DOI:
10.1039/D2RA01858F
(Paper)
RSC Adv., 2022,
12, 25478-25486
Aptamer-based colorimetric detection of the DNA damage marker 8-oxo-dG using cysteamine-stabilised gold nanoparticles†
Received
22nd March 2022
, Accepted 23rd August 2022
First published on 7th September 2022
Abstract
8-Oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG) is a crucial biomarker for oxidative DNA damage and carcinogenesis. Current strategies for 8-oxo-dG detection often require sophisticated instruments and qualified personnel. In this study, cysteamine-stabilised gold nanoparticles (cyst-AuNPs) were synthesised and used for colorimetric detection of 8-oxo-dG in urine. Sensing of 8-oxo-dG is based on the anti-aggregation of cyst-AuNPs, mediated by the specific recognition of 8-oxo-dG and its aptamer. In the absence of 8-oxo-dG, the aptamer was adsorbed onto the surface of cyst-AuNPs, resulting in aggregation and the development of a purple colour solution. Upon addition of the target molecule 8-oxo-dG, the aptamer specifically bound to it and could not induce the aggregation of cyst-AuNPs, leading to the dispersion of cyst-AuNPs in the solution. Simple visual examination could be used to monitor the purple-to-red colour change that started at 12 nM, a threshold concentration for visual analysis. The absorbance at 525 nm increased in direct relation to the number of the target molecule 8-oxo-dG. This aptamer/cyst-AuNPs system showed excellent sensing ability for the 8-oxo-dG concentration in the range of 15–100 nM, with a detection limit as low as 10.3 nM and a detection time of 30 min. Interference experiments showed that the developed colorimetric strategy had a good sensitivity. This simple and rapid colorimetric method has successfully been applied to inspect 8-oxo-dG concentration in real urine samples and provided recoveries between 93.6 and 94.1%, with a limit of quantification (LOQ) of 34.3 nM, which was comparable with an enzyme-linked immunosorbent-based detection of 8-oxo-dG. This new, easy-to-use, and rapid method could be used as an alternative and initiative strategy for the development of an on-site analysis of 8-oxo-dG in urine.
Introduction
The imbalance between antioxidant defence systems and free radical generation, referred to as oxidative stress, is known to cause oxidative damage to cellular DNA, proteins, and lipids. Mutations, which occur when the DNA base sequence is altered unexpectedly, can take place in a single base pair or throughout a large part of the chromosome, including numerous genes. Guanine (G) is the most susceptible to oxidation among the four bases in DNA due to its low redox potential.1 8-Oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG), also known as 8-hydroxy-2′-deoxyguanosine (8-OHdG), is the most frequently occurring oxidative DNA base modification. If the change is not corrected, 8-oxo-dG may mismatch with adenine (A), resulting in G:C to A:T transversion.2 DNA damage is involved in the pathophysiology of various disorders, including neurological and cancer diseases.3,4 As a result, 8-oxo-dG is a very important biomarker for oxidative DNA damage. It can be found in both intact DNA and as a “free” product that forms during repair processes.
The detection of 8-oxo-dG in the urine is regarded as the best non-invasive approach for detecting oxidative DNA damage. Numerous quantitative methods for the detection of 8-oxo-dG in urine have been developed, including high-performance liquid chromatography (HPLC) equipped with electrochemical detection, gas chromatography-mass spectrometry (GC-MS), liquid chromatography-mass spectrometry-mass spectrometry (LC-MS/MS), enzyme-linked immunosorbent assay (ELISA), HPLC-tandem mass spectrometry and capillary electrophoresis.5–10 Despite their various advantages, quantitative techniques have a number of drawbacks, including the requirement for specialised and expensive equipment, a highly trained operator, and a significant time commitment. Several rapid nanomaterial-based sensing approaches have been developed to overcome such limitations. For example, a biosensor based on graphene-modified glassy carbon electrode functionalised with single-stranded DNA was developed and demonstrated a promising electrochemical performance in its detection of 8-oxo-dG in real samples.11 Another electrochemical biosensor based on a molecularly imprinted polymer used an 8-oxo-dG antibody labelled with fluorescein isothiocyanate (FITC) for detecting 8-oxo-dG at a detection limit of 3.5 pM.12 Additionally, a cellulose paper-based biosensor coated with conductive carbon-based ink was produced for the measurement of 8-oxo-dG.13 Despite the fact that previously manufactured 8-oxo-dG sensors based on electrochemistry/electropolymerisation were very selective and sensitive, they had limitations such as laborious synthesis and modification of materials/electrodes, expensive equipment, and the need for skilled staff. This suggested 8-oxo-dG detection approach is simple, quick, cost-effective, and visible to the naked eye; it does not require expensive equipment.
Gold nanoparticles (AuNPs) have been extensively used as colorimetric biosensors because of their unique optical properties. When AuNPs are dispersed, they appear red; when they undergo aggregation, the colour of the solution changes from red to blue, which is visible with the naked eye.14 When spherical-shaped AuNPs are dispersed, the characteristic absorption at approximately 525 nm can be observed, causing the solution to appear red. Once AuNPs undergo aggregation until the interparticle distance (d) is less than 1 nm, a quantum tunneling effect begins to disturb the interaction between surface plasmon, leading to the redshift absorption. This results in the solution's colour changing from red to blue, which is visible to the naked eye. The advantages of the AuNP-based sensors are as follows: (i) it is simple to use due to the straightforward measuring process, which may be determined by the colour change of the solution; (ii) it is cost effective since only a trace amount of Au is required to cause a change in the colour of the solution; and (iii) the analytical technique is simple. As a result, it is accessible to everybody without requiring measurement skills. Biosensor development requires, in addition to AuNPs, recognition components, i.e., molecules capable of binding to other compounds in a specific manner, to detect a target analyte with high sensitivity. Aptamers are an intriguing class of recognition components that are attracting considerable attention in sensor applications. Aptamers are constructed from DNA, RNA, or peptide sequences and can be produced in large quantities in a regulated manner by Systematic Evolution of Ligands by Exponential Enrichment (SELEX) and at a reduced cost when compared to antibodies. Aptamers can rearrange themselves into a three-dimensional unique shape for highly specific binding to their specific targets, which can range from small molecules to cells. Aptamers possess features that make them ideal for sensor applications, including high temperature tolerance, resistance to acid–base environments and selectivity for target molecules. Numerous optical aptasensors have been created for the purpose of binding to and identifying various biomarkers. Combining the benefits of aptamers and AuNPs, aptamer- and AuNPs-based colorimetric sensors have already demonstrated outstanding performance for the identification of small molecules, such as bisphenol A, ochratoxin A and 7β-estradiol.15–18 Various AuNPs and aptamers have been developed as biosensors for the detection of 8-oxo-dG, including colorimetry,19 CD spectroscopy19,20 and fluorometric assay.21 Our group previously fabricated an aptasensor based on the high salt-induced aggregation of citrate-capped AuNPs and truncated 8-oxo-dG aptamers.22 While the limit of detection (LOD) was good, an additional step involving sodium chloride (NaCl) addition to the sensor system was required. We, therefore, sought to develop a new, efficient, and easy-to-use approach for 8-oxo-dG detection using positively charged cysteamine-capped AuNPs (cysteamine-AuNPs; cyst-AuNPs) as a colorimetric probe and the anti-8-oxo-dG aptamer as a recognition element. In comparison to the prior method, this developed aptamer-based colorimetric assay utilised AuNPs with a different type of capping agent and did not require the presence of NaCl in the system, thus eliminating a step. This method was also shown to be feasible for determining the levels of 8-oxo-dG in human urine samples.
