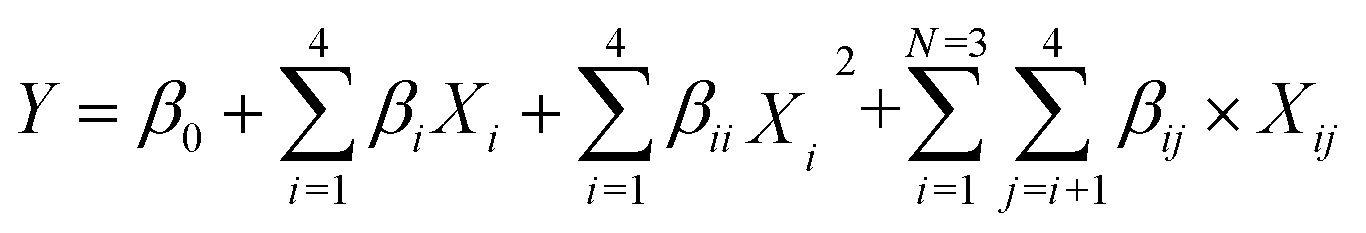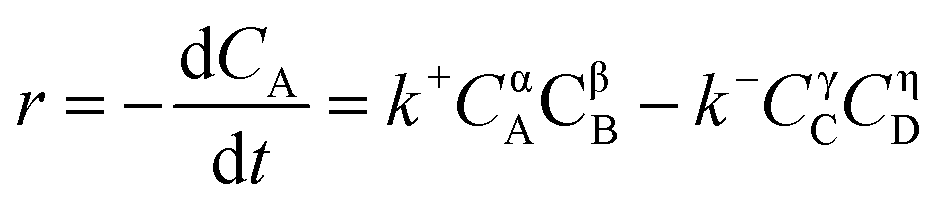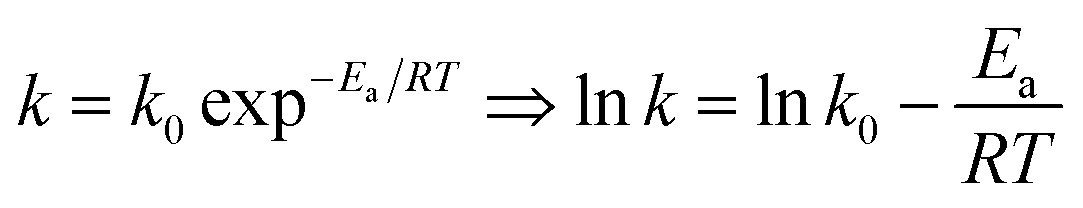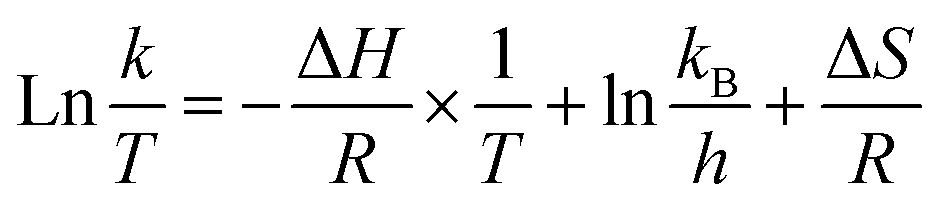DOI:
10.1039/D2RA01683D
(Paper)
RSC Adv., 2022,
12, 9744-9754
Lipophilic antioxidant dodecyl caffeate preparation by the esterification of caffeic acid with dodecanol using ionic liquid [Hnmp]HSO4 as a catalyst
Received
15th March 2022
, Accepted 22nd March 2022
First published on 28th March 2022
Abstract
Caffeic acid (CA) is widely found in nature, and has a broad spectrum of biological activities. However, the low hydrophilicity and lipophilicity of CA limited its application. Dodecyl caffeate (DC) is the lipophilic ester of caffeic acid (CA), and also has high antioxidant activity. In this work, CA, used as a substrate, and three ionic liquids with different acidities and H2SO4 were used as economic catalysts for DC preparation. The effects of variables on DC yield were investigated and optimized by response surface methodology (RSM). And the kinetic and thermodynamic parameters of the esterification of CA and dodecanol were evaluated. Results showed that lipophilic DC was successfully synthesized using ionic liquid ([Hnmp]HSO4) as a catalyst. And the optimal conditions by RSM were substrate ratio of 10.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, IL dosage of 9.8% at 87 °C for 118 min. Under the optional conditions, the maximum DC yield was 94.67 ± 1.32%. The k0, Ea, ΔH, ΔS, and ΔG were 7.18 × 107 mol (L min)−1, 65.77 kJ mol−1, 63.10 kJ (mol K)−1, −103.80 J (mol K)−1, and 99.78 kJ mol−1 at 363 K, respectively. DC prepared in this work showed a good DPPH radical scavenging activity, which indicated that DC can be used as a potential antioxidant in food and cosmetics.
:1, IL dosage of 9.8% at 87 °C for 118 min. Under the optional conditions, the maximum DC yield was 94.67 ± 1.32%. The k0, Ea, ΔH, ΔS, and ΔG were 7.18 × 107 mol (L min)−1, 65.77 kJ mol−1, 63.10 kJ (mol K)−1, −103.80 J (mol K)−1, and 99.78 kJ mol−1 at 363 K, respectively. DC prepared in this work showed a good DPPH radical scavenging activity, which indicated that DC can be used as a potential antioxidant in food and cosmetics.
1 Introduction
Caffeic acid (CA, 3,4-dihydroxycinnamic acid), a phenolic acid of plant secondary metabolites, is widely present in fruits, vegetables, and grains.1–3 CA has many biological activities such as anti-inflammatory, anti-diabetic, anticancer, antiviral and antioxidant activity in vitro.4–6 Therefore, CA has a broad prospective applications in food, pharmaceutical and cosmetic industries. And the global market and production quantity of CA were about $4.5 × 107 and 220 t in 2020.7,8 However, the low oil solubility of CA limits its application.4,6 These issues can be resolved by the modification of the side chain of CA using long-chain alkyl groups.9,10 In addition, the reported literatures showed that, the antioxidant activity of alkyl caffeates had a “cut-off” effect, and among these esters, butyl caffeate and dodecyl caffeate (DC) showed the maximum antioxidant activity in oil or emulsion systems.11–13 Therefore, DC was synthesized in this work.
In the previous reports, some catalysts were used to prepare alkyl caffeate by transesterification or esterification.14–17 Chen et al.15 and Xiao et al.18 synthesized octyl caffeate using Novozym 435 (2000$ per kg) in an isooctane system for a long time (>52 h). Pang et al.14 reported chemoenzymatic synthesis of propyl caffeate using lipase-catalyzed the transesterification of 1-propanol and intermediate (methyl caffeate). Fiuza et al.16 and Han et al.19 reported that propyl caffeate synthesis was catalyzed by H2SO4 and p-toluenesulfonic acid. These previous reports mainly focused on the preparation of short chain ester of alkyl caffeate. However, due to the great steric hindrance, the long chain ester of alkyl caffeate synthesis became very difficult with the increase of carbon numbers of alkyl chains. In our previous report,20 we prepared octodecyl caffeate by the esterification of caffeic acid-based deep eutectic solvent and octadecanol using cation exchange resin (A-35) as a catalyst, which firstly required the preparation of caffeic acid-based deep eutectic solvent. In this work, we want to develop another cheap catalyst for long chain alkyl caffeate preparation by the simple esterification of CA and dodecanol.
Recently, ILs, due to their negligible volatility, high catalytic activity, low corrosion and combustibility, excellent thermal and chemical stability, and potential for recyclability, had been used as environmentally benign reaction solvents and catalysts.21–23 In the previous reports, some ILs had also been used as reaction solvents and catalysts for the short chain esters of alkyl caffeate synthesis.14,24,25 Han et al.26 reported that propyl caffeate was synthesized using IL [BSO3HMIM]TS as a catalyst, and the maximum of propyl caffeate yield was 84%. Recently, due to the strong water absorbability, IL [Hnmp]HSO4 showed good catalytic performance for some esterifications.27,28 For example, methyl laurate was synthesized using [Hnmp]HSO4.27 However, IL as a catalyst for the preparation of long chain alkyl caffeate was not available.
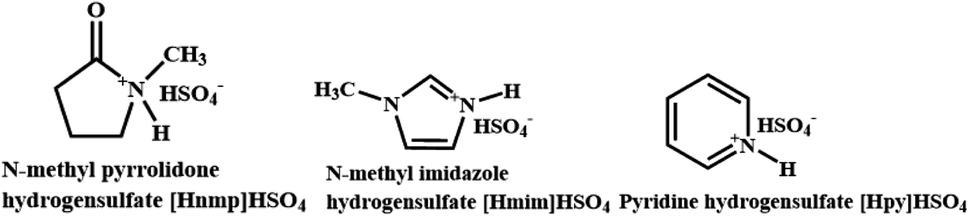
In this work, some ILs (Scheme 1) were prepared by one-step neutralization reaction between cheap heterocyclic compounds (N-methylpyrrolidone (NMP, 20$ per kg), N-methylimidazole (100$ per kg) and pyridine (15$ per kg)) and H2SO4. And the ILs were firstly used as dual solvent-catalysts for lipophilic DC preparation by the esterification of CA with dodecanol. The effect of the esterification conditions (temperature, substrate ratio, IL dosage, reaction time, and water content) were optimized by response surface methodology (RSM). The esterification kinetics and thermodynamics also were explored.
 |
| | Scheme 1 Structural formula and abbreviations of ILs. | |
2 Experimental section
2.1 Materials
NMP, N-methylimidazole and pyridine were supplied by Aladdin Biochemical Technology Co. Ltd (Shanghai, China). CA was supplied by Zelang Biological Technology Co., Ltd (Nanjing, China). Dodecanol was purchased from Macklin Biological Technology Co., Ltd (Shanghai, China).
2.2 IL preparation and characteristics
2.2.1 IL preparation. According to the previous methods,29,30 ILs were prepared as follows. Firstly, H2SO4 solution (water:H2SO4, 1:1, mol:mol) was added dropwise in N-methylpyrrolidone at 5 °C, and the mixture (H2SO4:H2O:NMP = 1:1:1, mol:mol:mol) was stirred at room temperature for 24 h. Secondly, the remained NMP in the mixture was removed by washing with ethyl acetate. And then the mixture was dried under vacuum to remove water and ethyl acetate. Finally, N-methylpyrrolidone bisulfate ionic liquid ([Hnmp]HSO4) was obtained (yield 98.5%). The preparation of N-methyl imidazole bisulfate ionic liquid ([Hmim]HSO4, yield 97.8%) and pyridine bisulfate ionic liquid ([Hpy]HSO4, yield 98.2%) were similar to [Hnmp]HSO4.
