
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalytic conversion of sunlight and carbon dioxide to solar fuels and chemicals
Mandy Ching Man Yau
a,
Martin Hayes
 b and
Shafeer Kalathil
*a
b and
Shafeer Kalathil
*a
aHub for Biotechnology in the Built Environment, Department of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle, NE1 8ST, UK. E-mail: shafeer.kalathil@northumbria.ac.uk
bJohnson Matthey Technology Centre, Cambridge Science Park, Milton Road, Cambridge, CB4 0FP, UK
First published on 6th June 2022
Abstract
This review discusses the progress in the assembly of photosynthetic biohybrid systems using enzymes and microbes as the biocatalysts which are capable of utilising light to reduce carbon dioxide to solar fuels. We begin by outlining natural photosynthesis, an inspired biomachinery to develop artificial photosystems, and the rationale and motivation to advance and introduce biological substrates to create more novel, and efficient, photosystems. The case studies of various approaches to the development of CO2-reducing microbial semi-artificial photosystems are also summarised, showcasing a variety of methods for hybrid microbial photosystems and their potential. Finally, approaches to investigate the relatively ambiguous electron transfer mechanisms in such photosystems are discussed through the presentation of spectroscopic techniques, eventually leading to what this will mean for the future of microbial hybrid photosystems.
 Mandy Ching Man Yau | Mandy Ching Man Yau obtained her MSc in Chemistry from Newcastle University. During her year abroad at Kyushu University, she conducted research on guest responsive luminescence of tetracyano Re(V) complexes under the supervision of Dr Masaaki Ohba. Working alongside Dr Fabio Cucinotta, she completed her thesis on tuning dye loadings in mesoporous silica. She is currently undertaking a PhD at Northumbria University under the supervision of Dr Shafeer Kalathil on integrating light harvesting materials with microbes to generate biofuels from solar energy and waste carbon. |
 Martin Hayes | Martin Hayes is Global Technology Manager for the Life Science Technologies business at Johnson Matthey plc (JM). Martin has worked in a variety of R&D and customer facing technical roles across JM's catalyst businesses over the course of his career. He holds a PhD in heterogeneous catalysis from the University of Reading and recently graduated with an MBA from Durham University. His interests lie in the application of catalytic technologies in sustainable processes for making the high value materials that enhance our quality of life. Martin was admitted as a Fellow of the Royal Society of Chemistry (FRSC) in 2017. |
 Shafeer Kalathil | Shafeer Kalathil received his PhD degree from Yeungnam University (with Prof. Moo Hwan Cho), and his doctoral research focused on electromicrobiology. He was a Japan Society for Promotion of Science (JSPS) Fellow at the University of Tokyo (with Prof. Kazuhito Hashimoto) and a Postdoctoral Fellow at KAUST (with Prof. Pascal Saikaly). He was a Marie Curie Individual Fellow at the University of Cambridge (with Prof. Erwin Reisner) before moving to Northumbria University where he is currently a Vice Chancellor’s Senior Fellow. His laboratory explores electrically active microbes to produce high-value chemicals and fuels from waste carbon. |
Introduction
Initiated by the industrial revolution in the eighteenth century, our world has made major strides in several industries such as transport, research and development, and agriculture. However, coupled with the increase in human population, there is now a greater energy demand, consequently generating the current global warming crisis from rising carbon dioxide (CO2) emissions. Hydrogen (H2) gas, a traditional energy carrier most commonly used in the production of fertilisers and refinement of oil, is a green alternative to reduce carbon emissions.1–3 It has a high energy density and a variety of applications but, along with carbon monoxide (CO), it is normally generated through the combustion of fossil fuels. Moreover, the biofuel is limited to H2 i.e., more complex biofuels such as ethanol and acetate cannot be made. Other current renewable technologies, such as solar and wind power, suffer from low efficiencies due to the fluctuations in the energy source input and this has created a large disparity between the supply and demand.4–6There are several pathways toward the synthesis of green, synthetic fuels to aid in the reduction of carbon emissions. This review aims to explore how biological hybrid systems can be exploited to utilise CO2 reduction to produce solar fuels and chemicals. While Reisner's article discusses similar topics in great detail,7 this updated review gives precedence to microbial hybrid photosystems and in particular, carbon dioxide fixation. This article also places the global importance of hydrogen as a fuel and touches upon the endeavours in the bioengineering, kinetics and proteomic and metabolomic aspect of microbial hybrid photosystems. Moreover, the potential for these microbial photosystems to be implemented on a commercial scale and how these biotechnologies are being supported is discussed.
Traditionally, artificial photosystems have high solar-to-chemical conversion efficiencies compared to natural photosynthetic systems, but they still have several cons – low stability, high costs, and more importantly, they struggle to produce multicarbon products such as acetate.8,9 Microbial photohybrid system, on the other hand, is an emerging green technology and have great promise to overcome those challenges and become a viable alternative to current artificial photosystems.
To first understand how the idea of assimilating biocatalysts with inorganic semiconductors was first conceived, we must look at natural photosynthesis. Photoautotrophs, such as cyanobacteria and plants, utilise photosynthesis where solar energy and CO2 are used to produce biomass. There are two phases to this reaction: light-dependent reaction and dark reaction. In the light-dependent reaction, light energy is used to make the reducing equivalents before proceeding to the dark reaction where they convert CO2 into carbohydrates and other organic molecules via the Calvin cycle (Fig. 1).10 The overall efficiency for the conversion of solar energy to biomass is low due to energy losses occurring throughout the system.11 However, the quantum efficiencies are high.12,13 Therefore, natural photosystems have been used as a blueprint for system design in current artificial and semi-artificial photosystems.
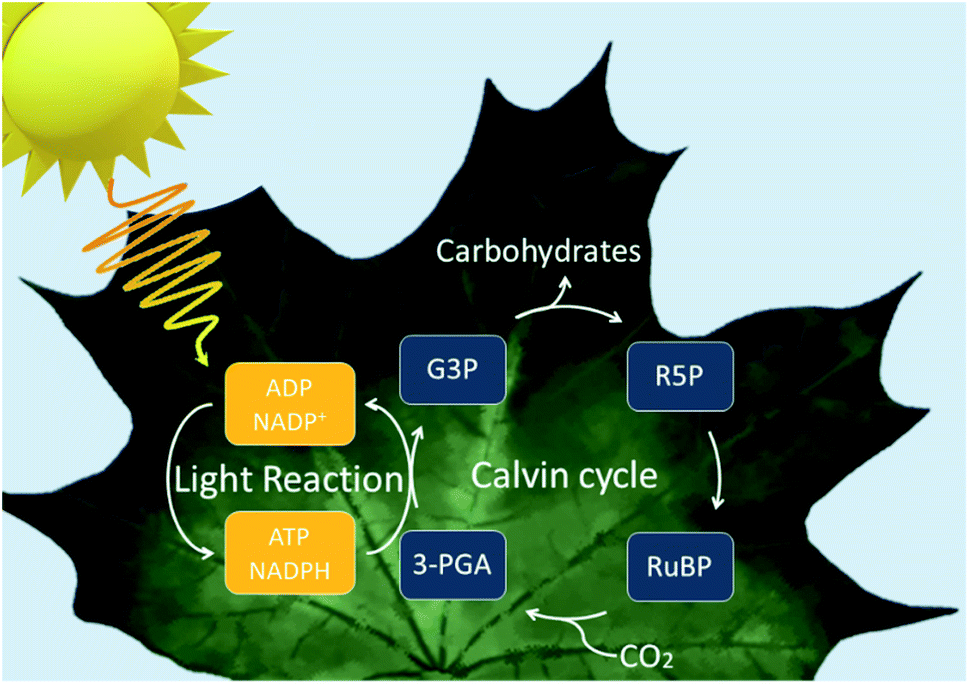 | ||
| Fig. 1 Overview of the reactions within a natural photosynthetic system. Upon light absorption, electrons are excited twice by the photosystems to reduce nicotinamide adenine dinucleotide phosphate (NADP+) to NADPH. Adenosine triphosphate (ATP) is also generated upon water oxidation and electron transport which resulted in a proton gradient which drive ATP synthesis. In the dark reaction, CO2 (and water) are combined with ribulose-1,5-biphosphate (RuBP) to yield 3-phosphoglycerate which is reduced to glyceraldehyde-3-phosphate with ATP and NADPH. | ||
Artificial photosystems
Artificial photosystems have been commonly used to convert solar energy and feedstock chemicals such as CO2 into fuels (Fig. 2). For example, Fujishima and Honda first demonstrated water splitting using solar energy and a photoelectrochemical (PEC) cell consisting of a titanium dioxide (TiO2) electrode.14 Upon exposure to light, if the photon energy is equal to or greater than the bandgap of the material, electron–hole pairs are generated, and they can either recombine to generate heat or take part in photoreactions. Although PEC cells are theoretically able to achieve fairly high solar-to-chemical (STC) efficiencies, in the case of water oxidation, four electrons and four protons are lost to generate a O–O double bond (498.36 ± 0.17 kJ mol−1).15 Thus, solar water splitting can be very energetically demanding with high kinetic and thermodynamic barriers. Nevertheless, to achieve high solar water-splitting efficiency, it is essential to develop a proton reduction catalyst capable of catalysing the production of H2 via the conduction band edge potential of the semiconductor.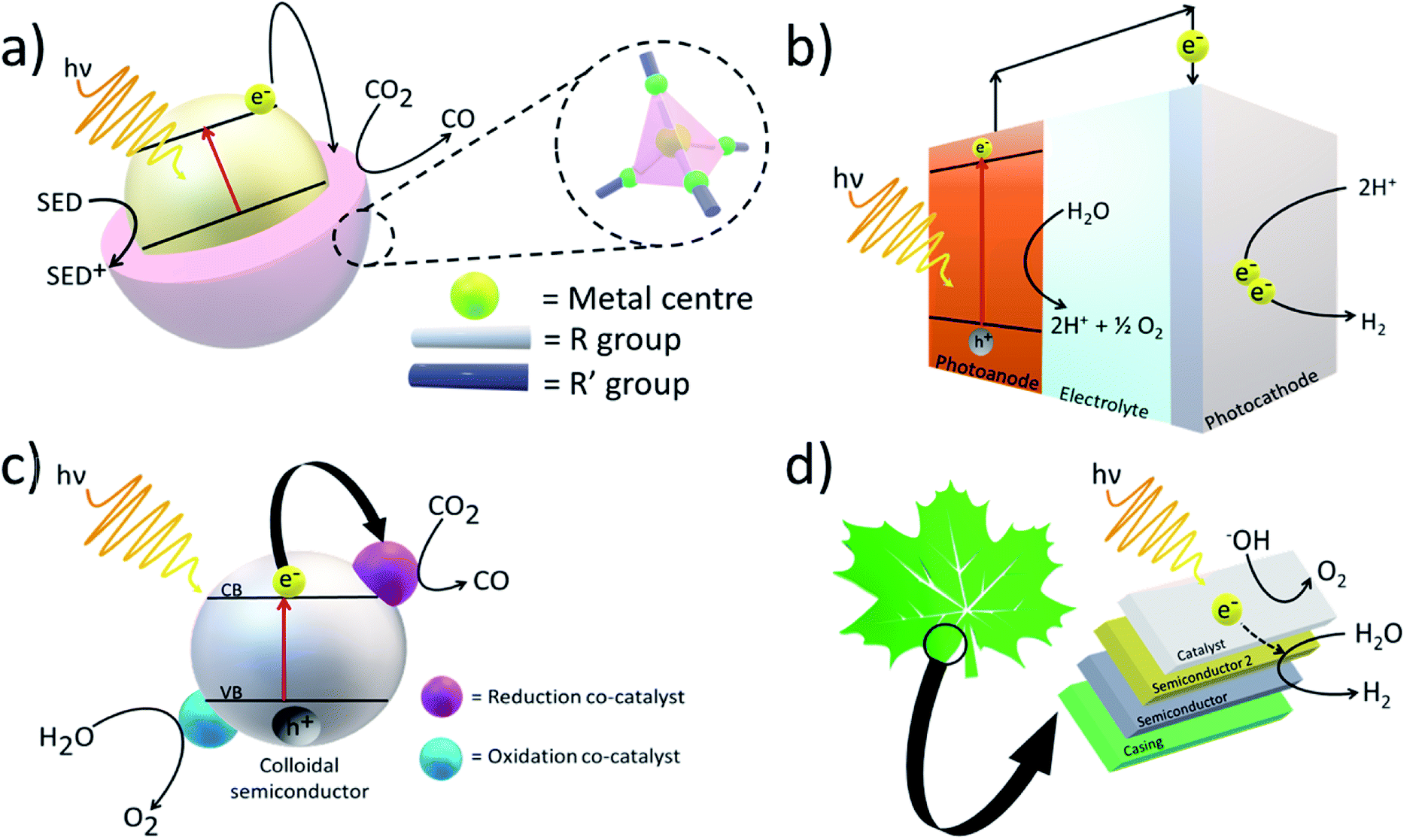 | ||
| Fig. 2 Example artificial photosystems showing (a) a zeolitic imidazolate framework (ZIF) composed of tetrahedrally-coordinated transition metals and imidazolate linkers. ZIFs showing high adsorption and complexation towards CO2 are combined with semiconductors with more appropriate bandgaps to generate electron–hole pairs for the photocatalytic reduction of CO2. (b) a photoelectrochemical cell producing H2 from solar energy (c) a general nanoparticle-based semiconductor for photocatalytic conversion of CO2 to CO (d) artificial leaf composed of a tandem construct to produce H2 from solar energy. | ||
