
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of Ni–Ag–ZnO solid solution nanoparticles for photoreduction and antimicrobial applications†
Sania Naseera,
Muhammad Aamir *a,
Muhammad Aslam Mirzaa,
Uzma Jabeenb,
Raja Tahirc,
Muhammad Najam Khan Malghanid and
Qamar Walie
*a,
Muhammad Aslam Mirzaa,
Uzma Jabeenb,
Raja Tahirc,
Muhammad Najam Khan Malghanid and
Qamar Walie
aMaterials Laboratory, Department of Chemistry, Mirpur University of Science and Technology (MUST), Mirpur 10250, AJK, Pakistan. E-mail: aamirorg@gmail.com; amir.chem@must.edu.pk
bFaculty of Basic Sciences, Sardar Bahadur Khan Women's University, Quetta 87300, Pakistan
cDepartment of Biotechnology, Mirpur University of Science and Technology (MUST), Mirpur 10250, AJK, Pakistan
dFaculty of Engineering and Architecture, Takatu Campus BUITEMS, Quetta, Pakistan
eSchool of Applied Sciences and Humanities, National University of Technology, I-12, Islamabad, 42000, Pakistan
First published on 8th March 2022
Abstract
ZnO is one of the most promising and efficient semiconductor materials for various light-harvesting applications. Herein, we reported the tuning of optical properties of ZnO nanoparticles (NPs) by co-incorporation of Ni and Ag ions in the ZnO lattice. A sonochemical approach was used to synthesize pure ZnO NPs, Ni–ZnO, Ag–ZnO and Ag/Ni–ZnO with different concentrations of Ni and Ag (0.5%, 2%, 4%, 8%, and 15%) and Ni doped Ag–ZnO solid solutions with 0.25%, 0.5%, and 5% Ni ions. The as-synthesized Ni–Ag–ZnO solid solution NPs were characterized by powdered X-ray diffraction (pXRD), FT-IR spectroscopy, scanning electron microscopy (SEM), energy dispersive spectroscopy (EDS), UV-vis (UV) spectroscopy, and photoluminescence (PL) spectroscopy. Ni–Ag co-incorporation into a ZnO lattice reduces charge recombination by inducing charge trap states between the valence and conduction bands of ZnO and interfacial transfer of electrons. The Ni doped Ag–ZnO solid solution NPs have shown superior 4-nitrophenol reduction compared to pure ZnO NPs which do not show this reaction. Furthermore, a methylene blue (MB) clock reaction was also performed. Antibacterial activity against E. coli and S. aureus has inhibited the growth pattern of both strains depending on the concentration of catalysts.
Introduction
Recently, metals and metal oxide nanomaterials have gained enormous interest in various fields.1 Among other metal oxides, ZnO has been a material of interest due to its effective biological, unique physical, and chemical properties.2 ZnO is an n-type semiconductor with a high exciton binding energy of 60 meV and has a similar band gap of 3.37 eV and similar mode action to TiO2.3 However, the major limitations of ZnO as a photocatalyst include the rapid recombination of charge carriers, low quantum yields, poor photostability, poor adsorption ability, and limited reusability.4–6 The catalytic activity of ZnO is largely affected by fast recombination of charge carriers and low interfacial charge carrier transfer rate.7 To avoid these limitations, various methods such as compositing with carbon materials,8 inducing surface defects,9 and doping with metal or non-metals.10 Among these strategies, the doping of metals is an effective approach and has gained enormous attention for tuning the optoelectronic properties of the materials. In the case of ZnO, metal ion doping increases its light absorption in the visible region. Moreover, metal doping also changes the optical, electrical, and magnetic properties allowing the tuning of band gap and better charge separation by inducing trap states.11A variety of transition and non-transition metals have been doped into the ZnO to improve its optical properties. The nickel offers a fine-tuning of optical properties by inducing the impurity level between the valence band and conduction band which can promote the visible light absorption and reduces the charge carrier recombination. Codoping has gained enormous attention as it offers better optoelectronic performance compared to single metal doping. Recently, Ag, Fe codoped ZnO,12 Au, Sn codoped ZnO,13 Ga, Al, Co co-doped ZnO,14 Zr, Ag co-doped ZnO,15 Al, Y co-doped ZnO,16 Ag, Au co-doped ZnO,17 Cr, In co-doped ZnO,18 Ce, Ag co-doped ZnO,19 Al, Ni codoped ZnO,7 and Ni, Th codoped ZnO20 have been reported. Liu et al.21 reported synthesis of Pd/ZnO nanoribbons which shows 99.3% 4-nitrophenol reduction efficiency. Likewise, Au/ZnO nanoparticles were used to reduce 4-nitrophenol.22 Bhatia et al.23 synthesized the NiO/ZnO nanocomposites by sol–gel method and have achieved the 100% reduction efficiency of 4-nitrophenol. Ag/ZnO nanoparticles were also reported for photoreduction and antibacterial activities.24 Whereas, Jeyachitra et al.25 have reported the synthesis of Ni and Ag codoped ZnO NPs for dye degradation applications. Azfar et al.26 have also reported the synthesis of Ni and Ag codoped ZnO NPs by using the sol–gel method. Recently, Sachi et al.27 reported the synthesis of NiAg@ZnO nanocomposites by in situ chemical co-reduction method and their applications in nitro-compounds reduction reactions. However, to the best of our knowledge, Ni and Ag co-incorporated ZnO solid solutions NPs have not been reported. In this system, the Ni reduces the charge carrier recombination by imparting trap state,20 where Ag will act as electron donor and reduces the recombination of electrons in ZnO lattices.28
We herein reported the synthesis of Ni and Ag co-incorporated ZnO solid solution NPs by sonochemical approach and their characterization by using various techniques. Ag dopant on the surface of ZnO and Ni2+ inducing the trap states, capture electrons to enhance charge separation for photocatalytic reduction of 4-NP to 4-AP. Moreover, reduction of methylene blue and antimicrobial studies were also investigated against E. coli and S. aureus by using as-synthesized materials. Finally, the possible mechanisms were proposed to explain the synergic effect of Ni–Ag–ZnO solid solutions.