Experimental
Materials
Sodium borohydride (≥98%) and glycine (≥98%) were purchased from Merck Chemicals GmbH (Darmstadt, Germany). Hydrogen tetrachloroaurate(III) trihydrate (≥99.9%), 2′-deoxyguanosine monohydrate (≥99%), 8-hydroxy-2′-deoxyguanosine (≥98.5%), guanosine (98%), guanine, cysteamine hydrochloride (≥98%), potassium chloride (≥99%), sodium hydrogen phosphate (≥99%), sodium phosphate monobasic monohydrate (≥99%), nitric acid (≥99%), hydrochloric acid (≥99%), and sodium hydroxide (≥99%), histidine (≥98%), uric acid (≥99%), ammonium chloride (≥99%) and urea (≥99%) were supplied by Sigma-Aldrich (St. Louis, MO, USA). Sodium chloride (≥99%) was supplied from QRëC™ (Quality Reagent Chemical, New Zealand). Phosphoric acid (≥99%) was purchased from Univar USA Inc. (Bedford Park, IL, USA). Creatinine anhydrous (≥99%) was purchased from Himedia (Mumbai, India). 8-Oxo-dG-binding aptamer23,24 with the following sequence was obtained as a lyophilised solid from Integrated DNA Technologies, Inc. (Coralville, IA, USA): 5′-GCG GGC GAT CGG CGG GGG GTG CGT GCG CTC TGT GCC AGG GGG TGG GAC AGA TCA TAT GGG GGT GCT-3′, 68 mer. Stock solutions of 8-oxo-dG (1 mM) and 8-oxo-dG-binding aptamer (50 μM) were prepared and stored at −20 °C. All reagents were of analytical grade and were used as received without further purification. Aqueous solutions were prepared with purified water using a Milli-Q system with ≥18.2 MΩ cm resistivity.
Apparatus
UV-vis spectra were recorded on a UV-1800 spectrophotometer (Shimadzu, Tokyo, Japan). The morphology of synthesised cyst-AuNPs was studied using transmission electron microscopy (TEM; FEI 5022/22 Tecnai G2 20 S-Twin, Czech Republic). Particle size analysis was performed using the ImageJ software. Zeta potential was measured using a SZ-100 nanoparticle analyser (Horiba Ltd, Tokyo, Japan). A Mettler Toledo LE438 pH meter (Switzerland) was used for pH measurements.
Synthesis of cysteamine-stabilised AuNPs
Cyst-AuNPs were prepared by sodium borohydride reduction of hydrogen tetrachloroaurate(III) trihydrate in the presence of cysteamine hydrochloride following a previously described procedure.25 Briefly, an aqueous solution of cysteamine hydrochloride (213 mM, 400 μL) was added to an aqueous solution of hydrogen tetrachloroaurate(III) trihydrate (1.5 mM, 39.6 mL). The mixture was stirred vigorously for 20 min at room temperature in the dark. Subsequently, freshly prepared sodium borohydride (10 mM, 10 μL) was added to the mixture, which was vigorously stirred for a further 60 min. The resulting wine-red solution was filtered through a 0.22 mm filter paper and stored at 4 °C until use. The obtained cysteamine-capped AuNPs were characterised by UV-vis, TEM and zeta potential measurements.
Optimisation of sensing parameters
To develop an aptamer-based colorimetric assay capable of detecting the presence of 8-oxo-dG with high sensitivity, it is necessary to carefully optimise all reaction parameters. The optimisation of the sensing parameters were performed by fixing the 8-oxo-dG concentrations at 12 nM and 50 nM, respectively. The following key experimental parameters of the aptamer–cyst-AuNPs-based sensing system were optimised by monitoring changes in the maximum absorbance values at 525 nm (ΔA525): (i) the pH was investigated from 2 to 8; (ii) the cyst-AuNPs concentration from 0.5 to 3 nM; (iii) the concentration of anti-8-oxo-dG aptamer from 0 to 10 nM; (vi) the reaction time between cyst-AuNPs and the aptamer from 2 to 30 min; and (v) the binding time of 8-oxo-dG and its aptamer from 2 to 30 min. All experiments were performed in phosphate buffer at pH 7.0, except for the optimisation of pH.
Naked-eye detection of 8-oxo-dG
To assess the detection system's ability for visualising the colour change of a known quantity of 8-oxo-dG (12 nM), the optimal parameters which were obtained when using 12 nM 8-oxo-dG (pH 7.0, 2.5 nM cyst-AuNPs, 5 nM anti-8-oxo-dG aptamer, 14 min incubation time, and 12 min binding time), different concentrations of 8-oxo-dG were used.
Sensitivity of the colorimetric assay for the detection of 8-oxo-dG
To detect 8-oxo-dG using the aptamer–cyst-AuNPs-based colorimetric method, UV-vis spectra of the sensing system were recorded with different concentrations (15–100 nM) of 8-oxo-dG under the optimised experimental conditions when fixing 8-oxo-dG concentration at 50 nM. In brief, a mixture of aptamer solution (5 nM, 1.5 μL) and different concentrations (15–100 nM) of 8-oxo-dG solution (3 μL) was equilibrated for 14 min at room temperature. Subsequently, a final concentration of 2.5 nM cyst-AuNPs was added into the previous mixture containing anti-8-oxo-dG aptamer and 8-oxo-dG, mixed, settled for a further 12 min at room temperature. To reach a final volume of 300 μL, phosphate buffer (1 mM, pH 7.0) was added to the solution. Absorption spectra of the solutions were recorded on a UV-1800 spectrophotometer (Shimadzu, Tokyo, Japan) using a 1 cm path length quartz cuvette. A calibration curve with known concentrations of 8-oxo-dG was established according to the absorbance of 525 nm of the system.
Recovery and precision studies
Recovery and precision studies for 8-oxo-dG determination in spiked urine and water samples at different concentrations were analysed using developed aptasensor. The precision of the methods was calculated in terms of intra-day and inter-day. Two different concentrations of 8-oxo-dG in urine and water were analysed in three replicates during the same day (intra-day precision) and three consecutive days (inter-day precision).
Specificity for detecting 8-oxo-dG
The specificity of the sensing system was determined by testing 8-oxo-dG against non-target molecules/ions (i.e., 2′-deoxyguanosine, 8′-hydroxyguanosine, 8′-hydroxyguanine, guanosine, guanine, uric acid, Ca2+, K+, Mg2+, Na+, PO43− and Zn2+, NH4+, creatinine, glycine, histidine, and urea). Each solution was prepared by incubating a specific analyte (3 μL, 100 nM) with the 8-oxo-dG-binding aptamer (1.5 μL, 5 nM) for 14 min at room temperature. Cyst-AuNPs solution (50 μL, 2.5 nM) was subsequently added to the solution which was incubated for a further 12 min at room temperature. The resulting mixture was diluted with phosphate buffer (1 mM, pH 7.0) to reach a final volume of 300 μL (245.5 μL). The UV-vis absorption spectra of each solution at 525 nm were recorded.
