
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous K-doped NiCo2O4 derived from a Prussian blue analog: high-yielding synthesis and assessment as oxygen evolution reaction catalyst†
Nam Woon Kim a,
Hyunung Yu*b and
Jihun Oh*cd
a,
Hyunung Yu*b and
Jihun Oh*cd
aDepartment of Nature-Inspired Nano Convergence Systems, Korea Institute of Machinery and Materials (KIMM), Daejeon 34103, Republic of Korea
bSurface Analysis Team, Korea Research Institute of Standards and Science (KRISS), Daejeon 34113, Republic of Korea. E-mail: peacewithu@kriss.re.kr
cDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: jihun.oh@kaist.ac.kr
dKAIST Institute for NanoCentury, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
First published on 25th April 2022
Abstract
The conversion and storage of clean renewable energy can be achieved using water splitting. However, water splitting exhibits sluggish kinetics because of the high overpotentials of the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER) and should therefore be promoted by OER and/or HER electrocatalysts. As the kinetic barrier of the former reaction exceeds that of the latter, high-performance OER catalysts are highly sought after. Herein, K-doped NiCo2O4 (HK-NCO) was hydrothermally prepared from a Prussian blue analog with a metal–organic framework structure and assessed as an OER catalyst. Extensive K doping increased the number of active oxygen vacancies and changed their intrinsic properties (e.g., binding energy), thus increasing conductivity. As a result, HK-NCO exhibited a Tafel slope of 49.9 mV dec−1 and a low overpotential of 292 mV at 10 mA cm−2, outperforming a commercial OER catalyst (Ir) and thus holding great promise as a component of high-performance electrode materials for metal-oxide batteries and supercapacitors.
Introduction
The increasing global energy demand necessitates the development of sustainable, clean and efficient ways of harvesting renewable energy,1,2 as exemplified by the production of hydrogen by electrocatalytic water splitting. However, water splitting suffers from sluggish kinetics due to the high overpotentials (i.e., kinetic barriers) of the anodic oxygen evolution reaction (OER; 2H2O → O2 + 4H+ + 4e−) and the cathodic hydrogen evolution reaction (HER; 2H+ + 2e− → H2). As the former reaction involves the transfer of four electrons, whereas the latter involves the transfer of only two electrons, the OER typically has a higher kinetic barrier than the HER and is therefore a key process determining the efficiency of water splitting.3 Noble metals such as Ir and Ru exhibit high OER activities but are of limited use in large-scale electrochemical water splitting because of their high cost and limited availability.2,4 Therefore, cheap and highly efficient OER catalysts with good stabilities in different electrolytes are highly sought after.Metal oxides hold great promise as water splitting catalysts, e.g., transition metal (Mn, Fe, Co, Ni, etc.) oxides have drawn much attention because of their high efficiencies and stabilities in alkaline solutions.5–7 In particular, NiCo2O4 (NCO) outperforms pure Ni or Co oxides as an OER catalyst.5,8–12 The OER activity of NCO in alkaline media is greatly influenced by the content of Ni, which affects both the energy of oxygen species adsorption at the active Co4+ surface sites and the energy barrier of the Co3+/Co4+ redox transition. The practical applications of NCO are hindered by its moderate conductivity and the insufficient number of active sites,13,14 which can be mitigated through hybridization with carbon materials or metal doping.15–18 Other factors influencing the performance of these catalysts include their synthetic route, morphology, and particle size. In this regard, it is critical to control the material morphology, number of active sites, specific surface area, active site accessibility, and the transport of electrolyte ions while ensuring sufficient catalyst stability.10
Prussian blue analogs (PBAs) and materials derived therefrom efficiently promote the OER because of their large specific surface areas, abundant active sites, short ion diffusion lengths, and high stabilities.1,19–23 In addition, PBAs adopt the M3II[MIII(CN)6]2·nH2O (M = Fe, Ni, Co, Mn, Cu, Zn, etc.) structure and can be used to prepare various materials (e.g., NCO) through M-site control.19,20,23–25 Herein, we hydrothermally prepare K-doped NCO with an elevated number of active sites (and, hence, with increased OER activity) from a metal–organic framework (MOF)-structured PBA with a large specific surface area and examine the effect of K doping on the oxygen vacancies of NCO and its OER overpotential.
