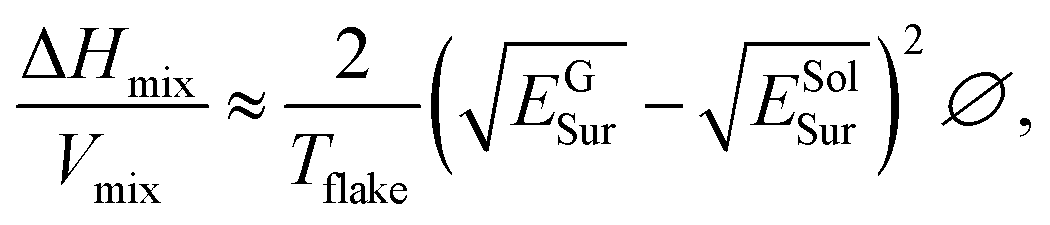DOI:
10.1039/D2RA01668K
(Paper)
RSC Adv., 2022,
12, 17990-18003
Facial fabrication of few-layer functionalized graphene with sole functional group through Diels–Alder reaction by ball milling†
Received
15th March 2022
, Accepted 2nd June 2022
First published on 17th June 2022
Abstract
The widespread use of graphene as a next-generation material in various applications requires developing an environmentally friendly and efficient method for fabricating functionalized graphene. Chemically, graphene can be used as an electron donor or attractor. Here, graphite was successfully exfoliated, and an in situ Diels–Alder reaction (D–A) was carried out to fabricate functionalized graphene with sole functional groups via mechanochemical ball milling. The reactivities of graphene acting as a diene or a dienophile were investigated. Few-layer (≤2 layers) graphene specimens were obtained by wet ball milling, heating in a nitrogen atmosphere, and solvent ultrasonic treatment. The ball-milling method was more effective than heating in a nitrogen atmosphere, and the [2 + 4] D–A of graphene was more dominant than the [4 + 2] D–A in the ball-milling process. The surface tension of functionalized graphene decreased, which provided a theoretical basis for the dispersion and exfoliation of graphite in a suitable solvent. Functionalized graphene still had a high electrical conductivity, which has far-reaching significance for functionalized graphene to be applied in electronic semiconductors and related applications. Meanwhile, functionalized graphene was applied to polymer composite fibers, the tensile strength and the Young's modulus could reach 780 MPa and 19 GPa. The volume resistivity was two orders of magnitude lower than that of pure fiber. Thus, the use of ball milling to efficiently exfoliate and in situ functionalize graphite will help to develop a strategy that can be widely used to manufacture nanomaterials for various application fields.
1. Introduction
Graphene, a 2D sheet of sp2-hybridized, networked carbon, has received considerable attention due to its unique properties, such as its high surface area, and unique mechanical, thermal, and structural properties, since its discovery in 2004.1 The use of graphene is promising in various applications, such as in supercapacitors,2 Li-ion batteries,3,4 sensors,5 composite additives,6 transparent conducting electrodes,7 fuel cells,8 field-effect transistors,9 and photovoltaic cells.10 Graphene is produced by two types of methods: bottom-up and top-down methods.11 The representative bottom-up methods are chemical vapor deposition (CVD) and epitaxial growth, and they offer a large area (up to 7.5 m2) of high-quality graphene with minor defects. However, these methods are costly due to the multistep processes involved, and they lack bulk production protocols. Hence, they are not suitable for scaling up for industrial-scale production.12,13 Compared with bottom-up technology, graphite can be exfoliated into graphene (top-down), which has the potential for large-scale production of graphene nanoplatelets due to the cheap and abundant graphite resources.14 In this regard, shear exfoliation technology using liquid-phase exfoliation or ball milling, which can exfoliate untreated graphite into graphene, has become an efficient and versatile technique.15,16 However, liquid-phase exfoliated graphite has the problem of waste liquid recovery, which has become a barrier to its efficient production. Therefore, exfoliating graphite into graphene through ball milling may be a more effective way to produce graphene on a large scale.17
Despite its unique physical and chemical properties, graphene is hydrophobic and easily agglomerates due to its strong van der Waals forces and the absence of an energy gap, which has limited its widespread application. Meanwhile, the regular atomic structure of graphene prevents it from exhibiting excellent interfacial effects in polymer composites. To engineer an energy band into graphene, the emergence of graphene oxide (GO), which is a derivative of graphene, has solved these problems.18 There are a large number of oxygen-containing groups, i.e., carboxylic, hydroxyl, ketone, and epoxide groups, connected to the infinitely extending base surface of a two-dimensional space composed of a layer of carbon atoms. The ratio and content of oxygen-containing functional groups in GO vary with the fabrication parameters. However, in the oxidation process, the original graphite is treated with strong inorganic protic acids (such as concentrated sulfuric acid, nitric acid, or their mixture), and then it is oxidized by a strong oxidant (such as KMnO4, KClO4, or H2O2).19 These processes are dangerous and generate serious waste water pollution. Furthermore, GO begins to gradually lose its oxygen-containing groups when the temperature is above approximately 130 °C, which is lower than most polymer processing temperatures.20 Therefore, it is very important to develop an environmentally friendly and efficient method for fabricating functionalized graphene.
Graphene can be used as a diene in the [4 + 2] cycloaddition reaction or as a dienophile in the [2 + 4] cycloaddition reaction.21–23 However, there are still disagreements in some aspects of these processes. First, the grafting sites of the Diels–Alder reaction (D–A) on graphene have been extensively studied. Cao used density functional theory (DFT) calculation results to show that the in-plane D–A activity of graphene is extremely low. The reaction occurs at the edge of graphene, especially in the Armchair position.24 In contrast, Daukiya suggested that the reaction occurs in-plane, especially at the Dirac point or defect location. The non-defective graphene and 3,5-bis(trifluoromethyl)phenyl-substituted maleimide derivatives have also been confirmed to react in the plane of graphene without any defects, and the reaction site was visually observed using scanning tunneling microscopy (STM).25 Meanwhile, some scholars suggested that the D–A sites can be both at the edge and in the plane, depending on the reaction conditions.26,27 Second, the D–A grafting ratio and the reaction conditions of graphene vary greatly in various reports. The pretreated graphite and furfuryl alcohol (FAL) reaction system was heated to 130 °C in an N2 atmosphere for 72 h, and the grafting ratio onto graphene reached up to 1.52 mmol g−1.28 However, in a solvent-free process, maleic anhydride (MA) can be grafted onto the edge-localized electron-rich active sites of graphite nanoplatelets resulting from high collision forces at room temperature with a grafting ratio of 0.94 mmol g−1.29 Third, the D–A is a reversible reaction. The reaction temperature is higher in some reports than in others, e.g., the reaction temperature of MA-grafted graphite reported by Seo was 220 °C.22 which was much higher than the reverse reaction temperature of 150 °C reported by Sarkar.30 All these discrepancies were likely due to various differences, such as the types of reactants and the environmental and engineering parameters. Therefore, the D–A of graphene has not been applied in large-scale industrial production, and its reaction mechanism needs to be further explored.
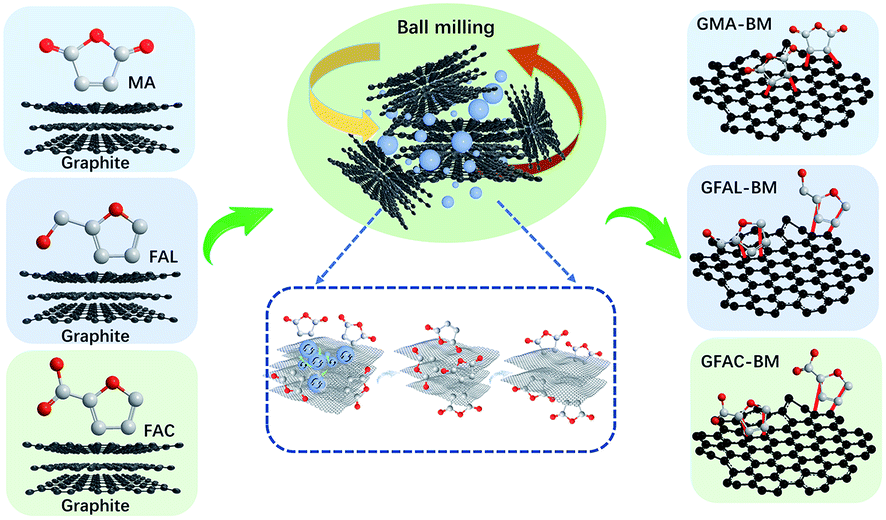
In this study, we demonstrated the exfoliation of graphite into few-layer graphene while performing in situ D–A via ball-milling. The simultaneous graphite exfoliation and D–A could be driven by the mechanical forces of the high-speed ball milling and the generated collision heat. Through the D–A of graphene, the functionalized graphene with sole functional groups was accurately introduced. Three layers of functionalized graphene or fewer with sole functional groups were fabricated via ball milling. The grafting ratio, dispersion, and thermoelectric properties of the functionalized graphene were analyzed. This work indicates that the fabrication of functionalized graphene with sole functional groups by ball milling may become a powerful alternative process for the fabrication of functionalized graphene by the graphite oxidation route.
2. Experimental section
2.1 Materials
Graphite powder (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mesh, lateral size in the range of 1–5 μm, layer number > 100) was supplied by Qing Dao Teng Sheng Da Tan Su Ji Xie Co., Ltd., Shandong Province, China. FAL (C5H6O2, 98%), 2-furoic acid (FAC, C5H4O3, 98%), MA (C4H2O3, 99%), N,N′-dimethylformamide (DMF, 99%), N-methyl-2-pyrrolidone (NMP, 99%), dimethyl sulfoxide (DMSO, 99%), N-N′-dimethylacetamide (DMAc, 99%), and acetone were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd., Shanghai, China.
000 mesh, lateral size in the range of 1–5 μm, layer number > 100) was supplied by Qing Dao Teng Sheng Da Tan Su Ji Xie Co., Ltd., Shandong Province, China. FAL (C5H6O2, 98%), 2-furoic acid (FAC, C5H4O3, 98%), MA (C4H2O3, 99%), N,N′-dimethylformamide (DMF, 99%), N-methyl-2-pyrrolidone (NMP, 99%), dimethyl sulfoxide (DMSO, 99%), N-N′-dimethylacetamide (DMAc, 99%), and acetone were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd., Shanghai, China.
2.2 In situ Diels–Alder reaction (D–A) via ball milling
The multi-directional planetary ball-milling machine used in this work differed from the conventional one. In addition to the normal operation in the radial direction, the planetary disc was also flipped 360°, achieving multi-dimensional and multi-directional motion of the balls and grinding tank to promote the full grinding between the balls and graphite. The D–A of FAL and graphite is described as an example. Pristine graphite (25 g) and FAL (250 g, 2.548 mol) were placed in a zirconia container with zirconia balls (2750 g, 1, 3, and 5 mm in diameter, mass ratio: 1 mm/3 mm/5 mm = 5/3/2). The container was then fixed in the planetary ball-milling machine (QXQM-4L, 0.75 kW, TENCAN POWDER, Changsha, Hunan Province) and rotated at 560 rpm (flipping speed of 20 rpm) for 12 h. Then, the solid powder was washed separately with water (2000 mL per time) and ethanol (2000 mL per time) five times, after which it was dried in a vacuum oven at 80 °C for 12 h. In the ball-milling process, the mass ratio of graphite to reactive organic monomers was 1/10, and the ratio of zirconia balls to the total mass of reactants was 10. The fabricated FAL-functionalized graphene is abbreviated as GFAL-BM. The FAC- and MA-functionalized graphene fabricated by ball milling is abbreviated as GFAC-BM and GMA-BM, respectively. The ball-milled graphite without any additives is abbreviated as G-BM. The processes of ball milling graphite are shown in Scheme 1.
