
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupling nucleic acid circuitry with the CRISPR-Cas12a system for universal and signal-on detection†
Rujian Zhaoab,
Chunxu Yuab,
Baiyang Lu*a and
Bingling Li *ab
*ab
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: bylv@ciac.ac.cn; binglingli@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 4th April 2022
Abstract
We report a universal and signal-on HCR based detection platform via innovatively coupling the CRISPR-Cas12a system with HCR. By using this CRISPR-HCR pathway, we can detect different targets by only changing the crRNA. The CRISPR-HCR platform coupling with an upstream amplifier can achieve a practical sensitivity as low as ∼aM of ASFV gene in serum.
A series of non-enzymatic isothermal nucleic acid circuiting reactions, known as enzyme-free DNA circuits,1–4 have caught increasing attention because they are able to execute nucleic acid assembly with low cost, high flexibility, programmability, and readout comparability. Hybridization chain reaction (HCR), first reported by the Piece group,5 is one of the most important examples of enzyme-free DNA circuits.6–8 It relates to a pair of complementary DNA hairpins with sticky ends (H1, H2) and an initiator (I0). During the pathway, the pair of complementary DNA hairpins is utilized as fuel packets to achieve self-assembly, forming a long-nicked double-stranded DNA (dsDNA) polymer. Due to the reaction can provide both size and concentration amplification, HCR has been deep-studied and extensively-utilized as an intelligent sensing unit during molecular recognition, amplification, and transduction. The reactions have been engineered and designed for various different targets, such as nucleic acids,9,10 proteins,11 small molecules,12 ions,13 bacteria,14 cells,15 etc.16
However, there are still problems in the existing HCR technology, which often limit its practical applications. For example, how to export an effective signal for an HCR reaction or products is always a serious challenge. The most-used signal is fluorescence resonance energy transfer (FRET).17 It is a common agreement that such a “signal-off” response may get easily confused with the false positives and insufficient intuitiveness. Therefore, continuous efforts have been made to develop “signal-on” responses. In many cases, classic HCR hairpin substrates have to be extended with additional oligonucleotides (named tails) to form an assembly-probe that can generate downstream signals.6–8,18–21 Taking an example from our previous publications,22 after HCR two adjacent tails can integrate into a full G-quadruplex structure. A porphyrin derivative (abbreviated as PPIX) can insert into the G-quadruplex and get much enhanced fluorescence emission. By similar concept, many other readouts, such as colorimetric, luminescent, and enzymatic signals have also been successfully realized.23,24 Even though, experimental evidence has revealed once the HCR hairpin is added with additional tail sequences, the assembly efficiency or signal-to-background ratio may be seriously exhibited, which makes the HCR difficult to control, design, and apply.22 In addition, because the HCR initiator is always the target itself or a sequence highly relevant to the target, the hairpin sequences have to be re-built, re-designed, and/or re-optimized once a new target is detected, adding the time, cost, and risk for failure.
Above issues are common for most HCR-based detection and require further advance to overcome them. With this purpose, here we report a universal and signal-on HCR based detection platform via innovatively coupling CRISPR25 (clustered regularly interspaced short palindromic repeats)-Cas12a system with the enzyme-free signal amplification circuits. The as-termed CRISPR-HCR detection (Scheme 1) turns the traditional “Off signal” into an “On signal”. Meanwhile, the essential HCR components don't have to be changed. That says, no matter which target is to be detected, an efficient, non-tailed HCR reaction can be adapted without laborious and challenging rebuilding. We have experimentally demonstrated the high efficiency and universality of CRISPR-HCR strategy through applying it for detection of three different targets just by changing crRNA sequences, which are short segments from African swine fever virus (ASFV) p72 gene (dsDNA-T1) and Human Papilloma Virus (HPV) DNA (dsDNA-T2) as well as a random ssDNA (ssDNA-T), which are 21 bp, 20 bp and 20 nt respectively. By coupling loop-mediated isothermal amplification (LAMP) as an upstream amplifier, the CRISPR-HCR strategy can further achieve a practical sensitivity of ∼aM of ASFV gene in serum. Finally, by successfully replacing HCR with another nucleic acid circuits of general interest, catalytic hairpin assembly (CHA), further demonstrating the flexibility of the platform.