Experimental
Fabrication of K-doped NCO
All chemicals were used as received without further purification. The composition of the precursor solution was determined by the stoichiometry of the final compound, NiCo2O4. Ni(NO3)2·6H2O (Sigma Aldrich, 99.999%) and Co(NO3)2·6H2O (Sigma Aldrich, ≥99%) were dissolved in distilled water (40 mL) to concentrations of 0.333 M (3.877 g) and 0.267 M (3.104 g), respectively, and this solution was slowly (over 3 min) poured into a 0.400 M (5.317 g) solution of K3[Co(CN)6] (Sigma Aldrich, ≥97%) in distilled water (40 mL) at room temperature (∼25 °C). After 10 min stirring, the reaction mixture was transferred to an autoclave, heated at 100 °C for 1 h, cooled to room temperature, and filtered. The K–Ni–Co-PBA precipitate was washed with distilled water (5 × 100 mL), dried overnight at 60 °C, heated to 250 °C at 1 °C min−1, and maintained at this temperature for 3 h to induce the formation of high-concentration K-doped NCO (HK-NCO). For comparison, low-concentration K-doped NCO (LK-NCO) was prepared without the hydrothermal process. When synthesis was performed using 80 mL of distilled water, the catalyst was obtained in a high yield of 3.46 g (0.4325 g/10 mL distilled water), which demonstrated the upscalability of our method.Material characterization
Microstructure and intrinsic crystallographic properties were probed by high-resolution transmission electron microscopy (HRTEM; JEM-ARM200F 200 kV, JEOL), while chemical composition was probed using energy-dispersive X-ray spectroscopy (EDS; Quantax 400, Bruker). The morphology of K-doped NCO nanoparticles was observed by environmental scanning electron microscopy (Quanta FEG 650, FEI Co.) at an accelerating voltage of 30 kV. Nanoparticle crystallinity was determined by X-ray diffraction (XRD; Smart Lab, Rigaku Co.) using Cu Kα radiation (λ = 1.5406 Å) and a scan rate of 0.2° s−1. X-ray photoelectron spectroscopy (XPS; Sigma Probe, Thermo VG Scientific) was employed for valence state analysis.Electrochemical measurements
Electrocatalytic activity was assessed in a three-electrode setup using a potentiostat (Bio-Logic, SP-150). A graphite rod, Ag/AgCl (saturated), and Ni foam (99.5%, 0.45 g cm−3) were used as counter, reference, and working electrodes, respectively. For catalyst ink preparation, the NCO catalyst (7 mg) and carbon black (1.2 mg) were dispersed in a pretreated Nafion solution (0.73 mL; 0.53 mL isopropyl alcohol + 0.13 mL distilled water + 27 μL 5 wt% Nafion), and the mixture was ultrasonicated for 3 h to obtain a homogeneous suspension. Ni foam was sequentially washed with acetone, methanol, and isopropyl alcohol upon ultrasonication, coated with the catalyst ink (1 cm2) on one side, and dried at 120 °C using a heating plate. The opposite side of the foam was coated with epoxy resin. Each synthesized NCO catalyst fabricated with the NCO/Ni foam electrode exhibited a catalytically active area of 494.29 cm2 (61.79 cm2/10 mL distilled water). Ir (Alfa Aesar, Product No. 039684, Iridium powder, Sponge, ∼325 mesh, 99.9%) was used as a benchmark OER catalyst for comparison. All evaluations were performed without gas purging and rotation in 1 M aqueous KOH (pH 14; Sigma Aldrich, 99.99%) at 0–800 mV vs. the saturated calomel electrode (SCE) using a scan rate of 1 mV s−1. The measured potentials were referenced to the reversible hydrogen electrode (RHE) as ERHE = ESCE + 0.197 + 0.059 pH. iR compensation was applied to all electrochemical data. Prior to measurements, the working electrode was activated in a steady state using 30 voltammetry cycles at 50 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were performed at a bias of 147 mV vs. Ag/AgCl, while the frequency was swept from 106 to 1 Hz at an AC amplitude of 10 mV. Chronoamperometric measurements were carried out to verify catalyst stability. Specifically, the catalyst-coated Ni foam was activated by cyclic voltammetry (CV) (∼30 cycles) in 1 M aqueous KOH, and measurements were then conducted for 12 h at a constant current density of 50 or 100 mA cm−2 to achieve reasonable OER progress.Results and discussion