In comparison, CO2 is more thermodynamically stable due to the presence of the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond (532.2 ± 0.4 kJ mol−1).15 Hence, more amount of energy will be required to overcome the activation barrier. Similar to water splitting, upon light absorption, an electron–hole pair is generated and to favourably reduce CO2, the choice of photocatalyst is extremely important. The photocatalyst should contain a suitable band structure where the conduction band edge must be more negative than the redox potential of CO2 (−1.9 V vs. normal hydrogen electrode at pH 7) and the valence band edge more positive than water oxidation.16,17 If not, surplus photon energy will be lost as heat. Currently, there are limited materials with the suitable alignment of the energy band positions. Different strategies to alter the band gap have been employed including doping and surface modifications. Compared to plain bismuth oxide (Bi2O3), α-Bi2O3 doped with selenium had a smaller bandgap and increased photocatalytic activity.18 In a more novel approach, tuning the stoichiometric amount of formic acid added to a dicyandiamide polymerisation reaction led to the tailoring of the O- and N- link chains in the polymer which determined the band position shifts.19
O double bond (532.2 ± 0.4 kJ mol−1).15 Hence, more amount of energy will be required to overcome the activation barrier. Similar to water splitting, upon light absorption, an electron–hole pair is generated and to favourably reduce CO2, the choice of photocatalyst is extremely important. The photocatalyst should contain a suitable band structure where the conduction band edge must be more negative than the redox potential of CO2 (−1.9 V vs. normal hydrogen electrode at pH 7) and the valence band edge more positive than water oxidation.16,17 If not, surplus photon energy will be lost as heat. Currently, there are limited materials with the suitable alignment of the energy band positions. Different strategies to alter the band gap have been employed including doping and surface modifications. Compared to plain bismuth oxide (Bi2O3), α-Bi2O3 doped with selenium had a smaller bandgap and increased photocatalytic activity.18 In a more novel approach, tuning the stoichiometric amount of formic acid added to a dicyandiamide polymerisation reaction led to the tailoring of the O- and N- link chains in the polymer which determined the band position shifts.19
The use of quantum dots (QDs) has also attracted lots of attention due to the quantum confinement effect giving rise to its distinct electronic properties, particularly its size-dependent bandgap.20 H2, CO, and methane (CH4) were all able to be selectively retrieved in the photoreduction of CO2 by tuning the size of the cadmium selenium (CdSe) crystals that had been doped onto nickel oxide.21 This in turn allowed for the control of the electron transfer kinetics. In another example, a zinc/cobalt-based ZIF was used to coat caesium lead bromide (CsPbBr3) QDs to greatly enhance the photocatalytic reduction of CO2 into CO and CH4.22 Compared to bare CsPbBr3, the electron consumption rate of CsPbBr3-ZIF67 and CsPbBr3-ZIF8 was 2.66 and 1.39 greater, respectively. In situ photoluminescence measurements on cobalt-containing ZIF revealed its role in promoting electron to cadmium sulfide (CdS).23 In other studies, the potential of ZIFs in the activation of CO2 has been demonstrated through enhanced activity.24–27
Layered double hydroxides (LDH) have previously been investigated for the use of CO2 reduction catalysts but the reported efficiencies have been low.28,29 By introducing palladium as a co-catalyst, a carbon nitride and LDH assembly proceeded to show an increase in reduction efficiency.30 This was due to the distortion of CO2 upon interaction with the photocatalyst surface, thus allowing for high reactivity of the photocatalyst for CO2 conversion. A similar improvement was also seen in the reduction of CO2 to CH4 through the use of sodium niobate (NaNbO3) nanowires in which higher photocatalytic activity was observed compared to its bulk counterpart.31 However, the reason for the increase in photocatalytic activity in this system is most likely due to the surface-to-volume ratio and crystallinity.
Whilst artificial photosystems have greater solar to fuel efficiency compared to natural photosystems, there are still several drawbacks including high costs, toxicity, relatively low efficiencies, and inability to produce higher fuels – all of which are bottlenecks for commercialisation.6,32–34 Over the last few years, there has been a shift in focus towards the development of biological-based photosystems, i.e., photobiohybrid systems. Their intrinsic metabolic pathways and numerous enzyme cascades enable the efficient conversion of simple feedstock into complex solar fuels and chemicals with greater product selectivity and stability. Photobiohybrid systems can be classified as enzymatic or microbial according to the biocatalyst.
Enzymatic photosystems
In a typical enzymatic photosystem, to which artificial photosystems are inspired, photocatalysts use solar energy to convert and generate photoexcited electrons which are then used to convert reducing equivalents or are directly supplied to the enzyme (Fig. 3). The selective nature of enzymes limits the breadth of CO2 transformations one can carry out. Therefore, although high rates and yields are achieved, sourcing the most suitable enzyme may result in a rise in costs. Redox enzymes play an important role in industrially relevant bioreactions and to carry out their jobs, reducing equivalents are generally required. Typically, nicotinamide adenine dinucleotide (NADH) and NADPH are generated by a secondary enzyme within the system. However, as mentioned above, photocatalysts can also provide reducing equivalents or directly transfer photoelectrons to active redox reactions. Photohybrid systems utilising enzymes have shown success in not only CO2 reduction processes but also nitrogen-fixation and the synthesis of value-added products.35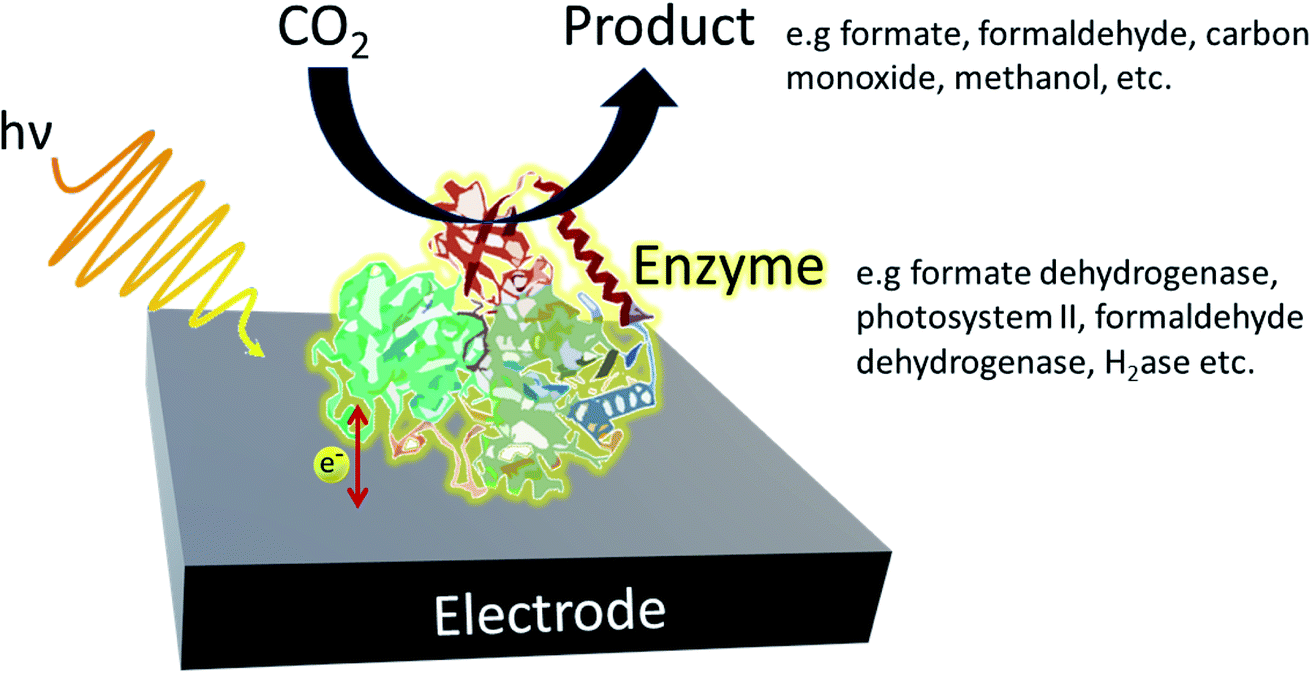 | ||
| Fig. 3 Example of a typical enzymatic photohybrid system in which CO2 is reduced by the enzyme to produce the appropriate biofuel. | ||
Hydrogenases, a type of metalloenzyme, are often used in the study of H2 evolution due to their remarkable catalytic activity. In some cases, turnover frequencies of greater than 1000 s−1 have been reported.36–38 [FeFe]-based hydrogenase was one of the first reported to be able to receive electrons from photocatalysts such as complexes involving rare metals and carbon nanotubes.39,40 Taking [FeFe]-hydrogenase from Clostridium acetobutylicum, Brown et al. integrated the enzyme with cadmium telluride (CdTe) nanocrystals but the random nature of absorption throughout the complex limited the efficiency of electron transfer complexes. This led to limited H2 production.41
A common approach to producing formic acid from CO2 is through the use of formate dehydrogenase (FDH) as a biocatalyst. Using FDH derived from Saccharomyces cerevisiae and zinc tetrakis(4-methylpyridyl)porphyrin as the light harvester, 62 μM formic acid was produced after 3 hours of irradiation.42 This was carried out in the presence of methyl viologen (MV) as the electron mediator. On the other hand, Noji et al. used tri(bipyridine)ruthenium(II) (Ru(bpy)32+) as the photosensitiser to produce formic acid from CO2.43 The use of a porous glass plate increased the conversion efficiencies of Ru(bpy)32+/MV2+/FDH from 1% to 22%. The nanopores within the porous glass plates allowed for the faster accumulation of MV+˙ than in the solution, leading to greater interactions between FDH and MV+˙ due to the higher density. This is also previously seen when the rate of photoreduction was 16 times higher in a nanopore than in solution when photosystem II was employed as the photocatalyst.44
In a hybrid system containing anatase/rutile TiO2 mixture as the semiconductor, a ruthenium bipyridyl photosensitiser, and the enzyme carbon monoxide dehydrogenase, CO2 was reduced to CO.45 However, a SED was required to quench the hole left by the ruthenium complex and to regenerate the photosensitiser. Moreover, the average turnover rate with P25 TiO2 was only 0.14 s−1 at 20 °C which was lower than with a MV radical (100 s−1). Even the addition of the electron donor ethylenediaminetetraacetic acid (EDTA) did not show any noticeable improvement, suggesting inefficient photosensitiser regeneration. A range of surface linkers was also tested to study the effects of enzyme binding to the TiO2 surface with regard to electron transfer efficiency. However, those tested, including polymyxin B sulfate and glutamic acid (which had been previously described to strongly coordinate with TiO2)46 showed very little change in activity and stability. In one case, the addition of o-phosphorylethanol amine even resulted in a lower enzyme uptake and thus, decreased the rate of catalysis.
