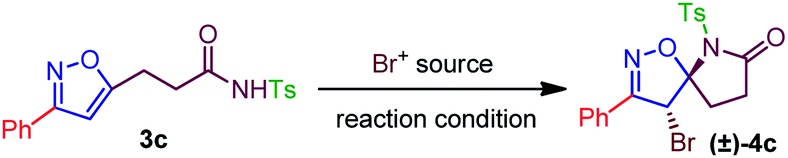DOI:
10.1039/D2RA01070D
(Paper)
RSC Adv., 2022,
12, 9628-9636
Bromo-lactamization of isoxazole via neighboring group participation: toward spiro-isoxazoline γ- and δ-lactams†
Received
17th February 2022
, Accepted 21st March 2022
First published on 28th March 2022
Abstract
Spiro-heterocycles containing natural products and synthetic analogues have a broader biomedicinal application due to their rigid 3D conformation and structural implications. In this context, constructing spiro-isoxazoline systems have continued our interest in natural products and synthetic units to investigate their novel biological activities. Herein, a bromo-lactamization mediated neighboring group participation approach has been utilized on various isoxazole-amides to construct an array of spiro-isoxazoline-lactams. The easy synthesis with diverse functionalization in the periphery of a novel 3D framework could be interesting for biomedical investigation.
Introduction
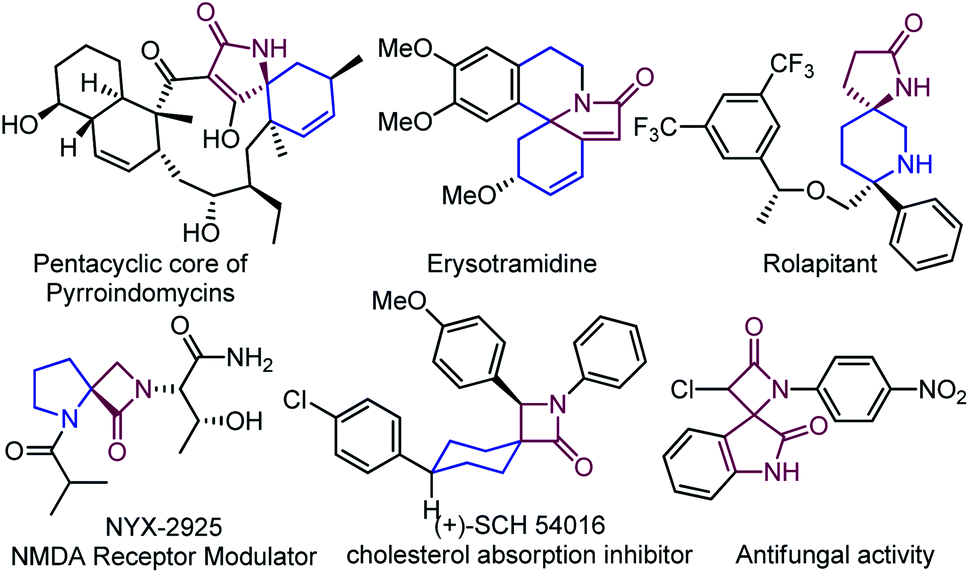
The spirocyclic compounds are the essential building blocks in natural products and synthetic compounds, delivering a wide range of biological activities and diverse applications in organic synthesis.1 The structural rigidity and three-dimensional feature of spiro-frameworks significantly impact ligand binding entropy, facilitating the spiro-unit to be an effective pharmacophore in drug discovery compared to two-dimensional aromatic compounds.1a,2 Among a wide range of spirocyclic compounds, spiro-lactams have received much interest due to their structural novelty and diversity in drug discovery, emerging as a prominent class.3 Herein, a few representative examples3a,b,4 of heterocyclic, carbocyclic, and dimeric spiro-lactams have been presented that show significant biological properties (Fig. 1).
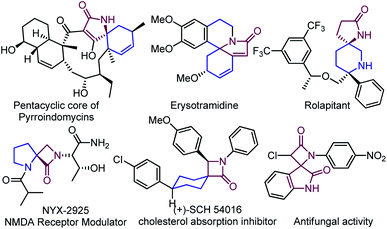 |
| | Fig. 1 Spiro-lactam containing natural products and synthetic compounds. | |
While the presence of β-, γ-, and δ-lactams are widely found with various spiro-carbocycles and -heterocycles,3 the spiro-isoxazoline-lactams are very uncommon in natural or synthetic sources; consequently, its synthesis is highly desirable for biological intervention. In the past, several impressive protocols were explored using bromonium-ion-mediated cyclization to synthesize related spiro units. For example, d’Alcontres et al. reported5 the synthesis of the spiro-5-isoxazoline system from an internal amide functionality where O-cyclization via a possible five-member bromo cyclization provided the desired compound; however, no trace of N-cyclization was reported (Scheme 1).
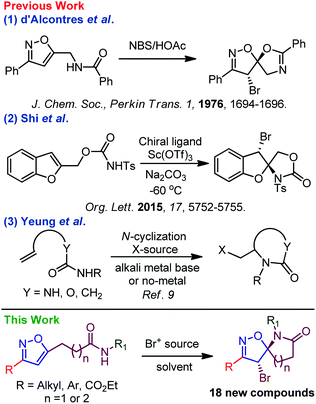 |
| | Scheme 1 Previous and current approach towards spiro-isoxazoline-lactams. | |
In 2015, Shi et al. reported an enantioselective bromo amino cyclization strategy for the synthesis of spiro system but oxazolidinones (Scheme 1).6 For N-heterocyclic compounds, such as pyrrolidines and piperidines, haloaminocyclization of olefinic amines has been the topic of significant research.7 However, halo-lactamization of an unactivated olefinic amide remained a formidable challenge because hard-O and soft-N nucleophiles compete with each other.8 Despite continued success with halo-N-cyclization of olefinic urea, olefinic carbamate, and olefinic amide,9 halo-N-lactamization of spiocyclic compounds is limited;10 to our knowledge, no report on spiro-isoxazoline-lactams exists. As a result, the desire to understand and address the issues associated with spiro-lactamization of isoxazole drove these findings in reaction discovery.
Based on our previous outcomes on spiroisoxaolines10 and proceeding interest in developing novel pharmacophore, spiro-isoxazoline-lactam was our immediate target. We detailed an effective bromo-lactamization on an isoxazoles framework to get diverse spiro-isoxazoline-lactams in this context. It is essential to highlight that sulfonamide is a crucial functional group in many pharmaceutical drugs, widely known as “sulfa drugs”, to exhibit antimicrobial activity.11 Therefore, we postulated that a sulfonamide, O- and N-heteroatoms, and substituents in the periphery of the 3D scaffold could potentially induce the drug nature of the molecule. Moreover, the selectivity of bromonium-ion formation and subsequent nucleophilic attack via neighboring group participation will generate two adjacent stereocenters; however, a non-stereoselective mode of reactivity has been demonstrated to primarily obtain the desire spiro-lactams.
Results and discussion
The precursor isoxazole amide was synthesized following two methods. Our first effort was to transform a previously synthesized isoxazole acid 2 (ref. 10a,c) to its corresponding amid 3, using a conventional method.12 Conversely, an alkynoic acid 1 was first converted to its corresponding amide 2′, which was later used as a precursor for 1,3-dipolar cycloaddition to obtain 3. Subsequently, a suitable bromo-lactamization of isoxazole amide 3 provided spiro-isoxazoline-amide 4 as a single diastereomer. The isoxazole ring was constructed using in situ nitrile oxide-mediated 1,3-dipolar cycloaddition.10a,c As depicted in the scheme, an array of substituted isoxazoles was obtained using a range of nitrile oxide precursors.10a,c Moreover, we have chosen the first method as most of the isoxazole acids10a,c were available to us (Scheme 2).
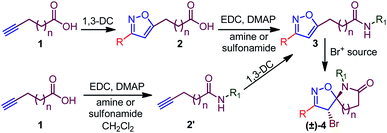 |
| | Scheme 2 General scheme for the synthesis of spiro-isoxazoline-lactam. | |
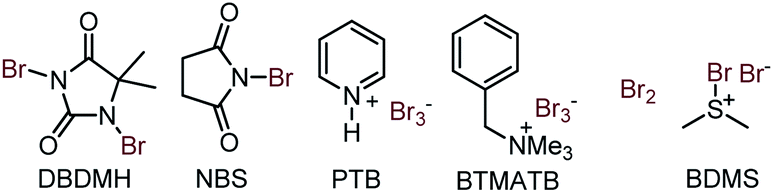
To validate the synthetic feasibility of isoxazoline-lactam (4c), our initial optimization for the bromo-lactamization was carried out with N-tosylamide (3c) as the test substrate and 1,3-Dibromo-5,5-Dimethylhydantoin (DBDMH) as the bromine source (Table 1). Other electrophilic brominating agents like N-bromosuccinimide (NBS), pyridinium perbromide (PTB), benzyltrimethylammonium bromide (BTMATB), and molecular bromine (Br2) were subsequently screened during optimization (Table 1). To our delight, spiro-isoxazoline-lactam 4c was isolated as a racemic mixture, albeit in 30% yield, when 3c was treated with DBDMH in CH2Cl2 at 0 °C (entry 1, Table 1). The reaction rate significantly improved at room and reflux temperatures compared to entry 1, producing 4c 85% and 80% yield. We speculate that a destabilization of bromonium ion formation at reflux temperature could possibly retard the reaction rate. The presence of K2CO3 as an additive was not surprising as compound 4c was isolated in 75% yield in entry 4, Table 1. Other chlorinated solvents like CHCl3 and ClCH2CH2Cl were suitable for bromo-lactamization to generate 4c in 84% and 82% yield. So far, we preferred to use the CH2Cl2 under the reaction condition.
