
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst stereoselective total synthesis of 4(S),5(S)-oxido-17(S)-hydroxy-6(E),8(E),10(Z),13(Z),15(E),19(Z)-docosahexaenoic acid, the biosynthetic precursor of resolvins D3 and D4†
Robert Nshimiyimana a,
Ting Fung Lama,
Shubhangi Aggarwala,
Charles N. Serhanb and
Nicos A. Petasis*a
a,
Ting Fung Lama,
Shubhangi Aggarwala,
Charles N. Serhanb and
Nicos A. Petasis*a
aDepartment of Chemistry and Loker Hydrocarbon Research Institute, University of Southern California, Los Angeles, California 90089, USA. E-mail: petasis@usc.edu
bCenter for Experimental Therapeutics and Reperfusion Injury, Department of Anesthesiology, Perioperative and Pain Medicine, Harvard Institutes of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts 02115, USA
First published on 19th April 2022
Abstract
The first total convergent synthesis of 4(S),5(S)-oxido-17(S)-hydroxy-6(E),8(E),10(Z),13(Z),15(E),19(Z)-docosahexaenoic acid (1) is described. The reported synthesis led to confirmation of the native epoxydocosahexaenoic acid as the biosynthetic precursor of lipid mediators resolvin D3 and resolvin D4. These potent enzymatic products of docosahexaenoic acid (DHA) are important signaling molecules in the resolution of inflammation. A stereocontrolled and chiral pool-based synthetic strategy was employed, with key features including epoxide transposition under basic conditions to form the oxirane ring, and a cis-selective Wittig reaction to secure the target docosahexaenoate backbone.
Introduction
Acute inflammation is an immunological response necessary to restore normal function in the face of injurious stimuli. Although this phenomenon is ideally self-limited, it can give rise to excessive inflammation if left unresolved.1 Research efforts involving the enzymatic oxygenation of polyunsaturated fatty acids (PUFAs) during the acute inflammatory process have led to the discovery of several classes of lipid mediators (LMs) that switch on resolution responses and promote tissue healing.1These studies uncovered a superfamily of pro-resolution lipid mediators, including the lipoxins2 and the E-series resolvins3 derived from arachidonic acid (AA) and eicosapentaenoic acid (EPA), respectively, as well as the D-series resolvins,4 the protectins,5 and the maresins6 produced from docosahexaenoic acid (DHA). A recent addition to this class of bioactive metabolites are the LMs originated from docosapentaenoic acid (DPA), including T-series resolvins and n-3 DPA series of resolvins, protectins, and maresins.7 Together, these hormone-like autacoids are termed specialized pro-resolving mediators (SPMs).8
SPMs counter-regulate proinflammatory signals and govern leukocytic movement, e.g., neutrophils and macrophages, at the inflamed site in order to protect the host.9 Notably, these molecules are generated in minuscule quantities in vivo and are enzymatically formed by lipoxygenases (LOX), cyclooxygenases (COX), and cytochrome P450s, often exhibiting potent bioactions in the low nM to pM range.9 Structurally, each SPM contains carbon–carbon double bonds with defined olefin geometry, as well as hydroxyl and peptidyl substituents with specified R- or S-configurations of tetrahedral centers. Due to their limited endogenous production,9 complete structural elucidation of LMs demands direct matching of endogenous metabolites with stereochemically pure synthetic materials.
