
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric total synthesis of (1S,2S,4S)-β-elemene†
Wei Chenab,
Zhun Feng*a and
Qiang Liu*b
aGuangdong-Macao Traditional Chinese Medicine Technology Industrial Park Development Co., Ltd., Zhuhai 519031, P. R. China. E-mail: zhun.feng@gmtcmpark.com
bSchool of Chinese Medicine, Southern Medicinal University, Guangzhou 510515, P. R. China. E-mail: gzlq2002@163.com
First published on 15th March 2022
Abstract
Elemenes are sesquiterpene natural products extracted from Chinese medicinal herbs and have been used as an important antitumor drug in China. Here, we report the first stereoselective total synthesis of (1S,2S,4S)-β-elemene using (R)-carvone as a chiral pool starting material. The isopropenyl moiety was achieved in a highly stereoselective manner through 1,4-Michael conjugate addition. The following transformations like regio- and stereoselective aldol condensation, Wittig olefination have been employed as the key steps, resulting in a concise total synthesis of (1S,2S,4S)-β-elemene. Our accomplishment will allow further biological investigations of this natural product and open opportunities for developing a new potentially promising antitumor drug.
Introduction
Traditional Chinese medicine (TCM) has been used to prevent and treat diseases for thousands of years, and now plays a more and more important role in the small molecule drug discovery.1 One of the most significant and recent developments is the use of elemenes in the treatment of various malignant tumors.2 Elemenes are a family of sesquiterpenes extracted from the Chinese medicinal herb Curcumae Rhizoma, and are classified into α-elemene (A), β-elemene (B), γ-elemene (C), δ-elemene (D) according to the position of the double bond (Fig. 1).3 Owing to their extensive and potent in vitro and in vivo antitumor activities, elemene oral emulsion and elemene injection have been approved by the China National Medical Products Administration (NMPA) as noncytotoxic broad-spectrum antitumor drugs,2a,e and are currently under consideration for clinical studies in the United States.4 | ||
| Fig. 1 Representative elemene sesquiterpenes. | ||
(1S,2S,4R)-β-Elemene (1, Fig. 1) is the predominant active ingredient of clinically administrated elemene antitumor drugs, with a recommended content of not less than 85%.2e Previous studies have indicated that 1 not only inhibits the growth of tumor but also enhances the activity of immune system,5 and even can reverse multiple-drug resistance,6 making it unique to the conventional chemotherapeutic drugs. Interestingly, (1R,2R,4S)-β-elemene (2), the enantiomer of 1, was isolated from lower plants or microorganisms, such as the gorgonian Eunicea mammosa,7 while 1 is commonly widespread in higher plants. Very recently, a new isomer of β-elemene, namely (1S,2S,4S)-β-elemene (3), was isolated and characterized by Shi and co-workers from the essential oil of Curcuma wenyujin.8 As compared to 1, 3 was found to possess higher antitumor activities for all cancer cell lines tested and lower toxicity in mice,8 demonstrating its potential importance in the development of a new promising anticancer agent. However, the exceptionally low content of 3 in the Chinese medicinal herb greatly restricted its systematic pharmacological evaluation. Therefore, development of an efficient and practical approach to synthesize 3 using readily available reagents under mild reaction conditions is highly desirable.
The intriguing pharmacological properties and structural diversities of β-elemene have attracted significant attention from the synthetic community.9 Indeed, several elegant asymmetric total syntheses of β-elemene have been described in the literature or patents.4a,10–12 In 1967, Rao and co-workers described the first asymmetric total synthesis of 1 in 9 steps using ozonolysis of α-santonin 4 as the key transformation (Scheme 1a).10 In 1995, Corey and co-workers reported the first asymmetric total synthesis of 2 in 7 steps via enantioselective Ireland-Claisen rearrangement of β,γ-unsaturated ester from geraniol 6 (Scheme 1b).11 To the best of our knowledge, the total synthesis of (1S,2S,4S)-β-elemene (3) has yet to be reported. Herein, we present the first and asymmetric total synthesis of 3 in five or eight steps from (R)-carvone 8, a commercially available chiral pool starting material (Scheme 1c).