Experimental
Materials and method
Zinc acetate dihydrate (Zn(CH3CO2)2·2H2O), sodium hydroxide (NaOH), silver nitrate (AgNO3), nickel acetate tetrahydrate (Ni(CH3CO2)2·4H2O), triethanolamine (TEA) and oleylamine were used to prepare solutions in 40% distilled water in ethanol. All the reagents used were purchased from Sigma-Aldrich and were used without further purification.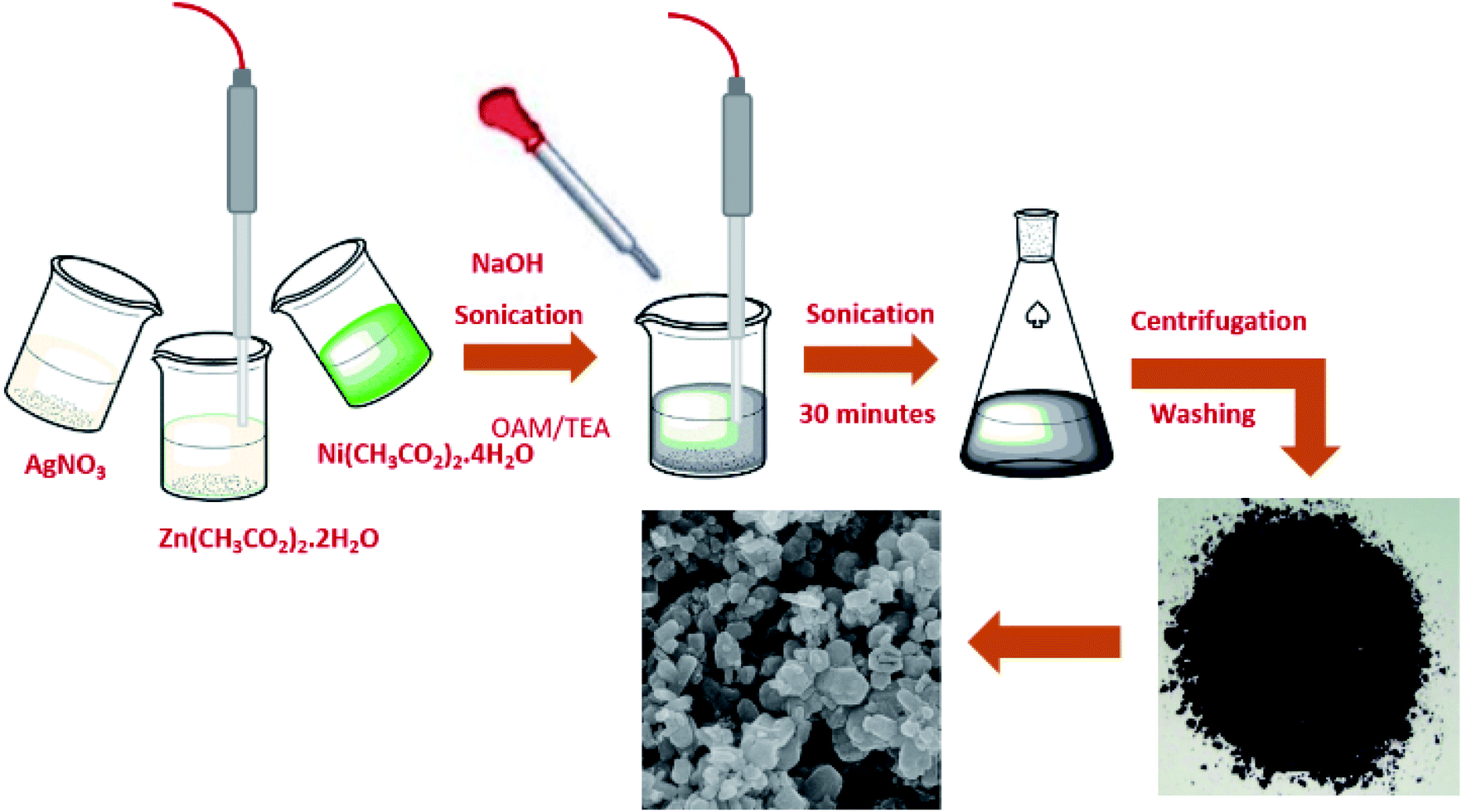 | ||
| Fig. 1 Schematic representation of the synthesis of solid solutions. | ||
Synthesis of Ni–Ag–ZnO solid solution NPs
The sonochemical method was used to synthesize the pure ZnO NPS and Ni–ZnO, Ag–ZnO, Ni–Ag–ZnO solid solution NPs. Briefly, 0.1 M of zinc acetate was dissolved in 10 mL of de-ionized water. Afterward, 1.0 mL of TEA and 10 μL of oleylamine were added as stabilizers to the above-mentioned solution under sonication. Then 1.0 M of NaOH solution in de-ionized water was added dropwise to maintain the pH at 8 under sonication for 30 minutes. The prepared precipitates were collected by centrifugation at 4000 rpm for 20 minutes, washed three times each with ethanol, distilled water, and dried at 100 °C. The dried product was calcined at 350 °C for 2 hours (Fig. 1). Satdeve et al.29 synthesized Ag/ZnO nanocomposites by using a sonication approach without any surfactants or capping agents. In the present work, Ni and Ag incorporated ZnO was prepared by using a similar methodology but with the use of oleylamine and triethanolamine as a stabilizer to control the size of nanoparticles. All the photocatalysts were prepared by using the same methodology with different molar concentrations of dopants. For incorporated samples, the molar ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for Ni and Zn to obtain the Ni–ZnO sample. While for Ag–ZnO, a 1:1 molar ratio was adjusted for Ag and Zn ions. In the case of co-incorporated samples, the different molar ratios (0.5%, 2%, 4%, 8%, and 15%) were used for Ni and Ag ions precursors. Moreover, the nickel (0.25%, 0.5% and 5% molar ratio) was doped in Ag–ZnO solid solution.
:1 for Ni and Zn to obtain the Ni–ZnO sample. While for Ag–ZnO, a 1:1 molar ratio was adjusted for Ag and Zn ions. In the case of co-incorporated samples, the different molar ratios (0.5%, 2%, 4%, 8%, and 15%) were used for Ni and Ag ions precursors. Moreover, the nickel (0.25%, 0.5% and 5% molar ratio) was doped in Ag–ZnO solid solution.
Photocatalytic reduction of 4-nitrophenol (4-NP)
Photocatalytic reduction of 4-NP was performed by taking 30 mL 4-nitrophenol solution and an excess amount of NaBH4 in a 100 mL beaker, under visible light. The 1.0 mg mL−1 of the catalyst was added to the nitrophenol solution. The absorption spectrum of the solution was measured at regular intervals and the solution was filtered to remove the catalyst to reuse. By using a UV-vis spectrophotometer, characteristic absorption peaks of 4-NP and 4-AP were observed at 317 nm and 300 nm, respectively.Reduction of methylene blue (clock reaction)
Clock reaction was performed by investigating the conversion of methylene blue (MB) into leuco-methylene blue (LMB) in the presence of NaBH4 and catalyst which then oxidizes into MB by adding NaOH. The clock cycle was carried out by adding 15 mg of NaBH4 in 5 ppm methylene blue solution (30 mL). Afterward, 15 mg of catalyst was added to the LMB solution. To perform the clock, 0.01 M NaOH solution (5 mL) was added after the removal of the catalyst to recover the MB in the solution.Determination of anti-bacterial activity
Antimicrobial activities of the prepared catalysts were checked against the Gram-positive (S. aureus) and Gram-negative (E. coli) bacterial strains, by turbidity method. Lysogeny broth (LB) media was used for the bacterial growth, it was prepared by mixing tryptone (1.0%), NaCl (0.5%), and yeast extract (0.5%) in distilled water. The 7 mL of sterilized media (autoclaved at 121 °C and 15 psi for 20 minutes) was taken in each test tube and aseptically inoculated with 100 μL of bacterial inoculum (having OD = 0.1 at 600 nm) of both strains. The tubes for test samples have media, bacterial inoculum, and 1 mL of catalysts of various concentrations (100 μg mL−1, 300 μg mL−1 & 500 μg mL−1) while the control tubes contain inoculum and media only. The tubes were placed in the incubator at 37 °C and 150 rpm for 24 hours. To monitor the growth inhibitory activity of the samples against both bacterial strains, optical density was measured at 600 nm for each sample and compared with that of the control. All the experiments were performed in triplicate and mean values were calculated. The comparison of ODs of samples containing catalysts with control will help to find the dose and time-dependent inhibitory effect of these on both bacterial strains.Characterization
Powdered X-ray diffraction (pXRD) analysis was used to determine the crystallographic structure of the materials by using JEOL JDX-3530. Fourier transform infrared spectroscopy (FT-IR) was performed to identify functional groups using Nicolet, Avatar 360 system. UV-vis absorption studies were performed by using Shimadzu UV-1800 spectrophotometer at room temperature. Shimadzu-RF-600 was used to determine the photoluminescence emission of as-synthesized materials at room temperature. Scanning electron microscopy (SEM) was performed by using FEI Nova Nano SEM 450 equipped with the Oxford EDX detector.Results and discussion