Compositional and structural characterization
Fig. 1 shows the TEM images and corresponding EDS mappings of HK-NCO, revealing the presence of uniform NCO nanoparticles with sizes of 4–9 nm and the occasional incorporation of KNO3 agglomerates (Fig. 1b, red rectangle). The fast Fourier transform (FFT) analysis of HK-NCO demonstrated the presence of NiCo2O4 polycrystallites and KNO3 (Fig. S1†). EDS analysis (Fig. 1c–g) showed that K, Ni, Co, and O were uniformly distributed throughout the composite. Thus, the calcination of K–Ni–Co-PBA was concluded to successfully induce K doping and thus produce K-doped NiCo2O4 and KNO3. As shown in Fig. S2,† we obtained 3.46 g of HK-NCO catalyst powder using a high concentration of precursor and 80 mL of distilled water. About 3.21 g of pure NiCo2O4 catalyst powder should theoretically be produced under the experimental conditions presented by us; however, we produced 0.25 g more powder. We believe that the higher mass is due to potassium doping in NiCo2O4 and the generation of KNO3 products. We obtained catalyst products with higher-than-theoretical masses using high precursor concentrations. In this way, we present a high-yielding synthesis method that is very production-scale ideal. | ||
| Fig. 1 (a) and (b) HRTEM images and (c)–(g) EDX mappings of HK-NCO. (c) Overall map and the (d) Ni, (e) Co, (f) O, and (g) K components. The FFT patterns of NiCo2O4 (white square) and KNO3 (red square) are presented in Fig. S1.† | ||
Fig. 2 shows the XRD patterns of HK-NCO and LK-NCO, revealing that all peaks could be indexed to a cubic spinel structure with a = 8.114 Å, in good agreement with the literature (PDF No. 01-073-1702). The crystallite size of HK-NCO (Dc) was calculated using the Debye–Scherrer equation as Dc = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where β is the full width at half maximum of the diffraction line, K is the shape factor (typically equaling 0.94), and λ (1.5406 Å) is the wavelength of the X-ray source. The average HK-NCO crystallite size was estimated as 5.27 nm (2θ = 36.8, β = 0.02898) and agreed with the value estimated from Fig. 1a. Despite the agglomeration of HK-NCO particles, each individual particle approximately featured the size deduced from XRD data.
cosθ, where β is the full width at half maximum of the diffraction line, K is the shape factor (typically equaling 0.94), and λ (1.5406 Å) is the wavelength of the X-ray source. The average HK-NCO crystallite size was estimated as 5.27 nm (2θ = 36.8, β = 0.02898) and agreed with the value estimated from Fig. 1a. Despite the agglomeration of HK-NCO particles, each individual particle approximately featured the size deduced from XRD data.
 | ||
| Fig. 2 (a) XRD spectra of HK-NCO and LK-NCO. (b) Expansion of the spectra in (a) highlighting the (104), (006), and (110) peaks of KNO3. | ||
Fig. S3† shows SEM images of HK-NCO and LK-NCO nanoparticles, revealing that doping extent had no significant effect on morphology. However, XRD data (Fig. 2) suggested that HK-NCO featured a higher KNO3 content than LK-NCO. Specifically, in addition to typical NCO peaks (PDF No. 01-073-1702, Fig. 2a), we observed peaks attributable to the (104), (006), and (110) planes of KNO3 (PDF No. 01-074-1198, Fig. 2b), the intensities of which were higher for HK-NCO. As shown in Fig. S4,† no KNO3 crystals were present in K–Ni–Co-PBA, i.e., KNO3 was produced during calcination. Previous reports have shown that the electrical conductivities of such systems increase with an increase in the number of K+ ions because of the concomitant enhancement of ionic properties.26–28 Therefore, the KNO3 formed during the calcination of K–Ni–Co-PBA was expected to provide K+ ions and thus increase ion mobility and electrical conductivity. It should be noted that we successfully observed a calcination-induced phase change during the preparation of PBA-derived NCO nanoparticles.
