
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew ent-kaurane diterpenoid acids from Nouelia insignis Franch and their anti-inflammatory activity†
Zhi-li Wu ab,
Jia-yu Lib,
Peng-li Huangb,
Ze-shi Sunb,
Hui-liang Li*ab and
Wei-dong Zhang*ab
ab,
Jia-yu Lib,
Peng-li Huangb,
Ze-shi Sunb,
Hui-liang Li*ab and
Wei-dong Zhang*ab
aSchool of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing, Jiangsu 211198, P. R. China. E-mail: faranli@hotmail.com; wdzhangy@hotmail.com
bSchool of Pharmacy, Second Military Medical University, Shanghai 200433, P. R. China
First published on 11th April 2022
Abstract
Eleven undescribed ent-kaurane-type diterpenoid acids, namely noueinsiancins A–K (1–11), together with sixteen related known analogs (12–27) were isolated from Nouelia insignis Franch. The chemical structures and absolute configurations of the new compounds were confirmed by the extensive spectroscopic data, electronic circular dichroism (ECD) data analysis and single crystal X-ray diffraction. Additionally, the anti-inflammatory assay was applied to estimate the nitric oxide (NO) inhibitory activities of all compounds by using lipopolysaccharide (LPS)-induced RAW 264.7 cells in vitro. The results revealed that 4–7 and 13–17 significantly inhibited NO production at the concentrations of 2.5 μM, 5.0 μM and 10.0 μM. Meanwhile, compounds 6 and 7 were found to down-regulate the protein expression levels of IL-6 and TNF-α in RAW 264.7 cells induced by LPS in a dose-dependent manner. In conclusion, these findings provided the reference values for exploring the new chemicals with biological activities from this genus.
1. Introduction
Nouelia insignis Franch belonging to the genus Nouelia is one of the infrequent woody plants from the Asteraceae family. It is widely distributed at an altitude of 1000–2500 meters in the mountainous areas of the Yunnan and Sichuan Provinces, especially in Panzhihua City in China. It is an endemic and endangered woody remnant species in our country due to the limited distribution and human deforestation.1–4 Meanwhile, the roots and leaves of Nouelia insignis Franch have been widely applied in traditional Chinese medicine (TCM), and have a long history of use to treat acute icteric hepatitis, chronic hepatitis, bacterial infections, inflammation, etc.5,6 At present, we find that there are few studies on the chemical ingredients of this plant based on the previous literature and querying a database (CA). Consequently, only a few biological activity studies were reported. In this manuscript, our phytochemical investigation of the MeOH extract of Nouelia insignis Franch led to the isolation and the identification of 27 ent-kaurane diterpenoid acids, including eleven new ones (1–11), together with sixteen related known analogs (12–27). Herein, the details of the isolation, structural elucidation and anti-inflammatory activities of the new compounds are reported (Fig. 1).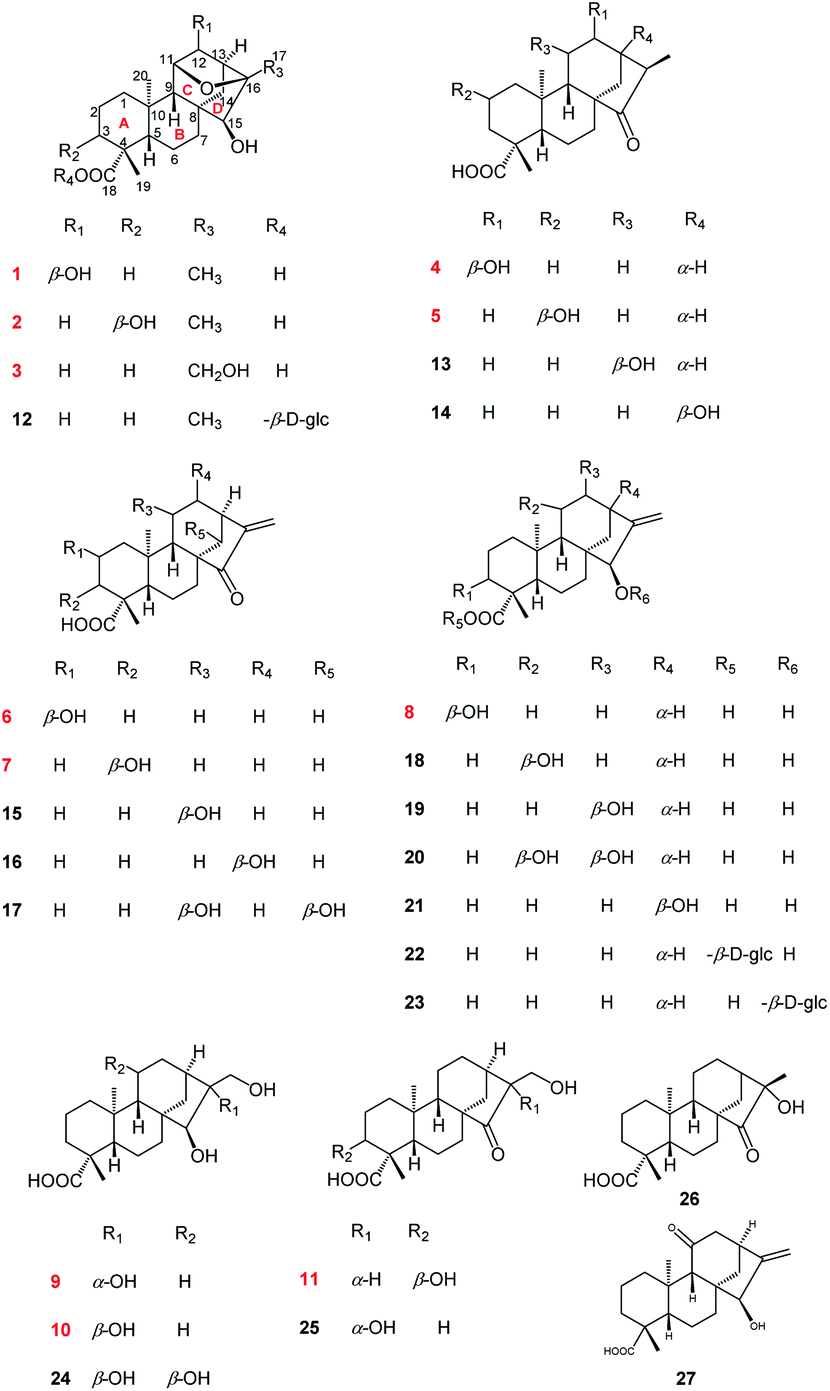 | ||
| Fig. 1 Chemical structures of 1–27. | ||
2. Experimental
2.1 General experimental procedure
Column chromatography (CC): silica gel H (10–40 μm) and silica gel (200–300 mesh) (Marine Chemical Factory, Qingdao, P. R. China). MCI gel CHP-20P: (Daiso, Co., Japan) and RP-C18 gel (40–63 μm; Daiso, Co., Japan) were used for column chromatography. Sephadex LH-20 (Pharmacia Fine Chemicals, Piscataway, NJ, USA); TLC: silica gel plates, visualization by spraying with 10% H2SO4 in EtOH and Dragendorff's reagent. Semi-preparative HPLC: Agilent 1260 series (Agilent Technologies, US) with a Zorbax SB-C18 (5 μM, 9.4 mm × 25 cm) column. NMR spectra: Bruker Avance III-500 spectrometer (Bruker, Switzerland). MS: Agilent MSD-Trap-XCT (for ESI) and Agilent-6520 Q-TOF mass spectrometer (for HR-ESI). IR: Thermo Scientific Nicolet 6700 (Thermo Scientific, US). UV spectra: Agilent 1260 series DAD detector (Agilent Technologies, US). CD spectrum: Brighttime Chirascan (Applied Photophysics Ltd, UK). Optical rotation: Rudolph Autopo V (Rudolph Research Analytical, Hackettstown, NJ).2.2 Plant material
Nouelia insignis Franch were collected from Sichun province of China in August 2020. It was identified by Prof. Bao-kang Huang (Department of Pharmacognosy, Second Military Medical University, Shanghai, China). A voucher specimen (no. 202008-VS) is deposited in the department of pharmacognosy, Second Military Medical University.2.3 Extraction and isolation