Table 1 Optimization of bromo-lactamizationa
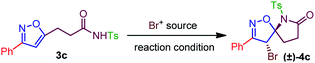
|
| Entryy |
Br+ Source |
Additive |
Solvent |
Temp (°C) |
Time (h) |
Yield (%)b |
| The reactions were carried out with substrate 3c (0.25 mmol) and Br+-source (0.3 mmol) in solvent (2.0 mL) at rt for 24h. Isolated yield based on 3c. K2CO3 (0.24 mmol) for entries 4, 9, and 10. Reaction carried out at reflux temperature for entries 3 and 7. |
| 1 |
DBDMH |
— |
CH2Cl2 |
0 |
24 |
30 |
| 2 |
DBDMH |
— |
CH2Cl2 |
rt |
24 |
85 |
| 3d |
DBDMH |
— |
CH2Cl2 |
50 |
6 |
80 |
| 4c |
DBDMH |
K2CO3 |
CH2Cl2 |
rt |
24 |
75 |
| 5 |
DBDMH |
— |
CHCl3 |
rt |
24 |
84 |
| 6 |
DBDMH |
— |
Cl(CH2)2Cl |
rt |
24 |
82 |
| 7 |
DBDMH |
— |
Toluene |
120 |
6 |
75 |
| 8 |
DBDMH |
— |
Ether |
rt |
24 |
50 |
| 9c |
DBDMH |
K2CO3 |
DME |
rt |
24 |
40 |
| 10c |
DBDMH |
K2CO3 |
MeOH |
rt |
24 |
45 |
| 11 |
NBS |
— |
CH2Cl2 |
rt |
24 |
80 |
| 12 |
PTB |
— |
CH2Cl2 |
rt |
24 |
30 |
| 13 |
BTMATB |
— |
CH2Cl2 |
rt |
24 |
10 |
| 14 |
Br2 |
— |
CH2Cl2 |
rt |
24 |
50 |
| 15 |
BDMS |
— |
CH2Cl2 |
rt |
24 |
65 |
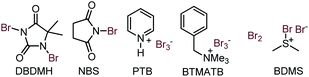 |
To scan with other solvents, using toluene under reflux condition, provided 75% of lactam 4c. While using ether as a solvent, 50% of conversion was realized. The presence of polar solvents like MeOH and DMF with K2CO3 were not surprised as we observed 40–45% transformation in entries 9 and 10. Keeping CH2Cl2 as the best solvent, we next examined other brominating agents. NBS efficiently delivered the lactamized product 4c in 80% yield. However, PTB and BTMATB were not good brominating agents for our system as we isolated 4c in 10–30% yield in entries 12 and 13. Moreover, molecular bromine was not successful either as we have isolated 4c with 50% yield. Finally, BDMS (Bromodimethylsulfonium bromide) in CH2Cl2 at rt provided 65% of the desired product 4c (Table 3, entry 15). Therefore, we decided to use DBDMH/CH2Cl2/rt as an optimized condition for our system to obtain the spiro-lactam satisfactorily.
We next synthesized a list of isoxazole-amides using our previously synthesized acids (Table 2).
Table 2 Synthesis of isoxazole-lactams 3(a–r)a
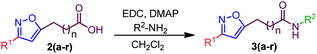
|
| Entry |
Acids (R1) |
Amines (R2) |
Time (h) |
Yield (%)b |
| DMAP (1.4 equiv.) was added to a suspension of EDC (1.3 equiv.) in DCM (1 M). The corresponding carboxylic acid (1 equiv.) and amine (1.2 equiv.) were then added at 0 °C. The mixture was stirred at room temperature for (12–24) hours. Isolated yield based on 2. n = 2. (4-Cs = 4-ClC6H4SO2). |
| 1 |
Me (2a) |
4-Ts |
24 |
78 (3a) |
| 2 |
nPr (2b) |
4-Ts |
24 |
80 (3b) |
| 3 |
Ph (2c) |
4-Ts |
18 |
82 (3c) |
| 4 |
4-MeC6H4 (2d) |
4-Ts |
12 |
82 (3d) |
| 5 |
4-OMeC6H4 (2e) |
4-Ts |
12 |
79 (3e) |
| 6 |
4-FC6H4 (2f) |
4-Ts |
12 |
85 (3f) |
| 7 |
4-ClC6H4 (2g) |
4-Ts |
12 |
88 (3g) |
| 8 |
2,6-di-Cl-C6H3 (2h) |
4-Ts |
12 |
80 (3h) |
| 9 |
4-BrC6H4 (2i) |
4-Ts |
12 |
86 (3i) |
| 10 |
4-lC6H4 (2j) |
4-Ts |
12 |
79 (3j) |
| 11 |
4-CF3C6H4 (2k) |
4-Ts |
12 |
88 (3k) |
| 12 |
CO2Et (2l) |
4-Ts |
24 |
78 (3l) |
| 13 |
Me (2a) |
PMB |
18 |
80 (3m) |
| 14 |
Ph (2c) |
PMB |
18 |
85 (3n) |
| 15c |
Ph (2o) |
PMB |
18 |
87 (3o) |
| 16 |
Me (2a) |
4-Csd |
24 |
89 (3p) |
| 17 |
nPr (2b) |
4-Csd |
24 |
89 (3q) |
| 18 |
Ph (2c) |
4-Csd |
12 |
88 (3r) |
Table 3 Synthesis of spiro-isoxazoline-lactams (±)-4(a–r)
Having isoxazole-amides and optimized condition in hand, the scopes of the spiro-lactamization were investigated (Table 3). Initially, Ts-amide containing various substituted isoxazoles were examined. It was found that alkyl group on isoxazole ring 3, like methyl (3a) and propyl (3b), were well tolerated, giving products 4a and 4b in 88% and 85% yield (Table 3); however, a mono- and di-bromination on Ts-functionality were identified, probably due to the reactive methyl group.13 Next various aromatic substituted isoxazoles were tested against optimized conditions, where phenyl substituted isoxazole-amide was found to produce the spirolactam 4c in 85% yield. Furthermore, the electron-donating groups such as 4-Me and 4-OMe on phenyl ring were also competent, delivering the corresponding product 4d and 4e in 89% and 75% yield. However, over bromination was identified in 4e due to an unavoidable α-bromination.14 Halogen substituents (–F, –Cl, –Br, and –I) on phenyl ring were also compatible, leading to the formation of 4f–j in 85–95% yield. Electron withdrawing groups such as –CF3 and –CO2Et on the isoxazole ring were also competent, producing the corresponding 4k and 4l in 90% and 80% yield. We next investigated the effect of other amides over Ts-amide to evaluate the efficiency of bromo-lactamization. In this regard, while treated 3m, 3n, and 3o DBDMH/CH2Cl2/rt, we isolated 4m, 4n, and 4o in a 75–79% yield. Regardless of focusing on limited examples, it is worth mentioning that 4o comprises a six-membered lactam ring, which also expands the scope toward a higher membered spiro-lactam. While investigating the reactivity of Cs-amide, we found –Me, –Pr, and –Ph containing isoxazoles were equally efficient in delivering the desired products 4p-4r in 89–95% yield (Table 3). Among three types of amides, the PMB-amides were relatively less reactive. We reasoned that the less reactivity of PMB-amide could be due to the less acidic nature of PMB amide compared to Ts- and Cs-amides. Seemingly, we synthesized alkyl, aromatic, and ester substituted spiro-lactams. Regardless of investigating the synthetic scopes, we also envisaged that various functionality in the periphery of a 3D molecule could infuse biophysical properties in the molecules to deliver the drug-like nature.
While the alkyl functionality is responsible for introducing the hydrophobicity in the molecule, the aromatic substitution containing various functionalizations is responsible for the molecular electronic properties. Moreover, an ester functionality could be transformed into acid and amide derivatives to investigate different biological properties. Overall, this study displayed a possible route toward a novel 3D-molecular architect with a diverse peripheral functionality of biological interest.
