DOI:
10.1039/D2RA01199A
(Paper)
RSC Adv., 2022,
12, 10749-10754
High capacity all-solid-state lithium battery enabled by in situ formation of an ionic conduction path by lithiation of MgH2†
Received
22nd February 2022
, Accepted 30th March 2022
First published on 6th April 2022
Abstract
All-solid-state Li batteries have attracted significant attention because of their high energy density and high level of safety. In a solid-state Li-ion battery, the electrodes contain a solid electrolyte that does not contribute directly to the capacity. Therefore, a battery that does not require a solid electrolyte in its electrode mixture should exhibit a higher energy density. In this study, a MgH2 electrode was used as the negative electrode material without a solid electrolyte in its mixture. The resultant battery demonstrated excellent performance because of the formation of an ionic conduction path based on LiH in the electrode mixture. LiH and Mg clearly formed upon lithiation and returned to MgH2 upon delithiation as revealed by TEM-EELS analysis. This mechanism of in situ electrolyte formation enables the development of a solid-state battery with a high energy density.
Introduction
All-solid-state Li batteries have attracted significant attention because they can provide high energy densities and high levels of safety.1,2 In recent years, sulfide- and oxide-based electrolytes have mainly been used in solid-state cells. However, hydride-based electrolytes are also well known for their high ionic conductivity and high compatibility with Li metal.3,4 Several studies on the electrochemical formation of electrodes from electrolyte materials have recently been reported. Yamamoto et al. reported the in situ formation of an anode from a sodium superionic conductor (NASICON)-type solid electrolyte via electrochemical lithiation at 2.3 V vs. Li/Li+, mainly via the redox reaction of Ti4+/Ti3+.5 Han et al. reported a battery made from a single material using a Li10GeP2S12 solid electrolyte to eliminate the high interfacial resistance of a solid-state battery.6 Hakari reported all-solid-state batteries with Li3PS4 glass as an active material.7,8 Our group recently studied an all-solid-state Li battery based on electrochemical Li+ insertion into a Mg(BH4)2 anode; the battery exhibited a suitable operating voltage (0.9 V vs. Li/Li+) and a large capacity.9 The insertion of Li+ into Mg(BH4)2 occurs via the following electrochemical reaction:| | |
Mg(BH4)2 + 2Li+ + 2e− → 2LiBH4 + Mg
| (1) |
In this case, LiBH4, a hydride with high Li+ ionic conductivity, is generated and an ionic conduction path is formed during the electrochemical reaction. The LiBH4 undergoes a structural transformation from a low-conductivity (orthorhombic) phase to a high-conductivity (hexagonal) phase at ∼115 °C, and the high-temperature phase exhibits high Li+ ionic conductivity.10,11 Surprisingly, the electrode mixture does not require a solid electrolyte; reasonable battery performance was observed with an electrode composed of Mg(BH4)2 and acetylene black (AB) without LiBH4 as an electrolyte. This performance is achieved via the in situ formation of an electrolyte, which contributes to an increase in the actual capacity of the battery. In a typical solid-state Li+-ion battery, the electrodes contain a solid electrolyte and conductive carbon, which do not contribute directly to the battery's capacity; therefore, the new concept of in situ electrolyte formation enables the development of a battery with a high energy density. The reversible capacity of the Mg(BH4)2 electrode was found to be 563 mA h g−1 in a previous study.9 However, the literature contains numerous reports on the electrochemical properties of Li batteries with hydride-based electrolytes and electrodes,12–25 including MgH2 electrodes, in particular.12–22 The following electrochemical reaction occurs upon the insertion of Li+ into MgH2:
| | |
MgH2 + 2Li+ + 2e− → 2LiH + Mg
| (2) |
According to one report, a MgH2 electrode demonstrated a reversible capacity of 1650 mA h g−1.12 In addition, the working potential of MgH2 is ∼0.5 V (vs. Li/Li+), which provides an adequate margin from the potential of Li metal. Therefore, from the viewpoint of capacity and potential, MgH2 has a large advantage as an electrode material. MgH2 is transformed into LiH upon insertion of Li+ ions. Although the ionic conductivity of LiH is not as high as that of LiBH4, LiH is a Li+ ionic conductor.26–28 Therefore, a MgH2 electrode can undergo in situ electrolyte formation upon the insertion of Li+ ions. Because the theoretical capacity of MgH2 (2036 mA h g−1) is much larger than that of Mg(BH4)2 (992 mA h g−1), which was previously studied as an electrode material that forms an electrolyte in situ, MgH2 should be a promising electrode material without a solid electrolyte incorporated into the electrode mixture. In the present study, the electrochemical in situ formation of a Li+ ionic conduction path in MgH2 is investigated and transmission electron microscopy with electron energy-loss spectroscopy (TEM-EELS) analysis is used to visualize the formation of the LiH and Mg in the electrode mixture during the electrochemical reactions.