Urine sample preparation and analytical procedure for 8-oxo-dG
Urine samples used in this study were leftovers from our previous research project on opisthorchiasis, of which its protocols were approved by the Ethics Committee of Khon Kaen University, Faculty of Medicine, Khon Kaen, Thailand (HE581330 and HE621151). Frozen urine samples were thawed to room temperature and centrifuged at 1500 rpm for 5 min before use. The levels of 8-oxo-dG (nM) in the urine samples were measured using the developed detection method based on cyst-AuNPs–aptamer under optimised experimental conditions. Briefly, an aqueous solution of anti-8-oxo-dG aptamer (1.5 μL, 5 nM) and diluted urine (3 μL; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32 in ultrapure water) was equilibrated for 14 min at room temperature before adding a final concentration of 2.5 nM cyst-AuNPs. The resulting solution was mixed, settled for further 12 min at room temperature. To reach a final volume of 300 μL, phosphate buffer (1 mM, pH 7) was added to the solution. The absorbance at 525 nm was recorded. Two urine samples were examined by ELISA using an 8-oxo-dG kit (MBS808265, MyBioSource, Inc., San Diego, CA, USA).
:32 in ultrapure water) was equilibrated for 14 min at room temperature before adding a final concentration of 2.5 nM cyst-AuNPs. The resulting solution was mixed, settled for further 12 min at room temperature. To reach a final volume of 300 μL, phosphate buffer (1 mM, pH 7) was added to the solution. The absorbance at 525 nm was recorded. Two urine samples were examined by ELISA using an 8-oxo-dG kit (MBS808265, MyBioSource, Inc., San Diego, CA, USA).
Results and discussion
Mechanism of 8-oxo-dG-induced anti-aggregation of cyst-AuNPs and aptamer
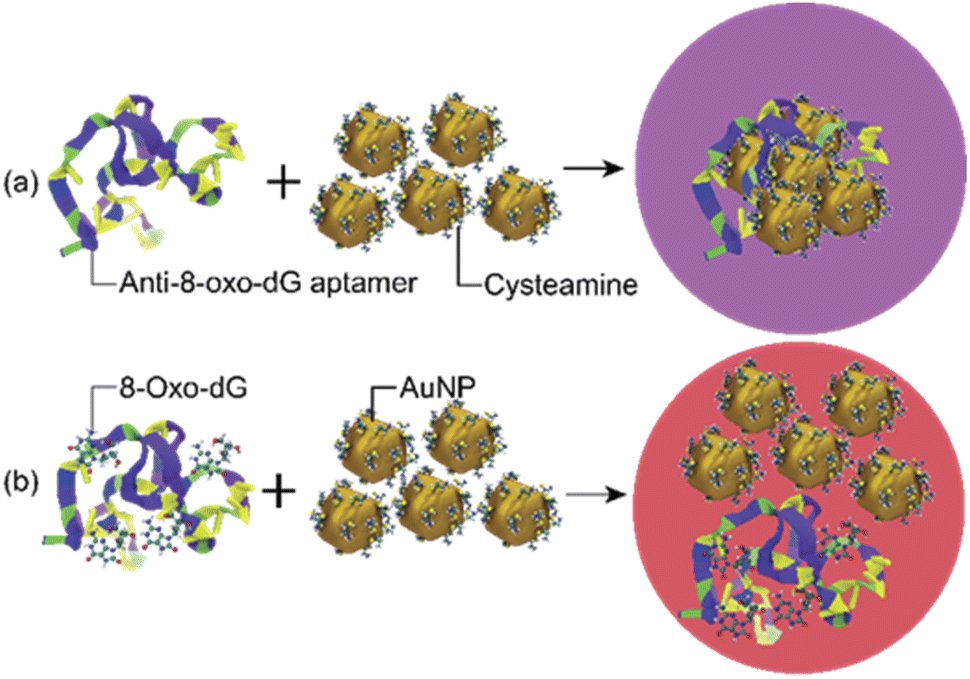
The sensing mechanism of the developed colorimetric assay is based on the anti-aggregation of cyst-AuNPs and the binding with high affinity and specificity between the anti-8-oxo-dG aptamer, which was previously established through SELEX technique, and 8-oxo-dG molecules.24 In the absence of the target molecule 8-oxo-dG, the solution containing cyst-AuNPs and the aptamer was purple in colour (Fig. 1(a)).
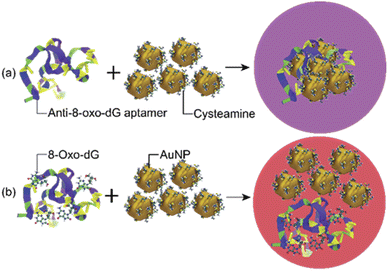 |
| | Fig. 1 Schematic illustration of the sensing mechanism for the detection of 8-oxo-dG based on the aptamer-mediated aggregation of cyst-AuNPs: (a) without 8-oxo-dG and (b) with 8-oxo-dG. | |
In the presence of 8-oxo-dG, the system appeared red due to the ability of 8-oxo-dG to bind to the aptamer, resulting in the dispersion of cyst-AuNPs in the solution (Fig. 1(b)). We recently reported an overview of the sensing mechanism based on molecular dynamics (MD) simulation.25 The rapid assembly of cyst-AuNPs was determined to be due primarily owing to electrostatic interactions and van der Waals stacking interactions. Due to the presence of cyst molecules on the surface of AuNPs, the surface was positively charged. AuNPs aggregated rapidly due to the favourable interaction between positively charged AuNPs and the negatively charged backbone of the aptamer. Furthermore, noncovalent interactions between the nitrogenous base and Au atoms were responsible for the aptamer's adsorption onto the AuNPs surface. Meanwhile, in the presence of the target molecule 8-oxo-dG, the aptamer's specific binding to 8-oxo-dG resulted in the development of a complex, which could prevent AuNPs from rapidly aggregating. This could also increase the steric hindrance. Therefore, the aptamer could no longer induce the aggregation of AuNPs. After the complex formation between 8-oxo-dG and the aptamer, the dispersion of cyst-AuNPs remained unaffected. The colour change of the proposed biosensor presented in this work is based upon the unique optical properties of AuNPs, due to localised surface plasmon resonance (LSPR). This phenomenon can be understood as a collective resonant oscillation of free electrons within a metal nanoparticle in response to an incident electromagnetic radiation. When the NPs are in proximity, the individual surface plasmon can couple, leading to a shift in the optical response. Surface plasmon coupling depends on the way the NPs are assembled. Important factors affecting the formation or stability of coloured product are (i) particle size; and (ii) interparticle distance.26 Although the detection sensitivity of the aptamer-based sensors can be improved by the use of relatively large nanoparticles as shown in the plasmon ruler equation Δλ/Δλ0 ≈ 0.18 exp(–(s/D)/0.23), where Δλ/Δλ0 is the fractional plasmon shift, s is the interparticle edge-to-edge separation, and D is the particle diameter, the long-term stability of sensors tends to be inversely proportional to nanoparticle sizes. Hence, small nanoparticles (e.g., 10–40 nm in diameter) have been most commonly used to achieve the best compromise between sensitivity and stability.26 The sensing mechanism of the present aptasensor relies on the gap between cyst-AuNPs, which is proportionally changed with the concentration of 8-oxo-dG. Based on this strategy, a label-free aptamer-based colorimetric sensing assay was developed for 8-oxo-dG determination.