The dried and crushed branch of Nouelia insignis Franch (20.0 kg) were ultrasonically extracted with 95% methanol overnight at room temperature (20 L × 3). After remove of solvent, the methanol extract (550.5 g) was extracted with water, petroleum ether (PE) and ethyl acetate (EtOAc) (3 times with 5 L each). The EtOAc extract (124.3 g) was segmented by silica gel H (10–40 μm) column chromatography eluting with petroleum ether–ethyl acetate (PE/EtOAc, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 0:1 v/v) to afford 7 fractions (Fr. 1–7). Fraction 4 (21.6 g) was separated by MCI column chromatography (MeOH/H2O, 10:90 to 100:0 v/v) to provide 5 subfractions (Fr. 4.1–4.5). Then, Fr. 4.3 was separated by ODS (CH3CN/H2O, 10:90 to 40:60 v/v) to afford 6 subfractions (Fr. 4.3.1–4.3.6). Fr. 4.3.2 and Fr. 4.3.4 were purified by semi-preparative RP-C18 HPLC (CH3OH/H2O, 63:37 v/v 1.0 mL min−1) to give compounds 4 (12.3 mg), 5 (10.2 mg), 13 (9.4 mg), 14 (13.6 mg) and 8 (9.5 mg), 18 (10.6 mg), 19 (9.2 mg), 21 (8.2 mg). Fr. 4.5 was also separated by ODS (CH3OH/H2O, 10:90 to 40:60 v/v) to afford compounds 22 (15.2 mg), 23 (9.4 mg). Fraction 5 was initially separated by Sephadex LH-20 CC eluting with CH3OH/H2O (55:45) to yield 5 subfractions (Fr. 5.1–5.5). Compounds 1 (14.3 mg) and 2 (10.3 mg) were obtained from Fr. 5.2 by ODS column chromatography (CH3OH/H2O, 30:70 to 55:45 v/v). Fr. 5.3–5.4 were purified by ODS column chromatography (CH3OH/H2O, 30:70 to 80:20 v/v) and further purified by semi-preparative RP-C18 HPLC (CH3OH/H2O, 65:35 v/v 1.0 mL min−1) to afford compounds 3 (13.3 mg), 12 (9.8 mg) and 9 (8.3 mg), 10 (11.8 mg), 24 (14.1 mg), 25 (9.7 mg). Fraction 6 was separated on a Sephadex LH-20 CC eluted with MeOH/H2O (60:40, v/v) to yield 7 subfractions (Fr. 6.1–6.7), compound 11 (8.3 mg) was obtained from Fr. 6.1 through the ODS column chromatography (CH3OH/H2O, 30:70 to 55:45 v/v). Meanwhile, purifications of Fr. 6.3 and 6.4 by RP-HPLC (CH3OH/H2O, 60:40 v/v 1.0 mL min−1) to yielded compounds 6 (9.8 mg), 7 (13.1 mg) and 17 (9.1 mg). Fr. 6.5 was separated with ODS column chromatography (CH3OH/H2O, 10:90 to 50:50 v/v) and further purified by semi-preparative RP-C18 HPLC (CH3OH/H2O, 60:34 v/v 1.0 mL min−1) to get compounds 15 (8.8 mg), 16 (12.7 mg) and 26 (13.3 mg). Fraction 7 was separated with ODS column chromatography (CH3OH/H2O, 10:90 to 40:60 v/v) and semi-preparative RP-C18 HPLC (CH3CN/H2O, 62:38 v/v 1.0 mL min−1) to give compounds 20 (9.3 mg) and 27 (8.6 mg). Above all, the whole compounds were obtained (Fig. 1).
:1 to 0:1 v/v) to afford 7 fractions (Fr. 1–7). Fraction 4 (21.6 g) was separated by MCI column chromatography (MeOH/H2O, 10:90 to 100:0 v/v) to provide 5 subfractions (Fr. 4.1–4.5). Then, Fr. 4.3 was separated by ODS (CH3CN/H2O, 10:90 to 40:60 v/v) to afford 6 subfractions (Fr. 4.3.1–4.3.6). Fr. 4.3.2 and Fr. 4.3.4 were purified by semi-preparative RP-C18 HPLC (CH3OH/H2O, 63:37 v/v 1.0 mL min−1) to give compounds 4 (12.3 mg), 5 (10.2 mg), 13 (9.4 mg), 14 (13.6 mg) and 8 (9.5 mg), 18 (10.6 mg), 19 (9.2 mg), 21 (8.2 mg). Fr. 4.5 was also separated by ODS (CH3OH/H2O, 10:90 to 40:60 v/v) to afford compounds 22 (15.2 mg), 23 (9.4 mg). Fraction 5 was initially separated by Sephadex LH-20 CC eluting with CH3OH/H2O (55:45) to yield 5 subfractions (Fr. 5.1–5.5). Compounds 1 (14.3 mg) and 2 (10.3 mg) were obtained from Fr. 5.2 by ODS column chromatography (CH3OH/H2O, 30:70 to 55:45 v/v). Fr. 5.3–5.4 were purified by ODS column chromatography (CH3OH/H2O, 30:70 to 80:20 v/v) and further purified by semi-preparative RP-C18 HPLC (CH3OH/H2O, 65:35 v/v 1.0 mL min−1) to afford compounds 3 (13.3 mg), 12 (9.8 mg) and 9 (8.3 mg), 10 (11.8 mg), 24 (14.1 mg), 25 (9.7 mg). Fraction 6 was separated on a Sephadex LH-20 CC eluted with MeOH/H2O (60:40, v/v) to yield 7 subfractions (Fr. 6.1–6.7), compound 11 (8.3 mg) was obtained from Fr. 6.1 through the ODS column chromatography (CH3OH/H2O, 30:70 to 55:45 v/v). Meanwhile, purifications of Fr. 6.3 and 6.4 by RP-HPLC (CH3OH/H2O, 60:40 v/v 1.0 mL min−1) to yielded compounds 6 (9.8 mg), 7 (13.1 mg) and 17 (9.1 mg). Fr. 6.5 was separated with ODS column chromatography (CH3OH/H2O, 10:90 to 50:50 v/v) and further purified by semi-preparative RP-C18 HPLC (CH3OH/H2O, 60:34 v/v 1.0 mL min−1) to get compounds 15 (8.8 mg), 16 (12.7 mg) and 26 (13.3 mg). Fraction 7 was separated with ODS column chromatography (CH3OH/H2O, 10:90 to 40:60 v/v) and semi-preparative RP-C18 HPLC (CH3CN/H2O, 62:38 v/v 1.0 mL min−1) to give compounds 20 (9.3 mg) and 27 (8.6 mg). Above all, the whole compounds were obtained (Fig. 1).