Experimental
All operations were carried out in a glove box filled with Ar gas. Commercially available LiBH4 (≥95%, Sigma-Aldrich) was hand milled for 5 min before use. The LiBH4 was used as a solid electrolyte. Two composite powders containing MgH2 (95%, Sigma-Aldrich) were prepared by a mechanical milling method: one containing a solid electrolyte and the other without the solid electrolyte. A planetary-type mill (FRITSCH Pulverisette 6) was used for mechanical milling. The composite powders were obtained by the mechanical milling of MgH2 with LiBH4 and AB in a 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:30 mass ratio or MgH2 and AB in a 70:30 mass ratio at 400 rpm for 30 min. For the electrode without LiBH4, other compositions (MgH2:AB = 99:1 and 95:5) were also evaluated. A cell set with a sleeve made of polyether ether ketone (PEEK, EQ-PSC, AA Portable Power Corp.) was used to fabricate a battery with an inside diameter of 10 mm. LiBH4 was pressed at 255 MPa. A portion of either the MgH2 + LiBH4 + AB or MgH2 + AB mixture was placed on top of the LiBH4 layer and pressed at 320 MPa. Li foil (Honjo Metal Co.) was pressed by hand between an SUS plate and a LiBH4 electrolyte layer as a counter electrode. Herein, batteries with a structure of Li/LiBH4/MgH2 + LiBH4 + AB (40:30:30 by weight) and Li/LiBH4/MgH2 + AB (70:30, 95:5, 99:1 by weight) were prepared. The discharge/charge properties of the batteries were examined using a galvanostat (Hokuto Denko Co.) at a current density of 0.5 mA cm−2 and at 120 °C. The voltage range was set between 0.3 and 1.0 V (vs. Li/Li+). The capacity was calculated using two methods. First, the capacity of each device was calculated on the basis of the mass of MgH2 in the electrode mixture, not including the masses of the solid electrolyte and AB. Second, the capacity was calculated from the mass of the electrode mixture including MgH2, the solid electrolyte, and the AB. Scanning Electron Microscope (SEM) images were obtained by JCM-7000 (JEOL Ltd). Nanoscopic structural analysis was performed using transmission electron microscopy (Titan Cubed 60-300 G2, Thermo Fisher Scientific) with a monochromator for the electron source, a spherical aberration corrector (DCOR, CEOS) for the probe-forming lens system, detectors for energy-dispersive X-ray spectroscopy (Super X), and an energy filter for electron energy loss spectroscopy (EELS; GIF Quantum 965 spectrometer, Gatan). The microscope was operated in scanning transmission electron microscopy (STEM) mode at an accelerating voltage of 300 kV. Three samples were prepared for the STEM analysis: MgH2 and AB composite powders in their initial state, and the corresponding layer in the charged and discharged states. These samples were extracted from the batteries and were mechanically crushed in a glove box filled with Ar gas. The resultant submicrometer grains were dispersed onto TEM grids with a carbon supporting film and were loaded into a dedicated cryo-holder (Mel-Build) for transfer from the glove box to the transmission electron microscope without exposure to the atmosphere. During the STEM analysis, the temperature at the top end of the sample holder was maintained at −100 °C by liquid N2.
:30:30 mass ratio or MgH2 and AB in a 70:30 mass ratio at 400 rpm for 30 min. For the electrode without LiBH4, other compositions (MgH2:AB = 99:1 and 95:5) were also evaluated. A cell set with a sleeve made of polyether ether ketone (PEEK, EQ-PSC, AA Portable Power Corp.) was used to fabricate a battery with an inside diameter of 10 mm. LiBH4 was pressed at 255 MPa. A portion of either the MgH2 + LiBH4 + AB or MgH2 + AB mixture was placed on top of the LiBH4 layer and pressed at 320 MPa. Li foil (Honjo Metal Co.) was pressed by hand between an SUS plate and a LiBH4 electrolyte layer as a counter electrode. Herein, batteries with a structure of Li/LiBH4/MgH2 + LiBH4 + AB (40:30:30 by weight) and Li/LiBH4/MgH2 + AB (70:30, 95:5, 99:1 by weight) were prepared. The discharge/charge properties of the batteries were examined using a galvanostat (Hokuto Denko Co.) at a current density of 0.5 mA cm−2 and at 120 °C. The voltage range was set between 0.3 and 1.0 V (vs. Li/Li+). The capacity was calculated using two methods. First, the capacity of each device was calculated on the basis of the mass of MgH2 in the electrode mixture, not including the masses of the solid electrolyte and AB. Second, the capacity was calculated from the mass of the electrode mixture including MgH2, the solid electrolyte, and the AB. Scanning Electron Microscope (SEM) images were obtained by JCM-7000 (JEOL Ltd). Nanoscopic structural analysis was performed using transmission electron microscopy (Titan Cubed 60-300 G2, Thermo Fisher Scientific) with a monochromator for the electron source, a spherical aberration corrector (DCOR, CEOS) for the probe-forming lens system, detectors for energy-dispersive X-ray spectroscopy (Super X), and an energy filter for electron energy loss spectroscopy (EELS; GIF Quantum 965 spectrometer, Gatan). The microscope was operated in scanning transmission electron microscopy (STEM) mode at an accelerating voltage of 300 kV. Three samples were prepared for the STEM analysis: MgH2 and AB composite powders in their initial state, and the corresponding layer in the charged and discharged states. These samples were extracted from the batteries and were mechanically crushed in a glove box filled with Ar gas. The resultant submicrometer grains were dispersed onto TEM grids with a carbon supporting film and were loaded into a dedicated cryo-holder (Mel-Build) for transfer from the glove box to the transmission electron microscope without exposure to the atmosphere. During the STEM analysis, the temperature at the top end of the sample holder was maintained at −100 °C by liquid N2.