The synthesis of pure ZnO, Ni–ZnO, Ag–ZnO, Ni–Ag co-incorporated ZnO, and Ni-doped Ag–ZnO solid solution NPs were synthesized by the sonochemical method. Fig. 2 shows the schematic presentation of synthesis solid solutions. Fig. 2(a) shows pXRD spectra of pure ZnO, Ni–ZnO, and Ag–ZnO solid solutions NPs. The pure ZnO shows the diffraction peaks at 2θ of 31.2°, 33.8°, 35.8°, 47.0°, 55.9°, 62.4°, 65.6°, 67.5°, 68.5°, 71.9°, and 76.4° which were indexed to the (100), (002), (101), (102), (110), (200), (103), (112), (201), (004), and (202) diffraction planes of hexagonal wurtzite structure (JCPDF 00-005-0664). All the diffraction peaks remained in Ni–ZnO solid solution NPs (1:1), but a new diffraction peak (200) at 42.9° was appeared suggesting the successful substitution of Ni2+ into the ZnO lattice (JCPDF 00-001-1239). Moreover, the slight shift in the peak positions to the higher diffraction angles and decrease in the peak intensities reflects the lattice contractions due to the smaller ionic radius of Ni2+ compared to Zn2+ ions (Fig. 2(b)).30 The disappearance of diffraction peaks at higher angles is due to the lattice strain.31 In Ag–ZnO solid solution NPs (1:1 molar ratio), the diffraction peaks (111), (200), (220), (311) located at 37.66°, 43.9°, 64.2°, and 77.5° belongs to the face-centred cubic (fcc) structure of silver (JCPDF 00-001-1164). The slight shift in peaks to the lower angle indicates the lattice expansion due to partial substitution of Zn2+ ions by Ag+ in ZnO lattice, as ionic radii of Ag+ (1.26 Å) is larger than Zn2+ (0.74 Å) as shown in Fig. 2(b).32 In Ni–ZnO, the appearance of sharp peak (200) confirms the presence of NiO, while in Ag–ZnO, it confirms the presence of reduced Ag. Moreover, the appearance of additional peaks of Ag in ZnO indicates the formation of plasmonic Ag NPs formed by the partial reduction of Ag+ ions.33
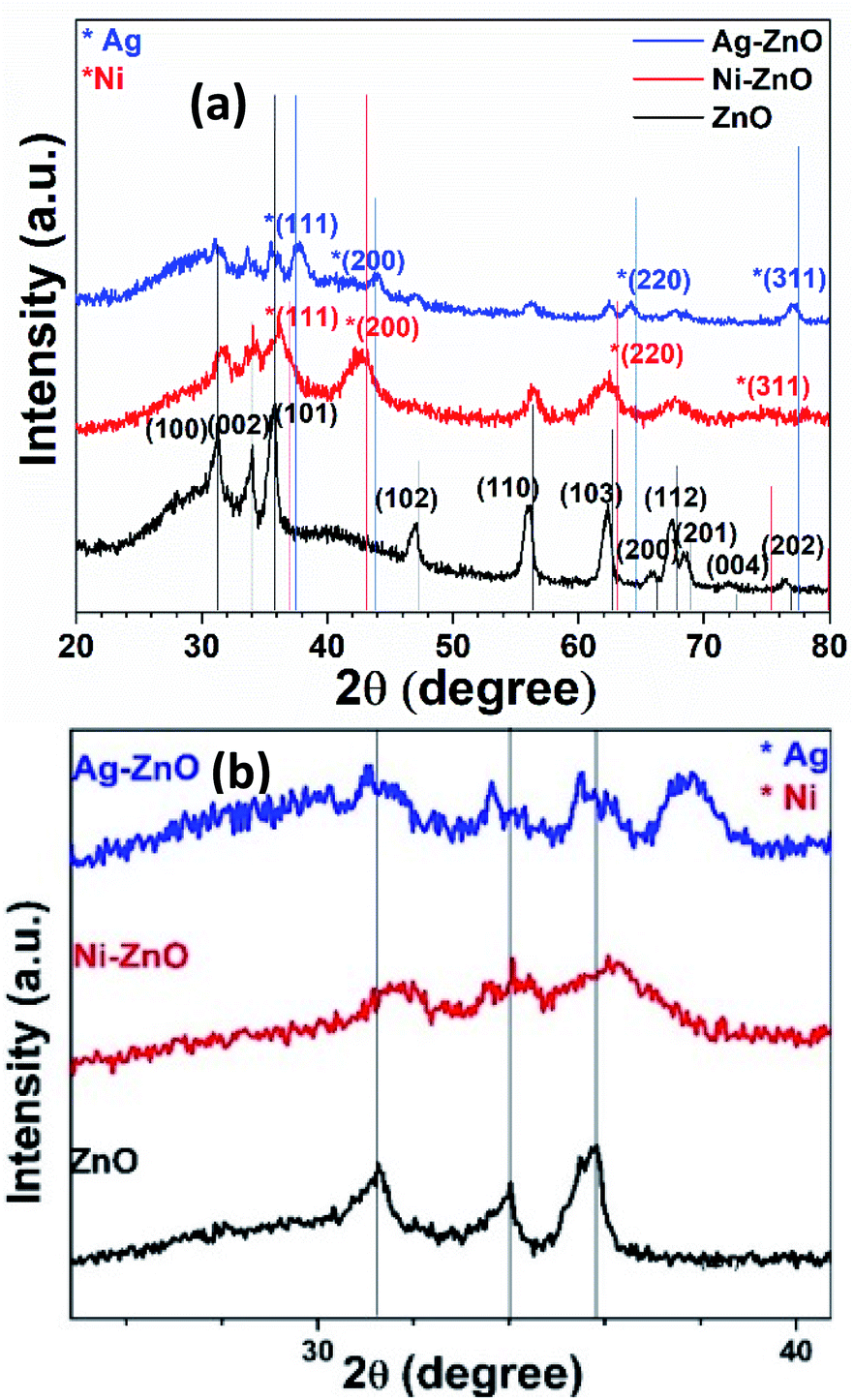 | ||
| Fig. 2 Comparative XRD spectra of (a) pure ZnO, Ni–ZnO (1:1 molar ratio) and Ag–ZnO (1:1 molar ratio) NPs samples. (b) Focused spectra indicate the shift in diffraction peak by the Ni and Ag incorporation in ZnO. | ||
To explore the co-effect of Ni and Ag in ZnO NPs, we have incorporated the equal molar amounts of Ni and Ag (0.5%, 2%, 4%, 8%, and 15%) in ZnO solid solution NPs. In Ni–Ag–ZnO (0.5%), the diffraction peak intensity was dropped compared to pure ZnO NPs as shown in Fig. S1(a).† However, the absence of characteristic diffraction peaks of Ni and Ag was due to the smaller concentrations in ZnO crystalline lattice. These diffraction peaks have appeared in 2% and 4% Ag and Ni co-incorporated ZnO NPs. Whereas the diffraction peak broadening and shifting appeared in 8% and 15% Ni and Ag co-incorporated ZnO lattices as shown in Fig. S1(a).†
Furthermore, we determined the effect of Ni2+ ions doping in Ag–ZnO solid solution (1:1). The sharp diffraction peak at 2θ ∼ 42.9° (200) was observed in Ni doped Ag–ZnO solid solution samples indicate the successful doping of Ni2+ ions in the Ag–ZnO (Fig. S1(b)†).
Lattice parameters of pure Ni/Ag–ZnO solid solutions were calculated by using following equations:
|
2dsinθ = nλ
| (1) |
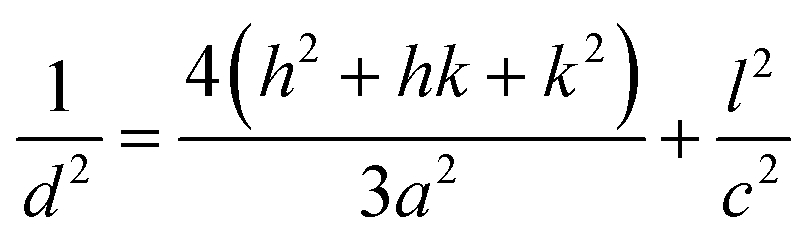 | (2) |
The crystallinity of as-synthesized ZnO NPs was found to be 84.2%. The introduction of Ni and Ag in ZnO NPs decreases the crystallinity to 77.2% and 82.1%, respectively. The crystallinity of ZnO is further reduced by the simultaneous incorporation of Ni and Ag into ZnO. With the increase in the concentration of Ni and Ag, the crystallinity of ZnO is found to decrease from 62.5–50.5%. Crystallinity for Ni/Ag–ZnO solid solution with an increasing ratio of Ni from 0.25% to 5% was found to be 41.8%, 62.5%, 46.7%, respectively. The Ni and Ag incorporation in ZnO NPs also reduces the growth of crystalline planes of ZnO lattice.
Fig. 3(a–c) shows the FESEM images of pure ZnO, Ni–ZnO, and Ag–ZnO solid solution NPs. The pure ZnO NPs appeared as nanospheres with an average particle size of 48 ± 1 nm (the histogram is shown in ESI Fig. S2†). The incorporation of Ni in ZnO (Ni–ZnO) didn't largely affect the shapes of NPs, however, the average particle size of Ni–ZnO solid solution NPs was dropped to 35 ± 1 nm. The formation of small-sized particles in the Ni–ZnO solid solution sample was due to the lattice strains.35 In the case of Ag–ZnO NPs, the average particle size was increased (60 ± 1 nm) with the incorporation of Ag ions in ZnO which is consistent with the reported literature (the histogram is shown in ESI Fig. S2†).36 Average size distribution of Ag on ZnO surface was found to be 33 ± 1 nm.
 | ||
| Fig. 3 FESEM images of as-prepared pure (a) ZnO NPs, (b) Ni–ZnO (1:1) and (c) Ag–ZnO (1:1) solid solution NPs. Likewise, (d) Ni–Ag–ZnO (0.25% Ni), (e) Ni–Ag–ZnO (0.5% Ni), (f) Ni–Ag–ZnO (5% Ni) NPs. | ||
The morphologies of Ni2+ and Ag1+ ions co-incorporated ZnO (Ni–Ag–ZnO) NPs (0.5%, 2%, 4%, 8%, and 15%) are shown in Fig. S3(a–e).† The mixed morphologies of spherical and rod-like structures were formed in 0.5% Ni–Ag–ZnO NPs was due to Ni2+ and Ag1+ diffusion into ZnO lattice. The average particle size for 0.5% co-incorporate ZnO NPs was found to be 38 ± 1 nm with 25 ± 1 nm average size of Ag on ZnO (Fig. S4(a)†), which was further decreased to 32 ± 1 nm for 2% incorporated sample with 12.5 ± 1 nm Ag (Fig. S4(b)†). Further, an increase in Ni and Ag concentration in the ZnO lattice increases the particle size from 70 ± 1 nm (13 ± 1 nm Ag) to 48 ± 1 nm (5 ± 1 nm Ag), and 90 ± 1 nm (26 ± 1 nm Ag) for 4%, 8%, and 15%, respectively (Fig. S4(c–e)†). Whereas the metallic Ag NPs can be observed on the surface of ZnO NPs as shown in Fig. S3(d).†
Fig. 2(e and f) shows the FESEM images of Ni doped Ag–ZnO solid solution NPs. It can be observed that Ni–Ag–ZnO containing 0.25% Ni2+ ions result in large-sized chunks mixed with small-sized rectangular morphologies. The average particle size was found to be 150 ± 1 nm with 21 ± 1 nm Ag on ZnO (size histogram is presented in ESI, Fig. S5†). Further increase in Ni2+ ions (0.5%) concentration in Ag–ZnO decreases the average particle size to 90 ± 1 nm (11.5 ± 1 nm Ag). However, in the case of 5% Ni2+ ions doping, morphology was changed predominantly to rectangular shapes with average particle size was further reduced to 47 ± 1 nm with 14.5 ± 1 nm average size of Ag. The increase in Ni2+ ions incorporation distorts Ni/Ag–ZnO (5% Ni) lattice and results in a change in morphology and size.