2.4 Compound characterizations of 1–11
527/3520 [R(int) = 0.0500], h (−8/8), k (−14/14), l (−26/24), final R indices R1 = 0.0389 and wR2 = 0.1043 (I > 2σ(I)), R1 = 0.0404 and wR2 = 0.1057 (all data), GOF = 1.093, largest diff. peak/hole, 0.387/−0.236 e Å−3. Absolute structure parameter: 0.04(8). CCDC: 2127069.†475/3259 [R(int) = 0.0428], h (−8/8), k (−23/23), l (−8/8), final R indices R1 = 0.0492 and wR2 = 0.1288 (I > 2σ(I)), R1 = 0.0503 and wR2 = 0.1304 (all data), GOF = 1.040, largest diff. peak/hole, 0.459/−0.227 e Å−3. Absolute structure parameter 0.02(9), CCDC: 2127070.†977/3447 [R(int) = 0.0331], h (−13/13), k (−8/9), l (−13/14), final R indices R1 = 0.0302 and wR2 = 0.0800 (I > 2σ(I)), R1 = 0.0310 and wR2 = 0.0911 (all data), GOF = 1.047, largest diff. peak/hole, 0.124/−0.094 e Å−3. Absolute structure parameter 0.09(6), CCDC: 2090338.†755/2977 [R(int) = 0.0359], h (−7/7), k (−9/9), l (−11/11), final R indices R1 = 0.0304 and wR2 = 0.0799 (I > 2σ(I)), R1 = 0.0305 and wR2 = 0.0800 (all data), GOF = 1.069, largest diff. peak/hole, 0.138/−0.097 e Å−3. Absolute structure parameter 0.07(4), CCDC: 2090337.†155/3409 [R(int) = 0.0564], h (−7/7), k (−17/17), l (−12/12), final R indices R1 = 0.0407 and wR2 = 0.1083 (I > 2σ(I)), R1 = 0.0431 and wR2 = 0.1090 (all data), GOF = 1.080, largest diff. peak/hole, 0.138/−0.097 e Å−3. Absolute structure parameter 0.01(11), CCDC: 2156441.†313420 [R(int) = 0.0399], h (−8/9), k (−12/12), l (−24/28), final R indices R1 = 0.0576 and wR2 = 0.0799 (I > 2σ(I)), R1 = 0.0305 and wR2 = 0.0800 (all data), GOF = 1.069, largest diff. peak/hole, 0.138/−0.097 e Å−3. Absolute structure parameter 0.07(4), CCDC: 2090340.†478/6401 [R(int) = 0.0438], h (−10/10), k (−23/23), l (−12/13), final R indices R1 = 0.0322 and wR2 = 0.0841 (I > 2σ(I)), R1 = 0.0327 and wR2 = 0.0846 (all data), GOF = 1.052, largest diff. peak/hole, 0.151/−0.138 e Å−3. Absolute structure parameter −0.02(5), CCDC: 2127071.†2.5 Cell culture and cytotoxicity assay
The RAW 264.7 cells were purchased from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM supplemented with 10% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C, 5% CO2. All the test compounds were dissolved in dimethyl sulfoxide (DMSO) and freshly prepared each time before use. Then, RAW 264.7 cells were seed at 96-well plates at a density of 5 × 104 cells per well cultured overnight. The cells were treated with the test compounds (2.5, 5.0 and 10.0 μM) and LPS (1 μg mL−1), respectively. After 24 h incubation, 10 μL CCK-8 solution was added to each well and incubated at 37 °C for 30 min. Finally, the optical OD-value was measured at 450 nm through a microplate reader, respectively (BioTek Instruments, Inc.).2.6 Nitric oxide production inhibitory activity assay
The nitric oxide (NO) level was determined through measuring the concentration of nitrite in culture medium using Griess assay. RAW 264.7 cells were seeded in 96-well plates (5 × 104 cells per well) for 24 h, then, the cells were treated with or without the test compounds (2.5, 5.0 and 10.0 μM) for 2 h prior to LPS (1 μg mL−1). After incubation for 24 h, 100 μL supernatant mixed with 100 μL Griess reagent and incubated for 20 min at 37 °C in the dark. Finally, the absorbance was measured at 540 nm through a microplate reader, respectively (BioTek Instruments, Inc.).2.7 The detection of cytokines production
RAW 264.7 cells were seeded in 12-well plates (2 × 105 cells per well) for 24 h and the cells were also treated with or without the test compounds at the concentration of 2.5, 5.0 and 10.0 μM for 2 h before LPS (1 μg mL−1) stimulation. After incubation for 24 h, the culture media were collected. Then, the levels of TNF-α and IL-6 were measured using the ELISA kits (Beyotime Institute of Biotechnology, Nantong, China) according to the manufacturer's instruction.2.8 Statistical analysis
Prism 8 (GraphPad, San Diego, CA, USA) were used for statistical analysis. Dates of three separate studies were analysed by the one-way ANOVA test and the Tukey post hoc test. Meanwhile, all date expressed as mean ± standard deviation (SD), which collected from three independent experiments. The significant difference was determined with P < 0.05.3. Results and discussion
3.1 Structural identification