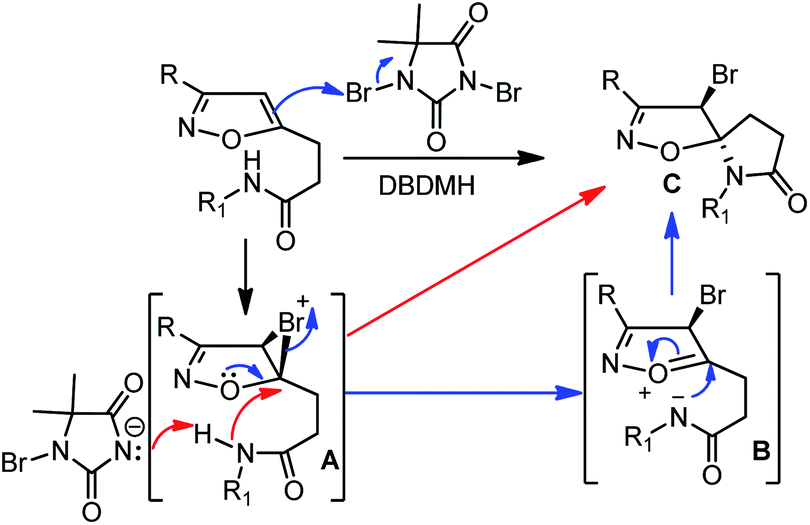
A possible mechanistic pathway has been depicted in Scheme 3. The selective bromonium ion formation on isoxazoline double bond generates the intermediate A. Subsequently, a facile proton abstraction from the pendent amide by hydantoin anion produces N-nucleophile. As a result, the neighbouring nucleophile can attack the bromonium ion intermediate A in two possible pathways. Following the red-line pathway, the N-nucleophile can directly attack on the bromonium ion A to produce the desired lactam C. Alternatively, a blue-line-pathway could follow an oxonium-ion mediated opening of bromonium ion to generate an intermediated B, followed by favourable 5-exo-trig cyclization, leading to the development of spiro-lactam C (Scheme 3). Furthermore, based on our previous bromo-lactonization, we hypothesized that the Br-atom and N-atom are opposite, resulting in racemization for 4.
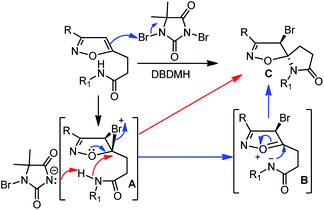 |
| | Scheme 3 Plausible mechanism for bromo-lactamization. | |
Conclusion
In conclusion, we have developed a straightforward method for synthesizing a wide range of spiro-isoxazoline-lactams as a novel source of 3D molecules with diverse functionalization in the periphery. Along with alkyl and aryl functionalities, an ester group could be expanded to access a wide range of amides for biological investigation. Moreover, the successful incorporation of a couple of sulfonamides and amide functionalities provided a novel class of spiro-sulfa compounds to investigate. Knowing the diverse application of our previously synthesized spiro-compounds, we speculate that the desired spiro-isoxazoline-lactams could exhibit a seminal scope to exhibit novel biological activities.
Experimental section
(a)Materials and methods
Unless otherwise stated, all solvents and reagents were commercially obtained and used without prior purification. The reaction progress was monitored by analytical thin-layer chromatography using 60 Å silica gel medium and 250 μm layer thickness. Compounds were visualized by 254 nm light, basic KMnO4 (40 g of K2CO3 + 6 g of KMnO4 in 600 mL of water, then 5 mL of 10% NaOH added), and subsequent development either no or gentle heating. The crude products were purified using hexanes and ethyl acetate ratio as eluent by flash column chromatography over silica gel (60 Å, 0.060–0.200 mm).
(b) Instrumental information
All NMR spectra were measured at 25 °C in the indicated deuterated solvents. 1H-NMR and 13C-NMR spectra were recorded in 500 MHz and 125 MHz, respectively. The NMR data are reported as follows: proton and carbon chemical shifts (δ) in ppm using tetramethylsilane as an internal standard, coupling constants (J) in hertz (Hz), and resonance multiplicities (br = broad, s = singlet, d = doublet, t = triplet, and m = multiplet). The residual protic solvent of CDCl3 (1H, δ 7.26 ppm); (13C, δ 77.0 ppm central resonance of the triplet), and C3D6O (1H, δ 2.05 ppm; 13C, δ 29.84 ppm) were used as the internal references in the 1H and 13C-NMR spectra. Melting points are uncorrected. Fourier transform infrared (FTIR) spectra were measured on neat NaCl. The absorptions are given in wavenumbers (cm−1). High-resolution mass spectrometry (HRMS) analyses were performed based on positive electrospray ionization on a Bruker 12 Tesla APEX – Qe FTICR-MS with an Apollo II ion source. Either protonated molecular ions [M + nH]n+ or sodium adducts [M + Na]+ were used for empirical formula confirmation.
General procedure for nitrone preparation
A mixture of hydroxylamine hydrochloride (1 mmol) and sodium bicarbonate (1.5 mmol), aldehyde (1 mmol), and anhydrous MgSO4 (1.5 mmol) in either dichloromethane or ether was stirred till the consumption of the starting materials (confirmed by TLC analysis). Upon completion, the mixture was filtered. The filtrate was concentrated in vacuo to yield the crude product, chromatographer on silica gel using ethyl acetate/hexane mixture as an eluant to afford the pure nitrones.
General procedure for the synthesis of isoxazole amide 3(a–r)
The isoxazole acid 2 (1.0 mmol, 1.0 equiv.) was added into a solution of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride, EDCI (201.7 mg, 1.3 mmol, 1.3 equiv.), and DMAP (171.0 mg, 1.4 mmol, 1.4 equiv.) in CH2Cl2 (5 mL) at 0 °C. 4-Methoxybenzylamine (PMBNH2) (164.6 mg) or p-toluene sulfonamide (pTsNH2) (205.5 mg) or 4-chlorobenzenesulfonamide (4-CsNH2) (230.0 mg) (1.2 mmol, 1.2 equiv.) was then added in one portion to the mixture. The mixture was allowed to warm slowly to room temperature and stirred for 24 h. Upon completion, the mixture was diluted with CH2Cl2 (10 mL) and washed with 1 N HCl solution (10 mL). The organic layer was removed, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were dried with MgSO4 and concentrated under a vacuum. The crude residue was subjected to flash chromatography on silica gel (hexanes/ethyl acetate = 4/1–2/1) to deliver the product 3.
3-(3-methylisoxazol-5-yl)-N-tosylpropanamide (3a). White solid, yield: 240.5 mg, 78% (hexane/ethyl acetate = 4/1); Mp: 178–180 °C; IR: νmax 3129, 3056, 2954, 2925, 2857, 2798, 1715, 1613, 1469, 1814, 1339, 1172, 1128, 1089, 867, 812, 658, 551, 536 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.89 (d, J = 7.6 Hz, 2H), 7.42 (d, J = 7.7 Hz, 2H), 5.83 (s, 1H), 2.92 (t, J = 7.0 Hz, 2H), 2.74 (t, J = 6.9 Hz, 2H), 2.43 (s, 3H), 2.14 (s, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 171.1, 169.3, 159.3, 144.5, 136.9, 129.3 (2C), 128.1 (2C), 101.6, 33.1, 20.9, 20.6, 10.3; HRMS (ESI) m/z: calcd for (C14H16N2O4S)Na+: 331.0723; found 331.0721.
3-(3-Propylisoxazol-5-yl)-N-tosylpropanamide (3b). White solid, yield: 269.1 mg, 80% (hexane/ethyl acetate = 5/1); Mp: 150–152 °C; IR: νmax 3354, 3255, 3117, 3072, 2958, 2925, 2871, 2856, 2798, 1712, 1601, 1465, 1417, 1378, 1342, 1288, 1168, 1132, 1090, 865, 808, 661, 553, 535 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.89 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 7.8 Hz, 2H), 5.87 (s, 1H), 2.94 (t, J = 7.0 Hz, 2H), 2.75 (t, J = 7.1 Hz, 2H), 2.50 (t, J = 7.4 Hz, 2H), 2.43 (s, 3H), 1.59 (dd, J = 14.8, 7.3 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 171.1, 169.3, 163.4, 144.5, 136.9, 129.3 (2C), 128.1 (2C), 100.6, 33.1, 27.5, 21.3, 20.9, 20.6, 13.1; HRMS (ESI) m/z: calcd for (C16H20N2O4S)Na+: 337.1216; found 337.1215.
3-(3-Phenylisoxazol-5-yl)-N-tosylpropanamide (3c). White solid, yield: 303.7 mg, 82% (hexane/ethyl acetate = 4/1); IR: νmax 3356, 3241, 3066, 3040, 2923, 1732, 1604, 1444, 1343, 1296, 1166, 1119, 1083, 859, 768, 666, 533, 557 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.89 (d, J = 8.4 Hz, 2H), 7.80–7.78 (m, 2H), 7.51–7.47 (m, 3H), 7.35 (d, J = 8.0 Hz, 2H), 6.48 (s, 1H), 3.05 (t, J = 7.2 Hz, 1H), 2.85 (t, J = 7.2 Hz, 1H), 2.35 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 172.3, 169.3, 162.0, 144.5, 136.8, 129.4 (2C), 129.3 (2C), 128.8, 127.9 (2C), 126.6 (2C), 126.1, 99.0, 33.0, 22.1, 20.5; HRMS (ESI) m/z: calcd for (C19H18N2O4S)H+: 371.1060; found 371.1059.