Synthesis of cysteamine-stabilised AuNPs
The synthesis of cyst-AuNPs was achieved by reducing HAuCl4 with NaBH4 in the presence of cyst. Cyst-AuNPs are positively charged due to the –NH3+ group in cyst. To maintain the protonated form of the –NH3+ group in cyst, phosphate buffer at pH 7.0 was used. Due to the electrostatic repulsion between cyst-decorated AuNPs, positively charged AuNPs produced are stabilised against aggregation. The concentration of the as-prepared AuNPs was determined to be 14 nM, based on the known initial concentration of the gold solution.27 The protocol yielded AuNPs, which appeared red, with an average diameter of 33.8 ± 3.5 nm, a surface potential of 13.9 ± 0.5 mV, a characteristic absorption peak at 525 nm, and which appeared red (Fig. S1 in the ESI†).
Optimisation of the sensing parameters
Different experimental parameters for the detection of 8-oxo-dG, i.e., pH, the concentration of cyst-AuNPs, the concentration of anti-8-oxo-dG aptamer, incubation time and binding time, were optimised. Two different sets of experiment were performed by fixing the 8-oxo-dG concentration at 12 and 50 nM, respectively. In both cases, the same optimal conditions were obtained: 2.5 nM cyst-AuNPs, 5 nM anti-8-oxo-dG aptamer, 14 min-incubation time and 12 min binding time of the aptamer with cyst-AuNPs. The optimisation data for 12 nM 8-oxo-dG are shown in Fig. S2–S5.† The optimisation data for 50 nM 8-oxo-dG are demonstrated in Fig. S6–S10.† The optimal pH was at 7.0. The obtained optimised conditions were used for the development of the cyst-AuNPs–aptamer-based colorimetric assay.
Performance of the colorimetric 8-oxo-dG assay
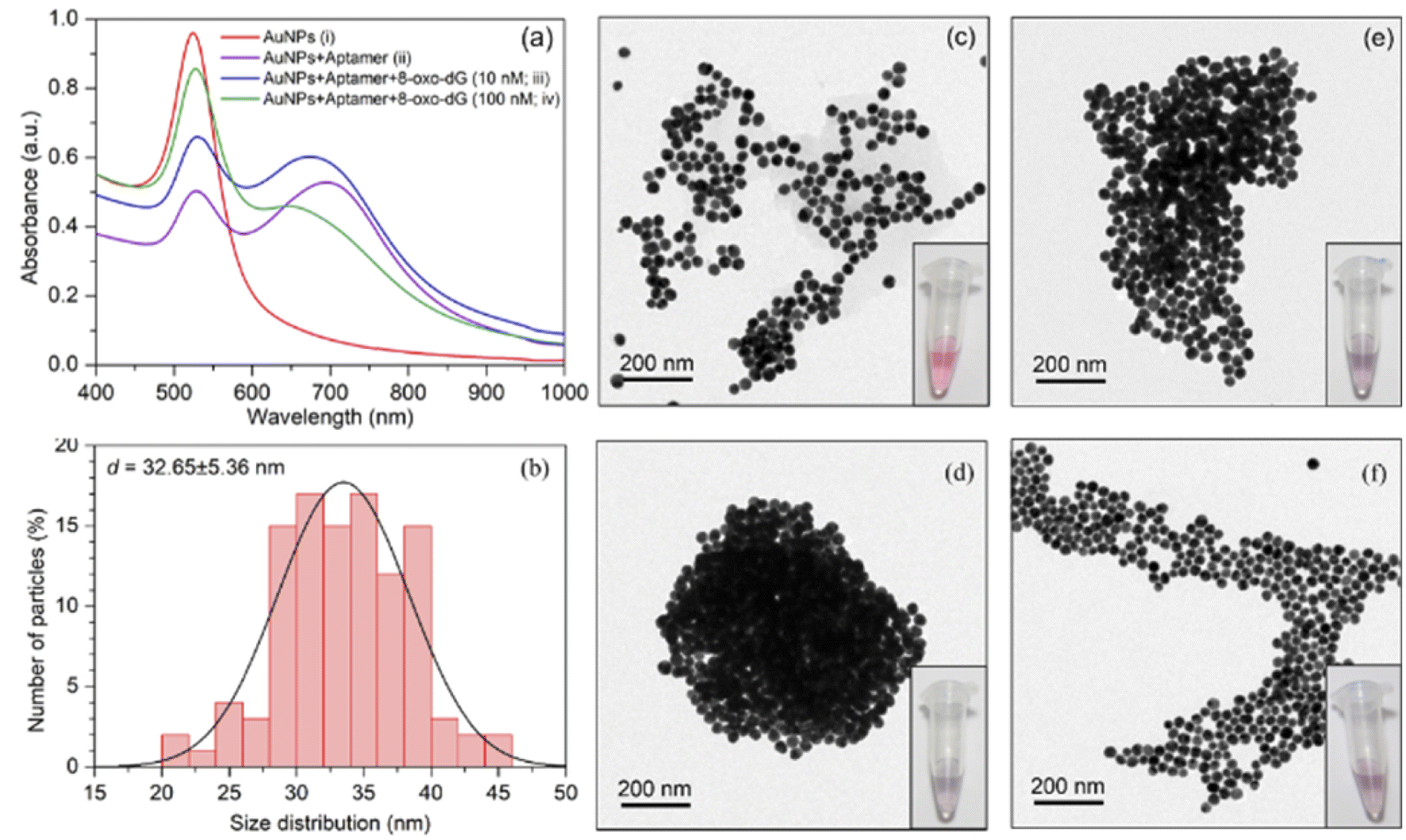
The increased concentration of 8-oxo-dG was evidenced by a rise in the absorption intensity at the wavelength of 525 nm. UV-vis spectrum of cyst-AuNPs showed one absorption peak at 525 nm (Fig. 2(a)). The average size of the synthesised cyst-AuNPs was 32.65 ± 5.36 nm (Fig. 2(b)). Upon addition of the aptamer, the absorption peak at 525 decreased and a new absorption band at 680 nm was observed, demonstrating the aptamer-induced aggregation of cyst-AuNPs. With increasing concentrations of the target molecule 8-oxo-dG, the absorbance at 525 nm increased in a dose-dependent manner, suggesting that the degree of re-dispersion depended on the concentration of 8-oxo-dG. Prior to the addition of the anti-8-oxo-dG aptamer, the colour of the cyst-AuNPs solution was red (Fig. 2(c)). In the presence of the anti-8-oxo-dG aptamer, the colour of the solution changed from red to purple, indicating that the aptamer was adsorbed onto the surface of cyst-AuNPs (Fig. 2(d)). As the concentrations of 8-oxo-dG were increased (10 and 100 nM), the solution changed from purple to red, which was clearly visible to the naked eye, affirming the complex formation of the aptamer and 8-oxo-dG which could not induce the aggregation of cyst-AuNPs. TEM images revealed dispersing cyst-AuNPs produced by the anti-8-oxo-dG aptamer (Fig. 2(e) and (f)). Visual detection of the target chemical 8-oxo-dG could be accomplished using the optimum sensing condition. Zeta potential and size of the sensing systems under different conditions are shown in Table S1.† It was found that when the aptamer molecules were added to the cyst-AuNPs solution, cyst-AuNPs aggregated and increased in size. In contrast, when 8-oxo-dG was present, the particle sizes decreased as the amount of 8-oxo-dG increased.