Compound 1 was isolated as colorless needle crystals from MeOH/H2O. Its molecular formula, C20H30O5, was deduced from the positive HRESIMS ion peak at m/z 373.1962 ([M + Na]+, calcd for C20H30O5Na, 373.1985), corresponding to six indices of hydrogen deficiency. Analysis of the 1H NMR spectra of 1 (Table 2) showed characteristic signals assignable to three methyl singlets at δH 1.34 (s, H3-17), 1.19 (s, H3-19) and 0.99 (s, H3-20), as well as three oxymethine protons at δH 4.09 (1H, H-11), 4.14 (1H, H-12) and 2.82 (1H, H-15). The 13C NMR and DEPT spectra (Table 1) indicated the presence of 20 carbon signals, including three methyls (δC 18.8, 21.0, 29.6); six methylenes (δC 20.2, 22.2, 35.1, 37.3, 39.1 and 42.6); six methines (including three oxygenated at δC 79.8, 82.3 and 82.5, three sp3 carbon signals at δC 50.7, 52.3 and 57.9) and five quaternary carbons [δC 37.8, 44.5, 45.6, 85.4 and 181.6 (a carboxyl group)]. Additionally, according to the 1H and 13C NMR spectra of 1, six indices of hydrogen deficiency were attributed to one carbonyl group (δC 181.6) and one epoxy group [δH 4.09, H-11 and δC 82.5, C-11; 85.4, C-16]. The remaining four ones exhibited the existence of a tetracyclic system in 1.7 Consequently, the four-ring was inferred to be a 6/6/6/5 fused rings A, B, C and D, a characteristic ent-kaurane diterpenoid skeleton,8–10 based on the 2D NMR experiments (Fig. 2): the 1H–1H COSY correlations of H2-1/H2-2/H2-3, H-5/H2-6/H2-7 and H-9/H-11/H-12/H-13/H2-14; the HMBC correlations from H3-19 to C-3, C-4, C-5 and C-18; from H3-20 to C-1, C-5, C-9 and C-10; from H2-7 to C-6 and C-8; from H-9 to C-5, C-8, C-10, C-11 and C-12; from H2-14 to C-8, C-9, C-13, C-15 and C-16; and from H3-17 to C-13, C-15 and C-16. In addition, the occurrence of a 11,16-epoxy motif in 1 was determined by the HMBC correlations of H-11 with C-16. Thus, the planar structure of 1 was determined.| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a NMR data were measured at 125 MHz in CD3OD for 1–11. | |||||||||||
| 1 | 42.6 | 35.7 | 41.2 | 41.1 | 49.7 | 49.9 | 35.0 | 33.9 | 42.0 | 40.3 | 34.4 |
| 2 | 20.2 | 27.1 | 18.8 | 20.1 | 64.7 | 64.8 | 27.2 | 25.9 | 20.3 | 18.9 | 27.0 |
| 3 | 39.1 | 71.5 | 37.7 | 39.0 | 47.7 | 47.7 | 72.0 | 70.3 | 39.3 | 37.8 | 71.5 |
| 4 | 44.5 | 48.3 | 43.0 | 44.7 | 45.8 | 45.9 | 45.0 | 47.7 | 44.7 | 43.3 | 48.6 |
| 5 | 57.9 | 50.3 | 56.7 | 57.3 | 56.9 | 56.9 | 49.6 | 48.3 | 57.7 | 56.4 | 49.3 |
| 6 | 22.2 | 21.9 | 20.9 | 21.4 | 21.4 | 21.2 | 21.1 | 21.0 | 23.1 | 21.4 | 21.1 |
| 7 | 35.1 | 35.9 | 34.4 | 34.5 | 35.4 | 34.9 | 34.9 | 38.7 | 36.0 | 34.3 | 35.2 |
| 8 | 45.6 | 46.6 | 45.3 | 53.5 | 53.9 | 53.7 | 54.0 | 45.6 | 49.3 | 45.9 | 54.5 |
| 9 | 52.3 | 51.6 | 51.0 | 52.3 | 53.1 | 53.1 | 53.2 | 45.4 | 47.6 | 46.0 | 53.1 |
| 10 | 37.8 | 37.6 | 36.5 | 40.8 | 42.4 | 42.7 | 41.4 | 38.8 | 40.4 | 39.0 | 41.0 |
| 11 | 82.5 | 77.8 | 76.3 | 29.0 | 19.4 | 19.6 | 19.5 | 17.5 | 19.7 | 18.5 | 19.5 |
| 12 | 79.8 | 39.7 | 38.2 | 71.9 | 25.6 | 33.2 | 33.4 | 33.1 | 27.5 | 26.2 | 25.8 |
| 13 | 50.7 | 43.3 | 38.3 | 49.4 | 36.3 | 39.4 | 39.5 | 40.1 | 45.0 | 39.5 | 33.6 |
| 14 | 37.3 | 38.9 | 37.5 | 37.5 | 38.2 | 37.7 | 37.7 | 36.1 | 41.6 | 40.6 | 38.0 |
| 15 | 82.3 | 82.9 | 77.7 | 226.2 | 227.0 | 212.3 | 212.8 | 81.8 | 90.4 | 79.9 | 224.0 |
| 16 | 85.4 | 85.6 | 86.8 | 42.9 | 49.0 | 151.2 | 151.4 | 157.6 | 84.1 | 77.1 | 57.3 |
| 17 | 21.0 | 20.7 | 61.8 | 11.7 | 10.3 | 115.4 | 115.1 | 103.7 | 64.6 | 69.1 | 59.9 |
| 18 | 181.6 | 181.3 | 180.2 | 181.3 | 180.7 | 181.2 | 183.0 | 180.2 | 181.7 | 180.3 | 181.0 |
| 19 | 29.6 | 25.1 | 28.2 | 29.4 | 29.3 | 29.5 | 25.3 | 23.6 | 29.5 | 28.1 | 24.9 |
| 20 | 18.8 | 18.2 | 17.0 | 16.3 | 17.2 | 17.3 | 16.2 | 14.8 | 16.3 | 14.7 | 15.8 |
 | ||
| Fig. 2 The 1H–1H COSY and HMBC correlations of 1–11. | ||
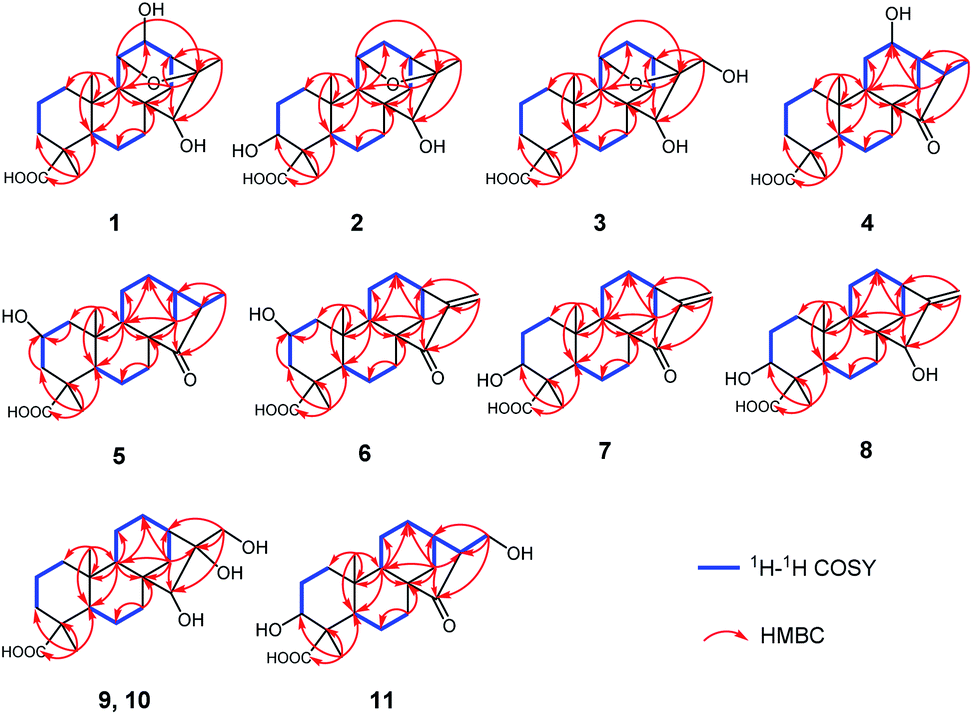
The relative configuration of 1 was determined through the NOESY spectrum (Fig. 3). The NOESY correlations of H3-19/H-5/H-9 indicated they were assigned the β-orientation. Meanwhile, H3-20, H-12 and H-11, H-13, H-15 were shown to be in a α-orientation according to the NOESY interactions from H3-20 to H-12 and H3-20 to H-11, H-13 and H-15. In order to determine its structure and absolute configuration, an appropriate crystal of 1 was obtained from a solvent system of MeOH/H2O after many single crystal cultures.
 | ||
| Fig. 3 The key NOESY correlations of 1–11. | ||
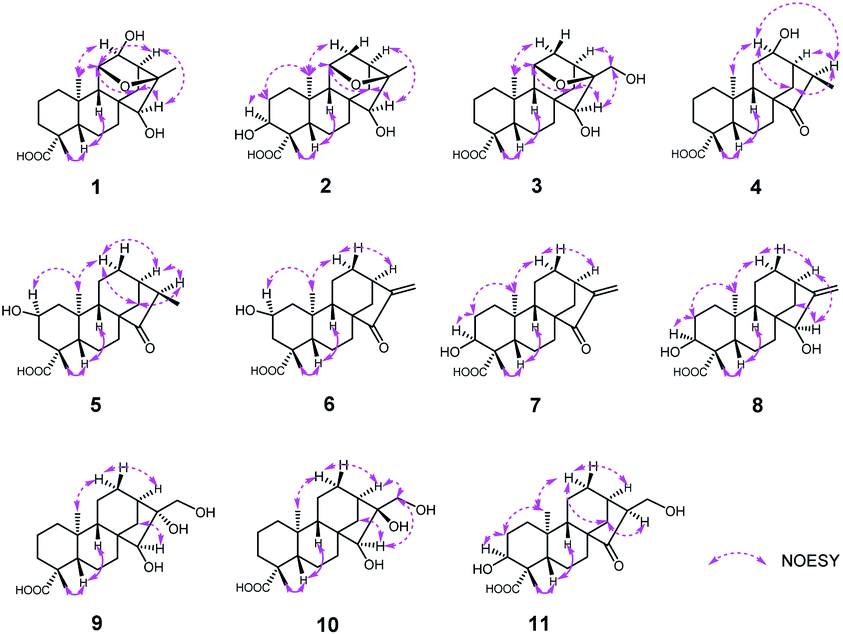
Therefore, the X-ray crystallographic analysis with Cu Kα radiation unambiguously confirmed the absolute configuration of 1 was 4R, 5S, 8R, 9S, 10R, 11R, 12S, 13S, 15S, 16R (Fig. 4). Finally, the structure of 1 was ascertained completely and named noueinsiancin A.