Results and discussion
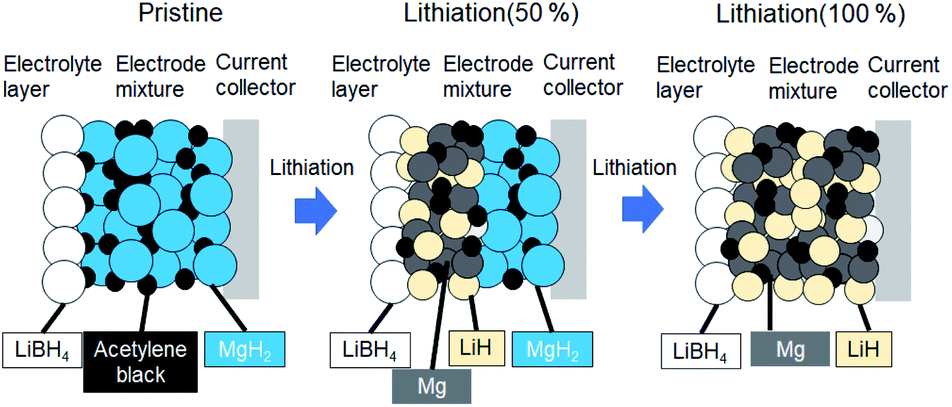
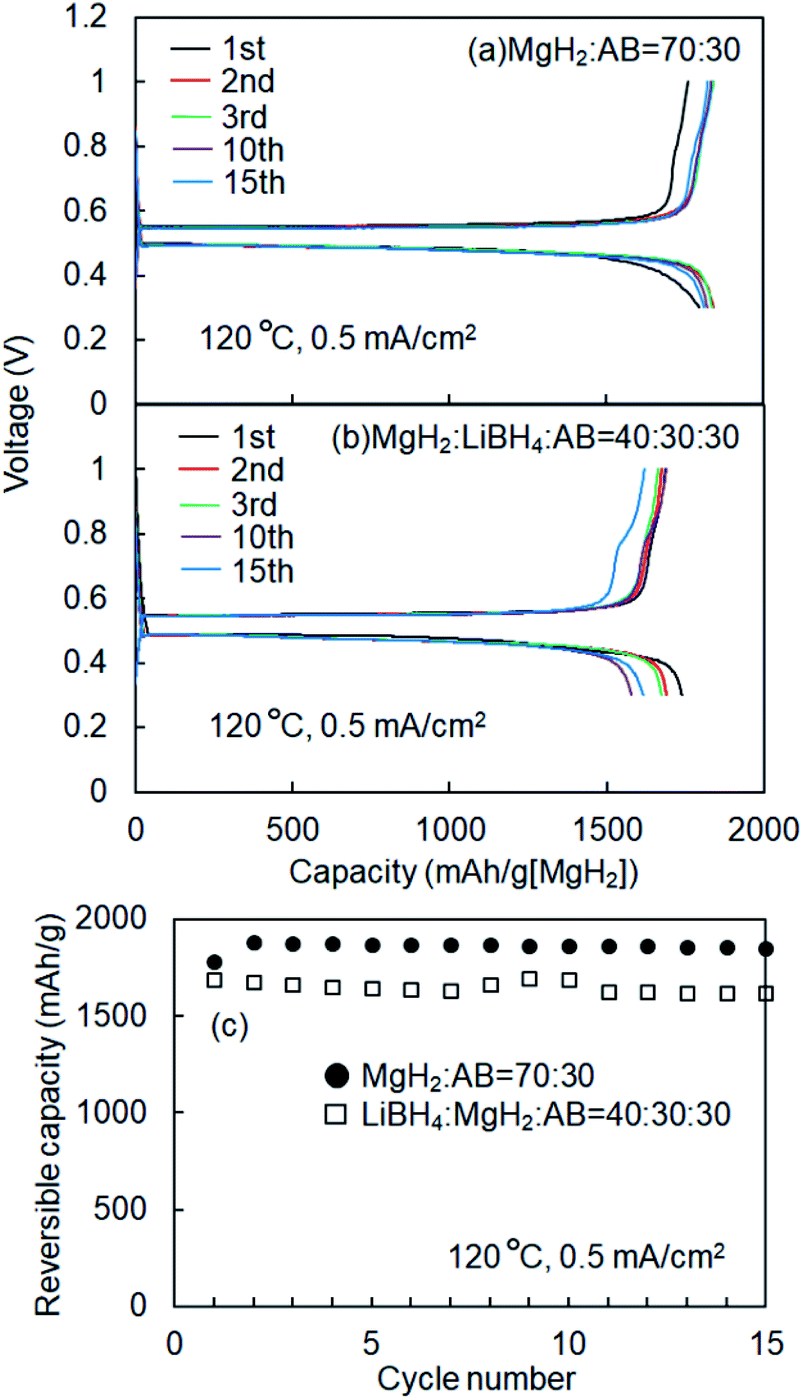
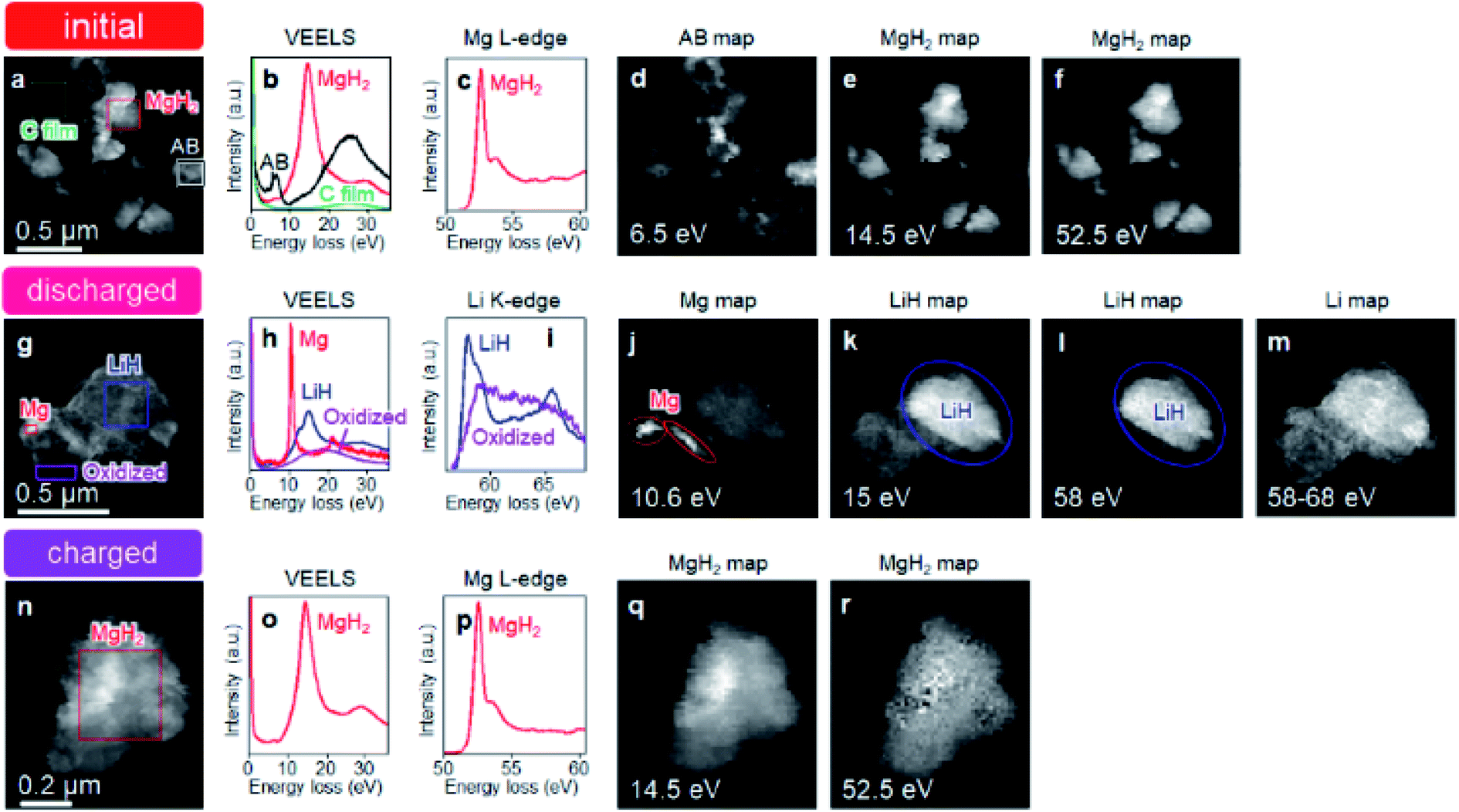
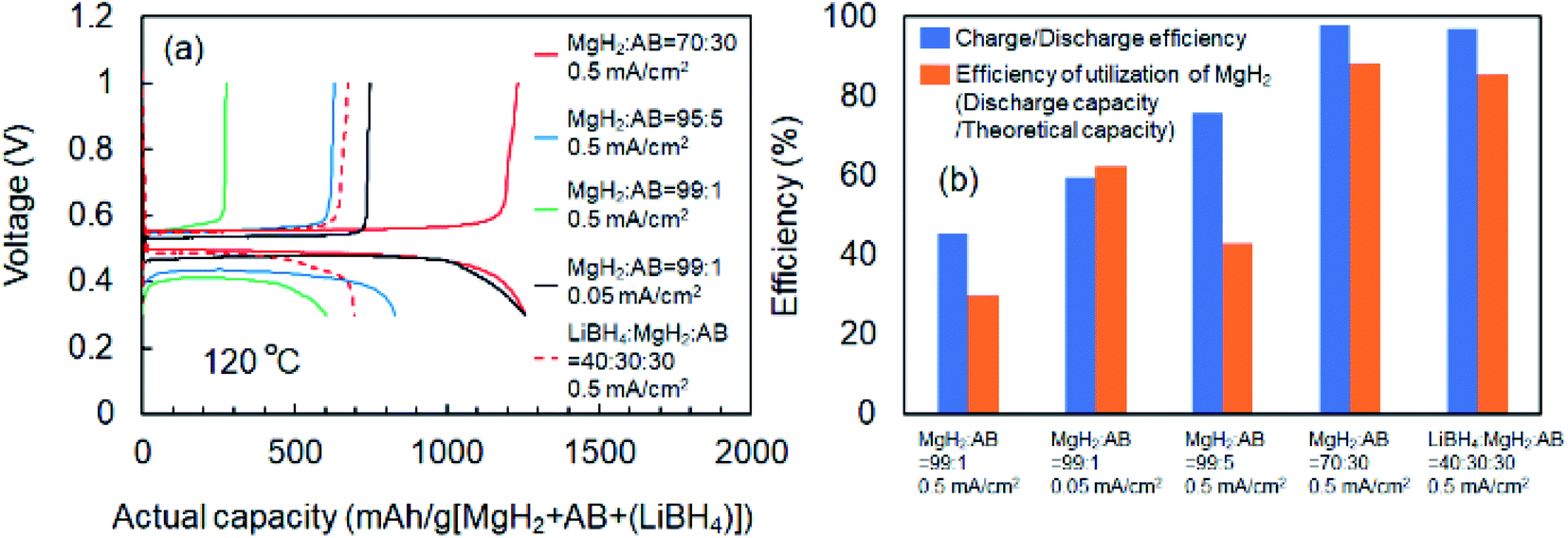
Fig. 1 shows schematics of the solid-state battery without a LiBH4 solid electrolyte in the electrode mixture. A solid electrolyte is typically required in the electrode mixture; however, the electrode reaction can be promoted by the in situ formation of a LiH solid electrolyte. In the case of the MgH2 electrode, LiH is formed during lithiation. If the conductivity of LiH is sufficiently high, the electrochemical reaction is considered to proceed over the entire electrode mixture (Fig. 1). Initially, the formation of LiH and Mg will proceed from the interface between the solid electrolyte layer and the electrode mixture and the electrode reaction will proceed toward the current collector through the LiH to form an ionic conduction pathway. In the present study, the electrochemical properties of cells with and without the LiBH4 solid electrolyte in the electrode mixture were compared. The morphology of the electrode mixture (MgH2:AB = 70:30) was compared using scanning electron microscopy (SEM) observations (ESI, Fig. S1†). Before the observations, the powder samples were pressed at 320 MPa, which is the same pressure used to construct the solid-state batteries. An SEM micrograph and energy-dispersive X-ray (EDS) mapping images (magnification: 3000×) of the electrode mixture are shown in Fig. S1(a) and S2(b),† respectively. The results confirm that AB particles with sizes of ∼0.5 μm were uniformly dispersed on the sample surfaces. Fig. S1(c)† shows an SEM image at higher magnification (10000×). Numerous 2–3 μm wide regions with a high density were observed, along with a large number of grain boundaries, indicating that the density after pelletization was not high. Fig. 2a and b show the discharge–charge curves for the Li/LiBH4/MgH2 cell with and without LiBH4 in the electrode mixture, respectively. For the cell with LiBH4 in the electrode mixture, the initial lithiation and delithiation capacities were 1738 and 1685 mA h g−1, respectively. These capacities are similar to those previously reported.12 However, for the electrode mixture without LiBH4, the initial capacity was 1818 mA h g−1 and increased to 1885 mA h g−1 in the second cycle. The difference in cell voltage between the first charge and first discharge (at 50 mA h g−1) with and without LiBH4 was 60 mV and 53 