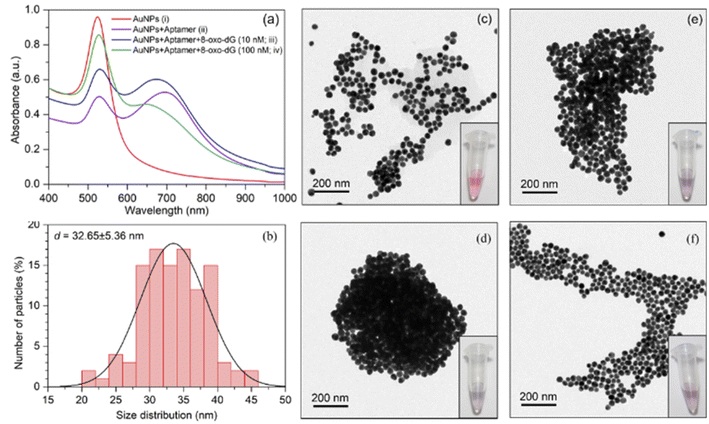 |
| | Fig. 2 (a) Absorption spectra of cyst-AuNPs obtained from the solutions containing: (i) cyst-AuNPs; (ii) cyst-AuNPs after addition of the anti-8-oxo-dG aptamers; (iii) cyst-AuNPs after addition of the anti-8-oxo-dG aptamers and 8-oxo-dG (10 nM); and (iv) cyst-AuNPs after addition of the anti-8-oxo-dG aptamers and 8-oxo-dG (100 nM). (b) Size distribution of cyst-AuNPs. (c)–(f) TEM images and visual observation of cyst-AuNPs corresponding to the conditions in (i)–(iv), respectively. | |
Naked-eye detection of 8-oxo-dG
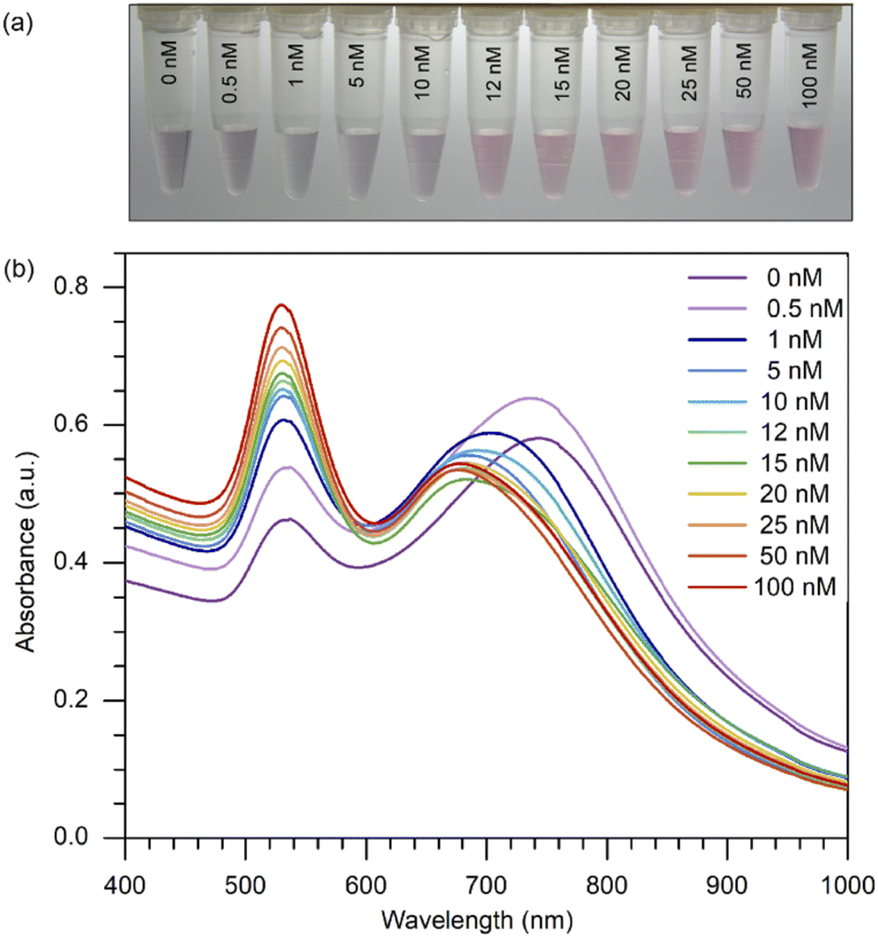
By setting the concentration of 8-oxo-dG at 12 nM, which was regarded as a low concentration, we were able to demonstrate that this method could be employed for naked-eye detection and that a distinct change from 10 to 12 nM could be noticed. Producing a change apparent to the unaided eye on a small scale implies that this approach can be performed on a larger scale by raising the concentration. This proved that even at a very low concentration, such as 12 nM, we could notice a change in colour. As shown in (Fig. 3(a)), when various concentrations of 8-oxo-dG (0–100 nM) were applied, the solution changed colour from purple to red beginning at a 12 nM-concentration of 8-oxo-dG.
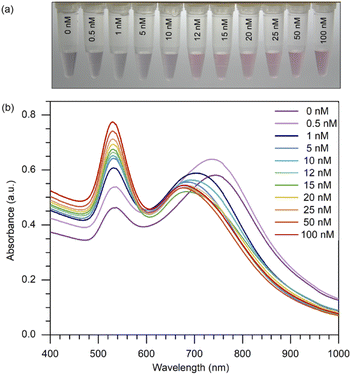 |
| | Fig. 3 (a) Naked-eye detection of 8-oxo-dG at different concentrations of 8-oxo-dG ranging from 0 to 100 nM can be accomplished by generating a red-coloured solution starting from 12 nM. Experimental conditions: cyst-AuNPs, 2.5 nM; aptamer, 5 nM; phosphate buffer, 1 mM; pH 7.0. (b) UV-vis absorption spectra of cyst-AuNPs for the detection of 8-oxo-dG at the concentrations of 0, 0.5, 1, 5, 10, 12, 15, 20, 25, 50 and 100 nM. | |
One can certainly assert that detection with the naked eye is feasible at large concentrations of the target molecule. Moreover, a progressive increase in the absorbance at 525 nm was observed with increasing concentration of 8-oxo-dG in the solution (Fig. 3(b)). This indicates that the colour change observed by the naked eye corresponds to a change in the absorption at 525 nm of the colorimetric assay.
