, M. Nolan and S. C. Pillai, Appl. Catal., B, 2019, 246, 266–276 CrossRef CAS.
- H. Liu, Z. Xu, Z. Zhang and D. Ao, Appl. Catal., B, 2016, 192, 234–241 CrossRef CAS.
- M. Hong, L. Zhang, H. Fang, X. Feng and Z. Li, Mater. Sci. Semicond. Process., 2021, 136, 106134 CrossRef CAS.
- H. Yan and H. Yang, J. Alloys Compd., 2011, 509, L26–L29 CrossRef CAS.
- G. Li, H. Liang, G. Xu, C. Li and J. Bai, J. Phys. Chem. Solids, 2020, 145, 109549 CrossRef CAS.
- N. Tian, H. Huang, C. Liu, F. Dong, T. Zhang, X. Du, S. Yu and Y. Zhang, J. Mater. Chem. A, 2015, 3, 17120–17129 RSC.
- M. K. Hussain, N. R. Khalid, M. Tanveer, I. Kebaili and H. Alrobei, Int. J. Hydrogen Energy, 2022, 47, 15491–15504 CrossRef CAS.
- J. Wang, Q. Zhu, Y. Liao, H. Fu, J. Chang, Y. Zhang, T. Kan, H. Gao and W. Huang, J. Alloys Compd., 2021, 883, 160832 CrossRef CAS.
- M. Haddad, A. Belhadi, L. Boudjellal and M. Trari, Int. J. Hydrogen Energy, 2021, 46, 37556–37563 CrossRef CAS.
- D. Wang, J. Liu, M. Zhang, Y. Song, Z. Zhang and J. Wang, Appl. Surf. Sci., 2019, 498, 143843 CrossRef CAS.
- S. Sharma, M. R. Pai, G. Kaur, Divya, V. R. Satsangi, S. Dass and R. Shrivastav, Renewable Energy, 2019, 136, 1202–1216 CrossRef CAS.
- K.-R. Park, H.-B. Cho, J. Lee, Y. Song, W.-B. Kim and Y.-H. Choa, Sens. Actuators, B, 2020, 302, 127179 CrossRef CAS.
- P. H. Phuoc, N. N. Viet, L. V. Thong, C. M. Hung, N. D. Hoa, N. V. Duy, H. S. Hong and N. V. Hieu, Sens. Actuators, B, 2021, 334, 129606 CrossRef CAS.
- L. Zhu, H. Li, Z. Liu, P. Xia, Y. Xie and D. Xiong, J. Phys. Chem. C, 2018, 122, 9531–9539 CrossRef CAS.
- H. Sudrajat and S. Hartuti, J. Colloid Interface Sci., 2018, 524, 227–235 CrossRef CAS PubMed.
- Z. Jiang, K. Qian, C. Zhu, H. Sun, W. Wan, J. Xie, H. Li, P. K. Wong and S. Yuan, Appl. Catal., B, 2017, 210, 194–204 CrossRef CAS.
- Y. Bu, Z. Chen and W. Li, Appl. Catal., B, 2014, 144, 622–630 CrossRef CAS.
- D. Zhang, S. Cui and J. Yang, J. Alloys Compd., 2017, 708, 1141–1149 CrossRef CAS.
- K. Perumal, S. Shanavas, A. Karthigeyan, T. Ahamad, S. M. Alshehri and P. Murugakoothan, Diamond Relat. Mater., 2020, 110, 108091 CrossRef CAS.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Hetong Lia
*a and
Hetong Lia