2.2.2 IL characteristics. FT-IR spectrum of IL was measured by PerkinElmer Frontier absorption spectrometer for KBr matrix in the wavelength range 4000–400 cm−1. SDT Q600 V20.9 Build 20 thermo-gravimetric analyzer was used for thermogravimetric analysis (TGA) of IL at 5 °C min−1 under the protection of N2. The Hammett acidity of IL was analyzed as follows: different ILs and H2SO4 (0.20 mmol L−1) were firstly dissolved in dimethyl yellow-ethanol solution (20 mg L−1), and then the Hammett acidities were determined using a UV-visible spectrophotometer at room temperature.
2.3 Esterification of CA and dodecanol
CA (0.2 g) and dodecanol (2.07 g) were mixed in 10 mL round bottom flasks at 500 rpm and different temperatures (70–100 °C). Then, the IL catalyst was added into the mixture, and the esterification was initiated. Samples (5 μL) were withdrawn at specified times intervals and dissolved in 10 mL methanol. Finally, the mixture was filtered using a microfilter (0.45 nm) for HPLC analysis. DC product was purified by column chromatogram (n-hexane/ethyl acetate, 6:4).
2.4 Product analysis
Reactant and product were analyzed using a HPLC system. Reaction mixture was eluted by using a reverse-phase C18 column (4.6 × 250 mm, 5 μm) at 35 °C, and UV detector was 325 nm. A binary ingredient of water (0.5% acetic acid) and solvent methanol (1:19, v:v) was used and eluted at 0.8 mL min−1. DC yield and purify were quantified by external standard method. The structure characterization of DC has been analyzed and was confirmed by the previous reports.20,31
2.5 Experimental design
RSM was employed in this study for the verification and prediction of the model equation and was used to optimize the esterification of CA and dodecanol. The independent variables (temperature, time, substrate ratio, and IL dosage) were varied in different levels (−1, 0, and +1) as presented in Table 1.
Table 1 Results of response surface experiments
| Run |
Temperature X1 (°C) |
Time X2 (min) |
Substrate ratio X3 (mol mol−1) |
IL dosage X4 (%) |
DC yield Y (%) |
| 1 |
90(0) |
120(0) |
10(0) |
10(0) |
92.21 ± 0.52 |
| 2 |
90(0) |
120(0) |
5(−1) |
15(1) |
71.18 ± 2.13 |
| 3 |
90(0) |
120(0) |
5(−1) |
5(−1) |
43.18 ± 1.41 |
| 4 |
90(0) |
90(−1) |
10(0) |
5(−1) |
58.05 ± 0.72 |
| 5 |
90(0) |
90(−1) |
5(−1) |
10(0) |
57.28 ± 1.56 |
| 6 |
80(−1) |
120(0) |
10(0) |
5(−1) |
53.74 ± 1.20 |
| 7 |
80(−1) |
150(1) |
10(0) |
10(0) |
85.49 ± 1.84 |
| 8 |
90(0) |
90(−1) |
10(0) |
15(1) |
80.38 ± 2.77 |
| 9 |
90(0) |
90(−1) |
15(1) |
10(0) |
82.33 ± 1.84 |
| 10 |
100(1) |
120(0) |
15(1) |
10(0) |
98.71 ± 0.14 |
| 11 |
90(0) |
120(0) |
10(0) |
10(0) |
95.41 ± 0.07 |
| 12 |
90(0) |
150(1) |
15(1) |
10(0) |
94.95 ± 1.03 |
| 13 |
90(0) |
120(0) |
10(0) |
10(0) |
95.03 ± 0.79 |
| 14 |
90(0) |
150(1) |
10(0) |
15(1) |
94.13 ± 0.10 |
| 15 |
100(1) |
90(−1) |
10(0) |
10(0) |
89.65 ± 0.87 |
| 16 |
90(0) |
150(1) |
10(0) |
5(−1) |
65.78 ± 0.29 |
| 17 |
100(1) |
120(0) |
5(−1) |
10(0) |
79.78 ± 0.12 |
| 18 |
80(−1) |
120(0) |
10(0) |
15(1) |
90.29 ± 2.23 |
| 19 |
90(0) |
120(0) |
15(1) |
15(1) |
93.04 ± 1.96 |
| 20 |
90(0) |
120(0) |
10(0) |
10(0) |
95.85 ± 1.18 |
| 21 |
80(−1) |
120(0) |
5(−1) |
10(0) |
56.85 ± 0.94 |
| 22 |
90(0) |
120(0) |
15(1) |
5(−1) |
74.25 ± 0.44 |
| 23 |
100(1) |
120(0) |
10(0) |
5(−1) |
82.01 ± 4.20 |
| 24 |
100(1) |
120(0) |
10(0) |
15(1) |
94.85 ± 0.27 |
| 25 |
100(1) |
150(1) |
10(0) |
10(0) |
95.27 ± 0.11 |
| 26 |
90(0) |
120(0) |
10(0) |
10(0) |
94.42 ± 1.81 |
| 27 |
80(−1) |
90(−1) |
10(0) |
10(0) |
67.56 ± 2.98 |
| 28 |
80(−1) |
120(0) |
15(1) |
10(0) |
85.49 ± 2.31 |
| 29 |
90(0) |
150(1) |
5(−1) |
10(0) |
67.92 ± 2.74 |
2.6 Antioxidant activity determination
Antioxidant activity of DC was evaluated by 1, 1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging activity. In brief, 0.1 mL of DC solutions with different concentrations were added to 3.9 mL of 100 μmol L−1 DPPH solution in ethanol. The mixtures were shaken vigorously and then placed in darkness for 30 min. The absorbance was measured at 517 nm in a 1 cm quartz cell, and to calculate IC50.
2.7 Statistical analysis
In this study, multiple regression coefficients were employed to predict the linear and interaction effects of the esterification variables. The mathematical relationship between the response and variables was as follows:| |
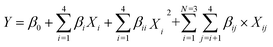 | (1) |
where Y is DC yield; Xi represent variables; all β are constants.
3 Results and discussion
3.1 Screening catalysts
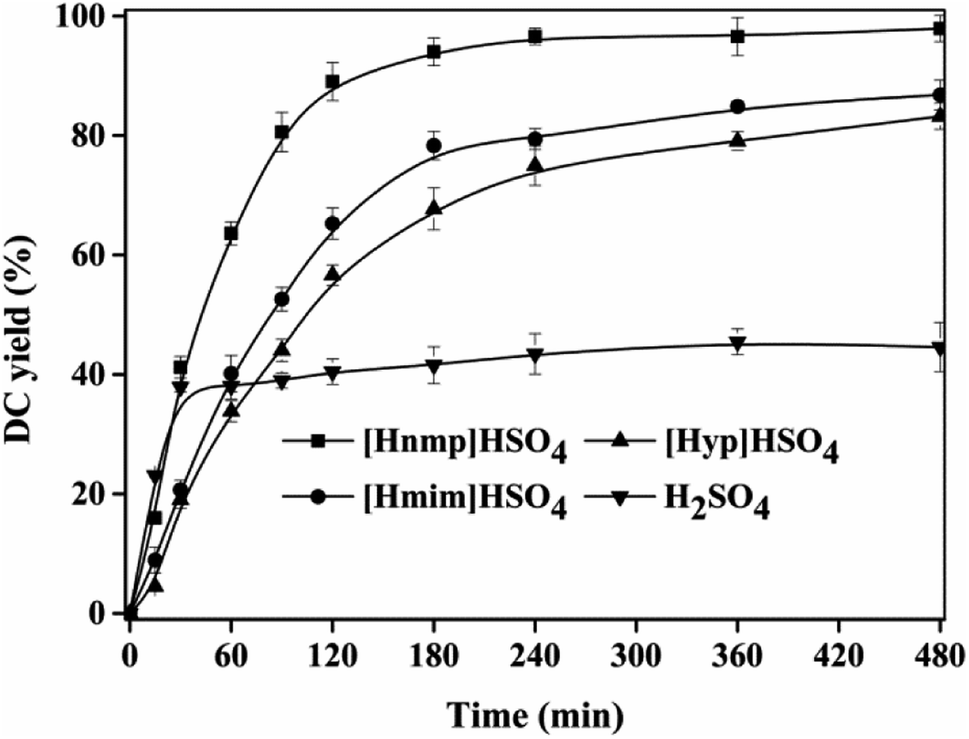
Fig. 1 showed the effect of different ILs on DC yield. Among these ILs, the maximum DC yield (93.03 ± 1.56%) was achieved using [Hnmp]HSO4 at 180 min, which was higher than other ILs ([Hmim]HSO4 78.33 ± 2.35% and [Hpy]HSO4 67.71 ± 3.52%) and H2SO4 (41.56 ± 3.10%). Therefore, the catalytic activity (DC yield) of ILs and H2SO4 decreased in an order of [Hnmp]HSO4 > [Hmim]HSO4 > [Hpy]HSO4 > H2SO4. The catalytic activity (DC yield) of H2SO4 was lower than ILs, which was due to that the oxidation of CA in the presence of H2SO4 (<30 min). This phenomenon has also been found in other reactions.26,29 These indicated that, among all tested ILs, [Hnmp]HSO4 was the best efficient catalyst for the esterification of CA with dodecanol, which was due to that the catalytic activities of ILs were relative to their acidities.22,26,29 Compared with octadecyl caffeate preparation using cation-exchange resins (24 h, 95.17 ± 2.76%),20 the time of DC preparation at 85 °C using [Hnmp]HSO4 was reduced to 3 h, which showed that [Hnmp]HSO4 had good performance for DC preparation by the esterification. In our previous work, we also used some immobilized lipases, Novozym 40086, Novozym TLIM, Novozym 435, and Candida rugose lipase, to catalyze the esterification to prepare DC. However, no DC product was found. Therefore, [Hnmp]HSO4 was the best catalyst for DC preparation.