Sensitivity of the colorimetric assay for the detection of 8-oxo-dG
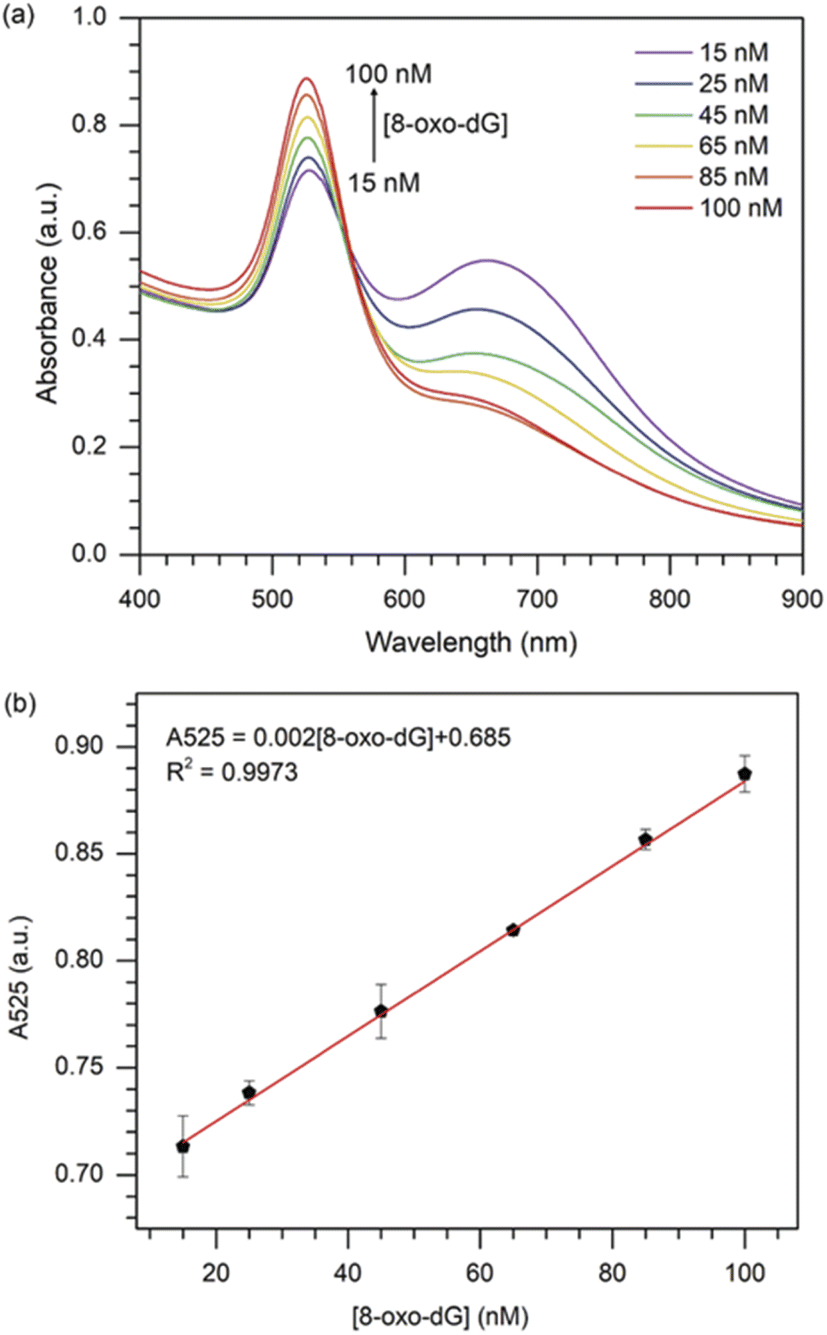
Under the optimised sensing conditions discussed in the previous section, the absorbance at 525 nm was used to determine the concentration of 8-oxo-dG. Fig. 4(a) shows the UV-vis absorption spectra of cyst-AuNPs at different concentrations of 8-oxo-dG. The absorbance at 525 nm increased as the concentration of 8-oxo-dG in the system augmented, demonstrating that cyst-AuNPs were gradually dispersed.
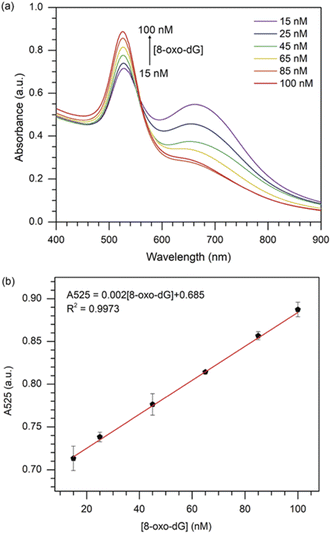 |
| | Fig. 4 (a) UV-vis absorption spectra of the colorimetric assay in the presence of various concentrations of 8-oxo-dG; (b) plot of the absorbance at 525 nm against the 8-oxo-dG concentration in the range of 15–100 nM. Error bars were obtained from three experiments. | |
Furthermore, a great linear relationship was demonstrated between A525 and 8-oxo-dG concentrations in the range of 15–100 nM, which was fitted as A525 = 0.002[8-oxo-dG] + 0.685 with a correlation coefficient of R2 = 0.9973 (Fig. 4(b)). The LOD and the limit of qualification (LOQ) of the colorimetric assay were determined to be 10.3 nM (3σ/slope) and 34.3 nM (10σ/slope), respectively. The LOD obtained in this study was superior or comparable to the previously reported assays (Table 1).
Table 1 Comparison between the colorimetric assay and previous reported methods for 8-oxo-dG detectiona
| Method |
System |
Complex matrix |
LOD |
Linear range |
Ref. |
| LOD: limit of detection, HPLC–ECD: high-performance liquid chromatography/electrochemical detection, LC-MS/MS: liquid chromatography-tandem mass spectrometry, CE: capillary electrophoresis, PtNPs: platinum nanoparticles, rGO: reduced graphene oxide, MIP: molecular imprinted polymer, CPE: carbon-paste electrode, PVDF: polyvinylidene fluoride, ABTS2+: 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt. |
| HPLC-ECD |
— |
Urine |
0.35 nM L−1 |
7.0–700 nM L−1 |
28 |
| LC-MS/MS |
— |
Saliva and urine |
10.0 ng mL−1 |
10.0–250.0 ng mL−1 |
29 |
| CE |
— |
Urine |
0.19 μg mL−1 |
0.1–50.0 μg mL−1 |
30 |
| Resonance light scattering |
Citrate-AuNPs/NaCl/aptamer |
Urine |
27.3 pM |
90.8 pM–14.1 nM |
20 |
| Fluorometry |
N-Methyl mesoporphyrin IX/K+/aptamer |
Urine |
1.19 nM |
3.96–211 nM |
31 |
| Amperometry |
PtNPs-rGO@guanosine poly-dopamine MIP/CPE |
Urine and serum |
0.0008 μM |
0.005–50 μM |
32 |
| Colorimetry and fluorometry |
PVDF membrane/polythiophene/aptamer |
Artificial urine |
∼350 pM |
50 pM–500 nM |
33 |
| Colorimetry |
Hemin/aptamer/ABTS2+/H2O2 |
Urine |
141 pM |
466 pM–247 nM |
23 |
| Colorimetry |
Citrate-AuNPs/NaCl/aptamer |
Urine |
1.7 nM |
5.6–282 nM |
34 |
| Colorimetry |
Citrate-AuNPs/NaCl/aptamer |
Urine |
13.2 nM |
15–100 nM |
22 |
| Colorimetry |
Cyst-AuNPs/aptamer |
Urine |
10.3 nM |
15–100 nM |
This work |
Specificity of the colorimetric assay for the detection of 8-oxo-dG
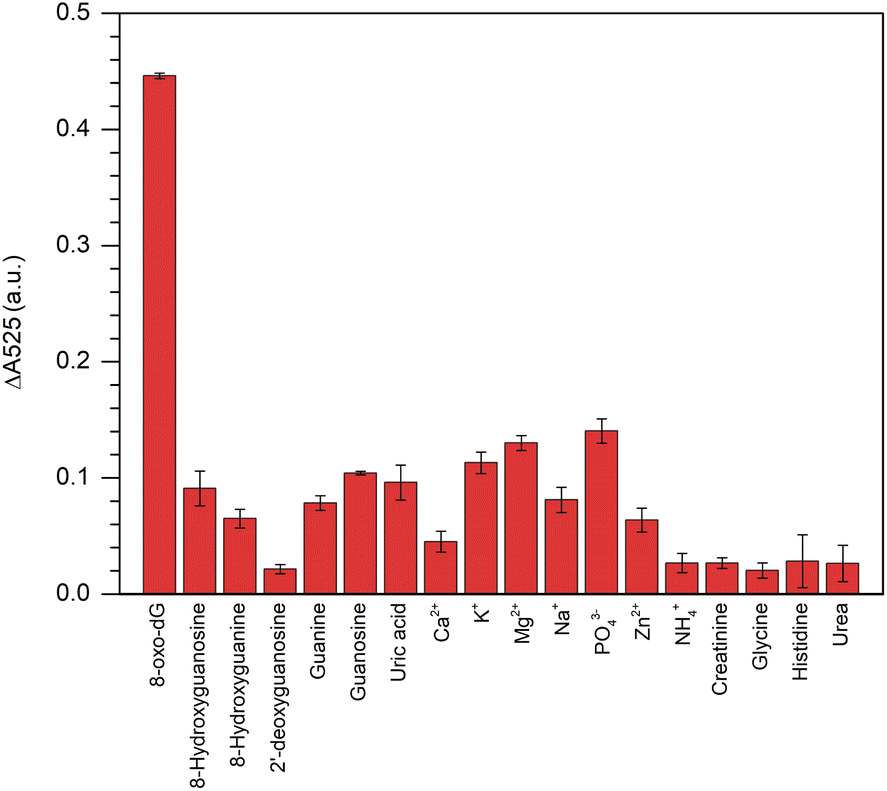
This colorimetric assay showed excellent specificity in its response to 8-oxo-dG, but not to its structural analogues, i.e., 2′-deoxyguanosine, 8′-hydroxyguanosine, 8′-hydroxyguanine, guanosine, guanine, uric acid, creatinine, glycine, histidine and urea or for other ions, i.e., Ca2+, K+, Mg2+, Na+, PO43−, Zn2+, and NH4+ when all the concentrations were fixed at 100 nm (Fig. 5).