The energy dispersive X-ray (EDX) analysis was performed to determine the presence of Ni, Ag, and Zn in the as-prepared Ag–Ni–ZnO solid solutions (Fig. S6–S8 ESI†). The presence of Zn in the as-prepared samples was determined by the characteristic Lα, Kα, and Kβ peaks located at 1.0 keV, 8.6 keV, and 8.6 keV, respectively. Whereas, Ni was characterized by peaks, Lα at 0.8 keV, and Kα at 9.4 keV. The Ag was characterized by Lα at 2.0 keV. The presence of respective peaks in the as-synthesized Ni/Ag–ZnO solid solutions indicated the successful formation of desired samples. The observed atomic ratio of Ni and Ag in ZnO NPs slightly varies from the theoretical ratios as shown in Fig. S6–S8 (ESI).†
Fig. S9(a–c)† shows FTIR spectra of as-prepared samples. As shown in Fig. S9(a),† the stretching vibrations located at 3417 cm−1 and bending vibrations at 1612 cm−1 represent the –OH group present on the surface of ZnO NPs.37 The peak at 2353 cm−1 belongs to the atmospheric CO2.38 Symmetric and asymmetric peaks of C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O can be observed at 1394 and 1516 cm−1.39 The Zn–O stretching vibration peaks can be observed between 500 and 600 cm−1.40 In the case of Ni–ZnO, the peak for ZnO shifts to ∼555 cm−1 due to the lattice distortion attributes the substitution of Zn2+ by Ni2+ ions. Whereas the peak at ∼657 cm−1 exhibits the stretching vibrations for NiO. In Ag–ZnO, Zn–O stretching vibration was shifted to 545 cm−1 and a weak absorption peak at ∼900 cm−1 indicates the stretching and bending vibrations of Ag and Zn–O.41 It can be observed that the Zn–O and Ni–O signals are present in all as-prepared samples as shown in Fig. S9(b and c).† However, the Ag signals are broad and overlap with the Zn–O signal.
O can be observed at 1394 and 1516 cm−1.39 The Zn–O stretching vibration peaks can be observed between 500 and 600 cm−1.40 In the case of Ni–ZnO, the peak for ZnO shifts to ∼555 cm−1 due to the lattice distortion attributes the substitution of Zn2+ by Ni2+ ions. Whereas the peak at ∼657 cm−1 exhibits the stretching vibrations for NiO. In Ag–ZnO, Zn–O stretching vibration was shifted to 545 cm−1 and a weak absorption peak at ∼900 cm−1 indicates the stretching and bending vibrations of Ag and Zn–O.41 It can be observed that the Zn–O and Ni–O signals are present in all as-prepared samples as shown in Fig. S9(b and c).† However, the Ag signals are broad and overlap with the Zn–O signal.
The optical properties of as-prepared samples were explored by UV-vis spectroscopy at room temperature. Fig. S10(a and b)† shows UV-vis spectra and Tauc's plot of pure ZnO, Ni–ZnO (1:1), and Ag–ZnO (1:1) solid solution. Pure ZnO shows an absorption band at 372 nm with a bandgap of 3.1 eV. The Ni–ZnO solution NPs show the blue shift in an absorption band edge to 317 nm with a bandgap of 3.2 eV compared to the pure ZnO. The increase in bandgap is due to the excessive incorporation of 3d states of Ni ions (50%) into ZnO lattice by filling excessive energy levels on the edge of the conduction level. The Ag–ZnO solid solution NPs exhibited band-to-band absorption at 308 nm with a bandgap of 3.59 eV and a plasmonic peak at 397 nm (2.0 eV). The blue shift in bandgap energy could be ascribed to the Moss–Burstein effect.42
Fig. S10(c)† shows the absorption spectra of Ni–Ag co-incorporated ZnO NPs. The plasmonic peaks were absent in 0.5% Ni–Ag–ZnO solid solutions. The excitonic peak was observed in 0.5% and 4% Ni–Ag–ZnO NPs. In the case of 2% Ni–Ag–ZnO solid solutions, a blue shift in absorption was observed (427 nm) with a band gap of 2.9 eV. This could be due to the bandgap renormalization effect.43 Excitonic peaks for 8% and 15% Ni–Ag–ZnO appeared at 368 nm and 363 nm, with band gaps of 2.9 eV and 3.1 eV respectively as shown in Fig. S10(b).† The 0.25% and 0.5% Ni doped Ag–ZnO solid solution NPs show absorption peak edges at 320 nm and 323 nm with band gaps of 3.3 eV, and 3.18 eV respectively, while 5% Ni depicted absorption band edge at 326 nm with the band gap of 2.8 eV. Plasmonic peaks were observed at 515 nm, 450 nm, and 538 nm, respectively.
Photoluminescence (PL) studies were performed to investigate the effect of Ni and Ag co-incorporation on the electronic structure of ZnO lattices. Fig. 4(a) shows the PL emission spectrum of pure ZnO, Ni–ZnO, and Ag–ZnO excited at 350 nm wavelength at room temperature. Pure ZnO displayed two PL emission peaks at 394 nm and 470 nm. The emission peak at 394 nm can be attributed to excitonic recombination from the localized state below CB to the VB,44 while the peak at 470 nm belongs to the deep level blue-green emission peak due to transition between zinc interstitials and photoinduced holes.45 PL peak intensity located at 394 nm for pure ZnO was decreased and blue-shifted to 381 nm for Ni–ZnO and Ag–ZnO solid solution NPs. The reduced PL intensities in Ni and Ag incorporated are due to the trapping of free electrons by the introduction of energy states by Ni2+ and Ag1+ doping and by interfacial carrier transfer from the conduction band of ZnO to Ag on the surface, which enhance charge separation by reducing excitonic recombination. Blueshift in emission peak of Ni and Ag-doped ZnO is attributed to Moss–Burstein effect. Fig. 4(b) exhibits PL spectra of Ni–Ag–ZnO (0.5, 2, 4, 8, and 15%) solid solutions. It can be observed that in 0.5% and 2% Ni–Ag co-incorporated ZnO displayed a slight decrease in PL intensity at 390 nm due to enhanced charge separation by electron capturing by Ni2+ defect sites and interfacial charge transfer from ZnO to Ag. A mild blue-green PL peak at 471 nm originated from the defect sites. This PL emission peak was the blue shift to 387 nm in 4% and 8% Ni–Ag–ZnO solid solutions. However, 15% Ni–Ag–ZnO NPs exhibited anomalous behavior by displaying intense peaks at 394 nm and 470 nm. This peak could be attributed to enhanced charge recombination due to Ag aggregates and acting as recombination center46 and shift in peak could be due to sp–d interaction of Ni2+ and band electrons.47,48 Fig. 4(c) depicts PL spectra of Ni-doped Ag–ZnO solid solutions (0.25%, 0.5%, and 5% Ni). With the increase in the concentration of Ni from 0.25% to 5%, the emission peak at 381 nm was shifted to 383 nm and 466 nm to 470 nm, respectively. As discussed above, the decrease in PL intensity was also observed in all these samples due to the introduction of impurity states between CB and VB of ZnO.