mV, respectively. Surprisingly, these results suggest that similar performance can be achieved in the cell without LiBH4 in the electrode mixture. Fig. 3 shows annular dark field (ADF) STEM images, EELS spectra, and energy-filtered maps obtained from the initial MgH2 and AB composite powders (Fig. 3a–f), the powders in the discharged state (Fig. 3g–m), and the powders in the charged state (Fig. 3n–r). For the initial state, several plasmon peaks were observed in valence EELS (VEELS) spectra collected from different areas (Fig. 3b). A plasmon peak was detected at 14.5 eV for a large fraction of the observed particles, as shown in a typical spectrum (red spectrum in Fig. 3b); an L-edge of Mg was also detected at 52.5 eV in the spectrum corresponding to the same area (Fig. 3c). The plasmon peak energy and the shape of the energy-loss near-edge structure (ELNES) at the Mg L-edge agree well with EELS and X-ray Raman scattering (XRS) spectra previously reported for MgH2.29–31 AB particles were also identified on the basis of π plasmon and π + σ plasmon peaks observed at 6.5 eV and 27 eV, respectively. These plasmon peaks are specific to graphite-like materials.32,33 For the discharged states (lithiated state), two plasmon peaks were detected at 10.6 eV and 15 eV; these peaks appear in different areas in the energy-filtered maps (Fig. 3j and k). The peak at 10.6 eV originates from metallic Mg,29,30 as also supported by the Mg map obtained by EDS (Fig. S2†). The peak at 15 eV indicates the existence of LiH, and is also supported by the ELNES results at the Li K-edge (Fig. 3i). The plasmon peak energy and the shape of the ELNES agree well with EELS or XRS spectra reported for LiH in previous studies.34,35 A Li K-edge was also detected in another area not assigned to either metallic Mg or LiH (Fig. 3i). However, the shape of the corresponding ELNES differs dramatically from those for LiH and other Li compounds;34 in addition, the plasmon peak energy and shape also differ from those for LiH (Fig. 3h). EDS analysis indicates that the surface of the particles includes a larger amount of oxygen relative to the LiH area inside the particle (Fig. S3†), suggesting that the surface was oxidized by residual O2 gas or vapor after the sample was crushed. For the charged state (delithiated state), a plasmon peak and an L-edge of Mg were again detected at 14.5 eV and 52.5 eV (Fig. 3o and p). The obtained spectra fully reproduce those of the initial state; that is, MgH2 particles were