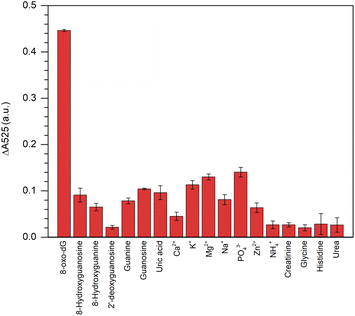 |
| | Fig. 5 Specificity of the aptamer-cyst–AuNPs-based colorimetric assay for 8-oxo-dG detection. The concentration of 8-oxo-dG and other interference molecules/ions were 100 nM. | |
The results indicated that the fabricated aptamer–AuNPs-based detection method had a good sensitivity for 8-oxo-dG because the different absorbance at 525 nm of the cyst-AuNPs solution with or without the analyte (ΔA525 = A525cyst-AuNPs − A525cyst-AuNPs+analyte). The ΔA525 of the detection system having 8-oxo-dG was found to be higher than those obtained from other non-target molecules/species. However, it could be noted that PO43− was more likely to induce a change in A525 than other ions/substances, which could be due to the fact that PO43− could cause electrostatic screening of cyst-AuNPs, similar to what was previously reported for the effect of Mg2+ on the dispersion of cyst-AuNPs.35 In addition, a greater concentration of PO43− was employed to assess the specificity of the developed sensor. The PO43− at a concentration of 438 nM did not significantly affect ΔA525 (Fig. S7†), suggesting that PO43− would no longer influence the sensing performance of the proposed assay. It can be concluded that the interaction between the aptamer and the 8-oxo-dG molecule is specific, causing the system to change and a high sensitivity of the colorimetric assay for 8-oxo-dG can be attained.
Determination of spiked 8-oxo-dG in water and urine samples
The recovery of the 8-oxo-dG colorimetric assay was tested by the proposed colorimetric method. Standard 8-oxo-dG solution was spiked into ultrapure water to obtain three different concentrations of 40, 60 and 80 nM. The recoveries were calculated to be ranged from 84.72 to 102.50% (n = 3; Table 2). Two different concentrations of spiked normal urine were prepared (39.17 and 49.17 nM) with the recoveries of 93.61% and 94.05%, respectively. Good recovery indicates that the 8-oxo-dG sensor is promising for practical analysis. It should be noted that, in normal urine, 8-oxo-dG can be detected, which explained why non-spiked urine contained 24.17 nM 8-oxo-dG.
Table 2 Determination of spiked 8-oxo-dG in water and urine
| Matrix |
Spiked (nM) |
Final 8-oxo-dG (nM) |
8-oxo-dG found (nM) |
Recovery (%) |
| Water |
40 |
40 |
37.17 |
92.92 |
| 60 |
60 |
50.83 |
84.72 |
| 80 |
80 |
82.00 |
102.50 |
| Urine |
0 |
24.17 |
— |
— |
| 15 |
39.17 |
36.67 |
93.61 |
| 25 |
49.17 |
47.83 |
94.05 |
Application of the 8-oxo-dG aptamer–cyst-AuNPs-based colorimetric assay in human urine
To determine the feasibility of the aptamer–cyst-AuNPs-based colorimetric assay, it was used to detect two urine samples containing varying quantities of 8-oxo-dG and the findings were compared to the reference values obtained using the conventional ELISA. The results in Table 3 were substantially identical to those obtained using the ELISA approach, with no significant differences. These findings strongly suggest that the developed detection assay may be used to identify 8-oxo-dG in urine samples.
Table 3 Comparison of urinary 8-oxo-dG levels detected by the developed detection assay and ELISA method
| Sample |
This method (nM) |
ELISA (nM) |
| 1 |
557.57 |
654.43 |
| 2 |
678.78 |
669.44 |
Conclusions
In summary, a colorimetric assay for the detection of 8-oxo-dG with good sensitivity and specificity was successfully developed. The detection mechanism is based on the dispersion of cyst-AuNPs, which is regulated by the interactions of the specific anti-8-oxo-dG aptamer and 8-oxo-dG. When cyst-AuNPs and the aptamer were combined, aggregation of cyst-AuNPs ensued. By contrast, when 8-oxo-dG was added to the assay solution, the colour of the solution changed from purple to red due to the complex formation between the aptamer and 8-oxo-dG which no longer induced the assembly of cyst-AuNPs. After optimisation, the produced colorimetric assay demonstrated a linear connection between UV-vis absorbance in the concentration range of 15–100 nM, and a high selectivity over other molecules/species. Good recoveries of 8-oxo-dG from spiked ultrapure water and urine samples were obtained. 8-Oxo-dG values determined by this detection assay were comparable with those by the ELISA technique,demonstrating the established colorimetric assay's high accuracy for the detection of 8-oxo-dG. Based on the benefits of low cost, high sensitivity, simple working principle, shortened detection time, and practical application, this approach would be expected for on-site and real-time screening of the oxidative DNA damage marker 8-oxo-dG in urine.