 | ||
| Fig. 4 The X-ray crystallographic structures of 1, 2, 4–6, 8 and 9. | ||
Compound 2, obtained as colorless needle crystals, possessed the same molecular formula of C20H30O5 as 1 based on the ion peak at m/z 373.1963 ([M + Na]+, calcd for C20H30O5Na, 373.1985) in the positive HRESIMS spectrum. Overall analysis of the 1D NMR (Tables 1 and 2) and 2D NMR (1H–1H COSY and HMBC) spectroscopic spectrum indicated that the gross structure of 2 was nearly close to that of 1. In fact, the only difference between them was the presence of a hydroxy group at C-3 (δC 71.5) position in 2 instead of the hydroxy group at C-12 (δC 79.8) position in 1. This was verified by the 1H–1H COSY correlations of H2-1/H2-2/H-3 (δH 3.99) and H-9/H-11/H2-12 (δH 2.00, 1.82)/H-13/H2-14, together with the HMBC correlations from H3-19 to C-3 (δC 71.5), C-4 (δC 48.3) and C-5 (δC 50.3) (Fig. 2). The analysis of NOESY data revealed that the molecular conformation of 2 was identical to that of 1 (Fig. 3). Similarly, to further determine the structure and absolute configuration of 2, a suitable crystal of 2 for the single-crystal X-ray diffraction experiment (Cu Kα) was obtained. Finally, its absolute configuration was confirmed as 3S, 4S, 5S, 8R, 9S, 10R, 11S, 13S, 15S, 16R (Fig. 4). Hence, the structure of 2 was defined and named noueinsiancin B.
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz in CD3OD for 1–6. | ||||||
| 1 | 1.88, 1.19 | 1.58 | 1.85, 1.18 | 1.86, 0.85 | 2.17, 0.67, t, (12.0) | 2.19, 0.70, t, (12.0) |
| 2 | 1.85, 1.43, m | 2.13, 1.57, m | 1.86, 1.42, m | 1.91, 1.42, m | 4.08, m | 4.09, m |
| 3 | 2.16, 1.05, m | 3.99, overlap | 2.16, 1.04, m | 2.13, 1.04, m | 2.41, 0.96, t, (12.2) | 2.41, 0.97, t, (12.3) |
| 5 | 1.05, m | 1.47, m | 1.06, m | 1.17, m | 1.11, m | 1.11, m |
| 6 | 1.84, 1.70, m | 1.76, m | 1.85, 1.75, m | 1.90, 1.78, m | 1.93, 1.81, m | 1.95, m |
| 7 | 1.54, 1.25, m | 1.78, 1.21, m | 1.56, 1.22, m | 1.74, 1.39, m | 1.67, 1.36, m | 1.82, 1.34, m |
| 9 | 1.72 | 1.68 | 1.70 | 1.16 | 1.06 | 1.19 |
| 11 | 4.09, overlap | 4.28, overlap | 4.33, overlap | 1.93, m | 1.71, 1.23, m | 1.77, 1.47, m |
| 12 | 4.14, overlap | 2.00, d, (11.4), 1.82 | 2.00, d, (11.2), 1.80 | 4.09, m | 1.69, m | 1.89, 1.69, m |
| 13 | 2.14 | 2.19 | 2.45 | 2.41, m | 2.44, m | 3.07, m |
| 14 | 1.95, d, (12.7), 1.19 | 1.95, d, (12.3), 1.14 | 1.97, d, (12.3), 1.11 | 2.33, d, (12.5), 1.48 | 2.45, overlap, 1.43 | 2.43, overlap, 1.40 |
| 15 | 2.82, s | 2.87, s | 3.06, s | |||
| 16 | 2.59, m | 2.30, m | ||||
| 17 | 1.34, s | 1.27, s | 3.59 dd, (11.5, 5.8) | 1.25, d, (7.2) | 1.09, d, (7.2) | 5.89, 5.32, s |
| 19 | 1.19, s | 1.24, s | 1.19, s | 1.21, s | 1.27, s | 1.27, s |
| 20 | 0.99, s | 1.01, s | 1.02, s | 0.98, s | 1.04, s | 1.06, s |
Compound 3 was isolated as a white amorphous powder and showed a molecular formula of C20H30O5 according to the positive HRESIMS data: 373.1960 ([M + Na]+, calcd for C20H30O5Na, 373.1985). Detailed comparison of the 1H and 13C NMR spectra of 3 and 1 (Tables 1 and 2) revealed that the structure of 3 was similar to that of 1. The main difference was that the absence of a hydroxy group at C-12 position in 3 and the replacement of the methyl group in 1 at C-16 position by a hydroxy-methylene group in 3. This conclusion was supported by the hydroxyl-methylene signal at δC 61.8 (C-17) and the HMBC correlations from H2-17 (δH 3.59) to C-13 (δC 38.3), C-16 (δC 86.8) and C-15 (δC 77.7) (Fig. 2). Additionally, detailed analysis of the NOESY data of 3 indicated its relative configuration was also the same as 1. Aim to elucidated its absolute configuration, ECD calculations, a method regarded as a powerful and effective method to confirm the absolute configuration of the natural products,11,12 was applied. Subsequently, its absolute configuration was established as 4R, 5S, 8R, 9S, 10R, 11S, 13S, 15S, 16S by comparing the calculated and experimental ECD spectrum (Fig. 5). Consequently, the structure of 3 was obtained and named noueinsiancin C.
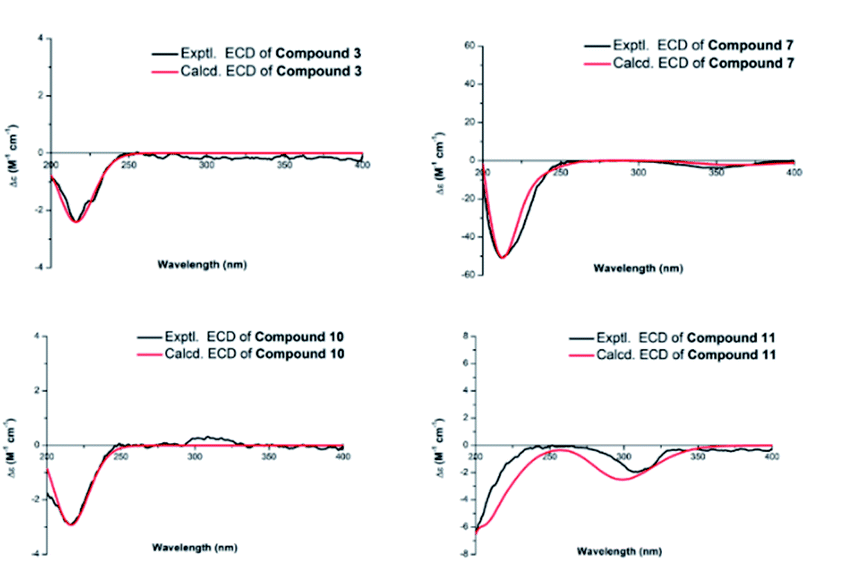 | ||
| Fig. 5 Experimental and calculated ECD spectra of compounds 3, 7, 10 and 11. | ||
Compound 4 was initially purified as colorless needle crystals and assigned a molecular formula of C20H30O4 from the [M + Na]+ ion peak at m/z 357.2023 (calcd for C20H30O4Na, 357.2036) in positive HRESIMS. The 1D NMR data of 4 (Tables 1 and 2) were closely similar to those of 1, except for the absence of the 11,16-epoxy group in 4 and the substitution of C-15 position (the hydroxy group for the former, and the carbonyl group for the latter). These above deductions were revealed by the 1H–1H COSY correlations of H-9/H2-11/H-12/H-13/H2-14 and H-13/H-16/H3-17, together with the HMBC correlations from H3-17 (δH 1.25, d, J = 7.2 Hz) to C-13 (δC 49.4), C-16 (δC 42.9) and C-15 (δC 226.2) (Fig. 2). In the NOESY spectrum (Fig. 3), the correlations of H3-19/H-5/H-9 and H3-20/H-12/H-16/H2-14 demonstrated the β-configuration and α-configuration of these protons, respectively. Finally, by means of the single-crystal X-ray diffraction analysis with Cu Kα radiation (Fig. 4), the absolute configuration of 4 was assigned as 4R, 5S, 8R, 9S, 10S, 12R, 13R, 16R. Consequently, the structure of 4 was determined named noueinsiancin D.