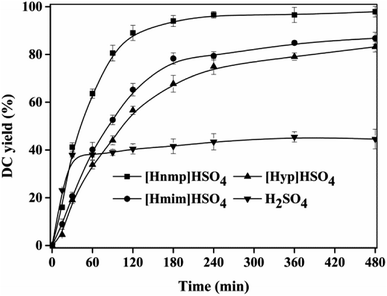 |
| | Fig. 1 Effect of different ILs ([Hnmp]HSO4, [Hpy]HSO4, [Hmim]HSO4 and H2SO4) on DC yield. Reactions were conducted with substrate ratio of 10:1 (dodecanol to CA, mo mol−1), IL dosage 10% (w/w) with different ILs at 85 °C. | |
3.2 IL characteristics
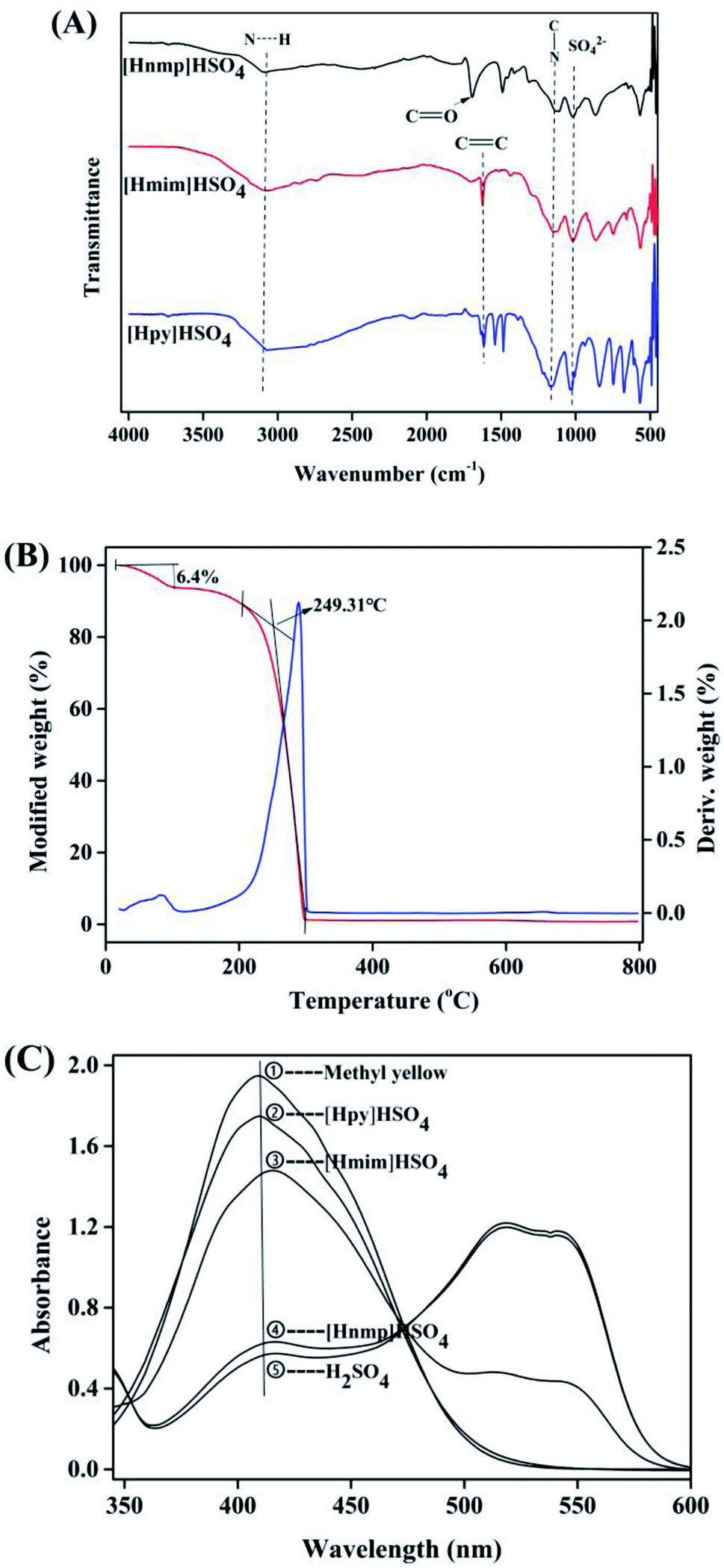
According to the FT-IR spectra of three ILs, the stretching vibration peaks of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N were found at 1037 cm−1 and 1151 cm−1, respectively (Fig. 2A). The bands at around 2900–3100 cm−1 were related to N–H stretching vibration. However, in [Hnmp]HSO4, a sharp CO stretching vibration peak was found at 1696 cm−1. The CC vibration peaks were found at 1615 cm−1 in the spectra of [Hmim]HSO4 and [Hpy]HSO4, respectively. These results showed that the three ILs were successfully prepared.29,32,33
O and C–N were found at 1037 cm−1 and 1151 cm−1, respectively (Fig. 2A). The bands at around 2900–3100 cm−1 were related to N–H stretching vibration. However, in [Hnmp]HSO4, a sharp CO stretching vibration peak was found at 1696 cm−1. The CC vibration peaks were found at 1615 cm−1 in the spectra of [Hmim]HSO4 and [Hpy]HSO4, respectively. These results showed that the three ILs were successfully prepared.29,32,33
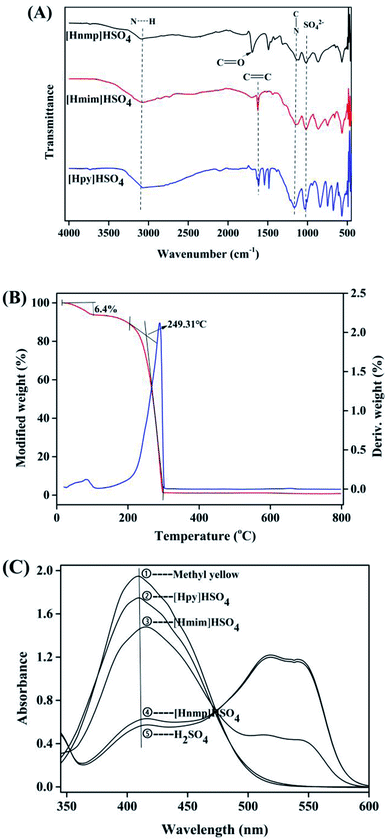 |
| | Fig. 2 Characterization of FT-IR ILs; (A) spectra of the three ILs; (B) TG curves of [Hnmp]HSO4; (C) acidity curves of the Brønsted ILs. | |
As shown in Fig. 2B, according to TGA analysis, two weight loss regions of [Hnmp]HSO4 were found at 54 °C and 249 °C, respectively. There were three stages for the thermal decomposition of [Hnmp]HSO4. The first stage was from 25 °C to 82 °C and ∼6% weight loss was found, which was mainly due to the evaporation of water. In the second stage from 82 °C to 153 °C, no significant weight loss (<1%) was found. The 3rd stage of the thermal decomposition of [Hnmp]HSO4 was from 153 °C to 296 °C, the great weight loss (93%) was found, which was due to the decomposition of [Hnmp]HSO4. After 296 °C, no significant weight loss of [Hnmp]HSO4 was found.
According to the previous reports,21,22,29 the acidities of different ILs and H2SO4 were investigated (Fig. 2C). The protonated structure of dimethyl yellow was formed by the combination of H+ from ILs and dimethyl yellow. The more protonated structures formed indicated the stronger acidity of ILs.22 Therefore, when the concentrations of different ILs and H2SO4 were same, the acidities decreased as H2SO4 > [Hnmp]HSO4 > [Hmim]HSO4 > [Hpy]HSO4. In the structure of [Hnmp]HSO4, oxygen atom of CO had high electronegativity, resulting in induction effect. Electrons were pulled to CO, and the formed hydrogen bond was easily broken and H+ was released. Which resulted in the higher acidity of [Hnmp]HSO4 than other ILs. The acidity and catalytic activity of different ILs were consistent, which indicated that the esterification reaction rate of CA and dodecanol was related to the rate of ILs release H+. This phenomenon has also been found in other esterification reactions catalyzed by ILs.21,22,29
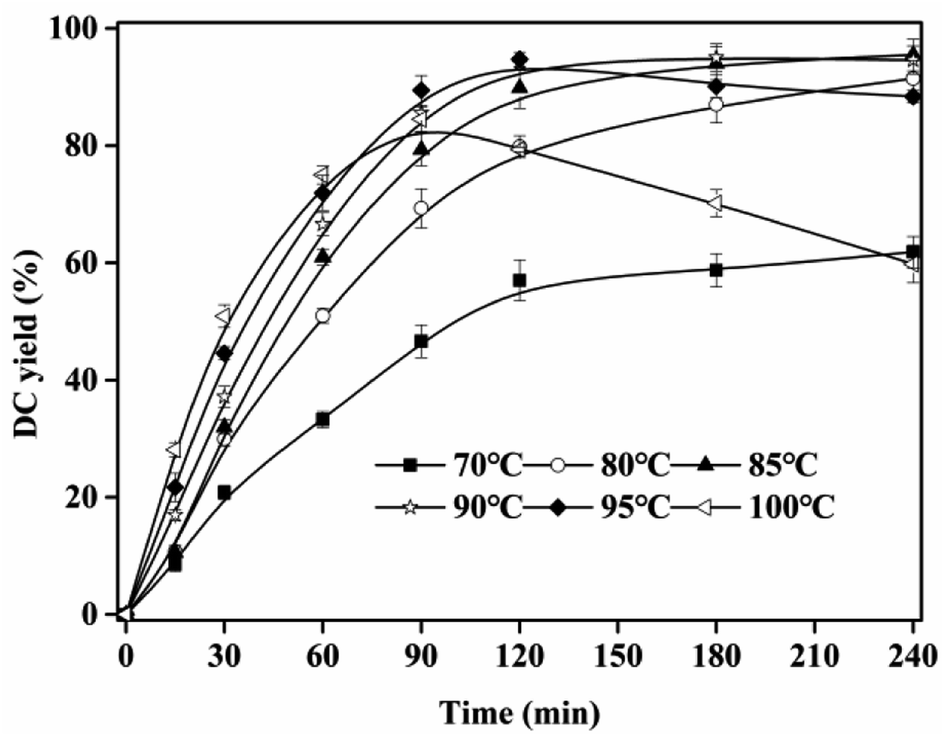
3.3 Effect of temperature
The high melting point of CA and great viscosity of IL resulted in the high viscosity of the esterification system. Suitable temperature can decrease the viscosity, and increase DC yield. As shown in Fig. 3, DC yield rapidly increased with temperature at <90 °C. At 120 min, DC yield increased from 54.01 ± 2.48% (70 °C) to 93.03 ± 1.01% (90 °C). This result was ascribed to the fact that, more substrate molecules were activated, and the viscosity of the reaction system decreased at high temperature, which resulted in the high reaction rate. When temperature was higher than 90 °C, DC yield decreased, and the color of reaction system was enhanced, which was due to the oxidization of CA and DC at high temperature (>90 °C). This phenomenon can also be found in another esterification of gallic acid and n-propanol catalyzed by [Hnmp]HSO4.29
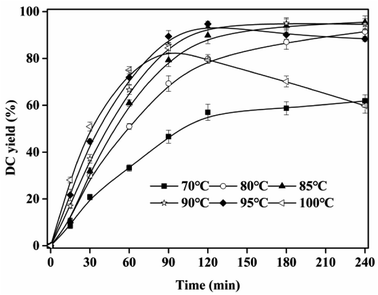 |
| | Fig. 3 Effect of reaction temperature on DC yield. Reactions were conducted with IL dosage of 10% and substrate ratio of 10:1. | |
3.4 Effect of substrate ratio
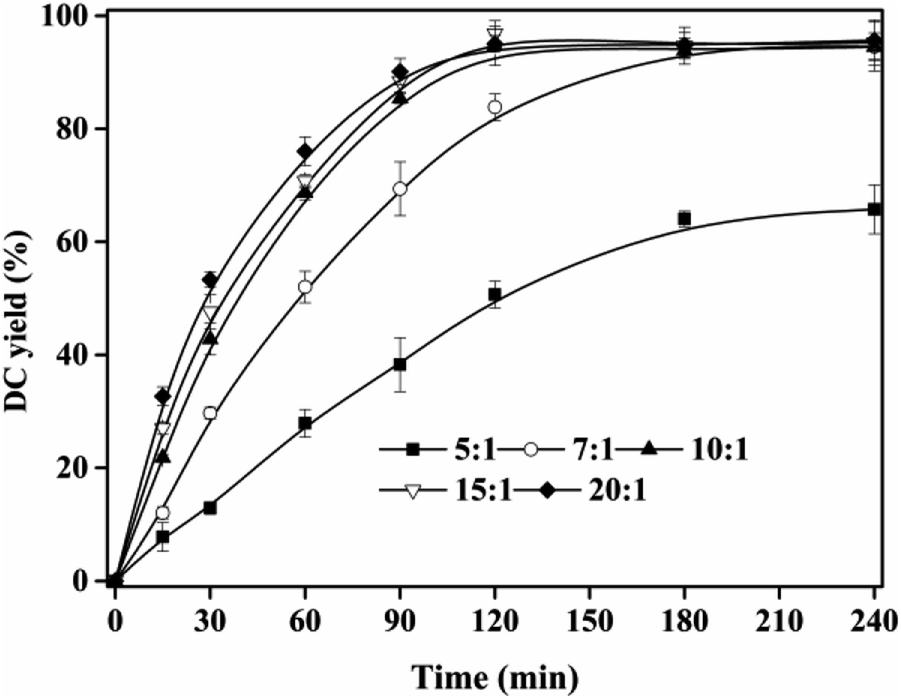
The esterification of CA with dodecanol was a reversible reaction. High DC yield could be obtained if the reverse reaction was inhibited. Therefore, in this reaction system, excessive dodecanol was used, which moved the reaction equilibrium to the formation of DC.