In the present report, we disclose the first stereocontrolled total synthesis of 4(S),5(S)-epoxy-17(S)-hydroxy-6(E),8(E),10(Z),13(Z),15(E),19(Z)-docosahexaenoic acid (1). For storage and handling purposes, 1 was kept in the methyl ester form owing to its increased stability vis-à-vis the free acid and was readily hydrolysed as needed. The naturally occurring 4(S),5(S)-epoxy-docosanoid was originally postulated as the biosynthetic precursor to both resolvins D3 (RvD3) and D4 (RvD4).4 The synthetic accessibility of this critical intermediate enabled biological investigations that corroborated this hypothesis, revealing that human leukocytes convert this transient epoxide to both RvD3 and RvD4.10
As autacoids acting on local tissues, both RvD3 and RvD4 are involved in restoring normal cellular functions following inflammatory insults. RvD3 evokes protective actions by reducing pro-inflammatory cytokines, and enhancing human macrophage efferocytosis and neutrophil-bacterial phagocytosis; RvD3 and its 17R-epimeric isomer also initiate leukocytic actions, and relieve dermal inflammation.11 Furthermore, RvD4 stimulates clearance of activated neutrophils by human macrophages at nM doses.12 This chemical mediator also alleviates deep vein thrombosis in mice,13 protects against secondary organ injury, inhibits PMN infiltration during skin infection, and enhances macrophage efferocytosis of cellular debris.12
The biosynthetic pathways involved in the conversion of DHA to the 4(S),5(S)-epoxy-17(S)-hydroxy-DHA, 4(S),5(S)-epoxy-17(S)-HDHA (1), and its derived pro-resolving products are illustrated in Scheme 1.4,12 Enzymatic lipoxygenation of DHA catalyzed by 15-lipoxygenase (15-LOX) yields 17S-hydroperoxy-docosahexaenoic acid (17S-HpDHA). A second oxygenation by 5-LOX generates a hydroperoxide at the C4 position, and this intermediate is further transformed to the 4(S),5(S)-epoxide (1),4 a direct precursor to both RvD3 and RvD4.10
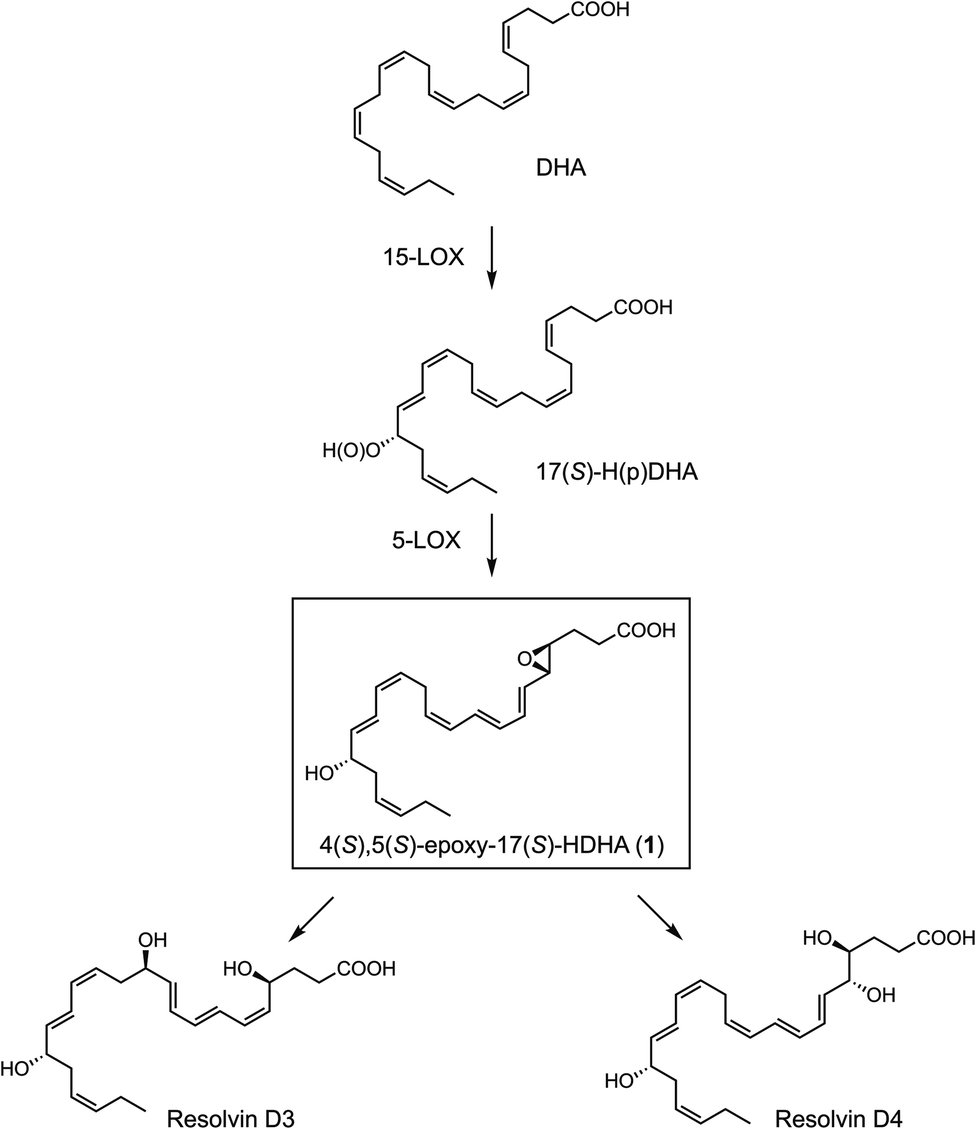 | ||
| Scheme 1 Biosynthesis of resolvin D3 and resolvin D4 via 4(S),5(S)-epoxy-17(S)-HDHA (1). | ||
Results and discussion
Retrosynthetic analysis
An overview of retrosynthetic disconnections leading to the target compound (1) is outlined in Scheme 2. Following our previous strategies, we approached the current synthesis with similar guiding principles, by employing nonracemic and commercially available building fragments and relying on highly stereoselective transformations to establish desired configurations of the target molecule—viz., 4(S),5(S)-epoxy-17(S)-hydroxy-6(E),8(E),10(Z),13(Z),15(E),19(Z)-docosahexaenoic acid (1). | ||
| Scheme 2 Retrosynthetic disconnections made for the target compound (1). | ||
Given the extreme lability of this polyunsaturated fatty acid epoxide, we devised and conducted the synthesis in a convergent manner. The retrograde analysis identified compounds 2, 11, and 12 as the key intermediates to be connected, with commercially available synthon 11 playing the role of a linker allowing the access to a phosphorane required for the final coupling.