 | ||
| Scheme 1 Asymmetric total syntheses of β-elemene 1–3. | ||
Results and discussion
Intrigued by the fascinating pharmacological properties of (1S,2S,4S)-β-elemene (3), we embarked on the first total synthesis of this sesquiterpene natural product. Our conceptual route to 3 is shown in Scheme 2. Retrosynthetically, 3 could be traced back to primary alcohol 9 via Wittig olefination and common oxidation reaction. We then envisioned that 9 could be generated from the chiral pool starting materials 8 by employing chemoselective and diastereoselective 1,4-Michael conjugate addition of Grignard reagent, aldol condensation and simple reduction of ketone. | ||
| Scheme 2 Retrosynthetic analysis of (1S,2S,4S)-β-elemene. | ||
Chiral pool synthesis represents a useful approach to synthesize enantiopure organic molecules from readily available enantiomerically pure compounds, which features low cost, high abundance, and general renewability.13 Therefore, our synthesis began with the preparation of optically pure intermediate 12 from the chiral pool building block (R)-carvone 8 ($0.39 per g, Merck), as shown in Scheme 3. Stimulated by Knölker's pioneering work,14a it was anticipated that the new generated stereogenic center in cyclohexanone 11 could be secured by diastereoselective 1,4-Michael addition of isopropenyl Grignard reagent 10 to 8.14b To our delight, we were able to isolate desired product 11 exclusively in 70% yield. Notably, the steric and electronic influence in the diastereoselective transformations of chiral enolates have been systematically studied by Krause's group.15 Treatment of 11 with formaldehyde in the presence of KOH afforded primary alcohol 12 as a single diastereomer in 50% isolated yield for three cycles.14b,16 Subsequently, oxidation of 12 with Dess–Martin periodinane (DMP)17 gave aldehyde 13 in 70% yield. In an attempt to obtain advanced intermediate 14 via Wittig olefination, several reaction conditions (e.g., PPh3PCH2Br/n-BuLi, TMSCH2MgBr)18 were screened, but none afforded the desired product. Instead, the undesired retro-aldol reaction would occur preferentially to yield 11 in quantitative yield.
 | ||
| Scheme 3 Attempts to synthesize advanced intermediate 14. | ||
To avoid this undesired side reaction, we next turned our attention to the protection of alcohol 12 at an early stage (Scheme 4). First, compound 12 was treated with Ac2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 in the presence of triethylamine (TEA) in DCM afforded 15 in 90% yield. Subsequent treatment of 15 with sodium borohydride (NaBH4)20 in MeOH gave alcohol 16 in 90% yield (d.r. = 3:2). As planned ahead, removal of the hydroxyl group in 16 using Barton–McCombie radical deoxygenation21 and a subsequent KOH (dissolved in MeOH solution) workup delivered desired product 9 in 50% yield for two steps. Next, the oxidation of primary hydroxyl group in 9 using pyridinium chlorochromate (PCC)22 in DCM afforded desired aldehyde 17 in 75% yield. With 17 in hand, we continued toward the final stage of the total synthesis of (1S,2S,4S)-β-elemene (3) via Wittig olefination. Gratifyingly, treatment of 17 with PPh3PCH2Br and n-BuLi in THF,18 gave 3 in 82% yield, completing the first and asymmetric total synthesis of 3. The 1H and 13C NMR spectra of synthetic 3 and its optical rotation (synthetic, [α]20D = +24.5° (c 1.0, EtOH); natural, [α]20D = +36.2° (c 1.4, EtOH)) were in good agreement with those of the isolated natural product.8 Taken together, an eight-step asymmetric total synthesis of 3 was accomplished in 8.7% overall yield from readily available starting material 8. Moreover, the structure and absolute configuration of naturally occurring 3 were unambiguously confirmed by our asymmetric total synthesis.
19 in the presence of triethylamine (TEA) in DCM afforded 15 in 90% yield. Subsequent treatment of 15 with sodium borohydride (NaBH4)20 in MeOH gave alcohol 16 in 90% yield (d.r. = 3:2). As planned ahead, removal of the hydroxyl group in 16 using Barton–McCombie radical deoxygenation21 and a subsequent KOH (dissolved in MeOH solution) workup delivered desired product 9 in 50% yield for two steps. Next, the oxidation of primary hydroxyl group in 9 using pyridinium chlorochromate (PCC)22 in DCM afforded desired aldehyde 17 in 75% yield. With 17 in hand, we continued toward the final stage of the total synthesis of (1S,2S,4S)-β-elemene (3) via Wittig olefination. Gratifyingly, treatment of 17 with PPh3PCH2Br and n-BuLi in THF,18 gave 3 in 82% yield, completing the first and asymmetric total synthesis of 3. The 1H and 13C NMR spectra of synthetic 3 and its optical rotation (synthetic, [α]20D = +24.5° (c 1.0, EtOH); natural, [α]20D = +36.2° (c 1.4, EtOH)) were in good agreement with those of the isolated natural product.8 Taken together, an eight-step asymmetric total synthesis of 3 was accomplished in 8.7% overall yield from readily available starting material 8. Moreover, the structure and absolute configuration of naturally occurring 3 were unambiguously confirmed by our asymmetric total synthesis.
 | ||
| Scheme 4 Eight-step total synthesis of (1S,2S,4S)-β-elemene. | ||
Although (1S,2S,4S)-β-elemene (3) has been accomplished in a linear sequence of eight steps from 8, however, the yield of this protocol was relatively low (8.7% overall yield), and the use of stannyl reagent would put off its acceptability in the pharmaceutical industry. Consequently, we next sought to develop a more practical route to access 3, which would be more attractive for pharmaceutical manufacturing.