3-(3-(p-tolyl)isoxazol-5-yl)-N-tosylpropanamide (3d). White solid, yield: 315.2 mg, 82% (hexane/ethyl acetate = 4/1); Mp: 162–164 °C; IR: νmax 3356, 3257, 3235, 1725, 1713, 1605, 1435, 1300, 1159, 1087, 908, 864, 811, 544 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.89 (d, J = 7.5 Hz, 2H), 7.67 (d, J = 7.3 Hz, 2H), 7.34 (d, J = 7.5 Hz, 2H), 7.29 (d, J = 7.5 Hz, 2H), 6.44 (s, 1H), 3.03 (t, J = 6.9 Hz, 2H), 2.83 (t, J = 6.9 Hz, 2H), 2.37 (s, 3H), 2.34 (s, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 172.2, 169.6, 161.9, 144.4, 139.9, 137.1, 129.6, 129.5, 129.4, 129.3, 128.0, 127.9, 126.6, 126.5, 126.4, 98.9, 33.2, 21.2 (2C), 20.5; HRMS (ESI) m/z: calcd for (C20H20N2O4S)H+: 385.1216; found 385.1219.
3-(3-(4-methoxyphenyl)isoxazol-5-yl)-N-tosylpropanamide (3e). White solid, yield: 316.3 mg, 79% (hexane/ethyl acetate = 4/1); Mp: 96–98 °C; IR: νmax 3102, 2954, 2922, 2854, 1716, 1613, 1528, 1457, 1435, 1349, 1297, 1169, 1124, 1086, 1026, 905, 852, 812, 681, 659, 599, 533 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.88 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 9.0 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 7.04 (d, J = 8.9 Hz, 2H), 6.42 (s, 1H), 3.85 (s, 4H), 3.02 (t, J = 7.2 Hz, 2H), 2.81 (t, J = 7.2 Hz, 3H), 2.36 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 172.1, 170.1, 161.6, 161.1, 144.1, 137.4, 129.2 (2C), 127.9 (4C), 121.8, 114.2 (2C), 98.8, 54.8, 33.3, 21.2, 20.6; HRMS (ESI) m/z: calcd for (C20H20N2O5S)Na+: 423.0985; found 423.0985.
3-(3-(4-fluorophenyl)isoxazol-5-yl)-N-tosylpropanamide (3f). White solid, yield: 330.1 mg, 85% (hexane/ethyl acetate = 4/1); Mp: 164–166 °C; IR: νmax 3354, 3260, 3091, 2926, 2858, 1717, 1594, 1526, 1437, 1347, 1222, 1174, 1127, 1086, 913, 849, 802, 657, 557 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.88 (d, J = 8.4 Hz, 2H), 7.85 (dd, J = 9.0, 5.4 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 7.27 (t, J = 8.9 Hz, 2H), 6.50 (s, 1H), 3.05 (t, J = 7.2 Hz, 2H), 2.83 (t, J = 7.2 Hz, 2H), 2.36 (s, 2H); 13C{1H} NMR (125 MHz, CDCl3): δ 172.7, 169.7, 163.6 (d, J = 247.6), 161.1, 144.2, 137.2, 129.3 (2C) 128,7 (t, J = 9.1 Hz), 128.0 (2C), 125.86 (d, J = 3.3 Hz), 115.8 (d, J = 21.8 Hz), 99.0 33.2, 21.2, 20.6; HRMS (ESI) m/z: calcd for (C19H17FN2O4S)Na+: 411.0785; found 411.0783.
3-(3-(4-Chlorophenyl)isoxazol-5-yl)-N-tosylpropanamide (3g). White solid, yield: 356.3 mg, 88% (hexane/ethyl acetate = 4/1); Mp 166–168 °C; IR: νmax 3357, 3299, 3260, 2918, 1715, 1603, 1446, 1427, 1336, 1170, 1133, 1085, 861, 847, 815, 668, 544 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.88 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.7 Hz, 2H), 7.54 (d, J = 8.7 Hz, 2H), 7.36 (d, J = 7.9 Hz, 2H), 6.52 (s, 1H), 3.06 (t, J = 7.2 Hz, 2H), 2.85 (t, J = 7.2 Hz, 2H), 2.36 (s, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 172.7, 169.3, 161.1, 144.5, 136.9, 135.2, 129.3 (2C), 129.1 (2C), 128.2 (2C), 128.0 (2C), 126.03, 99.2, 33.0, 21.1, 20.6; HRMS (ESI) m/z: calcd for (C19H17ClN2O4S)Na+: 427.0489; found 427.0487.
3-(3-(2,6-dichlorophenyl)isoxazol-5-yl)-N-tosylpropanamide (3h). White solid, yield: 351.4 mg, 80% (hexane/ethyl acetate = 4/1); Mp: 138–140 °C; IR: νmax 3354, 3235, 2967, 2923, 1732, 1597, 1558, 1430, 1388, 1331, 1172, 1129, 1085, 863, 812, 786, 662, 538 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.91 (d, J = 8.4 Hz, 2H), 7.60–7.51 (m, 3H), 7.35 (d, J = 8.5 Hz, 2H), 6.11 (s, 1H), 3.11 (t, J = 7.1 Hz, 2H), 2.86 (t, J = 7.2 Hz, 2H), 2.37 (s, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 172.3, 169.6, 158.6, 144.4, 137.1, 134.9 (2C), 131.7, 129.4 (2C), 128.2 (2C), 128.1 (2C), 126.1, 102.7, 33.2, 21.1, 20.7; HRMS (ESI) m/z: calcd for (C19H16Cl2N2O4S)H+: 439.0281; found 439.0286.
3-(3-(4-bromophenyl)isoxazol-5-yl)-N-tosylpropanamide (3i). White solid, yield: 386.4 mg, 86% (hexane/ethyl acetate = 4/1); Mp: 192–194 °C; IR: νmax 3357, 3308, 3260, 2972, 2918, 1716, 1602, 1443, 1426, 1336, 1132, 1085, 1009, 815, 667, 544 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.88 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.3 Hz, 2H), 7.75 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.7 Hz, 2H), 6.52 (s, 1H), 3.05 (t, J = 7.2 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H), 2.40 (s, 3H), 2.36 (s, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 172.8, 169.4, 161.1, 144.4, 136.9, 132.1 (2C), 129.3 (2C), 128.4 (2C), 128.1 (2C), 126.1, 123.5, 99.1, 33.1, 21.1, 20.5; HRMS (ESI) m/z: calcd for (C19H17BrN2O4S)Na+: 470.9985; found 470.9985.
3-(3-(4-iodophenyl)isoxazol-5-yl)-N-tosylpropanamide (3j). White solid, yield: 392.1 mg, 79% (hexane/ethyl acetate = 4/1); Mp: 214–216 °C; IR: νmax 3082, 2055, 2951, 2922, 2851, 1717, 1601, 1436, 1427, 1376, 1335, 1169, 1131, 1083, 1003, 861, 814, 669, 541 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.89 (t, J = 7.8 Hz, 4H), 7.60 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.1 Hz, 2H), 6.52 (s, 1H), 3.05 (t, J = 7.2 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H), 2.36 (s, 3H); 13C NMR (125 MHz, CD3COCD3): δ 172.8, 169.3, 161.3, 144.4, 138.2 (2C), 136.9, 129.3 (2C), 129.0, 128.4 (2C), 128.1 (2C), 99.1, 95.4, 33.0, 21.11 20.6; HRMS (ESI) m/z: calcd for (C19H17IN2O4S)Na+: 518.9846; found 518.9848.
N-tosyl-3-(3-(4-(trifluoromethyl)phenyl)isoxazol-5-yl)propan-amide (3k). White solid, yield: 385.8 mg, 88% (hexane/ethyl acetate = 4/1); Mp: 148–150 °C; IR: νmax 3357, 3257, 3229, 3109, 2967, 2923, 1714, 1599, 1460, 1331, 1160, 1118, 1085, 1019, 845, 811, 673, 536 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 8.03 (d, J = 8.1 Hz, 2H), 7.89 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 8.6 Hz, 2H), 6.62 (s, 1H), 3.08 (t, J = 7.4 Hz, 2H), 2.87 (t, J = 7.2 Hz, 2H), 2.34 (s, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 173.2, 169.4, 160.9, 144.4, 137.0, 133.2, 130.9 (q, J = 32.5 Hz), 129.3 (2C), 128.0 (2C), 127.2 (d, J = 6.1 Hz, 2C), 126.0 (d, J = 13.9 Hz, 2C), 124.2 (q, J = 271.4 Hz), 99.4, 33.0, 21.1, 20.5; HRMS (ESI) m/z: calcd for (C20H17F3N2O4S)Na+: 461.0753; found 461.0749.