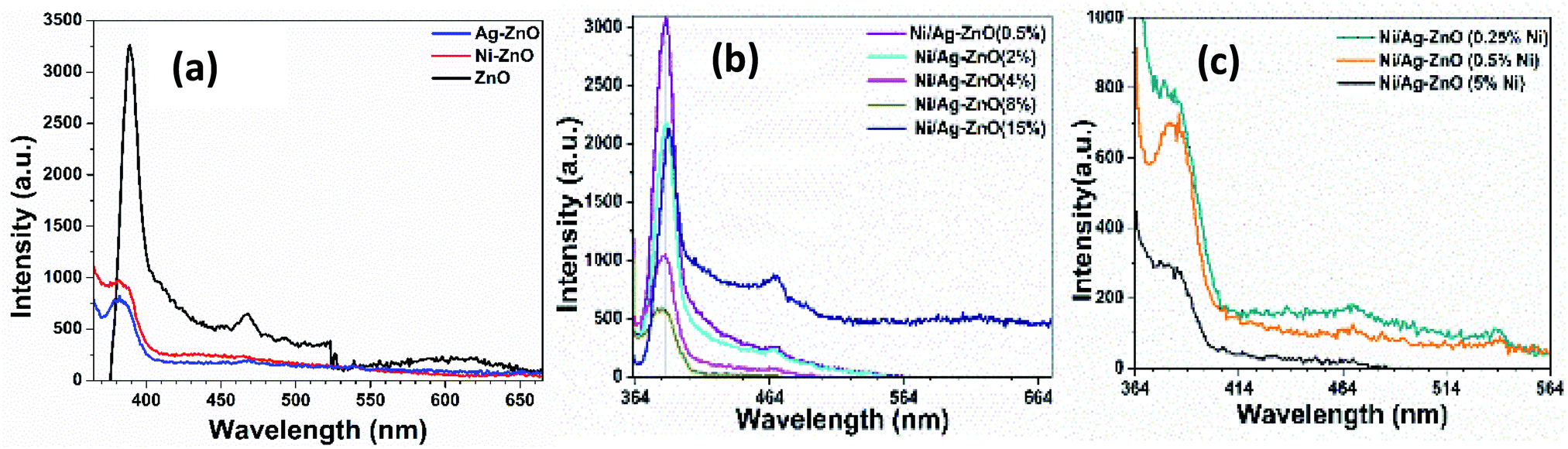 | ||
| Fig. 4 PL spectra of (a) pure, Ni, and Ag–ZnO (b) Ni/Ag–ZnO (0.5, 2, 4,8, 15%) (c) Ni/Ag–ZnO (0.25, 0.5, 5% Ni). | ||
The Ni–Ag–ZnO solid solutions were utilized in the heterogeneous photocatalysis process to convert 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in aqueous media as a model reaction. The reduction of 4-NP into 4-Ap (E0(4-NP/4-AP) = −0.76 V) is thermodynamically feasible in the presence of NaBH4 (E0(H3BO3/BH4−) = −1.33 V), but it is kinetically restricted in the absence of photocatalyst.49 Therefore, the addition of NaBH4 in the 4-NP produces yellowish color due to the formation of nitrophenolate ion which shows an absorption peak at 400 nm. Moreover, 4-NP without NaBH4 and photocatalyst did not show a reduction reaction (Fig. S11(a–c)†). However, in the presence of photocatalyst, reduction of 4-NP to colorless 4-AP was observed (Fig. S11(d)†).
The complete suppression of the peak at 400 nm indicates the completion of the reduction reaction. It was observed that the pure ZnO NPs did not reduce the 4-NP solution (Fig. S11(c)†), rather it shows photocatalytic degradation of 4-NP. It was observed that the color of pure ZnO NPs was changed to yellow due to the adsorption of 4-nitrophenolate ions. Moreover, the FTIR spectrum of pure ZnO and after reduction reaction process shows the formation of a complex with ZnO through phenolate ion (Fig. S12†).50 The linkage of ZnO with nitrophenolate ion through phenolate linkage keeps the nitro (–NO2) group of 4-NP away from hydrogen adsorbed onto the surface of ZnO, preventing reduction reaction. Performance comparison of reduction activities of bare ZnO, Ag and Ni NPs, and Ag and Ni doped ZnO NPs based catalyst is given in Table S1 (ESI).† The Ni and Ag ZnO solid solutions have shown comparable activity with reference to Ag NPs and Ni NPs.
The Ni–ZnO (1:1) and Ag–ZnO (1:1) solid solution photocatalysts have shown 74.6% and 97% in 27 minutes and 17 minutes, respectively (Fig. S13(a)†). The enhanced photocatalytic reduction 0f 4-NP by Ag–ZnO is attributed to the local field produced by the localized surface plasmon resonance (LSPR) effect of metallic Ag which contributed to interfacial charge transfer between metallic Ag and ZnO and enhance the charge separation.51 A comparison of catalytic efficiencies of as-synthesized photocatalysts is shown in Fig. S13(a–c).† The mean reaction conversions with standard deviation is given in Fig. S13(a–c).† The linear correlation between reaction time and ln(Ct/C0). The reaction follows the pseudo-first-order due to excess of NaBH4, rate constants for each concentration was calculated by following eqn (1):
| ln(Ct/C0) = −kt | (3) |
:1) and Ag–ZnO (1:1) were found to be 3.01 s−1, and 11.165 s−1, respectively (Fig. S13(a)†). It can be observed that Ag–ZnO (1:1) has shown a higher rate constant due to greater interfacial charge transfer compared to Ni incorporated ZnO photocatalysts.52,53 As expected, the activity factor of Ag–ZnO (1:1) photocatalysts was 372 s−1 g−1 which was higher than Ni–ZnO (1:1) photocatalyst (102 s−1 g−1). The rate constant and activity factors of all the photocatalysts are given in Table 1.
| Catalyst | Rate constants (s−1) | Activity factor (s−1 g−1) |
|---|---|---|
| ZnO | — | — |
| Ni–ZnO | 3.06 | 102 |
| Ag–ZnO | 11.165 | 372 |
| Ni/Ag–ZnO (0.5%) | — | — |
| Ni/Ag–ZnO (2%) | — | — |
| Ni/Ag–ZnO (4%) | 7.2 | 240 |
| Ni/Ag–ZnO (8%) | 9.6 | 320 |
| Ni/Ag–ZnO (15%) | 6.24 | 208 |
| Ni/Ag–ZnO(0.25% Ni) | 12 | 400 |
| Ni/Ag–ZnO(0.5% Ni) | 15 | 500 |
| Ni/Ag–ZnO (5% Ni) | 12 | 400 |
To determine the synergic effect of Ni and Ag concentrations, we have used 0.5%, 2%, 4%, 8% and 15% Ni and Ag incorporated ZnO NPs photocatalysts. It was observed that the 0.5% and 2% Ni–Ag–ZnO photocatalysts have not shown any catalytic activity due to the smaller concentration of Ni and Ag in ZnO lattices. However, when the concentration of Ni and Ag was increased to 4%, 8%, and 15%, the reduction percentage of 85%, 97%, and 81% was observed in 17, 21, and 15 minutes, respectively.
Moreover, the rate constants were found to be 7.2, 9.6, and 6.24 s−1 for 4%, 8%, and 15% Ni–Ag–ZnO, respectively (Fig. S13(b)†). The decrease in catalytic efficiency of the photocatalyst with 15% Ni–Ag–ZnO catalyst was due to an enhanced recombination rate as per PL studies. The activity factor also follows the same trend as given in Table 1. These results suggest that the high Ni ion concentration in the ZnO lattice is not effective for the reduction reaction. The higher Ag incorporation is more effective for the photocatalytic reduction reaction. To further evaluate the effect of Ni doping in Ag–ZnO solid solution NPs. We determined the catalytic efficiencies of Ni-doped Ag–ZnO solid solution NPs (0.25%, 0.5%, 5% Ni). The rate constants for Ni-doped Ag–ZnO (0.25%, 0.5%, 5% Ni) photocatalyst were 12, 15, 12 s−1 and activity factors were 400 s−1 g−1, 500 s−1 g−1 and 400 s−1 g−1 respectively (Fig. S13(c)†). The sonication of catalyst before performing the reduction reaction was have produced a dramatic effect. The reaction was completed just in one-minute duration as shown in Fig. 5(a and b). The higher rate of reaction in the sonicated sample was due to the better dispersion of catalyst in the 4-NP solution.
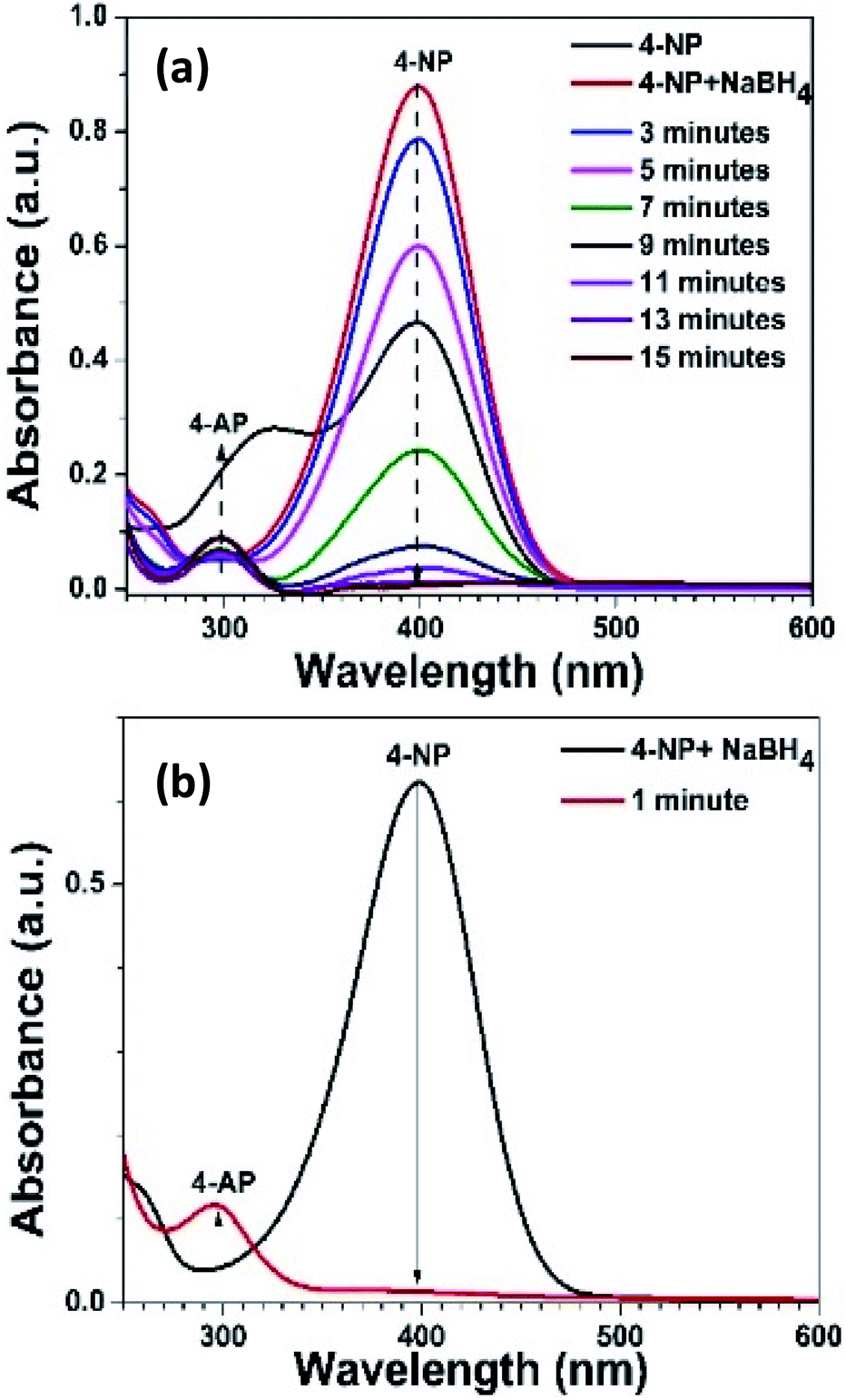 | ||
| Fig. 5 4-NP reduction (a) without sonic energy (b) with sonic energy. | ||
These results have shown that the 0.5% Ni-doped Ag–ZnO photocatalysts were the highly efficient photocatalysts for the nitrophenol reduction reaction. These results indicate that the 5% Ni doped Ag–ZnO solid solution is an efficient photocatalyst for nitrophenol reduction. Moreover, the synergic effect of Ni and Ag is an effective approach to induce photocatalytic activity in ZnO materials.
A recyclability test was performed for 0.5% Ni-doped Ag–ZnO solid solution NPs photocatalyst for five consecutive cycles. After each cycle, the photocatalysts were recovered by centrifugation, washed with deionized water, and dried before reuse for the next catalytic cycle. Conversion (%) of 4-nitrophenol to 4-aminophenol was determined by using the following relationship (4).
| Conversion (%) = 1 − (A/A0) × 100 | (4) |
To determine the possible cause of a decrease in efficiency, the amount of catalyst was weighed. After the 5th cycle, the 0.9 mg of catalyst was left which was the reason for the decrease in reduction efficiency. Leaching of the catalyst is caused during washing, centrifugation, and drying. To validate this study, all experiments were conducted in triplicate.
Scheme 1 presents the possible mechanism for the reduction of 4-nitrophenol by using the as-synthesized photocatalysts in the presence of NaBH4. In the current study, we proposed the reduction of 4-NP takes place through Langmuir–Hinshelwood model. According to this model, 4-nitrophenolate ions and BH4− ions are adsorbed on the surface of the catalyst. BH4− reacts with water to produce BO3− and release hydrogen in solution. Ag on the surface of ZnO captures electrons from BH4− and transfers them to 4-NP ions. The nitro group of 4-NP tends to take hydrogen and electrons from the catalyst.54 After several steps of hydrodeoxygenation reactions, 4-aminophenol is generated as the end product.
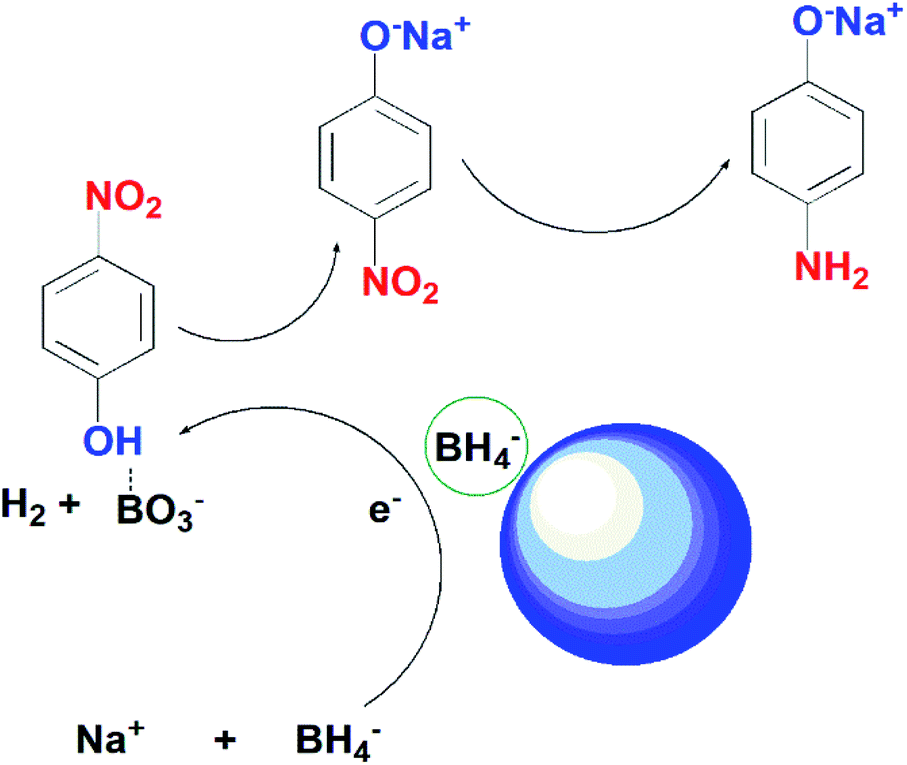 | ||
| Scheme 1 Proposed mechanism of action of ZnO NPs on reduction of 4-NP and MB. | ||
Reduction of methylene blue (clock reaction)
The photocatalytic reduction of methylene blue (MB) solution was also performed by using 0.5% Ni doped Ag–ZnO. The MB shows a characteristic absorption peak located at 664 nm originated from n–π* transition and a shoulder peak at 615 nm.55 The reduction of MB in the presence of NaBH4 was monitored using UV-vis spectroscopy and it was observed that only 30% absorption intensity of MB solution was dropped in the presence of NaBH4 without catalyst even after 30 minutes as shown in Fig. 6(a). However, the blue color was faded into light blue, indicating the incomplete reduction of MB in the presence of NaBH4.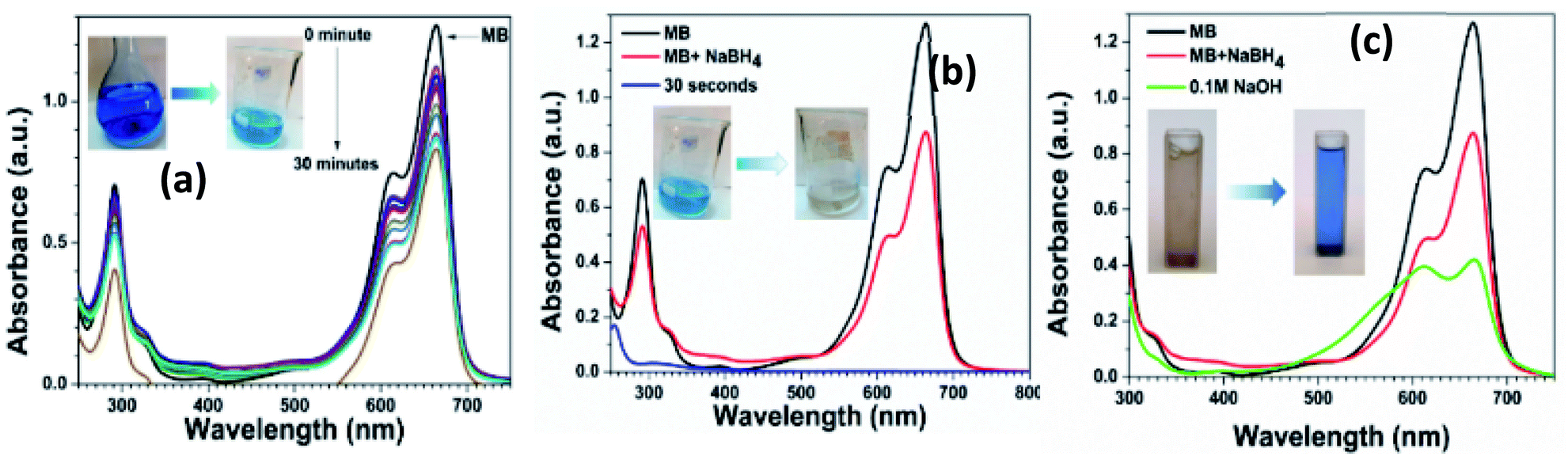 | ||
| Fig. 6 Clock cycle of MB (a) MB with NaBH4 (b) reduction of MB to LMB (c) oxidation of LMB to MB. | ||
After adding 0.5% Ni-doped Ag–ZnO catalyst, the characteristic peak of MB decreased rapidly within 30 seconds and the blue color was completely disappeared as shown in Fig. 6(b). The color disappearance was due to the formation of leuco-methylene blue (LMB) in the presence of the catalyst. After the formation of LMB, the catalyst was recovered from the solution by centrifugation. We added 0.1 M NaOH solution into the LMB solution which turns blue indicating the oxidation of LMB into MB (Fig. 6(c)). The number of hydroxyl ions in the solution influenced the oxidation process. Oxidation of MB increased with an increase in the number of hydroxyl ions in the solution.56 The regeneration of the blue color of MB indicates that the catalyst was not directly involved during the reduction reaction.57 In this reduction reaction, the catalyst transfers the electrons from BH4− to MB+ to produce colorless LMB.58 These results indicate the efficient transfer of the electron from the as-synthesized catalysts.
Antimicrobial analysis
Time and dose-dependent antibacterial properties of synthetic photocatalysts against E. coli and S. aureus were evaluated by monitoring optical density (OD) for 24 hours and its comparison with that of control. The antibacterial activity of five synthesized catalysts including Ni–ZnO, Ag–ZnO, Ni/Ag–ZnO (8%), and Ni/Ag–ZnO (5% Ni) was performed. The inhibitory effect of pure ZnO, Ag–ZnO, Ni–ZnO, Ni, and Ag co-incorporated ZnO and Ni doped Ag–ZnO NPs on the rate of bacterial growth was explored by taking OD of 600 nm at different intervals (2, 4, 6, 10, 18, 22, and 24 hours).59 Impact of various concentrations of synthesized catalysts against E. coli and S. aureus is illustrated in Fig. S15 and S16.† The growth inhibition of both bacterial strains increased with an increase in the concentration of catalysts from 100 μg mL−1 to −500 μg mL−1 and time.The maximum growth inhibitory activity against E. coli was shown by the Ni–ZnO (Fig. S15a†) and 0.5% Ni doped Ag–ZnO (Fig. S15d†) indicated by the lower value of absorbance at 600 nm compared to the control. Increasing the dose of the catalysts up to 500 μg mL−1 increases the growth inhibition of E. coil in all cases. The physical examination of the tubes confirmed the decrease in the turbidity of the samples with increasing concentration of catalysts due to the inhibition of the bacterial growth (Fig. 7). The maximum growth inhibitory activity against S. aureus was shown by the 0.5% Ni doped Ag–ZnO solid solution at concentrations of 500 μg mL−1 (Fig. S16d†) and Ag–ZnO up to 18 hours (Fig. S16b†). An increase in the doses of catalysts increases the growth inhibitory activity against S. aureus except that of Ni–ZnO, where 300 μg mL−1 has more growth inhibitory activity.
 | ||
| Fig. 7 Antibacterial action of prepared nanoparticles. | ||
The growth inhibitory activity of the as-synthetic catalysts is either due to oxidative stress or changes in the electric charge on the bacterial cell wall as shown in Fig. 8. Possible mechanism for generation of ROS could be disruption of mitochondrial redox state by Zn2+ ions released from ZnO NPs.60,61 ZnO NPs can create oxidative stress by generating reactive oxygen species (ROS) at the cell surface and these highly reactive species cause oxidative damage to the biomolecules in the surrounding.45,62 Bacterial cell membrane is mechanically degraded by the release of ROS which also suppresses the antioxidant defense system of bacteria and limits ATP production. The production of hydrogen peroxide from the zinc oxide at the surface has been previously postulated as an effective approach to restrict bacterial growth.63,64 Ag or AgO nanoparticles neutralized the electric charge at the surface of the cell wall and affect the penetrability of the bacterial cell which ultimately leads to its death.
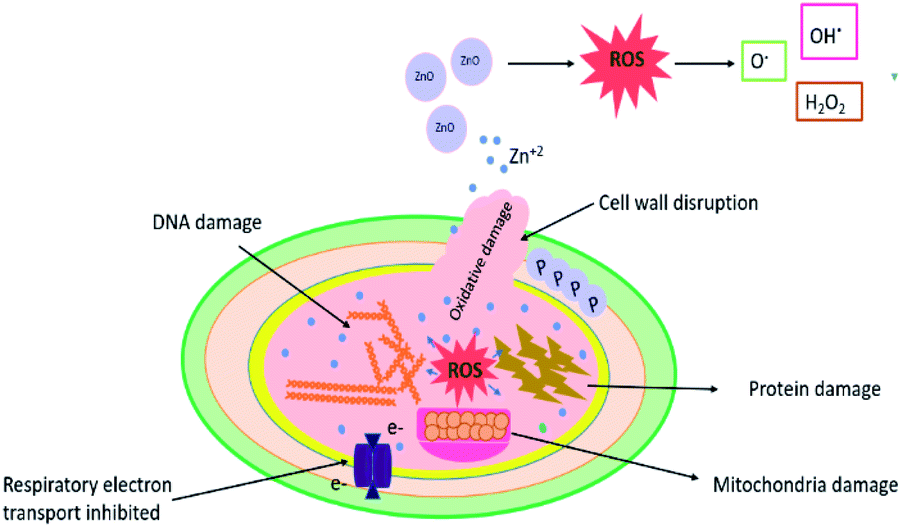 | ||
| Fig. 8 Schematic illustration of the antimicrobial mechanism of ZnO NPs against bacterial cells. | ||
Conclusions
In summary, we presented the Ni–Ag co-incorporated ZnO solid solution NPs as a facile route for inducing the combined effect of both these metals to prepare an efficient catalyst for the reduction of 4-nitrophenol to 4-aminophenol, which is otherwise not possible by pure ZnO NPs. The synergic effect of Ni–Ag–ZnO solid solution on structure, optical, and morphology was also determined. Our comparative studies have shown that the 0.5% Ni doped Ag–ZnO solid solution NPs are an effective catalyst for nitrophenol reduction with a fast rate of reaction. Pure ZnO NPs have not shown a reduction reaction due to the fast recombination of charge careers. The methylene blue clock reaction was also performed to further explore the utility of the synergic effect of Ni–Ag–ZnO solid solution. Antimicrobial analysis shows that Ni–Ag–ZnO solid solution NPs efficiently inhibit the growth of E. coli and S. aureus bacterial strains. Based on the results in the present study, we believe that this facile engineering to tune the optoelectronic properties of ZnO opens new opportunities for the synthesis of co-incorporated metal oxides as efficient photocatalysts for environmental remediations.Author contributions
Sania Naseer: performed all the experimental work, paper write-up. Muhammad Aamir: conceived the idea, supervised the experimental work, paper write-up. Muhammad Aslam Mirza: supervised the experimental work and facilitate some characterization tools. Uzma Jabeen: provided the pXRD facilities and SEM facilities. Raja Tahir: performed biological activities and write-up. Muhammad Najam Khan Malghani: performed the pXRD analysis and write-up. Qamar Wali: performed the analyses and write-up.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. acknowledged Dr Javeed Akhtar, Associate Professor at Department of Chemistry, Mirpur University of Science and Technology (MUST), Mirpur, AJK, Pakistan for guidelines during the experimental work.References
- M. Aamir, T. Adhikari, M. Sher, N. Revaprasadu, W. Khalid, J. Akhtar and J.-M. Nunzi, New J. Chem., 2018, 42, 14104–14110 RSC.
- E. Mohammadi, M. Aliofkhazraei, M. Hasanpoor and M. J. Chipara, Crit. Rev. Solid State Mater. Sci., 2018, 43, 475–541 CrossRef CAS.
- W. A. Doolittle, G. Namkoong, A. G. Carver and A. S. Brown, Solid State Electron., 2003, 47, 2143–2147 CrossRef CAS.
- W. Yu, J. Zhang and T. Peng, Appl. Catal., B, 2016, 181, 220–227 CrossRef CAS.
- S. G. Kumar and K. K. Rao, Appl. Surf. Sci., 2017, 391, 124–148 CrossRef CAS.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4, 1–8 CrossRef.
- I. N. Reddy, C. V. Reddy, J. Shim, B. Akkinepally, M. Cho, K. Yoo and D. Kim, Catal. Today, 2020, 340, 277–285 CrossRef CAS.
- X. Bai, L. Wang, R. Zong, Y. Lv, Y. Sun and Y. Zhu, Langmuir, 2013, 29, 3097–3105 CrossRef CAS PubMed.
- Y. Leung, X. Chen, A. Ng, M. Guo, F. Liu, A. Djurišić, W. Chan, X. Shi and M. Van Hove, Appl. Surf. Sci., 2013, 271, 202–209 CrossRef CAS.
- B. M. Rajbongshi and S. Samdarshi, Appl. Catal., B, 2014, 144, 435–441 CrossRef CAS.
- M. Rezaei and A. Habibi-Yangjeh, Mater. Lett., 2013, 110, 53–56 CrossRef CAS.
- Q. Zhang, J.-K. Liu, J.-D. Wang, H.-X. Luo, Y. Lu and X.-H. Yang, Ind. Eng. Chem. Res., 2014, 53, 13236–13246 CrossRef CAS.
- A. Senthilraja, B. Krishnakumar, B. Subash, A. J. Sobral, M. Swaminathan and M. Shanthi, J. Ind. Eng. Chem., 2016, 33, 51–58 CrossRef CAS.
- H. Serier, O. Toulemonde, D. Bernard, A. Demourgues, J. Majimel and M. Gaudon, Mater. Res. Bull., 2012, 47, 755–762 CrossRef CAS.
- B. Subash, B. Krishnakumar, M. Swaminathan and M. Shanthi, Langmuir, 2013, 29, 939–949 CrossRef CAS PubMed.
- J. Huo, L. Fang, Y. Lei, G. Zeng and H. Zeng, J. Mater. Chem. A, 2014, 2, 11040–11044 RSC.
- A. Senthilraja, B. Subash, B. Krishnakumar, D. Rajamanickam, M. Swaminathan and M. Shanthi, Mater. Sci. Semicond. Process., 2014, 22, 83–91 CrossRef CAS.
- R. Mimouni, A. Souissi, A. Madouri, K. Boubaker and M. Amlouk, Curr. Appl. Phys., 2017, 17, 1058–1065 CrossRef.
- B. Subash, B. Krishnakumar, R. Velmurugan, M. Swaminathan and M. Shanthi, Catal. Sci. Technol., 2012, 2, 2319–2326 RSC.
- K. Vignesh, M. Rajarajan and A. Suganthi, J. Ind. Eng. Chem., 2014, 20, 3826–3833 CrossRef CAS.
- M. Liu, F. Cui, Q. Ma, L. Xu, J. Zhang, R. Zhang and T. Cui, New J. Chem., 2020, 44, 4042–4048 RSC.
- R. Biswas, B. Banerjee, M. Saha, I. Ahmed, S. Mete, R. A. Patil, Y.-R. Ma and K. K. J. T. Haldar, J. Phys. Chem. C, 2021, 125, 6619–6631 CrossRef CAS.
- P. Bhatia and M. J. Nath, J. Water Process Eng., 2020, 33, 101017 CrossRef.
- H. Abou Oualid, O. Amadine, Y. Essamlali, I. M. Kadmiri, H. El Arroussi and M. J. Zahouily, Nanoscale Adv., 2019, 1, 3151–3163 RSC.
- R. Jeyachitra, S. Kalpana, T. S. Senthil and M. Kang, Water Sci. Technol., 2020, 81, 1296–1307 CrossRef CAS PubMed.
- A. Azfar, M. Kasim, I. Lokman, H. Rafaie and M. Mastuli, R. Soc. Open Sci., 2020, 7, 191590 CrossRef CAS PubMed.
- Sachi, A. P. Singh and M. Thirumal, ACS Omega, 2021, 6, 34771–34782 CrossRef CAS PubMed.
- R. Wang, J. H. Xin, Y. Yang, H. Liu, L. Xu and J. Hu, Appl. Surf. Sci., 2004, 227, 312–317 CrossRef CAS.
- N. Satdeve, R. Ugwekar and B. J. Bhanvase, J. Mol. Liq., 2019, 291, 111313 CrossRef CAS.
- W. Chen, T. Gao, H. Gan, L. Xu and L. Jin, Mater. Technol., 2015, 30, 356–361 CrossRef CAS.
- S. Rana and R. P. Singh, J. Mater. Sci.: Mater. Electron., 2016, 27, 9346–9355 CrossRef CAS.
- D. Thatikayala, V. Banothu, J. Kim, D. S. Shin, S. Vijayalakshmi and J. Park, J. Mater. Sci.: Mater. Electron., 2020, 31, 5324–5335 CrossRef CAS.
- B. D. Ahn, H. S. Kang, J. H. Kim, G. H. Kim, H. W. Chang and S. Y. Lee, J. Appl. Phys., 2006, 100, 093701 CrossRef.
- G. Srinivasan, R. R. Kumar and J. Kumar, J. Sol-Gel Sci. Technol., 2007, 43, 171–177 CrossRef CAS.
- K. Raja, P. S. Ramesh and D. Geetha, Spectrochim. Acta, Part A, 2014, 120, 19–24 CrossRef CAS PubMed.
- S. Khosravi-Gandomani, R. Yousefi, F. Jamali-Sheini and N. M. Huang, Ceram. Int., 2014, 40, 7957–7963 CrossRef CAS.
- M. Anbuvannan, M. Ramesh, G. Viruthagiri, N. Shanmugam and N. Kannadasan, Spectrochim. Acta, Part A, 2015, 143, 304–308 CrossRef CAS PubMed.
- G.-X. Tong, F.-F. Du, Y. Liang, Q. Hu, R.-N. Wu, J.-G. Guan and X. Hu, J. Mater. Chem. B, 2013, 1, 454–463 RSC.
- G. Vijayaprasath, R. Murugan, T. Mahalingam and G. Ravi, J. Mater. Sci.: Mater. Electron., 2015, 26, 7205–7213 CrossRef CAS.
- S. Fabbiyola, V. Sailaja, L. J. Kennedy, M. Bououdina and J. J. Vijaya, J. Alloys Compd., 2017, 694, 522–531 CrossRef CAS.
- F. Khan, S.-H. Baek and J. H. Kim, J. Alloys Compd., 2016, 682, 232–237 CrossRef CAS.
- S. M. Shah, H. Naz, R. N. Ali, A. Ali, M. Farooq, A. Shah, A. Badshah, M. Siddiq and A. Waseem, Arabian J. Chem., 2017, 10, 1118–1124 CrossRef CAS.
- H. Lv, D. Sang, H. Li, X. Du, D. Li and G. Zou, Nanoscale Res. Lett., 2010, 5, 620–624 CrossRef CAS PubMed.
- S. Cho, J. Ma, Y. Kim, Y. Sun, G. K. Wong and J. B. Ketterson, Appl. Phys. Lett., 1999, 75, 2761–2763 CrossRef CAS.
- J. I. Pankove, Opt. Process. Semicond., Courier Corporation, 1975 Search PubMed.
- R. S. Sabry, W. J. Aziz and M. I. Rahmah, J. Inorg. Organomet. Polym. Mater., 2020, 30, 4533–4543 CrossRef CAS.
- R. Bylsma, W. Becker, J. Kossut, U. Debska and D. Yoder-Short, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8207 CrossRef CAS PubMed.
- Y. Lee, A. Ramdas and R. Aggarwal, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 10600 CrossRef CAS PubMed.
- B. Baruah, G. J. Gabriel, M. J. Akbashev and M. E. Booher, Langmuir, 2013, 29, 4225–4234 CrossRef CAS PubMed.
- U. Stafford, K. A. Gray, P. V. Kamat and A. Varma, Chem. Phys. Lett., 1993, 205, 55–61 CrossRef CAS.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS.
- J. Ma, X. Guo, Y. Zhang and H. Ge, Chem. Eng. J., 2014, 258, 247–253 CrossRef CAS.
- Y. Yin, Q. Lin, H. Sun, D. Chen, Q. Wu, X. Chen and S. Li, Nanoscale Res. Lett., 2012, 7, 1–8 CrossRef PubMed.
- A. K. Abay, X. Chen and D.-H. Kuo, New J. Chem., 2017, 41, 5628–5638 RSC.
- M. Basu, A. K. Sinha, M. Pradhan, S. Sarkar, A. Pal, C. Mondal and T. Pal, J. Phys. Chem. C, 2012, 116, 25741–25747 CrossRef CAS.
- D. Yang, G. Guo, J. Hu, C. Wang and D. Jiang, J. Mater. Chem., 2008, 18, 350–354 RSC.
- B. Das, M. Sharma, J. C. Sarmah and K. K. Bania, J. Environ. Chem. Eng., 2017, 5, 4212–4219 CrossRef CAS.
- C. Ray, S. Dutta, S. Sarkar, R. Sahoo, A. Roy and T. Pal, RSC Adv., 2013, 3, 24313–24320 RSC.
- D. K. Tiwari, J. Behari and P. Sen, Curr. Sci., 2008, 647–655 CAS.
- W. Song, J. Zhang, J. Guo, J. Zhang, F. Ding, L. Li and Z. J. Sun, Toxicol. Lett., 2010, 199, 389–397 CrossRef CAS PubMed.
- M. Saliani, R. Jalal and E. K. J. Goharshadi, Nanomed. J., 2016, 3, 1–14 CAS.
- K. Raja, P. Ramesh and D. Geetha, Spectrochim. Acta, Part A, 2014, 120, 19–24 CrossRef CAS.
- H. Shivapratap, T. Philip and D. Sharma, Indian J. Seric., 1996, 35, 107–110 Search PubMed.
- H. Yang, Y.-y. Ren, T. Wang and C. Wang, Results Phys., 2016, 6, 299–304 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00717g |
| This journal is © The Royal Society of Chemistry 2022 |