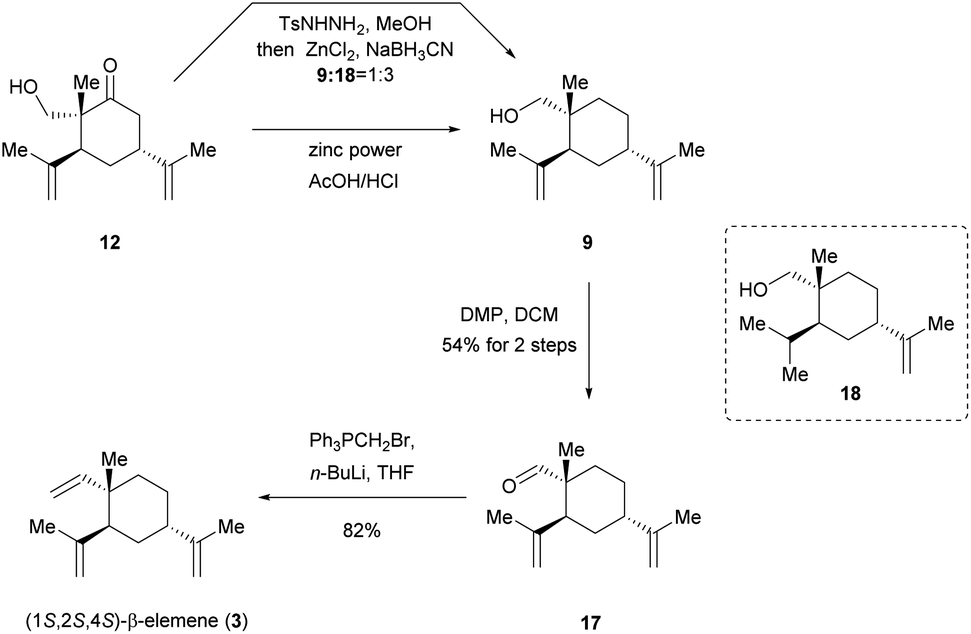
With this target in mind, our new protocol for the total synthesis of (1S,2S,4S)-β-elemene (3) is depicted in Scheme 5. In an attempt to the reduction of ketone 12 under standard conditions of Wolff–Kishner–Huang reduction (NH2NH2, KOH),23 decomposition of starting material was observed at the high temperature. Inspired by the elegant work of Kim's group,24 a zinc-modified cyanoborohydride (TsNHNH2/ZnCl2/NaBH3CN) was developed to be a versatile reducing agent under milder reaction conditions. Pleasingly, the desired product 9 could in fact be achieved, but along with another over-reduced byproduct 18 (9:18 = 1:3). We attributed this result to the possible coordination of hydroxyl group and neighbouring double bond with the electrophilic zinc chloride. It was noticed that separation of 9 and 18 by column purification was proved to be difficult. Therefore, we continued to optimize this reaction. Encouraged by Kappe's remarkable work,25 by using zinc powder (particle size < 45 μm) in acetic acid/hydrochloric acid, ketone 12 was successfully reduced to desired product 9, which was subsequently subjected to DMP oxidation without further chromatographic purifications, affording aldehyde 17 in 54% yield for two steps. Finally, the Wittig olefination was employed to provide the targeted (1S,2S,4S)-β-elemene (3) in 82% yield, completing a five-step asymmetric total synthesis of 3 in 15.5% overall yield. We believed that such a process would be commercially relevant and potentially more practical for pharmaceutical manufacturing.