Compound 5, isolated as colorless needle crystals, provided the same molecular formula as that of 4 by its positive HRESIMS. Comparison of the 1D NMR spectra (1H, 13C and DEPT) of 5 (Tables 1 and 2) with those of 4, followed by the detailed analysis the 2D NMR spectra (1H–1H COSY and HMBC) established that the structure of 5 was close to that of 4. The main difference between 5 and 4 was in the positioning of a hydroxy group, which in 5 was at C-2 position and in 4 was at C-12 position. This ratiocination was corroborated by the 1H–1H COSY correlations of H2-1 (δH 2.17, 0.67)/H-2 (δH 4.08)/H2-3 (δH 2.41, 0.96), as well as the HMBC cross-peaks from H3-19 (δH 1.27, s) to C-3 (δC 47.7) methylene, from H3-20 (δH 1.04, s) to C-1 (δC 49.7) methylene and from H2-3 (δH 2.41, 0.96) and H2-1 (δH 2.17, 0.67) both to C-2 (δC 64.7) (Fig. 2). Moreover, the crucial NOESY interactions of H-2/H3-20 and H3-20/H-12α/H2-14/H-16 (Fig. 3) confirmed that these protons were both α-oriented and the configuration of 2-OH was β-oriented. Its absolute configuration was 2S, 4R, 5S, 8R, 9S, 10S, 13R, 16R as consolidated by the single crystal X-ray diffraction with Cu Kα radiation (Fig. 4) Finally, the structure of 5 was defined and named noueinsiancin E.
Compound 6 was isolated as colorless needle crystals. Its molecular formula (C20H28O4) was deduced by analysing the positive HRESIMS (m/z 355.1861 [M + Na]+, calcd for C20H28O4Na, 355.1880). By detailed comparison of the 1H and 13C NMR spectra (Tables 1 and 2) between 6 and 5 indicated that the closely related structures of them, except for the exocyclic Δ16(17) double bond in 6 instead of the methine (C-16, δC 49.0) and the methyl (CH3-17, δC 10.3) in 5. This conclusion was evidenced by the HMBC correlations from H2-17 (δH 5.89, 5.32, 2H) to C-13 (δC 39.4), C-16 (δC 151.2) and C-15 (δC 212.3). In the NOESY data (Fig. 3), the correlations of H3-19/H-5/H-9 verified the β-orientation of Me-19, H-5 and H-9, while the NOESY correlations of H-2/H3-20/H-12α/H-13 assigned H-2, Me-20 and H-13 as α-orientated. Then, the absolute configuration of 6 was confirmed as 2S, 4R, 5S, 8R, 9S, 10S, 13R on the basis of the single crystal X-ray diffraction experiment with Cu Kα radiation (Fig. 4). Hence, the structure of 6 was obtained and named noueinsiancin F.
Compound 7 was assigned an equal molecular formula (C20H28O4) to that of 6 from the ion peak at m/z 355.1860 ([M + Na]+, calcd for C20H28O4Na, 355.1880) in the positive HRESIMS spectrum and 13C NMR data. Detailed interpretation of its 1D NMR (Tables 1 and 3) and 2D NMR data indicated that the structure of 6 was highly resembled that of 5 with different positioning of a hydroxy group (the hydroxy group at the C-3 (δC 72.0) position for 7, and the hydroxy group at the C-2 (δC 64.8) position in 6). This deduction was determined by the 1H–1H COSY correlations of H2-1 (δH 1.54, 1.24)/H2-2 (δH 2.24, 1.53)/H-3 (δH 4.00) and the HMBC correlations from H3-19 (δH 1.24, 3H) to C-3 (δC 72.0), C-4 (δC 45.0) and C-5 (δC 49.6). Additionally, the NOESY spectrum (Fig. 3) suggested that 7 had the same relative configuration as 6, where the H-3 and H3-20 assigned the α-orientation by the NOESY correlations from H-3 to H2-2 and H3-20. Its absolute configuration was determined to be 3S, 4S, 5S, 8R, 9S, 10S, 13R based on the comparison of the calculated and experimental ECD spectra (Fig. 5). Thus, the structure of compound 7 was established and named noueinsiancin G.
| No. | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|
| a NMR data were measured at 500 MHz in CD3OD for 7–11. | |||||
| 1 | 1.54, 1.24 | 1.63, 1.32 | 1.90, 0.89 | 1.92, 0.89 | 1.55, 1.23 |
| 2 | 2.24, 1.53, m | 2.19, 1.55, m | 1.91, 1.40, m | 1.91, 1.40, m | 2.18, 1.54, m |
| 3 | 4.00 | 3.98 | 2.12, 1.02, m | 2.12, 1.01, m | 3.97, overlap |
| 5 | 1.54, m | 1.48, m | 1.05, m | 1.04, m | 1.57, m |
| 6 | 1.92, 1.80, m | 1.86, 1.73, m | 1.83, m | 1.82, 1.60, m | 1.86, 1.76, m |
| 7 | 1.82, 1.31, m | 1.65, 1.29, m | 1.91, 1.45, m | 1.94, 0.88, m | 1.72, 1.34, m |
| 9 | 1.22 | 1.44 | 1.40 | 1.40 | 1.14 |
| 11 | 1.72, 1.43, m | 1.63, 1.49, m | 1.61, 1.53, m | 1.69, 1.47, m | 1.69, 1.21, m |
| 12 | 1.89, 1.67, m | 1.63, 1.45, m | 1.63, 1.47, m | 1.76, 1.40, m | 1.75, m |
| 13 | 3.06, m | 2.59, m | 1.92, m | 1.96, m | 2.65, m |
| 14 | 2.46, d, (12.1), 1.38 | 2.00, d, (11.9), 1.01 | 1.58, 1.42 | 1.60, 1.33 | 2.50, d, (12.2), 1.42 |
| 15 | 3.68 | 3.42, s | 3.06, s | ||
| 16 | 2.50 | ||||
| 17 | 5.88, 5.30, s | 5.04, s, 4.91, overlap | 3.75 dd, (11.9, 10.2) | 3.42, d, (11.2), 3.27 d, (11.3) | 3.94, dd, (11.3, 6.6), 3.59, dd, (9.2, 2.1) |
| 19 | 1.24, s | 1.23, s | 1.18, s | 1.18, s | 1.24, s |
| 20 | 1.08, s | 0.99, s | 0.99, s | 0.97, s | 1.05, s |
The molecular formula of 8 was C20H30O4 as deduced from the ion peak at m/z 357.2023 ([M + Na]+, calcd for C20H30O4Na, 357.2036) in the positive HRESIMS spectrum. The 1H and 13C NMR spectroscopic data (Tables 1 and 3) of 8 were found to be very identical with those of 7, indicating that the structure of 8 was nearly similar to that of 7. Unlike compound 7, the signal ascribed to the carbonyl group at C-15 position were not observed for 8. Instead, a hydroxy group (δH 3.68; δC 81.8) was present at C-15 position in 8. This was determined by the HMBC correlations from H2-17 (δH 5.04, 4.91) to C-15 (δC 81.8), C-13 (δC 40.1) and C-16 (δC 157.6) (Fig. 2). Similarly, the NOESY data was also showed their same relative configuration, and the OH-15 was assigned as β-oriented verified by the NOESY cross-peaks from H3-20 to H-12α, H-13 and H-15 (Fig. 3). The absolute configuration of 8 was determined as 3S, 4S, 5S, 8R, 9S, 10S, 13R, 15R by the single crystal X-ray diffraction experiment with Cu Kα radiation (Fig. 4). Consequently, its structure was defined and named noueinsiancin H.