Author contributions
Chadamas Sakonsinsiri: conceptualisation, methodology, funding acquisition, visualisation, data curation, validation, formal analysis, writing – original draft preparation, writing – review and editing, funding acquisition. Theerapong Puangmali: conceptualisation, methodology, software, validation, data curation, writing – review and editing. Kaniknun Srijivangsa and Sireemas Koowattanasuchat: investigation and visualisation. Raynoo Thanan: writing – review and editing, resources. Apiwat Chompoosor: writing – review and editing, resources. Sirinan Kulchat: writing – review and editing and Paiboon Sithithaworn: writing – review and editing. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research has received funding support from the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (Grant number #B05F630053/5856) and partial support from the Cholangiocarcinoma Screening and Care Program, Khon Kaen University.
References
- S. S. David, V. L. O'Shea and S. Kundu, Nature, 2007, 447, 941–950 CrossRef CAS PubMed
 .
. - P. T. Henderson, J. C. Delaney, F. Gu, S. R. Tannenbaum and J. M. Essigmann, Biochemistry, 2002, 41, 914–921 CrossRef CAS PubMed .
- J. E. Klaunig and L. M. Kamendulis, Annu. Rev. Pharmacol. Toxicol., 2004, 44, 239–267 CrossRef CAS .
- K. Roszkowski, W. Jozwicki, P. Blaszczyk, A. Mucha-Malecka and A. Siomek, Med. Sci. Monit., 2011, 17, CR329–CR333 CrossRef CAS PubMed .
- C. Zhang, G. Nestorova, R. A. Rissman and J. Feng, Electrophoresis, 2013, 34, 2268–2274 CrossRef CAS PubMed .
- C. Guo, X. Li, R. Wang, J. Yu, M. Ye, L. Mao, S. Zhang and S. Zheng, Sci. Rep., 2016, 6, 32581 CrossRef CAS .
- A. Nanashima, H. Izumino, Y. Sumida, T. Tominaga, K. Wakata, S. Hidaka, T. Tsuchiya and T. Nagayasu, Anticancer Res., 2016, 36, 3899–3903 CAS .
- L. L. Wu, C. C. Chiou, P. Y. Chang and J. T. Wu, Clin. Chim. Acta, 2004, 339, 1–9 CrossRef CAS .
- A. Valavanidis, T. Vlachogianni and C. Fiotakis, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2009, 27, 120–139 CrossRef CAS PubMed .
- R. Thanan, M. Murata, S. Pinlaor, P. Sithithaworn, N. Khuntikeo, W. Tangkanakul, Y. Hiraku, S. Oikawa, P. Yongvanit and S. Kawanishi, Cancer Epidemiol., Biomarkers Prev., 2008, 17, 518–524 CrossRef CAS .
- L. P. Jia, J. F. Liu and H. S. Wang, Biosens. Bioelectron., 2015, 67, 139–145 CrossRef CAS PubMed .
- G. V. Martins, A. C. Marques, E. Fortunato and M. G. F. Sales, Biosens. Bioelectron., 2016, 86, 225–234 CrossRef CAS PubMed .
- G. V. Martins, A. P. M. Tavares, E. Fortunato and M. G. F. Sales, Sci. Rep., 2017, 7, 14558 CrossRef PubMed .
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS PubMed .
- H. Pu, Z. Huang, D.-W. Sun, X. Xie and W. Zhou, Water, Air, Soil Pollut., 2019, 230, 124 CrossRef .
- Y. Luan, J. Chen, C. Li, G. Xie, H. Fu, Z. Ma and A. Lu, Toxins, 2015, 7, 5377–5385 CrossRef CAS .
- W. Phanchai, U. Srikulwong, A. Chompoosor, C. Sakonsinsiri and T. Puangmali, Langmuir, 2018, 34, 6161–6169 CrossRef CAS .
- Z. Mei, H. Chu, W. Chen, F. Xue, J. Liu, H. Xu, R. Zhang and L. Zheng, Biosens. Bioelectron., 2013, 39, 26–30 CrossRef CAS PubMed .
- Y. Liu, M. Wei, L. Zhang, Y. Zhang, W. Wei, L. Yin, Y. Pu and S. Liu, Anal. Chem., 2016, 88, 6509–6514 CrossRef CAS PubMed .
- J.-C. Wang, Y.-S. Wang, J.-H. Xue, B. Zhou, Q.-M. Qian, Y.-S. Wang, J.-C. Yin, H. Zhao, H. Liu and S.-D. Liu, Biosens. Bioelectron., 2014, 58, 22–26 CrossRef CAS PubMed .
- W. Wei, M. Wei, L. Yin, Y. Pu and S. Liu, Microchim. Acta, 2018, 185, 494 CrossRef PubMed .
- P. Matulakul, D. Vongpramate, S. Kulchat, A. Chompoosor, R. Thanan, P. Sithithaworn, C. Sakonsinsiri and T. Puangmali, ACS Omega, 2020, 5, 17423–17430 CrossRef CAS PubMed .
- H. Liu, Y.-S. Wang, J.-C. Wang, J.-H. Xue, B. Zhou, H. Zhao, S.-D. Liu, X. Tang, S.-H. Chen, M.-H. Li and J.-X. Cao, Anal. Biochem., 2014, 458, 4–10 CrossRef CAS PubMed .
- Y. Miyachi, N. Shimizu, C. Ogino, H. Fukuda and A. Kondo, Bioorg. Med. Chem. Lett., 2009, 19, 3619–3622 CrossRef CAS PubMed .
- T. Niidome, K. Nakashima, H. Takahashi and Y. Niidome, Chem. Commun., 2004, 1978–1979, 10.1039/b406189f .
- P. K. Jain, W. Huang and M. A. El-Sayed, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS .
- W. Haiss, N. T. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed .
- O. Zengi, A. Karakas, U. Ergun, M. Senes, L. Inan and D. Yucel, Clin. Chem. Lab. Med., 2012, 50, 529–534 CAS .
- K. Kahremanoglu, E. R. Temel, T. E. Korkut, A. A. Nalbant, B. B. Azer, C. Durucan, M. Volkan and E. Boyaci, J. Sep. Sci., 2020, 43, 1925–1933 CrossRef CAS PubMed .
- M. J. Li, J. B. Zhang, W. L. Li, Q. C. Chu and J. N. Ye, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3818–3822 CrossRef CAS .
- H. Liu, Y. S. Wang, X. Tang, H. X. Yang, S. H. Chen, H. Zhao, S. D. Liu, Y. F. Zhu, X. F. Wang and Y. Q. Huang, J. Pharm. Biomed. Anal., 2016, 118, 177–182 CrossRef CAS .
- N. Nontawong, M. Amatatongchai, P. Jarujamrus, D. Nacapricha and P. A. Lieberzeit, Sens. Actuators, B, 2021, 334, 129636 CrossRef CAS .
- G. Ammanath, U. H. Yildiz, A. Palaniappan and B. Liedberg, ACS Appl. Mater. Interfaces, 2018, 10, 7730–7736 CrossRef CAS .
- J.-C. Wang, Y.-S. Wang, W.-Q. Rang, J.-H. Xue, B. Zhou, L. Liu, Q.-M. Qian, Y.-S. Wang and J.-C. Yin, Microchim. Acta, 2014, 181, 903–910 CrossRef CAS .
- W. Phanchai, U. Srikulwong, A. Chuaephon, S. Koowattanasuchat, J. Assawakhajornsak, R. Thanan, C. Sakonsinsiri and T. Puangmali, ACS Appl. Nano Mater., 2022, 5, 9042–9052 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Theerapong Puangmali
*ab,
Theerapong Puangmali