 | ||
| Scheme 5 Five-step total synthesis of (1S,2S,4S)-β-elemene. | ||
Conclusions
In summary, the first total synthesis of (1S,2S,4S)-β-elemene (3) was achieved in five steps (15.5% overall yield) or eight steps (8.7% overall yield) from the chiral pool starting material 8. The stereocenters in 3 were constructed in simple and diastereoselective manners based on the chiral pool strategy. Notably, the transformations involved in our current synthetic routes, including 1,4-Michael conjugate addition, aldol condensation and Wittig olefination are well-developed and viable. Moreover, this work will allow the diverse syntheses of other β-elemene and their analogs to enable further biological research, which is underway and will be reported in due course.Experimental section
General methods
Reagents were purchased at the highest commercial quality (>95%) and used without further purification, unless otherwise stated. Anhydrous tetrahydrofuran (THF), toluene (PhMe) were distilled from sodium-benzophenone, dichloromethane (DCM), methanol (MeOH) were distilled from calcium hydride. All reactions were carried out with magnetic stirring, and if moisture or air sensitive, under an argon atmosphere with dry solvents. External bath temperatures were used to record all reaction temperatures. Low temperature reactions were carried out in a Dewar vessel filled with acetone/dry ice (−78 °C) or distilled water/ice (0 °C). Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm Tsingdao silica gel glass-backed plates (60F-254) and visualized under UV light at 254 nm. Staining was performed with an ethanolic solution of phosphomolybdic acid and heat as developing agents. Tsingdao silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography. Yields refer to chromatographically, unless otherwise specified. NMR spectra were recorded on Brüker Advance 500 (1H: 500 MHz, 13C 125 MHz) and Brüker Advance 400 (1H: 400 MHz, 13C 100 MHz). The spectra were reported as followed: chemical shift δ in ppm (multiplicity, coupling constant J in Hz, number of protons) for 1H NMR spectra and chemical shift δ in ppm for 13C NMR spectra. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or combinations thereof. Residual solvent peaks of CDCl3 (δ H = 7.26 ppm, δ C = 77.16 ppm) were used as an internal reference, unless otherwise stated. High resolution mass spectrometric (HRMS) data were recorded on a Brüker Apex IV RTMS instrument and a VG Auto Spec-3000 spectrometer, respectively. Optical rotation values were measured with a Jasco P-1020 polarimeter. The specific rotation was calculated as followed: [α]20D = (α × 100)/(c × d).:20 → 1:15) to give compound 11 (8.9 g) as a pale yellow oil in 70% yield. Rf = 0.45 (silica gel, EtOAc/hexanes = 1:6); [α]20D = −4.5°(c 0.50, CDCl3); 1H NMR (500 MHz, CDCl3) δ 4.93 (s, 1H), 4.80 (s, 1H), 4.73 (s, 1H), 4.52 (s, 1H), 2.71 (d, J = 5.1 Hz, 1H), 2.65 (d, J = 4.6 Hz, 1H), 2.62–2.57 (m, 1H), 2.53 (m, 1H), 2.34–2.27 (m, 1H), 2.03–1.96 (m, 1H), 1.86–1.79 (m, 1H), 1.73 (s, 3H), 1.70 (s, 3H), 1.07 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 213.7, 147.5, 144.1, 112.9, 110.6, 47.2, 46.6, 45.1, 40.5, 32.1, 23.9, 21.3, 12.1; IR νmax (film) 3082, 2986, 2935, 1706, 1646, 1377, 901 cm−1; HRMS (ESI) calcd for C13H21O [M + H]+: 193.1587; found: 193.1589.:20 → 1:10) to give compound 12 (4.6 g) as a pale yellow oil in 50% cumulative yield (the recovery of starting material 11 was ran for three cycles). Rf = 0.35 (silica gel, EtOAc/hexanes = 1:6); [α]20D = +3.5°(c 0.45, CDCl3); 1H NMR (400 MHz, CDCl3) δ 4.95 (s, 1H), 4.87 (s, 1H), 4.74 (s, 1H), 4.70 (s, 1H), 3.67 (d, J = 11.5 Hz, 1H), 3.43 (d, J = 11.5 Hz, 1H), 2.75–2.69 (m, 1H), 2.69–2.63 (m, 2H), 2.60–2.51 (m, 1H), 2.45 (s, 1H), 2.14 (m, 1H), 1.84 (d, J = 14.3 Hz, 1H), 1.76 (s, 3H), 1.71 (s, 3H), 1.