generated during the charge process. Accordingly, the STEM-EELS analysis supports the proposed concept of an electrochemical reaction that includes in situ electrolyte formation during discharge–charge cycling. We found that the MgH2 electrode does not require that a LiBH4 solid electrolyte be incorporated into the electrode mixture because LiH is formed during Li+ insertion. In industrial applications, reducing the amount of carbon is also important for increasing the actual capacity of the electrode. We therefore attempted to decrease the amount of carbon in the electrode mixture (Mg + AB). Fig. S3† shows the discharge–charge curves for cells with different amounts of AB. Surprisingly, even for the composition MgH2:AB = 99:1, the discharge–charge reaction proceeded at 0.5 mA cm−2. When the current density was lowered to 0.05 mA cm−2, a lithiation capacity of 1268 mA h g−1 was observed even for the electrode with a composition of MgH2:AB = 99:1. However, the capacity decreased with decreasing AB content. Assuming that the density of carbon is 2 g cm−3, the volume percentages of carbon at MgH2:AB = 70:30 and 99:1 are 15.8% and 1.5%, respectively. Therefore, when the low-carbon-content electrode (MgH2:AB = 99:1) was used, the electron conduction path was not well connected. Fig. 4a shows the discharge–charge curve for the “actual capacity” on the x-axis. Here, “actual capacity” refers to the capacity per weight of the whole electrode mixture, including the carbon, active material, and the solid electrolyte. The actual lithiation capacity without LiBH4 (MgH2:AB = 70:30) was more than twice as large as the capacity with LiBH4 (LiBH4:MgH2:AB = 40:30:30). However, the actual Li insertion capacity was approximately the same when the current density was 0.05 mA cm−2 at MgH2:AB = 99:1 and when the current density was 0.5 mA cm−2 at MgH2:AB = 70:30. Fig. 4b shows the discharge–charge efficiency in the initial cycle and the utilization efficiency of the MgH2 in the initial discharge capacity in the cell with a different electrode composition. The charge–discharge efficiency increased with increasing carbon content. When the carbon content was low, the reversible capacity was drastically diminished. We speculate that, when a large discharge (lithiation) capacity cannot be attained, the charge (delithiation) capacity also decreases because sufficient ionic and electronic conduction paths are not formed in the electrode mixture. When the carbon content is low, a greater