Compound 9 was obtained as colorless needle crystals, with a molecular formula of C20H32O5 based on the quasi-molecular ion peak at m/z 375.2123 ([M + Na]+, calcd for C20H32O5Na, 375.2142) in its positive HRESIMS data. A detailed analysis revealed the 1H and 13C NMR spectra (Tables 1 and 3) of 9 were similar to those of 8, except for the absence of a hydroxyl group at C-3 position and a 16, 17-diol functional group13 instead of an exocyclic double bond at C-16 and C-17 position in 9. This deduction was subsequently verified the 1H–1H COSY correlations from H2-1/H2-2/H2-3, together with the HMBC correlations from H2-17 (δH 3.75) to C-13 (δC 45.0) C-15 (δC 90.4) and C-16 (δC 84.1) (Fig. 2). In the NOESY spectrum (Fig. 3), the correlations of H3-19/H-5/H-9 and H3-20/H-12α/H-13 verified the β- and α-orientation of these protons. However, the relative configuration of the remaining chiral carbons could not be determined due to the lack of NOESY correlations. Then, a suitable crystal of 9 was obtained followed by the single-crystal X-ray diffraction experiment using the Cu Kα radiation (Fig. 4). Finally, its absolute configuration was confirmed as 4R, 5S, 8R, 9S, 10S, 13R, 15S, 16R. Consequently, the structure of 9 was defined and named as noueinsiancin I.
The molecular formula of 10 (C20H32O5) was possessed to be identical to that of 9 on the basis of the (+)-HRESIMS ion peak at m/z 375.2121 ([M + Na]+, calcd for C20H32O5Na, 375.2142). The planar structure of 10 was conjectured to be the same as 9 by the detailed analysis its NMR data (1D and 2D) (Fig. 2). However, further upon comparison of their 13C NMR data, the main difference was the change in the chemical shifts of C-15, C-16, and C-17 from δC 90.4, 84.1 and 64.6 in 9 to 79.9, 77.1 and 69.1 in 10. The shieldings of C-15 (Δδ −10.5) and C-16 (Δδ −7.0) in 10 were clearly implied the configurational change of C-16. This deduction was determined by the NOESY correlations of H-15/H2-17/H-13 (Fig. 3), suggesting these protons were on the same face and confirmed as α-orientation. Therefore, compound 10 was deduced as the 16-epimer of compound 9. By comparing the calculated and experimental ECD spectra (Fig. 5), the absolute configuration of 10 was confirmed as 4R, 5S, 8R, 9S, 10S, 13R, 15S, 16S. Hence, its structure was established and named noueinsiancin J.
Compound 11 was obtain as white amorphous powder and possessed a ion peak at m/z 373.1966 ([M + Na]+, calcd for C20H30O5Na, 373.1985) in the positive HRESIMS, corresponding to the molecular formula of C20H30O5. The 1H and 13C NMR spectra (Tables 1 and 3) of 11 were closely similar to those of 7, with the difference being the presence of a hydroxy-methylene group (δH 3.94, 3.59, δC 59.9) instead of an exocyclic Δ16(17) double bond at C-16 position in 11. This was corroborated by the clear 1H–1H COSY correlations of H-13 (δH 2.65)/H-16 (δH 2.50)/H2-17 (δH 3.94, 3.59), as well as the HMBC correlations from H2-17 (δH 3.94, 3.59) to C-15 (δC 224.0), C-16 (δC 57.3) and C-13 (δC 33.6) (Fig. 3). In addition, the NOESY spectrum suggested that the relative configuration of 11 was identical with 7. H-16 was assigned to be α-orientation based on the NOESY correlations from H3-20 to H-12α, H2-14 and H-16 (Fig. 3). The absolute configuration of 11 was assigned to be 3S, 4S, 5S, 8R, 9S, 10S, 13R, 16S based on by the high agreement between the calculated and experimental ECD curves (Fig. 5), accordingly, its structure was determined and named noueinsiancin K.
3.2 Nitric oxide production inhibitory activity
All the isolated compounds (1–27) were assayed for their inhibitory activities against NO production in LPS-stimulated RAW 264.7 cells. Compared with the LPS group, compounds 4–7 and 13–17 evidently decreased the NO production in LPS-induced RAW 264.7 cells (Fig. 6A). And, they possessed no significant cytotoxicity at their effective concentration for the inhibition of NO production (Fig. 6B). Notably, the new compounds 6 and 7 showed stronger inhibitory activities at 2.5, 5.0 and 10.0 μM dose concentrations. Thus, we chose 6 and 7 as the material to further study on the anti-inflammatory activity.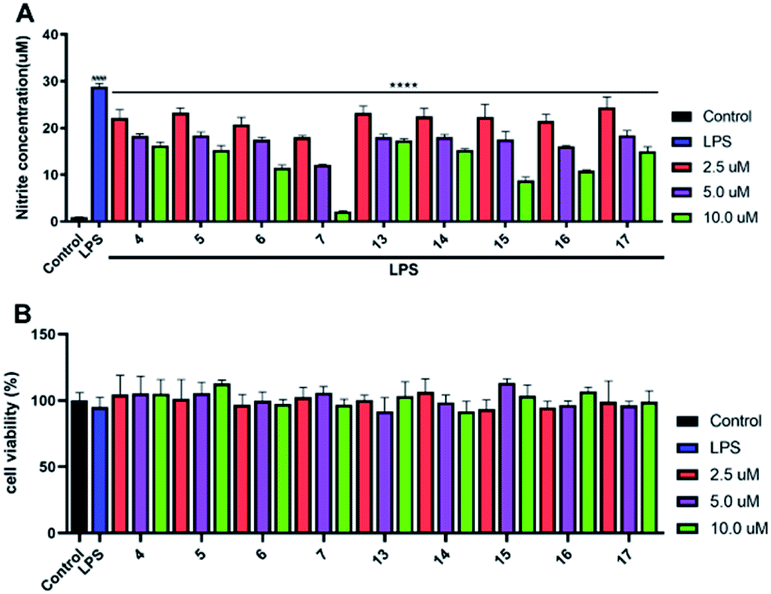 | ||
| Fig. 6 NO inhibitory activities study in vitro, (A) the inhibitory effects of 4–7 and 13–17 on NO production in LPS-induced RAW 264.7 cells at 2.5, 5.0 and 10.0 μM dose concentrations. (B) The cytotoxic effect of these compounds on RAW 264.7 cells by CCK8 assay. All results were expressed as mean ± S.D. (n = 3). ####p < 0.0001 vs. control group. ****p < 0.0001 vs. the LPS group. | ||
The preliminary analysis of the structure–activity relationship on inhibitory activities against NO production in LPS-induced RAW 264.7 cells were discussed as follows: in all the tested compounds, we found that the five compounds (6, 7, 15–17) possessed the α-methylene-γ-carbonyl group at C-16 and C-15 positions, displaying much better inhibitory activities than others. Additionally, in compounds 4, 5, 13 and 14, the methyl group instead of the α-methylene group at C-16 position would weaken the activity (6, 7, 15–17 > 4, 5, 13, 14). Consequently, we inferred that the α-methylene-γ-carbonyl group was a functional structure for inhibiting NO production in LPS-induced RAW 264.7 cells.
3.3 6 and 7 decreased the protein levels of TNF-α and IL-6 in LPS-induced RAW 264.7 cells
Due to the TNF-α and IL-6 were the important factors of inflammation.14–16 Therefore, the effects of 6 and 7 on the protein levels of IL-6 and TNF-α in LPS-induced RAW 264.7 cells were examined. After RAW 264.7 cells were induced by LPS (1 μg mL−1) for 24 h with or without 6 and 7, the culture media were collected. Then, the levels of TNF-α and IL-6 were measured using the ELISA kits. As shown in Fig. 7A–D. Compared with the normal group, treated LPS could evidently up-regulated the protein expressions of TNF-α and IL-6. Then, compounds 6 and 7 treatment led to an obvious downregulation in the abovementioned protein expressions in a dose-dependent manner, comparing to the LPS-stimulated group.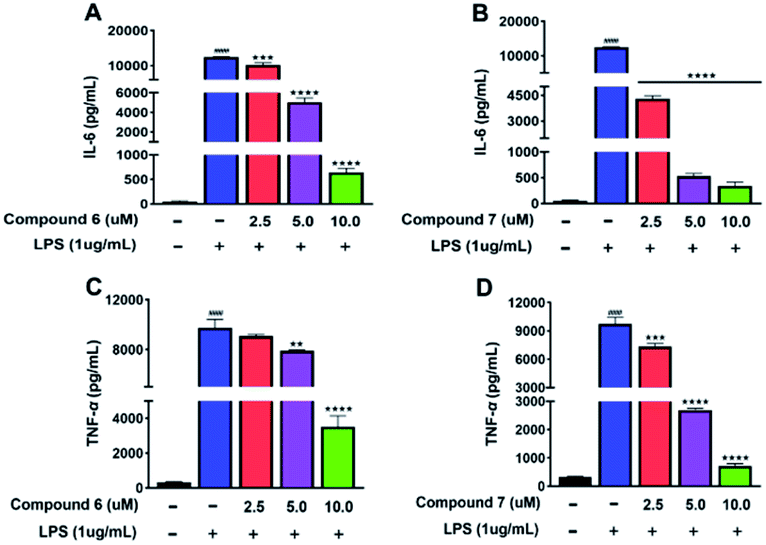 | ||
| Fig. 7 (A and C) The effects of 6 on the protein expression levels of IL-6 and TNF-α in LPS-induced RAW 264.7 cells. (B and D) The effects of 7 on the protein expression levels of IL-6 and TNF-α in LPS-induced RAW 264.7 cells. All results were expressed as mean ± S.D. (n = 3). ####p < 0.0001 vs. control group. **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. the LPS group. | ||
4. Conclusions
In conclusion, noueinsiancins A–K (1–11), eleven new ent-kaurane diterpenoid acids, together with sixteen related known analogs (12–27) were isolated from Nouelia insignis Franch. The chemical structures including absolute configurations of the eleven new compounds were identified by the analyses of their NMR spectra, HRESIMS spectra, ECD calculation and single crystal X-ray diffraction. Moreover, the anti-inflammatory activity evaluation research revealed that compounds 4–7 and 13–17 significantly inhibited NO production at the concentration of 2.5 μM, 5.0 μM and 10.0 μM in LPS-stimulated RAW 264.7 cells. At the same time, compounds 6 and 7 evidently reduced the protein expression levels of IL-6 and TNF-α in RAW 264.7 cells induced by LPS. Especially, the analysis of the structure–activity relationship indicated that the α-methylene-γ-carbonyl group was a functional structure for anti-inflammatory activity. Above all, our findings provide the idea that the administration of Nouelia insignis Franch may be beneficial for the treatment of inflammation disease.Conflicts of interest
The authors declare no conflicts of interest in this paper.Acknowledgements
This study was supported by the National Natural Science Foundation of China (82141203, 82004003, 82004215), Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTDD-202004), Shanghai Sailing Program (20YF1459000), Shanghai Municipal Health Commission Project (20204Y0326) and Shanghai Frontiers Science Center of TCM Chemical Biology.References
- F. Y. Liu, C. J. Gao, M. Chen, G. Y. Tang and Y. Y. Sun, Ecol. Evol., 2021, 11, 9396–9409 CrossRef PubMed.
- Y. L. Peng, Y. Q. Hu and H. Sun, Acta Bot. Yunnanica, 2003, 25, 563–571 CAS.
- F. Y. Liu, C. J. Gao, M. Chen, Y. Y. Sun and K. Li, Nord. J. Bot., 2003, 2543 Search PubMed.
- C. Y. Wang, J. Liu, H. G. Xiao, J. W. Zhou and D. L. Du, An. Acad. Bras. Cienc., 2016, 88, 1791–1797 CrossRef PubMed.
- X. Y. Hu, Y. G. Luo, X. Z. Chen, L. Zhou and G. L. Zhang, J. Asian Nat. Prod. Res., 2008, 10, 125–131 CrossRef CAS PubMed.
- Y. J. Zhao and X. Guo, BMC Evol. Biol., 2015, 15, 134 CrossRef PubMed.
- N. Lin, H. Li, J. R. Wang, W. Tang, M. Y. Zheng, H. Wang, C. S. Jiang and Y. W. Guo, Chin. J. Chem., 2022, 40, 28–38 CrossRef CAS.
- J. M. Dai, K. Hu, B. C. Yan, X. R. Li, X. N. Li and H. D. Sun, J. Nat. Prod., 2020, 83, 3717–3725 CrossRef CAS PubMed.
- M. S. Qiu, B. Yang and D. Cao, Phytochem. Lett., 2016, 16, 156–162 CrossRef CAS.
- H. H. Xing, L. J. An, Z. T. Song, S. S. Li, H. M. Wang, J. Zhang, M. Tuerhong, M. Abudukeremu, D. H. Li, D. H. Lee, J. Xu and Y. Q. Guo, J. Nat. Prod., 2019, 83, 2844–2853 CrossRef PubMed.
- F. L. Li, Z. Ye, Z. Y. Huang, X. Chen, W. G. Sun, W. X. Gao, S. T. Zhang, F. Cao, J. P. Wang, Z. X. Hu and Y. H. Zhang, Bioorg. Chem., 2020, 117, 105452 CrossRef PubMed.
- X. F. He, J. J. Chen, T. Z. Li, J. Hu, X. Y. Huang, X. M. Zhang, Y. Q. Guo and C. A. Geng, Chin. J. Chem., 2021, 39, 3051–3063 CrossRef CAS.
- B. Alessandra, A. Annahil, M. Ivano, M. Jeannette, H. B. Chai, S. Steven, K. Dougals and T. Nunziatina, Planta Med., 2004, 70, 540–550 CrossRef PubMed.
- B. Y. Zhang, D. Liu, W. Y. Ji, K. Otsuki, F. Zhao, W. Li and F. Qiu, Front. Chem., 2021, 9, 649287 CrossRef CAS PubMed.
- A. Raksat, S. Choodej, T. Aree, S. N. Ebrahimi and K. Pudhom, Phytochemistry, 2022, 196, 113074 CrossRef CAS PubMed.
- Y. Chen, J. Y. Ruan, F. Sun, H. M. Wang, S. S. Yang, Y. Zhang, J. J. Yan, H. Y. Yu, Y. Q. Guo and Y. Zhang, Front. Chem., 2020, 8, 73 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2127069–2127071, 2090337–2090338, 2090340 and 2156441. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra01684b |
| This journal is © The Royal Society of Chemistry 2022 |