Ethyl-5-(3-(4-methylphenylsulfonamido)-3-oxopropyl)-isoxazole-3-carboxylate (3l). White solid, yield: 285.8 mg, 78% (hexane/ethyl acetate = 4/1); Mp: 106–108 °C; IR: νmax 3171, 3138, 3073, 3028, 2984, 2928, 1729, 1690, 1592, 1456, 1253, 1139, 1086, 1018, 932, 843, 811, 787, 660, 550 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 7.88 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 6.45 (s, 1H), 4.37 (q, J = 7.1 Hz, 2H), 3.07 (t, J = 7.2 Hz, 2H), 2.87 (t, J = 7.2 Hz, 2H), 2.42 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CD3COCD3): δ 173.9, 169.3, 159.6, 156.3, 144.5, 136.8, 129.3 (2C), 128.1 (2C), 101.7, 61.5, 32.8, 21.0, 20.7, 13.5; HRMS (ESI) m/z: calcd for (C16H18N2O6S)H+: 367.0958; found 367.0959.
N-(4-methoxybenzyl)-3-(3-methylisoxazol-5-yl)propenamide (3m). Colorless oil, yield: 219.5 mg, 80% (hexane/ethyl acetate = 4/1); IR: νmax 3299, 3119, 2920, 2838, 1637, 1606, 1547, 1511, 1437, 1416, 1231, 1173, 1031, 1001, 813, 524 cm−1; 1H NMR (500 MHz, CD3OD): δ 7.14 (d, J = 8.2 Hz, 2H), 6.83 (d, J = 8.2 Hz, 2H), 5.94 (s, 1H), 4.26 (br s, 2H), 3.75 (s, 3H), 3.02 (t, J = 7.1 Hz, 2H), 2.57 (t, J = 7.1 Hz, 2H), 2.18 (s, 3H); 13C{1H} NMR (125 MHz, CD3OD): δ 171.9, 159.9, 158.9 (2C), 130.4 (2C), 128.5, 113.4 (2C), 101.7, 54.2, 42.2, 32.9, 22.1, 9.8; HRMS (ESI) m/z: calcd for (C15H18N2O3)H+: 275.1390; found 275.1389.
N-(4-methoxybenzyl)-3-(3-phenylisoxazol-5-yl)propanamide (3n). Colorless oil, yield: 285.9 mg, 85% (hexane/ethyl acetate = 4/1); IR: νmax 3297, 3109, 3064, 2949, 2913, 2839, 1640, 1552, 1513, 1470, 1435, 1255, 1174, 1032, 919, 812, 765, 689 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.76–7.75 (m, 2H), 7.44 (br m, 3H), 7.13 (d, J = 7.6 Hz, 2H), 6.78 (d, J = 7.6 Hz, 2H), 6.34 (s, 1H), 5.78 (s, 1H, NH), 4.36 (d, J = 4.9 Hz, 2H), 3.73 (s, 3H), 3.20 (t, J = 7.0 Hz, 2H), 2.64 (t, J = 7.0 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3): δ 172.1, 170.4, 162.5, 159.1, 130.0, 129.9, 129.1 (2C), 129.09, 128.9 (2C), 126.7 (2C), 114.1 (2C), 99.8, 55.2, 43.2, 34.0, 22.8; HRMS (ESI) m/z: calcd for (C20H20N2O3)Na+: 359.1366; found 359.1365.
N-(4-methoxybenzyl)-4-(3-phenylisoxazol-5-yl)butanamide (3o). Colorless oil, yield: 304.8 mg, 87% (hexane/ethyl acetate = 4/1); Mp: 141–143 °C; IR: νmax 3294, 3125, 3064, 3007, 2950, 2930, 2834, 1638, 1550, 1513, 1469, 1410, 1032, 952, 911, 811, 765, 729, 692 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 3.89 Hz, 2H), 7.43 (br m, 3H), 7.20 (d, J = 8.1 Hz, 2H), 6.85 (d, J = 8.1 Hz, 2H), 6.30 (s, 1H), 5.82 (s, 1H), 4.37 (d, J = 5.2 Hz, 2H), 3.79 (s, 3H), 2.86 (t, J = 7.1 Hz, 2H), 2.28 (t, J = 7.1 Hz, 2H), 2.14–2.09 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 173.1, 171.6, 162.4, 159.0, 130.3, 129.9, 129.2 (2C), 129.2, 128.8 (2C), 126.7 (2C), 114.1 (2C), 99.3, 55.3, 43.1, 35.2, 26.0, 23.4; HRMS (ESI) m/z: calcd for (C21H22N2O3)Na+: 373.15226; found 373.15221.
N-((4-chlorophenyl)sulfonyl)-3-(3-methylisoxazol-5-yl)prop-anamide (3p). Colorless oil, yield: 292.6 mg, 89% (hexane/ethyl acetate = 4/1); IR: νmax 3330, 3237, 3135, 29,50, 2803, 1722, 1611, 1476, 1421, 1346, 1326, 1278, 1169, 1126, 1089, 871, 820, 753, 622 cm−1; 1H NMR (500 MHz, CD3OD): δ 7.94 (d, J = 8.7 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 5.83 (s, 1H), 2.92 (t, J = 7.2 Hz, 2H), 2.62 (t, J = 7.2 Hz, 2H), 2.17 (s, 3H); 13C{1H} NMR (125 MHz, CD3OD): δ 171.5, 161.2, 159.9, 142.4, 137.8, 129.4 (2C), 128.8 (2C), 101.6, 33.1, 20.9, 9.7; HRMS (ESI) m/z: calcd for (C13H13ClN2O4S)H+: 329.0357; found 329.0356; HRMS (ESI) m/z: calcd for (C13H13ClN2O4S)H+: 329.0357; found 329.0356.
N-((4-chlorophenyl)sulfonyl)-3-(3-propylisoxazol-5-yl)pro-panamide (3q). Colorless oil, yield: 282.6 mg, 89% (hexane/ethyl acetate = 4/1); IR: νmax 3119, 3066, 2964, 2932, 2875, 2810, 1716, 1606, 1587, 1476, 1349, 1132, 1083, 863, 824, 753, 626, cm−1; 1H NMR (500 MHz, CD3COCD3): δ 8.01 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 5.88 (s, 1H), 2.95 (t, J = 6.8 Hz, 2H), 2.77 (t, J = 7.0 Hz, 2H), 2.51 (t, J = 7.4 Hz, 2H), 1.60 (dd, J = 14.6, 7.3 Hz, 2H), 0.92 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 171.1, 169.8, 163.4, 139.2, 138.7, 129.8 (2C), 128.9 (2C), 100.5, 33.2, 27.5, 21.3, 21.0, 13.1; HRMS (ESI) m/z: calcd for (C15H17ClN2O4S)Na+: 379.0489; found 379.04916.
N-((4-chlorophenyl)sulfonyl)-3-(3-phenylisoxazol-5-yl)pro-panamide (3r). Colorless oil, yield: 343.9 mg, 88% (hexane/ethyl acetate = 4/1); IR: νmax 3329, 3231, 2961, 2924, 2855, 1717, 1598, 1579, 1462, 1351, 1168, 1120, 1081, 906, 852, 820, 752, 621 cm−1; 1H NMR (500 MHz, CD3COCD3): δ 8.02 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 6.7 Hz, 2H), 7.61 (d, J = 8.2 Hz, 2H), 7.50–7.48 (m, 3H), 6.72 (br s, 1H, NH), 6.51 (s, 1H), 3.06 (t, J = 7.0 Hz, 2H), 2.87 (t, J = 7.0 Hz, 2H); 13C{1H} NMR (125 MHz, CD3COCD3): δ 172.3, 169.6, 162.0, 139.3, 138.5, 129.9 (2C), 129.3, 129.0 (2C), 128.9 (2C), 127.9, 126.5 (2C), 99.1, 33.1, 21.1; HRMS (ESI) m/z: calcd for (C18H15ClN2O4S)Na+: 413.0333; found 413.0331.
General procedure for the synthesis of isoxazole amide (±)- 4(a–r).
To a stirred solution of isoxazole amides 3 (0.5 mmol) in anhydrous CH2Cl2 (5 mL) was added DBDMH (171.6 mg, 0.6 mmol) at rt. The solution was stirred for 24 h before quenching with a saturated Na2SO3 aqueous solution (3 mL) and diluted with CH2Cl2 (10 mL). The organic layer was extracted with CH2Cl2, washed with brine, dried over MgSO4, and concentrated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate = 4/1, v/v) to afford the product 4.
4-Bromo-3-methyl-6-tosyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4a). Colorless oil, yield: 102.1 mg, 88% (hexanes/ethyl acetate = 4/1). IR: νmax 3031, 2957, 2924, 2853, 1751, 1718, 1406, 1364, 1240, 1174, 1115, 1085, 1059, 941, 874, 818, 765, 670, 613, 566, 539 cm−1; 1H NMR (500 MHz, CDCl3): δ 8.00 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 5.87 (s, 1H), 4.48 (s, 2H), 2.86–2.80 (m, 1H), 2.61–2.44 (m, 3H), 2.27 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.1, 158.4, 144.4, 137.1, 129.5 (2C), 129.4 (2C), 103.5, 58.2, 33.4, 31.2, 29.5, 11.9; HRMS (ESI) m/z: calcd for (C14H14Br2N2O4S)Na+: 486.8933; found 486.8939.
4-Bromo-3-propyl-6-tosyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4b). Colorless oil, yield: 243.6 mg, 85% (hexanes/ethyl acetate = 4/1). IR: νmax 3117, 3037, 2998, 2919, 2850, 1769, 1454, 1404, 1375, 1275, 1175, 1120, 1082, 904, 872, 750, 685, 638, 585 cm−1; 1H NMR (500 MHz, CDCl3): δ 8.03 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 8.5 Hz, 2H), 6.63 (s, 1H), 6.30 (s, 1H), 3.03–2.90 (m, 2H), 2.87–2.80 (m, 1H), 2.64–2.51 (m, 3H), 1.66–1.54 (m, 2H), 1.39 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 172.5, 162.3, 147.6, 138.2, 129.6 (2C), 127.2 (2C), 105.7, 57.6, 55.3, 42.8, 38.5, 33.9, 29.3, 12.1; HRMS (ESI) m/z: calcd for (C16H17Br3N2O4S)Na+: 592.8351; found 592.8358.
4-Bromo-3-phenyl-6-tosyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4c). White solid, yield: 202.2 mg, 90% (hexanes/ethyl acetate = 4/1). Mp: 84–86 °C; IR: νmax 3021, 2967, 2918, 2850, 1739, 1365, 1229, 1216, 1085, 891, 673, 575 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.89 (d, J = 8.4 Hz, 2H), 7.86–7.83 (m, 2H), 7.51–7.50 (m, 3H), 7.31 (d, J = 8.0 Hz, 2H), 6.43 (s, 1H), 2.96–2.88 (m, 1H), 2.67–2.51 (m, 3H), 2.42 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.1, 159.8, 145.8, 131.0, 129.5, 129.4 (2C), 129.0 (2C), 128.9 (2C), 127.6 (2C), 126.9, 104.5, 54.6, 33.6, 29.6, 21.7; HRMS (ESI) m/z: calcd for (C19H17BrN2O4S)Na+: 470.9985; found 470.9985.
4-Bromo-3-(p-tolyl)-6-tosyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4d). Colorless oil, yield: 206.2 mg, 89% (hexanes/ethyl acetate = 4/1); IR: νmax 3035, 2970, 2921, 2850, 1741, 1365, 1229, 1216, 1087, 890, 813, 672, 571, 528 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.88 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 7.3 Hz, 4H), 6.42 (s, 1H), 2.94–2.87 (m, 1H), 2.67–2.50 (m, 3H), 2.42 (s, 3H), 2.41 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.2, 159.8, 145.8, 141.5, 134.4, 129.7 (2C), 129.4 (2C), 129.0 (2C), 127.6 (2C), 124.0, 104.5, 54.8, 33.6, 29.6, 21.7, 21.6; HRMS (ESI) m/z: calcd for (C20H19Br2N2O4)Na+: 485.0141; found 485.0138.
4-Bromo-3-(4-methoxyphenyl)-6-tosyl-1-oxa-2,6-diazaspiro-[4.4]non-2-en-7-one (±)-(4e). Colorless oil, yield: 238.9 mg, 75% (hexanes/ethyl acetate = 4/1). IR: νmax 3104, 2955, 2924, 2854, 1717, 1613, 1529, 1458, 1437, 1350, 1297, 1169, 1124, 1085, 1025, 905, 854, 812, 680, 660, 600, 530 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.09 (s, 1H), 7.97 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 7.5 Hz, 1H), 7.75 (d, J = 8.1 Hz, 2H), 7.00 (d, J = 8.6 Hz, 1H), 6.33 (s, 1H), 4.47 (s, 2H), 3.97 (s, 3H), 2.94–2.98 (m, 1H), 2.69–2.53 (m, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 173.1, 158.5, 157.9, 144.5, 136.9, 132.4, 129.5 (2C), 129.4 (2C), 128.3, 127.6, 120.4, 111.8, 104.6, 56.4, 54.4, 33.5, 31.2, 29.5; HRMS (ESI): m/z calcd for (C20H17Br3N2O5S)Na+: 656.8300; found 656.8308.
4-Bromo-3-(4-fluorophenyl)-6-tosyl-1-oxa-2,6-diazaspiro-[4.4]non-2-en-7-one (±)-(4f). Colorless oil, yield: 222.0 mg, 95% (hexanes/ethyl acetate = 4/1). IR: νmax 3026, 2967, 2922, 2851, 1744, 1602, 1512, 1364, 1229, 1158, 1050, 840, 671, 570 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.87 (d, J = 8.4 Hz, 2H), 7.84 (dd, J = 8.8, 5.2 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.19 (t, J = 8.6 Hz, 2H), 6.38 (s, 1H), 2.96–2.87 (m, 1H), 2.65–2.51 (m, 3H), 2.42 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.2, 164.3 (d, J = 252.1 Hz), 158.9, 145.9, 134.3, 129.8 (d, J = 8.6 Hz, 2C), 129.5 (2C), 128.9 (2C), 123.2 (d, J = 3.4 Hz), 116.2 (d, J = 22.1 Hz, 2C), 104.6, 54.7, 33.5, 29.6, 21.8; HRMS (ESI) m/z: calcd for (C19H16BrFN2O4S)Na+: 488.9890; found 488.9888.
4-Bromo-3-(4-chlorophenyl)-6-tosyl-1-oxa-2,6-diazaspiro-[4.4]non-2-en-7-one (±)-(4g). White solid, yield: 222.5 mg, 92% (hexanes/ethyl acetate = 4/1). Mp: 178–180 °C; IR: νmax 3029, 3002, 2970, 2923, 2845, 1738, 1366, 1216, 1229, 901, 667, 544 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.87 (d, J = 8.2 Hz, 2H), 7.79 (d, J = 8.5 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 6.39 (s, 1H), 2.91 (dt, J = 12.7, 9.4 Hz, 1H), 2.66–2.50 (m, 3H), 2.42 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.1, 159.0, 145.9, 137.1, 134.3, 129.5 (2C), 129.3 (2C), 128.9 (2C), 128.9 (2C), 126.4, 104.7, 54.4, 33.5, 29.5, 21.8; HRMS (ESI) m/z: calcd for (C19H16BrClN2O4S)Na+: 504.9595; found 504.9592.
4-Bromo-3-(2,6-dichlorophenyl)-6-tosyl-1-oxa-2,6-diazaspiro-[4.4]non-2-en-7-one (±)-(4h). White solid, yield: 230.6 mg, 89% (hexanes/ethyl acetate = 4/1). Mp: 206–208 °C; IR: νmax 3030, 2997, 2921, 2853, 1746, 1430, 1376, 1290, 1235, 1180, 1167, 1898, 811, 784, 660, 550 cm−1; 1H NMR (500 MHz, CDCl3): δ 8.04 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 1.0 Hz, 1H), 7.44 (s, 1H), 7.39–7.36 (m, 1H), 7.34 (d, J = 8.1 Hz, 2H), 6.96 (s, 1H), 3.14–3.09 (m, 1H), 2.58–2.48 (m, 3H), 2.44 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 172.4, 155.6, 145.8, 144.6, 134.8, 131.8 (2C), 129.5 (2C), 129.4 (2C), 128.6 (2C), 126.2, 105.4, 55.8, 34.2, 29.3, 21.8; HRMS (ESI) m/z: calcd for (C19H15BrCl2N2O4S)Na+: 538.9205; found 538.9855.
4-Bromo-3-(4-bromophenyl)-6-tosyl-1-oxa-2,6-diazaspiro-[4.4]non-2-en-7-one (±)-(4i). White solid, yield: 243.0 mg, 92% (hexanes/ethyl acetate = 4/1). Mp: 198–200 °C; IR: νmax 3091, 3059, 3004, 2921, 2853, 1736, 1592, 1489, 1367, 1245, 1179, 1164, 1087, 902, 812, 666, 609, 540 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.86 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.64 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 6.37 (s, 1H), 2.95–2.87 (m, 1H), 2.63–2.51 (m, 3H), 2.42 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.0, 159.1, 145.9, 134.3, 132.3 (2C), 129.5 (2C), 129.1 (2C), 128.9 (2C), 125.8, 125.6, 104.6, 54.2, 33.5, 29.5, 21.8; HRMS (ESI) m/z: calcd for (C19H16Br2N2O4S)Na+: 548.9089; found 548.9087.
4-Bromo-3-(4-iodophenyl)-6-tosyl-1-oxa-2,6-diazaspiro-[4.4]non-2-en-7-one (±)-(4j). White solid, yield: 244.5 mg, 85% (hexanes/ethyl acetate = 4/1). Mp: 160–162 °C; IR: νmax 3034, 2954, 2921, 2854, 1797, 1753, 1591, 1489, 1398, 1361, 1248, 1174, 1121, 1086, 1008, 894, 861, 812, 671, 607, 571![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 536 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.85 (t, J = 7.5 Hz, 3H), 7.71 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 8.6 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.31 (d, J = 8.1 Hz, 2H), 6.37 (s, 1H), 2.93–2.87 (m, 1H), 2.66–2.51 (m, 3H), 2.42 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.0, 159.1, 145.9, 138.2 (2C), 134.3, 132.3, 129.5 (2C), 129.1 (2C), 128.9 (2C), 104.6, 97.7, 54.2, 33.5, 29.5, 21.8; HRMS (ESI) m/z: calcd for (C19H16BrIN2O4S)Na+: 596.8951; found 596.8960.
536 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.85 (t, J = 7.5 Hz, 3H), 7.71 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 8.6 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.31 (d, J = 8.1 Hz, 2H), 6.37 (s, 1H), 2.93–2.87 (m, 1H), 2.66–2.51 (m, 3H), 2.42 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.0, 159.1, 145.9, 138.2 (2C), 134.3, 132.3, 129.5 (2C), 129.1 (2C), 128.9 (2C), 104.6, 97.7, 54.2, 33.5, 29.5, 21.8; HRMS (ESI) m/z: calcd for (C19H16BrIN2O4S)Na+: 596.8951; found 596.8960.
4-Bromo-6-tosyl-3-(4-(trifluoromethyl)phenyl)-1-oxa-2,6-di-azaspiro[4.4]non-2-en-7-one (±)-(4k). White solid, yield: 232.8 mg, 90% (hexanes/ethyl acetate = 4/1). Mp: 208–210 °C; IR: νmax 3029, 2921, 2856, 1744, 1592, 1411, 1323, 1249, 1165, 1126, 1069, 943, 850, 813, 668, 552 cm−1; 1H NMR (50 0 MHz, CDCl3): δ 7.97 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 6.42 (s, 1H), 2.98–2.90 (m, 1H), 2.64–2.55 (m, 3H), 2.43 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 172.9, 158.8, 146.0, 132.6 (d, J = 32.9 Hz), 130.4 (d, J = 1.3 Hz), 129.5 (2C), 128.9 (2C), 127.9 (2C), 125.8 (q, J = 3.7 Hz), 104.8, 54.0, 33.5, 29.5, 21.7; HRMS (ESI) m/z: calcd for (C20H16BrF3N2O4S)Na+: 538.9858; found 538.9857.
Ethyl-4-bromo-7-oxo-6-tosyl-1-oxa-2,6-diazaspiro-[4.4]non-2-ene-3-carboxylate (±)-(4l). Colorless semi solid, yield: 178.1 mg, 80% (hexanes/ethyl acetate = 4/1). IR: νmax 3170, 3136, 3071, 3028, 2984, 2928, 1730, 1690, 1592, 1455, 1250, 1139, 1086, 1018, 932, 840, 812, 787, 660, 551 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.89 (d, J = 8.0 HZ, 2H), 7.33 (d, J = 8.0 Hz, 2H), 6.21 (s, 1H), 4.41 (br m, 2H), 2.44 (s, 3H), 2.34 (d, J = 14.1 Hz, 2H), 2.21 (t, J = 14.1 Hz, 1H), 1.81 (t, J = 13.6 Hz, 1H), 1.4 (t, J = 6.6 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 169.2, 158.6, 151.0, 145.4, 135.2, 129.4 (2C), 129.2 (2C), 109.8, 67.5, 63.1, 30.9, 30.3, 21.7, 13.9; HRMS (ESI) m/z: calcd for (C16H17BrF3N2O6S)Na+: 466.9883; found 466.9889.
4-Bromo-6-(4-methoxybenzyl)-3-methyl-1-oxa-2,6-diazaspiro-[4.4]non-2-en-7-one (±)-(4m). White solid, yield: 139.5 mg, 79% (hexanes/ethyl acetate = 4/1). Mp: 198–200 °C; IR: νmax 3295, 3062, 2929, 2836, 1630, 1546, 1513, 1440, 1303, 1247, 1214, 1072, 1036, 818, 751 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.16 (d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 5.85 (s, 1H), 4.34 (d, J = 5.6 Hz, 2H), 3.78 (s, 3H), 3.11 (t, J = 7.6 Hz, 2H), 2.60 (dd, J = 16.3, 8.3 Hz 2H), 2.23 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 170.1, 168.0, 159.5, 129.2 (2C), 128.2, 114.1 (2C), 92.7, 55.3, 43.2, 42.6, 32.7, 21.8, 10.6; HRMS (ESI) m/z: calcd for (C15H17BrN2O3)H+: 353.0495; found 353.0495.
4-Bromo-6-(3-bromo-4-methoxybenzyl)-3-phenyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4n). White solid, yield: 192.7 mg, 78% (hexanes/ethyl acetate = 4/1). Mp: 100–102 °C; IR: νmax 3307, 3066, 3009, 2929, 2838, 1669, 1541, 1497, 1440, 1398, 1279, 1258, 1136, 1054, 1021, 923, 811, 769, 695, 667 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.82 (d, J = 4.9 Hz, 2H), 7.49–7.44 (m, 4H), 7.15 (d, J = 8.3 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 5.93 (s, 1H), 4.36 (d, J = 5.2 Hz, 2H), 3.83 (s, 3H), 3.23 (t, J = 7.3 Hz, 2H), 2.69 (d, J = 7.3 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3): δ 170.2, 160.5, 155.3, 132.7, 131.6, 130.2, 128.7 (2C), 128.1 (2C), 128.0, 127.7, 112.0, 111.7, 91.0, 56.2, 42.6 (2C), 32.7, 22.0; HRMS (ESI) m/z: calcd for (C20H18Br2N2O3)Na+: 514.95764; found 514.9583.
4-Bromo-6-(3-bromo-4-methoxybenzyl)-3-phenyl-1-oxa-2,6-diazaspiro[4.5]dec-2-en-7-one (±)-(4o). White solid, yield: 161.0 mg, 75% (hexanes/ethyl acetate = 4/1). Mp: 96–98 °C; IR: νmax 3310, 3066, 3008, 2929, 2851, 1731, 1667, 1497, 1405, 1258, 1153, 1055, 1021, 912, 810, 769, 695, 667 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.83–7.78 (m, 2H), 7.49–7.44 (m, 4H), 7.20 (d, J = 8.2 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 5.81 (s, 1H), 4.36 (d, J = 5.3 Hz, 2H), 3.88 (s, 3H), 2.93 (t, J = 6.9 Hz, 2H), 2.30 (t, J = 6.9 Hz, 2H), 2.18–2.13 (m 2H); 13C{1H} NMR (125 MHz, CDCl3): δ 171.5, 160.4, 155.3, 132.8 (2C), 131.8, 130.2 (2C), 128.7 (2C), 128.1 (2C), 111.9, 101.7, 90.9, 56.3, 42.6 (2C), 35.1, 25.2, 22.7; HRMS (ESI) m/z: calcd for (C21H20Br2N2O3)Na+: 528.9733; found 528.9741.
4-Bromo-6-((4-chlorophenyl)sulfonyl)-3-methyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4p). White solid, yield: 181.4 mg, 89% (hexanes/ethyl acetate = 4/1). Mp: 150–152 °C; IR: νmax 3018, 2918, 2849, 1739, 1574, 1475, 1367, 1241, 1187, 1084, 872, 755, 622 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.97 (d, J = 8.9 Hz, 2H), 7.50 (d, J = 8.9 Hz, 2H), 5.85 (s, 1H), 2.86–2.80 (m, 1H), 2.60–2.44 (m, 3H), 2.27 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.1, 158.4, 141.4, 135.8, 130.5 (2C), 129.2 (2C), 103.6, 58.1, 33.4, 29.5, 11.9; HRMS (ESI) m/z: calcd for (C13H12BrClN2O4S)Na+: 428.9282; found 428.9281.
4-Bromo-6-((4-chlorophenyl)sulfonyl)-3-propyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4q). Colorless oil, yield: 193.9 mg, 89% (hexanes/ethyl acetate = 4/1). IR: νmax 3092, 2961, 2927, 2873, 2854, 1756, 1585, 1477, 1370, 1248, 1175, 1084, 875, 756, 626 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.97 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 8.9 Hz, 2H), 5.91 (s, 1H), 2.88–2.80 (m, 1H), 2.63–2.58 (m, 1H), 2.57–2.55 (m, 1H), 2.53–2.49 (m, 2H), 2.44–2.41 (m, 1H), 1.84–1.77 (m, 2H), 1.08 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.1, 161.2, 141.3, 135.8, 130.5 (2C), 129.2 (2C), 103.4, 57.8, 33.7, 29.5, 28.7, 19.0, 14.0; HRMS (ESI) m/z: calcd for (C15H16BrClN2O4S)H+: 434.9775; found 434.9773.