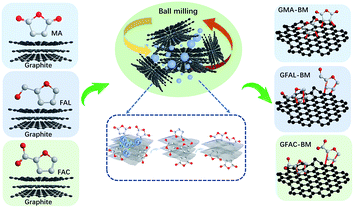 |
| | Scheme 1 Graphite ball-milling processes. | |
2.3 D–A in dimethylformamide (DMF)
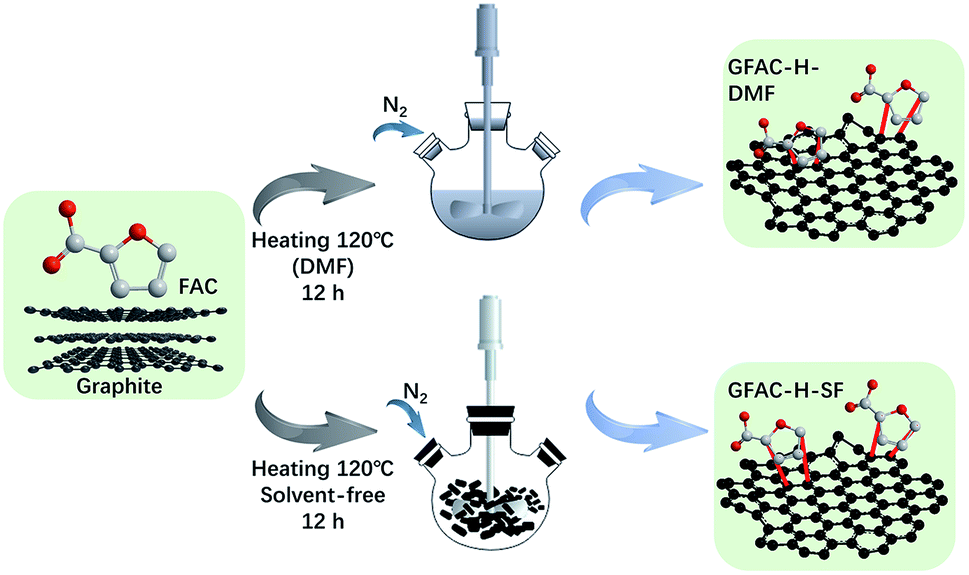
Graphite was used as a dienophile, and FAC was used as a diene. A typical D–A reaction system was carried out. Pristine graphite (2 g) was dispersed in 500 mL of DMF with an ultrasonic cleaner for 20 min. FAC (20 g, 0.178 mol) was then added to the graphite dispersion liquid. The reaction system was heated to 120 °C in an N2 atmosphere for 12 h. Then, the solid powder was collected by centrifugation, washed with excess ethanol five times, washed separately with excess water and acetone three times, and dried in a vacuum oven at 80 °C for 12 h. The fabricated FAC-functionalized graphene in the DMF heating reaction system is abbreviated as GFAC-H-DMF. The process is shown in Scheme 2.
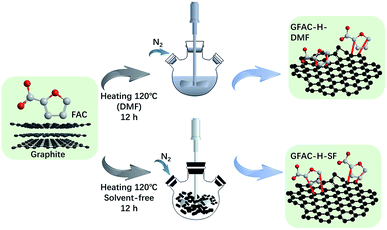 |
| | Scheme 2 Diels–Alder reaction (D–A) in dimethyl formamide (DMF). | |
2.4 Solvent-free D–A
Graphite was used as a dienophile, FAC was used as a diene, and a typical D–A was carried out. A mixture of pristine graphite (2 g) and FAC (20 g, 0.178 mmol) was placed in a glass ampoule. The reaction system was then heated to 120 °C in an N2 atmosphere for 12 h. After this, the solid power was washed with excess ethanol five times, washed separately with excess water and acetone three times by an ultrasonic cleaner, and dried in a vacuum drying oven at 80 °C for 12 h. The fabricated FAC-functionalized graphene in solvent-free D–A is abbreviated as GFAC-H-SF. The fabricating process is shown in Scheme 2.
2.5 Fabrication of polyethylene terephthalate (PET) nanocomposite fibers
The fabrication methods of PET pellets and composite fibers refer to the previous report.20
2.6 Characterization
The apparent morphology was examined using transmission electron microscopy (TEM, Hitachi H7650, Japan) and field-emission scanning electron microscopy (FE-SEM, Hitachi S-4800, Japan). Before the TEM test, the samples were dispersed in ethanol and sonicated for 20 min. The samples were dispersed on the microgrid film with a 1 mL disposable syringe, and placed in a vacuum oven at 45 °C for drying. Atomic force microscopy (AFM, Bruker Icon, USA) was used in tapping mode. The ethanol dispersion of samples were fully diluted, then dropped on the mica sheet, and spin-coated to obtain the AFM test sample. The Brunauer–Emmett–Teller (BET) specific surface area of graphite was measured with an Autosorb-iQ-C instrument (Quantachrome Instruments, USA) with N2 as an adsorbent. The samples were analyzed under nitrogen atmosphere (adsorption–desorption isotherms at 77 K) in a volumetric working device. The moisture content in the samples was removed by drying them at 80 °C for 36 h prior to the analysis. Fourier-transform infrared spectroscopy (FT-IR, Bruker TERSOR37, Germany) spectra of the specimens were obtained in the range of 4000–400 cm−1. The test samples were ground and pressed with KBr for testing. KBr was baked in a vacuum oven at 120 °C for 4 h in advance to remove moisture. The ultraviolet absorption coefficient (α) was obtained by an ultraviolet-visible-near infrared spectrometer (Hitachi UH4150, Japan). X-ray diffraction (XRD, Rigaku D/MAX-gA, Japan) with filtered Cu Kα radiation (λ = 0.15406 nm) was used in the range from 3° to 40° (2θ) at a scan rate of 8° min−1. X-ray photoelectron spectroscopy (XPS) measurements were obtained using a Genesis 60 spectrometer (Edax, USA) equipped with an Al Kα radiation source (hv = 1486.4 eV). Micro-Raman mapping spectra were recorded on an XPLORA PLUS Raman microscope equipped with a 532 nm laser source. Thermogravimetric analysis (TGA) and derivative thermogravimetric analysis (DTGA) (NETZSCH STA449F3, Germany) were also performed in an N2 atmosphere with the temperature increasing from 25 °C to 800 °C (heating rate = 10 °C min−1). The thermal conductivity of graphite and PET composite, which was formed into a round slice with a diameter of 1 cm (thickness over 1 mm), was measured by a thermal conductivity instrument (DRL-III thermal conductivity meter, hot surface temperature 60 °C). The electrical resistivity of the round slice was tested by a four-dimension four-point probe system (Model 280PI, Four Dimensions, USA). Sample thickness in electrical resistivity measurement were between 0.5–1 mm. The mechanical properties of the composite fibers were tested using an electronic single fiber strength tester (LLY-06AD, China) operated at a clamp interval of 10 mm and a rate of 10 mm min−1. The relative humidity and temperature in the test environment were 60 ± 10% and 20 ± 2 °C, respectively. The results presented in Section 3 are the averaged values of 10 repeated tests.
3. Results and discussion
3.1 Morphologies and structures of pristine graphite and exfoliated functionalized graphene
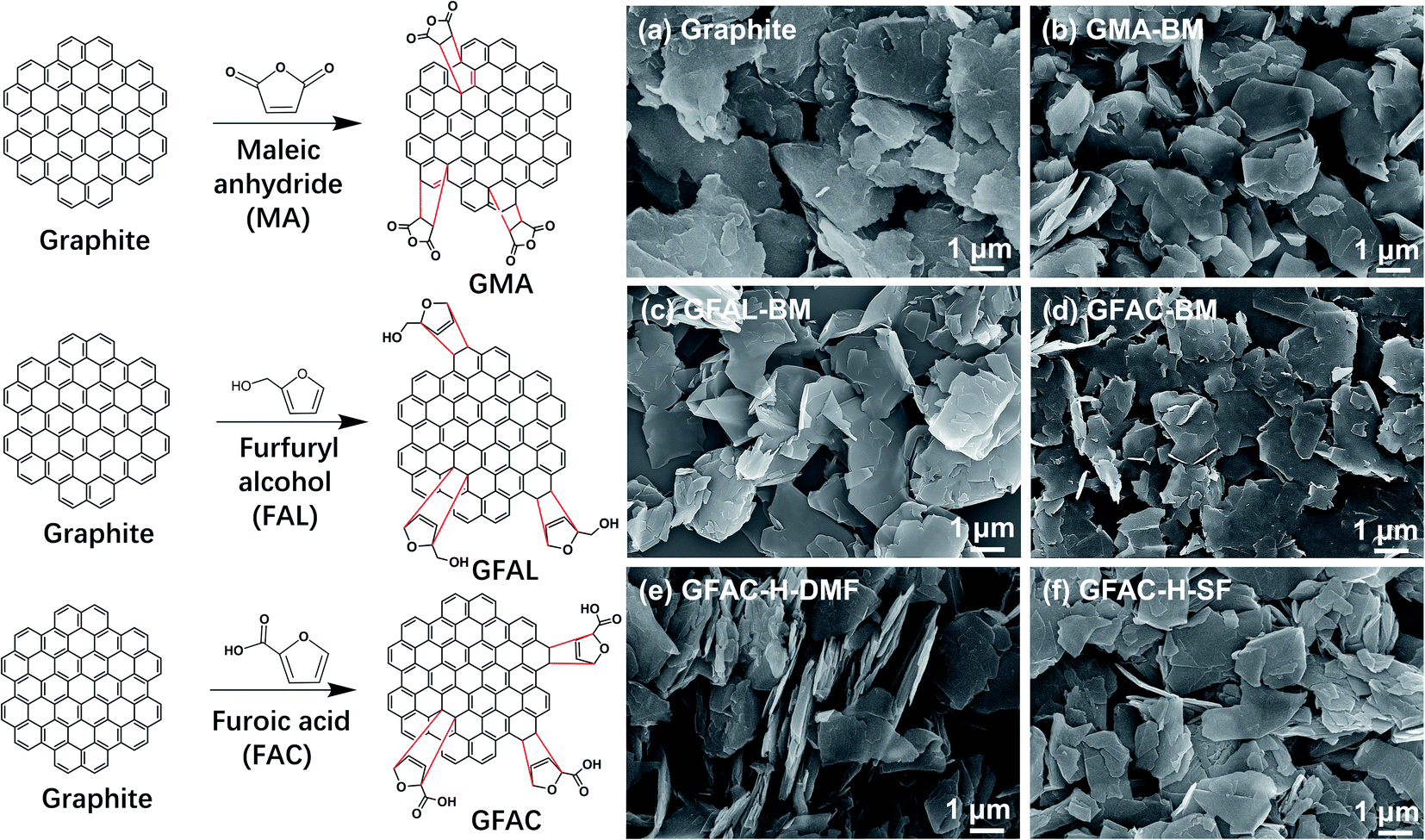
According to the current reports on the D–A of graphite or graphene, graphite acts as the 4π component reacting with MA and as the 2π component reacting with FAL (FAC), as shown in Fig. 1 (left).21,25,26,31 The formation of various functionalized graphene samples with various procedures can be discerned unambiguously in Fig. 1 (right). The pristine graphite appeared as a stack of massive sheets (Fig. 1a). After ball milling, the stack thicknesses of the graphite flakes were smaller (Fig. 1a–d). The graphite also exhibited a certain exfoliation effect under ultrasonic treatment (Fig. 1e and f). Graphite and processed graphite could be more clearly identified by TEM (Fig. 2), and it was found that the graphite after ball milling was well exfoliated (Fig. S1†). A significant decrease in transparency was observed, indicating a significant decrease in the number of graphite layers. After ball milling, the stacks of single-layer graphene were clearly visible (Fig. 2b–d). The lateral size of the graphite was reduced from ∼4 μm of pristine graphite to ∼1.5 μm of GFAL-BM, indicating that the shearing and impact effects of ball milling on graphite coexisted. After the heat treatment (Fig. 2e and f), the peeling effect of graphite was weakened, especially for GFAC-H-SF (Fig. 2f). During the implementation of the solvent-free heating method, due to the high temperature, furoic acid was more likely to agglomerate, which would affect the intercalation content of furoic acid in the graphite, thereby limiting its exfoliation. The lateral sizes of GFAC-H-DMF and GFAC-H-SF were about 2 and 3 μm, respectively, indicating that the ultrasonic treatment could tear the graphite sheet structure.