The effect of substrate ratio on DC yield was shown in Fig. 4. When substrate ratio (dodecanol to CA) increased from 5:1 to 10:1, DC yield rapidly increased from 50.67 ± 2.37% to 94.71 ± 3.50% at 120 min, and the equilibrium time was shortened from >240 min to 120 min. In theory, during the esterification, 1 mol of CA is required to react with 1 mol dodecanol to form 1 mol DC and 1 mol water. However, in this work, excessive dodecanol was used to move the esterification towards the formation of DC and avoid the hydrolysis.34,35 With further increase of substrate ratio up to more than 10:1, DC yields were maintained at ∼95%. Compared with other enzymatic or chemical synthesis of CA alkyl esters,14,15,25,26 the equilibrium time for DC preparation was shorter and the substrate ratio was more economic. Therefore, substrate ratio of 10:1 was used in the next experiments.
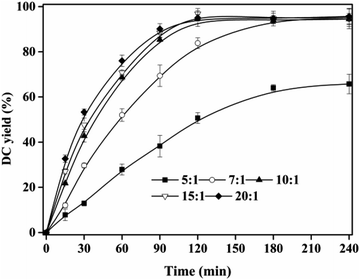 |
| | Fig. 4 Effect of substrate ratio on DC yield. Reactions were conducted with IL dosage of 10% at 90 °C. | |
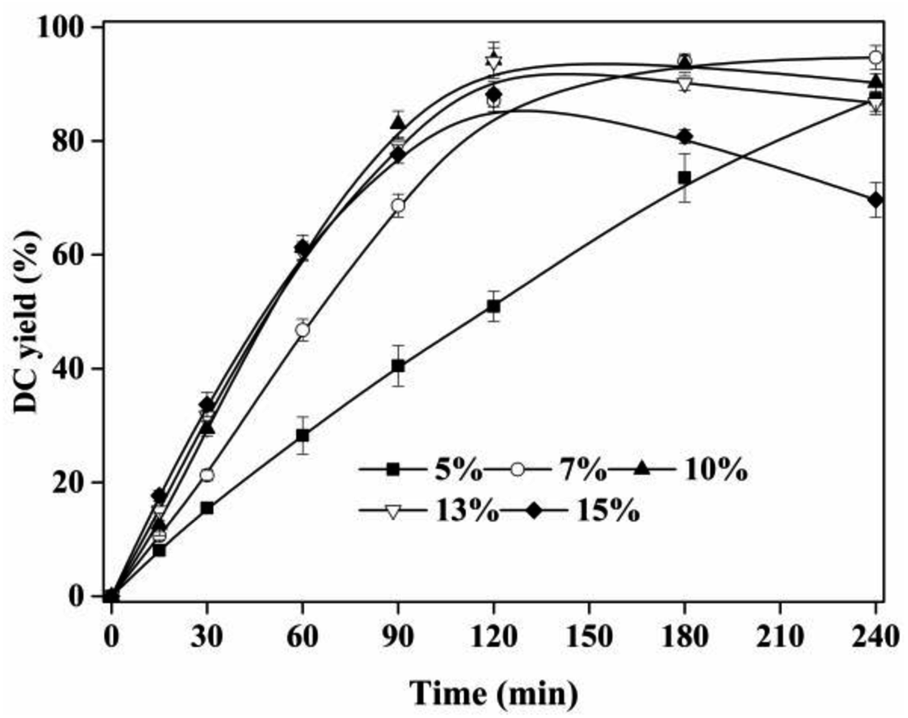
3.5 Effect of IL dosage
The esterification of CA and dodecanol was a heterogeneous system. The amount of active site of IL catalyst in the reaction system had a significant effect of DC yield. As shown in Fig. 5, with the increase of IL dosage from 3% to 10%, DC yield increased from 35.01 ± 3.32% to 96.22 ± 3.18% at 120 min. However, with further increase of IL dosage from 10% to 15%, DC yields decreased, which was due to the increase of DC oxidation with the increase of acidity of reaction system. These indicated that 10% IL dosage was enough for the esterification reaction of CA and dodecanol. Similar results that, the catalyst efficiency per unit of ILs decreased when excess ILs was presented in reaction system, can also be found in other IL-catalyzed reactions.21,36 Compared with propyl caffeate preparation using [BSO3HMIM]TS (40% IL dosage),26 the catalytic activity of [Hnmp]HSO4 (10% IL dosage) to prepare DC was more efficient. Therefore, 10% IL dosage was used in the next experiments.
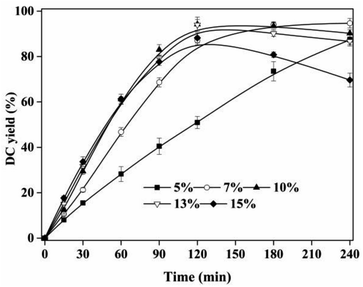 |
| | Fig. 5 Effect of IL dosage on DC yield. Reactions were conducted with substrate ratio of 10:1 at 90 °C. | |
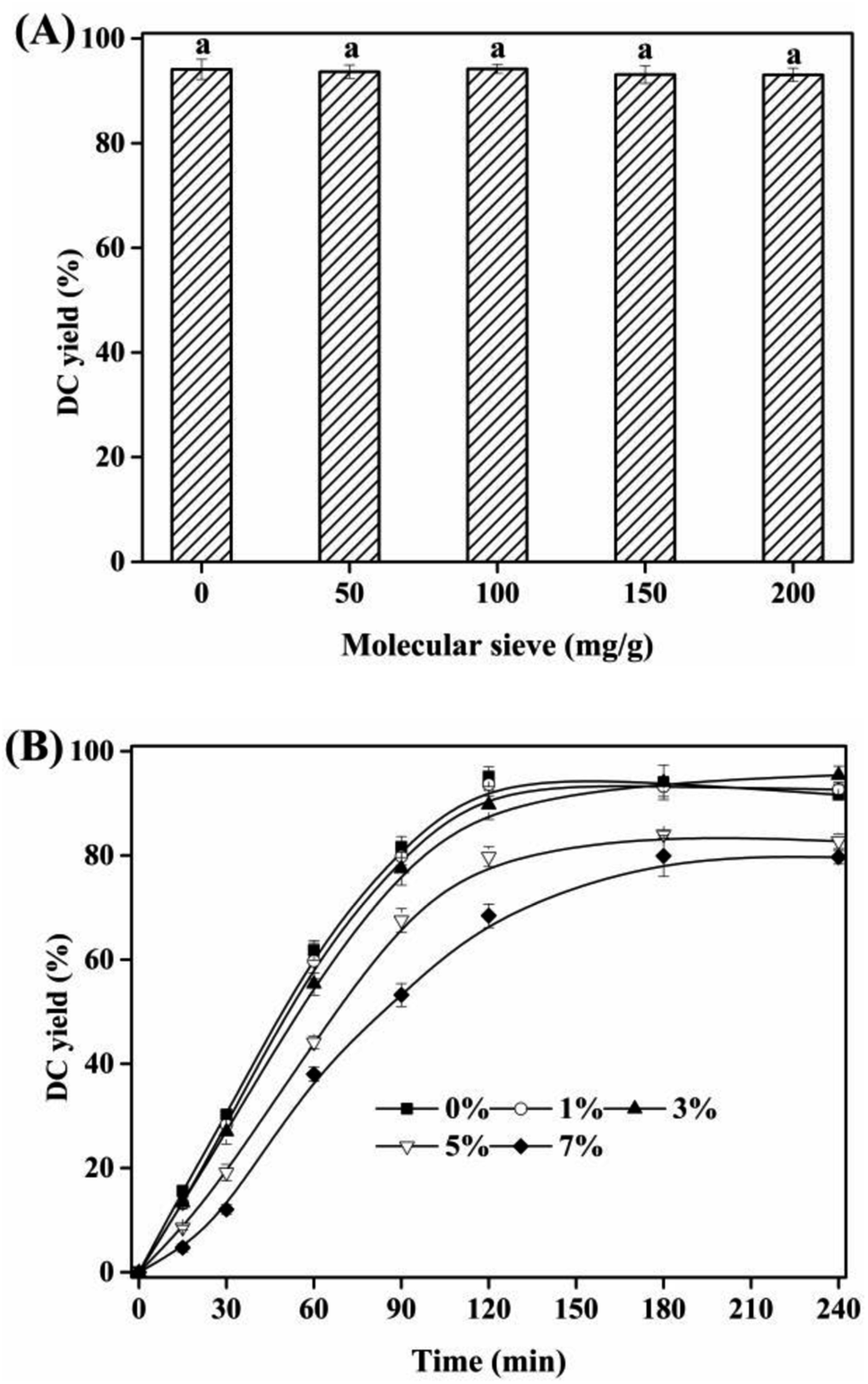
3.6 Effect of water content
Water was the by-product of the esterification of CA with dodecanol. The effects of molecular sieve added and initial water content on the esterification were shown in Fig. 6. With the increase of molecular sieve load added from 0 to 200 mg g−1, no significant effect of molecular sieve load on DC yield was found (Fig. 6A), which was due to that, (i) the little water formed by the esterification, (ii) reaction substrate was volatilized out of the reaction system at 90 °C, (iii) [Hnmp]HSO4 was a water-tolerable acidic IL.26 Considering the water content in reaction substrates (dodecanol) and catalyst ([Hnmp]HSO4), the effects of initial water content on DC yield and reaction rate were investigated (Fig. 6B). With the increase of initial water content (<3%), minor influence on DC yield was found. However, the increase of initial water content (>3%), DC yield decreased rapidly from 89.74 ± 2.81% to 68.49 ± 2.45%, which was due to which was due to the inhibition of esterification of CA and dodecanol in the presence of more water. Therefore, the lower initial water content (<3%) was minor influence on the esterification of CA with dodecanol.