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 217.2, 146.4, 144.4, 114.8, 112.5, 66.5, 53.7, 42.7, 42.1, 40.4, 28.8, 24.0, 22.1, 16.7; IR νmax (film) 3445, 2975, 2875, 2364, 1714, 1456, 999 cm−1; HRMS (ESI) calcd for C14H23O2Na [M + Na]+: 245.1512; found: 245.1512.:40 → 1:20) to give compound 13 (346 mg) as a colorless oil in 70% yield. Rf = 0.55 (silica gel, EtOAc/hexanes = 1:6); [α]20D = +2.5°(c 0.50, CDCl3); 1H NMR (500 MHz, CDCl3) δ 9.39 (d, J = 1.1 Hz, 1H), 4.97 (s, 1H), 4.80 (s, 1H), 4.70 (s, 1H), 4.51 (s, 1H), 2.99 (t, J = 5.0 Hz, 1H), 2.76–2.67 (m, 1H), 2.55 (dd, J = 14.9, 4.9 Hz, 1H), 2.49–2.40 (m, 1H), 1.92 (dt, J = 13.9, 4.7 Hz, 1H), 1.86–1.77 (m, 1H), 1.70 (s, 3H), 1.69 (s, 3H), 1.19 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 209.8, 199.8, 146.6, 142.4, 114.8, 111.3, 64.0, 45.8, 44.5, 40.0, 30.1, 24.4, 21.0, 14.4; IR νmax (film) 2931, 1745, 1715, 1373, 1178, 990 cm−1; HRMS (ESI) calcd for C14H21O2 [M + H]+: 221.1536; found: 221.1538.:50 → 1:30) to give compounds 15 (4.9 g) as a colorless oil in 90% yield. Rf = 0.75 (silica gel, EtOAc/hexanes = 1:6); [α]20D = +1.5°(c 0.65, CDCl3); 1H NMR (500 MHz, CDCl3) δ 4.92–4.88 (m, 1H), 4.80 (s, 1H), 4.64 (s, 1H), 4.57 (s, 1H), 4.21 (d, J = 10.9 Hz, 1H), 3.96 (d, J = 10.9 Hz, 1H), 2.73–2.63 (m, 2H), 2.52 (t, J = 5.6 Hz, 2H), 2.02–1.95 (m, 1H), 1.94 (s, 3H), 1.85 (m, 1H), 1.68 (s, 6H), 1.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 212.5, 170.6, 146.6, 144.0, 114.8, 111.5, 67.1, 51.2, 44.2, 42.4, 40.1, 28.9, 23.9, 21.6, 20.7, 17.2; IR νmax (film) 2925, 2854, 1768, 1377, 1024, 999 cm−1; HRMS (ESI) calcd for C16H24O3Na [M + Na]+: 287.1618; found: 287.1615.:20 → 1:15) to give mixture of diastereomers 16 (2.7 g, d.r. = 3:2) as a pale yellow oil in 90% yield. Rf = 0.40 (silica gel, EtOAc/hexanes = 1:6); 1H NMR (500 MHz, CDCl3) δ 4.90 (s, 2H), 4.87 (s, 0.8H), 4.85–4.79 (m, 2.5H), 4.73 (d, J = 9.9 Hz, 2H), 4.36 (d, J = 11.4 Hz, 1H), 4.20 (d, J = 10.9 Hz, 0.7H), 4.00 (d, J = 10.9 Hz, 0.7H), 3.76 (dd, J = 6.5, 3.9 Hz, 0.7H), 3.55 (dd, J = 12.0, 3.8 Hz, 2H), 2.71 (s, 1H), 2.61–2.53 (m, 0.7H), 2.43 (s, 1H), 2.41–2.37 (m, 0.7H), 2.30 (dd, J = 13.4, 3.0 Hz, 1H), 2.06 (s, 3H), 2.04 (s, 2.3H), 2.03–1.98 (m, 1H), 1.96–1.81 (m, 3H), 1.80 (s, 2H), 1.77 (s, 2H), 1.73 (s, 3H), 1.71–1.67 (m, 1H), 1.03 (s, 2H), 0.84 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 172.2, 171.5, 149.45, 146.47, 146.45, 145.48, 114.1, 114.0, 110.7, 109.5, 73.4, 69.0, 67.4, 67.3, 43.5, 42.5, 41.9, 41.3, 38.3, 38.1, 31.6, 30.96, 29.7, 28.6, 24.6, 23.2, 22.8, 22.1, 21.1, 21.0, 18.2, 9.8; IR νmax (film) 3435, 2930, 2856, 1427,1083, 859 cm−1; HRMS (ESI) calcd for C16H26O3Na [M + Na]+: 289.1774; found: 289.1772.:EtOAc). The organic solvent was removed under reduced pressure and the resultant residue was dissolved in toluene (20.0 mL). Then the reaction mixture was heated to reflux and tributyltin hydride (3.30 g, 11.28 mmol, 2.0 equiv.) and AIBN (370 mg, 11.28 mmol, 0.2 equiv.) was added. After being stirring for 8 h, a solution of KOH (1.0 g) in MeOH (20.0 mL) was added and stirred for 2 h. The reaction mixture was diluted by water (20.0 mL) and the aqueous layer was extracted with