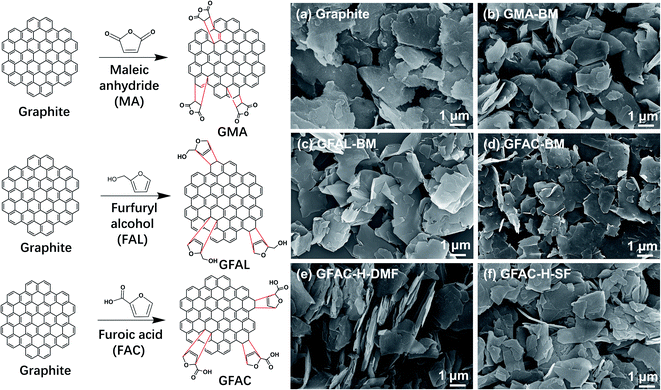 |
| | Fig. 1 D–A schematic representation of graphite (left). Scanning electron microscopy (SEM) images (right) of (a) graphite, (b) GMA-BM, (c) GFAL-BM, (d) GFAC-BM, (e) GFAC-H-DMF, and (f) GFAC-H-SF. | |
 |
| | Fig. 2 Transmission electron microscopy (TEM) images of (a) graphite, (b) GMA-BM, (c) GFAL-BM, (d) GFAC-BM, (e) GFAC-H-DMF, and (f) GFAC-H-SF. | |
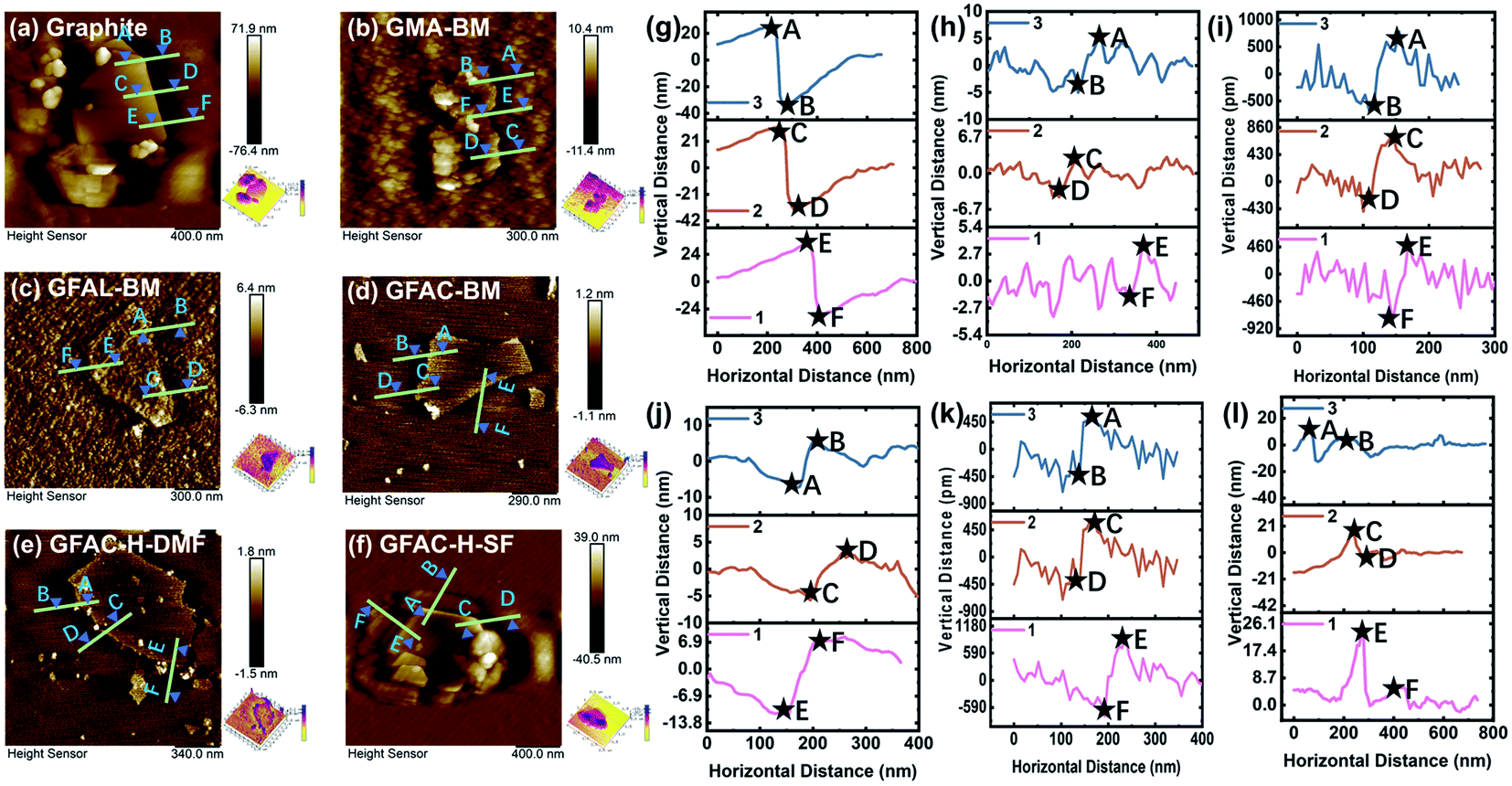
Fig. 3 and S2† show the AFM tapping-mode images of the graphite and functionalized graphene. Three groups at various positions were selected in each image for thickness measurements. The measurement results are listed in Table 1. The thickness measurements of the graphite flakes by AFM verified the SEM and TEM results described above. The pristine graphite was 63.2 nm thick and was composed of about 186 layers of single-layer graphene, corresponding to a single-layer graphene thickness of 0.34 nm.32 For G-BM, the number of layers decreased somewhat after ball milling. As several publications have shown that the apparent height of the monolayers measured by the tapping-mode AFM ranged from 0.4 to 1 nm.33,34 The smallest number of monolayer stacks was found in the GFAL-BM, with an average thickness of about 1.2 nm, which should be considered as monolayer/bilayer graphene. This number of layers (three layers) was the lowest of the graphene sheets fabricated by the ball-milling method reported previously.35 For GFAC-H-DMF, the average thickness was about 1.4 nm, the corresponding number of layers could also be regarded as monolayer/bilayer. This indicated that by using either ball milling or ultrasonic treatment, graphite could be successfully exfoliated into a few layers or even a single layer of graphene in a liquid environment. Liquid molecules could be more fully intercalated between the graphite flakes. In particular, for GFAL-BM, FAL provided a liquid environment and acted as a reactant for the D–A. According to the AFM test results (Table 1), the exfoliation ability of graphite was ranked as follows: GFAL-BM > GFAC-H-DMF > GMA-BM > GFAC-BM > GFAC-H-SF.
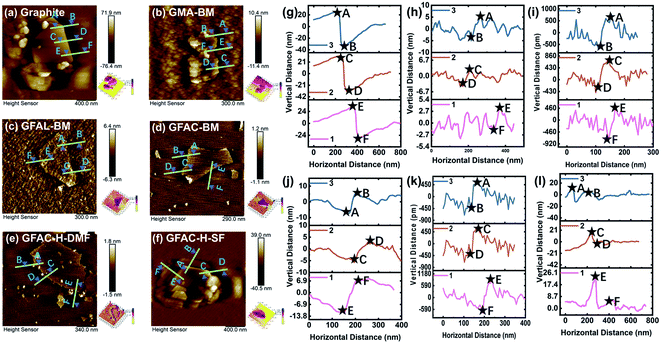 |
| | Fig. 3 Atomic force microscopy (AFM) tapping-mode images of (a, g) graphite, (b, h) GMA-BM, (c, i) GFAL-BM, (d, j) GFAC-BM, (e, k) GFAC-H-DMF, and (f, l) GFAC-H-SF. | |
Table 1 Thickness of graphite and functionalized graphene
| |
Thickness (nm) |
| Line 1 |
Line 2 |
Line 3 |
Average |
| Graphite |
64.2 |
65.8 |
59.6 |
63.2 |
| G-BM |
33.2 |
32.4 |
38.2 |
34.6 |
| GMA-BM |
5.9 |
6.8 |
6.9 |
6.5 |
| GFAL-BM |
1.3 |
1.1 |
1.2 |
1.2 |
| GFAC-BM |
10.6 |
7 |
12.2 |
9.9 |
| GFAC-H-DMF |
1.7 |
1.3 |
1.2 |
1.4 |
| GFAC-H-SF |
23.1 |
24.1 |
13.7 |
20.3 |
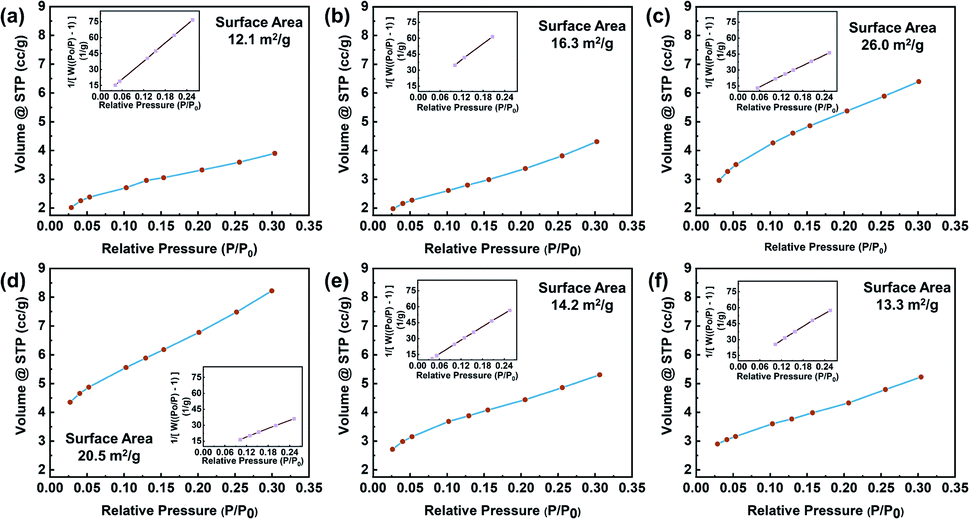
To observe the graphite structures under various treatment conditions, the surface microstructures were analyzed by a nitrogen adsorption instrument, and the adsorption and desorption data were obtained, as shown in Fig. 4. The specific surface areas of the specimens were calculated by the BET method from the adsorption isotherms recorded at relative pressures (P/P0) of 0.025–0.30. For the control, the BET surface area was 12.1 m2 g−1, which was higher than that of typical natural graphite due to the low particle size.36 For G-BM (Fig. S3†), the surface area was increased (15.1 m2 g−1), indicating the exfoliation effect of ball milling. After ball-milling functionalization, the specific surface area of graphite increased. In particular, for the GFAL-BM, the specific surface area increased to 26 m2 g−1. During the ball-milling process, the three-dimensional structure of graphite underwent two changes. One was the intercalation and exfoliation of the graphite flakes, and the other was the fracture of the graphite flakes by mechanical forces. For the heat-treated graphite, the increase in the specific surface area was lower than ball-milled graphite, which was caused by the re-stacking of the graphite sheets after drying treatment. Therefore, based on the analysis of the number of exfoliated graphite layers in the AFM results, among the methods considered, the combination of ball-milled graphite and FAL was the best for the production of few-layer graphene.
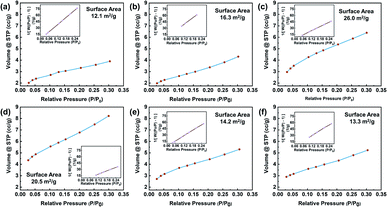 |
| | Fig. 4 Nitrogen adsorption/desorption isotherm of (a) graphite, (b) GMA-BM, (c) GFAL-BM, (d) GFAC-BM, (e) GFAC-H-DMF, and (f) GFAC-H-SF. | |
3.2 Degree of in situ D–A of functionalized graphene
The energy-dispersive X-ray spectroscopy (EDS) images and the elemental contents of graphite are shown in Fig. 5 and Table 2, respectively. According to Elemental Analyzer data, the oxygen content of pristine graphite was only 2.4%, which was not much different from the oxygen content (2.5%) of G-BM, indicating that ball milling does not affect the chemical structure of graphite. The oxygen content of the GFAC-BM was the highest at 7.5%, which was greater than those of the GMA-BM and GFAL-BM. Furthermore, it was 1.8 times that of the GFAC-H-DMF and 2.3 times that of the GFAC-H-SF. The oxygen content can be used to roughly judge the grafting ratio (Table 2). GFAC-BM had a high oxygen content, which was due to the high oxygen content of the FAC molecule itself. The actual number of grafted molecules should be the highest for GFAL-BM (1.17 mmol g−1). In related reports, the grafting rate of graphene grafted with FAL was only 1.52 mmol g−1, and the reaction was carried out at 130 °C for 72 h.28 Therefore, compared with other related methods (Table 2), the methods used in this report have great energy and efficiency advantages. The grafting ratio of graphene can also be calculated in subsequent analysis. In the EDS images, the O generated by the functionalization was not only visible on the edge of the graphene sheet but also on the surface. Therefore, it can be roughly judged that the [2 + 4] and [4 + 2] D–A processes of graphene may have occurred both in the plane and on the edges of graphene sheets.