Epoxy aldehyde 2 was disconnected back to D-erythronolactone (7), with a base-mediated epoxide rearrangement planned as the critical step to install the epoxide functionality with absolute stereoselectivity. On the other hand, (E)-alkenyl halide 12 was deconstructed to cis-4-heptenal (15), with a highly enantioselective organocatalytic α-oxidation14 and a Takai olefination being key transforms in the forward direction.
The Sonogashira cross-coupling between linchpin 11 and vinyl iodide 12 was determined as the key reaction for furnishing a 1,3-enyne-containing intermediate, with the intent to create phosphonium salt 8 after further straight-forward manipulations.
Taking advantage of a cis-selective Wittig condensation in the final stages, we envisioned the union between the two segments, C1–C10 (2) and C11–C22 (8), to yield the desired docosahexaenoate carbon skeleton of compound (1). This approach was chosen over cross-coupling chemistry to avoid tampering with the labile epoxide group, as well as prevent possible double bond isomerization.
Synthesis of C1–C10 α-fragment (2)
The synthesis commenced with the construction of the α-segment 2. As indicated in Scheme 3 (A), we initially envisioned installing the oxirane ring via a Sharpless asymmetric epoxidation,16 borrowing from our previous strategies. To this end, we projected to generate a prochiral allylic alcohol substrate first. | ||
| Scheme 3 Initial approaches toward the C1–C10 fragment (2). | ||
A straight-forward esterification of γ-butyrolactone 18 in basic methanol and a subsequent Dess–Martin oxidation of the resulting alcohol gave aldehyde 17 in fair yields. A two-carbon Wittig reaction extension followed by NaBH4-based reduction of the intermediary allylic aldehyde favorably led to allylic alcohol 16 in good yields. With the said substrate at hand, the epoxidation was executed with (+)-diethyl L-tartrate in methylene chloride under the prescribed conditions.16
Surprisingly, however, TLC and 1H NMR analyses of the crude mixture, before workup, revealed no formation of epoxy alcohol 3, despite clear consumption of the starting material. We suddenly learned that the close proximity of the methyl ester to the epoxide induced an intramolecular epoxy-ester decomposition involving these functionalities and the Lewis-acid, titanium-based catalyst employed in the procedure. In fact, Corey17 reported a similar obstacle on an analogous substrate, in which the crude products did not seem to contain epoxide or ester units (1H NMR analysis).
In addition, Sharpless18 himself also observed this phenomenon where the ester internally attacked the epoxide, cyclizing to a diol lactone. Although the use of catalytic amounts of Ti(O-iPr)4 has successfully yielded the epoxy alcohol in the case of the one methylene extended analogue,19 this modification proved futile and thence we resorted to a different building block, but-2-yne-1,4-diol (21), for the synthesis as depicted in Scheme 3 (B).