ethyl acetate (3 × 30.0 mL), and the combined organic layers were dried over Na2SO4. The extracts were filtered off and then evaporated under vacuum. The residue was purified by flash chromatography on silica gel (EtOAc/hexanes = 1:50 → 1:20) to give compound 9 (500 mg) as a white solid in 50% yield for two steps. Rf = 0.30 (silica gel, EtOAc/hexanes = 1:10); [α]20D = +7.5°(c 0.60, CDCl3); 1H NMR (500 MHz, CDCl3) δ 4.88 (d, J = 1.3 Hz, 1H), 4.82 (s, 1H), 4.79 (s, 1H), 4.76 (d, J = 1.5 Hz, 1H), 3.39–3.30 (m, 2H), 2.35 (s, 1H), 2.28 (dd, J = 10.9, 3.7 Hz, 1H), 1.81 (m, 2H), 1.76 (s, 3H), 1.72 (s, 3H), 1.71–1.62 (m, 3H), 1.56–1.48 (m, 1H), 1.17 (dd, J = 9.1, 4.1 Hz, 1H), 0.94 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.2, 147.2, 112.9, 110.5, 72.3, 44.8, 39.0, 38.9, 31.8, 29.9, 23.9, 23.3, 22.5, 17.9; IR νmax (film) 2995, 1755, 1249, 1240, 764, 748 cm−1; IR νmax (film) 3427, 2933, 2857, 1427, 1063, 990 cm−1; HRMS (ESI) calcd for C14H25O [M + H]+: 209.1900; found: 209.1900.:CH2Cl2). The organic solvent was removed under reduced pressure and purified by flash chromatography on silica gel (EtOAc/hexanes = 1:80 → 1:50) to give compound 17 (314 mg) as a white solid in 75% yield. Rf = 0.60 (silica gel, EtOAc/hexanes = 1:10); [α]20D = +8.5°(c 0.25, CDCl3); 1H NMR (400 MHz, CDCl3) δ 9.39 (s, 1H), 4.91–4.88 (m, 1H), 4.82 (d, J = 1.2 Hz, 1H), 4.77 (s, 1H), 4.74 (s, 1H), 2.67 (dd, J = 7.2, 4.8 Hz, 1H), 2.38–2.28 (m, 1H), 1.93–1.84 (m, 1H), 1.74 (s, 3H), 1.70 (s, 3H), 1.68–1.70 (m, 4H), 1.39 (m, 1H), 1.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.4, 147.9, 146.3, 113.6, 110.1, 49.5, 42.5, 38.5, 30.5, 29.4, 25.4, 24.9, 21.8, 17.1; IR νmax (film) 2915, 1735, 1680, 1435, 1202, 980 cm−1; HRMS (ESI) calcd for C14H23O [M + H]+: 207.1743; found: 207.1742.:80 → 1:50) to give compound 17 (200 mg) as a white solid in 54% yield for two steps.:50 → 1:30) to give a mixture of 9 and 18 (200 mg) as a pale yellow oil, the 1H NMR spectrum indicated their ratio is 1:3 (9:18), Rf = 0.50 (silica gel, EtOAc/hexanes = 1:8).Compound 18 was isolated for characterization purpose by taking an aliquot from the combined organic layers, followed by concentration under reduced pressure and the residue was purified by PTLC (silica gel, EtOAc/hexanes = 1:10) to give 18 as a colorless oil. Rf = 0.60 (silica gel, EtOAc/hexanes = 1:8); [α]20D = +3.5°(c 0.35, CDCl3); 1H NMR (400 MHz, CDCl3) δ 4.87–4.78 (m, 1H), 4.78–4.70 (m, 1H), 3.35 (s, 2H), 2.27 (dd, J = 11.6, 3.6 Hz, 1H), 1.74 (s, 3H), 1.73–1.61 (m, 2H), 1.61–1.46 (m, 5H), 1.31–1.25 (m, 1H), 1.09 (d, J = 12.3 Hz, 1H), 0.92 (s, 3H), 0.87 (t, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 149.4, 112.8, 72.4, 44.1, 40.4, 38.8, 31.4, 29.7, 26.8, 23.6, 23.2, 21.4, 21.2, 17.8; IR νmax (film) 2925, 2855, 1732, 1250, 1024, 986 cm−1; HRMS (ESI) calcd for C14H26NaO [M + Na]+: 233.1876; found: 233.1876.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (No. 2020M682925). We would also like to thank Dr C. G. Jiang at XtalPi for his kindly help and useful discussions.Notes and references
- (a) W. Tan, J. J. Lu, M. Q. Huang, Y. B. Li, M. W. Chen, G. S. Wu, J. Gong, Z. F. Zhong, Z. T. Xu, Y. Y. Dang, J. J. Guo, X. P. Chen and Y. T. Wang, Chin. Med., 2011, 6, 27 CrossRef CAS PubMed; (b) C.-Y. Shen, J.-G. Jiang, L. Yang, D.-W. Wang and W. Zhu, Br. J. Pharmacol., 2017, 174, 1395–1425 CrossRef CAS PubMed; (c) C.-L. Yao, J.-Q. Zhang, J.-Y. Li, W.-L. Wei, S.-F. Wu and D.-A. Guo, Nat. Prod. Rep., 2021, 38, 1618–1633 RSC.