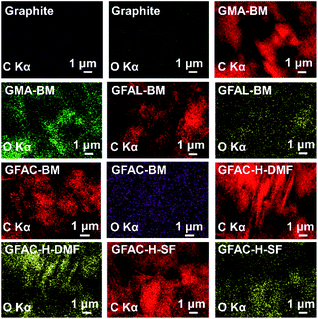 |
| | Fig. 5 Energy-dispersive X-ray spectroscopy (EDS) images of graphite and functionalized graphene. | |
Table 2 Analysis of elemental contents
| Specimen |
Element content (%) |
Grafting ratio (mmol g−1) |
Reactant |
Reference for comparison |
Ref. |
| C |
O |
H |
Grafting ratio (mmol g−1) |
Reaction condition |
| MI: maleimide; HPM: N-(4-hydroxyl phenyl) maleimide; mPC: cyclopentadienyl-capped poly(ethylene glycol) monomethyl ether; HAD: hexadecyl acrylate; MWCNT: multi-walled carbon nanotubes. |
| Graphite |
96.4 |
2.4 |
1.2 |
0 |
MA |
Graphite |
0.19 |
220 °C 12 h |
22 |
| MIa |
Graphite |
0.10 |
160 °C 12 h |
22 |
| GMA-BM |
93.4 |
5.1 |
1.5 |
0.58 |
FAL |
Graphene |
1.52 |
130 °C 72 h |
28 |
| HPMa |
Graphene |
0.22 |
90 °C 12 h |
37 |
| GFAL-BM |
92.7 |
2.0 |
1.3 |
1.17 |
MA |
Graphene |
0.94 |
Kitchen blender (120 min) |
29 |
| mPCa |
Graphene |
0.45 |
80 °C 24 h |
38 |
| GFAC-BM |
91.3 |
7.5 |
1.2 |
1.12 |
MA |
Graphene |
0.64 |
120 °C 12 h |
39 |
| HDAa |
Graphene |
4.54 |
180 °C 12 h |
40 |
| GFAC-H-DMF |
94.5 |
4.2 |
1.3 |
0.38 |
MA |
GO |
0.35 |
60 °C 12 h |
41 |
| MA |
GO |
3.85 |
Ultrasonic 75 °C 10 h |
20 |
| GFAC-H-SF |
95.5 |
3.2 |
1.3 |
0.17 |
FAL |
MWCNTa |
0.32 |
150 °C 24 h |
42 |
| FAC |
MWCNT |
0.23 |
150 °C 24 h |
42 |
In addition, in terms of yield and energy consumption, for the preparation of GFAL-BM, for example, 100 g of graphite and 1000 g of FAL were required to produce 103 g of GFAL-BM. Based on the grafting rate of FAL, the loss of graphite was 7.6% and FAL was 98.9%. In terms of energy efficiency, the 0.087 kW h of electricity was required to produce 1 g of GFAL-BM, or 314 kJ of energy required (ignoring energy efficiency), which was the lower value of energy required for exfoliating graphite previously reported (300 kJ g−1).16 In the comparative literature in Table 2, mostly milligram level functionalized graphene was produced, which would lead to high energy consumption per unit yield. Therefore, the power utilization rate was better than previous reports, which was mainly due to the use of more large-scale ball milling equipment in this work.
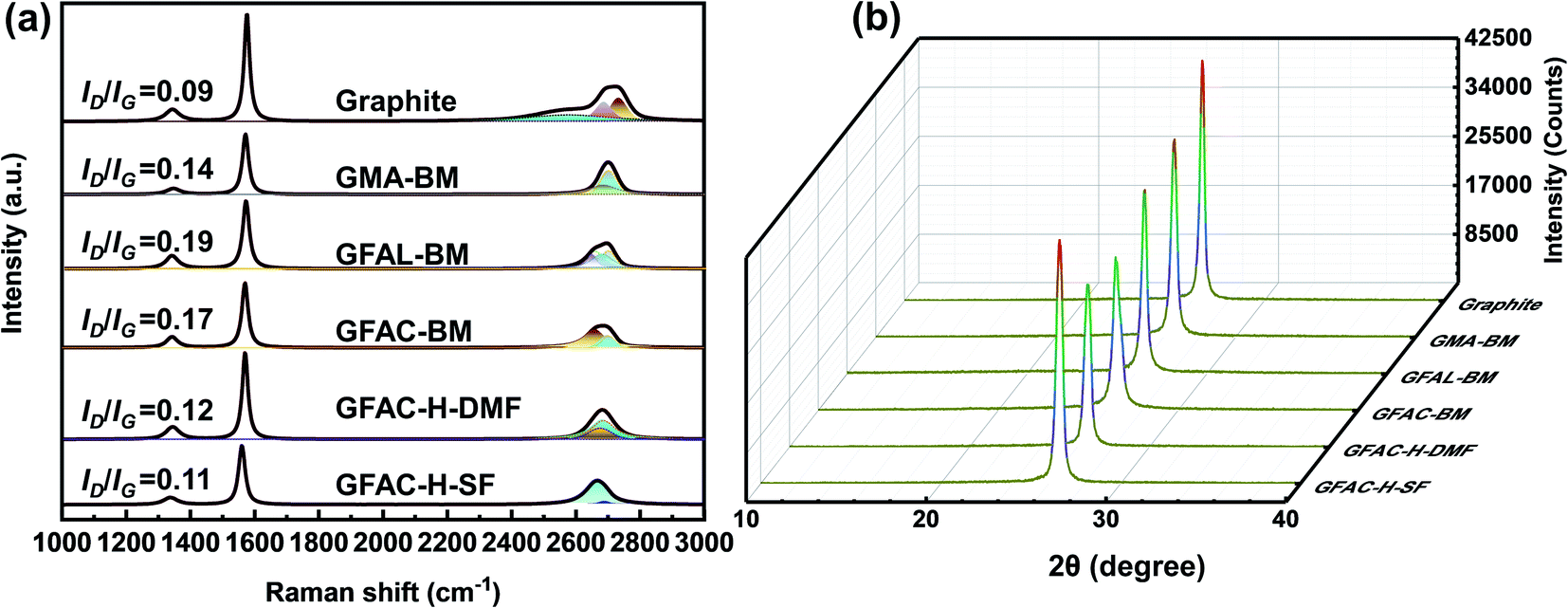
Raman spectroscopy is an effective and non-destructive tool to explore the degree of graphene functionalization.43 The graphene exhibited the following characteristic peaks in the region of 1000–3000 cm−1: D, G, and 2D peaks, which were located at around 1350, 1560, and 2700 cm−1, respectively. The D band was due to the vibration of the local sp3 disordered carbon atoms of disordered graphene. The G peak corresponded to the E2g phonon in-plane vibrations of the sp2 carbon lattice of the graphitic domain at the Brillouin zone center reflecting the symmetry and order of the material. The 2D peak is a two-phonon resonance Raman peak, and its intensity, shape, and position reflect the stacking degree of graphene.44 The intensity ratio of the D band to the G band, ID/IG, can be used to estimate the contents of functional groups. Fig. 6a shows the Raman spectra of graphite and functionalized graphene. Pristine graphite and G-BM (Fig. S4†) have an ID/IG ratio of only 0.09, indicating that there were only a few defects in the graphite sheets. The ID/IG values of the GMA-BM, GFAL-BM, and GFAC-BM were 0.14, 0.19, and 0.17, respectively. The increase in the ID/IG value indicated the formation of covalent bonds. Under the ball-milling conditions, based on the value of ID/IG, it was preliminarily determined that the degree of the [2 + 4] D–A of graphite was higher than that of [4 + 2] D–A during ball milling. The ID/IG values of the GFAL-BM and GFAC-BM showed that the GFAL-BM had a higher degree of functionalization. The number of layers of graphene sheets could be determined by the strength, shape, and position of the 2D band. However, when there are more than five layers, the specific number of graphite layers becomes difficult to distinguish by Raman spectroscopy.43 The Raman spectroscopy data showed that the 2D peak of functionalized graphene shifted to a lower wavenumber relative to that of the original graphite (2712 cm−1), indicating the effective preparation of a fewer layers of graphene. At the same time, the full width at half maximum (FWHM) of the 2D peak was reduced from about 80 cm−1 of graphite to about 50 cm−1 of functionalized graphene, which also proved that a few layers of graphene were produced. The 2D peak of the GFAL-BM could be decomposed into four components (Lorentzian-shaped peaks), which is a characteristic feature of few-layer (fewer than five layers) graphene Raman spectra, as reported previously.43–45
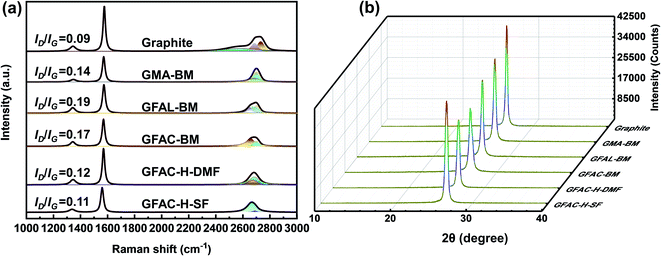 |
| | Fig. 6 (a) Raman and (b) X-ray diffraction (XRD) spectra of graphite and functionalized graphene. | |
The in-plane crystallite sizes (La, the size of the sp2 carbon clusters) of the graphite specimens were calculated using the following equation:
| | |
La = (2.4 × 10−10) × λlaser4 × (ID/IG)−1,
| (1) |
where
λlaser is the laser excitation wavelength (532 nm).
The results of La are listed in Table 3. La reflects the defect density or crystallite boundary along the base surface of the graphite material. Covalent functionalization can lead to a significant defect density (decreasing grain size), which further supports the conclusion that covalent bonds were generated after the functionalization reactions. The La value (213.6 nm) of graphite was the largest, while the La value (101.2 nm) of GFAL-BM was the smallest, indicating that GFAL-BM contained more defects in the graphite, that is, a higher degree of functionalization. The La values of the GFAL-BM (101.2 nm) and GFAC-BM (113.1 nm) were both smaller than that of the GMA-BM (137.3 nm), which proved that the [2 + 4] D–A of graphite was more dominant under the action of ball milling. The La values of the functionalized graphene fabricated by ball milling were generally lower than those obtained by the D–A via heating, indicating that ball milling was more conducive to promoting the D–A of graphite. In particular, the La value of the GFAC-BM was smaller than those of the GFAC-H-DMF and GFAC-H-SF, which also proved the advantage of ball milling for graphite functionalization.
Table 3 XRD and Raman analysis data of graphite and functionalized graphene
| Specimen |
2θ (°) |
d (nm) |
Lc (nm) |
La (nm) |
| Graphite |
26.6 |
0.33 |
26.9 |
213.6 |
| GMA-BM |
25.8 |
0.35 |
20.3 |
137.3 |
| GFAL-BM |
24.5 |
0.37 |
17.3 |
101.2 |
| GFAC-BM |
25.6 |
0.35 |
19.9 |
113.1 |
| GFAC-H-DMF |
25.4 |
0.35 |
21.6 |
160.2 |
| GFAC-H-SF |
26.3 |
0.33 |
22.7 |
174.8 |
The XRD spectra of the pristine graphite and functionalized graphene are shown in Fig. 6b. The grain size (Lc) was assessed based on the Scherrer formula.46 Evidently, a strong diffraction signal appeared at the 2θ value of ∼26°, which could be classified as a (002) diffraction signal. The variation trend of Lc (Table 3) was the same as that of La, which further proved the conclusion of the Raman analysis. The diffraction intensity decreased, indicating that the crystallinity of the functionalized graphene decreased. Interestingly, the interplanar spacing (d) slightly increased, indicating that although the surface functionalization increased the original d value of graphite, more functionalization could occur at the edge of the graphite sheet. Although the interlayer spacing after functionalization only increased by a small amount, it was sufficiently favorable for the organic molecules to enter the graphite lattice and react with graphite.