The catalyst surface compositions as well as valence and K-doping states were probed by XPS (Fig. 3). The spectra of both HK-NCO and LK-NCO were calibrated using the C 1s peak of adventitious carbon at 284.8 eV (Fig. S5a†).29,30 High-resolution core-level Ni 2p (Fig. 3a) and Co 2p (Fig. 3b) spectra featured two typical twin spin-orbital doublets flanked by two satellite peaks. The Ni 2p3/2 peak of HK-NCO was deconvoluted into the signals of Ni3+ (855.77 eV) and Ni2+ (854.14 eV), while the corresponding Co 2p3/2 peak was deconvoluted into the signals of Co3+ (779.76 eV) and Co2+ (781.58 eV). The M3+/M2+ ratio on the surface of spinel-structured species such as NCO affects their electronic structure and catalytic performance, as these points act as OER active sites.31–34 The Ni3+/Ni2+ ratios of HK-NCO and LK-NCO were determined as 0.948 and 0.704, respectively, whereas the respective Co3+/Co2+ ratios equaled 2.972 and 2.635. Thus, trivalent metal ions were more abundant on the surface of HK-NCO. Previous reports have suggested that the OER activity of NCO catalysts is influenced by the presence of Ni at octahedral sites, indicated that Ni valence mainly determines the OER activity of inverse-spinel NCO.34 Therefore, the OER performance of HK-NCO was expected to be superior to that of LK-NCO. The intensities of the N 1s peaks (Fig. S5b†) were used to account (Fig. S5c†) for the contributions of KNO3 to O 1s and K 2p spectra (Fig. 3c and d, respectively) and thus single out the contributions of K-doped NCO, with the data used for this calculation presented in Table S1.† The O 1s spectra could be deconvoluted into the peaks of metal–oxygen bonds (O1; M–O–M, 529.38 eV), oxygen vacancies (O2; M–O–H, 531.37 eV), and water chemically or physically adsorbed on the surface (O3; H–O–H, 532.87 eV).33,35,36 It should be noted that oxygen vacancy adjustment significantly changes the surface electronic state, thereby increasing the activity of catalytically active states. In particular, catalysts with adjusted oxygen vacancies maintain highly oxidizing conditions in the OER process and thus exhibit elevated activities.34,37,38 Consequently, techniques such as pulse laser ablation,34 NaBH4 treatment,38,39 Ar plasma treatment,40 and calcination in an Ar or N2 atmosphere29,37,41 have been used to form oxygen vacancies in OER catalysts. Compared to these methods, our approach offers the benefits of simplicity and high speed. The oxygen vacancy fraction in K-doped catalysts was determined as the O2/(O1 + O2) peak area ratio, which equaled 0.447 for HK-NCO and 0.344 for LK-NCO. As HK-NCO had a 29.9% greater oxygen vacancy fraction than LK-NCO, it was expected to exhibit a higher OER activity. The K 2p3/2 peak of HK-NCO (292.63 eV) was located at a lower energy than that of LK-NCO (292.84 eV), which means that K doped into HK-NCO had a higher electron abundance than that doped into LK-NCO. As a result, HK-NCO lost more electrons in Ni and Co than LK-NCO, increasing the number of ions in Ni3+ and Co3+. Due to this effect, we think that the peaks of O1 and K of HK-NCO shift to low binding energy levels by charge-transferring. The extent of K doping, expressed as KNCO/(Nitotal + Cototal + KNCO), was 3.13 times higher for HK-NCO than for LK-NCO (0.02964 vs. 0.00946). As a result, more M3+ and oxygen vacancies were available in HK-NCO, as confirmed by XPS analysis, which is believed to contribute to the enhanced OER activity of this catalyst.
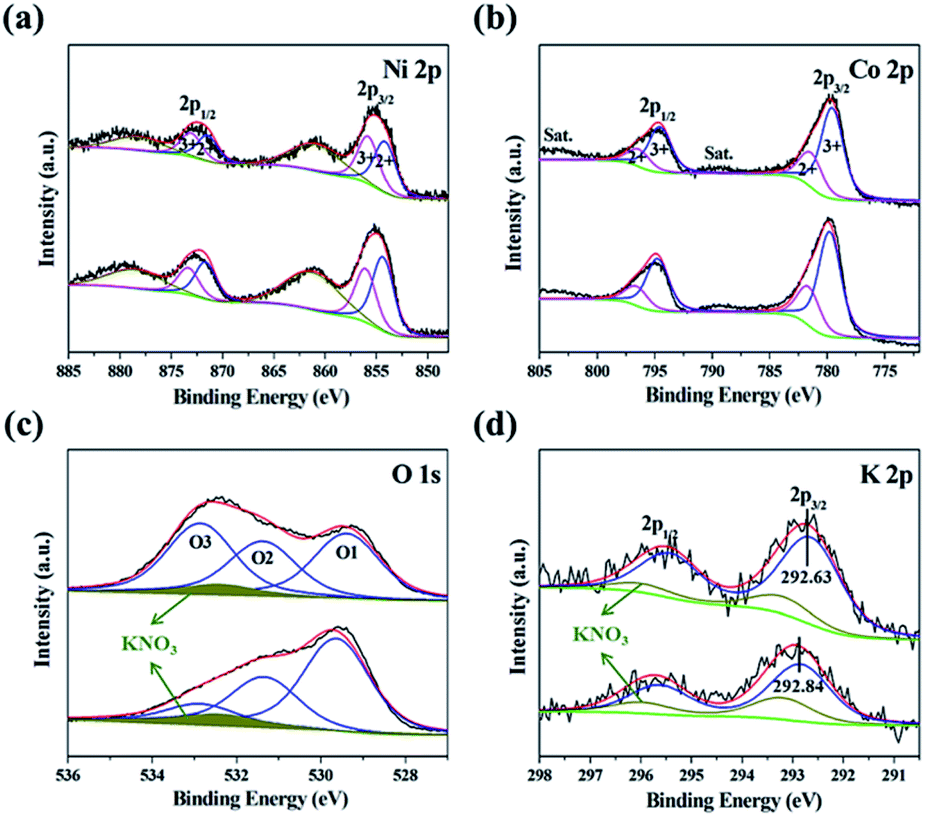 | ||
| Fig. 3 (a) Ni 2p, (b) Co 2p, (c) O 1s, and (d) K 2p spectra of NCO catalysts. The O 1s and K 2p spectra were fitted excluding the peaks due to KNO3. The spectra of HK-NCO are shown at the top, while those of LK-NCO are shown at the bottom. Black lines indicate raw data, while red lines indicate the fitting curve, with green lines indicating baselines. | ||
Electrochemical activity and stability
The iR-corrected results of catalyst activity and stability evaluation are shown in Fig. 4. Fig. 4a shows the results of linear sweep voltammetry (LSV) measurements performed at a scan rate of 1 mV s−1. The oxidation peak was mainly attributed to the reactions associated with M–O/M–O–OH (M = Ni or Co). In the case of NCO, Ni3+/Ni4+ and Co3+/Co4+ oxidation peaks are usually observed at 1.43 and 1.45 V, respectively.42 As the oxidation peak in Fig. 4a appeared at <1.43 V, it corresponded to an electrochemical reaction involving other ions.42–45 Therefore, we considered that the more negative oxidation peak observed for NCO may promote the production of Ni4+ and Co4+ species and consequently improve OER performance.46 HK-NCO exhibited a significantly larger oxidation peak area than LK-NCO and was therefore concluded to have a greater number of available active sites and a greater degree of Ni4+ and Co4+ production, which resulted in enhanced OER performance.46 The overpotentials of HK-NCO and LK-NCO at a current density of 100 mA cm−2 were determined as 369 and 410 mV, respectively. The results of reverse-current CV scanning (Fig. S6†) also indicated a low overpotential of 292 mV at a current density of 10 mA cm−2. | ||
| Fig. 4 (a) OER polarization curves and (b) ΔJ (=Ja − Jc) vs. scan rate plots of the investigated catalysts. ECSAs were calculated from Cdl values. (c) Tafel slopes obtained using reverse-current CV measurements and (d) Nyquist plots of the two catalysts. | ||
The role of ions within the electrochemical surface boundaries of OER catalysts was probed by double-layer capacitance (Cdl) measurements. Specifically, Cdl values were extracted from CV curves in non-faradaic potential regions (1.15–1.2 V vs. RHE) by plotting the current density at 1.175 V vs. RHE against the scan rate. The Cdl values of HK-NCO and LK-NCO were determined from the slopes of the corresponding linear fits as 0.369 and 0.263 F cm−2, respectively (Fig. 4b), indicating that the double layer of HK-NCO contained 40% more OH− ions than that of LK-NCO and showing that these functional groups were adsorbed on the catalyst surface.4 In other words, HK-NCO possessed a greater number of electrochemically active sites capable of triggering the OER, which is consistent with the results inferred from XPS data.
Reverse-current CV scanning (Fig. S6†) was performed in view of the difficulty of obtaining the correct Tafel slopes of HK-NCO and LK-NCO due to the wide Ni and Co oxidation peaks observed at a scan rate of 1 mV s−1. As a result, the Tafel slopes of these catalysts were determined as 49.9 and 58.6 mV dec−1, respectively. These results indicate that the HK-NCO electrode exhibited slightly faster charge transfer kinetics than the LK-NCO electrode.43,47
Fig. 4d shows the Nyquist plots of HK-NCO and LK-NCO. In these plots, the x-axis intercept in the high-frequency region is related to the equivalent series resistance (Rs), which includes the contributions of the ionic resistance of the electrolyte, the intrinsic resistivity of the substrate, and the contact resistance of the active material/current collector. The diameter of the semicircle corresponds to ionic charge transfer resistance (Rct), which is caused by the electric double-layer capacitor (Cdl) and faradaic reactions. The Warburg resistance (ZW), which corresponds to the 45° portion of the Nyquist plot, represents the diffusion resistance of OH− ions within the K-doped NiCo2O4/Ni foam collector, and CPE is the constant-phase-angle element related to ZW.48,49 The fitting result is shown in Fig. S7.† The series resistances (Rs) of HK-NCO and LK-NCO were determined as 1.132 and 1.161 Ω, respectively, while the corresponding Rct values equaled 0.458 and 0.630 Ω, respectively. These values indicate faster interfacial charge and electron transfer in HK-NCO and are directly related to OER performance. According to previous reports,26,27,50,51 the electrical conductivity can be increased by the incorporation of KNO3. Therefore, we concluded that the KNO3 present in both catalysts contributed to their increased conductivity and decreased resistance. Subsequently, we obtained electrochemical active surface area (ECSA)-normalized LSV curves (Fig. S8†) to investigate the effect of increased conductivity on intrinsic catalytic activity. ECSAs were calculated from Cdl and the specific capacitance (Cs) of NCO (i.e., 0.026 mF cm−2) as ECSA = Cdl/Cs, as has previously been reported for a flat substrate.4 In the obtained curves, the current increase factor of HK-NCO results from its increased conductivity and elevated ECSA.
Next, we compared the LSV performances of Ir nanoparticles, a representative OER catalyst, and Ni foam bearing HK-NCO. All catalysts were fabricated as electrodes under the same conditions, and the results are shown in Fig. 5a. Notably, HK-NCO exhibited a steep current increase and proved to be an outstanding OER catalyst with an overpotential 76 mV lower than that of the Ir catalyst at 100 mV cm−2. We also demonstrated the superiority of HK-NCO catalysts compared to other literature electrocatalysts shown in Table S2.† Finally, the stability of HK-NCO was evaluated at high current densities of 50 and 100 mA cm−2. As can be seen from Fig. 5b, this catalyst exhibited good stability over 12 h, thus holding great promise for future applications.
 | ||
| Fig. 5 (a) OER polarization curves of HK-NCO/Ni foam, pure Ni foam, and Ir/Ni foam electrodes. (b) Chronoamperograms of HK-NCO recorded over 12 h at current densities of 50 and 100 mA cm−2 for stability evaluation. | ||
Conclusions
High-concentration K-doped NCO (HK-NCO) hydrothermally prepared from a PBA analog exhibited high performance as an OER catalyst, outperforming a benchmark Ir catalyst and low-concentration K-doped NCO prepared in the absence of hydrothermal treatment. The enhanced OER performance of HK-NCO (overpotential of 369 mV at 100 mV cm−2 and a Tafel slope of 49.9 mV dec−1) was ascribed to the increased number of active sites due to extensive K doping. These results indicate that K-doped NCO nanoparticles efficiently promote the OER and are well suited for practical applications as high-performance electrode materials of metal oxide batteries, supercapacitors, and electrochemical catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Council of Science & Technology (NST) grant by the Korean Government (MSIT) (No. CAP-18-04-KRISS). This work was supported by the National Research Foundation of Korea (NRF-2019M1A2A2065612). The authors gratefully acknowledge financial support provided by the “Carbon to X Project” (NRF-2020M3H7A1096388) through the National Research Foundation (NRF), funded by the Ministry of Science and ICT, Republic of Korea.Notes and references
- W. Ahn, M. G. Park, D. U. Lee, M. H. Seo, G. Jiang, Z. P. Cano, F. M. Hassan and Z. Chen, Adv. Funct. Mater., 2018, 28, 1 CrossRef.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290 CrossRef CAS.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488 CrossRef CAS.
- C. Broicher, F. Zeng, J. Artz, H. Hartmann, A. Besmehn, S. Palkovits and R. Palkovits, ChemCatChem, 2019, 11, 412 CrossRef CAS.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337 RSC.
- J. R. Galán-Mascarós, ChemElectroChem, 2015, 2, 37 CrossRef.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- H. Wang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 6255 CrossRef CAS PubMed.
- C. Zhu, D. Wen, S. Leubner, M. Oschatz, W. Liu, M. Holzschuh, F. Simon, S. Kaskel and A. Eychmüller, Chem. Commun., 2015, 51, 7851 RSC.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926 CrossRef CAS PubMed.
- H. N. Nong, H. S. Oh, T. Reier, E. Willinger, M. G. Willinger, V. Petkov, D. Teschner and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 2975 CrossRef CAS PubMed.
- X. Wang, Y. Fang, B. Shi, F. Huang, F. Rong and R. Que, Chem. Eng. J., 2018, 344, 311 CrossRef CAS.
- M. Li, L. Tao, X. Xiao, X. Jiang, M. Wang and Y. Shen, ACS Sustainable Chem. Eng., 2019, 7, 4784 CrossRef CAS.
- P. Wu, S. Cheng, M. Yao, L. Yang, Y. Zhu, P. Liu, O. Xing, J. Zhou, M. Wang, H. Luo and M. Liu, Adv. Funct. Mater., 2017, 27, 1 Search PubMed.
- Y. Zhu, Z. Wu, M. Jing, H. Hou, Y. Yang, Y. Zhang, X. Yang, W. Song, X. Jia and X. Ji, J. Mater. Chem. A, 2015, 3, 866 RSC.
- C. Zhang, J. Wei, L. Chen, S. Tang, M. Deng and Y. Du, Nanoscale, 2017, 9, 15423 RSC.
- Q. Zhang, B. Zhao, J. Wang, C. Qu, H. Sun, K. Zhang and M. Liu, Nano Energy, 2016, 28, 475 CrossRef.
- L. Han, X. Y. Yu and X. W. (David) Lou, Adv. Mater., 2016, 28, 4601 CrossRef CAS PubMed.
- Q. Ma, R. Dong, H. Liu, A. Zhu, L. Qiao, Y. Ma, J. Wang, J. Xie and J. Pan, J. Alloys Compd., 2020, 820, 153438 CrossRef CAS.
- H. Xu, H. Shang, L. Jin, C. Chen, C. Wang and Y. Du, J. Mater. Chem. A, 2019, 7, 26905 RSC.
- X. Gu, Z. Liu, H. Liu, C. Pei and L. Feng, Chem. Eng. J., 2021, 403, 126371 CrossRef CAS.
- S. Lei, Q. H. Li, Y. Kang, Z. G. Gu and J. Zhang, Appl. Catal., B, 2019, 245, 1 CrossRef CAS.
- X. Y. Yu, L. Yu, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 5331 CrossRef CAS PubMed.
- J. Nai and X. W. Lou, Adv. Mater., 2019, 31, 1 CrossRef.
- K. Sreelalitha and K. Thyagarajan, International Journal of Thin Film Science and Technology, 2014, 3, 39 CrossRef.
- C. K. Mahadevan, Phys. B, 2008, 403, 57 CrossRef CAS.
- E. Rysiakiewicz-Pasek, A. Cizman, I. Drozdova, I. Polyakova and T. Antropova, Composites, Part B, 2016, 91, 291 CrossRef CAS.
- D. Sun, Y. Li, X. Cheng, H. Shi, S. Jaffer, K. Wang, X. Liu, J. Lu and Y. Zhang, Electrochim. Acta, 2018, 279, 269 CrossRef CAS.
- X. Wang, T. Gao, X. Fan, F. Han, Y. Wu, Z. Zhang, J. Li and C. Wang, Adv. Funct. Mater., 2016, 26, 7164 CrossRef CAS.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1 Search PubMed.
- S. Liu, L. Hu, X. Xu, A. A. Al-Ghamdi and X. Fang, Small, 2015, 11, 4267 CrossRef CAS PubMed.
- J. Wang, R. Gao, D. Zhou, Z. Chen, Z. Wu, G. Schumacher, Z. Hu and X. Liu, ACS Catal., 2017, 7, 6533 CrossRef CAS.
- Y. Liu, P. Liu, W. Qin, X. Wu and G. Yang, Electrochim. Acta, 2019, 297, 623 CrossRef CAS.
- E. Umeshbabu and G. R. Rao, Electrochim. Acta, 2016, 213, 717 CrossRef CAS.
- J. F. Marco, J. R. Gancedo, M. Gracia, J. L. Gautier, E. Ríos and F. J. Berry, J. Solid State Chem., 2000, 153, 74 CrossRef CAS.
- R. Gao, L. Liu, Z. Hu, P. Zhang, X. Cao, B. Wang and X. Liu, J. Mater. Chem. A, 2015, 3, 17598 RSC.
- D. Yan, W. Wang, X. Luo, C. Chen, Y. Zeng and Z. Zhu, Chem. Eng. J., 2018, 334, 864 CrossRef CAS.
- C. Zhu, S. Fu, D. Du and Y. Lin, Chem.–Eur. J., 2016, 22, 4000 CrossRef CAS PubMed.
- Z. Xiao, Y. Wang, Y. C. Huang, Z. Wei, C. L. Dong, J. Ma, S. Shen, Y. Li and S. Wang, Energy Environ. Sci., 2017, 10, 2563 RSC.
- W. Liu, J. Bao, L. Xu, M. Guan, Z. Wang, J. Qiu, Y. Huang, J. Xia, Y. Lei and H. Li, Appl. Surf. Sci., 2019, 478, 552 CrossRef CAS.
- P. Rasiyah and A. C. C. Tseung, J. Electrochem. Soc., 1982, 129, 1724 CrossRef CAS.
- L. Zhang, Y. Li, J. Peng and K. Peng, Electrochim. Acta, 2019, 318, 762 CrossRef CAS.
- J. Haenen, W. Visscher and E. Barendrecht, J. Electroanal. Chem., 1986, 208, 273 CrossRef CAS.
- X. Zhang, W. Sun, H. Du, R. M. Kong and F. Qu, Inorg. Chem. Front., 2018, 5, 344 RSC.
- C. Xiao, Y. Li, X. Lu and C. Zhao, Adv. Funct. Mater., 2016, 26, 3515 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- M. J. Pang, S. Jiang, G. H. Long, Y. Ji, W. Han, B. Wang, X. L. Liu, Y. L. Xi, F. Z. Xu and G. D. Wei, RSC Adv., 2016, 6, 67839 RSC.
- F. O. Ochai-Ejeh, A. Bello, J. Dangbegnon, A. A. Khaleed, M. J. Madito, F. Bazegar and N. Manyala, J. Mater. Sci., 2017, 52, 10600 CrossRef CAS.
- M. C. Rao, C. S. Rao and T. Srikumar, Mater. Today: Proc., 2018, 5, 26365 CAS.
- P. Kesharwani, D. K. Sahu, Y. K. Mahipal and R. C. Agrawal, Mater. Chem. Phys., 2017, 193, 524 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional FFT patterns, optical image, SEM images, XRD patterns, XPS data, and electrochemical characterization results. See https://doi.org/10.1039/d2ra01235a |
| This journal is © The Royal Society of Chemistry 2022 |