- For selected reviews on elemenes, see: (a) J.-J. Lu, Y.-Y. Dang, M. Huang, W.-S. Xu, X.-P. Chen and Y.-T. Wang, J. Ethnopharmacol., 2012, 143, 406–411 CrossRef CAS PubMed; (b) Z. Y. Jiang, J. A. Jacob, D. S. Loganathachetti, P. Nainangu and B. A. Chen, Front. Pharmacol., 2017, 8, 105 Search PubMed; (c) B. T. Zhai, Y. Y. Zeng, Z. W. Zeng, N. N. Zhang, C. X. Li, Y. J. Zeng, Y. You, S. L. Wang, X. B. Chen, X. B. Sui and T. Xie, Int. J. Nanomed., 2018, 13, 6279–6296 CrossRef CAS PubMed; (d) B. T. Zhai, N. N. Zhang, X. M. Han, Q. J. Li, M. M. Zhang, X. Y. Chen, G. H. Li, R. N. Zhang, P. Chen, W. G. Wang, C. X. Li, Y. Xiang, S. P. Liu, T. Duan, J. S. Lou, T. Xie and X. B. Sui, Biomed. Pharmacother., 2019, 114, 108812 CrossRef CAS PubMed; (e) Z. Q. Bai, C. S. Yao, J. L. Zhu, Y. Y. Xie, X.-Y. Ye, R. R. Bai and T. Xie, Molecules, 2021, 26, 1499 CrossRef CAS PubMed.
- (a) M. Adio, Tetrahedron, 2009, 65, 5145–5159 CrossRef; (b) W. Chen, J.-B. Wang, Z. Feng and Q. Liu, Chin. J. Syn. Chem., 2021, 29, 802–812 Search PubMed.
- (a) L. Huang, US Pat., US8507562B2, 2013 Search PubMed; (b) H. Cai, L. L. Ren, Y. Wang and Y. J. Zhang, Front. Oncol., 2021, 11, 571476 CrossRef PubMed.
- (a) X. M. Yu, M. Y. Xu, N. Li, Z. J. Li, H. Y. Li, S. J. Shao, K. Zou and L. J. Zou, Biochem. Biophys. Res. Commun., 2017, 490, 514–520 CrossRef CAS PubMed; (b) Y. Zhou, T. Takano, Y. Wang, X. Li, R. Wang, Y. Wakatsuki, H. Nakajima-Adachi, M. Tanokura, T. Miyakawa and S. Hachimura, iScience, 2021, 24, 101883 CrossRef CAS PubMed; (c) Y. Y. Zhou, W. Y. Qiu, Y. M. Wang, R. Wang, T. Takano, X. Y. Li, Z. L. Zhu, H. Nakajima-Adachi, M. Tanokura, S. Hachimura and T. Miyakawa, Biomedicines, 2021, 9, 704 CrossRef CAS PubMed.
- (a) B. Wang, J. Guo, J. Di and Q. Shi, Chin. Clin. Cancer, 1999, 26, 10–13 Search PubMed; (b) T.-Z. Zhu, X.-M. Li, L. H. Luo, Y.-H. Xu, P. Cao, Y. Liu and G.-B. Liang, Mol. Med. Rep., 2014, 10, 1122–1128 CrossRef CAS PubMed; (c) G. N. Zhang, C. R. A. Jr, Y. K. Zhang, Z. S. Chen and H. Guo, Chin. J. Cancer, 2015, 34, 488–495 CAS.
- A. J. Weinheimer, W. W. Youngblood, P. H. Washecheck, T. K. B. Karns and L. S. Ciereszko, Tetrahedron Lett., 1970, 11, 497–500 CrossRef.
- Y. M. Shi, R. Y. Wang, X. Feng, X. T. Chen, J. Li, A. L. He, J. Shi, Y. M. Yuan, Y. C. Wang and S. Liu, CN Pat., CN109438166B, 2021 Search PubMed.