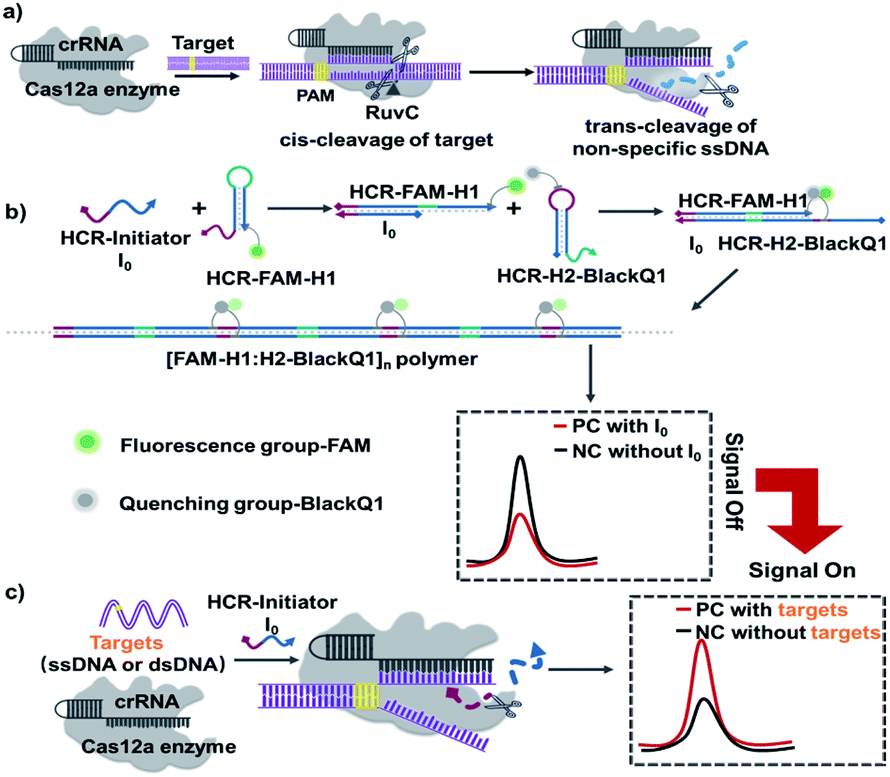 | ||
| Scheme 1 Schematic illustration of the CRISPR-HCR platform. (a) Principle of CRISPR-Cas12a-mediated cis and trans cleavage of DNA. (b and c) CRISPR-HCR platform. Without the target sequence, I0 activates the HCR assembly and produces low fluorescence background due to FRET (b). After target binds with crRNA, Cas12a cleaves I0 and remains HCR unreacted, producing increased fluorescence targeting signal (c). | ||
The detailed CRISPR-HCR pathway is illustrated in Scheme 1. As well-reported, Cas12a can be programmed with CRISPR RNAs (crRNAs) to specifically recognize the target DNA.26 Its collateral cleavage of the nonspecific single stranded DNA (ssDNA) reporter (trans-cleavage), initiated by the recognition and cleavage of the target DNA (cis-cleavage), has been used for the detection of nucleic acids (Scheme 1a).27,28 Based on our preliminary experiment, it is proved that ssDNA I0 can be cleaved by CRISPR Cas12a in Fig. S1.† In our CRISPR-HCR platform (Scheme 1b and c), H1, H2, initiator I0 (a 24 nt ssDNA), crRNA and Cas12a are the primitive elements. A fluorophore (FAM) and a quencher is labelled on H1 (FAM-H1) and H2 (H2-BlackQ1) respectively. In the absence of target (Scheme 1b), the I0 can initial a classic HCR reaction to generate [FAM-H1:H2-BlackQ1]n polymer. Because of the adjacent position of the fluorophore and quencher, the fluorescence of FAM is quenched, getting low background signal compared with the reaction without I0. While in presence of the targeting sequence (either ssDNA or dsDNA, Scheme 1c), the Cas12a can recognize the target through the bind of antisense guide RNA (crRNA), forming a specific hairpin scaffold structure. Then the Cas12a undergoes a series of conformational changes in its RuvC active site allowing for cis-cleavage of the target. Trans-cleavage activity is subsequently activated, and indiscriminately cleaves all ssDNA in the surrounding.29 In this way, I0 is non-specifically cleaved and loses its ability to start the HCR reaction, the FAM and BlackQ1 molecules are still apart far from each other, producing a target dependent high fluorescence intensity compared with the non-target background.