Microbial photosystems
Enzymatic inorganic hybrid systems appear to overcome the challenges presented by artificial photosystems. However, as described by the above studies, they suffer from several disadvantages including instability issues, poor durability, complicated experimental conditions, and high costs (Table 1). Out of the many carbon fixation pathways known, the Wood–Ljungdahl pathway is a classic for CO2 fixation. In this approach, CO2 is reduced to acetyl-coenzyme A and eventually converted to acetic acid which can be further used to make more complex products.47–51 By utilising the countless metabolic pathways present within the microbe and combining it with the high solar-to-energy efficiencies achieved by artificial semiconductors, products more complex and selective than what enzymes are capable of producing can be generated.| Type of photosystem | Advantages | Disadvantages |
|---|---|---|
| Non-biological | High solar energy conversion efficiencies | Low product selectivity from CO2 reduction |
| Ease of design, tunability, and characterisation | Tends to produce CO only pH gradients result in transfer limitations and may subject the electrodes to corrosion | |
| Broad absorption bands | Internal resistance of the system may require an external bias to overcome | |
| Use of limited raw materials e.g., rare earth metals | ||
| Enzymatic | High rates and yields | Expensive to isolate and purify |
| High selectivity towards CO2-to-chemicals | Sensitive to a variety of environmental conditions (e.g., O2, pH and temperature) | |
| Microbial | Utilises a myriad of enzyme cascades and metabolic pathways to synthesise complex products from simple feedstocks | Prioritises survival over efficiency |
| Resilient to environmental stress | Susceptible to electron losses due to slow kinetics between microbe and electrode | |
| Self-healing capabilities | ROS produced during photocatalysis may deactivate microbial activities | |
| Can be genetically engineered to increase conversion efficiencies, produce more complex chemicals, and improve product selectivity | ||
| Mild reaction conditions |
In the following discussion, we summarise the recent achievements of artificial microbial photohybrid systems utilising solar energy and waste carbon to produce solar fuels and chemicals. Microbial photohybrid systems typically rely on the photosensitisation from organic or inorganic light absorbers to provide the reducing equivalents that allow them to carry out CO2 reduction. Nanoparticles, in particular, are especially attractive and have drawn a lot of interest due to their quantum confinement effect. Essentially, if the size of the particle is comparable to that of the Bohr radius, then the optical and electronic properties become size-dependent.52,53 By altering the valence and conduction bands of the material, photocatalytic activity can be improved and in turn, greater solar-to-fuel efficiencies can be achieved.
Semiconductors are a vital aspect when striving towards an efficient photosystem capable of converting solar energy to biofuels. When discussing semiconductors, it is natural to immediately think about all those made in the laboratory. Decades of science and technology have made probing into light-harvesting capabilities of countless photosystems possible. However, long before we had the technology to investigate these systems, and before man-made semiconductors came about, nature had its inherent semiconductor found, remarkably, in rocks. In rock samples taken from China, Li et al. found that most of the samples had thin iron and manganese oxide coatings.54 Presently, researchers often use Mn- and Fe- oxide in the study of photochemical systems due to their efficiency as photocatalysts and their intrinsic properties. As expected, it was found that photocurrent was only detectable in the Fe- and Mn- region on the coating and, upon exposure to solar energy, generated approximately 2.23 × 1016 photoelectrons. This is very meaningful as those photoelectrons could act as a major energy source to fuel the growth of photoelectronic bacteria which has been reported numerous times.55,56 The study conducted by Li et al. is an important precursor to the research into artificial microbial systems as it reinforces the current research on the stimulation of bacterial growth obtained through phototrophic experiments using semiconductors as photocatalysts.57
A pivotal development in the world of microbial photosystems was made by Sakimoto et al., in the study of Moorella thermoacetica with CdS QDs.47 From Tauc plots, a method of estimating bandgap energies, and absorption spectra, the quantum confinement effect was considered to have come into play when the direct bandgap was reported to be 2.51 ± 0.05 eV, as opposed to the expected lower value of 2.42 eV. The electrons generated by CdS when irradiated under visible light generated H+, a reducing equivalent, which was then used in the Wood–Ljungdahl pathway to produce acetic acid from CO2 with a maximum yield of 90%. Later, TiO2-manganese phthalocyanine (MnPc) was added to form a tandem system and cysteine/cystine (CySS/Cys) was chosen as the redox couple.58 In general, this expanded hybrid system demonstrated greater acetic acid production. The system containing bare TiO2 and M. thermoacetica–CdS had greater acetic acid production than M. thermoacetica–CdS but less than that of TiO2–MnPc. These results demonstrate the superior efficiency of MnPc as a catalyst for CySS reduction. However, a recent study has highlighted pitfalls in the usage of organic chemicals such as cysteine as the SED.59 The study revealed that bacteria can convert cysteine to acetate under dark condition which was largely neglected in previous studies due to poor control experiments. Hence, extreme care is needed in the selection of SED as bacteria can metabolize various substrates. Overall, this milestone, along with many other microbial photobiohybrid systems including the use of Clostridium ljungdahlii with CdS, Escherichia coli with Eosin Y, and Sporomusa ovata with silicon nanowires have led to the innovation of many new and improved microbial hybrid photosystems (Table 2).
| System | Biological substance | Light absorber | Reaction | Production rates/yields/activity | Comments | Reference |
|---|---|---|---|---|---|---|
| Inorganic | — | Magnesium–aluminium–LDH/carbon nitride | CO2 to CH4 | 3.7 μmol in 24 hours | — | 30 |
| — | NaNbO3 nanowire | CO2 to CH4 | 653 ppm h−1 g−1 | — | 31 | |
| NaNbO3 bulk | 32 ppm h−1 g−1 | — | ||||
| — | Cobalt-ZIF-CdS | CO2 to CO + H2 | 124.4 μmol in 3 hours | TEOA = SED, bipyridine = e− transfer assistant, 82% CO selectivity | 23 | |
| — | Lanthanum/rhodium-doped strontium titanium(III) oxide photosheet | CO2 to formate | 0.08% solar to formate conversion efficiency | 97% formate selectivity | 142 | |
| Enzymatic | Carbon monoxide dehydrogenase I | Anatase/rutile TiO2–RuP | CO2 to CO | 0.14 s−1 | 2-(N-Morpholino)ethanesulfonic acid = SED | 45 |
| Formate dehydrogenase | tetrakis(4-methylpyridyl)porphyrin | CO2 to HCOOH | 62 μM in 3 hours | TEOA = SED | 42 | |
| MV2+ = e− mediator | ||||||
| Microbial | E. coli | Silver indium sulfide/indium sulfide | H+ to H2 | 1660 μmol in 3 hours | Cysteine = SED | 68 |
| M. thermoacetica | Gold nanocluster | CO2 to acetate | 6.01 mmol g−1 over 7 days | Cysteine = SED | 67 | |
| S. ovata | Silicon nanowires | CO2 to acetate | 0.3 g L−1 over 7 days | — | 104 | |
| R. palustris | Graphitic carbon nitride | Fructose to PHB | 6.73 g L−1 after 96 hours | TEOA = SED | 69 | |
| R. eutropha | CdS nanorods | CO2 to PHB | 28 mg over 48 hours | 10% lactic acid = SED | 70 | |
| Graphitic carbon nitride catalyse | CO2 to PHB | 41.02 mg L−1 after 48 hours | — | 73 | ||
| Core–shell QDs | PHB | 100 mg g−1 d−1 | L-Ascorbic acid or HEPES = SED | 71 | ||
| 2,3-Butanediol | 10 mg g−1 d−1 | |||||
| Ethylene | 1 mg g−1 d−1 | |||||
| Isopropanol | 2 mg g−1 d−1 | |||||
| R. capsulata | Bi2O3 | H+ to H2 | ∼0.26 mL h−1 | MV2+ = e− mediator | 80 | |
| EDTA = SED | ||||||
| M. barkeri | n+/p-Si/NiMo | CO2 to CH4 | 17.6 mL over 72 hours | H2 = e− donor | 107 | |
| CdS | CO2 to CH4 | 0.19 μmol h−1/13.70 μmol | Cysteine = SED | 64 |
As can be inferred from the examples of microbial photobiohybrid systems listed above, there are two approaches in which microbes can be interfaced with synthetic light absorbers: colloidal and non-colloidal. We begin the discussion below with colloidal microbial photosystems before moving on to non-colloidal systems which involve the use of nanowires and electrodes.
Microbial colloidal photosystems
In a typical colloidal photosystem, the semiconductor is present as a suspension. In some cases, they may be deposited onto the surface of the microbe. Such is the case for the microbial photohybrid system consisting of CdS nanoparticles attached to the surface of Clostridium autoethanogenum.60 Under light exposure, the CdS–C. autoethanogenum system produced almost 4-fold greater acetate than under dark conditions, and a 3-fold increase was observed under CO2-only, confirming the H2-driven autotrophic conversion to acetate. The system used ethanol to enable the solubility of lipoic acid in the culture medium. Later, it was confirmed that C. autoethanogenum oxidised ethanol due to the metabolic shift in response to the change in the redox environment. Ethanol oxidation enabled the hybrid system to obtain NADPH from the oxidised redox environment and this led to greater biomass production. Acetate production and NAD(P)H/NAD(P)+ increased when paired with H2. NAD(P)H/NAD(P)+ play an important role in various biological processes, including biomass and biofuel production and often prevent efficient biotransformation processes. Therefore, by enhancing the production of NAD(P)H/NAD(P)+, increased biomass generation will take place. However, microbial systems often contain various intricate biological networks and physiological systems and can self-organise their metabolic system, such that even with bioengineering, manipulation of the NAD(P)H/NAD(P)+ production pathway will be met with unexpected barriers.61–63 Future research into strategies to increase NADH/NAD+ or NADPH/NAD+ production will be required to understand and optimise conditions for NADH/NAD+ and NADPH/NAD+ production for various biotransformation processes. The CdS–C. autoethanogenum system demonstrates the viability of coupling microbes with semiconductors, and although further improvements are to be made in terms of electron capture for NADH and NADPH production, the system shows great potential for increased CO2-fixing efficiency in nanoparticle-based hybrid photosystems through electron transfer processes.The use of CdS nanoparticles has also been implemented in a hybrid system with M. barkeri which resulted in a CH4 production rate of 0.19 μmol h−1 with 0.34% quantum efficiency.64 This system was further improved when doped with nickel, as well as CdS, which increased the photoelectron transfer within the system and led to an approximate rate of 0.24 μmol h−1 CH4.65 Investigation into the proteomics of the system revealed that the addition of nickel caused a higher expression of proteins related to electron transfer, energy conversion and CO2 fixation which contributed to the increased photoelectron transfer rate. Increasing protein expression is one method to increase electron transfer rates. Another viable method is through pi conjugation. Using the organic semiconductor poly(fluorene-co-phenylene) and perylene diimide derivative to photosensitise M. thermoacetica, the production of acetic acid from CO2 was successful.66 Efficient electron transfer was enabled through the pi conjugation present in the semiconductor which has been suggested to exhibit higher biocompatibility. Moreover, the quantum efficiency of the perylene diimide derivative was found to be comparable to inorganic semiconductors.
The production of acetic acid via CO2 reduction was also realised through the use of gold nanoclusters with M. thermoacetica.67 In a previous study carried out by Jiang et al., the microbe viability before and after the photocatalytic reaction was measured through cell density and the lack of change indicated good viability of the cells.68 In this study, the gold nanoclusters quenched reactive oxygen species (ROS) as well as acted as a light absorber. This enabled M. thermoacetica to ensure high bacterium viability over 6 days of continuous CO2-fixation, showcasing the system's promising potential as a sustainable system.
Semi-artificial microbial systems can also be used to make more complex biofuels. Proof-of-concept was demonstrated in the works of Xu and coworkers where graphitic nitride and R. eutropha H16 were used to increase polyhydroxybutyrate (PHB) production from fructose 1.4-fold with triethanolamine (TEOA) as an SED.69 Further works by the group include the use of CdS nanorods to produce PHB from CO2 and fructose by C. necator.70 Optimisation of CdS nanorods resulted in high photoactivity and achieved a yield of 28 mg over 48 hours PHB from CO2. On the other hand, Ding et al. employed different core–shell QDs to bind to specific enzyme binding sites in bacteria to improve product selectivity in photosynthetic biohybrid systems.71 Through this, not only was gram scale production of PHB achieved but also multi-carbon compounds. The production of PHB successfully increased using only a bioreactor with LED panels, illustrating the ease of scaling up beyond laboratory conditions and implementation on a commercial scale.
Many of the hybrid systems mentioned are limited by anaerobic conditions which may impede the assimilation of microbial hybrid photosystems in industry. The hybrid system presented by Liu et al. consists of CdS nanoparticles biologically precipitated onto the surface of Thiobacillus thioparus and is able to reduce CO2 to multicarbon glutamate synthase without being restricted by the need for an anaerobic environment.72 Interestingly, while chemically synthesised CdS predictably led to cell death in the later stages of cultivation, the biologically synthesised CdS showed a higher promoting effect for the growth of T. thioparus. For the system to sustain cell growth in the presence of biologically synthesised CdS, not only is the electron transfer between the semiconductor-microbe interface efficient but also CO2 fixation. Once CO2 has been fixed by NADPH, the resulting precursors which undergo chemical transformations within T. thioparus become biochemical components that support cell growth and replication. Hence, enabling the hybrid system to undergo self-replication. In not being limited by strict anaerobic conditions and its ability to sustain microbial growth with only simple, economical components, this work shows potential for the reduction of CO2 in terms of design and sustainability.
While enzymes and microbes have individually been interfaced with light absorbers, the hybrid system established by Tremblay et al. involves both R. eutropha and H2O2-degrading catalyse for bioplastic production and water-splitting, respectively.73 Although using the light absorber, graphitic nitride, and R. eutropha alone can successfully produce PHB, the yield was lower than with the addition of H2O2-degrading catalyse. Possible explanations for the increased yield include increased H2 for R. eutropha to maintain its activity and the use of O2 produced from water-splitting as an additional electron acceptor. The increased PHB production resulting from coupling water-splitting reactions and biochemical cascades within R. eutropha showcases a promising platform for efficient PHB production. However, compromises may have to be made with regards to performance and design due to the expense and temperament of enzymes.
H2 is becoming an increasingly popular energy carrier, as evident by the global initiatives to invest in the hydrogen economy, such as the UK's Hydrogen Strategy,74 USA's Hydrogen Program Plan,75 Canada's Hydrogen Strategy,76 Japan's Basic Hydrogen Strategy,77 and the European Clean Hydrogen Alliance which consists of industries and authorities from various countries.78 At present, the production of H2 is still mainly from fossil fuels but in recent years, research into the electrolysis of water from renewable electricity has garnered a lot of attention. The downfall of this method, however, is that rare and precious metals are used in the synthesis of the catalysts involved, meaning an increase in production costs and limited sustainability and scale-up potential.
As previously stated, current inorganic systems involving photovoltaic technology and semiconductors have surpassed natural biotransformation efficiencies. However, reaching similar conversion efficiencies is still a challenge yet to be achieved. Clostridium butyricum and Escherichia coli, along with semiconductors, were initially used to explore photocatalytic H2 production.79–84 To this day, the pool of microorganisms used to explore H2 production has expanded to include Shewanella oneidensis and more.85–89 In the works of Martins and coworkers, Desulfovibrio desulfuricans, Citrobacter freundii, and S. oneidensis were used in conjunction with CdS nanoparticles to investigate H2 production.84 D. desulfuricans was found to be the most efficient, owing to the high biological activity and/or efficient electron transfer. This was accompanied by the fact that it showed the highest hydrogenase activity compared to S. oneidensis, E. coli, and C. freundii (280 μmol gdcw−1 min−1, 8.4 μmol gdcw−1 min−1, 3.7 μmol gdcw−1 min−1, and 1.6 μmol gdcw−1 min−1 respectively). High levels of the enzyme hydrogenase correlate with greater H2 production as they catalyse H2 formation from protons or the oxidation to protons. The high levels of hydrogenase in D. desulfuricans were found to be from the [NiFe] and [FeFe] families. These enzymes tend to be located in the periplasm of the microorganism,90,91 leading to a more efficient transfer of electrons from the nanoparticle to the enzyme than those found in the intracellular compartment. Cyanide was used to inhibit the hydrogenases within D. desulfuricans, almost completely preventing H2 formation. This ultimately highlights the importance of hydrogenase in the pursuit of efficient H2 formation. H2 evolution was also possible through the use of a tandem involving silver indium sulfide/indium sulfide and E. coli.68 In this system, the quantum efficiency of 3.3% was achieved and showed very little change in cell viability after the photocatalytic reaction, demonstrating the stability of the hybrid system.
A major bottleneck of microbial hybrid photosystems is the photoelectron transfer from the inorganic component of the system to the biological cell. In a study to increase H2 production, reduced graphene oxide was chosen to be integrated with S. oneidensis MR-1.92 The reduced graphene oxide acted as a solid support surface and was able to efficiently collect electrons for the semiconductor, cuprous oxide (Cu2O). In addition, the reduced graphene oxide operated as a pathway for electrons to travel between. All the above allowed for efficient electron transfer from the Cu2O nanoparticles to S. oneidensis MR-1, thus contributing to efficient H2 production, as evident in the increased photocatalytic H2 production when compared to pristine Cu2O and Cu2O coupled with reduced graphene oxide (322.0 μmol gCu2O−1 vs. 7.0 μmol gCu2O−1 and 4.0 μmol gCu2O−1 within 4 hours respectively).
It is important to elucidate the electron transfer pathway to understand the mechanism behind H2 evolution within a hybrid system. Upon deletion of H2ase, it was clear that within S. oneidensis, H2ase was the active site for H2 evolution as there was no H2 production.92 Deletion of the proteins MtrC/OmcA was found to have inhibited the production of H2, thus establishing the fact that the cytochromes, including MtrA and MtrB, are involved in the electron transfer between the nanoparticle and S. oneidensis. While the work by Shen and coworkers concerns H2 production, the understanding of the electron transfer pathway in the photocatalytic conversion extends to the production of all biofuels, not just H2. In resolving and understanding the electron transfer pathway within a system, the overall design of hybrid systems can then be further improved, yielding greater electron transfer efficiency and ultimately, greater yields.
The kinetics of the electron transfer is extremely important in the conversion of solar energy to solar fuels. The previously mentioned work by Martin and coworkers on D. sulfuricans involved hydrogenases found in the periplasm of the microbe. Similarly, S. oneidensis also expresses periplasmic hydrogenases.93 This was exploited by Shi and coworkers, in which copper indium sulfide/zinc sulfide (CuInS2/ZnS) QDs were translocated into S. oneidensis, resulting in an increase in H2 production due to the decrease in electron transfer distance and energy loss.94 Through the deletion of certain genes, and the introduction of MV as a redox mediator, direct electron transfer from the QDs to S. oneidensis was inferred. As expected, single and double deletion of the periplasmic hydrogenase, [FeFe] and [NiFe], caused a decrease in the production of H2, which suggests the two coenzymes play a crucial role in H2 production. Deletion of the electron transport proteins had little to no effect on the production of H2, nor did the introduction of MV as a redox mediator.
Manipulation of the genetic material within the microbe is becoming an increasingly popular method of increasing the production of biofuels. Using genetically engineered E. coli with precipitated CdS, [NiFe]-hydrogenase HyaABCDEF plasmid was transformed into the bacteria to catalyse H2 formation.95 Compared to non-HydA-induced hybrids, the resulting output per cell was much greater. In the case involving CdS and E. coli, genetic engineering was used to emphasise the expression of [FeFe]-hydrogenase gene, consequently yielding a photoactive E. coli hybrid system capable of converting solar energy into H2 within a single cell.96 Although the yield was less than previously reported studies involving TiO2 and E. coli,97 the genetically engineered hybrid is a viable method for enhancing photocatalytic H2 production.
Microbial non-colloidal photosystems
Along with colloidal systems, microbes have also been interfaced with nanowires and PEC systems. Due to the large surface area, nanowires are extraordinary light harvesters. Initial studies on mammalian cells have demonstrated the ability to recognise and respond to nanoscale topographies.98–100 In response to this, a silicon nanowire array was interfaced with S. oneidensis MR-1.101 Through this model, it was discovered that not only attachment locations were influenced by the topography, but also the swimming patterns of the microbes. In a previous study conducted by Goto et al., the tendency of single flagellated bacteria to travel in a straight trajectory was revealed.102 However, S. oneidensis was shown to have circled the nanowire despite being a singly flagellated bacterium. Moreover, the reversible bacterial attachment was demonstrated after some of the bacteria were found to separate from the Si array following a short period. The swimming behaviour exhibited by S. oneidensis is also observed by E. coli K-12 strains.103 By highlighting the important role of topography and its interaction with bacteria, knowledge that will serve to aid in the design of future microbial-nanowire interfaces was realised.As expected from the above study by Jeong et al., the use of a nanowire array proved to be more advantageous than that of a single nanowire.101 The use of arrays has been widely implemented in several research studies. Liu et al. cultured Sporomusa ovata with a silicon nanowire array passivated by TiO2 protection layer, leading to the production of acetic acid from CO2 after the incubation period.57 Using dual light-absorbers allow the system to fully absorb solar energy, thus increasing photovoltage for the system. This is demonstrated by the high faradaic efficiency (90%). This system was taken further and extended by allowing genetically engineered E. coli to activate acetate into acetyl-CoA, ultimately partaking in the biosynthesis of various complex molecules such as cadinene, PHB, and n-butanol. The study is especially important as the system itself generated a local anaerobic environment at the bottom of the nanowire array, encouraging the reduction of CO2 under aerobic (21% O2) conditions. Su et al. later used highly doped p+ silicon nanowire arrays and platinum wire as the counter electrode to replace the TiO2 photoanode previously used.104 By adjusting the electrolyte pH and increasing the buffering capacity, the group successfully achieved the formation of closed-pack silicon nanowires. The resulting close-packed biohybrid system obtained a solar-to-acetate efficiency of 3.8% and a current density of 0.65 ± 0.11 mA cm−2, as opposed to the energy conversion of 0.38% for acetic acid production and 0.35 mA cm−2 photocurrent by Liu and colleagues.57
Using methanogens in a PEC cell setup, a one-step transformation from CO2 to CH4 with a faradaic efficiency of up to 96% with only solar energy as the only energy input was achieved.105 Solar-to-fuel conversion efficiency was found to be at 0.1%, approximately half of that of natural photosynthesis. The greatest limitation in the system is expected to be from the bandgap of the TiO2 photoanode employed. TiO2 is often used in designing photosystems, but its downfall is that it can only absorb a small range of wavelengths of solar energy. Thus, in expanding the absorption range of materials used in photosystems, the efficiency will likely increase. A combination of TiO2/CdS was also used as a photoanode in the reduction of CO2 by methanogens.106 Fig. 4 illustrates the system constructed by Xiao and coworkers which demonstrated 94.4% faradaic efficiency and solar-to-fuel conversion efficiency of 1.28%.106 Cu2ZnSn4 was later employed to sensitise the photoanode. Due to the lower charge separation resistance, the photogenerated electron–hole separation was much more favoured and this led to greater CH4 production. Suspending microorganisms in solution is also a viable method to catalyse redox reactions fuelled by solar energy. By interfacing the archaea Methanosarcina barkeri with a platinum electrode, Nichols et al. achieved a faradaic efficiency of up to 86% for electrochemical CO2 to CH4 conversion.107 Replacing the platinum electrode with synthetic α-nickel sulfide H2 evolution reaction catalyst resulted in similar faradaic efficiencies and CH4 yields. Furthermore, by using an indium phosphide photocathode and a TiO2 photoanode, the system proceeded to result in unassisted light-driven CH4 production from CO2 and this system yielded much greater CH4 than that with the platinum electrode and α-NiS catalyst.107 This proof-of-concept of bridging semiconductors with microbes proves efficient solar conversion to carbon-based fuels is attainable.
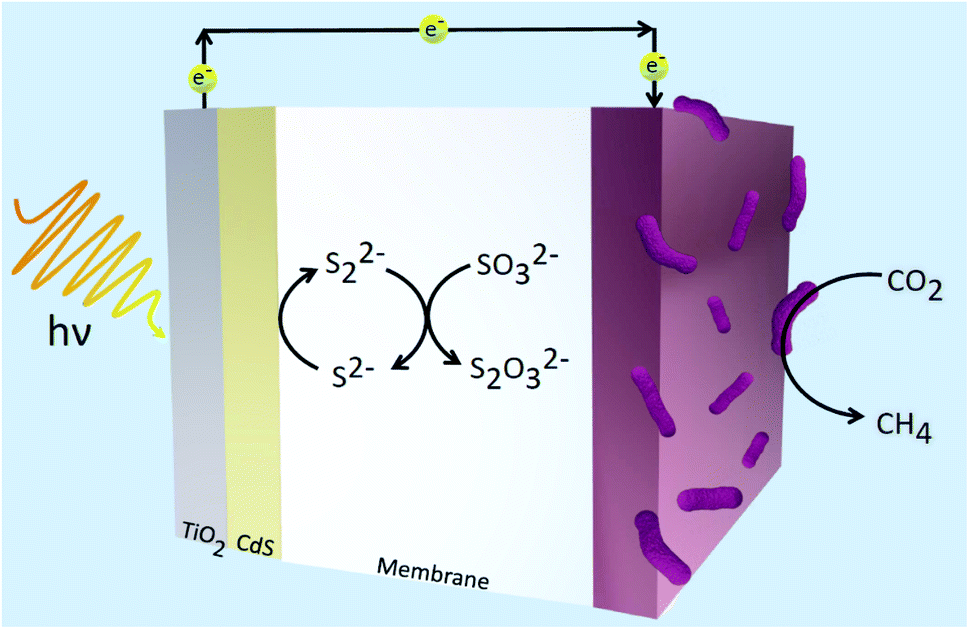 | ||
| Fig. 4 Hybrid microbial photosystem designed by Xiao et al. consisting of a TiO2/CdS photoanode and a biocathode for CO2 reduction to CH4.106 The sulfide electrolyte solution was used as sacrificial reagents to consume holes and prevent the corrosion of CdS. | ||
Elucidation of electron transfer mechanisms and microbial life in microbial photosystems
Although microbial hybrid systems have proven to be a promising approach to producing biofuels, achieving yields that can compete with current energy outputs from fossil fuels are still yet to be realised. Fig. 5 displays the general mechanisms taking place within a microbial hybrid photosystem but because of the knowledge gaps behind the mechanics of the hybrid system, the finer details are still relatively unknown. Some challenges involve the seamless integration of synthetic and biological products and increasing the efficiencies of light absorption and solar-to-biofuel conversion. To work towards a system that can be applied to the industry, it is important to investigate the electron transfer mechanisms between the inorganic substrate and the biological component as they are the major bottleneck in the system. Therefore, at present, many techniques have been employed to probe into such interactions within the hybrid system to allow us to gain a deeper understanding.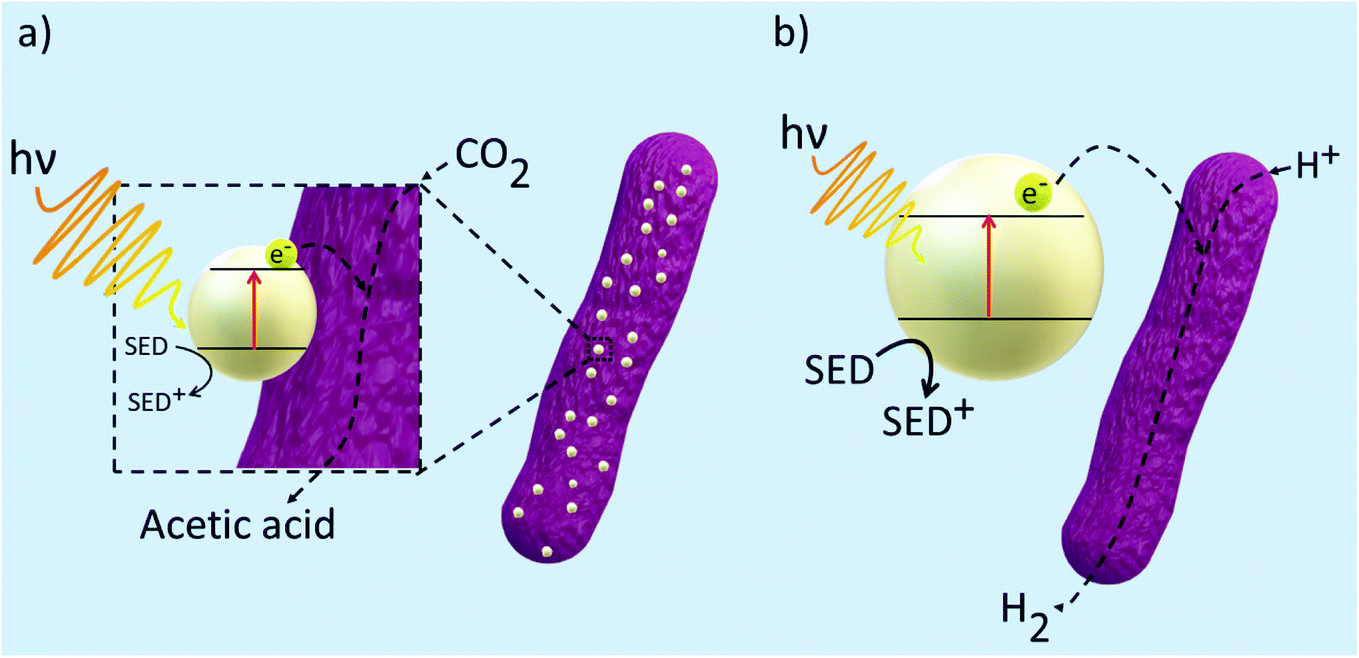 | ||
| Fig. 5 General mechanism for electron transfer between a semiconductor and microbe in a hybrid photosystem. (a) Direct photoreduction of CO2 to acetic acid in which the semiconductor is deposited onto the surface of the microbe (b) indirect photoreduction of CO2 to H2 by colloidal nanoparticles in the presence of electron mediators and SEDs. | ||
Attenuated total reflection infrared (ATR-IR) (Fig. 6a) has been used to probe into the attachment of microbes onto inorganic surfaces in several previous works.108–111 ATR-IR spectroscopy is an attractive method for being not only suitable for solid samples but also for biological liquid samples. In the works of Freitag and colleagues, an acoustic trap was combined with ATR-IR spectroscopy to enable the entrapment of enzyme-labelled beads without the need for mechanical retention components, such as optical fibres, which are normally found in typical bead injection systems.112 The set-up allowed for the conversion of p-nitrophenylphosphate into p-nitrophenol and phosphate under varying loadings of enzymes to be monitored, showcasing its great potential for enzymatic kinetic studies.
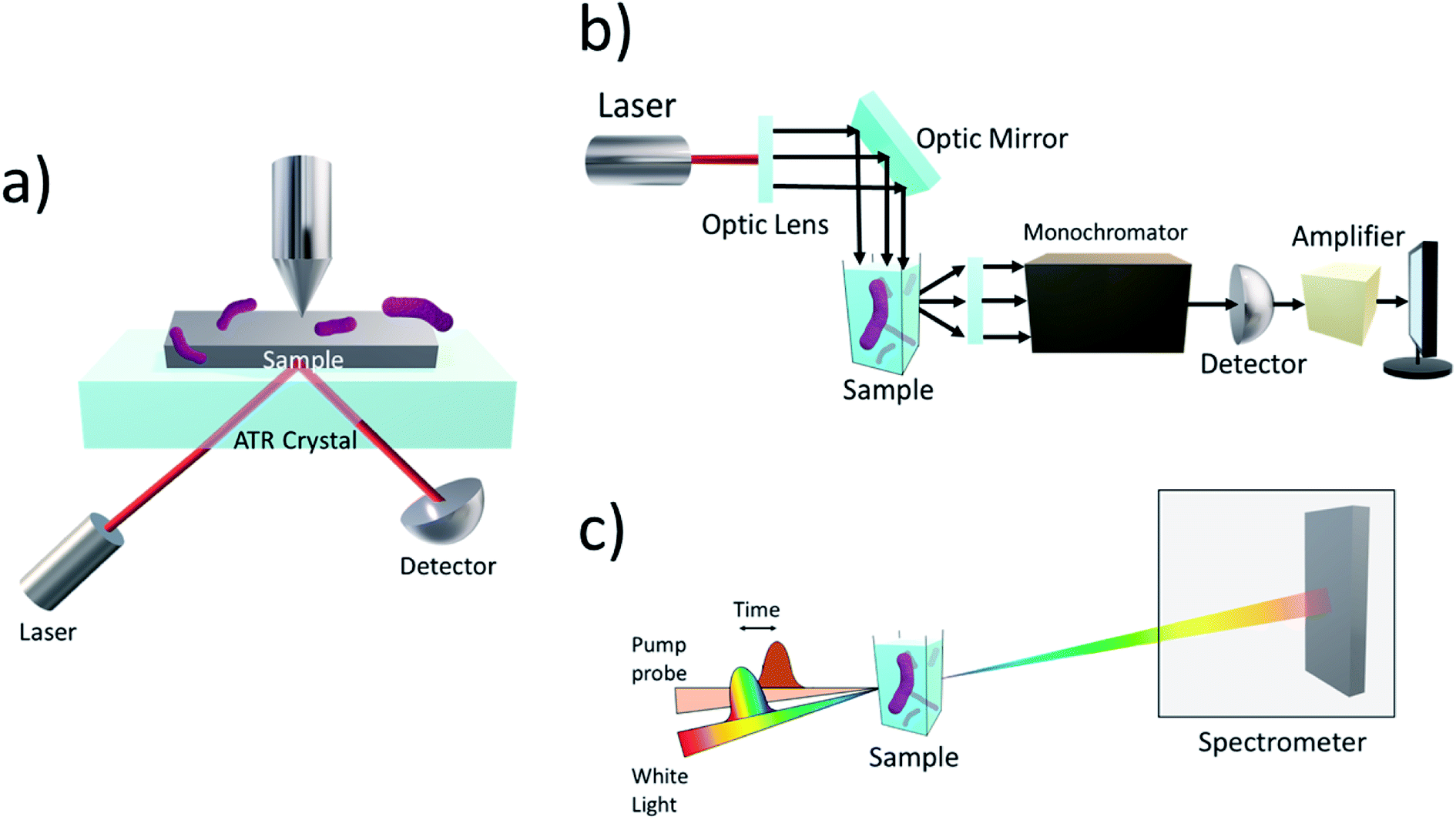 | ||
| Fig. 6 (a) Schematics of ATR. The infrared beam interacts with the biofilm sample at one point of reflection (or at multiple points with multi-bounce ATR). Some of the evanescent waves formed from total internal reflection interact with the sample, resulting in an attenuated total reflection. Reactions can be monitored in real-time and information about kinetics and key transient reaction intermediates can be obtained, aiding in the study of kinetics and mechanism of the system. (b) Experimental setup of photoluminescence spectroscopy. The laser is directed to the sample using optical lenses. Once the laser hits the sample, the electrons are excited to higher energy states and as they relax, radiation is emitted which is detected by the photoluminescence spectrometer at specific wavelengths which reflect the energy level differences. The decay kinetics of the photoluminescent curve obtain will give information about the processes taking place in the hybrid photosystem. (c) In transient absorption spectroscopy, a pump pulse is used to excite the sample containing microbes interfaced with nanoparticles. Changes in the optical absorption of the sample are measured as a function of time and thus, the sample's absorbance is measured before and after pumping. The resulting information regarding the system's decay and lifetimes will shed light onto the kinetics of the system. | ||
Steady-state photoluminescence (Fig. 6b) on the previously discussed work by Shen et al. was used to investigate the separation and transfer of photogenerated charge carriers in the system.92 In comparison with the absence of the Cu2O nanoparticle, the steady-state photoluminescence intensity of Cu2O/reduced graphene oxide decreased. This indicates that the separation of the photogenerated electron–hole pair was sufficient for a pathway between the Cu2O nanoparticle and S. oneidensis cells for electron transfer. Similar observations were made with CuInS2/ZnS QDs interfaced with genetically engineered S. oneidensis.94
A more commonly used technique is time-resolved infrared spectroscopy (TRIR). TRIR is often used to study the kinetics, formation of intermediate products, and intramolecular charge transport of both biological and chemical molecular species.113–115 For example, TRIR spectroscopy, coupled with transient absorption (TA) spectroscopy (Fig. 6c), was used in an M. thermoacetica–CdS hybrid system to explain the decreasing CO2 to acetic acid conversion rates with increasing H2 incubation in the first three hours of photosynthesis.116 Changes in the vibrational modes of CO and carbon nitride, along with other amino acid residues, were observed and due to the peaks decaying on the same timescale as the TA signal, it was speculated the picosecond e− transfer was from a molecular process, not from a purely physical source.
In the same model, TA was used to aid in the elucidation of the decay kinetics of M. thermoacetica–CdS. It was reported that M. thermoacetica–CdS incubated with H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 displayed faster decay kinetics compared to that incubated with glucose.47,116 The faster decay kinetics was attributed to the increased H2ase activity and hence, the greater e− transfer rates and/or the greater quantity of e− acceptors in the H2 incubated system. Unsurprisingly, multi-exponential decay in the timescale of picoseconds was found to be expressed in the hybrid system. Likewise, in the previously mentioned works of Luo et al., when CuInS2/ZnS QDs were integrated with S. oneidensis MR-1 through co-incubation, transient fluorescence and transient absorption lifetimes were shorter than that with CuInS2/ZnS than with the hybrid system, indicating enzymes are efficient acceptors of photogenerated charge.94 Decay kinetics in the same timescale as the M. thermoacetica–CdS hybrid was reported in a hybrid metal–semiconductor nanoparticle system consisting of CdSe–Au.117 However, lifetimes reported in the range of nanoseconds were found in Fe–Fe H2ase–CdS systems,118 possibly owing to hydrogen bonding, solvent effects, and charge transfer barriers. From the evidence gathered via TA and TRIR spectroscopy, two proposed pathways for charge and energy transfer were proposed: the non-H2ase-mediated pathway in glucose incubated cells and the membrane-bound H2ase mediated pathways in H2 incubated cells. Overall, the existence of a charge-transfer pathway was supported with TA and TRIR spectroscopy.
:CO2 displayed faster decay kinetics compared to that incubated with glucose.47,116 The faster decay kinetics was attributed to the increased H2ase activity and hence, the greater e− transfer rates and/or the greater quantity of e− acceptors in the H2 incubated system. Unsurprisingly, multi-exponential decay in the timescale of picoseconds was found to be expressed in the hybrid system. Likewise, in the previously mentioned works of Luo et al., when CuInS2/ZnS QDs were integrated with S. oneidensis MR-1 through co-incubation, transient fluorescence and transient absorption lifetimes were shorter than that with CuInS2/ZnS than with the hybrid system, indicating enzymes are efficient acceptors of photogenerated charge.94 Decay kinetics in the same timescale as the M. thermoacetica–CdS hybrid was reported in a hybrid metal–semiconductor nanoparticle system consisting of CdSe–Au.117 However, lifetimes reported in the range of nanoseconds were found in Fe–Fe H2ase–CdS systems,118 possibly owing to hydrogen bonding, solvent effects, and charge transfer barriers. From the evidence gathered via TA and TRIR spectroscopy, two proposed pathways for charge and energy transfer were proposed: the non-H2ase-mediated pathway in glucose incubated cells and the membrane-bound H2ase mediated pathways in H2 incubated cells. Overall, the existence of a charge-transfer pathway was supported with TA and TRIR spectroscopy.
Due to the extremely fast nature of the charge separation and transfer, (from milliseconds to picoseconds), TA is also capable of providing information on parameters such as triplet excitons, exciton energy, and decay kinetics which can be used to speculate charge transfer processes.119–121 The previously mentioned system consisting of cadmium selenium–gold (CdSe–Au) had undergone photoexcitation of the local surface plasmon resonance of the metal. By using ultrafast spectroscopy, along with high temporal resolution and near-infrared excitation, the plasmon-induced charge transfer (PICT) pathway was resolved, and it was discovered that hot-electron transfer to CdSe occurred in the timescale of fewer than 30 femtoseconds, whereas back transfer from the semiconductor to the metal was found to be within 210 fs.117 To compete with the extremely fast back-transfer to the metal, extraction of the electrons needs to be greater than the rate of back-transfer. This issue needs to be realised when designing hybrid systems to avoid inefficient and undesired transfer pathways. With suitable band alignments, such issues may be mitigated.
The kinetic study of this hybrid construct using high time resolution TA, along with the aforementioned studies, has highlighted the importance of spectroscopic techniques in the pursuit of elucidating electron transfer kinetics in photoactive systems. In establishing the requirements for resolving ultrafast photophysics of hybrid systems, there will be a greater understanding of the kinetic pathways within a system, resulting in a more intelligent design of hybrid systems which will allow for more diverse photocatalytic applications.
The involvement of proteomic and metabolomic analysis in the study of hybrid systems has recently become more prominent. Whilst the above-mentioned spectroscopic techniques will help clarify kinetics and electron transfer processes, proteomics and metabolomics will enable a greater understanding of the biocatalytic mechanisms, energy conservation in microbes, and physiological state of the organism. Taking the model M. thermoacetica–CdS hybrid system developed by Sakimoto and coworkers, Zhang et al. studied the energy conservation processes within the system using untargeted and targeted protein and metabolite quantification processes.122 Through this approach, it was discovered that the energy-metabolism-associated glycolysis and tricarboxylic acid cycle were active alongside the well-known Wood–Ljungdahl pathway. M. thermoacetica was also implied to contain an increased expression of proteins capable of protecting M. thermoacetica from oxidative stress but the damage far outweighed the protection provided.
Although the current biohybrid photosystems are still in their infancy, with the study presented by Zhang et al.,122 the aforementioned spectroscopic techniques, and more importantly, with technological advancements, the means to fabricate highly efficient, low cost, and industrially competitive biohybrid photosystems will become more tangible in the nearby future.
Conclusion
To conclude, while artificial photosystems and enzymatic photosystems have many advantages, artificial microbial photosystems present themselves as a much more powerful tool for converting sunlight and CO2 to biofuels. However, there are still significant developments to be made. The commercialisation of hybrid photosystems continuously proves to be a challenge and several considerations including, but not limited to, cost, toxicity, environmental impact, and the transition from laboratory to industrial scale still need to be addressed further.With emerging technologies able to elucidate the electron transfer mechanisms in microbial hybrid photosystems, our understanding of the interactions occurring within the system will deepen, allowing for maximisation of solar-to-chemical yields, more complex solar fuel production, and smarter system design which will pave the way towards a revolutionised CO2-utilisation technology sector. This will be further aided by the advancement of the synthetic biology sector, allowing for a new class of responsive, functionalised, and customisable hybrid systems to drive the future of green energy.
Future perspectives
The microbial conversion of CO2 to fuels is promising but the variants of CO2-fixation pathways all have different kinetic and ATP requirements. For example, the Calvin cycle is deemed inefficient due to its slow kinetics and high demand for ATP. For this reason, it is essential to design and develop pathways that can support greater carbon fixation. An example of this would be the pyruvate synthase-pyruvate carboxylate-glyoxylate pathway.123 Genome-resolved metagenomics has also been used to identify potential enzymes that will show greater efficiency once integrated into CO2-fixation pathways. Using metagenomics, Figueroa et al. identified genes responsible for phosphite oxidation and CO2 reduction within Candidatus Phosphitivorax anaerolimi strain Phox-21.124 It was discovered that while Ca. P anaerolimi contained the genes necessary for Wood–Ljungdahl pathway, the genes for other carbon fixation pathways were absent. Moreover, CO2 reduction via the Wood–Ljungdahl pathway was not feasible for Phox-21 as certain genes for particular carbonyl steps were missing. In light of these discoveries, Figeuroa et al. proposed an approach as to how Phox-21 could reduce CO2 to formate before proceeding to formate assimilation pathways. With the rapid development of analytical and biological techniques, carbon fixation may be enhanced to the standards of practical applications.Expanding the library of microbes may also lead to unfamiliar reaction pathways that can be exploited to give greater STC efficiencies and more energy-dense solar fuels. The metabolic engineering of Clostridium autoethanogenum has already led to its large-scale industrial application for the production of ethanol from syngas.125 Microorganisms lacking CO2-fixing pathways, such as E. coli and S. cerevisiae, may be transplanted into heterotrophs like S. oneidensis and G. sulfurreducens. Pyrococcus furiosus is also another host candidate and attempts have been made to incorporate CO2-fixation pathways into the archaea.126,127 The use of thermophile such as P. furiosus in hybrid microbial photosystems will be highly beneficial as solar transformations of CO2 to volatile biofuels at higher temperatures will become more feasible.
Whilst the study and discovery of new carbon fixation pathways within the microbe is essential for more meaningful CO2 transformation, investigations into the energy modules should not be disregarded. As described at the beginning of the review, a lot of energy is required to convert CO2 to biofuels due to its high stability. To circumvent this, the study of energy modules has been undertaken regarding semiartificial microbial photosystems. Owing to stoichiometric modelling, and computational analyses we now have a comparative analysis of autotrophic growth and production based on different natural and synthetic CO2 fixation pathways and energy uptake.128,129 Advances in technology have now allowed for the multi-omics study of not only ATP production, but also electron transfer from semiconductor to microbe and CO2 reduction which was carried out by Zhang et al.122
Understanding the electron transfer between the semiconductor and microbe will also be essential for the future development of hybrid photosystems. Optimisation of the charge transfer processes through genetic engineering can take place, ensuring superior interaction between the biological and semiconductor interface. This in turn will theoretically give us greater STC efficiencies as well as a deeper understanding of how more complex biofuels can be generated.
Metal–organic frameworks (MOF) are becoming an emerging contender for light harvesting. MOFs are composed of transition metal ions that are connected to organic linkers and have crystalline, porous, extended structures. They are structurally tuneable, allowing for the assembly of higher surface areas for the capture of solar energy and its self-assembly into crystalline structures enables the MOF to be computationally analysed.130,131 MOFs have shown their capability through successfully harvesting water from desert air with only ambient sunlight as the energy input.132,133 For the solar conversion of CO2 to biofuels, when interfaced with anaerobic microbes, MOFs have the potential to offer protection from oxygen and ROS, ultimately prolonging the lifetime of the bacteria and ultimately allowing for continuous biofuel production.134 In using a zirconium nanocluster-based monolayer to wrap around M. thermoacetica, CO2 transformation to acetate was able to last approximately 2.5 days whereas, without the MOF protection, CO2 fixation was only able to last 1 day.135 This was owed to the zirconium nanoclusters present within the MOF as it decomposed the hydrogen peroxide formed on the cell membrane, preventing ROS accumulation. Hydrogels have also been used to shield photosensitised M. thermoacetica, extending the viability of the bacterium and increasing acetate production.136
Despite the benefits of microbial hybrid photosystems, commercialisation of large-scale production is still in the distant future. The major factors to consider are cost and efficiency, not to mention more research into novel artificial semiconductors that will bolster STC is needed. Hence, fossil fuels are still the most widely used source of fuel production.137 However, initiatives such as the Paris Agreement and investments into green energy will help further the advancement of microbial biohybrid photosystems, especially as world leaders initiate the phasing out of fossil fuel industries. Like hybrid photosystems, next-generation materials consisting of smart, functional properties are being heavily invested in. In particular, synthetic biological materials are attracting attention for their potential in the construction of smart living due to their environmental responsiveness, adaptive abilities, self-healing capabilities, and self-regeneration, just to name a few. Several successful engineering efforts have taken place to mimic these biological materials on a large scale. Buildings like the OME, an experimental biological house in the North-East of England which aims to test new, experimental biotechnologies for sustainable living138 will help bridge the transition from laboratory to industry, and eventually, to real, everyday life. By monitoring the biological materials' response to the environment, the integration of microbial photosystems into these ‘smart’ properties will become more feasible, alleviating the population's dependency on fossil fuels.
In the 2000s, the invention of the genetic toggle switch first implemented within the plasmid of E. coli stimulated the synthetic biology sector and has now reached the fields of material science and engineering.139 Rather than relying on bioengineering, the genetic toggle switch is controlled via manipulation of the network architecture. Demonstrating that the principles of engineering may be exploited to revolutionize simple cells into living machines, manipulation of the network architecture will enable the control of cell function and maximise STC efficiency. Moreover, by interfacing with inorganic materials such as gold nanoparticles and QDs, numerous environmentally responsive and controllable functional composite materials were developed. As previously mentioned, analysis of these environmental responses and adaption of the cellular functions will be important if the integration of microbial photosystems in buildings is to take place.
In recent years, the ability to transform waste carbon into biomass has become more and more important. Studies in this area include the works of Gleizer et al. who reported the engineering of E. coli to produce biomass from only CO2.140 To do this, the glycolysis and pentose-phosphate pathways, were disrupted whilst key enzymes in the Calvin cycle were expressed. A vital step in this study was the introduction of NAD-dependent FDH which would later be used to generate NADH to serve as the reducing power to drive carbon fixation. The strain was left to optimise for CO2 fixation by allowing it to adapt and evolve for over 350 days. Genomic sequence analysis confirmed the presence of mutations that regulate gene expression and pathway regulations, overall achieving 100% biomass origin from CO2 fixation. Similarly, Gassler and coworkers rewrote the xylulose monophosphate cycle to introduce the Calvin cycle.141 Methanol assimilating genes within the peroxisomes of Pichia pastoris were deleted and the energy and reducing power were supplied by methanol oxidation. These modifications resulted in the fungi utilising CO2 as its sole carbon source, increasing its maximum growth rate on CO2 more than two-fold. These studies highlight the importance of not only carbon fixation pathways but also the energy and reducing power on cell growth.
The works of Gleizer and co-workers have set an important milestone towards future works in the sustainable production of biofuels from CO2 only, especially in allowing for a more in-depth study of how synthetic CO2-fixation pathways can be assimilated into non-native hosts, as well as opening the likelihood of increasing the repertoire of microbes able to undergo solar reduction of CO2 where new metabolic pathways may be discovered. The accomplishments of Gleizer and more have stimulated the interdisciplinary field of synthetic biology and the various research currently in motion have demonstrated the great potential of smart materials in the application of sustainable production of biofuels in photohybrid systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Research England (E3) Grant.References
- A. H. Awad and T. N. Veziroǧlu, Int. J. Hydrogen Energy, 1984, 9, 355–366 CrossRef
.
- M. Reese, C. Marquart, M. Malmali, K. Wagner, E. Buchanan, A. McCormick and E. L. Cussler, Ind. Eng. Chem. Res., 2016, 55, 3742–3750 CrossRef CAS
- M. Balat, Int. J. Hydrogen Energy, 2008, 33, 4013–4029 CrossRef CAS
- P. Li, IEEE Nanotechnol. Mag., 2008, 2, 13–18 Search PubMed
- H. Lund, Energy, 2007, 32, 912–919 CrossRef
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed
- X. Fang, S. Kalathil and E. Reisner, Chem. Soc. Rev., 2020, 49, 4926–4952 RSC
- H. Lu, Z. Wang and L. Wang, ACS ES&T Engg, 2021 DOI:10.1021/acsestengg.1c00336
- C. Du, X. Wang, W. Chen, S. Feng, J. Wen and Y. A. Wu, Mater. Today Adv., 2020, 6, 100071 CrossRef
- J. Barber and B. Andersson, Nature, 1994, 370, 31–34 CrossRef CAS
- J. R. Bolton and D. O. Hall, Photochem. Photobiol., 1991, 53, 545–548 CrossRef CAS
- A. Melis, Plant Sci., 2009, 177, 272–280 CrossRef CAS
- J. U. Grobbelaar, J. Appl. Phycol., 2009, 21, 519–522 CrossRef
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 1 edn, 2007 Search PubMed
- A. A. Khan and M. Tahir, J. CO2 Util., 2019, 29, 205–239 CrossRef CAS
- X. Jiao, K. Zheng, Z. Hu, Y. Sun and Y. Xie, ACS Cent. Sci., 2020, 6, 653–660 CrossRef CAS PubMed
- R. Sharma, M. Khanuja, S. N. Sharma and O. P. Sinha, Int. J. Hydrogen Energy, 2017, 42, 20638–20648 CrossRef CAS
- Y. Wang, F. Silveri, M. K. Bayazit, Q. Ruan, Y. Li, J. Xie, C. R. A. Catlow and J. Tang, Adv. Energy Mater., 2018, 8, 1801084 CrossRef
- S. Baskoutas and A. F. Terzis, J. Appl. Phys., 2006, 99, 013708 CrossRef
- H. Cho, W. D. Kim, K. Lee, S. Lee, G.-S. Kang, H.-I. Joh and D. C. Lee, Appl. Surf. Sci., 2018, 429, 2–8 CrossRef CAS
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS
- S. Wang and X. Wang, Appl. Catal., B, 2015, 162, 494–500 CrossRef CAS
- Y. Guo, H. Yang, X. Zhou, K. Liu, C. Zhang, Z. Zhou, C. Wang and W. Lin, J. Mater. Chem. A, 2017, 5, 24867–24873 RSC
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS
- S. Dou, J. Song, S. Xi, Y. Du, J. Wang, Z.-F. Huang, Z. J. Xu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 CrossRef CAS PubMed
- T. Jose, Y. Hwang, D.-W. Kim, M.-I. Kim and D.-W. Park, Catal. Today, 2015, 245, 61–67 CrossRef CAS
- N. Ahmed, Y. Shibata, T. Taniguchi and Y. Izumi, J. Catal., 2011, 279, 123–135 CrossRef CAS
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed. Engl., 2012, 51, 8008–8011 CrossRef CAS PubMed
- J. Hong, W. Zhang, Y. Wang, T. Zhou and R. Xu, ChemCatChem, 2014, 6, 2315–2321 CrossRef CAS
- H. Shi, T. Wang, J. Chen, C. Zhu, J. Ye and Z. Zou, Catal. Lett., 2011, 141, 525–530 CrossRef CAS
- M. A. Quader and S. Ahmed, in Clean Energy for Sustainable Development, ed. M. G. Rasul, A. k. Azad and S. C. Sharma, Academic Press, 2017, pp. 91–140, DOI:10.1016/B978-0-12-805423-9.00004-1
- M. Sandroni, R. Gueret, K. D. Wegner, P. Reiss, J. Fortage, D. Aldakov and M. N. Collomb, Energy Environ. Sci., 2018, 11, 1752–1761 RSC
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Interface Focus, 2015, 5, 20150009 CrossRef PubMed
- S. Sultana, P. Chandra Sahoo, S. Martha and K. Parida, RSC Adv., 2016, 6, 44170–44194 RSC
- M. W. Ratzloff, M. B. Wilker, D. W. Mulder, C. E. Lubner, H. Hamby, K. A. Brown, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2017, 139, 12879–12882 CrossRef CAS PubMed
- H. Land, P. Ceccaldi, L. S. Mészáros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2019, 10, 9941–9948 RSC
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed
- W. Wang, T. Yu, Y. Zeng, J. Chen, G. Yang and Y. Li, Photochem. Photobiol. Sci., 2014, 13, 1590–1597 CrossRef CAS PubMed
- D. Svedruzic Chang, T. McDonald, Y.-H. Kim, J. Blackburn, M. Heben and P. King, Interaction of [FeFe]-hydrogenases with single-walled carbon nanotubes, SPIE, 2007 Search PubMed
- K. A. Brown, S. Dayal, X. Ai, G. Rumbles and P. W. King, J. Am. Chem. Soc., 2010, 132, 9672–9680 CrossRef CAS PubMed
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931–1934 CrossRef CAS
- T. Noji, T. Jin, M. Nango, N. Kamiya and Y. Amao, ACS Appl. Mater. Interfaces, 2017, 9, 3260–3265 CrossRef CAS
- T. Noji, K. Kawakami, J.-R. Shen, T. Dewa, M. Nango, N. Kamiya, S. Itoh and T. Jin, Langmuir, 2016, 32, 7796–7805 CrossRef CAS PubMed
- T. W. Woolerton, S. Sheard, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Energy Environ. Sci., 2011, 4, 2393–2399 RSC
- A. D. Roddick-Lanzilotta and A. J. McQuillan, J. Colloid Interface Sci., 2000, 227, 48–54 CrossRef CAS PubMed
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS PubMed
- K. K. Sakimoto, Biochemist, 2017, 39, 30–33 CrossRef CAS
- K. K. Sakimoto, N. Kornienko, S. Cestellos-Blanco, J. Lim, C. Liu and P. Yang, J. Am. Chem. Soc., 2018, 140, 1978–1985 CrossRef CAS PubMed
- G. Borrel, P. S. Adam and S. Gribaldo, Genome Biol. Evol., 2016, 8, 1706–1711 CrossRef CAS PubMed
- S. W. Ragsdale and E. Pierce, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1873–1898 CrossRef CAS PubMed
- M. A. El-Sayed, Acc. Chem. Res., 2004, 37, 326–333 CrossRef CAS PubMed
- J. Jasieniak, M. Califano and S. E. Watkins, ACS Nano, 2011, 5, 5888–5902 CrossRef CAS PubMed
- A. Lu, Y. Li, H. Ding, X. Xu, Y. Li, G. Ren, J. Liang, Y. Liu, H. Hong, N. Chen, S. Chu, F. Liu, Y. Li, H. Wang, C. Ding, C. Wang, Y. Lai, J. Liu, J. Dick, K. Liu and M. F. Hochella, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9741 CrossRef CAS PubMed
- A. Lu, Y. Li, S. Jin, X. Wang, X.-L. Wu, C. Zeng, Y. Li, H. Ding, R. Hao, M. Lv, C. Wang, Y. Tang and H. Dong, Nat. Commun., 2012, 3, 768 CrossRef PubMed
- A. Lu, Y. Li, X. Wang, H. Ding, C. Zeng, X. Yang, R. Hao, C. Wang and M. Santosh, Precambrian Res., 2013, 231, 401–408 CrossRef CAS
- C. Liu, J. J. Gallagher, K. K. Sakimoto, E. M. Nichols, C. J. Chang, M. C. Y. Chang and P. Yang, Nano Lett., 2015, 15, 3634–3639 CrossRef CAS PubMed
- K. K. Sakimoto, S. J. Zhang and P. Yang, Nano Lett., 2016, 16, 5883–5887 CrossRef CAS PubMed
- L. Göbbels, A. Poehlein, A. Dumnitch, R. Egelkamp, C. Kröger, J. Haerdter, T. Hackl, A. Feld, H. Weller, R. Daniel, W. R. Streit and M. C. Schoelmerich, Sci. Rep., 2021, 11, 2139 CrossRef PubMed
- S. Jin, Y. Jeon, S. Jeon Min, J. Shin, Y. Song, S. Kang, J. Bae, S. Cho, J.-K. Lee, R. Kim Dong and B.-K. Cho, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020552118 CrossRef CAS PubMed
- A. Kondo, J. Ishii, K. Y. Hara, T. Hasunuma and F. Matsuda, J. Biotechnol., 2013, 163, 204–216 CrossRef CAS PubMed
- S. Aslan, E. Noor and A. Bar-Even, Biochem. J., 2017, 474, 3935–3950 CrossRef CAS PubMed
- S. Stephens, R. Mahadevan and D. G. Allen, Front. Bioeng. Biotechnol., 2021, 8 DOI:10.3389/fbioe.2020.610723
- J. Ye, J. Yu, Y. Zhang, M. Chen, X. Liu, S. Zhou and Z. He, Appl. Catal., B, 2019, 257, 117916 CrossRef CAS
- J. Ye, G. Ren, L. Kang, Y. Zhang, X. Liu, S. Zhou and Z. He, iScience, 2020, 23, 101287 CrossRef CAS PubMed
- P. Gai, W. Yu, H. Zhao, R. Qi, F. Li, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 7224–7229 CrossRef CAS PubMed
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed
- Z. Jiang, B. Wang, J. C. Yu, J. Wang, T. An, H. Zhao, H. Li, S. Yuan and P. K. Wong, Nano Energy, 2018, 46, 234–240 CrossRef CAS
- M. Xu, P.-L. Tremblay, L. Jiang and T. Zhang, Green Chem., 2019, 21, 2392–2400 RSC
- M. Xu, P.-L. Tremblay, R. Ding, J. Xiao, J. Wang, Y. Kang and T. Zhang, Sci. Total Environ., 2021, 753, 142050 CrossRef CAS PubMed
- Y. Ding, J. R. Bertram, C. Eckert, R. R. Bommareddy, R. Patel, A. Conradie, S. Bryan and P. Nagpal, J. Am. Chem. Soc., 2019, 141, 10272–10282 CrossRef CAS PubMed
- G. Liu, F. Gao, H. Zhang, L. Wang, C. Gao and Y. Xiong, Nano Res., 2021 DOI:10.1007/s12274-021-3883-0
- P.-L. Tremblay, M. Xu, Y. Chen and T. Zhang, iScience, 2020, 23, 100784 CrossRef CAS PubMed
- UK hydrogen strategy, https://www.gov.uk/government/publications/uk-hydrogen-strategy/uk-hydrogen-strategy-accessible-html-version, accessed 28/03/22 Search PubMed.
- S. McQueen, J. Stanford, S. Satyapal, E. Miller, N. Stetson, D. Papageorgopoulos, N. Rustagi, V. Arjona, J. Adams, K. Randolph, D. Peterson, L. Hill, M. Koleva, T. Reinhardt, E. Frye, R. Schrecengost, A. Kokkinos, J. Litynski, R. Conrad, G. Soloveichik, D. Tew, S. Litzelman, J. Vetrano, R. Onuschak, A. Hahn, E. Hsieh and R. Costa, Department of Energy Hydrogen Program Plan, United States, 2020 Search PubMed
- Canada's Hydrogen Strategy, https://nrcan.gc.ca/climate-change-adapting-impacts-and-reducing-emissions/canadas-green-future/the-hydrogen-strategy/23080#shr-pg0, accessed 28/03/22 Search PubMed.
- Basic Hydrogen Strategy, https://www.meti.go.jp/english/press/2017/<?pdb_no 1226?>1226<?pdb END?>_003.html, accessed 28/03/22 Search PubMed.
- European Clean Hydrogen Alliance Members, https://ec.europa.eu/docsroom/documents/48994, accessed 28/03/22 Search PubMed.
- A. A. N. Krasnovskii, V. V. Nikandrov and S. A. Nikiforova, Dokl. Akad. Nauk SSSR, 1985, 285, 6 Search PubMed
- P. Maruthamuthu, S. Muthu, K. Gurunathan, M. Ashokkumar and M. V. C. Sastri, Int. J. Hydrogen Energy, 1992, 17, 863–866 CrossRef CAS
- B. Wang, C. Zeng, K. H. Chu, D. Wu, H. Y. Yip, L. Ye and P. K. Wong, Adv. Energy Mater., 2017, 7, 1700611 CrossRef
- K. Xiao, T. H. Tsang, D. Sun, J. Liang, H. Zhao, Z. Jiang, B. Wang, J. C. Yu and P. K. Wong, Adv. Energy Mater., 2021, 11, 2100291 CrossRef CAS
- Y. Honda, H. Hagiwara, S. Ida and T. Ishihara, Angew. Chem., Int. Ed., 2016, 55, 8045–8048 CrossRef CAS PubMed
- M. Martins, C. Toste and I. A. C. Pereira, Angew. Chem., Int. Ed., 2021, 60, 9055–9062 CrossRef CAS PubMed
- F. Grein, A. R. Ramos, S. S. Venceslau and I. A. C. Pereira, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 145–160 CrossRef CAS PubMed
- P.-H. Hsieh, Y.-C. Lai, K.-Y. Chen and C.-H. Hung, Int. J. Hydrogen Energy, 2016, 41, 21685–21691 CrossRef CAS
- H. Koku, İ. Eroğlu, U. Gündüz, M. Yücel and L. Türker, Int. J. Hydrogen Energy, 2002, 27, 1315–1329 CrossRef CAS
- S. Mohanraj, K. Anbalagan, P. Rajaguru and V. Pugalenthi, Int. J. Hydrogen Energy, 2016, 41, 10639–10645 CrossRef CAS
- A. Selvaggi, C. Tosi, U. Barberini, E. Franchi, F. Rodriguez and P. Pedroni, J. Photochem. Photobiol., A, 1999, 125, 107–112 CrossRef CAS
- J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. W. Adams, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1350–1369 CrossRef CAS PubMed
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed
- H. Shen, Y.-Z. Wang, G. Liu, L. Li, R. Xia, B. Luo, J. Wang, D. Suo, W. Shi and Y.-C. Yong, ACS Catal., 2020, 10, 13290–13295 CrossRef CAS
- G. Meshulam-Simon, S. Behrens, A. D. Choo and A. M. Spormann, Appl. Environ. Microbiol., 2007, 73, 1153–1165 CrossRef CAS PubMed
- B. Luo, Y.-Z. Wang, D. Li, H. Shen, L.-X. Xu, Z. Fang, Z. Xia, J. Ren, W. Shi and Y.-C. Yong, Adv. Energy Mater., 2021, 11, 2100256 CrossRef CAS
- W. Wei, P. Sun, Z. Li, K. Song, W. Su, B. Wang, Y. Liu and J. Zhao, Sci. Adv., 2018, 4, eaap9253 CrossRef PubMed
- Y. Honda, Y. Shinohara, M. Watanabe, T. Ishihara and H. Fujii, ChemBioChem, 2020, 21, 3389–3397 CrossRef CAS PubMed
- Y. Honda, M. Watanabe, H. Hagiwara, S. Ida and T. Ishihara, Appl. Catal., B, 2017, 210, 400–406 CrossRef CAS
- R. Yan, J.-H. Park, Y. Choi, C.-J. Heo, S.-M. Yang, L. P. Lee and P. Yang, Nat. Nanotechnol., 2012, 7, 191–196 CrossRef CAS PubMed
- W. Kim, J. K. Ng, M. E. Kunitake, B. R. Conklin and P. Yang, J. Am. Chem. Soc., 2007, 129, 7228–7229 CrossRef CAS PubMed
- K. Chung, J. A. DeQuach and K. L. Christman, Nano LIFE, 2010, 1, 63–77 CrossRef CAS PubMed
- H. E. Jeong, I. Kim, P. Karam, H.-J. Choi and P. Yang, Nano Lett., 2013, 13, 2864–2869 CrossRef CAS
- T. Goto, K. Nakata, K. Baba, M. Nishimura and Y. Magariyama, Biophys. J., 2005, 89, 3771–3779 CrossRef CAS PubMed
- Y. Kinosita, T. Ishida, M. Yoshida, R. Ito, Y. V. Morimoto, K. Goto, R. M. Berry, T. Nishizaka and Y. Sowa, Sci. Rep., 2020, 10, 15887 CrossRef CAS PubMed
- Y. Su, S. Cestellos-Blanco, J. M. Kim, Y.-x. Shen, Q. Kong, D. Lu, C. Liu, H. Zhang, Y. Cao and P. Yang, Joule, 2020, 4, 800–811 CrossRef CAS
- Q. Fu, S. Xiao, Z. Li, Y. Li, H. Kobayashi, J. Li, Y. Yang, Q. Liao, X. Zhu, X. He, D. Ye, L. Zhang and M. Zhong, Nano Energy, 2018, 53, 232–239 CrossRef CAS
- S. Xiao, Z. Li, Q. Fu, Y. Li, J. Li, L. Zhang, Q. Liao and X. Zhu, Chem. Eng. J., 2020, 390, 124530 CrossRef CAS
- E. M. Nichols, J. J. Gallagher, C. Liu, Y. Su, J. Resasco, Y. Yu, Y. Sun, P. Yang, M. C. Y. Chang and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11461 CrossRef CAS
- S. J. Parikh, F. N. D. Mukome and X. Zhang, Colloids Surf., B, 2014, 119, 38–46 CrossRef CAS PubMed
- M. J. McWhirter, A. J. McQuillan and P. J. Bremer, Colloids Surf., B, 2002, 26, 365–372 CrossRef CAS
- S.-Y. Kang, P. J. Bremer, K.-W. Kim and A. J. McQuillan, Langmuir, 2006, 22, 286–291 CrossRef CAS PubMed
- M. J. McWhirter, P. J. Bremer, I. L. Lamont and A. J. McQuillan, Langmuir, 2003, 19, 3575–3577 CrossRef CAS
- S. Freitag, B. Baumgartner, S. Tauber, C. Gasser, S. Radel, A. Schwaighofer and B. Lendl, Anal. Chem., 2019, 91, 7672–7678 CrossRef CAS PubMed
- S. Maiti, G. C. Walker, B. R. Cowen, R. Pippenger, C. C. Moser, P. L. Dutton and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10360 CrossRef CAS PubMed
- K. Wilke and H. D. Breuer, J. Photochem. Photobiol., A, 1999, 121, 49–53 CrossRef CAS
- A. Mezzetti and W. Leibl, Photosynth. Res., 2017, 131, 121–144 CrossRef CAS PubMed
- N. Kornienko, K. K. Sakimoto, D. M. Herlihy, S. C. Nguyen, A. P. Alivisatos, C. B. Harris, A. Schwartzberg and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11750 CrossRef CAS PubMed
- F. V. A. Camargo, Y. Ben-Shahar, T. Nagahara, Y. E. Panfil, M. Russo, U. Banin and G. Cerullo, Nano Lett., 2021, 21, 1461–1468 CrossRef CAS PubMed
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316–4324 CrossRef CAS PubMed
- H. Satzger and W. Zinth, Chem. Phys., 2003, 295, 287–295 CrossRef CAS
- S. A. Haque, S. Handa, K. Peter, E. Palomares, M. Thelakkat and J. R. Durrant, Angew. Chem., Int. Ed., 2005, 44, 5740–5744 CrossRef CAS PubMed
- B. S. Veldkamp, W.-S. Han, S. M. Dyar, S. W. Eaton, M. A. Ratner and M. R. Wasielewski, Energy Environ. Sci., 2013, 6, 1917–1928 RSC
- R. Zhang, Y. He, J. Yi, L. Zhang, C. Shen, S. Liu, L. Liu, B. Liu and L. Qiao, Chem, 2020, 6, 234–249 CAS
- A. Bar-Even, E. Noor, N. E. Lewis and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8889–8894 CrossRef CAS PubMed
- I. A. Figueroa, T. P. Barnum, P. Y. Somasekhar, C. I. Carlström, A. L. Engelbrektson and J. D. Coates, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E92–E101 CrossRef CAS PubMed
- P. Dürre and B. J. Eikmanns, Curr. Opin. Biotechnol., 2015, 35, 63–72 CrossRef PubMed
- H. Atomi, T. Sato and T. Kanai, Curr. Opin. Biotechnol., 2011, 22, 618–626 CrossRef CAS PubMed
- M. W. Keller, G. J. Schut, G. L. Lipscomb, A. L. Menon, I. J. Iwuchukwu, T. T. Leuko, M. P. Thorgersen, W. J. Nixon, A. S. Hawkins, R. M. Kelly and M. W. W. Adams, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5840–5845 CrossRef CAS PubMed
- A. G. Fast and E. T. Papoutsakis, Curr. Opin. Chem. Eng., 2012, 1, 380–395 CrossRef
- N. R. Boyle and J. A. Morgan, Metab. Eng., 2011, 13, 150–158 CrossRef CAS PubMed
- V. Bernales, M. A. Ortuño, D. G. Truhlar, C. J. Cramer and L. Gagliardi, ACS Cent. Sci., 2018, 4, 5–19 CrossRef CAS PubMed
- C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2012, 4, 83–89 CrossRef CAS PubMed
- N. Hanikel, M. S. Prévot, F. Fathieh, E. A. Kapustin, H. Lyu, H. Wang, N. J. Diercks, T. G. Glover and O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1699–1706 CrossRef CAS PubMed
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed
- Z. Ji, H. Zhang, H. Liu, O. M. Yaghi and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10582 CrossRef CAS PubMed
- Z. Ji, H. Zhang, H. Liu, O. M. Yaghi and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10582–10587 CrossRef CAS
- S. Cestellos-Blanco, H. Zhang and P. Yang, Faraday Discuss., 2019, 215, 54–65 RSC
- F. Perera, Int. J. Environ. Res. Public Health, 2017, 15, 16 CrossRef
- Hub for Biotechnology in the Built Environment, https://bbe.ac.uk/, accessed 30/03/22 Search PubMed.
- T. S. Gardner, C. R. Cantor and J. J. Collins, Nature, 2000, 403, 339–342 CrossRef CAS
- S. Gleizer, R. Ben-Nissan, Y. M. Bar-On, N. Antonovsky, E. Noor, Y. Zohar, G. Jona, E. Krieger, M. Shamshoum, A. Bar-Even and R. Milo, Cell, 2019, 179, 1255–1263 CrossRef CAS PubMed
- T. Gassler, M. Sauer, B. Gasser, M. Egermeier, C. Troyer, T. Causon, S. Hann, D. Mattanovich and M. G. Steiger, Nat. Biotechnol., 2020, 38, 210–216 CrossRef CAS PubMed
- Q. Wang, J. Warnan, S. Rodríguez-Jiménez, J. J. Leung, S. Kalathil, V. Andrei, K. Domen and E. Reisner, Nat. Energy, 2020, 5, 703–710 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2022 |