4-Bromo-6-((4-chlorophenyl)sulfonyl)-3-phenyl-1-oxa-2,6-diazaspiro[4.4]non-2-en-7-one (±)-(4r). White solid, yield: 223.1 mg, 95% (hexanes/ethyl acetate = 4/1). Mp: 92–94 °C; IR: νmax 329, 3235, 2969, 2923, 2847, 1737, 1570, 1356, 1326, 1216, 1149, 1087, 821, 754, 625, 528 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.86 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 6.1 Hz, 2H), 7.50–7.26 (m, 5H), 5.33 (s, 1H), 2.98–2.86 (m, 2H), 2.74–2.89 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3): δ 173.7, 158.9, 140.3, 139.3, 131.5, 129.4 (2C), 129.1 (2C), 127.9 (2C), 127.4 (2C), 126.0, 114.5, 50.0, 28.9, 28.46; HRMS (ESI) m/z: calcd for (C18H14BrClN2O4S)Na+: 490.9438; found 490.9437.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was supported by grants from the National Institute of General Medicinal Sciences (NIH/NIGMS) (2SC3GM094081-08) and the National Science Foundation (HBCU-EiR) (1900127). The Analytical and NMR CORE Facilities was supported by NIH/NIMHD (G12MD007581).
Notes and references
-
(a) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed;
(b) E. Chupakhin, O. Babich, A. Prosekov, L. Asyakina and M. Krasavin, Molecules, 2019, 24(1–37), 4165 CrossRef CAS PubMed;
(c) K. Hiesinger, D. Dar’in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef CAS PubMed;
(d) R. Rios, Chem. Soc. Rev., 2012, 41, 1060–1074 RSC;
(e) L. K. Smith and I. R. Baxendale, Org. Biomol. Chem., 2015, 13, 9907–9933 RSC;
(f) A. Quintavalla, Curr. Med. Chem., 2018, 25, 917–962 CrossRef CAS PubMed;
(g) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 12, 2209–2219 CrossRef;
(h) A. K. Franz, N. V. Hanhan and N. R. Ball-Jones, ACS Catal., 2013, 3, 540–553 CrossRef CAS.
-
(a) M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. E. Chan, Chem. Biol. Drug Des., 2014, 83, 450 CrossRef CAS PubMed;
(b) T. J. Richie, S. J. Macdonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2009, 14, 1011 CrossRef PubMed;
(c) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed;
(d) F. Lovering, Med. Chem. Commun., 2013, 4, 515 RSC.
-
(a) N. G. Alves, A. J. S. Alves, M. I. L. Soares and T. M. V. D. Pinho E Melo, Adv. Synth. Catal., 2021, 363, 2464–2501 CrossRef CAS;
(b) A. J. S. Alves, N. G. Alves, M. I. L. Soares and T. M. V. D. Pinho E Melo, Org. Chem. Front., 2021, 8, 3543–3593 RSC;
(c) M. A. Khan, D. R. Houck, A. L. Gross, X.-L. Zhan, C. Cearley, T. M. Madsen, R. A. Kroes, P. K. Stanton, J. Burgdorf and J. R. Moskal, Int. J. Neuropsychopharmacol., 2018, 21, 242–254 CrossRef CAS PubMed;
(d) P. Micuch, L. Fisera, V. Ondrus and P. Ertl, Molecules, 1997, 2, 57–61 CrossRef CAS;
(e) F. Golmohammadia, S. Balalaie, V. F. Vavsaria, M. U. Anwar and A. Al-Harrasi, J. Org. Chem., 2020, 85, 13141–13152 CrossRef PubMed;
(f) A. J. S. Alves, N. G. Alves, C. C. Caratão, M. I. M. Esteves, D. Fontinha, I. Bártolo, M. I. L. Soares, S. M. M. Lopes, M. Prudêncio, N. Taveira and T. M. V. D. Pinho E Melo, Curr. Top. Med. Chem., 2020, 20, 140–152 CrossRef CAS PubMed;
(g) G. S. Singh, M. D’hooghe and N. De Kimpe, Tetrahedron, 2011, 67, 1989–2012 CrossRef CAS.
- S. M. Glass, S. M. Leddy, M. C. Orwin, G. P. Miller, K. A. Furge and L. L. Furge, Drug Metab. Dispos., 2019, 47, 567–573 CrossRef CAS PubMed.
- G. S. d'Alcontres, C. Caristi, A. Ferlazzo and M. Gattuso, J. Chem. Soc., Perkin Trans. 1, 1976, 16, 1694–1696 RSC.
- Z. Li and Y. Shi, Org. Lett., 2015, 17, 5752–5755 CrossRef CAS PubMed.
-
(a) S. R. Chemler and M. T. Bovino, ACS Catal., 2013, 3, 1076 CrossRef CAS PubMed;
(b) L. Zhou, J. Chen, C. K. Tan and Y.-Y. Yeung, J. Am. Chem. Soc., 2011, 133, 9164 CrossRef CAS PubMed;
(c) W. Xie, G. Jiang, H. Liu, J. Hu, X. Pan, H. Zhang, X. Wan, Y. Lai and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12924 CrossRef CAS PubMed;
(d) D. Huang, H. Wang, F. Xue, H. Guan, L. Li, X. Peng and Y. Shi, Org. Lett., 2011, 13, 6350 CrossRef CAS PubMed;
(e) C. B. Tripathi and S. Mukherjee, Org. Lett., 2014, 16, 3368 CrossRef CAS PubMed;
(f) G.-O. Liu, Z.-Y. Ding, L. Zhang, T.-T. Li, L. Li, L. Duan and Y.-M. Li, Adv. Synth. Catal., 2014, 356, 2303 CrossRef CAS.
-
(a) G. Cardillo and M. Orena, Tetrahedron, 1990, 46, 3321 CrossRef CAS;
(b) K. E. Harding, and T. H. Tiner, In Comprehensive Organic Synthesis,ed. B. M. Trost, Pergamon Press, New York, 1991, vol. 4, p. 363 Search PubMed.
-
(a) O. Kitagawa, M. Fujita, H. Li and T. Taguchi, Tetrahedron Lett., 1997, 38, 615 CrossRef CAS;
(b) M. Fujita, O. Kitagawa, T. Suzuki and T. Taguchi, J. Org. Chem., 1997, 62, 7330 CrossRef CAS PubMed;
(c) O. Kitagawa and T. Taguchi, Synlett, 1999, 8, 1191 CrossRef;
(d) D. Huang, X. Liu, L. Li, Y. Cai, W. Liu and Y. Shi, J. Am. Chem. Soc., 2013, 135, 8101 CrossRef CAS PubMed;
(e) G.-T. Fan, M.-H. Sun, G. Gao, J. Chen and L. Zhou, Synlett, 2014, 25, 1921 CrossRef CAS;
(f) H. Li and R. A. Widenhoefer, Tetrahedron, 2010, 66, 4827 CrossRef CAS PubMed;
(g) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, J. Org. Chem., 2016, 81, 545–552 CrossRef CAS PubMed.
-
(a) P. Das, S. Boone, D. Mitra, L. Turner, R. Tandon, D. Raucher and A. T. Hamme II, RSC Adv., 2020, 10, 30223–30237 RSC;
(b) P. Das, M. H. Hasan, D. Mitra, R. Bollavarapu, E. J. Valente, R. Tandon, D. Raucher and A. T. Hamme II, J. Org. Chem., 2019, 84, 6992–7006 CrossRef CAS PubMed;
(c) P. Das, A. O. Omollo, L. J. Sitole, E. McClendon, E. J. Valente, D. Raucher, L. R. Walker and A. T. Hamme II, Tetrahedron Lett., 2015, 56, 1794–1797 CrossRef CAS PubMed.
-
(a) A. Kalgutkar, R. Jones, and A. Sawant, In Metabolism Pharmacokinetics and Toxicity of Functional Groups; D. A. Smith, Ed.; RSC: Cambridge, UK, 2010; RSC Drug Discovery Series No. vol. 1, Chapter 5 Search PubMed;
(b) C. Hansch, P. G. Sammes, and J. B. Taylor, Comprehensive Medicinal Chemistry; Pergamon Press: Oxford, UK, 1990; vol. 2, Chapter 7.1 Search PubMed;
(c) H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef CAS PubMed;
(d) M. Fenga, B. Tanga, S. H. Liangb and X. Jianga, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef PubMed;
(e) Y. Kwon, J. Song, H. Lee, E.-Y. Kim, K. Lee, S. K. Lee and S. Kim, J. Med. Chem., 2015, 58, 7749–7762 CrossRef CAS PubMed.
- M. R. Manzoni, T. P. Zabawa, D. Kasi and S. R. Chemler, Organometallics, 2004, 23, 5618–5621 CrossRef CAS.
-
(a) A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Tetrahedron Lett., 2006, 47, 1097–1099 CrossRef CAS;
(b) K. Ziegler, G. Schenck, E. W. Krockow, A. Siebert, A. Wenz and H. Weber, Justus Liebigs Ann. Chem., 1942, 551, 1–79 CrossRef CAS.
-
(a) W. P. Reeves, C. V. Lu, B. Schulmeier, L. Jonas and O. Hatlevik, Synth. Commun., 1998, 28, 499–505 CrossRef CAS;
(b) R. Cordoba and J. Plumet, Tetrahedron Lett., 2002, 43, 9303–9305 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H, and 13C NMR for synthesized compounds have been included. See DOI: 10.1039/d2ra01070d |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Cord Carter‡
,
Gulrukh Shaheen‡ and
Ashton T. Hamme II
*,
Cord Carter‡
,
Gulrukh Shaheen‡ and
Ashton T. Hamme II