As illustrated, the introduction of CRISPR-Cas12a system has successfully turns the HCR reaction from a “signal-off” to “signal-on” response. Also important, due to the non-specific cleavage function of Cas12a to I0, one set HCR components (I0, H1, and H2) can be suitable to any targeting sequence without rebuilding. Therefore, here we directly select the sequences from one of the most powerful HCR reaction.24
In initial principle verification and optimization, we use a piece of ds-DNA fragment within ASFV p72 (ref. 30) as the model target, being named dsDNA-T1. The sequence of dsDNA-T1 is rationally selected starting from a potential PAM site starting from “TTTA” at 5′ end. Its crRNA guide sequence (crRNA-T1) is designed to include antisense of dsDNA-T1 and a hairpin structure. Fig. 1a experimentally verifies the introduction of CRISPR-Cas12a system indeed turns the HCR from a “signal-off” response (Fig. 1a, left) to a “signal-on” one (Fig. 1a, right).
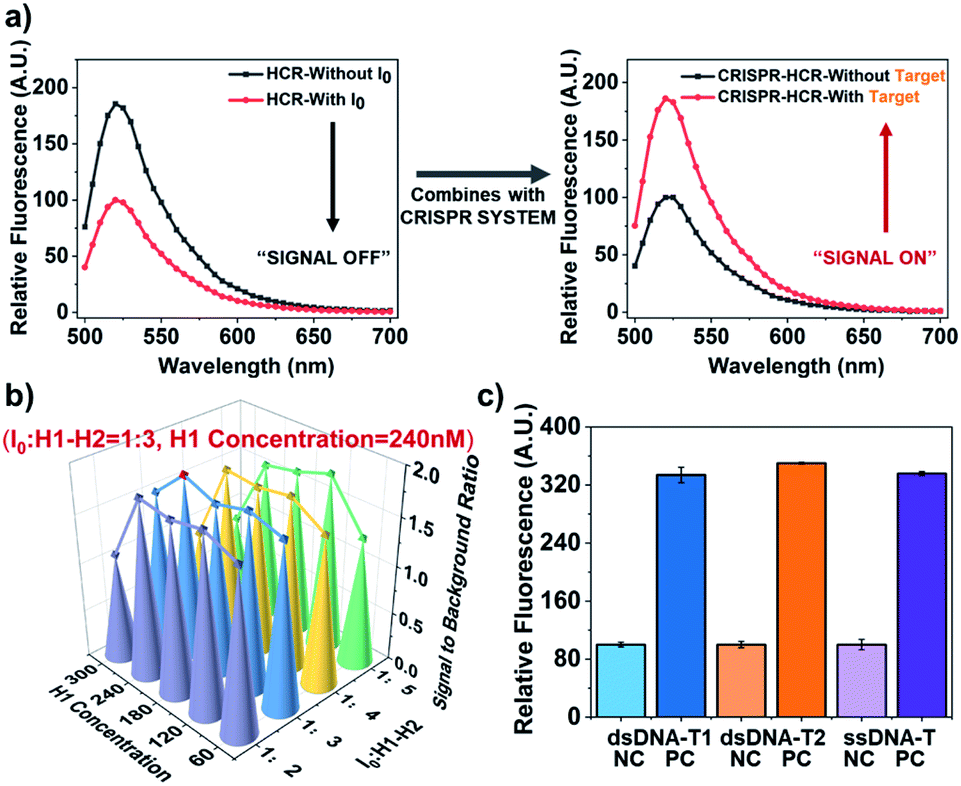 | ||
| Fig. 1 Performance verification for CRISPR-HCR platform. (a) Signal (fluorescence spectra) comparison between traditional HCR and CRISPR-HCR strategy. (b) 3D-Barograph for the optimization of the signal-to-background ratio of CRISPR-HCR. (c) Barograph of the fluorescence intensity to validate the universality of the CRISPR-HCR with different targets, including dsDNA-T1 (blue), dsDNA-T2 (orange) and ssDNA-T (purple), the real-time fluorescence signal has been shown in Fig. S2.† The error bars represent standard deviation from two independent tests. | ||
The experimental condition used in Fig. 1a is selected from a series of optimization. 300 nM Cas12a is used because its presences the highest signal-to-background ratio (Fig. S3†). We also investigate two critical factors that may usually affect the HCR module, the concentration ratio of I0 to H1 and H2 and the concentration of H1 (or H2). For convenience, the final concentrations of H1 and H2 are always equal. According to the 3D-bargraph shown in Fig. 1b, among all the conditions tested, the highest signal-to-background ratio appears when the concentration of H1 (or H2) is 240 nM, and the concentration ratio of I0 to H1 (or H2) is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.