At this point, we envisaged the alternative route to allow epoxide installation prior to methoxycarbonyl formation, thus bypassing said Lewis acid-mediated intramolecular attack. A similar strategy was employed to produce four isomeric epoxides critical in the synthesis of 2-nor-leukotriene analogs,20 therefore making this a promising strategy.
The first step involved a reduction of 21 by LAH to give the trans-butenediol intermediate, which was directly mono-protected as its p-bromobenzyl derivative 20. Notably, asymmetric Sharpless epoxidation16 of 20 proceeded seamlessly with 82% yield. Subsequent Dess–Martin oxidation of the free alcohol delivered the corresponding aldehyde 19 in adequate yield. A Wittig-type olefination of 19 with stabilized ylide Ph3PCHCO2Me gave the α,β-unsaturated intermediate, which was submitted to a mild and complete hydrogenation of the double bond without tempering with the oxirane moiety. This critical step was accomplished by diazene generated in situ from 2,4,6-triisopropylbenzenesulfonoyl hydrazide.
At this juncture, however, large-scale production of these building blocks was halted, creating a major hurdle to move in the forward direction.
In light of these outcomes, the synthetic routes were put to the side in favor of substitute and more robust strategies. Now we decided to build the C1–C10 epoxy aldehyde fragment (2) from chiral feedstocks instead of non-chiral materials.
Starting with D-erythronolactone (7) or D-erythrose (23), we envisioned to introduce the oxirane ring via epoxide transposition under basic conditions (Scheme 4).
 | ||
| Scheme 4 Successful synthesis of the C1–C10 fragment (2). | ||
Acetonide protection of 7 with excess 2,2-dimethoxypropane and catalytic p-TsOH·H2O in DMF, followed by smooth diisobutyl-aluminium hydride (DIBAL-H) reduction cleanly gave the isopropylidene lactol 6 in 95% and 93% yields, respectively. A Witting-type olefination of lactol 6 with methyl(triphenylphosphoranylidene)acetate in methylene chloride and catalytic amounts of benzoic acid at reflux afforded a cis/trans mixture of the α,β-unsaturated isopropylidene ester 5 in 89%, predominantly in the cis-isomeric form.21 Often without chromatographic purification, the isomeric mixture was submitted to complete hydrogenation using palladium on carbon, and the saturated primary alcohol was converted to the transient tosylate with p-TsCl in 85% yield, over two steps. Acid-catalyzed deisopropylidination cleanly gave 4 as a colorless oil after condensation under reduced pressure and without further purification. Treatment of the tosylate (4) with sodium methoxide in the presence of sodium sulfate evoked the base-catalyzed terminal epoxide migration,22 affording the target chiral epoxy alcohol 3 in appreciable yields.
This epoxide cascade initially proceeds via the intermediacy of a 1,2-epoxy alcohol which is then transposed to the internal 2,3-position under basic conditions, with the inversion of the configuration of carbon 2 and the retention at carbon 3, via SN2 nucleophilic substitutions (Scheme 4).22
Smooth Dess–Martin oxidation of epoxy alcohol 3, followed by a Wittig-type four-carbon chain homologation of the aldehyde intermediate with stabilized ylide(triphenylphosphoranylidene)acetaldehyde in toluene at 95 °C afforded the highly coveted C1–C10 segment (2) in fair yields. The latter transformation yielded a lower two-carbon vinylogue (data not shown), which was recycled back to furnish the desired intermediate.
In parallel, compound 2 was also constructed from the aldotetrose D-erythrose (23) and the oxirane stereochemistry was secured similarly to the erythronolactone strategy. However, this starting piece was disadvantageous due to its (i) physical state as a wet viscous syrup which, before use, required thorough drying under high vacuum for several hours and often resulting in significantly reduced residuum, as well as its (ii) relatively high price (∼US$355 per 8.3 mmol). Thus 7 was the preferred chiral synthon owing to its inexpensive commercial availability, and its highly efficient transformations in the onward sequence involving the protecting-group chemistry.