charge capacity (delithiation) can be attained in the second cycle than in the first cycle because the ionic and electronic conduction paths are gradually connected in the electrode mixture by cycling. The network of the ionic and electronic paths may change depending on the composition of the electrode mixture, current density, and the temperature. Details of the morphology of the electrode mixture will be revealed in a future study.
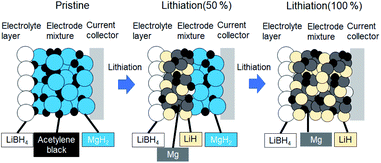 |
| | Fig. 1 Schematics of electrochemical reactions of solid-state battery with MgH2 electrode. | |
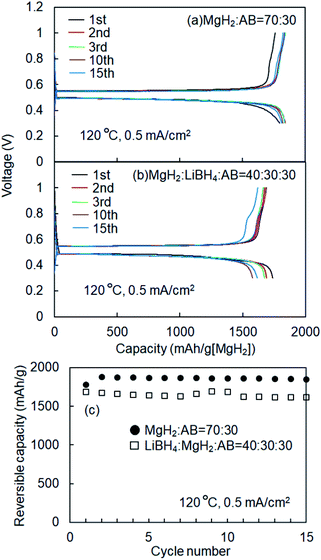 |
| | Fig. 2 (a) Discharge–charge curves for Li/LiBH4/MgH2 cell without LiBH4 in electrode mixture. (b) Charge–discharge curves for Li/LiBH4/MgH2 cell with LiBH4 in electrode mixture. (c) Cyclic properties of Li/LiBH4/MgH2 cell with and without LiBH4 in electrode mixture. | |
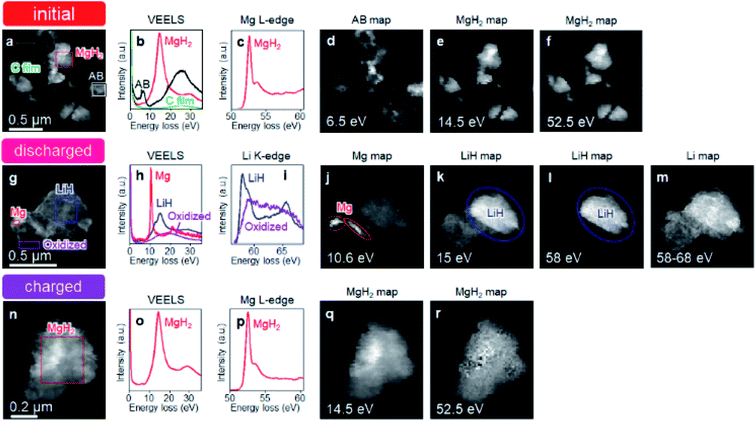 |
| | Fig. 3 STEM-EELS analysis of initial MgH2 and AB composite powders, discharged state, and charged state. (a) ADF-STEM image of initial MgH2 and AB composite powders. (b and c) Typical VEELS spectra and ELNES at Mg L-edge extracted from areas indicated in (a). The colour of the selection windows in (a) corresponds to the colour used for plotting each of the spectra in (b) and (c). (d–f) Energy-filtered maps of (a) at (d) 6.5 eV, (e) 14.5 eV, and (f) 52.5 eV. (g) ADF-STEM image of composite powders in discharged state. (h and i) Typical VEELS spectra and ELNES at Li K-edge extracted from areas indicated in (g). The colour of the selection windows in (g) corresponds to the colour used for plotting each of the spectra in (h) and (i). (j–m) Energy-filtered maps corresponding to (g) at (j) 10.6 eV, (k) 15 eV, (l) 58 eV, and (m) 58 to 68 eV. (n) ADF-STEM image of composite powders in charged state (after first discharge–charge cycle). (o and p) Typical VEELS spectrum and ELNES at Mg L-edge extracted from area indicated in (n). (q and r) Energy-filtered maps corresponding to (n) at (j) 14.5 eV and (m) 52.5 eV. The energy width for filtering in all the maps except that in (m) is 1 eV, where the aforementioned energy values for the maps are at the centers of the selected energy ranges of the 1 eV width. The plotted intensity in each of the energy-filtered maps was normalized by the intensity of the zero-loss peak so that it was proportional to the excitation probability. | |
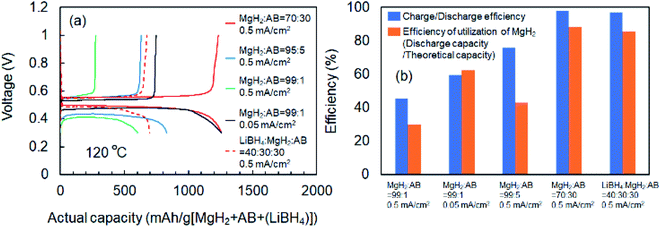 |
| | Fig. 4 (a) Discharge (lithiation)–charge (delithiation) curves for cells with different electrode compositions. (b) Discharge (lithiation)–charge (delithiation) efficiency in initial cycle and utilization efficiency of initial discharge capacity in cells with different electrode compositions. Results were obtained for different electrode mixture compositions and different current densities during discharge–charge measurements. | |
Conclusions
In summary, this work demonstrated the in situ formation of an ionic conducting path upon lithiation of MgH2. The electrochemical performance of the cell without LiBH4 in the electrode mixture matched that of the cell with LiBH4 electrolyte incorporated. LiH and Mg clearly formed upon lithiation and returned to MgH2 upon delithiation, as revealed by TEM-EELS analysis. Even when the composition was MgH2:AB = 99:1, the discharge–charge reaction proceeded. The electrochemical formation of LiH contributed to the superior electrochemical performance of the conversion reaction in the absence of an electrolyte in the electrode mixture. This concept of in situ electrolyte formation should be useful in designing novel high-capacity anodes for next-generation all-solid-state batteries.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was in part supported by JSPS KAKENHI Grant Number JP20H05297 (Grant-in-Aid for Scientific Research on Innovative Areas “Interface IONICS”) and JSPS KAKENHI Grant Number JP21K05243.
Notes and references
- O. Sheng, C. Jin, X. Ding, T. Liu, Y. Wan, Y. Liu, J. Nai, Y. Wang, C. Liu and X. Tao, Adv. Funct. Mater., 2021, 31, 2100891 CrossRef CAS.
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed.
- K. Kisu, S. Kim, H. Oguchi, N. Toyama and S. Orimo, J. Power Sources, 2019, 436, 226821 CrossRef CAS.
- S. Kim, H. Oguchi, N. Toyama, T. Sato, S. Takagi, T. Otomo, D. Arunkmar, N. Kuwata, J. Kawamura and S. Orimo, Nat. Commun., 2019, 10, 1081 CrossRef PubMed.
- T. Yamamoto, Y. Sugiura, H. Iwasaki, M. Motoyama and Y. Iriyama, Solid State Ionics, 2019, 337, 19–23 CrossRef CAS.
- F. Han, T. Gao, Y. Zhu, K. J. Gaskell and C. Wang, Adv. Mater., 2015, 27, 3473–3483 CrossRef CAS PubMed.
- T. Hakari, Y. Sato, S. Yoshimi, A. Hayashi and M. Tatsumisago, J. Electrochem. Soc., 2017, 164, A2804–A2811 CrossRef CAS.
- T. Hakari, M. Nagao, A. Hayashi and M. Tatsumisago, J. Power Sources, 2015, 293, 721–725 CrossRef CAS.
- H. Sato, R. Sakamoto, H. Minami, H. Izumi, K. Ideta, A. Inoishi and S. Okada, Chem. Commun., 2021, 57, 2605–2608 RSC.
- M. Matsuo, Y. Nakamori, S. Orimo, H. Maekawa and H. Takamura, Appl. Phys. Lett., 2007, 91, 224103–224105 CrossRef.
- H. Maekawa, M. Matsuo, H. Takamura, M. Ando, Y. Noda, T. Karahashi and S. Orimo, J. Am. Chem. Soc., 2009, 131, 894–895 CrossRef CAS PubMed.
- L. Zeng, K. Kawahito, S. Ikeda, T. Ichikawa, H. Miyaoka and Y. Kojima, Chem. Commun., 2015, 51, 9773–9776 RSC.
- S. Ikeda, T. Ichikawa, K. Kawahito, K. Hirabayashi, H. Miyaoka and Y. Kojima, Chem. Commun., 2013, 49, 7174–7176 RSC.
- F. Cano-Banda, A. Gallardo-Guierrez, L. Luviano-Ortiz, A. Hernandez-Guerrero, A. Jain and T. Ichikawa, Int. J. Hydrogen Energy, 2021, 46, 1030–1037 CrossRef CAS.
- L. Zeng, T. Ichikawa, K. Kawahito, H. Miyaoka and Y. Kojima, ACS Appl. Mater. Interfaces, 2017, 9, 2261–2266 CrossRef CAS PubMed.
- L. Huang, L. Aymard and J. Bonnet, J. Mater. Chem. A, 2015, 3, 15091–15096 RSC.
- A. E. Kharbachi, H. Uesato, H. Kawai, S. Wenner, H. Miyaoka, M. H. Sørby, H. Fjellvåg, T. Ichikawa and B. C. Hauback, RSC Adv., 2018, 8, 23468–23474 RSC.
- Y. Oumellal, A. Rougier, G. A. Nazri, J.-M. Tarascon and L. Aymard, Nat. Mater., 2008, 7, 916–921 CrossRef CAS PubMed.
- S. Brutti, G. Mulas, E. Piciollo, S. Panero and P. Reale, J. Mater. Chem., 2012, 22, 14531–14537 RSC.
- S. Ikeda, T. Ichikawa, K. Goshome, S. Yamaguchi, H. Miyaoka and Y. Kojima, J. Solid State Electrochem., 2015, 19, 3639–3644 CrossRef CAS.
- D. Meggiolaro, G. Gigli, A. Paolone, P. Reale, M. L. Doublet and S. Brutti, J. Phys. Chem. C, 2015, 119, 17044–17052 CrossRef CAS.
- Y. Oumellal, C. Zlotea, S. Bastide, C. Xachet-Vivier, E. Léonel, S. Sengmany, E. Leroy, L. Aymard, J. Bonnet and M. Latroch, Nanoscale, 2014, 6, 14459–14466 RSC.
- Y. Matsumura, K. Takagishi, H. Miyaoka and T. Ichikawa, Mater. Trans., 2019, 60, 2183–2187 CrossRef CAS.
- F. Mo, X. Chi, S. Yang, F. Wu, Y. Song, D. Sun, Y. Yao and F. Fang, Energy Storage Mater., 2019, 18, 423–428 CrossRef.
- S. Sartori, F. Cuevas and M. Latroche, Appl. Phys. A, 2016, 122, 135 CrossRef.
- V. B. Ptashnik, T. Y. Dunaeva and Y. M. Baikov, Phys. Status Solidi B, 1982, 110, K121–K124 CrossRef CAS.
- M. Ikeya, J. Phys. Soc. Jpn., 1977, 42, 168–174 CrossRef CAS.
- P. A. Varotsos and S. Mourikis, Phys. Rev. B: Solid State, 1974, 10, 5220–5224 CrossRef CAS.
- B. Paik, I. P. Jones, A. Walton, V. Mann, D. Book and I. R. Harris, Philos. Mag. Lett., 2010, 90, 1–7 CrossRef CAS.
- M. Hamm, M. D. Bongers, V. Roddatis, S. Dietrich, K. H. Lang and A. Pundt, Int. J. Hydrogen Energy, 2019, 44, 32112–32123 CrossRef CAS.
- C. J. Sahle, S. Kujawski, A. Remhof, Y. Yan, N. P. Stadie, A. Al-Zein, M. Tolan, S. Huotari, M. Krisch and C. Sternemann, Phys. Chem. Chem. Phys., 2016, 18, 5397–5403 RSC.
- L. Calliari, S. Fanchenko and M. Filippi, Carbon, 2007, 45, 1410–1418 CrossRef CAS.
- S. Urbonaite, S. Wachtmeister, C. Mirguet, E. Coronel, W. Y. Zou, S. Csillag and G. Svensson, Carbon, 2007, 45, 2047–2053 CrossRef CAS.
- P. S. Miedema, P. Ngene, A. M. J. van der Eerden, D. Sokaras, T. C. Weng, D. Nordlund, Y. S. Au and F. M. F. de Groot, Phys. Chem. Chem. Phys., 2014, 16, 22651–22658 RSC.
- M. J. Zachman, Z. Tu, S. Choudhury, L. A. Archer and L. F. Kourkoutis, Nature, 2018, 560, 345–349 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01199a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hiroki Satob,
Yixin Chenb,
Hikaru Saitoa,
Ryo Sakamoto
*a,
Hiroki Satob,
Yixin Chenb,
Hikaru Saitoa,
Ryo Sakamoto