:3.
Based on the optimized conditions, we evaluate the sensitivity of the CRISPR-HCR for detection of the dsDNA-T1. A broad dynamic range (pM to nM, Fig. S4 and S5†) of more than three orders of magnitude is achieved, with the lowest recognition limit around 1 pM. Furthermore, the universality of the CRISPR-HCR platform is verified by changing the crRNA guide sequence according to a new targeting sequence. Here another dsDNA fragment from Human Papilloma Virus26 (HPV, named dsDNA-T2) and a randomly designed ssDNA sequence (named ssDNA-T) are selected as the models. As shown in Fig. 1c, under the same condition the responses of all three targets (dsDNA-T1, dsDNA-T2, and ssDNA-T) are very close, presenting equally high signal-to-background ratios.
Through above proof-of-concept experiments, we have obtained a universal, flexible, and both dsDNA and ssDNA targets. In order to realize more practical performance which can be generalized to those real-world pathogen genes existing at very trace amounts, we further couple an ultra-isothermal nucleic acid amplification with CRISPR-HCR. LAMP reaction31,32 is selected as the model because it can amplify 109 to 1010-fold at a constant temperature (60–65 °C) within 1–2 hours.33,34 In details shown in Fig. 2a and b, the target in above demonstration (Scheme 1c) is replaced by the LAMP amplicons for a special targeting gene. At first, the ASFV gene including the dsDNA-T1 fragment (named dsDNA-ASFV), which is selected as the model-target, has been selected as our template. The LAMP primers are self-designed. The efficiency of the LAMP reaction is verified using traditional agarose gel electrophoresis (Fig. S6†). We think that the assay detection limit depended on the LAMP detection limit, based on our experiments, the detection limit of the LAMP reaction is 1.25 copies per μL when the detection rate is 95% (Fig. S7†). To further evaluate the sensitivity of the LAMP-CRISPR-HCR platform, the LAMP products amplified from different concentrations (0.5 copies per μL to 5000 copies per μL) of synthetic dsDNA-ASFV (named ASFV-LAMP-products) are used to start the downstream CRISPR-HCR platform. The fluorescence spectrum (Fig. 2b) clearly shows ASFV-LAMP-products amplified from as low as 0.5 copies per μL (∼aM) dsDNA-ASFV can already generate very high fluorescence enhancement compared with the background of water control (labelled with “0 copy”). It is notable, according to the Fig. S7,† the detection rate of 0.5 copies per μL is about 25%, and theoretically more than 5 copies per μL can achieve 100% detection rate. On contrast, the signals (Fig. 2c) for LAMP products amplified from genes of non-specific pathogens, for example, Middle East Respiratory Syndrome35 (named MERS-LAMP-products) and M13mp18 DNA32 (named M13mp18-LAMP-products), are almost same as that of water control (labelled with “NC”), which verifies the practically high sensitivity and specificity of the LAMP-CRISPR-HCR platform.
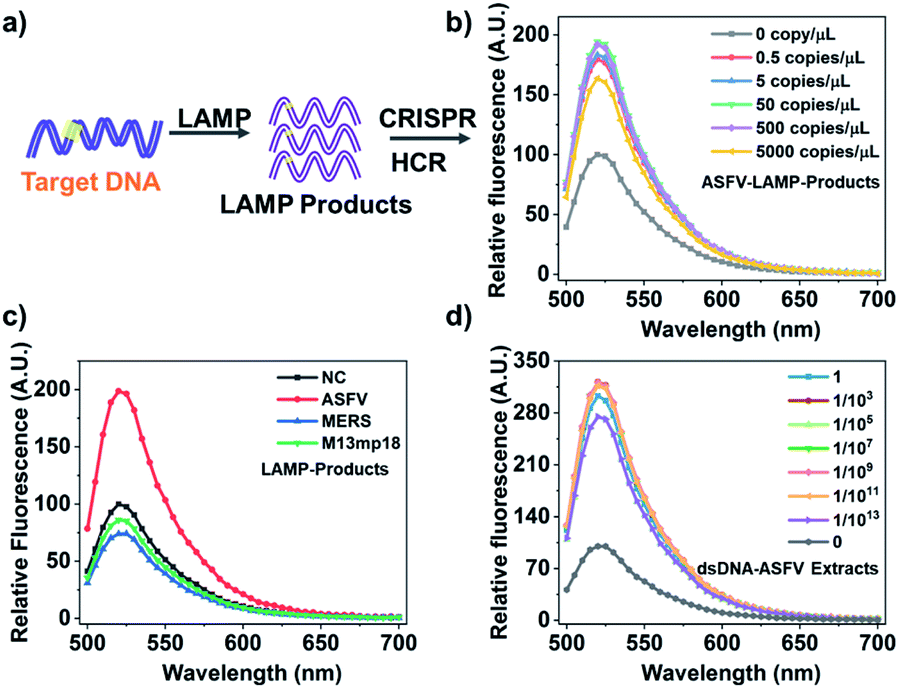 | ||
| Fig. 2 Coupling LAMP with CRISPR-HCR platform (LAMP-CRISPR-HCR). (a) Schematic illustration of CRISPR-HCR system coupling with LAMP reaction. (b) Fluorescence spectra of CRISPR-HCR for ASFV-LAMP-products amplified from different amounts of synthetic dsDNA-ASFV. (c) Fluorescence spectra of the CRISPR-HCR for LAMP products amplified from specific target (5000 copies per μL dsDNA-ASFV), and non-specific targets (water control “NC”, 20000 copies per μL synthetic dsDNA-MERS and dsDNA-M13mp18). dsDNA-MERS and dsDNA-M13mp18 is, in respective, the synthetic DNA sequence segmented from MERS-CoV and M13mp18 gene. (d) Fluorescence spectra of CRISPR-HCR for ASFV-LAMP-products amplified from water control “NC” and different dilutions of the dsDNA-ASFV extracts in pig serum sample. | ||
To test the feasibility of applying the LAMP-CRISPR-HCR strategy in real samples, we explore the detection of dsDNA-ASFV in pig serum, one of the most challenging matrices that contains many interfering molecules. We prepare the pig serum samples spiked with 1 × 1014 copies per μL of dsDNA-ASAV templates. After extracting the dsDNA-ASFV by the commercial extraction TIANamp genomic DNA kit, the extracts are diluted by different folds, being followed by LAMP-CRISPR-HCR detection. It is found when the sample is diluted by 1013 folds, the dsDNA-ASFV can still be detected with very high signal-to-background ratio (Fig. 2d). This result shows the components in real samples cannot affect the detection efficiency, which verifies the practicability of the platform.
In order to further evaluate the adaptability of the method, we replace the hybridization chain reaction (HCR) with catalytic hairpin assembly (CHA), which is another widely-used nucleic acid circuit.1,3,36 According to Fig. 3a, CHA also requires two hairpin substrates (named H3 and H4-FAM), in which H4 is FAM tagged. In the absence of the target, the CHA catalyst C1 (kind of I0 for HCR) initiates two strand-displacement (SD) reactions that forms H3:H4-FAM:C1 intermediate. Then C1 automatically dissociate from the intermediate and starts another reaction cycle, leaving H3:H4-FAM as the assembly product. At the same time, the tail of H3:H4-FAM is able to start another SD reaction to bind a probe sequence labelled with BlackQ1 (named P-BlackQ1), finally forming the H3:FAM-H4:P-BlackQ1 complex. Very similar to the CRISPR-HCR, once the targeting sequence exists, the Cas12a can recognize the target through the bind of antisense guide RNA (crRNA). Thus, C1 is non-specifically cleaved, remaining the CHA unreacted. An enhanced fluorescence signal is produced (Fig. 3b). Taking dsDNA-T1 as the model-target, the CRISPR-CHA conditions are optimized (Fig. S8–S10†). Then as low as 1.25 nM dsDNA-T1 can lead to saturated fluorescence enhancement compared with the negative background (labelled with “NC”), as shown in Fig. 3c. Further, via replacing the dsDNA-T1 with LAMP products, we also couple the LAMP with the CRISPR-CHA strategy taking dsDNA-ASFV as the model target. As expected, Fig. 3d shows the ASFV-LAMP-products from 500 copies of dsDNA-ASFV (labelled with “PC”) has successfully produce much higher signal compared that of negative control (labelled with “NC”).
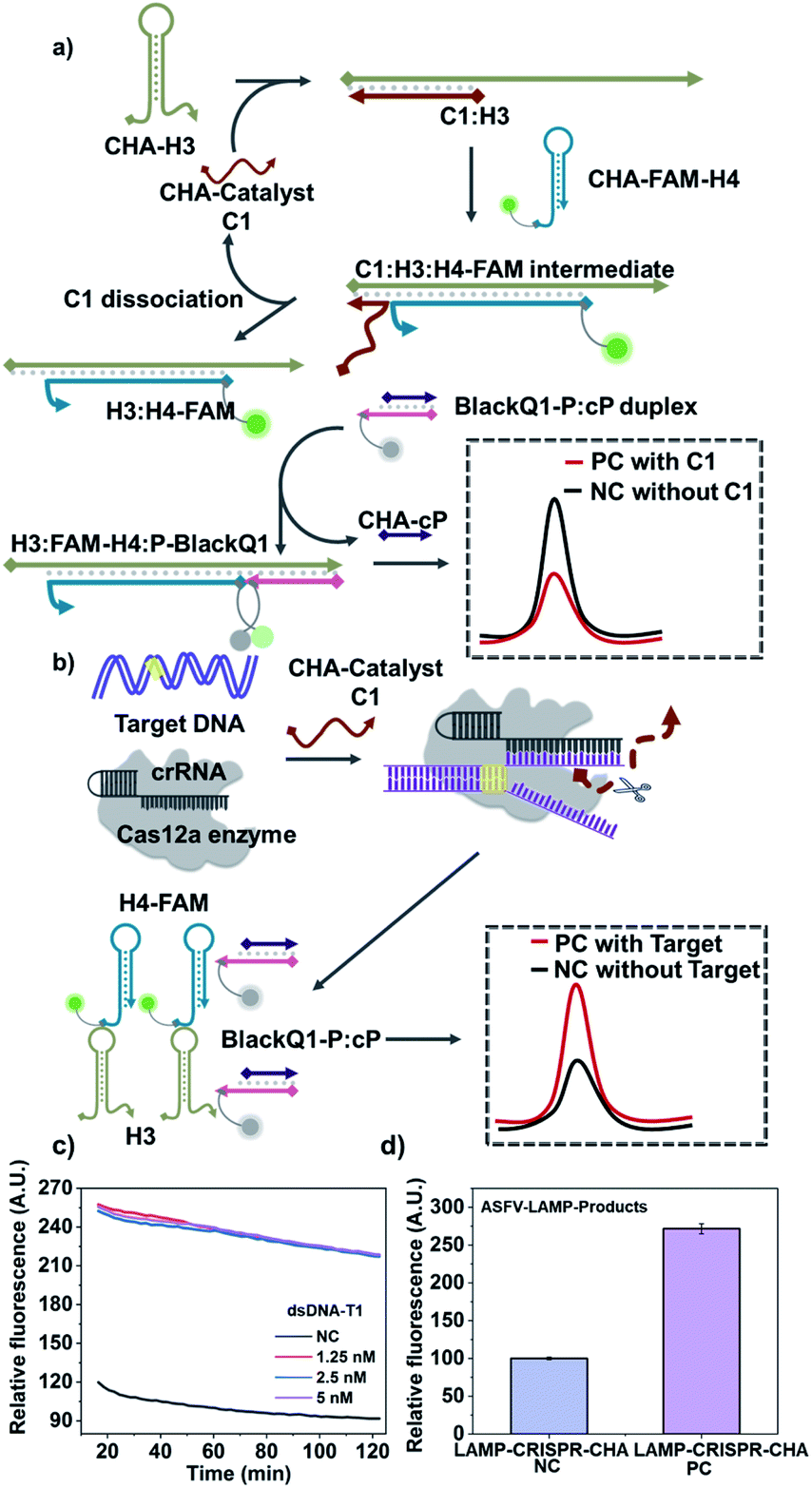 | ||
| Fig. 3 Verification of the flexibility via coupling CRISPR with CHA circuit (CRISPR-CHA). (a and b) Schematic illustration of CRISPR-CHA. Without the target sequence, C1 activates the CHA assembly and produces low fluorescence back-ground due to FRET (a). After target binds with crRNA, Cas12a cleaves C1 and remains CHA unreacted, producing increased fluorescence targeting signal (b). (c) Fluorescence kinetic curves of CRISPR-CHA without (“NC”) and with different concentrations of dsDNA-T1, in presence of C1, H3, H4-FAM and BlackQ1-P:cP. (d) Barograph of the fluorescence intensity of CRISPR-CHA detection for the ASFV-LAMP-products amplified from water control (“LAMP-CRSIPR-CHA NC”) and 500 copies per μL dsDNA-ASFV (“LAMP-CRSIPR-CHA PC”). The error bars represent standard deviation from two independent tests. | ||
In this communication, a specific, sensitive, and universal detection platform has been established via coupling CRISPR-Cas12a system and nucleic acid signal amplification circuits. The hybridization chain reaction, HCR, has been taken as the main model circuit. With the assistance of CRISPR-Cas12a system, HCR with traditional “off” signal has been improved to an “on” response. And a well-optimized, high-performance circuit can be generalized to any targeting sequence, either ssDNA or dsDNA. By combing an upstream ultra-amplifier (i.e., LAMP), the so-called CRISPR-HCR platform can detect pathogen genes down to aM level, meeting the high requirement of real-world applications. Finally, similar platform is proven also applicable to other circuits via replacing HCR with CHA circuit. The high adaptability is further confirmed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by National Key R&D Program of China (2019YFC1905400), Natural Science Foundation of China (22004118), Key R&D Program of Jilin Province (20200404178YY).Notes and references
- B. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110 CrossRef CAS PubMed.
- C. Jung and A. D. Ellington, Acc. Chem. Res., 2014, 47, 1825–1835 CrossRef CAS PubMed.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS PubMed.
- X. Chen, N. Briggs, J. R. McLain and A. D. Ellington, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5386–5391 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- J. Ikbal, G. S. Lim and Z. Gao, TrAC, Trends Anal. Chem., 2015, 64, 86–99 CrossRef CAS.
- S. Bi, S. Yue and S. Zhang, Chem. Soc. Rev., 2017, 46, 4281–4298 RSC.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- Q. Wu, H. Wang, K. K. Gong, J. H. Shang, X. Q. Liu and F. Wang, Anal. Chem., 2019, 91, 10172–10179 CrossRef CAS PubMed.
- S. G. Li, H. X. Li, X. Li, M. Zhu, H. Li and F. Xia, Anal. Chem., 2021, 93, 8354–8361 CrossRef CAS PubMed.
- K. Peng, H. Zhao, Y. Yuan, R. Yuan and X. Wu, Biosens. Bioelectron., 2014, 55, 366–371 CrossRef CAS PubMed.
- N. Liu, Y. Jiang, Y. Zhou, F. Xia, W. Guo and L. Jiang, Angew. Chem., Int. Ed., 2013, 52, 2007–2011 CrossRef CAS PubMed.
- J. Huang, X. Gao, J. Jia, J.-K. Kim and Z. Li, Anal. Chem., 2014, 86, 3209–3215 CrossRef CAS PubMed.
- J. Chen, Z. Huang, Z. Luo, Q. Yu, Y. Xu, X. Wang, Y. Li and Y. Duan, Anal. Chem., 2018, 90, 12019–12026 CrossRef CAS PubMed.
- H. Chu, J. Zhao, Y. Mi, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2019, 58, 14877–14881 CrossRef CAS PubMed.
- N. N. Liu, F. J. Huang, X. D. Lou and F. Xia, Sci. China: Chem., 2017, 60, 311–318 CrossRef CAS.
- J. Huang, Y. R. Wu, Y. Chen, Z. Zhu, X. H. Yang, C. J. Yang, K. M. Wang and W. H. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS PubMed.
- D. Yang, Y. Tang and P. Miao, TrAC, Trends Anal. Chem., 2017, 94, 1–13 CrossRef CAS.
- L. Yang, C. Liu, W. Ren and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 6450–6453 CrossRef CAS PubMed.
- Z.-Z. Yang, Z.-B. Wen, X. Peng, Y.-Q. Chai, W.-B. Liang and R. Yuan, Chem. Commun., 2019, 55, 6453–6456 RSC.
- J. Wei, X. Gong, Q. Wang, M. Pan, X. Liu, J. Liu, F. Xia and F. Wang, Chem. Sci., 2018, 9, 52–61 RSC.
- C. Yu, Y. Wang, R. Wu, Z. Zhu and B. Li, ACS Appl. Bio Mater., 2021, 4, 3649–3657 CrossRef CAS PubMed.
- F. Li, W. Yu, J. Zhang, Y. Dong, X. Ding, X. Ruan, Z. Gu and D. Yang, Nat. Commun., 2021, 12, 1138 CrossRef CAS PubMed.
- B. Li, Y. Jiang, X. Chen and A. D. Ellington, J. Am. Chem. Soc., 2012, 134, 13918–13921 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- Y. Xiong, J. Zhang, Z. Yang, Q. Mou, Y. Ma, Y. Xiong and Y. Lu, J. Am. Chem. Soc., 2020, 142, 207–213 CrossRef CAS PubMed.
- Y. Dai, R. A. Somoza, L. Wang, J. F. Welter, Y. Li, A. I. Caplan and C. C. Liu, Angew. Chem., Int. Ed., 2019, 58, 17399–17405 CrossRef CAS PubMed.
- D. C. Swarts and M. Jinek, Mol. Cell, 2019, 73, 589–600 CrossRef CAS PubMed.
- R. J. Yanez, J. M. Rodriguez, M. L. Nogal, L. Yuste, C. Enriquez, J. F. Rodriguez and E. Vinuela, Virology, 1995, 208, 249–278 CrossRef CAS PubMed.
- N. Tomita, Y. Mori, H. Kanda and T. Notomi, Nat. Protoc., 2008, 3, 877–882 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- Y. Tang, Z. Zhu, B. Lu and B. Li, Chem. Commun., 2016, 52, 13043–13046 RSC.
- Y. Tang, B. Lu, Z. Zhu and B. Li, Chem. Sci., 2018, 9, 760–769 RSC.
- Z. Zhu, Y. Tang, Y. S. Jiang, S. Bhadra, Y. Du, A. D. Ellington and B. Li, Sci. Rep., 2016, 6, 36605 CrossRef CAS PubMed.
- Z. Zhu, X. Duan, Q. Li, R. Wu, Y. Wang and B. Li, J. Am. Chem. Soc., 2020, 142, 4481–4492 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01332k |
| This journal is © The Royal Society of Chemistry 2022 |