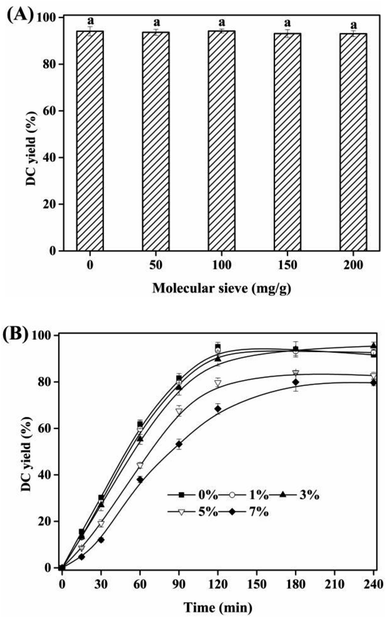 |
| | Fig. 6 (A) The comparison of DC yields of 2 h at different molecular sieve load; (B) effect of initial water content on DC yields. The reactions were conducted with substrate ratio of 10:1 and IL dosage of 10% at 90 °C. Different letters indicated significant differences (p < 0.05). | |
3.7 RSM model fitting
RSM was employed to evaluate the interactions of variables and predict optimal DC yield. The design of experiments and results were shown in Table 1. And the quadratic polynomial equation was as follows:| | |
DC yield (%) = 95.80 + 8.49X1 + 6.01X2 + 12.86X3 + 13.16X4 − 2.08X1X2 − 2.43X1X3 − 5.68X1X4 + 1.57X2X3 + 2.98X2X4 − 1.80X3X4 − 3.20X12 − 7.16X22 − 11.88X32 − 12.07X42
| (2) |
The regression coefficients and significance test of the model were verified by the standard analysis of variance (ANOVA) (Table 2). The quadratic polynomial regression model was extremely significant (p < 0.0001). The “Pred R2” (0.9892) was in reasonable accordance with the “Adj R2” (0.9959), and the “Lack of Fit” was not significant (F = 3.21, p > 0.05), which indicated that the model was reliable in predicting the response. The value of R2 was 0.9980, which was larger than the minimal R2 of 0.80 for adequate explanation on the variability in the experiments.34 According to these results, the prediction results of the quadratic model for DC yield were satisfactory.
Table 2 Variance analysis of the regression model
| Source |
Sum of squares |
df |
Mean square |
F value |
p-value prob > F |
|
| Model |
7238.41 |
14 |
517.03 |
491.03 |
<0.0001 |
Significant |
| X1-temperature |
575.56 |
1 |
575.56 |
546.61 |
<0.0001 |
|
| X2-time |
340.66 |
1 |
340.66 |
323.53 |
<0.0001 |
|
| X3-substrate ratio |
1357.43 |
1 |
1357.43 |
1289.17 |
<0.0001 |
|
| X4-IL dosage |
1755.57 |
1 |
1755.57 |
1667.28 |
<0.0001 |
|
| X1X2 |
17.31 |
1 |
17.31 |
16.44 |
0.0012 |
|
| X1X3 |
23.55 |
1 |
23.55 |
22.36 |
0.0003 |
|
| X1X4 |
128.88 |
1 |
128.88 |
122.40 |
<0.0001 |
|
| X2X3 |
9.84 |
1 |
9.84 |
9.35 |
0.0085 |
|
| X2X4 |
35.55 |
1 |
35.55 |
33.76 |
<0.0001 |
|
| X3X4 |
13.00 |
1 |
13.00 |
12.34 |
0.0034 |
|
| X12 |
66.56 |
1 |
66.56 |
63.21 |
<0.0001 |
|
| X22 |
332.96 |
1 |
332.96 |
316.22 |
<0.0001 |
|
| X32 |
915.79 |
1 |
915.79 |
869.73 |
<0.0001 |
|
| X42 |
944.33 |
1 |
944.33 |
896.84 |
<0.0001 |
|
| Residual |
14.74 |
14 |
1.05 |
|
|
|
| Lack of fit |
13.11 |
10 |
1.31 |
3.21 |
0.1364 |
Not significant |
| Pure error |
1.64 |
4 |
0.41 |
|
|
|
| Cor total |
7253.15 |
28 |
|
|
|
|
| R2 |
0.9980 |
|
|
|
|
|
The p-values of four independent factors indicated that a significant effect on DC yield (p < 0.0001) was found, and that the effect order of four independent factors was IL dosage > substrate ratio > temperature > reaction time. Moreover, quadratic terms were also great significant (p < 0.0001) for the esterification of CA with dodecanol (Table 2).
3.8 RSM optimization and model verification
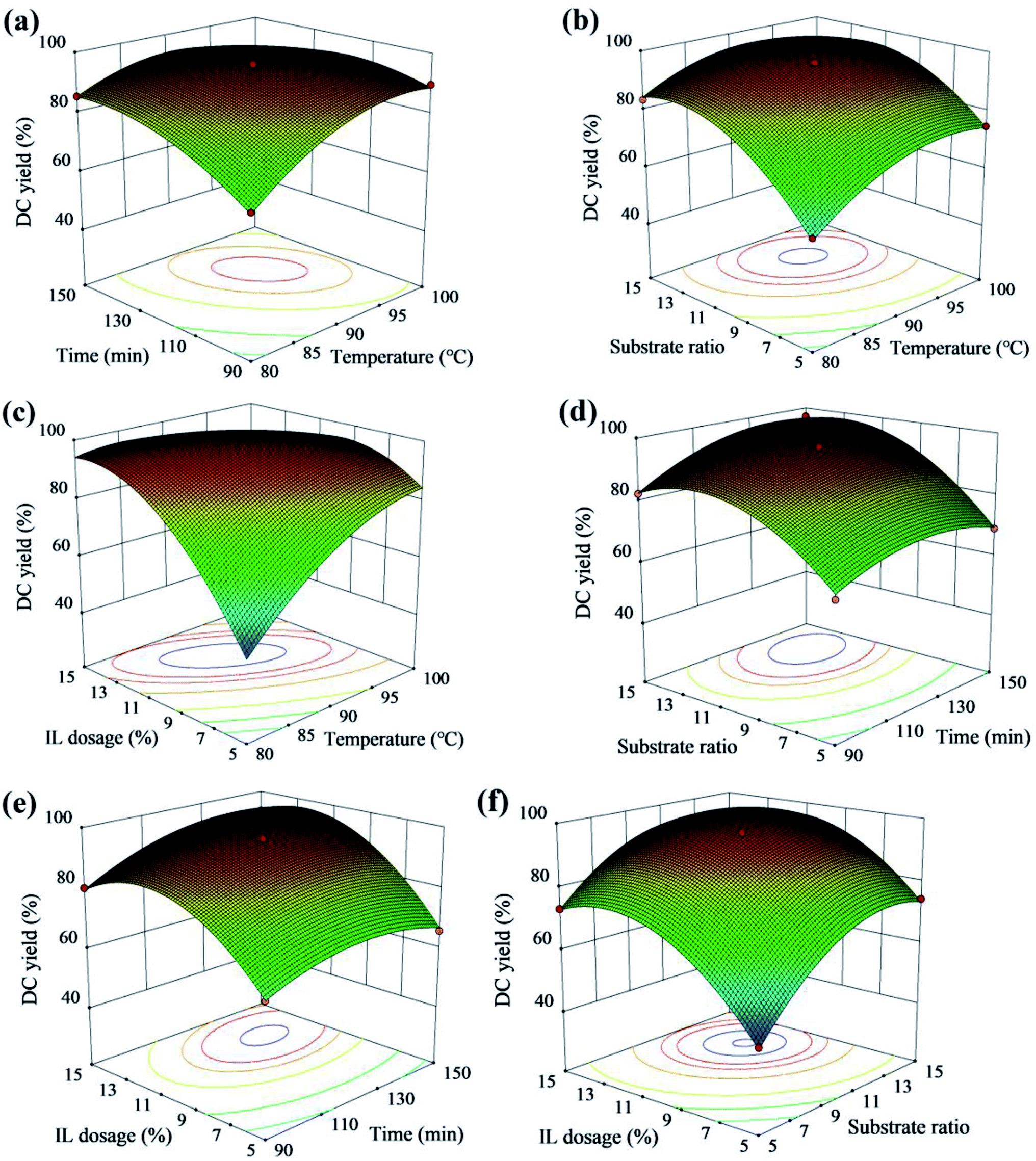
Fig. 7 showed the relationship between the interaction effect of variables and DC yield. The increase of temperature with substrate ratio from 9:1 to 12:1 and reaction time from 110 min to 135 min resulted in the increase of DC yield (Fig. 7a and b). As shown in Fig. 7c, the optimal DC yield appeared at 90 °C to 100 °C and IL dosage from 10% to 12%, and the effect of IL dosage on DC yield was greater than temperature. The optimal DC yield appeared at 110 min to 135 min and substrate from 11:1 to 14:1 with IL dosage from 12% to 14% (Fig. 7d and e). According to the variance analysis of the regression model, the effects of IL dosage and substrate ratio on DC yield were greater than reaction time (Table 2). The interaction of IL dosage and substrate ratio on DC yield was also significant (Fig. 7f).
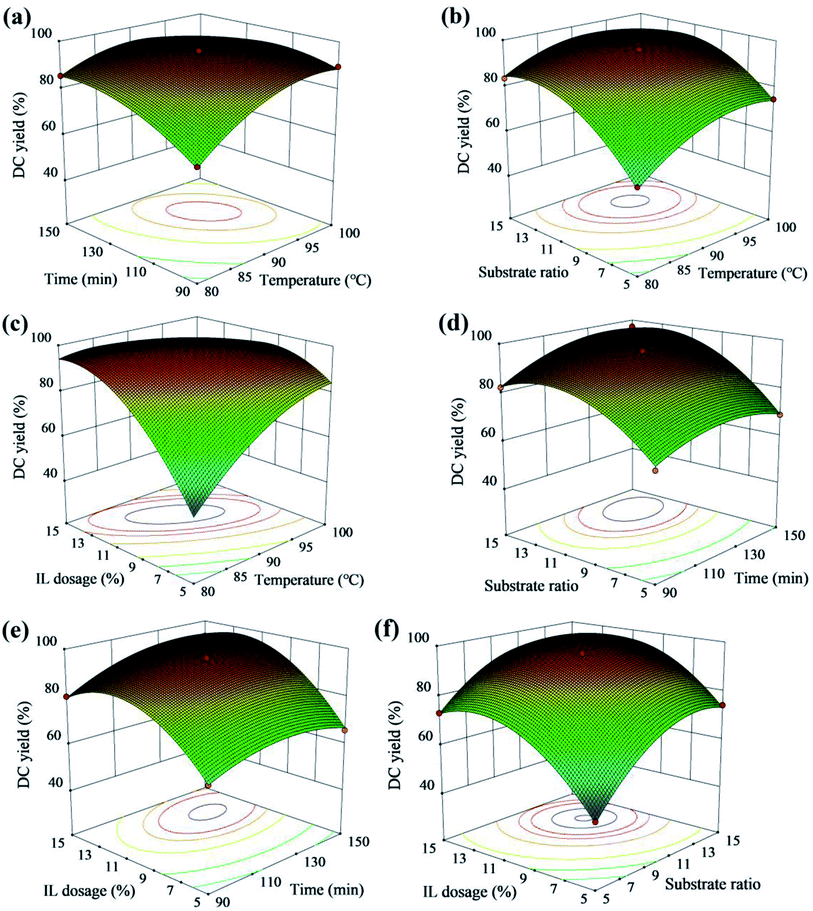 |
| | Fig. 7 3D surface of the combined effects of (a) reaction time and temperature; (b) temperature and substrate ratio; (c) temperature and IL dosage; (d) reaction time and substrate ratio; (e) reaction time and ILdosage; (f) substrate ratio and ILdosage on DC yield with the other variables held at a constant level. | |
RSM experiments were performed to find the optimum esterification conditions to maximize DC yield. A quadratic programming model was used to optimize reaction conditions, and the optimized reaction conditions were substrate ratio of 10.2:1, IL dosage of 9.8% at 87 °C for 118 min, respectively. Under the optimal variables, the maximum DC yield (94.67 ± 1.32%) was achieved, and it was well accorded with the predicted DC yield (93.75%), which indicated the validation of the model. Compared with CA alkyl esters preparation in previous reports (more than 4 h),14,15,20,24,27, [Hnmp]HSO4 as a catalyst to prepare DC was more efficient (118 min).
Under the optimal variables, other long chain alkyl caffeates, such as, tetradecanol caffeate, hexadecyl caffeate and octadecanol ceffeate, were also synthesized according to the method of DC synthesis, and the yields of these esters were more than 80% (90.16 ± 1.71%, 88.69 ± 0.23% and 80.01 ± 1.23%). These indicated the synthesis method used in this work was also applicable to other long chain alkyl caffeate preparation.
3.9 Kinetic study
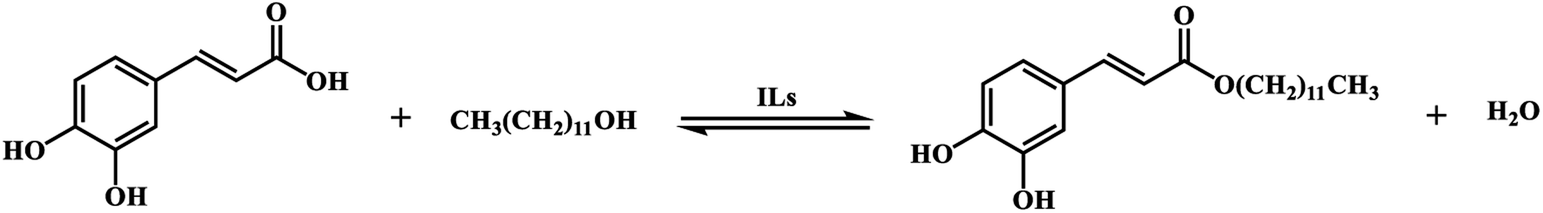
According to the relative reports,35,36 the esterification between CA with dodecanol was a reversible reaction in the presence of IL catalyst [Hnmp]HSO4 (Scheme 2). And the esterification reaction rate equation can be expressed as eqn (3).| |
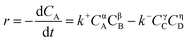 | (3) |
where CA, CB, CC, and CD represented the instant concentration of CA, dodecanol, DC, and water respectively. Where α, β, γ, and η were the reaction orders for A, B, C, and D, respectively. And k+ and k− were the forward and reverse reaction rate constants, respectively.
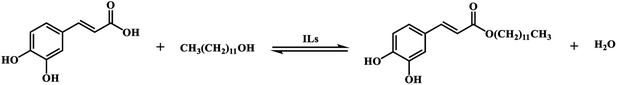 |
| | Scheme 2 Esterification reaction of CA with dodecanol. | |
In this work, excessive dodecanol was used to move the reaction equilibrium toward DC formation. Therefore, the concentration dodecanol was considered as a constant, and the reverse hydrolysis was neglected.37,38 The esterification of CA with dodecanol was considered as a pseudo 1st order.39 Eqn (3) was as follows:
where
k and
X are the reaction rate constant (mol (L min)
−1) and CA conversion (%), respectively.
The plots of ln((1 − X)CA0) vs. t at different temperatures (from 70 °C to 90 °C) were obtained. And k0 and Ea could also be obtained from eqn (5).
| |
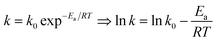 | (5) |
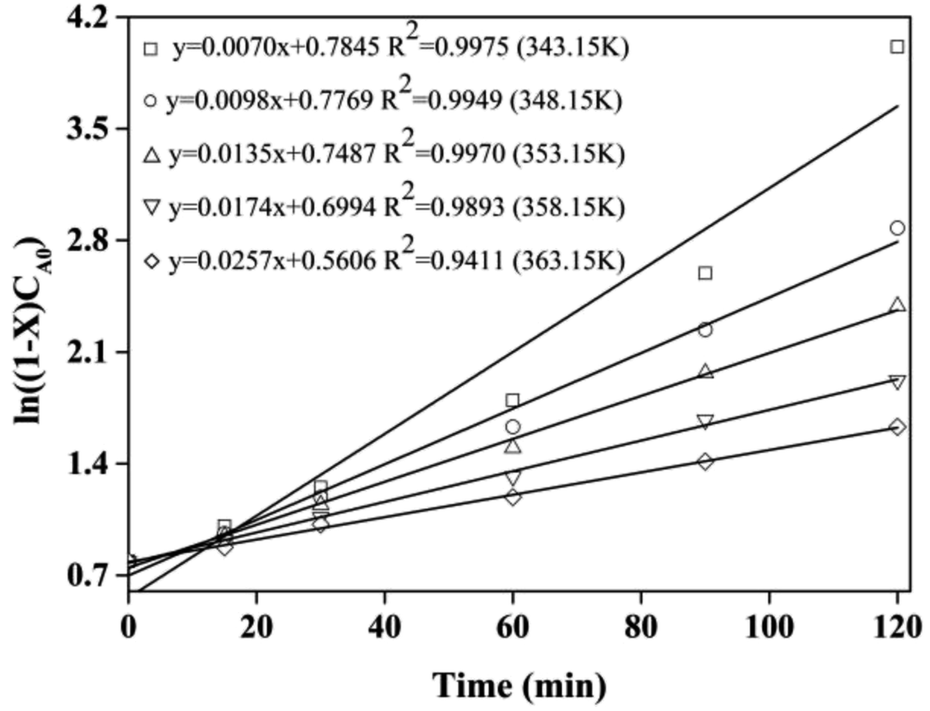
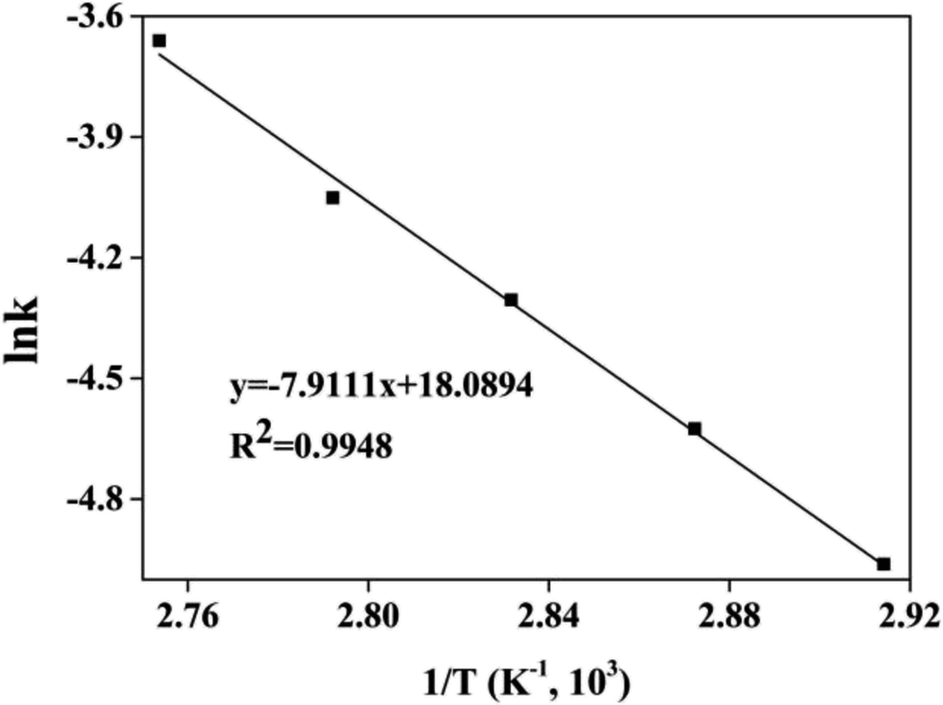
The kinetic and thermodynamic of the esterification of CA and dodecanol were investigated under the optimized conditions.27,40,41 Fig. 8 showed the curves of ln((1 − X)CA0) vs. t, and the k at different temperatures (from 70 °C to 90 °C) were 7.0 × 10−3, 9.8 × 10−3, 1.35 × 10−2, 1.74 × 10−2, and 2.51 × 10−2 mol (L min)−1, respectively. These were because the esterification was endothermic and the forward reaction rate was enhanced with the increase of temperature. The regression relationship of lnk vs. 1/T was a linear and the regression coefficient (R2) was 0.9948 (Fig. 9), which indicated that the esterification of CA with dodecanol was a pseudo 1st order reaction. According to Fig. 9, the k0 and Ea of esterification between CA with dodecanol was calculated as 7.18 × 107 mol (L min)−1 and 65.77 kJ mol−1, respectively. The k0 (7.18 × 107 mol (L min)−1) was notably high, which indicated that the reverse reaction could be ignored in the presence of excessive dodecanol.42 The Ea of DC synthesis catalyzed by [Hnmp]HSO4 (65.77 kJ mol−1) was lower than solid acid (A-35, 90.19 kJ mol−1).20 These indicated that [Hnmp]HSO4 catalyst was the best choice for the esterification of CA to dodecanol.
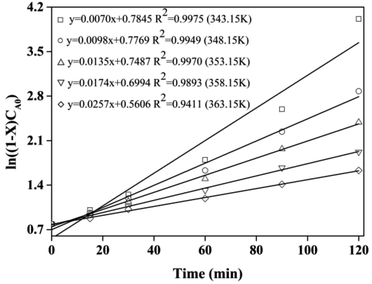 |
| | Fig. 8 The curves of ln((1 − X)CA0) versus t. | |
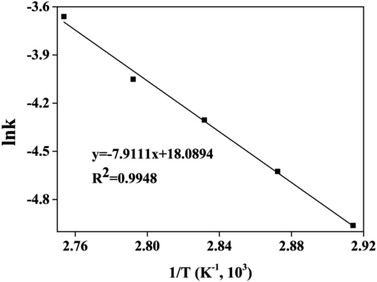 |
| | Fig. 9 The curve of lnk versus 1/T. | |
3.10 Thermodynamic study
Based on the transition state theory, Eyring equation (eqn (6)) was used to relate rate constant to temperature.40 ΔS and ΔH were calculated from the plot of ln(k/T) and 1/T (eqn (6)). And ΔG was calculated from eqn (7).| |
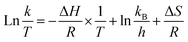 | (6) |
where kB and h are Boltzmann's constant (1.38 × 10−23 J k−1) and Plank's constant (6.626 × 10−23 J s), respectively.
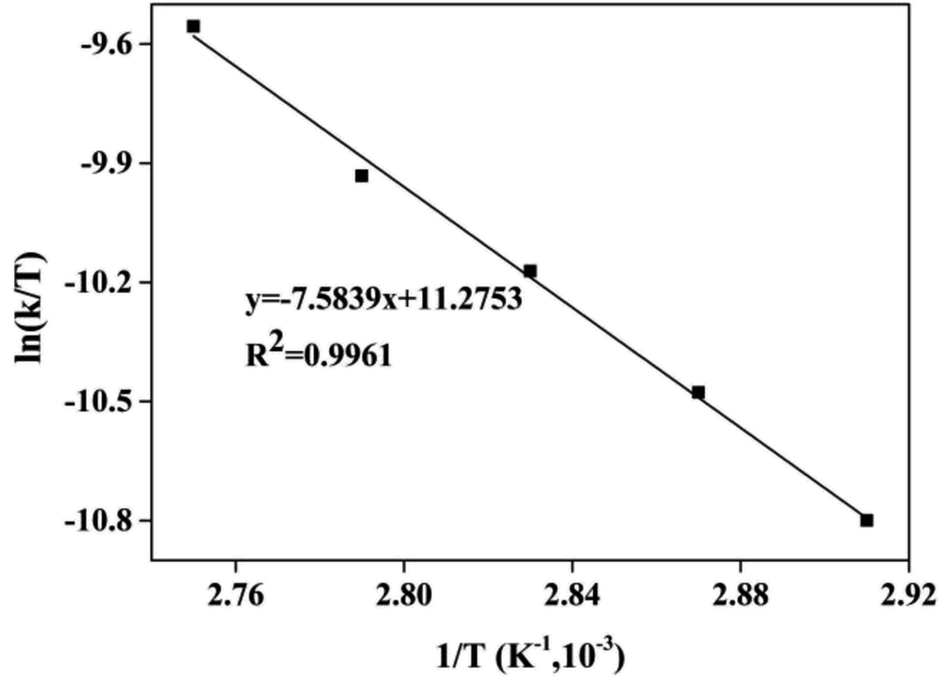
As shown in Fig. 10, the Eyring curve of ln(k/T) vs. 1/T had a good linear fit (R2 = 0.9961). The ΔH value of the esterification of CA with dodecanol from the Eyring curve was slightly lower than the Ea (65.77 kJ mol−1) (eqn (8)).
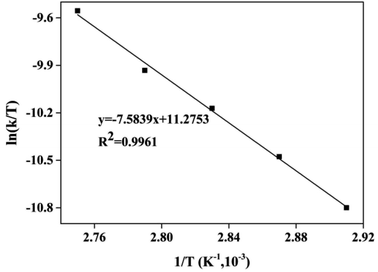 |
| | Fig. 10 The Eyring curve of ln(k/T) vs. 1/T. | |
In this work, because the value of RT (eqn (8)) was great lower than the Ea of 65.77 kJ mol−1, the esterification of CA with dodecanol was an endothermic reaction (ΔH > 0). Positive ΔH (63.10 kJ (mol K)−1) indicated that heat input was necessary for the esterification of CA with dodecanol to synthesize DC, and also indicated that DC yield increased with the temperature increasing.40,42
The negative ΔS (−103.80 J (mol K)−1) showed that the esterification of CA and dodecanol had a substantial entropy loss in the formation of IL–substrate complex, which was due to the loss of translational and rotational freedom degrees of reactant and catalytic groups. And negative ΔS (−103.80 J (mol K)−1) also indicated the reaction system was more disorder.42–44 Positive ΔG (99.78 kJ mol−1 at 363 K) revealed that the esterification of CA with dodecanol was a nonspontaneous process, and also showed that heat input was necessary for DC formation.
3.11 Esterification mechanism
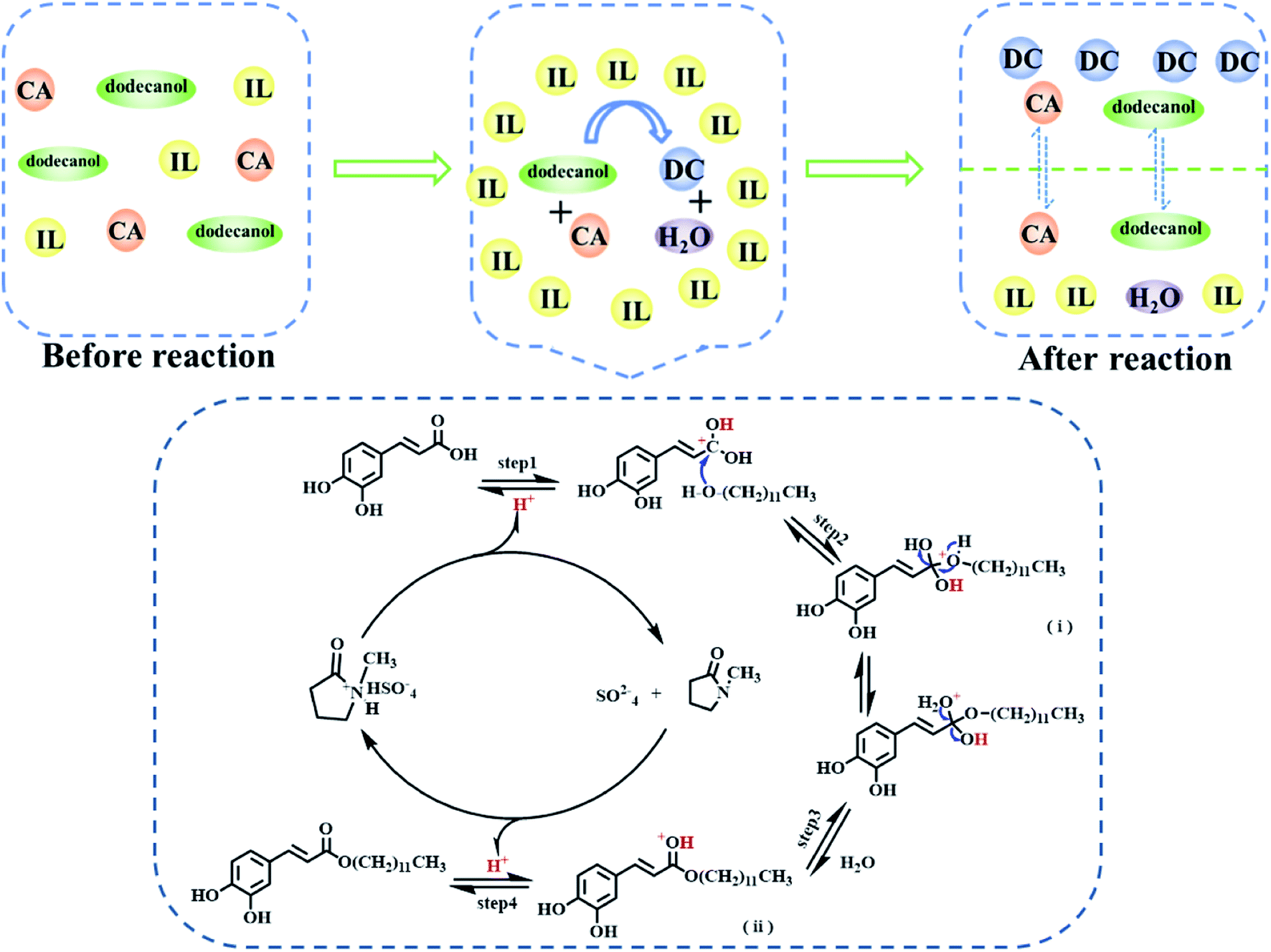
According to the section 3.2 and the previous reports,29,43,45–48 the esterification mechanism of [Hnmp]HSO4 as a catalyst for DC preparation was proposed in this work (Scheme 3). Firstly, the application of [Hnmp]HSO4 can improve the dissolution of CA in dodecanol to form homogeneous phase before reaction, which made the efficient contact of substrates and [Hnmp]HSO4. H+ was released from [Hnmp]HSO4, and then H+ was connected with a carbon atom of the carboxyl group of CA, which can improve the electrophilicity of the carbonyl carbon (step 1) and facilitate the attack of oxygen atom in dodecanol hydroxyl group to form an unstable active tetrahedral intermediate (i) (step 2). The unstable active tetrahedral intermediate (i) was rearranged, and one water and another intermediate (ii) were formed (step 3). Finally, one H+ was released from the intermediate (iii) to produce DC (step 4). Under this reaction system, product DC and by-product water were produced by the esterification reaction of CA and dodecanol. Due to different interaction, water and [Hnmp]HSO4 were maintained in one phase and DC was automatically transferred to another phase, which promoted the reaction toward DC formation. Therefore, high DC yield was achieved in this reaction system.
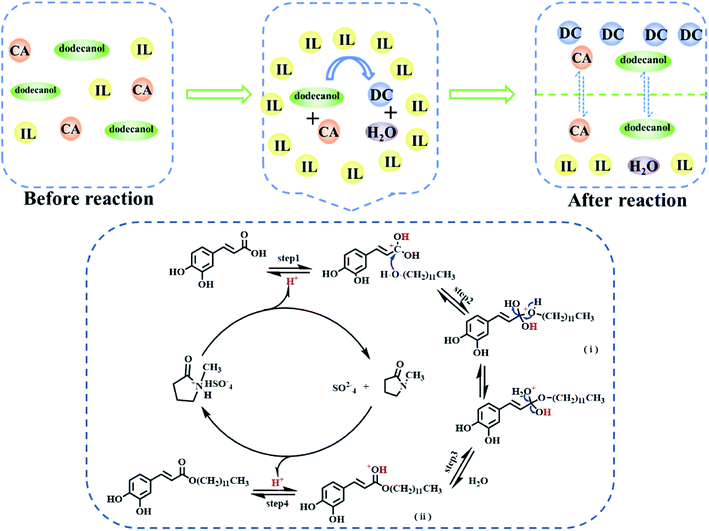 |
| | Scheme 3 The esterification reaction mechanism of CA and dodecanol. | |
3.12 Antioxidant activity of DC
The antioxidant activity of DC was evaluated at 517 nm by DPPH radical scavenging activity, which was compared with TBHQ, BHT, AP, and CA (Table 3). The IC50 of these antioxidants were calculated by the linear regressions of plots as follows: AP (37.48 ± 0.99e) > BHT (29.93 ± 0.37d) > DC (23.41 ± 0.10c) > TBHQ (20.43 ± 0.58b) > CA (15.18 ± 0.12a). These results indicated that the DPPH radical scavenging activity of DC was lower that of CA and TBHQ, which were higher than BHT and AP. The number and position of free phenolic hydroxyl groups in these antioxidants was relative to their DPPH radical scavenging activity,9,49 and the great hindrance of n-dodecyl in DC decreased the DPPH radical scavenging activity of DC, which was lower than of CA. These also indicated that DC can be used as a potential antioxidant in food and cosmetics.