Synthesis of C11–C22 ω-fragment (8)
Having successfully furnished the C1–C10 α-segment (2), the next objective was to build the C11–C22 ω-fragment (8) as illustrated in Scheme 5. The synthesis began with the commercially available cis-4-heptenal (15), proceeding via a highly enantioselective and organocatalytic MacMillan α-oxyamination of the substrate using D-proline and nitrosobenzene, followed by an in situ aldehyde reduction with NaBH4. Cleavage of the O–N bond was accomplished using Zn dust and acetic acid, furnishing the known23 diol 14 in 74% yield over 2 steps.14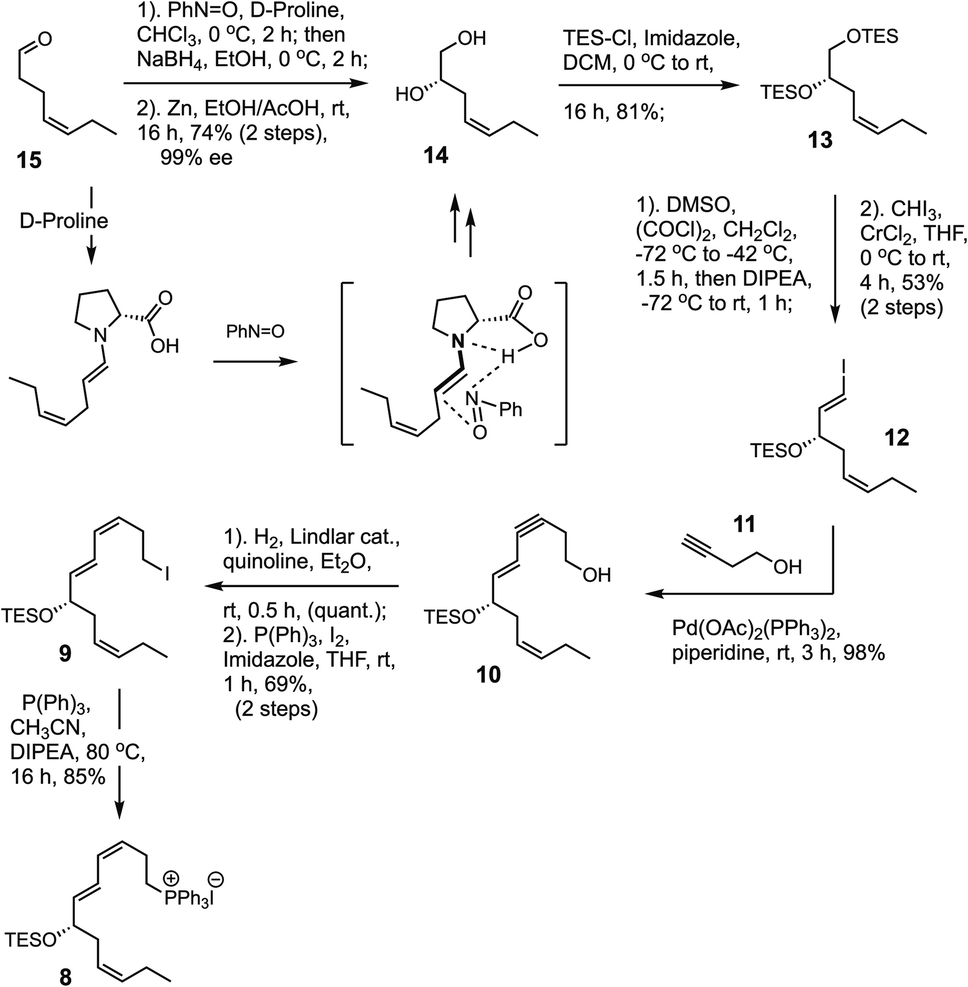 | ||
| Scheme 5 Synthesis of the C11–C22 fragment (8). | ||
The enantiopurity of this chiral intermediate was greater than 99% as determined by 400 MHz NMR on the Mosher's ester derivative (see, ESI†).14 Subsequently, 14 was silylated with triethylsilyl (TES) to give 13 in 81% yield. This intermediate facilitated a direct regio-selective Swern oxidation of the primary alcohol and, using a Takai olefination,24 the crude α-siloxy aldehyde was directly converted to the (E)-alkenyl iodide 12 in 53% over 2 steps.
Next, the combination between alkynyl lynchpin 11 with vinyl iodide 12 was accomplished using an efficient copper-free Sonogashira cross-coupling to afford the 1,3-enyne containing intermediate 10 in 98% yield.15 A mild cis-selective Lindlar hydrogenation of 10 in the presence of quinoline formed the 1,3-butadiene moiety, followed by conversion of the intermediate primary alcohol to iodide 9 in 69% over 2 steps. Finally, treatment of 9 with triphenylphosphine in the presence of DIPEA as a basic buffer afforded phosphonium salt 8 in 85% isolated yield.
Synthesis of the target compound (1)
With both the C1–C10 (2) and C11–C22 (8) segments at hand, the coupling of these building blocks was accomplished using a cis-selective Wittig olefination25 under closely monitored conditions (Scheme 6). | ||
| Scheme 6 Assembly of target compound (1). | ||
Reaction of aldehyde 2 with the ylide prepared in situ from phosphonium iodide 8 with KHMDS at −78 °C in THF afforded the silyl protected epoxy methyl ester in modest 21% yield (ε282 ≈ 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). Given the high sensitivity of this product, it was subjected to coarse and rapid column chromatography to avoid possible decomposition.
000 M−1 cm−1). Given the high sensitivity of this product, it was subjected to coarse and rapid column chromatography to avoid possible decomposition.
The silyl ether was cleaved in THF with nBu4NF buffered with acetic acid affording the methyl ester of 1 in 40–50% yield, after coarse purification. The structures of both the TES ether and methyl ester of 1 were confirmed by 600 MHz 1H NMR, including a wet1D pulse sequence to attenuate the dominant H2O and C6D6 protium signals, in addition to two-dimensional pulsed field gradient COSY experiments (ESI†).
The vinylic configuration of the direct building blocks of the target compound, namely the 13(Z), 15(E)-diene unit in 8 (J13,14 = 10.92 and J15,16 = 14.98) and the 6(E), 8(E)-diene in 2 (J6,7 = 15.32 and J8,9 = 15.36) were confirmed by proton coupling constants. Similarly, the 6(E), 8(E), and 15(E) olefinic regions (J6,7 = 15.26, J8,9 = 14.97 and J15,16 = 15.20) in the methyl ester of 1 were confirmed using 2D 1H NMR, while the other alkenyl protons exhibited overlapping resonances. Due to the inherent instability and limited amount of the target molecule and its protected versions, 13C NMR scans were not procured.
Since the methyl ester of 1 was the preferred form for storage, as the free acid is prone to facile decomposition, only a fraction of this compound was subjected to mild hydrolysis under basic conditions to yield epoxy resolvin 1 quantitatively (ε282 ≈ 40000 M−1 cm−1). In addition to high-resolution mass spectral information, this target material gave a UV signature with a triplet band of absorption, λMeOHmax, at 282 nm and shoulders at 273 nm and 295 nm, characteristic of a conjugated triene system. A single broad absorption at 229 nm was also observed, typical of a conjugated diene moiety present in 1 (see, ESI†). However, readable NMR absorption signals were, at this stage, unattainable.
With the synthetic epoxydocosanoid available, cell-type specific biological studies further validated its intact stereochemical structure via direct conversion to both resolvin D3 and resolvin D4,10 and provided evidence of its previously proposed central role in the biosynthesis of these biologically active chemical agents.4 In addition, when aqueous solutions of 1 were adjusted to physiological pH, epoxide hydrolysis proceeded to give a mixture of expected products10 resulting from non-stereoselective attack of water on the delocalized cation intermediate, yielding the all-trans triene isomers of RvD3 and vicinal diols of RvD4.
Conclusions
In summary, the first stereocontrolled total synthesis of 4(S),5(S)-epoxy-17(S)-hydroxy-6(E),8(E),10(Z),13(Z),15(E),19(Z)-docosahexaenoic acid (1) has been achieved. The reported synthetic strategy is convergent and stereospecific, allowing the production of this indispensable epoxydocosanoid in unambiguous manner. The synthetic availability of this metastable material provides the basis of recent10 and forthcoming investigations focused on its role in mediating the endogenous formation of chemical mediators that trigger inflammatory resolution processes, facilitating the studies of its biological properties and significance.Author contributions
N. A. P. and C. N. S. conceived the project. R. N. and N. A. P. designed the experiments. R. N. and T. F. L. conducted the experiments. S. A. acquired HRMS data. R. N. and N. A. P. wrote the manuscript. All authors discussed the results and gave approval to the final manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the NIH for financial support (Grant P01-GM095467 to N. A. P., and C. N. S.), and gratefully acknowledge the resources provided by Prof. Valery V. Fokin and the USC Agilent Center of Excellence in Biomolecular Characterization.References
- C. N. Serhan and N. A. Petasis, Resolvins and Protectins in Inflammation Resolution, Chem. Rev., 2011, 111(10), 5922–5943 CrossRef CAS PubMed.