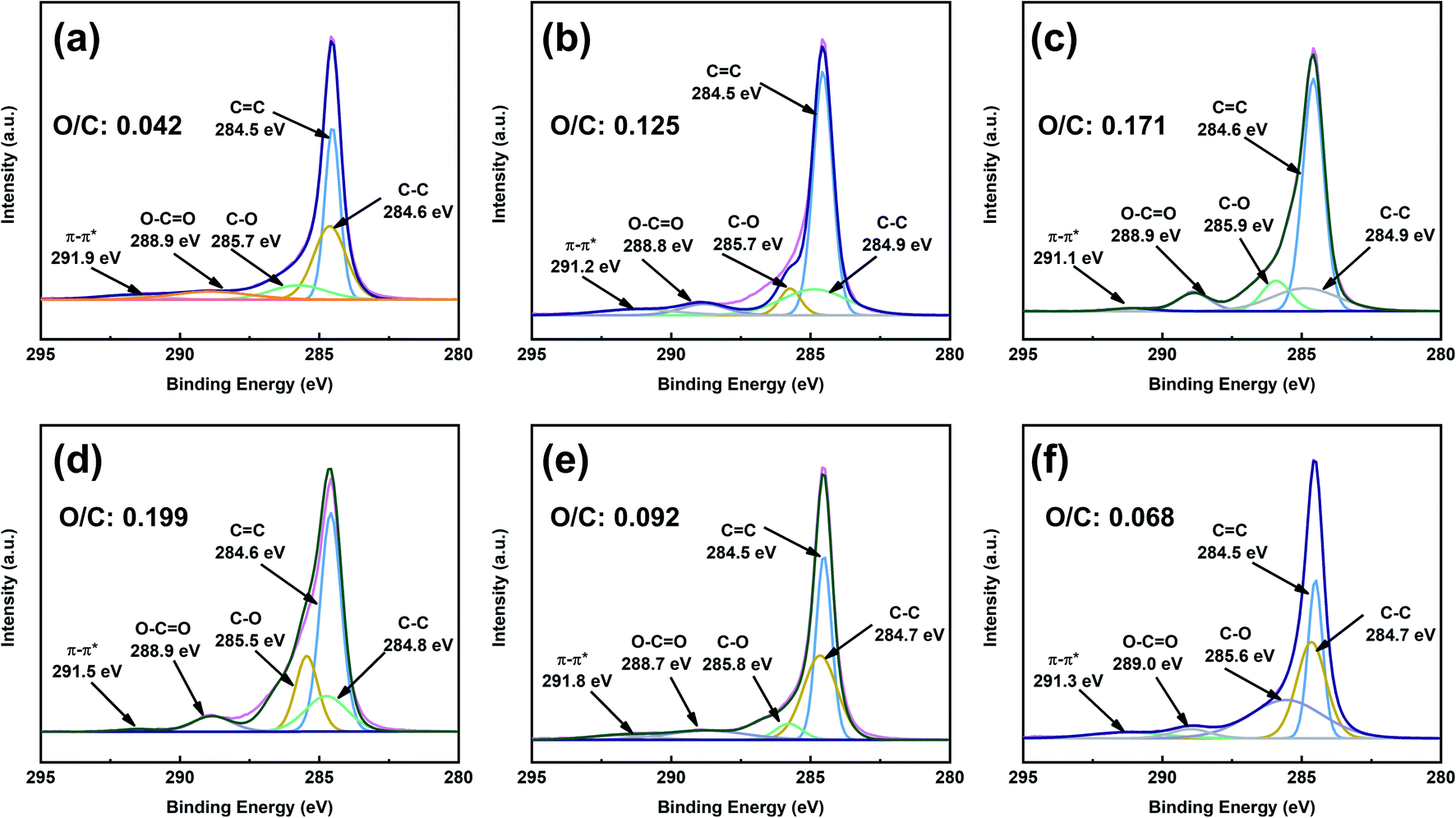
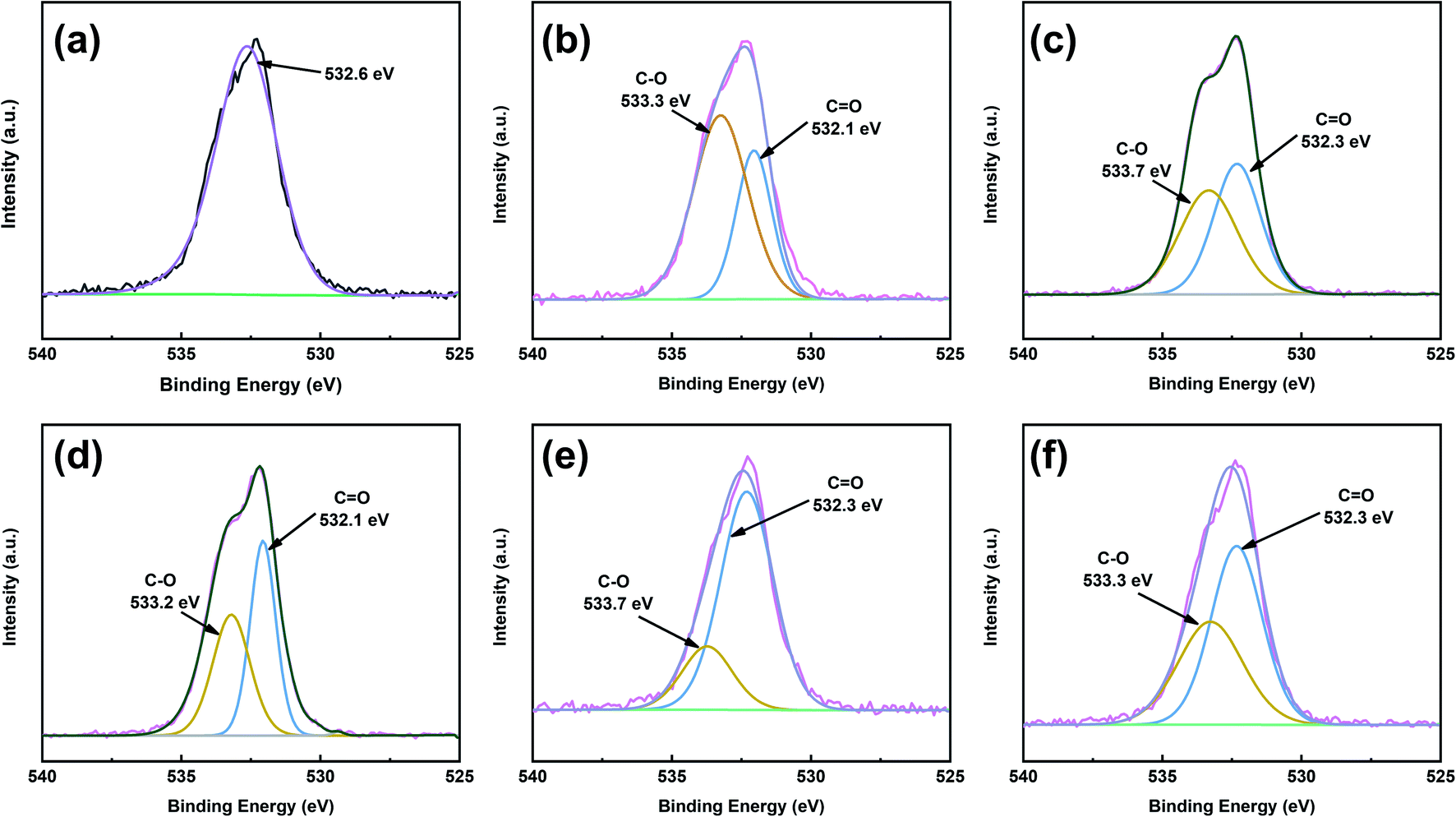
To obtain better insight into the surface chemistry of graphite, we performed XPS (Fig. 7 and 8). The C 1s region of pristine graphite could be fitted to five peaks (Fig. 7a), corresponding to C–C (284.6 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.5 eV), C–O (285.7 eV), O–CO (288.9 eV), and π–π* transitions (291.9 eV). Pristine graphite produced an O 1s signal at 532 eV (Fig. 8), which could be attributed to the physically absorbed oxygen on the graphite due to moisture, atmospheric O2, or CO2.47 The C 1s peaks of functionalized graphene were similar to those of graphite. The C 1s peak at ∼284 eV was attributed to the carbon skeleton. However, the C–O and O–CO peak intensities of functionalized graphene were significantly higher than those of pristine graphite, and the deconvolution of O 1s showed two electron binding energy peaks, indicating the presence of hydroxyl and carboxyl groups from the functionalized graphene. The O/C ratio of the functionalized graphene increased as the degree of functionalization increased. GFAC-BM had a high oxygen content, which was due to the high oxygen content of the FAC molecules. This was consistent with the EDS elemental analysis. The element analysis results of the EDS and XPS were different, mainly due to the differences in resolution caused by the working principles of the two techniques. XPS is more representative of the surface element information of graphite, while EDS is more representative of the overall element information of graphite. The trends of the elemental contents were the same, so they were mutually corroborated.
C (284.5 eV), C–O (285.7 eV), O–CO (288.9 eV), and π–π* transitions (291.9 eV). Pristine graphite produced an O 1s signal at 532 eV (Fig. 8), which could be attributed to the physically absorbed oxygen on the graphite due to moisture, atmospheric O2, or CO2.47 The C 1s peaks of functionalized graphene were similar to those of graphite. The C 1s peak at ∼284 eV was attributed to the carbon skeleton. However, the C–O and O–CO peak intensities of functionalized graphene were significantly higher than those of pristine graphite, and the deconvolution of O 1s showed two electron binding energy peaks, indicating the presence of hydroxyl and carboxyl groups from the functionalized graphene. The O/C ratio of the functionalized graphene increased as the degree of functionalization increased. GFAC-BM had a high oxygen content, which was due to the high oxygen content of the FAC molecules. This was consistent with the EDS elemental analysis. The element analysis results of the EDS and XPS were different, mainly due to the differences in resolution caused by the working principles of the two techniques. XPS is more representative of the surface element information of graphite, while EDS is more representative of the overall element information of graphite. The trends of the elemental contents were the same, so they were mutually corroborated.
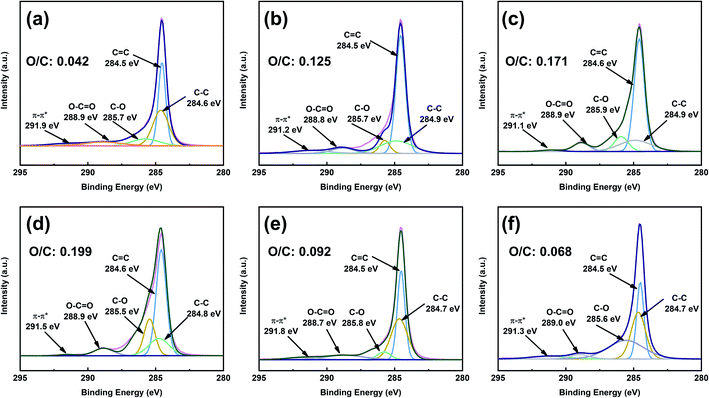 |
| | Fig. 7 C 1s X-ray photoelectron spectroscopy (XPS) spectra of graphite and functionalized graphene: (a) graphite, (b) GMA-BM, (c) GFAL-BM, (d) GFAC-BM, (e) GFAC-H-DMF, and (f) GFAC-H-SF. | |
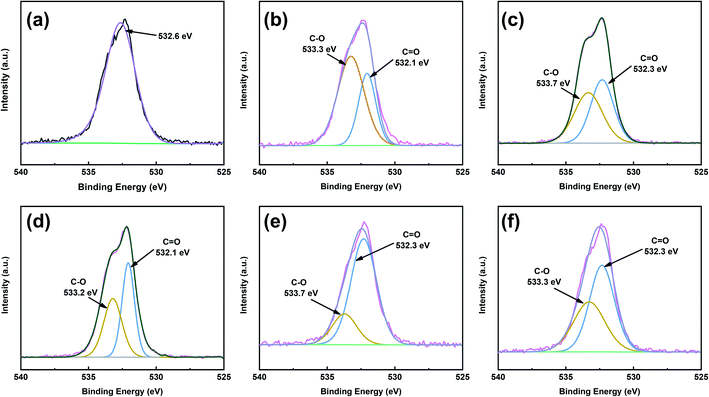 |
| | Fig. 8 O 1s XPS spectra patterns of graphite and functionalized graphene: (a) graphite, (b) GMA-BM, (c) GFAL-BM, (d) GFAC-BM, (e) GFAC-H-DMF, and (f) GFAC-H-SF. | |
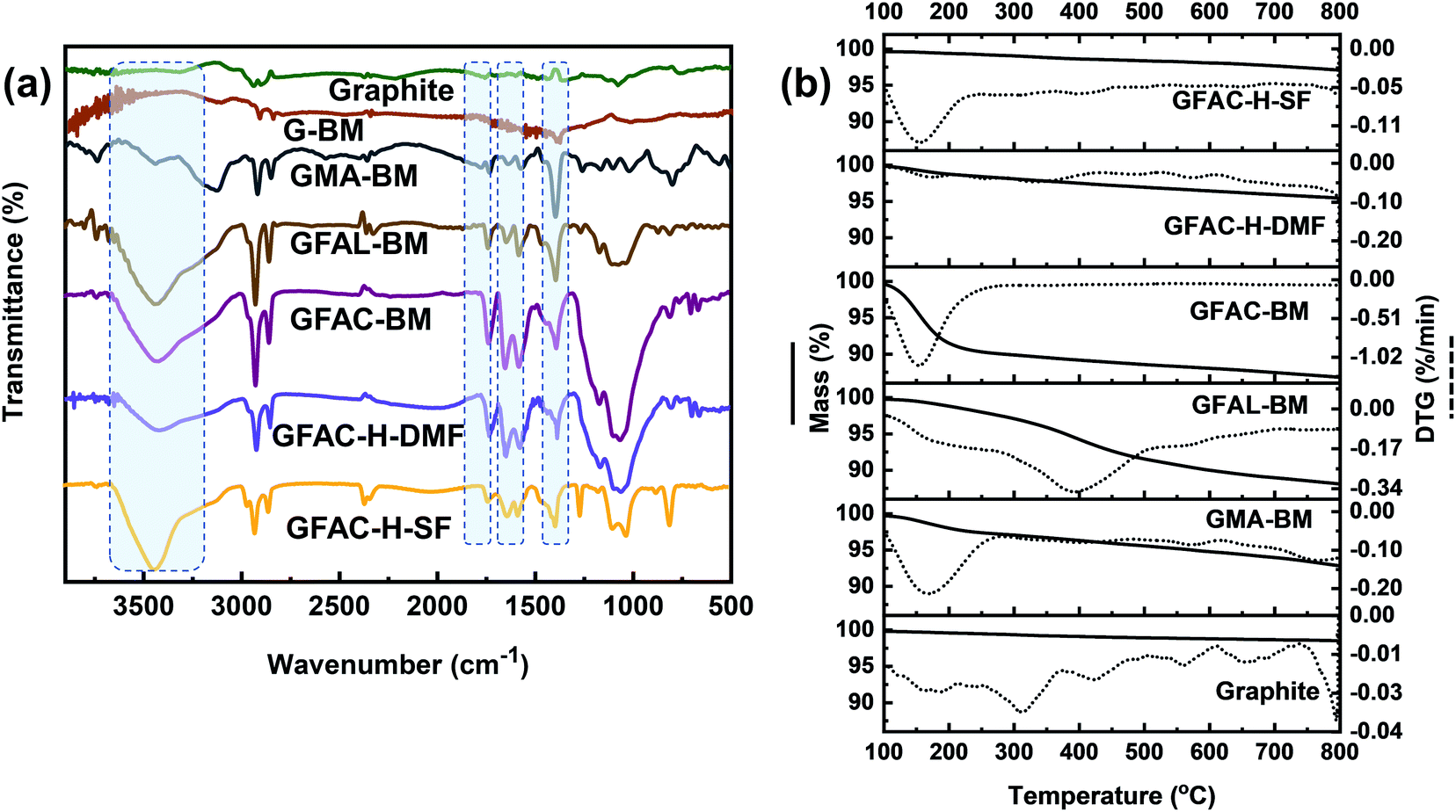
To further qualitatively identify D–A adducts, FT-IR was performed (Fig. 9a). The pristine graphite and G-BM showed a strong CC stretching band at 1590 cm−1 and other faint bands due to physically adsorbed moisture (3300 cm−1) and oxygen (1730 cm−1). Compared with the pristine graphite and G-BM, the FT-IR spectrum of the functionalized graphene showed several characteristic absorption peaks at 3300 cm−1, of which the strong peak came from the free stretching vibrations of the hydroxyl groups. The peaks at 1650 and 1560 cm−1 corresponded to the CC stretching vibrations of furan rings and substituted aromatic rings, respectively. The stretching vibration peaks of C-O and CO appeared at 1360 and 1730 cm−1, respectively, indicating the success of the organic functionalization by the [2 + 4] and [4 + 2] D–A processes of graphite. The difference in the FT-IR spectra between the GFAC-BM, GFAC-H-DMF, and GFAC-H-SF was that the vibration peaks of the GFAC-BM and GFAC-H-DMF were stronger than those of the GFAC-H-SF, which may have been contributed to partially by FAC molecules in the internal layer structure of graphene in the GFAC-H-SF, resulting in limited atom vibrations.