Table 3 Antioxidant activity of DC and well known antioxidantsa
| Antioxidant |
IC50 (μg mL−1) |
| Different letters meant significantly different using ANOVA followed by Duncan's. |
| TBHQ |
20.43 ± 0.58b |
| BHT |
29.93 ± 0.37d |
| AP |
37.48 ± 0.99e |
| CA |
15.18 ± 0.12a |
| DC |
23.41 ± 0.10c |
4 Conclusions
In this work, a low cost and easy synthesis [Hnmp]HSO4 showed the best catalytic activity for the esterification of CA with dodecanol to prepare DC. Esterification conditions were optimized by RSM as follows: temperature of 87 °C, substrate ratio of 10.2:1, IL dosage of 9.8% and reaction time of 118 min. The maximum DC yield of 94.67 ± 1.32% was achieved under these conditions. The k0, Ea, ΔH, ΔS, and ΔG were 7.18 × 107 mol (L min)−1, 65.77 kJ mol−1, 63.10 kJ (mol K)−1, −103.80 J (mol K)−1, and 99.78 kJ mol−1 at 363 K, respectively. DC prepared in this work showed a good DPPH radical scavenging activity, which indicated that DC can be used as a potential antioxidant in food and cosmetics.
Author contributions
Xuejing Liu: methodology, date curation, writing – original draft, writing – review & editing. Xiaowei Chen and Hao Zhang: characterization, writing – review & editing. Shangde Sun: methodology, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge financial support from the basic research project of the key scientific research projects of colleges and universities in Henan Province (21zx010) and top young talents in China's grain industry (LQ2020101).
References
- A. D. Meinhart, F. M. Damin, L. Caldeirao, M. de Jesus, L. C. da Silva, L. D. Constant, J. Teixeira, R. Wagner and H. T. Godoy, Food Chem., 2019, 286, 51–63 CrossRef CAS PubMed.
- M. Y. Ali, A. A. I. Sina, S. S. Khandker, L. Neesa, E. M. Tanvir, A. Kabir, M. I. Khalil and S. H. Gan, Foods, 2020, 10, 45 CrossRef PubMed.
- H. Punia, J. Tokas, A. Malik, Satpal and S. Sangwan, Cereal Res. Commun., 2021, 49, 343–353 CrossRef CAS.
- R. W. Jiang, K. M. Lau, P. M. Hon, T. C. W. Mak, K. S. Woo and K. P. Fung, Curr. Med. Chem., 2005, 12, 237–246 CrossRef CAS PubMed.
- W. Xu, Q. Luo, X. Wen, M. Xiao and Q. J. Mei, Trop. J. Pharm. Res., 2020, 19, 1227–1232 CrossRef CAS.
- F. A. Khan, A. Maalik and G. Murtaza, J. Food Drug Anal., 2016, 695–702 CrossRef CAS PubMed.
- China IECIC 2015, https://cosmetic.chemlinked.com/translation/inventory-existing-cosmetic-ingredients-china-iecic2015.
- Global and China caffeic acid market research and development trend forecast report.
- Q. Q. Chen, H. Pasdar and X. C. Weng, Grasas Aceites, 2020, 71, e352 CrossRef CAS.
- Y. Huang, Z. W. Jiang, X. Y. Liao, J. P. Hou and X. C. Weng, Czench J. Food Sci., 2014, 32, 348–353 CrossRef.
- M. Costa, S. Losa Da -Barreiro, F. Paiva-Martins, C. Bravo-Díaz and L. S. Romsted, Food Chem., 2015, 175, 233–242 CrossRef CAS PubMed.
- B. Yesiltas, P. J. Garcia-Moreno, A. D. M. Sorensen, S. Anankanbil, Z. Guo and C. Jacobsen, J. Agric. Food Chem., 2018, 66, 12512–12520 CrossRef CAS PubMed.
- O. Laguna, E. Durand, B. Barea, S. Dauguet, F. Fine, P. Villeneuve and J. Lecomte, J. Agric. Food Chem., 2020, 68, 9308–9318 CrossRef CAS PubMed.
- N. Pang, S. S. Gu, J. Wang, H. S. Cui, F. Q. Wang, X. Liu, X. Y. Zhao and F. A. Wu, Bioresour. Technol., 2013, 139, 337–342 CrossRef CAS PubMed.
- H. C. Chen, Y. K. Twu, C. M. J. Chang, Y. C. Liu and C. J. Shieh, Ind. Crops Prod., 2010, 32, 522–526 CrossRef CAS.
- S. M. Fiuza, C. Gomes, L. J. Teixeira, M. T. Girao da Cruz, M. N. D. S. Cordeiro, N. Milhazes, F. Borges and M. P. M. Marques, Bioorg. Med. Chem., 2004, 12, 3581–3589 CrossRef CAS PubMed.
- S. D. Sun and L. Y. Tian, RSC Adv., 2018, 8, 37184–37192 RSC.
- P. L. Xiao, A. J. Zhang, L. Y. Zheng and Y. Q. Song, Ind. Eng. Chem. Res., 2014, 53, 11638–11645 CrossRef CAS.
- L. Y. Han, W. Liu, H. L. Liu and Y. L. Bi, J. Henan Univ. Technol., Nat. Sci. Ed., 2014, 35, 40–44 CAS.
- X. Y. Wang, S. D. Sun and X. B. Hou, ACS Omega, 2020, 5, 11131–11137 CrossRef CAS PubMed.
- J. H. Sun, H. S. Li, H. R. Song, Q. Wu, Y. Zhao and Q. Z. Jiao, RSC Adv., 2015, 5, 87200–87205 RSC.
- J. Lunagariya, A. Dhar and R. L. Vekariya, RSC Adv., 2017, 7, 5412–5420 RSC.
- H. B. Zhang, F. Xu, X. H. Zhou, G. Y. Zhang and C. X. Wang, Green Chem., 2007, 9, 1208–1211 RSC.
- J. Wang, S. S. Wang, Z. J. Li, S. S. Gu, X. Y. Wu and F. Wu, J. Mol. Catal. B: Enzym., 2015, 111, 21–28 CrossRef CAS.
- J. Wang, S. S. Gu, H. S. Cui, L. Q. Yang and X. Y. Wu, Bioresour. Technol., 2013, 149, 367–374 CrossRef CAS PubMed.
- W. Liu and L. Y. Han, J. Oleo Sci., 2015, 64, 1297–1305 CrossRef CAS PubMed.
- B. Y. Han, W. D. Zhang, F. Yin, S. Q. Liu, X. L. Zhao, J. Liu, C. M. Wang and H. Yang, R. Soc. Open Sci., 2018, 5, 180672 CrossRef PubMed.
- B. H. Huang, Y. F. Wang, K. Mang, Y. X. Fang and B. L. Zhou, Chin. J. Catal., 2007, 28, 743–748 CAS.
- C. Zhang, X. Y. Pan, M. J. Yu, L. Jin and J. Wu, Chem. Eng. J., 2012, 209, 464–468 CrossRef CAS.
- D. J. Tao, Z. M. Li, Z. Cheng, H. Na and X. S. Chen, Ind. Eng. Chem. Res., 2013, 51, 16263–16269 CrossRef.
- B. Jayaprakasam, M. Vanisree, Y. J. Zhang, D. L. Dewitt and M. G. Muraleedharan, J. Agric. Food Chem., 2006, 54, 5375–5381 CrossRef CAS PubMed.
- A. Dhar, N. Siva Kumar, M. Khimani, A. S. Al-Fatesh, A. A. Ibrahim, A. H. Fakeeha, H. Patel and R. L. Vekariya, RSC Adv., 2020, 10, 15282–15292 RSC.
- D. J. Tao, X. M. Lu, J. F. Lu, K. Huang, Z. Zhou and Y. T. Wu, Chem. Eng. J., 2011, 171, 1333–1339 CrossRef CAS.
- Z. Hussain and R. Kumar, Int. J. Green Energy, 2018, 15, 629–640 CrossRef CAS.
- X. Y. Wang, X. W. Chen, S. D. Sun and R. Xu, Food Funct., 2021, 12, 9763–9772 RSC.
- D. J. Tao, J. Wu, Z. Z. Wang, Z. H. Lu, Z. Yang and X. S. Chen, RSC Adv., 2014, 4, 1–7 RSC.
- W. A. Jensen, J. Qual. Technol., 2017, 49, 186–187 CrossRef.
- A. H. M. Fauzi and N. A. S. Amin, Energy Convers. Manage., 2013, 76, 818–827 CrossRef.
- A. Naeem, I. W. Khan, M. Farooq, T. Mahmood, I. U. Din, Z. A. Ghazi and T. Saeed, Bioresour. Technol., 2021, 328, 124831 CrossRef CAS PubMed.
- Y. W. Tiong, C. L. Yap, S. Y. Gan and W. S. P. Yap, Renewable Energy, 2020, 146, 932–943 CrossRef CAS.
- W. C. Wang, F. S. Li and Y. Li, Green Process. Synth., 2019, 8, 776–785 CAS.
- P. Nautiyal, K. A. Subramanian and M. G. Dastidar, Fuel, 2014, 135, 228–234 CrossRef CAS.
- I. Tankov and R. Yankova, J. Mol. Liq., 2019, 277, 241–253 CrossRef CAS.
- K. R. Spacino, D. Borsato, G. M. Buosi and L. T. Chendynski, Fuel Process. Technol., 2015, 137, 366–370 CrossRef CAS.
- D. Y. Jiang, L. Chen, A. L. Wang and Z. C. Yan, RSC Adv., 2014, 4, 54427–54433 RSC.
- S. H. Zhao, X. J. Wang and L. W. Zhang, RSC Adv., 2013, 3, 11691–11696 RSC.
- J. T. Yu, Y. H. Wang, L. Q. Sun, Z. Xu, Y. D. Du, H. L. Sun, W. Li, S. Luo, C. H. Ma and S. X. Liu, ACS Omega, 2021, 6, 7896–7909 CrossRef CAS PubMed.
- L. Y. Jia, Y. X. Wang, Y. Qiao, Y. Q. Qi and X. L. Hou, RSC Adv., 2014, 4, 44253–44260 RSC.
- K. Sidoryk, A. Jaromin, N. Filipczak, P. Cmoch and M. Cybulski, Molecules, 2018, 23, 2199 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Hao Zhang and
Shangde Sun
,
Hao Zhang and
Shangde Sun