- For selected total syntheses of (±)-β-elemene, see: (a) O. P. Vig, B. Ram, K. S. Atwal and S. S. Bari, J. Indian Chem. Soc., 1975, 52, 257–260 CAS; (b) J. E. McMurry and P. Kočovský, Tetrahedron Lett., 1985, 26, 2171–2172 CrossRef CAS; (c) D. Kim, J. Lee, J. Chang and S. Kim, Tetrahedron, 2001, 57, 1247–1252 CrossRef CAS; (d) A. F. Barrero, M. M. Herrador, J. F. Quılez del Moral, P. Arteaga, N. Meine, M. C. Perez-Morales and J. V. Catalán, Org. Biomol. Chem., 2011, 9, 1118–1125 RSC; (e) D. P. Iglesias, P. H. Teijón, R. R. González and A. Fernán-dez-Mateos, Eur. J. Org. Chem., 2018, 35, 4926–4932 CrossRef.
- K. S. Kulkarni and A. S. Rao, Tetrahedron, 1965, 21, 1167–1173 CrossRef CAS.
- E. J. Corey, B. E. Roberts and B. R. Dixon, J. Am. Chem. Soc., 1995, 117, 193–196 CrossRef CAS.
- L. Q. Huang, X. Ke, Q. S. Gao, X. Wang, X. Y. Song, D. J. Wang, J. Yang, J. Guo, Y. N. Wang and Z. F. Shi, CN Pat., CN111423300A, 2020 Search PubMed.
- For selected books and reviews on chiral pool synthesis, see: (a) S. Hanessian, Total Synthesis of Natural Products: The ‘Chiron’ Approach, ed. J. E. Baldwin, Pergamon, Oxford, UK, 1983 Search PubMed; (b) Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS PubMed; (c) C. N. Stout and H. Renata, Acc. Chem. Res., 2021, 54(5), 1143–1156 CrossRef CAS PubMed; (d) A. Gogoi, S. Mezhubeinuo, S. Nongrum and G. Bez, Curr. Org. Chem., 2021, 25, 1566–1610 CrossRef CAS.
- (a) H.-J. Knölker, A. Ecker, P. Struwe, A. Steinmeyer, G. Müller and G. Neef, Tetrahedron, 1997, 53, 91–108 CrossRef; (b) H. Shi, C. H. Tan, W. B. Zhang, Z. C. Zhang, R. Long, J. X. Gong, T. P. Luo and Z. Yang, J. Org. Chem., 2016, 81, 751–771 CrossRef CAS PubMed.
- N. Krause, Angew. Chem., Int. Ed., 1994, 33, 1764–1765 CrossRef.
- L. Kürti, B. Czakó and E. J. Corey, Org. Lett., 2008, 10, 5247–5250 CrossRef PubMed.
- Z. Y. Zhou, A. X. Gao and S. A. Snyder, J. Am. Chem. Soc., 2019, 141, 7715–7720 CrossRef CAS PubMed.
- K. C. Nicolaou, S. Y. Pan, Y. Shelke, Q. J. Ye, D. Das and S. Rigol, J. Am. Chem. Soc., 2021, 143, 20970–20979 CrossRef CAS PubMed.
- C. C. Yuan, S. Y. Zhong, X. Y. Li, Y. H. Wang, M.-M. Xun, Y. L. Bai and K. K. Zhu, Org. Biomol. Chem., 2018, 16, 7843–7850 RSC.
- J. Y. Liu, J. L. Wu, J.-H. Fan, X. Yan, G. J. Mei and C.-C. Li, J. Am. Chem. Soc., 2018, 140, 5365–5369 CrossRef CAS PubMed.
- T. R. R. Pettus, M. Inoue, X.-T. Chen and S. J. Danishefsky, J. Am. Chem. Soc., 2000, 122, 6160–6168 CrossRef CAS.
- M. E. Fox, C. Li, J. P. Marino and L. E. Overman, J. Am. Chem. Soc., 1999, 121, 5467–5480 CrossRef CAS.
- M. Toyota, T. Wada and M. Ihara, J. Org. Chem., 2000, 65, 4565–4570 CrossRef CAS PubMed.
- S. Kim, H. C. Oh, J. S. Ko, K. H. Ahn and Y. J. Kim, J. Org. Chem., 1985, 50, 1927–1932 CrossRef CAS.
- T. Kappe, R. Aigner, P. Roschger, B. Schnell and W. Stadibauer, Tetrahedron, 1995, 51, 12923–12928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: copies of 1H and 13C NMR spectra. See DOI: 10.1039/d2ra01408d |
| This journal is © The Royal Society of Chemistry 2022 |