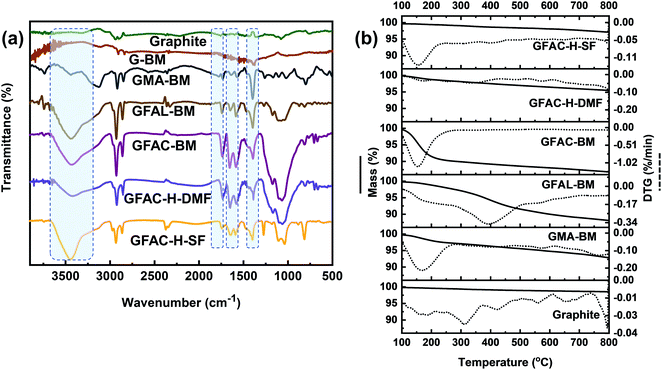 |
| | Fig. 9 (a) Fourier-transform infrared (FT-IR) spectra and (b) thermogravimetric analysis (TGA) curves of graphite and functionalized graphene. | |
The thermal stability of graphite and functionalized graphene was determined from the TGA thermograms (Fig. 9b). Compared with the functionalized graphene, there was no evident mass loss peak in the pristine graphite and G-BM (Fig. S5†). Below 200 °C, there were significant single mass loss plateaus in the GMA-BM, GFAC-BM, and GFAC-H-SF. Among all the specimens, GFAL-BM showed the most stable thermal resistance, with a maximum decomposition temperature of 388 °C. The residual masses of pristine graphite, G-BM,GMA-BM, GFAL-BM, GFAC-BM, GFAC-H-DMF, and GFAC-H-SF were 98.5%, 98.3%, 92.8%, 88.1%, 86.8%, 95.4%, and 97.1%, respectively, corresponding to graphite grafting ratios of 0, ∼0.58, ∼1.1, ∼1.0, ∼0.28, and ∼0.12 mmol g−1, respectively. In agreement with the data from Elemental Analyzer, FT-IR and Raman, the TGA decomposition curves of pristine graphite and G-BM were almost identical, which also indicated the consistency of the chemical structure of G-BM and graphite. The temperatures at which the masses of GMA-BM, GFAL-BM, and GFAC-BM were 95% of their original values were 574 °C, 379 °C, and 158 °C, respectively. The temperature of 574 °C was much higher than the maximum decomposition temperature of GMA-BM (167 °C), while the temperatures of 379 °C and 158 °C were similar to the maximum decomposition temperatures of GFAL-BM (388 °C) and GFAC-BM (150 °C), which further showed that the GFAL-BM had the highest thermal stability. The GFAL-BM had the highest grafting ratio. The wet ball milling may have been better at exfoliating the graphite and provided more reactive sites for the graphite with a larger specific surface area than dry ball milling. Therefore, considering the grafting ratio, the [2 + 4] D–A of graphene was more dominant than the [4 + 2] D–A by ball milling.
3.3 Dispersion performance of functionalized graphene
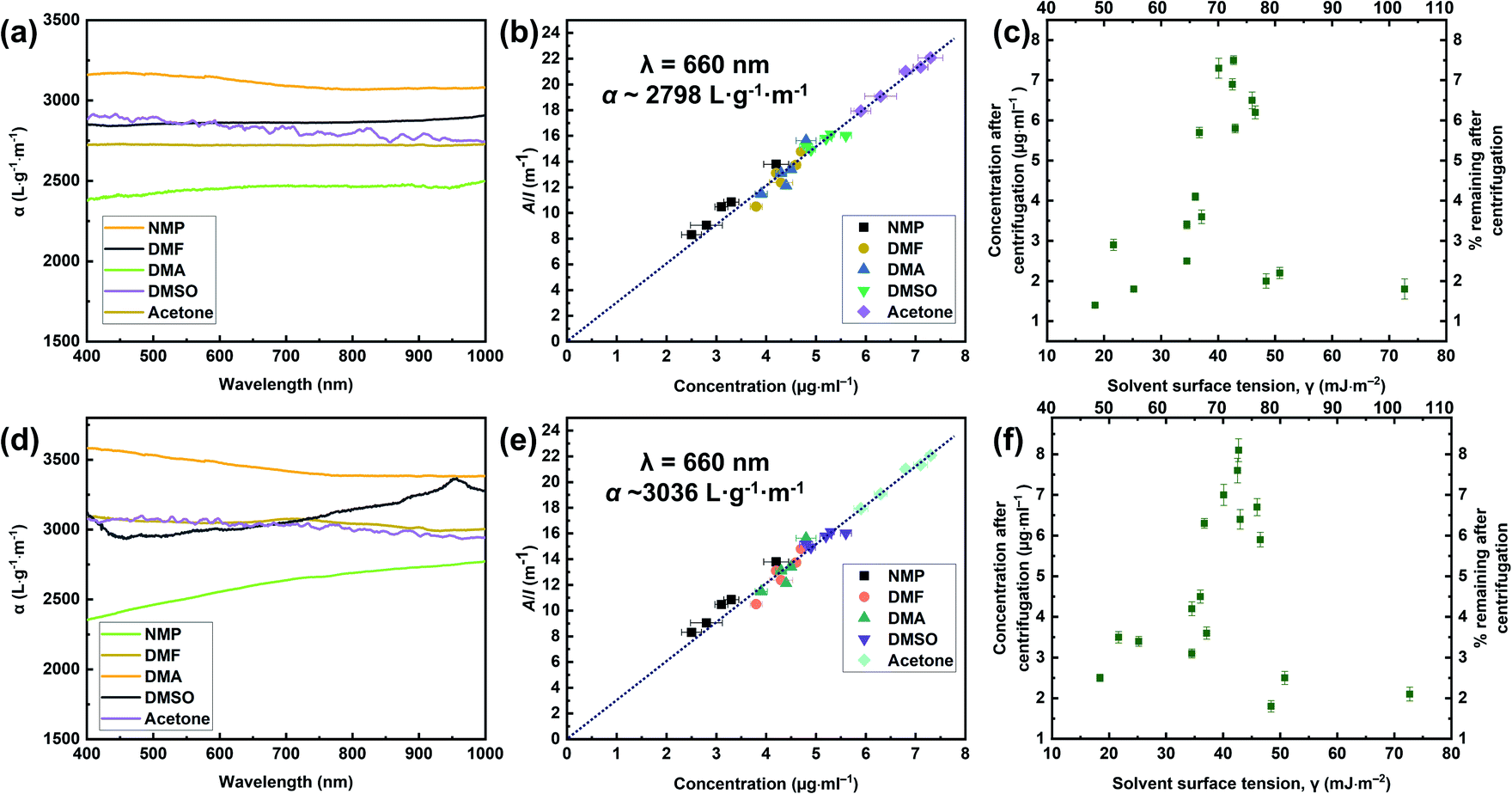
The exfoliation of graphite occurred because of the strong interactions between the solvent and the outside sheet, which meant that the energy loss of exfoliation and subsequent solvation was small. Through the above analysis and discussion, we found that the GFAL-BM had an advantage in terms of its functionalization or exfoliation effect compared to the specimens obtained by other processes. In the absorbance tests described in this section, the initial concentrations of graphite and GFAL-BM after 30 min of ultrasonic treatment, before centrifugation, were both 0.1 mg mL−1. The resultant dispersions were then centrifuged for 90 min at 500 rpm. To determine the concentrations after centrifugation, we passed the graphite and GFAL-BM dispersions through polyvinylidene fluoride filters. The filtered masses were carefully measured to obtain the dispersed phase concentrations after centrifugation. These dispersions were then characterized by UV absorption spectroscopy, and the absorption coefficients (α) varied with wavelength (Fig. 10a and d). These dispersions were diluted several times, and the absorption spectra at a wavelength of 660 nm were recorded (Fig. 10b and e). The absorption coefficients of the graphite and GFAL-BM specimens at a wavelength of 660 nm were calculated using the following equation:where A is the absorbance (660 nm), I is the cell length, and C is the graphite or GFAL-BM concentration.
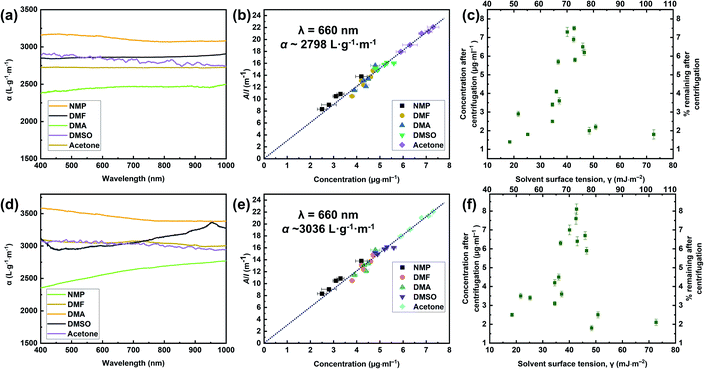 |
| | Fig. 10 Optical characterization of (a, b, c) pristine graphite and (d, e, f) GFAL-BM dispersions. (a, d) Absorption spectra for the graphite and GFAL-BM flakes dispersed in N-methyl-2-pyrrolidone (NMP), DMF, N-N′-dimethylacetamide (DMAc), dimethyl sulfoxide (DMSO), and acetone at concentrations ranging from 2 to 10 μg mL. (b, e) Optical absorbance divided by cell length (A/l) as a function of the concentration for graphite and GFAL-BM in the five solvents. (c, f) Graphite and GFAL-BM concentrations measured after centrifugation for a range of solvents plotted versus solvent surface tension. On the top axis of Fig. 10c and f, the number represents the surface energy (ESolSur, mJ m−2). | |
Fig. 10b and e shows that all the solvents used followed Lambert–Beer behavior. Furthermore, αgraphite = 2798 L g−1 m−1 and αGFAL-BM = 3036 L g−1 m−1. To study the dispersion behaviors of graphite and GFAL-BM in solvents under various surface tensions, the absorbances after centrifugation were measured. The corresponding concentrations after centrifugation were calculated using the absorption coefficients obtained from Fig. 10b and e. The exfoliation and good dispersion of graphite flakes could only occur when the net energy cost was small. In chemistry, this energy balance is expressed as the enthalpy of mixing (unit volume). In this case, we can approximate it as follows:15
| |
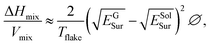 | (3) |
| | |
γ = ESolSur − TSSolSur,
| (4) |
where Δ
Hmix is the net energy cost,
Vmix is the volume of the mixture,
Tflake is the thickness of a graphene flake,
EGSur and
ESolSur are the surface energies of the phase graphite/GFAL-BM and solvent, respectively, ∅ is the graphite volume fraction,
γ is the surface tension of solvent, and
SSolSur is the solvent surface entropy (∼0.1 mJ m
−2 K
−1).
15
The enthalpy of mixing depended on the balance of the surface energy between graphite and the solvent. For graphite, the surface energy was defined as the energy per unit area required to overcome the van der Waals forces when two sheets of graphite were peeled off. Fig. 10c and f shows the dispersion behaviors of the graphite/GFAL-BM in solvents with various values of the surface tension/surface energy. The dispersion concentration showed a strong solvent peak, and its surface energy was close to that (∼65–80 mJ m−2) of graphite reported previously.48 In addition, the surface tensions of the graphite and GFAL-BM functionalized graphene had peaks in the range of 40–50 mJ m−2, which provides a theoretical basis for the dispersion and exfoliation of graphite and functionalized graphene in a suitable solvent.
To further verify the surface tension range of graphite and functionalized graphene, a series of contact angle tests were carried out on the specimens (Fig. 11). For pristine graphite, the steady-state contact angle under various solvents reached the lowest value in DMSO, indicating that the surface tension of graphite was closer to that of DMSO (∼43.0 mJ m−2). Unlike pristine graphite, the minimum contact angle of GFAL-BM appeared in DMAc, that is, the approximate surface tension was 36.7 mJ m−2, indicating that functionalized graphene would significantly reduce the original high surface energy characteristics of graphite. Moreover, the water contact angles of the other functionalized graphene samples were smaller than that of graphite, indicating that the functionalized graphene was more hydrophilic.
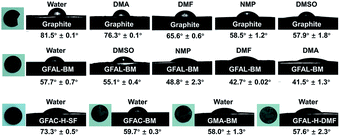 |
| | Fig. 11 Contact angles of graphite and functionalized graphene in various solvents. | |
3.4 Thermal and electrical properties of functionalized graphene
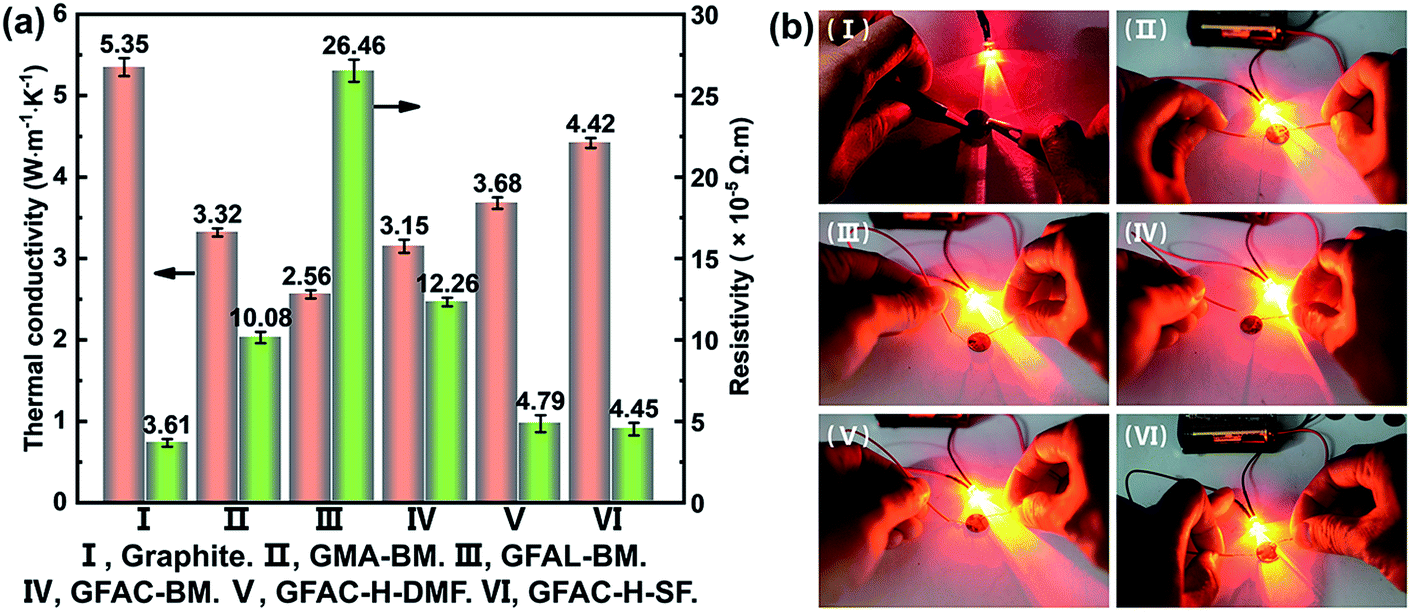
The thermal conductivity and electrical resistivity of graphite, which was formed into a circular disk with a diameter of 1 cm (thickness over 1 mm), were measured. Fig. 12a shows a histogram of the specific data, and Fig. 12b shows a photograph of the round slice as part of the circuit lit up by a light-emitting diode (LED). The thermal conductivity of the pristine graphite (5.35 W m−1 K−1) sheet was much lower than the theoretical value of single-layer graphene (5300 W m−1 K−1).49 This was because the thermal conductivity of graphite was mainly reflected in the horizontal plane of the graphite structure. The thermal conductivity perpendicular to the graphite plane structure was very low, and a large amount of graphite was pressed into a compact slice with a thermal conductivity of 5.35 W m−1 K−1, indicating that the graphite sheet structure was mostly stacked. When the graphite surface was organically functionalized, the thermal conductivity decreased, which was caused by the destruction of the regular electronic arrangement of graphite. Importantly, the GFAL-BM with the highest grafting ratio had the lowest thermal conductivity (2.56 W m−1 K−1). Similar to the thermal conductivity, the original resistivity of pristine graphite was 3.61 × 10−5 Ω m, which was close to the planar resistivity of graphite reported in the literature.50 Corresponding to the thermal conductivity results, as the degree of functionalization of the graphite surface increased, the resistivity increased from 3.61 × 10−5 Ω m of the pristine graphite to 26.46 × 10−5 Ω m of the GFAL-BM. Although the conductivity of graphite was increased by more than seven times at most, it still had an excellent conductive effect based on Fig. 12b, which has far-reaching significance for functionalized graphene in electronic semiconductors and related applications.
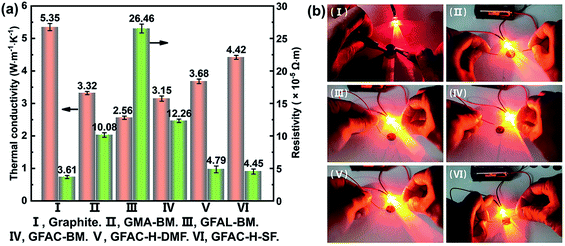 |
| | Fig. 12 (a) Thermal conductivity and electrical resistivity of graphite and functionalized graphene. (b) Digital photographs of the electrical conductivity experiments. | |
3.5 Mechanical properties of PET/GFAL-BM composite fiber
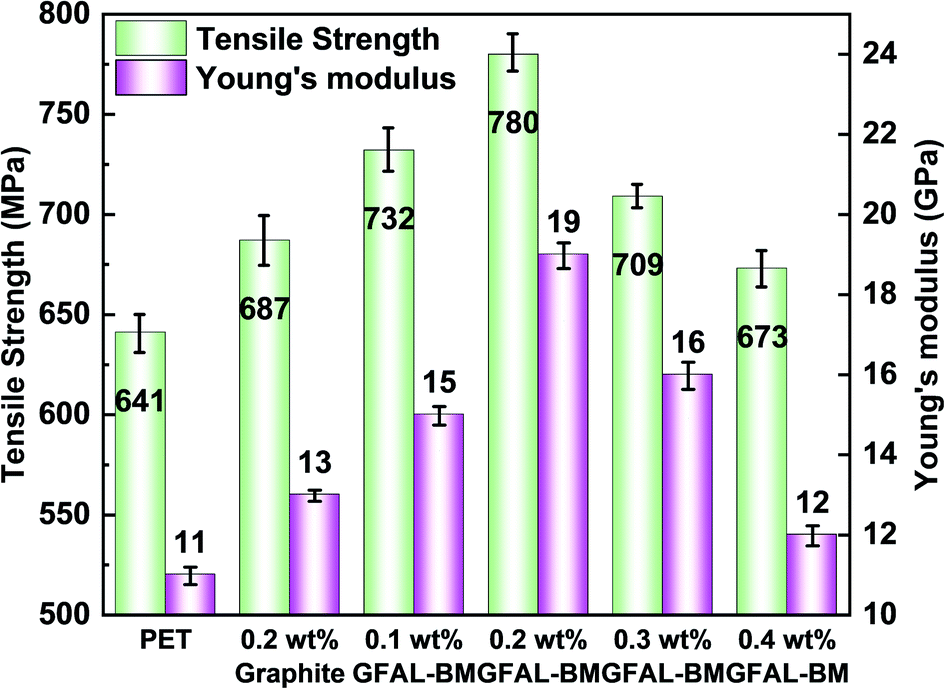
The dispersion of functionalized graphene in PET composite fibers and the effect on crystallinity of fibers are shown in the ESI (S2, S3†). Fig. 13 presents the influence of GFAL-BM on the mechanical properties of PET composite fibers. After adding graphite or GFAL-BM, the tensile strength and Young's modulus of the fiber were improved. When the loading of GFAL-BM was 0.2 wt%, the mechanical strength of the composite fiber was the best, the tensile strength and the Young's modulus could reach 780 MPa, 19 GPa. Compared with pure PET fiber, the tensile strength and the modulus were increased by 21.7% and 72.7%, respectively. The content of GFAL-BM exceeded 0.2 wt%, the mechanical properties of the fiber decreased slightly. When the addition amount was 0.4 wt%, the mechanical properties were even lower than that of 0.2 wt% graphite. This may be due to the agglomeration of excessive GFAL-BM in the PET matrix. In the process of fiber stretching and fracture, the aggregated sheet layer produced stress concentration inside the fiber. According to the analysis of DSC crystallization data, the higher the crystallinity of the fiber, the better the mechanical properties. The fiber with high crystallinity can withstand higher stress during the stretching process. Meanwhile, GFAL-BM was interspersed between PET crystals to form a mutual transmission network. During the stretching process, more stress could be transferred to the well-dispersed GFAL-BM.
