
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid detection of donor-dependent photocatalytic hydrogen evolution by NMR spectroscopy†
Tomohiro Fukushima ,
Daiki Ashizawa and
Kei Murakoshi*
,
Daiki Ashizawa and
Kei Murakoshi*
Department of Chemistry, Faculty of Science, Hokkaido University, Japan. E-mail: kei@sci.hokudai.ac.jp
First published on 28th April 2022
Abstract
Understanding molecular processes at nanoparticle surfaces is essential for designing active photocatalytic materials. Here, we utilize nuclear magnetic resonance (NMR) spectroscopy to track photocatalytic hydrogen evolution using donor molecules and water isotopologues. Pt–TiO2 catalysts were prepared and used for isotopic hydrogen evolution reactions using alcohols as electron donors. 1H NMR monitoring revealed that evolution of the H2 and HD species is accompanied by the oxidation of donor molecules. The isotopic selectivity in the hydrogen evolution reaction gives rise to formal overpotential. Based on a comparison of the rates of hydrogen evolution and donor oxidation, we propose the use of ethanol as an efficient electron donor for the hydrogen evolution reaction without re-oxidation of radical intermediates.
The conversion of light energy to chemical energy requires a combination of electronic excitation and sequential electron transfer.1–3 Efficient electronic excitation is achieved by choosing materials with suitable optical properties, while efficient electron transfer can be achieved by rational design of catalytically active surface sites.4 To achieve high catalytic performance, an understanding of the molecular processes occurring at the catalyst surface is required.
Photocatalytic hydrogen evolution is accompanied by oxidation of the electron donor. Most studies on this reaction have been conducted using in-line mass spectrometry measurements5 or oxygen-quenching methods.6 However, monitoring the whole reaction cycle using one methodology remains challenging.
Nuclear magnetic resonance (NMR) spectroscopy is a powerful tool for observing chemical reactions. This method is mainly used to confirm small-molecule conversions in organic synthesis. However, NMR spectroscopy can also be used to gain information of nanoparticle surfaces7 or even for the detection of photocatalytic reactions.8–10 Furthermore, NMR spectroscopy can be used to determine the nuclear spin states of product molecules.11 Nevertheless, there are very few reported studies on the in situ observation of photocatalytic hydrogen evolution using NMR spectroscopy.
Accordingly, in the present study, we utilized NMR spectroscopy to observe the photocatalytic hydrogen evolution reaction. We employed Pt–TiO2, which is frequently used for the photocatalytic hydrogen evolution reaction, as a model catalyst for this study. NMR spectroscopy enabled sub-micromole-scale detection of reaction products within one minute. We investigated the dependence of isotopic hydrogen evolution reactions on the donor molecules. The effects of efficient donors on the photocatalytic hydrogen evolution reaction are discussed.
Pt–TiO2 nanoparticles were prepared by a typical chemical reduction method (see ESI†). The morphologies of TiO2 and Pt–TiO2 nanoparticles were characterized by transmission electron microscopy, as shown in Fig. S1 and S2.† The average size of the Pt nanoparticles is approximately 5 nm. The average size of the TiO2 nanoparticles is within the range 20–30 nm. Typically, 5 mg of catalysts and 0.6 mL of reaction mixture were introduced to an NMR tube under Ar for observation of the photocatalytic hydrogen evolution reaction by NMR spectroscopy.
Fig. 1a shows the 1H NMR spectra of Pt–TiO2/2-propanol/D2O before and after light irradiation. Before light irradiation, three peaks are observed (Fig. 1a, black). The single peak observed at 4.81 ppm is assigned to HDO.12 1H signals from the methine and methyl groups of 2-propanol are observed at 4.01 and 1.20 ppm, respectively.
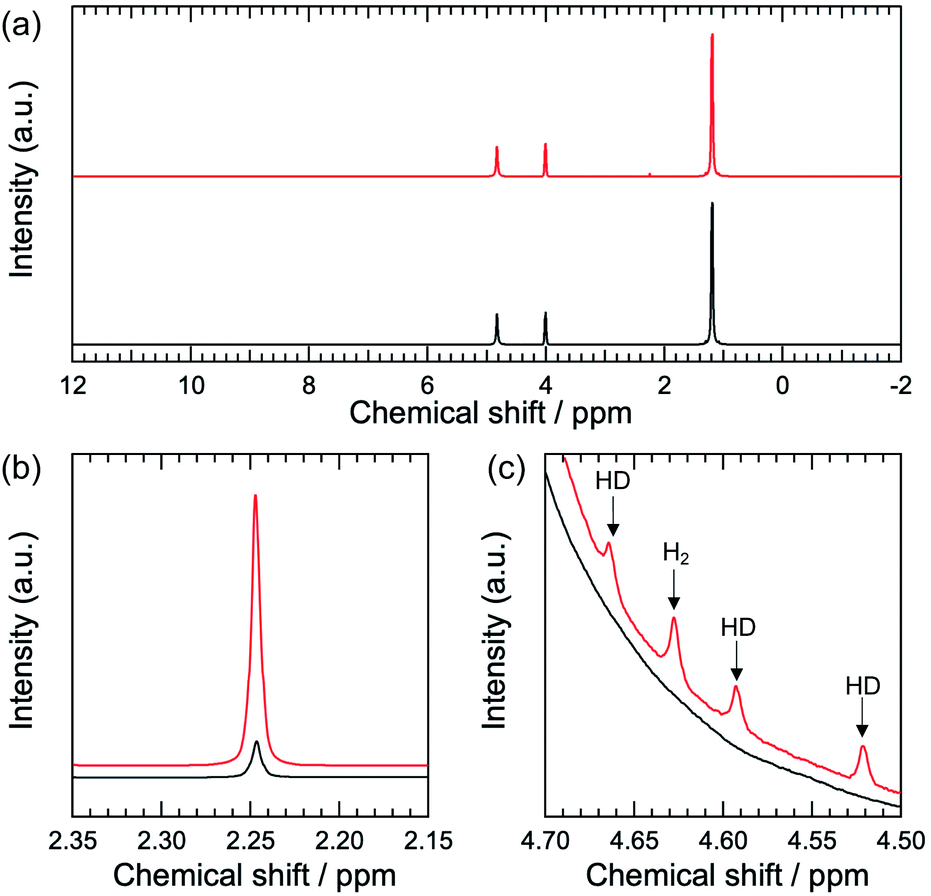 | ||
| Fig. 1 1H NMR spectra of Pt–TiO2/2-propanol/D2O. The black lines are the 1H NMR spectra before light irradiation. The red lines are the 1H NMR spectra after light irradiation for 15 min. (a) Full spectrum. (b) Enlargement of the oxidation product. (c) Enlargement of the area for the H2 and HD species. | ||
After light irradiation, additional species are observed (Fig. 1a, red). We confirmed the photocatalytic response of the Pt–TiO2 nanoparticles from ON–OFF experiments (Fig. S3†). The oxidation product is acetone resulting from two-electron and two-proton oxidation of 2-propanol. The signal at 2.25 ppm is assigned to the methyl group in the acetone (Fig. 1b).12 Hydrogen evolution is observed as a reduction reaction. Four peaks are observed between 4.5 and 4.7 ppm (Fig. 1c). The single peak at 4.63 ppm can be assigned to H2 dissolved in the solvent.13 Other peaks at 4.66, 4.59, and 4.52 ppm are assigned to the HD.14–16 The observed coupling constant for HD is 43 Hz, which is a typical value for HD.14–16 The difference in the chemical shifts of H2 and HD is due to variation of the nuclear magnetic screening constants with interatomic separation as a consequence of the zero-point energy in vibration.17,18
Importantly, NMR spectroscopy can detect H2 and HD species from the photocatalytic hydrogen evolution reaction. The observed peak splitting of the three peaks is due to the heteronuclear coupling between hydrogen and deuterium atoms.14–16 The observed chemical shift for H2 in methanol/D2O is 4.56 ppm. The observed chemical shifts of HD in methanol/D2O are 4.60, 4.53, and 4.46 ppm (Fig. S4†). These values are similar to those for 2-propanol/D2O. The observed chemical shift of H2 in ethanol/D2O is 4.61 ppm. Those for HD in ethanol/D2O are 4.65, 4.57, and 4.50 ppm (Fig. S5†). The slight shift in the H2 and HD signals is due to the difference in the shielding effect depending on the solvation environment.19–21 The coupling constant between hydrogen and deuterium in HD is 43 Hz, and it is 43 Hz in methanol/D2O and ethanol/D2O. The similarity in the coupling constants for the different solvents indicates that the chemical bonding between hydrogen and deuterium is consistent.14–16 Interestingly, the fullwidth at half maximum (FWHM) values for the H2 and HD signals are dependent on the solvent. The FWHM values for the H2 signal are 1.48, 2.24, and 3.51 Hz in methanol/D2O, ethanol/D2O, and 2-propanol/D2O, respectively. The FWHM values for the HD signal are 1.59, 2.00, and 3.32 Hz for methanol/D2O, ethanol/D2O, and 2-propanol/D2O, respectively. H2 and HD show similar FWHM values in the same solvent. However, the FWHM value is solvent-dependent. In general, a wider peak indicates lower mobility.16 Therefore, it is expected that 2-propanol induces lower mobility for the hydrogen, probably because of the rotation or diffusional freedom of hydrogen molecules. The solvation environment of hydrogen influences the molecular mobility of hydrogen species in the photocatalytic hydrogen evolution reaction.
Oxidation products of the donor molecules are observed in the NMR spectra, as shown in Fig. S4 and S5.† The number of product molecules is quantified on the basis of the hydrogen atoms in the alkyl chain groups in 2-propanol, ethanol, and methanol as reactants. For methanol, the signals for methylene glycol, 1-methoxymethanol, and methyl formate are observed as shown in Fig. S4.† For ethanol, acetaldehyde and acetic acid are observed as the products, as shown in Fig. S5.† As described above, the oxidation product of 2-propanol is limited to acetone. This is due to the unstable intermediate formed in the oxidation of 2-propanol.22 Conversely, the reaction products of methanol23–26 and ethanol27 are complicated owing to the sequential oxidation and/or hydration reactions.
We evaluated the isotopic selectivity of the hydrogen evolution reaction depending on the donor molecules. Fig. 2 shows the typical isotopic selectivity of the hydrogen evolution reaction. The amounts of H2 and HD were quantified from the NMR spectra. The HD/H2 ratios were calculated to be 4.1, 3.4, and 1.9 for 2-propanol, ethanol, and methanol, respectively, where the mixture ratio of D2O and alcohol is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. In both cases, attenuation of the hydrogen evolution reaction is specifically observed for methanol. This is probably due to poisoning of the Pt surface with carbon monoxide molecules evolved from the oxidation of methanol at the TiO2 surface.28
:1. In both cases, attenuation of the hydrogen evolution reaction is specifically observed for methanol. This is probably due to poisoning of the Pt surface with carbon monoxide molecules evolved from the oxidation of methanol at the TiO2 surface.28
 | ||
| Fig. 2 Isotopic selectivity for HD (black) and H2 (red) from the photocatalytic hydrogen evolution reaction using a 1:1 mixture of D2O and the corresponding alcohol upon light irradiation for 15 min. | ||
H2 is classified as o-H2 or p-H2 depending on the nuclear spin isomer.29,30 o-H2 is observable and p-H2 is not by NMR because of the Zeeman splitting of the nucleus spin momentum. Because of the spin statistic, the ratio of o-H2 and p-H2 is 3:1.29,30 D2 is not included in the observation because of the low sensitivity to D atoms, even in 2H NMR spectroscopy measurements. Similarly, we observed an increase in the oxidation products of methanol and ethanol. Importantly, the selectivity for the oxidation and hydrogen evolution reaction were continuously monitored, as shown in Fig. S6–S11.† In addition, the maximum concentration of H2 in this photocatalytic reaction is approximately 1 mmol L−1, which is below the solubility limits of water and alcohol.31–34 These results suggest that a robust photocatalytic process continues throughout the catalytic cycle.
Isotopic hydrogen evolution provides information about the reaction mechanism at the metal surface.35–39 The reaction follows an electrochemical adsorption and desorption cycle. The adsorption of atomic hydrogen from the proton donor (Volmer step)40 is followed by either desorption via recombination of adsorbed hydrogens (Tafel step)41 or desorption of atomic hydrogen with a proton donor (Heyrovsky step).42 The enrichment of hydrogen over deuterium is observed for the Heyrovsky, Tafel, and Volmer step sequence.40–42 Generally, the Tafel step is rate-limiting in the hydrogen evolution process for Pt surfaces. Therefore, isotopic selectivity is not dependent on electrochemical potential.
As shown in Fig. 2, the isotopic selectivity is similar for the reactions using 2-propanol and ethanol. This suggests that the formal potential of the hydrogen evolution reaction is similar for these two conditions. Additionally, we evaluated self-diffusion of water molecules and each alcohol molecule as shown in Table S1.† We determined diffusion coefficients for the alcohols and HDO. These results suggest that the diffusion of the reactant in the hydrogen evolution reaction is not the rate-determining step in the photocatalytic reaction cycle.43
Interestingly, the efficiency of the multi-electron transfer is dependent on the donor molecule. Fig. S12† shows the time-course of the oxidation and reduction reactions obtained by accounting for the half-reaction. Linearity in the time-course plot is observed, indicating stable photocatalysis. Therefore, the reaction rate was calculated from the slope of each reaction. Fig. 3 and S12† show the rates of oxidation and reduction obtained by accounting for the number of electrons in the half-reaction, defined as rox and rred. For a 3:1 ratio of D2O and alcohol, rred is nominally low. This is probably due to the small number of donor molecules in the catalytic reaction. Importantly, rred shows the highest value of 0.26 μmol min−1 for the combination of 2-propanol/D2O (1:1). This value is comparable with that for ethanol/D2O (1:1), which is 0.22 μmol min−1. Conversely, the rox values for 2-propanol and ethanol are not comparable. Indeed, rox for 2-propanol is seven times higher than that for ethanol.
 | ||
| Fig. 3 Rates of the oxidation reaction (black) and hydrogen evolution reaction (red) using a 1:1 mixture of D2O and the corresponding alcohol. | ||
Finally, we discuss the effect of donor molecules on the efficiency of the photocatalytic hydrogen evolution reaction. The stability of the radical derived from the alcohol plays an important role in the reaction efficiency. 2-Propanol is oxidized to the tertiary carbocation radical intermediate, which is consumed by spontaneous oxidation at the TiO2 surface (Fig. 4a).44–47 For ethanol (Fig. 4b), the oxidized carbocation radical species is expected to be unstable compared with that for 2-propanol. Therefore, the rate of the hydrogen evolution reaction is comparable with the rate of the oxidation reaction. For methanol (Fig. 4c), the carbon monoxide evolved is expected to attenuate the hydrogen evolution reaction.28 Thus, the efficiency of the redox reaction can be evaluated from the NMR spectroscopy results.
 | ||
| Fig. 4 Schematic representations of photocatalytic hydrogen evolution reactions over Pt–TiO2 using (a) 2-propanol, (b) ethanol, and (c) methanol. | ||
In conclusion, we used NMR spectroscopy to track the photocatalytic hydrogen evolution reaction using Pt–TiO2 as a model catalyst. We performed rapid detection of dissolved hydrogen molecules in the solvent and the oxidized product at the sub-micromole scale by 1H NMR. The method is useful for observation of the dynamic state of molecules in solution and product-based determination of the reaction mechanism. This method is also applicable to the screening of photocatalysts under given conditions. In addition, we found that an efficient multi-electron-transfer photocatalytic reaction is possible using ethanol as the donor molecule. This study demonstrates the utility of NMR for the clarification of the hydrogen evolution reaction mechanism as a means to evaluate potential catalysts, from organic molecular catalysts to inorganic nanocrystals.
Author contributions
T. F. and K. M. designed the project. T. F. and D. A. conducted experiments and analysed all the data. All the author commented and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr Atsuhiro Tanaka and Prof. Hiroshi Kominami (Kindai University) for the discussion on the photocatalytic experiments on the nanoparticle suspension. This work was supported by a JSPS KAKENHI (Grant Number: JP21K14596 and JP22H02023) and Grant-in-Aid for Scientific Research on Innovative Areas “Interface IONICS” (Grant Number: JP20H05281) and “Optical manipulation” (Grant Number: JP16H06506). This study was also supported by the JST-Mirai Program grant number JPMJMI21EB, the Frontier Photonic Sciences Project of National Institutes of Natural Sciences (NINS) grant number 01213010, and the Photo-excitonix Project in Hokkaido University.Notes and references
- M. Ashokkumar, Int. J. Hydrogen Energy, 1998, 23, 427–438 CrossRef CAS.
- H. Kisch, Angew. Chem., Int. Ed. Engl., 2013, 52, 812–847 CrossRef CAS PubMed.
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, J. Am. Chem. Soc., 2020, 142, 9752–9762 CAS.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- H. Minamimoto, R. Osaka and K. Murakoshi, Electrochim. Acta, 2019, 304, 87–93 CrossRef CAS.
- J. Kim, T. Fukushima, R. Zhou and K. Murakoshi, Materials, 2019, 12, 211 CrossRef CAS PubMed.
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721–2739 CrossRef CAS.
- X. L. Wang, W. Liu, Y. Y. Yu, Y. Song, W. Q. Fang, D. Wei, X. Q. Gong, Y. F. Yao and H. G. Yang, Nat. Commun., 2016, 7, 11918 CrossRef CAS PubMed.
- B. B. Xu, M. Zhou, R. Zhang, M. Ye, L. Y. Yang, R. Huang, H. F. Wang, X. L. Wang and Y. F. Yao, J. Phys. Chem. Lett., 2020, 11, 3738–3744 CrossRef CAS PubMed.
- R. Zhang, M. Ye, Y. N. Yang, R. Huang, X. L. Wang and Y. F. Yao, J. Catal., 2020, 382, 173–180 CrossRef CAS.
- I. F. Silvera, Rev. Mod. Phys., 1980, 52, 393–452 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- H. Gilboa, B. E. Chapman and P. W. Kuchel, J. Magn. Reson., Ser. A, 1996, 119, 1–5 CrossRef CAS.
- J. R. Beckett and H. Y. Carr, Phys. Rev. A: At., Mol., Opt. Phys., 1981, 24, 144–148 CrossRef CAS.
- J. R. Beckett and H. Y. Carr, Phys. Rev. A: At., Mol., Opt. Phys., 1984, 29, 1587 CrossRef.
- W. T. Raynes and N. Panteli, Chem. Phys. Lett., 1983, 94, 558–560 CrossRef CAS.
- T. W. Marshall, Mol. Phys., 1961, 4, 61–63 CrossRef CAS.
- H. S. Gutowsky, J. Chem. Phys., 1959, 31, 1683–1684 CrossRef CAS.
- H. Fujiwara, J. Yamabe and S. Nishimura, Chem. Phys. Lett., 2010, 498, 42–44 CrossRef CAS.
- P. Garbacz, K. Jackowski, W. Makulski and R. E. Wasylishen, J. Phys. Chem. A, 2012, 116, 11896–11904 CrossRef CAS PubMed.
- J. Y. Chen, A. A. Marti, N. J. Turro, K. Komatsu, Y. Murata and R. G. Lawler, J. Phys. Chem. B, 2010, 114, 14689–14695 CrossRef CAS PubMed.
- K. Domen, S. Naito, T. Onishi and K. Tamaru, Chem. Lett., 1982, 11, 555–558 CrossRef.
- M. Kawai, S. Naito, K. Tamaru and T. Kawai, Chem. Phys. Lett., 1983, 98, 377–380 CrossRef CAS.
- T. Kawai and T. Sakata, J. Chem. Soc., Chem. Commun., 1980, 694–695, 10.1039/c39800000694.
- M. Miyake, J. Catal., 1979, 58, 22–27 CrossRef CAS.
- M. Ohno, H. Uzawa, T. Miyazaki and K. Tarama, Chem. Lett., 1987, 16, 779–782 CrossRef.
- T. Sakata and T. Kawai, Chem. Phys. Lett., 1981, 80, 341–344 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2018, 237, 491–503 CrossRef CAS.
- K. Fukutani and T. Sugimoto, Prog. Surf. Sci., 2013, 88, 279–348 CrossRef CAS.
- K. Motizuki and T. Nagamiya, J. Phys. Soc. Jpn., 1956, 11, 93–104 CrossRef.
- R. Wiebe and V. L. Gaddy, J. Am. Chem. Soc., 1934, 56, 76–79 CrossRef CAS.
- R. W. Cargill, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 1444–1456 RSC.
- M. S. Wainwright, T. Ahn, D. L. Trimm and N. W. Cant, J. Chem. Eng. Data, 1987, 32, 22–24 CrossRef CAS.
- S. Jaatinen, J. Touronen, R. Karinen, P. Uusi-Kyyny and V. Alopaeus, J. Chem. Thermodyn., 2017, 112, 1–6 CrossRef CAS.
- P. Pichat, M. N. Mozzanega and H. Courbon, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 697–704 RSC.
- L. P. Krishtalik, Russ. J. Electrochem., 2001, 37, 102–106 CrossRef CAS.
- J. O. M. Bockris and S. Srinivasan, Electrochim. Acta, 1964, 9, 31–44 CrossRef CAS.
- B. E. Conway, Proc. R. Soc. London, Ser. A, 1960, 256, 128–144 CAS.
- H. Eyring, S. Glasstone and K. J. Laidler, J. Chem. Phys., 1939, 7, 1053–1065 CrossRef CAS.
- J. O. M. Bockris and S. Srinivasan, J. Electrochem. Soc., 1964, 111, 844 CrossRef CAS.
- J. O. M. Bockris and S. Srinivasan, J. Electrochem. Soc., 1964, 111, 858 CrossRef CAS.
- J. O. M. Bockris and S. Srinivasan, J. Electrochem. Soc., 1964, 111, 853 CrossRef CAS.
- B. B. Xu, M. Zhou, M. Ye, L. Y. Yang, H. F. Wang, X. L. Wang and Y. F. Yao, J. Am. Chem. Soc., 2021, 143, 10940–10947 CrossRef CAS PubMed.
- S. R. Morrison and T. Freund, J. Chem. Phys., 1967, 47, 1543–1551 CrossRef CAS.
- H. Gerischer, Surf. Sci., 1969, 13, 265–278 CrossRef CAS.
- M. Miyake, H. Yoneyama and H. Tamura, Chem. Lett., 1976, 5, 635–640 CrossRef.
- A. Y. Ahmed, T. A. Kandiel, I. Ivanova and D. Bahnemann, Appl. Surf. Sci., 2014, 319, 44–49 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01676a |
| This journal is © The Royal Society of Chemistry 2022 |