- N. A. Petasis, I. Akritopoulou-Zanze, V. V. Fokin, G. Bernasconi, R. Keledjian, R. Yang, J. Uddin, K. C. Nagulapalli and C. N. Serhan, Design, Synthesis and Bioactions of Novel Stable Mimetics of Lipoxins and Aspirin-Triggered Lipoxins, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2005, 73(3–4), 301–321 CrossRef CAS PubMed.
- C. N. Serhan, C. B. Clish, J. Brannon, S. P. Colgan, N. Chiang and K. Gronert, Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2-Nonsteroidal Antiinflammatory Drugs and Transcellular Processing, J. Exp. Med., 2000, 192(8), 1197–1204 CrossRef CAS PubMed.
- C. N. Serhan, S. Hong, K. Gronert, S. P. Colgan, P. R. Devchand, G. Mirick and R. L. Moussignac, Resolvins: A Family of Bioactive Products of Omega-3 Fatty Acid Transformation Circuits Initiated by Aspirin Treatment That Counter Proinflammation Signals, J. Exp. Med., 2002, 196(8), 1025–1037 CrossRef CAS PubMed.
- V. L. Marcheselli, S. Hong, W. J. Lukiw, X. H. Tian, K. Gronert, A. Musto, M. Hardy, J. M. Gimenez, N. Chiang, C. N. Serhan and N. G. Bazan, Novel Docosanoids Inhibit Brain Ischemia-Reperfusion-Mediated Leukocyte Infiltration and Pro-Inflammatory Gene Expression, J. Biol. Chem., 2003, 278(44), 43807–43817 CrossRef CAS PubMed.
- C. N. Serhan, R. Yang, K. Martinod, K. Kasuga, P. S. Pillai, T. F. Porter, S. F. Oh and M. Spite, Maresins: Novel Macrophage Mediators with Potent Antiinflammatory and Proresolving Actions, J. Exp. Med., 2009, 206(1), 15–23 CrossRef CAS PubMed.
- J. Dalli, R. A. Colas and C. N. Serhan, Novel n-3 Immunoresolvents: Structures and Actions, Sci. Rep., 2013, 3(1940), 1–13 Search PubMed.
- C. N. Serhan, Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology, Nature, 2014, 510(7503), 92–101 CrossRef CAS PubMed.
- C. N. Serhan and B. D. Levy, Resolvins in Inflammation: Emergence of the pro-Resolving Superfamily of Mediators, J. Clin. Invest., 2018, 128(7), 2657–2669 CrossRef PubMed.
- A. E. Shay, R. Nshimiyimana, B. Samuelsson, N. A. Petasis, J. Z. Haeggström and C. N. Serhan, Human Leukocytes Selectively Convert 4S,5S-Epoxy-Resolvin to Resolvin D3, Resolvin D4, and a Cys-Resolvin Isomer, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(51), 1–7 CrossRef PubMed.
- J. Dalli, J. W. Winkler, R. A. Colas, H. Arnardottir, C. Y. C. Cheng, N. Chiang, N. A. Petasis and C. N. Serhan, Resolvin D3 and Aspirin-Triggered Resolvin D3 Are Potent Immunoresolvents, Chem. Biol., 2013, 20(2), 188–201 CrossRef CAS PubMed.
- J. W. Winkler, S. K. Orr, J. Dalli, C. Y. C. Cheng, J. M. Sanger, N. Chiang, N. A. Petasis and C. N. Serhan, Resolvin D4 Stereoassignment and Its Novel Actions in Host Protection and Bacterial Clearance, Sci. Rep., 2016, 6(18972), 1–11 Search PubMed.
- D. Cherpokova, C. C. Jouvene, S. Libreros, E. P. DeRoo, L. Chu, X. De La Rosa, P. C. Norris, D. D. Wagner and C. N. Serhan, Resolvin D4 Attenuates the Severity of Pathological Thrombosis in Mice, Blood, 2019, 134(17), 1458–1468 CrossRef PubMed.
- S. P. Brown, M. P. Brochu, C. J. Sinz and D. W. C. MacMillan, The Direct and Enantioselective Organocatalytic α-Oxidation of Aldehydes, J. Am. Chem. Soc., 2003, 125(36), 10808–10809 CrossRef CAS PubMed.
- N. A. Petasis, R. Yang, J. W. Winkler, M. Zhu, J. Uddin, N. G. Bazan and C. N. Serhan, Stereocontrolled total synthesis of neuroprotectin D1/protectin D1 and its aspirin-triggered stereoisomer, Tetrahedron Lett., 2012, 53(14), 1695–1698 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, The First Practical Method for Asymmetric Epoxidation, J. Am. Chem. Soc., 1980, 102(18), 5974–5976 CrossRef CAS.
- E. J. Corey, S. Hashimoto and A. E. Barton, Chirally Directed Synthesis of (-)-Methyl 5(S),6(S)-Oxido-7-Hydroxyheptanoate (1), Key Intermediate for the Total Synthesis of Leukotrienes A, C, D, and E, J. Am. Chem. Soc., 1981, 103(3), 721–722 CrossRef CAS.
- B. E. Rossiter, T. Katsuki and K. B. Sharpless, Asymmetric Epoxidation Provides Shortest Routes to Four Chiral Epoxy Alcohols Which Are Key Intermediates in Syntheses of Methymycin, Erythromycin, Leukotriene C-1, and Disparlure, J. Am. Chem. Soc., 1981, 103(2), 464–465 CrossRef CAS.
- I. R. Ollmann, J. H. Hogg, B. Muñoz, J. Z. Haeggström, B. Samuelsson and C.-H. Wong, Investigation of the Inhibition of Leukotriene A4 Hydrolase, Bioorg. Med. Chem., 1995, 3(7), 969–995 CrossRef CAS PubMed.
- F. D. Bellamy, M. Bondoux, B. Boubia, P. Dodey and C. Mioskowski, Enantioselective Synthesis of Four Isomeric Building Blocks Useful in the Synthesis of 2-nor-Leukotriene Analogues, Tetrahedron: Asymmetry, 1992, 3(3), 355–358 CrossRef CAS.
- S. G. Davies, E. M. Foster, J. A. Lee, P. M. Roberts and J. E. Thomson, Doubly Diastereoselective Conjugate Additions of the Antipodes of Lithium N-Benzyl-N-(α-Methylbenzyl)Amide to Enantiopure ε-O-Protected α,β-Unsaturated Esters Derived from D-Ribose, Tetrahedron: Asymmetry, 2014, 25(6–7), 534–546 CrossRef CAS.
- A. R. Rodriguez and B. W. Spur, First total synthesis of pro-resolving and tissue-regenerative resolvin sulfido-conjugates, Tetrahedron Lett., 2017, 58, 1662–1668 CrossRef CAS.
- A. F. Reinertsen, K. G. Primdahl, A. E. Shay, C. N. Serhan, T. V. Hansen and M. Aursnes, Stereoselective Synthesis and Structural Confirmation of the Specialized Pro-Resolving Mediator Resolvin E4, J. Org. Chem., 2021, 86(4), 3535–3545 CrossRef CAS PubMed.
- J. W. Winkler, J. Uddin, C. N. Serhan and N. A. Petasis, Stereocontrolled Total Synthesis of the Potent Anti-Inflammatory and pro-Resolving Lipid Mediator Resolvin D3 and Its Aspirin-Triggered 17 R-Epimer, Org. Lett., 2013, 15(7), 1424–1427 CrossRef CAS PubMed.
- E. J. Corey and M. M. Mehrotra, A stereoselective and practical synthesis of 5,6(S,S)-epoxy-15(S)-hydroxy-7(E),9(E),11(Z),13(E)-eicosatetraenoic acid (4), possible precursor of the lipoxins, Tetrahedron Lett., 1986, 27(43), 5173–5176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01537d |
| This journal is © The Royal Society of Chemistry 2022 |