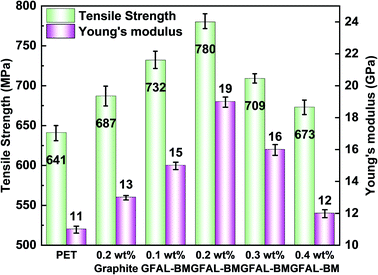 |
| | Fig. 13 The effect of GFAL-BM loading on the tensile strength and Young's modulus of PET composite fiber. | |
3.6 Effect of GFAL-BM on the electrical and thermal properties of composite fibers
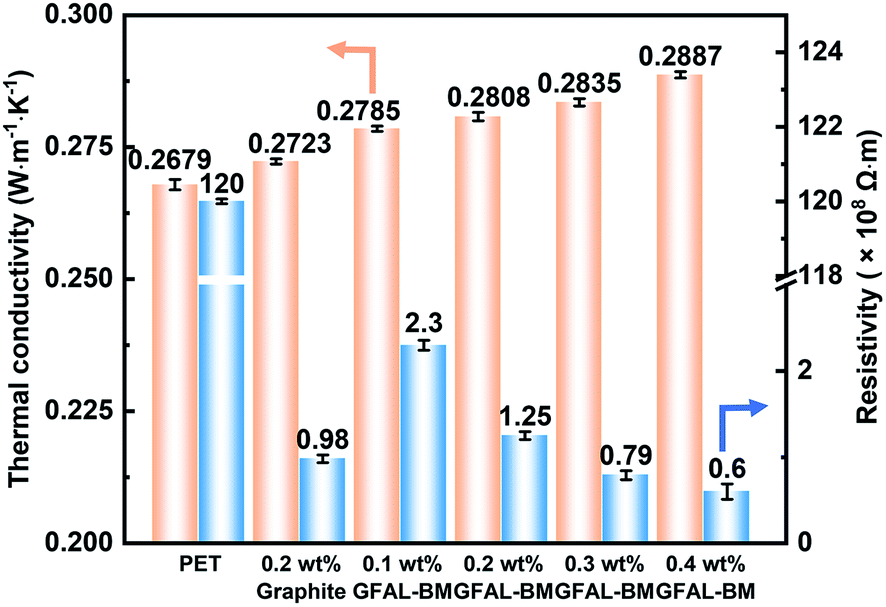
Fig. 14 shows the thermal conductivity and electrical properties of PET/GFAL-BM composite. After the graphite or GFAL-BM was added, the thermal conductivity of the composite increased. And it increased as the loading of GFAL-BM increased. The thermal conductivity of 0.2 wt% PET/GFAL-BM composite (0.2808 W m−1 K−1) was 4.8% higher than that of pure PET (0.2679 W m−1 K−1). At the same content, the effect of GFAL-BM on the thermal conductivity of the composite was significantly higher than that of graphite. This was due to the strong interaction between GFAL-BM and PET, which promoted heat transfer in the form of sound energy. In addition, the vibration coupling at the interface of PET and GFAL-BM significantly reduced the generation of thermal resistance, so thermal conductivity can be improved.
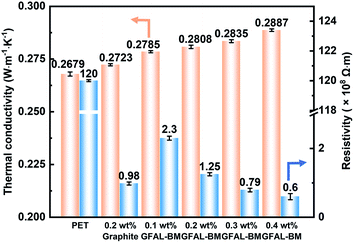 |
| | Fig. 14 Thermal conductivity and electrical properties of PET/GFAL-BM composite fiber. | |
After adding graphite and GFAL-BM, the volume resistivity of the fiber decreased significantly, and as the loading of GFAL-BM increased, the volume resistivity of the fiber decreased. As shown in Fig. 14, after adding 0.2 wt% graphite, the volume resistivity of the fiber decreased from 1.2 × 1010 Ω m of pure PET to 9.8 × 107 Ω m. Compared with adding the same loading of graphite, the volume resistivity of PET/GFAL-BM fiber (1.25 × 108 Ω m) was two orders of magnitude lower than that of pure PET. Although the dispersibility was better than graphite, there was still a gap in the structure of graphite. It could be seen that as the loading of GFAL-BM increased, the resistivity of fiber gradually decreased. When the loading exceeded 0.2 wt%, the decrease in volume resistivity gradually slowed down. This was because the content of GFAL-BM increased in the polymer matrix. The dispersibility of the polymer became poor, which was not conducive to the formation of the conductive network in the polymer. When the loading of GFAC-BM exceeds 0.3 wt%, the antistatic effect of the nanocomposite fibers were achieved.
4. Conclusions
Graphite as a diene or as a dienophile was functionally modified by a variety of D–A processes. Through a ball-milling method, in situ functionalization could be performed while exfoliating graphite into a few layers (≤2) of graphene simultaneously. Compared with the specimens functionalized with MA as a dienophile (0.58 mmol g−1), the functionalized graphene specimens fabricated by ball milling with FAL (1.17 mmol g−1) and furoic acid (1.12 mmol g−1) as the dienes had higher grafting yields. The results show that the [2 + 4] D–A of graphene was more dominant than the [4 + 2] D–A by ball milling. In addition, compared with heat treatment reactions, the fabrication of functionalized graphene by ball milling has a higher efficiency and greater application prospects.
Unlike pristine graphite, the minimum contact angle of GFAL-BM appeared in DMAc (the approximate surface tension was 36.7 mJ m−2), indicating that functionalized graphene significantly reduced the original high surface energy characteristics of graphite. When the graphite surface was organically functionalized, the thermal conductivity and the electrical conductivity decreased, which was caused by the destruction of the regular electronic arrangement of graphite. Despite this, it still had an excellent conductive effect, which has far-reaching significance for functionalized graphene, polymer composite materials, and related applications. In the field of polymer applications, GFAL-BM had the best dispersibility in PET composite fibers. When the addition amount of GFAL-BM was 0.2 wt%, the mechanical properties were the best, the tensile strength could reach 780 MPa, and the Young's modulus reached 19 GPa, the tensile strength and the modulus were increased by 21.7% and 72.7% compared with pure PET fiber. The thermal conductivity of composite fiber (0.2808 W m−1 K−1) was 4.8% higher than that of pure PET fiber (0.2679 W m−1 K−1). The volume resistivity was two orders of magnitude lower than that of pure PET fiber. We believe that this work provides an effective nanotechnology method, that is, the use of ball milling, to efficiently exfoliate and in situ functionalize graphite with sole functional groups. This could become a powerful alternative process for the fabrication of functionalized graphene by the graphite oxidation route and could help to develop a strategy that can be widely used to manufacture nanomaterials and nanocomposite.
Author contributions
Wenguang Yu (the first author): writing – original draft, conceptualization, data curation, investigation. Xuefeng Gao: investigation, validation. Zhicheng Yuan: investigation, validation. Haihui Liu: resources, project administration. Xuechen Wang: data curation, validation. Xingxiang Zhang (the corresponding author): writing – review & editing, conceptualization, investigation, funding acquisition, validation.
Conflicts of interest
We declare that we have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant No. 2016YFB0303000), the New Materials Research Key Program of Tianjin (Grant No. 16ZXCLGX00090) and the Tianjin Science and Technology Key Project (Grant No. 18ZXJMTG00110).
References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- T. Liu, S. Zhou, X. Yu, C. Mao, Y. Wei, X. Yu, L. Chen, X. Zhao, G. Tian and L. Chen, RSC Adv., 2022, 12, 4029–4041 RSC.
- J. He, Y. Liu, Y. Meng, X. Sun, S. Biswas, M. Shen, Z. Luo, R. Miao, L. Zhang, W. E. Mustain and S. L. Suib, RSC Adv., 2016, 6, 24320–24330 RSC.
- W. Guo, X. Li, D. H. L. Ng and J. Ma, RSC Adv., 2015, 5, 96681–96684 RSC.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed.
- X. Wang, D. Tan, Z. Chu, L. Chen, X. Chen, J. Zhao and G. Chen, RSC Adv., 2016, 6, 112486–112492 RSC.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- I. Meric, C. R. Dean, A. F. Young, N. Baklitskaya, N. J. Tremblay, C. Nuckolls, P. Kim and K. L. Shepard, Nano Lett., 2011, 11, 1093–1097 CrossRef CAS PubMed.
- Z. Y. Yin, S. X. Wu, X. Z. Zhou, X. Huang, Q. C. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307–312 CrossRef CAS PubMed.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- R. Q. Ye and J. M. Tour, ACS Nano, 2019, 13, 10872–10878 CrossRef CAS PubMed.
- W. Kong, H. Kum, S. H. Bae, J. Shim, H. Kim, L. P. Kong, Y. Meng, K. J. Wang, C. Kim and J. Kim, Nat. Nanotechnol., 2019, 14, 927–938 CrossRef CAS PubMed.
- W. C. Du, X. Q. Jiang and L. H. Zhu, J. Mater. Chem. A, 2013, 1, 10592–10606 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- M. Buzaglo, I. P. Bar, M. Varenik, L. Shunak, S. Pevzner and O. Regev, Adv. Mater., 2017, 29, 1603528 CrossRef PubMed.
- V. Leon, M. Quintana, M. A. Herrero, J. L. G. Fierro, A. de la Hoz, M. Prato and E. Vazquez, Chem. Commun., 2011, 47, 10936–10938 RSC.
- H. Y. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- W. G. Yu, X. F. Gao, Y. Z. Wang, X. C. Wang and X. X. Zhang, Macromol. Mater. Eng., 2021, 306, 2000661 CrossRef CAS.
- S. Sarkar, E. Bekyarova, S. Niyogi and R. C. Haddon, J. Am. Chem. Soc., 2011, 133, 3324–3327 CrossRef CAS PubMed.
- J. M. Seo and J. B. Baek, Chem. Commun., 2014, 50, 14651–14653 RSC.
- J. M. Seo, I. Y. Jeon and J. B. Baek, Chem. Sci., 2013, 4, 4273–4277 RSC.
- Y. Cao, S. Osuna, Y. Liang, R. C. Haddon and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 17643–17649 CrossRef CAS PubMed.
- L. Daukiya, C. Mattioli, D. Aubel, S. Hajjar-Garreau, F. Vonau, E. Denys, G. Reiter, J. Fransson, E. Perrin, M. L. Bocquet, C. Bena, A. Gourdon and L. Simon, ACS Nano, 2017, 11, 627–634 CrossRef CAS PubMed.
- L. V. Frolova, I. V. Magedov, A. Harper, S. K. Jha, M. Ovezmyradov, G. Chandler, J. Garcia, D. Bethke, E. A. Shaner, I. Vasiliev and N. G. Kalugin, Carbon, 2015, 81, 216–222 CrossRef CAS PubMed.
- A. Abdolmaleki, S. Mallakpour, M. Mahmoudian, S. Kamali, M. Zhiani, B. Rezaei and A. R. T. Jahromi, ChemistrySelect, 2018, 3, 13070–13075 CrossRef CAS.
- J. Zhang, W. Wang, H. Peng, J. Qian, E. Ou and W. Xu, J. Iran. Chem. Soc., 2016, 14, 89–93 CrossRef.
- O. Zabihi, M. Ahmadi, T. Abdollahi, S. Nikafshar and M. Naebe, Sci. Rep., 2017, 7, 3560 CrossRef PubMed.
- S. Sarkar, E. Bekyarova and R. C. Haddon, Acc. Chem. Res., 2012, 45, 673–682 CrossRef CAS PubMed.
- J. F. Xu, X. M. Zhao, F. X. Liu, L. Jin and G. H. Chen, New J. Chem., 2020, 44, 1236–1244 RSC.
- Z. H. Ni, H. M. Wang, J. Kasim, H. M. Fan, T. Yu, Y. H. Wu, Y. P. Feng and Z. X. Shen, Nano Lett., 2007, 7, 2758–2763 CrossRef CAS PubMed.
- W. Liu, V. A. Tanna, B. M. Yavitt, C. Dimitrakopoulos and H. H. Winter, ACS Appl. Mater. Interfaces, 2015, 7, 27027–27030 CrossRef CAS PubMed.
- A. Ciesielski and P. Samori, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- I. Y. Jeon, S. Y. Bae, J. M. Seo and J. B. Baek, Adv. Funct. Mater., 2015, 25, 6961–6975 CrossRef CAS.
- O. N. Shornikova, E. V. Kogan, N. E. Sorokina and V. V. Avdeev, Russ. J. Phys. Chem. A, 2009, 83, 1022–1025 CrossRef CAS.
- J. Zhang, K. Hu, Q. Ouyang, Q. Gui and X. Chen, Front. Mater. Sci., 2020, 14, 198–210 CrossRef.
- J. Yuan, G. Chen, W. Weng and Y. Xu, J. Mater. Chem., 2012, 22, 7929 RSC.
- Y. Wang, Q. Liang, S. Liu, Z. Yan, X. Yu and T. Wang, Plast. Rubber Compos., 2021, 50, 1–11 CrossRef.
- R. R. Cao, Y. Z. Wang, S. Chen, N. Han, H. H. Liu and X. X. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 8982–8991 CrossRef CAS PubMed.
- P. P. Brisebois, C. Kuss, S. B. Schougaard, R. Izquierdo and M. Siaj, Chemistry, 2016, 22, 5849–5852 CrossRef CAS PubMed.
- S. Munirasu, J. Albuerne, A. Boschetti-de-Fierro and V. Abetz, Macromol. Rapid Commun., 2010, 31, 574–579 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- W. G. Yu, X. Y. Zhang, X. F. Gao, H. H. Liu and X. X. Zhang, J. Mater. Sci., 2020, 55, 8940–8953 CrossRef CAS.
- C. M. Q. Le, X. T. Cao and K. T. Lim, Ultrason. Sonochem., 2017, 39, 321–329 CrossRef CAS PubMed.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- K. Sun, M. A. Stroscio and M. Dutta, Superlattices Microstruct., 2009, 45, 60–64 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab