
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iodine-mediated oxythiolation of o-vinylanilides with disulfides for the synthesis of benzoxazines†
Yu Yang‡
ab,
Liyan Liu‡b,
Kuiliang Lib,
Zhenggen Zha b,
Qi Sun*b and
Zhiyong Wang*b
b,
Qi Sun*b and
Zhiyong Wang*b
aSchool of Chemistry and Chemical Engineering, Hefei Normal University, Hefei, 230601, China
bHefei National Laboratory for Physical Sciences at Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 4th March 2022
Abstract
An efficient iodine-mediated oxythiolation of o-vinylanilides with disulfides was developed. By virtue of this method, a series of thio-tethered benzoxazine derivatives were synthesized in good to excellent yields. The reaction features high yields, is metal-free, and has a wide substrate scope.
As one of the most basic organic skeletons, alkenes are important starting materials in organic synthesis. It is convenient to introduce functional groups into the carbon chain by addition reaction.1,2 Furthermore, heterocyclic compounds also could be built efficiently from alkenes by addition–cyclization with reasonable design of starting materials.3–5 For example, benzo[d][1,3]oxazine, an essential heterocyclic skeleton which is widely found in natural products and biological active molecule,6–10 could be synthesized by the intramolecular cyclization of N-(2-(1-phenylvinyl)phenyl)-benzamides (Scheme 1).11–23 In the 1980s, Capozzi and co-workers synthesized methylthio-benzo[d][1,3]oxazine with this strategy for the first time, using Me3S3SbCl6 as the methylthiolating agent (Scheme 1a).19 Besides, this was the only way to generate this kind of compound for the next three decades. In 2018, Anbarasan and co-workers found that N-(arylthio)-succinimides could also be used as an electrophilic reagent to react with N-(2-vinylphenyl)amides, generating a series of arylthio benzo[d][1,3]-oxazine derivatives (Scheme 1b).20 Recently, different sulfonyl sources, such as sulfur dioxides,21 sodium sulfonates22 and sulfonylhydrazines,23 were developed in this methodology to afford sulfonated benzo-[d][1,3]oxazine. The studies mentioned above provided a series of tools to build sulfur-containing benzo[d][1,3]oxazines. However, there are still some limitations in these procedures: (1) the involvement of expensive metals or a large amount of acid; (2) limited substrate applicability. In view of the importance of sulfur-containing compounds,24–31 it is still highly desirable to develop a simple and efficient method to build thio-tethered compounds with good reaction compatibility under mild conditions.
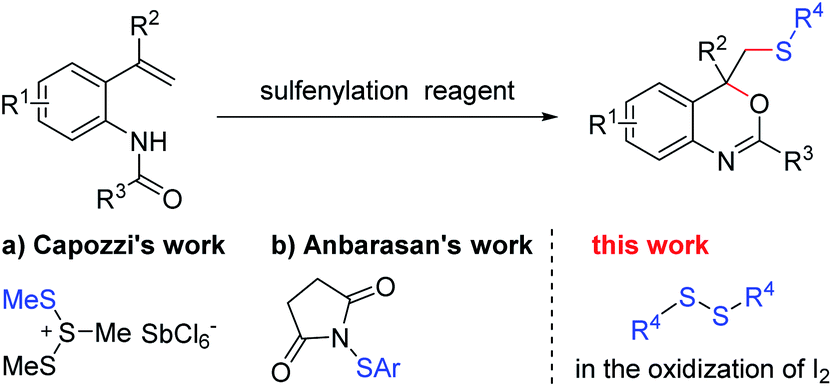 | ||
| Scheme 1 Building sulfur-containing benzo[d][1,3]oxazines from o-vinylanilides. | ||
Our group have been focusing on green synthetic methods for a long time.3,4 It was reported that disulfide, a sulfenylation reagent, could generate two sulfur synthons by the cleavage of S–S bond without any by-product.32–39 Based on these previous studies, we are going to develop a more efficient oxythiolation reaction to build sulfur-containing heterocyclic compounds. Herein we disclose an iodine mediated cyclization of o-vinylanilides with disulfides to form thio-benzo[d][1,3]oxazines, in which both aryl and alkyl disulfides could performed well to afford corresponding products with good yield under mild condition.
At the outset of this study, N-(2-(1-phenylvinyl)phenyl)-benzamide (1a) and 1,2-diphenyldisulfide (2a) were employed as the substrates to optimize the reaction conditions (Table 1). Initially, 1a (1 equiv.) and 2a (0.5 equiv.) were treated with I2 (1.0 equiv.) in DCE at 50 °C for 12 hours. To our delight, the cyclization product 3aa was obtained in 57% yield under the promotion of I2 (entry 1). When other oxidants, such as [bis(trifluoroacetoxy)iodo]benzene (PIFA) or (diacetoxyiodo)benzene (PIDA), were used instead of I2, no corresponding product was observed (entries 2 and 3). Next, the efficiency of different solvents was tested. Screening of solvent showed CH3CN to be the optimal solvent, affording 3aa in 63% yield (entries 4–9). It was found that the yield of product 3aa could be increased to 81% by increasing the dosage of 2a and I2 from 0.5 equiv. to 0.75 equiv. and 1.0 equiv. to 1.5 equiv., respectively (entry 10). However, continuously increasing the amount of 2a and I2 had no effect on the yield (entry 11). On the other hand, the reaction temperature had a great influence on the reaction. For instance, the yield of 3aa was enhanced to 87% by elevating the reaction temperature to 60 °C. Further increase in temperature led to a decrease in yield, perhaps due to the oxidation of 2a and the volatilization of molecular iodine at 70 or 80 °C (entries 13 and 14).
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Solvent | Temp. (°C) | Yieldb (%) |
| a Reaction condition: 1a (0.2 mmol, 1 equiv.), 2a (0.1 mmol, 0.5 equiv.), oxidant (0.2 mmol, 1 equiv.), 12 h.b Isolated yield.c n.d. = not detected.d 2a (0.75 equiv.) and I2 (1.5 equiv.) were used.e 2a (1.0 equiv.) and I2 (2.0 equiv.) were used. | ||||
| 1 | I2 | DCE | 50 | 57 |
| 2 | PIDA | DCE | 50 | n.d.c |
| 3 | PIFA | DCE | 50 | n.d. |
| 4 | I2 | DMSO | 50 | n.d. |
| 5 | I2 | DMF | 50 | n.d. |
| 6 | I2 | CH3CN | 50 | 63 |
| 7 | I2 | 1,4-Dioxane | 50 | 35 |
| 8 | I2 | EtOH | 50 | <5 |
| 9 | I2 | Toluene | 50 | <5 |
| 10d | I2 | CH3CN | 50 | 81 |
| 11e | I2 | CH3CN | 50 | 81 |
| 12d | I2 | CH3CN | 60 | 87 |
| 13d | I2 | CH3CN | 70 | 72 |
| 14d | I2 | CH3CN | 80 | 60 |

With a set of optimized conditions in hand, we studied the scope of this reaction. A series of N-(2-vinylphenyl) amides 1a–1q were subjected to the reaction with disulfide 2a (Scheme 2). Most of the N-(2-vinylphenyl) amides were found to be applicable, affording the corresponding products in good to excellent yields. The effect of the substituent R1 in the aryl moiety of substrates 1 was studied. When the R1 was halide, the reaction could be carried out smoothly (3ba–3da). When R1 was NO2, the corresponding product 3ea could also be isolated in 70% yield. Substrates 1 bearing electro-donating groups, such as OMe, Me and n-Bu, were suitable for this transformation, affording the cyclization product 3fa–3ha with excellent yields (90–95%). N-(4-(tert-Butyl)-2-(1-phenylvinyl)phenyl)-benzamide (1i) bearing a bulky tert-butyl group, moreover, also worked well in this reaction in spite of a slightly lower yield (3ia). In general, the substrates with electron-donating groups (R = Me, OMe, n-Bu, t-Bu) on the phenyl ring gave slightly higher yields than that bearing electron-withdrawing groups (R = F, Cl, Br, NO2). For different aromatic substituents R2 on the vinylic double bond, both halide and methyl were tolerated under the reaction conditions, affording the corresponding product 3ka–3na in good yields (68–83%). Substrate 1 with different protecting groups were also proceeded smoothly in this reaction to give the corresponding product 3oa and 3pa with the yields of 95% and 67%, respectively. It was noted that when R3 was methyl instead of aryl, the cyclization product 3pa was also obtained in 67% yield.
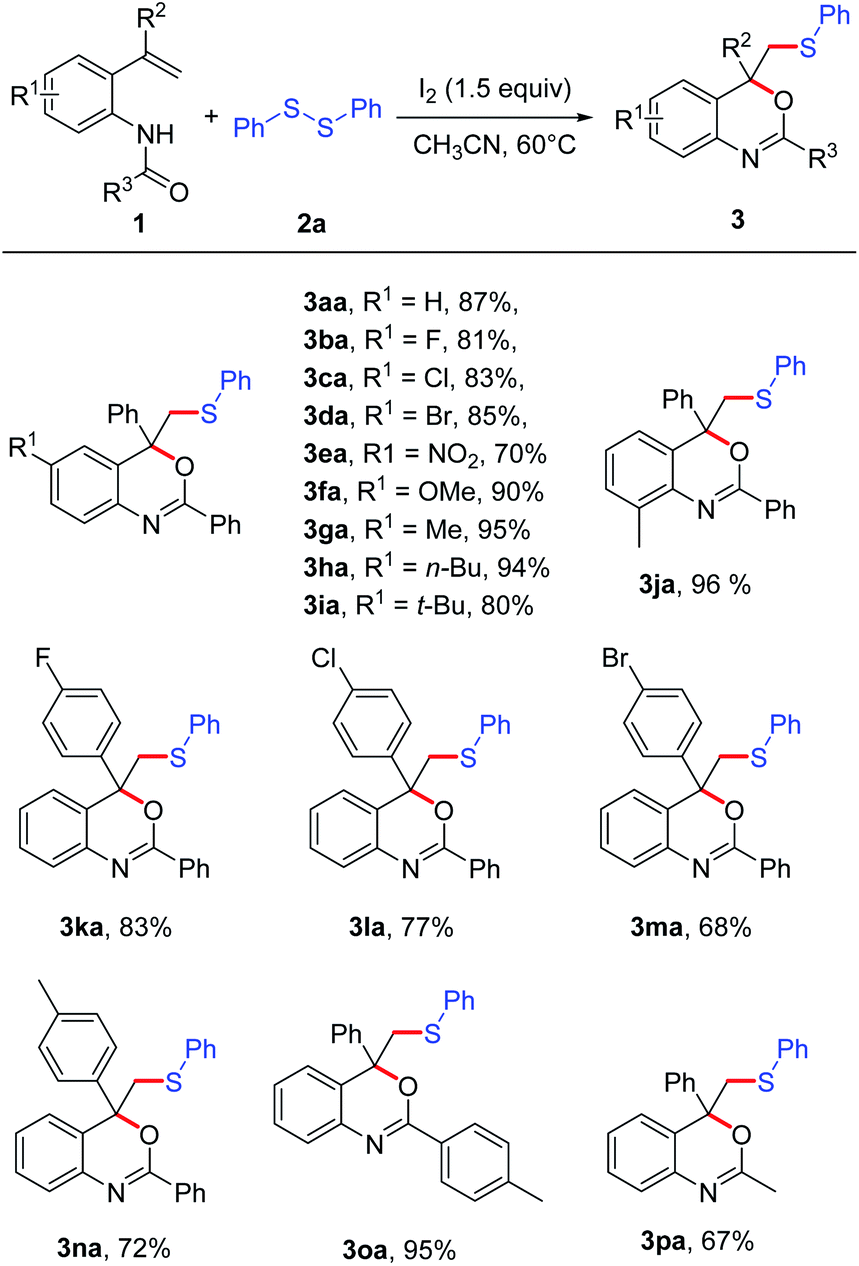 | ||
| Scheme 2 Substrate scope of amide derivatives. The reaction was carried out with 1 (0.2 mmol), 2a (0.75 equiv.), I2 (1.5 equiv.), in CH3CN (2 mL) at 60 °C for 12 h; isolated yields of all products. | ||
To further study the scope of the reaction, different disulfides were subsequently examined (Scheme 3). Generally, the diaryl disulfides bearing either electro-withdrawing groups or electro-donating groups were all suitable substrates in this reaction, affording the benzoxazine derivatives (3ab–3ai) in good yields to excellent yields (80–96%). Especially, the strong electro-deficient substrate, 1,2-bis(4-nitrophenyl)disulfide, could perform well in this transformation to afford 3ae in 80% yield. It was found that the position of methyl on the aromatic ring affected the yield slightly, which might be attributed to the steric hindrance (3ag–3ai). Furthermore, when dialkyl disulfides were used as the substrates, the corresponding products 3aj and 3ak could be isolated in good yields.
 | ||
| Scheme 3 Substrate scope of disulfides. The reaction was carried out with 1a (0.2 mmol), 2 (0.75 equiv.), I2 (1.5 equiv.), in CH3CN (2 mL) at 60 °C for 12 h; isolated yields of all products. | ||
In order to gain insight into the mechanism of the reaction, several control experiments were conducted (Scheme 4). First, radical trapping experiment was conducted to elucidate whether the reaction involved radical species. When the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO; 2 equiv.) was employed under the standard conditions (Scheme 4a), the yield of 3aa decreased to 45%, implying that part of product were formed involving a free radical process. Iodine was essential in this transformation since 3aa could not be obtained in the absence of I2 (Scheme 4b). To figure out the way iodine promoted this reaction, 1a was treated with molecular iodine under standard reaction condition, but no addition product was observed (Scheme 4c). Thus, it is reasonable to hypothesize that the reaction should not involve an iodonium ion intermediate. Further investigation showed that I2 could promoted the cleavage and formation of S–S bond. As show in Scheme 4d, mixing 2a and 2g could afford disulfide 5 slowly, with only 5% of product 5 obtained after 1 h. But this process could be accelerated significantly in the present of I2, affording 5 with the yields of 50% after 5 minutes.
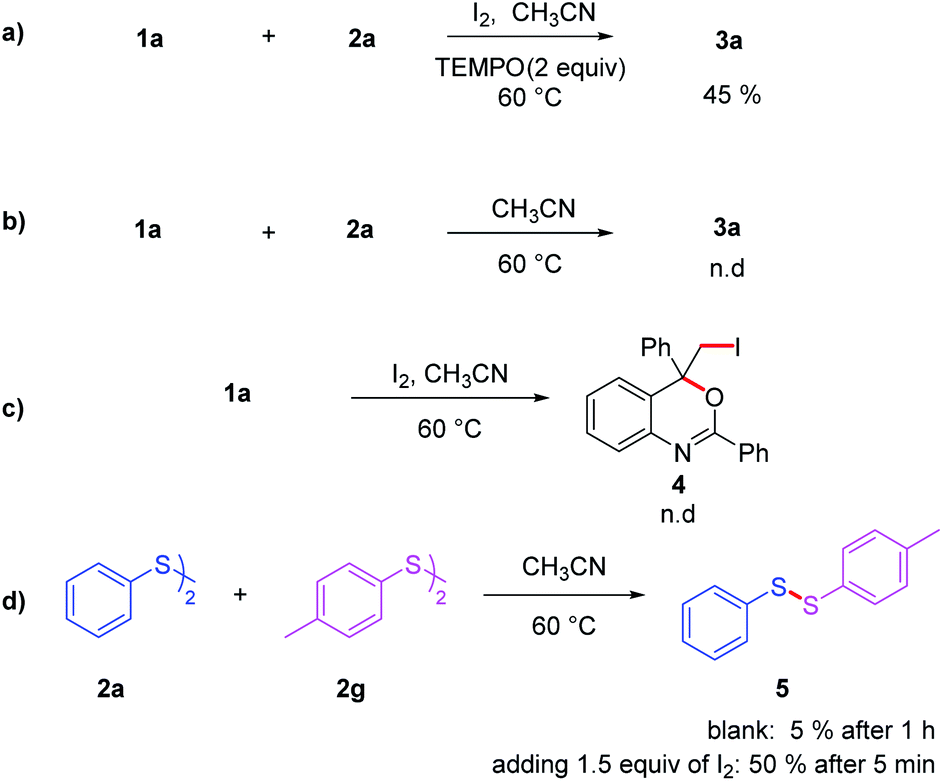 | ||
| Scheme 4 The reaction was carried out in CH3CN (2 mL) at 60 °C for 12 h. (a) 1a (0.25 mmol), 2a (2 eqiuv.), I2 (2 eqiuv.), TEMPO (2 equiv.); (b) 1a (0.25 mmol), 2a (2 eqiuv.); (c) 1a (0.25 mmol), 2a (2 eqiuv.), I2 (2 eqiuv.); (d) 2a (0.25 mmol), 2g (0.25 mmol). | ||
Based on the control experiments and previous studies,40–52 two plausible reaction paths were outlined in Scheme 5. In the promotion of I2, PhS· and PhSI could be generated in situ from 2a. Subsequently, PhS· performed with 1a via a radical addition to generate carbon radical intermediate A, which is oxidized into carbon cation intermediate B in path a. Meanwhile, intermediate B also could be generated from the electrophilic addition of 1a and PhSI directly in path b. Finally, an intramolecular nucleophilic attack of B occurs to form intermediate C, followed by loss of proton to afford the thio-tethered benzoxaine 3aa. Additionally, to investigate possible process of path b further, DFT calculation were carried out (Scheme 6). Calculation results show that the BED of S–I bond in PhSI is 31.1 kcal mol−1 while the energy barrier (ΔG) of the addition between 1a and PhSI is 15.2 kcal mol−1. It suggests that PhSI reacting with alkene via an electrophilic addition directly is more likely to happen than that of generating PhS· via the cleavage of S–I bond in path b. Two paths proposed and computational results agree with the control experimental data that the yield of 3aa decreases by nearly half when the radical scavenger is employed.
 | ||
| Scheme 5 Proposed mechanism. | ||
 | ||
| Scheme 6 Gibbs energy profiles for proposed mechanism. | ||
Conclusions
In summary, we developed an efficient synthesis of thio-benzo[d][1,3]oxazine derivatives from o-vinylanilides and disulfides mediated by iodine. A wide range of substrates were tolerated in this transformation. This reaction can be carried out smoothly under metal-free and mild conditions with a variety of thio-benzo[d][1,3]oxazine derivatives obtained in good to excellent yields by virtue of this developed conditions. Research to develop more efficient and green methods along this line is ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Natural Science Foundation of China (21772185, 21801233, 22001241), the assistance of the product characterization from the Chemistry Experiment Teaching Center of University of Science and Technology of China and Anhui Engineering Laboratory for Medicinal and Food Homologous Natural Resources Exploration. This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences, grant XDB20000000. This work was supported by the China Postdoctoral Science Foundation (2020TQ0303, 2018M632532), Department of Science and Technology of Anhui Province (S201901e11010120, 201903a07020003) and the high-level talent project of Hefei Normal University (2020rcjj32).Notes and references
- J. H. Lee, S. Choi and K. B. Hong, Molecules, 2019, 24, 2634 CrossRef PubMed.
- Y. Ping and W. Kong, Synthesis, 2020, 52, 979 CrossRef.
- L. Y. Liu, Q. Sun, Z. Yan, X. Liang, Z. Zha, Y. Yang and Z. Wang, Green Chem., 2018, 20, 3927 RSC.
- L. Y. Liu and Z. Wang, Green Chem., 2017, 19, 2076 RSC.
- J. Wolfe and D. Schultz, Synthesis, 2012, 44, 351 CrossRef PubMed.
- G. Fenton, C. G. Newton, B. M. Wyman, P. Bagge, D. I. Dron, D. Riddell and G. D. Jones, J. Med. Chem., 1989, 32, 265 CrossRef CAS PubMed.
- S. J. Hays, B. W. Caprathe, J. L. Gilmore, N. Amin, M. R. Emmerling, W. Michael, R. Nadimpalli, R. Nath, K. J. Raser and D. Stafford, J. Med. Chem., 1998, 41, 1060 CrossRef CAS PubMed.
- J. S. Plummer, C. Cai, S. J. Hays, J. L. Gilmore, M. R. Emmerling, W. Michael, L. S. Narasimhan, M. D. Watson, K. Wang and R. Nath, Bioorg. Med. Chem. Lett., 1999, 9, 815 CrossRef CAS PubMed.
- P. Zhang, E. A. Terefenko, A. Fensome, Z. Zhang, Y. Zhu, J. Cohen, R. Winneker, J. Wrobel and J. Yardley, Bioorg. Med. Chem. Lett., 2002, 12, 787 CrossRef CAS PubMed.
- A. Krantz, R. W. Spencer, T. F. Tam, T. J. Liak, L. J. Copp, E. M. Thomas and S. P. Rafferty, J. Med. Chem., 1990, 33, 464 CrossRef CAS PubMed.
- X.-Q. Chu, X.-P. Xu, H. Meng and S.-J. Ji, RSC Adv., 2015, 5, 67829 RSC.
- H. Yang, X. H. Duan, J. F. Zhao and L. N. Guo, Org. Lett., 2015, 17, 1998 CrossRef CAS PubMed.
- S. Jana, A. Ashokan, S. Kumar, A. Verma and S. Kumar, Org. Biomol. Chem., 2015, 13, 8411 RSC.
- J. F. Zhao, X. H. Duan, H. Yang and L. N. Guo, J. Org. Chem., 2015, 80, 11149 CrossRef CAS PubMed.
- Y. M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928 CrossRef CAS PubMed.
- J. Wang, R. Sang, X. Chong, Y. Zhao, W. Fan, Z. Li and J. Zhao, Chem. Commun., 2017, 53, 7961 RSC.
- W. Fu, X. Han, M. Zhu, C. Xu, Z. Wang, B. Ji, X. Q. Hao and M. P. Song, Chem. Commun., 2016, 52, 13413 RSC.
- Q. H. Deng, J. R. Chen, Q. Wei, Q. Q. Zhao, L. Q. Lu and W. J. Xiao, Chem. Commun., 2015, 51, 3537 RSC.
- G. Capozzi, R. Ottana’, G. Romeo and G. Valle, J. Chem. Res., 1986, 6, 200 Search PubMed.
- M. Chaitanya and P. Anbarasan, Org. Lett., 2018, 20, 1183 CrossRef CAS PubMed.
- T. Liu, D. Q. Zheng, Z. H. Li and J. Wu, Adv. Synth. Catal., 2018, 360, 865 CrossRef CAS.
- J. Wu, Y. Zong, C. Zhao, Q. Yan, L. Sun, Y. Li, J. Zhao, Y. Ge and Z. Li, Org. Biomol. Chem., 2019, 17, 794 RSC.
- T. J. He, W. Q. Zhong and J. M. Huang, Chem. Commun., 2020, 56, 2735 RSC.
- M. L. Alcaraz, S. Atkinson, P. Cornwall, A. C. Foster, D. M. Gill, L. A. Humphries, P. S. Keegan, R. Kemp, E. Merifield, R. A. Nixon, A. J. Noble, D. O'Beirne, Z. M. Patel, J. Perkins, P. Rowan, P. Sadler, J. T. Singleton, J. Tornos, A. J. Watts and I. A. Woodland, Org. Process Res. Dev., 2005, 9, 555 CrossRef CAS.
- Y. Harrak, G. Casula, J. Basset, G. Rosell, S. Plescia, D. Raffa, M. G. Cusimano, R. Pouplana and M. D. Pujol, J. Med. Chem., 2010, 53, 6560 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- S. Raghavan, V. Krishnaiah and B. Sridhar, J. Org. Chem., 2010, 75, 498–501 CrossRef CAS PubMed.
- M. Hamada, M. Kiuchi and K. Adachi, Synthesis, 2007, 1927 CrossRef CAS.
- J. C. Sheehan and K. R. Henerylogan, J. Am. Chem. Soc., 1959, 81, 3089 CrossRef CAS.
- S. Pasquini, C. Mugnaini, C. Tintori, M. Botta, A. Trejos, R. K. Arvela, M. Larhed, M. Witvrouw, M. Michiels, F. Christ, Z. Debyser and F. Corelli, J. Med. Chem., 2008, 51, 5125 CrossRef CAS PubMed.
- R. A. Bragg, S. Brocklehurst, F. Gustafsson, J. Goodman, K. Hickling, P. A. MacFaul, S. Swallow and J. Tugwood, Chem. Res. Toxicol., 2015, 28, 1991 Search PubMed.
- (a) P. D. Morse and D. A. Nicewicz, Chem. Sci., 2015, 6, 270 RSC; (b) X. Xu, J. Li and Z. Wang, Chin. J. Org. Chem., 2020, 40, 886 CrossRef CAS; (c) N. Mukherjee and T. Chatterjee, Green Chem., 2021, 23, 10006 RSC; (d) J. Qin, H. Zuo, Y. Ni, Q. Yu and F. Zhong, ACS Sustainable Chem. Eng., 2020, 8, 12342 CrossRef CAS.
- T. B. Grimaldi, G. Lutz, D. F. Back and G. Zeni, Org. Biomol. Chem., 2016, 14, 10415 RSC.
- A. M. Recchi, D. F. Back and G. Zeni, J. Org. Chem., 2017, 82, 2713 CrossRef CAS PubMed.
- K. Sun, Y. Lv, Z. Shi, S. Mu, C. Li and X. Wang, Org. Biomol. Chem., 2017, 15, 5258 RSC.
- R. An, L. Liao, X. Liu, S. Song and X. Zhao, Org. Chem. Front., 2018, 5, 3557 RSC.
- B. Q. Ni, Y. P. He, X. J. Rong and T. F. Niu, Synlett, 2019, 30, 1830 CrossRef CAS.
- C. R. Reddy, R. Ranjan and S. K. Prajapti, Org. Lett., 2019, 21, 623 CrossRef CAS PubMed.
- C. Wei, Y. He, J. Wang, X. Ye, L. Wojtas and X. Shi, Org. Lett., 2020, 22, 5462 CrossRef CAS PubMed.
- K. R. Sharma and R. M. Noyes, J. Am. Chem. Soc., 2002, 98, 4345 CrossRef.
- S. Serna, I. Tellitu, E. Dominguez, I. Moreno and R. Sanmartin, Org. Lett., 2005, 7, 3073 CrossRef CAS PubMed.
- A. Correa, I. Tellitu, E. Dominguez and R. SanMartin, J. Org. Chem., 2006, 71, 8316 CrossRef CAS PubMed.
- R. Fan, D. Pu, F. Wen and J. Wu, J. Org. Chem., 2007, 72, 8994 CrossRef CAS PubMed.
- M. Kirihara, Y. Asai, S. Ogawa, T. Noguchi, A. Hatano and Y. Hirai, Synthesis, 2007, 3286 CrossRef CAS.
- M. Uyanik, H. Okamoto, T. Yasui and K. Ishihara, Science, 2010, 328, 1376 CrossRef CAS PubMed.
- S. K. Alla, R. K. Kumar, P. Sadhu and T. Punniyamurthy, Org. Lett., 2013, 15, 1334 CrossRef CAS PubMed.
- D. Liang, Y. He, L. Liu and Q. Zhu, Org. Lett., 2013, 15, 3476 CrossRef CAS PubMed.
- Q. C. Xue, J. Xie, P. Xu, K. D. Hu, Y. X. Cheng and C. J. Zhu, ACS Catal., 2013, 3, 1365 CrossRef CAS.
- J. H. Chu, W. T. Hsu, Y. H. Wu, M. F. Chiang, N. H. Hsu, H. P. Huang and M. J. Wu, J. Org. Chem., 2014, 79, 11395 CrossRef CAS PubMed.
- S. Manna, K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2014, 53, 8163 CrossRef CAS PubMed.
- G. Y. Qian, B. X. Liu, Q. T. Tan, S. W. Zhang and B. Xu, Eur. J. Org. Chem., 2014, 2014, 4837 CrossRef CAS.
- M. Uyanik, H. Hayashi and K. Ishihara, Science, 2014, 345, 291 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01078j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |