
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical aspects of coinage metal nanoparticles for catalysis and spectroscopy
Deblina Roy†
a,
Anjali Pal
b and
Tarasankar Pal
 *c
*c
aDepartment of Chemistry, National Institute of Technology, Rourkela, Odisha, India
bDepartment of Civil Engineering, Indian Institute of Technology Kharagpur, Kharagpur 721302, India
cDepartment of Chemical Sciences, University of Johannesburg, P. O. Box 524, Auckland Park 2006, Kingsway Campus, South Africa. E-mail: tpal@chem.iitkgp.ac.in
First published on 21st April 2022
Abstract
Down scaling bulk materials can cause colloidal systems to evolve into microscopically dispersed insoluble particles. Herein, we describe the interesting applications of coinage metal nanoparticles (MNPs) as colloid dispersions especially gold and silver. The rich plasmon bands of gold and silver in the visible range are elaborated using the plasmon resonance and redox potential values of grown metal microelectrode (GME). The gradation of their standard reduction potential values (E0), as evaluated from the Gibbs free energy change for bulk metal, is ascribed to the variation in their size. Also, the effect of nucleophiles in the electrolytic cell with metal nanoparticles (MNPs) is described. The nucleophile-guided reduction potential value is considered, which is applicable even for bulk noble metals. Typically, a low value (as low as E0 = +0.40 V) causes the oxidation of metals at the O2 (air)/H2O interface. Under this condition, the oxidation of noble metal particles and dissolution of the noble metal in water are demonstrated. Thus, metal dissolution as a function of the size of metal nanoparticles becomes eventful and demonstrable with the addition of a surfactant to the solution. Interestingly, the reversal of the nobility of gold (Au) and silver (Ag) microelectrodes at the water/electrode interface is confirmed from the evolution of normal and inverted ‘core–shell’ structures, exploiting visible spectrophotometry and surface-enhanced Raman scattering (SERS) analysis. Subsequently, the effect of the size, shape, and facet- and support-selective catalysis of gold nanoparticles (NPs) and the effect of incident photons on current conversion without an applied potential are briefly discussed. Finally, the synergistic effect of the emissive behaviour of gold and silver clusters is productively exploited.
 Deblina Roy | Currently, Deblina Roy, MSc, is working on her PhD Degree at the Institute of Analytical and Environmental Sciences, National Tsing Hua University, Hsinchu 30013, Taiwan. She obtained her UG and PG Degrees in Chemistry from the National Institute of Technology (NIT), Rourkela, Odisha, India. Deblina has worked on “Binary Nano Metal Chalcogenides and their Heterostructures with Metals” and completed two internship programs from the Indian Institute of Technology (IIT), Kharagpur, West Bengal, India. During her internship tenures, she completed two projects: (1) liquid phase integration of metals for alloy formation and (2) determination of ion-pair stability out of solid phase reaction and subsequent solvent extraction methods. |
 Anjali Pal | Dr Anjali Pal, MSc, PhD is a chemistry graduate from the University of Calcutta and at present is a Professor in the Department of Civil Engineering, Indian Institute of Technology (IIT) Kharagpur. Dr Pal is actively engaged in teaching and research in the field of environmental engineering and science. She has published more than 200 research papers and supervised 12 students for PhD programme. Her field of research involves speciation of arsenic, modified Fenton and photo-Fenton reactions, photocatalysis, catalysis with metal nanoparticles, adsolubilization processes, and spectroscopy. Wastewater remediation by new adsorption technology for ‘zero waste’ technology is now her priority for field application. Dr Pal has received The Convention Award from the Indian Chemical Society, International Hall of Fame award and R & D-100 Award from USA. She has visited many countries as a Visiting Professor. |
 Tarasankar Pal | Professor Tarasankar Pal, MSc, PhD, DSc, FNASc, FWAST an alumnus of the University of Burdwan, worked in the Department of Chemistry, Indian Institute of Technology (IIT) Kharagpur from 1984 till 2017. He started nanoparticle research way back in 1983 and has published 340 research papers. His ‘arsenic detection kit’ for arsenic affects rural people and ‘bench mark model reaction for nitrophenol reduction’ to test any metal nanoparticle catalyst deserves special mention. His research interest includes the synthesis and applications of metal and semiconductor nanoparticles for catalytic, photocatalytic, electrocatalytic and spectroscopic applications. His original contribution to the surface-enhanced Raman scattering (SERS), synergistic fluorescence enhancement by Ag and Au particles and supercapacitor fields is noteworthy. He has received numerous national and international awards. Professor Pal has supervised 32 students for their PhD degrees. Currently, he is working as a Distinguished Visiting Professor of the University of Johannesburg, South Africa, where his research is focused on sensors and device fabrication. |
1. Introduction
Over the last 180 years, since their discovery, the applications of colloids have continuously increased. The pioneering work by Selmi (1845),1 Faraday (1857),2 Graham (1861),3 and Zsigmondy (1865)4 laid the foundation for the recent milestones in electrochemistry, catalysis and spectroscopy regarding Au and Ag colloid nanoparticles (NPs). On 1st March 1869, the Russian chemist Mendeleev created a roadmap to scientifically organize elements based on their atomic weight. The periodic table proposed by Mendeleev contained 63 elements including seven ancient elements Fe, Sn, Pb, Hg, Cu, Ag, and Au, which was submitted to the Russian Chemical Society. His quintessential draft of the table was based on coinage metals and predicted elements, which became the hallmark of the scientific community. Gold glitters with a yellow gloss and does not tarnish and is insoluble in simple acid and water. However, copper and silver metal tarnish in moist air. Therefore, humans have cherished gold since ancient times for many reasons. Three noble metals, i.e., copper, silver and gold, are known as coinage metals with positive reduction potential values. Among them, copper is the cheapest and most abundant metal in the Earth's crust. Cu exhibits remarkable catalytic performance towards various reactions owing to its intriguing physical and chemical properties. Considering its plasmon-driven light-harvesting ability and catalytic function, Cu serves as a promising platform for efficient light-driven chemical reactions such as plasmonic Au and Ag.5It is worth mentioning that for Au and Cu, a superimposed collective and discrete excitation occurs upon the absorption of light, and in the case of Ag, discrete electronic oscillation, i.e., scattering/reflection, happens due to the interband transition at the shorter wavelength. In the case of Cu, although it possesses a tunable localized surface plasmon resonance (LSPR) from the ultraviolet-visible to near infrared region, it is limited by its facile oxidation (E0, +0.34 V) due to its immediate surroundings. Therefore, our discussion is mainly focused on Au and Ag.6–9 Among the three coinage metals, gold is the noblest and it retains its golden yellow luster, and thus it is widely used to create jewelry. Additionally, among them, gold has the highest standard redox potential (E0 = +1.50 V) value, with the representative equation [aq. Au(III) + 3e− → Au(s)]. Alternatively, among them, silver is the highest electrical conducting metal, with an E0 value of +0.79 V, while that for Cu is +0.34 V.
Their representative redox reactions are [aq. Ag(I) + e− → Ag(s)] and [aq. Cu(II) + e− → Cu(s)], respectively.10 This gradation of the redox potential value of the coinage metals in the periodic table is a predictive group property, but has a far-reaching consequences compared to Mendeleev's periodic table. This value indicates that very stable metallic Au(0) is easily obtained from the thermodynamically unstable Au(III) ion, which is one reason why metallic gold is ubiquitous in some places in the Earth's crust. Here, it is pertinent to mention that Au is the softest base of all the metals in the periodic table.
The Indian River Subarnarekha (means golden line in Bengali) got its name because metallic gold is found in its sand banks. Alaskan soil is also rich in elemental gold. With the advent of quantum mechanics, the large relativistic effect of gold in the 6th period was observed. However, this value is lower for silver in the 5th period of the periodic table. In fact, the pattern of the accommodation of the elements is the power of Mendeleev's periodic table.
Aqua regia, a mixture of concentrated aq. hydrochloric acid and concentrated nitric acid in ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) can easily dissolve metallic gold. The simple case of the dissolution of bulk gold in aqua regia proceeds through surface oxidation of gold by HNO3 and the available Cl− from the concentrated HCl present in aqua regia results in the formation of the yellow chloroauric acid (HAuCl4). Huge amounts of gold are used to make gold ornaments and the number of published research papers has reached over 1862565 in the literature due to its higher stability than AgNPs. Thus, the innumerable applications of metal nanoparticles especially AuNPs have revolutionized the world of nanotechnology. This has become possible because of the spectacular increase in the resolution power of microscopy and spectroscopy. The imaging of small particles on the nanometer scale is possible due to the recognizable interaction of the electron beam of wavelength λ (in meter) with the negligible mass of nanoparticles, m, in kg, and velocity, v, in meters s−1. This became a reality only after understanding the wave-particle duality, as given by the de Broglie equation (h is Planck's constant in J Hz−1). Thus, colloid research, an adventure in the “neglected dimension” as designated by Ostwald11 entered the era of nanoparticle/nanotechnology research.
:1 (v/v) can easily dissolve metallic gold. The simple case of the dissolution of bulk gold in aqua regia proceeds through surface oxidation of gold by HNO3 and the available Cl− from the concentrated HCl present in aqua regia results in the formation of the yellow chloroauric acid (HAuCl4). Huge amounts of gold are used to make gold ornaments and the number of published research papers has reached over 1862565 in the literature due to its higher stability than AgNPs. Thus, the innumerable applications of metal nanoparticles especially AuNPs have revolutionized the world of nanotechnology. This has become possible because of the spectacular increase in the resolution power of microscopy and spectroscopy. The imaging of small particles on the nanometer scale is possible due to the recognizable interaction of the electron beam of wavelength λ (in meter) with the negligible mass of nanoparticles, m, in kg, and velocity, v, in meters s−1. This became a reality only after understanding the wave-particle duality, as given by the de Broglie equation (h is Planck's constant in J Hz−1). Thus, colloid research, an adventure in the “neglected dimension” as designated by Ostwald11 entered the era of nanoparticle/nanotechnology research.
In 1783, Carl Wilhelm Scheele discovered the reaction for metallic gold dissolution in an aqueous solution of cyanide.12 This reaction is termed cyanidation, which is represented as follows:
| 4Au(s) + 8NaCN(aq.) + O2(g) + 2H2O(l) → 4 Na[Au(CN)2](aq.) + 4NaOH(aq.) | (1) |
After that, Bagration, Elsner and even Michael Faraday independently demonstrated the binding stoichiometry of Au(III) with the cyanide ion. In this process, oxygen is consumed to form the complex Au(CN)−2 ion. Metallic gold is extracted from its reaction with aqueous NaCN or KCN. Finally, gold is precipitated from aqueous solution by Zn flakes.
| Zn + 2Na[Au(CN)2] → 2Au↓ + Na2[Zn(CN)4] | (2) |
It is worth mentioning that asphyxiation, a deadly condition that occurs in mammals and from cyanide poisoning, chokes the oxygen uptake capacity of the body. Surprisingly, HCN is produced as a metabolic product of some compounds present in apples, almonds, etc. It is now known that the O2/H2O or O2/OH− system can also dissolve bulk gold. Furthermore, the unique dissolution of gold can be expedited by employing AuNPs. This is because the agglomeration number of AuNPs is only a few atoms, whereas for bulk gold it is ∞ (Fig. 1),13 given that the formal potential of Au(III)/Aun is more negative than the E0 values of O2/H2O (+1.23 V) or O2/OH− (+0.40 V) system. In the cyanidation process, gold or silver redox couples attain a very low reduction potential value.
 | ||
| Fig. 1 Standard redox potential of the silver microelectrode as a function of agglomeration number. For a large value of n, the potential approaches that of the conventional silver electrode. Reprinted with permission from The Royal Society of Chemistry (ref. 13). | ||
Therefore, Au(I) forms more stable complexes with ligands containing soft donor atoms such as S, C, Se, and P, while Au(III) forms more stable complexes with more electronegative or hard donor atoms such as N, O, F, and Cl. This indicates that complexes of Au(III) with soft ligands are unstable, easily reduced to the Au(I) state, while complexes of Au(I) with a hard donor are unstable, which disproportionate to Au(III) and Au(0) according to the following reaction:
| 3AuCl2− = AuCl4− + 2Au + 2Cl− | (3) |
It is pertinent to mention that the standard reduction potential values of Au(III)/Au(0) and Au(I)/Au(0) are +1.50 and +1.69 V, respectively. Thus, Au(I) is more oxidizing than Au(III). Metals or any material with dimensions of <100 nm are generally called nanoparticles, whereas colloid particles are between 1 nm to 1 μm in size. A reduction in the size of a material can be achieved via physical and chemical methods, which are known as the ‘top down’ and ‘bottom up’ methods, respectively. On the nanoscale, all three coinage metal nanoparticles exhibit rich plasmon bands in the visible range of electromagnetic radiation. It is interesting to mention that metallic gold is yellow (because of its very large relativistic effect and absorption of more blue light) and AuNPs in the form of a dispersion appear pink because of plasmon absorption. However, sub-nanometer particles (with a size of <1 nm called metal cluster) of gold and silver have no plasmon band, instead they noticeably fluoresce because of interband transitions.14 Furthermore, the stability of metal clusters both in aqueous and organic solvents has recently been described in the literature.15
Herein, the dissolution of Au and Ag as bulk metal or nanoparticles (NPs) in water is unambiguously described. Thus, the importance of the reduction potential value for metal colloid dissolution in water is generalized, taking the agglomeration number in one case, and electron injection in the Fermi level of MNPs in the other. Therefore, the highlighted electrochemistry approach becomes demonstrable especially for microelectrodes of Au, and also for Ag in aqueous surfactant medium. This demonstration becomes striking simply because of their rich plasmon band in the visible range. Among the innumerable possible plasmonic applications of Au and Ag NPs, surface-enhanced Raman scattering (SERS) is also detailed here. The inertness of gold, the classical reactions for its extraction and its electrochemical behavior are explained at the atomic level. Furthermore, the formation of bimetallic NPs, alteration of nobility and exquisite properties related to supported AuNPs in catalysis, the effect of incident photon to current conversion efficiency (IPCE),16–19 and Au clusters (sub-nanometer particles) are briefly discussed for their future prospects. Also, a discussion is presented on a few other metals and compounds, but the main focus is Au and Ag nanoparticles.
2. Metal electrodes and interfaces with or without an external power source
Electrochemistry is a subject dealing with the quantitative relationship between electricity and the chemical properties of substances. Alternatively, colloidal microelectrodes or grown microelectrodes (GME)20 are considered to have some similar functions as that of conventional electrodes, though former are more versatile, new and popular. GME do not have any external battery connection, which will be discussed in detail as a major part herein. The electrolysis of water was conducted about 200 years ago for the 1st time by William Nicholson and Anthony Carlisle with an applied potential. After 34 years, Michael Faraday (1834) announced the laws of electrolysis but after the mechanism of conductance was suggested by von Grotthuss in 1805. The static double layer concept at the electrode–solution interface was conceived by Helmholtz, and subsequently a new idea of the diffuse layer model was introduced by Gouy and Chapman. Stern combined the two above-mentioned concepts and suggested a hybrid model to explain the existing equilibrium at the electrode–solution interface.The electrode potential originates from the potential difference developed at the interface, similar to GME between the electrode and electrolyte. It is developed due to the transfer of charged species across the interface. The succeeding part relates to the normal idea of electrolytic cells. When a metal rod is partially immersed into an electrolytic solution of ions of the same metal, the metal atoms have a tendency to enter into the solution in the form of positive ions. This develops an electrostatic potential (called electrode potential) at the surface of the separation of the metal rod and the electrolytic solution. This escaping tendency of the metal is known as the ‘solution pressure’ or ‘solution tension’ of the metal and is constant at a given temperature. The metallic rod develops a negative charge when its positive ions leave and enter the solution and an electrical double layer is created at the interface. The reverse tendency of metal ions to interact with metal rods is known as the osmotic pressure of the ions in the solution. Thus, the electrode potential of a metal rod arises due to the difference between its solution pressure and the osmotic pressure of its ions.
For a redox reaction at an electrode surface, one can fix the concentrations of the oxidized and reduced species in solution. Subsequently, a Galvani potential difference is imposed; otherwise, using a potentiostat, the Galvani potential difference can be fixed experimentally to fix the ratio of the activities of the respective species in solution. Alternatively, it is not possible to independently fix both the Galvani potential difference and the concentrations in solutions.
Undoubtedly, the kinetics of chemical processes at electrodes cannot be explained using the concept of osmosis. It is admissible and useful to compare the properties of osmotic and electrochemical equilibrium but better to consider the exchange currents and even small deviations from equilibrium. However, the influence of activation barriers has to be explicitly excluded for the electrodic processes to remain reversible in the thermodynamic sense compared to osmotic processes, which is a serious limitation. A parallel between osmotic and electrochemical potentials does not exist for many important types of electrodes. Nevertheless, all the applications of (classical) thermodynamics in electrochemistry are governed by this limitation. Accordingly, this limitation can be simplified considering that the reaction proceeds in continuous electrochemical equilibrium. Here, we deal with this unrealistic relation to link thermodynamics and a part of electrochemistry. Modestly, the concepts of osmotic and electrochemical potential under the conditions of continuous osmotic and electrochemical equilibrium may be loosely considered as being parallel.21 When the solution pressure of a metal is more than the osmotic pressure of the ions, the cations pass into the solution more rapidly, leaving the electrode negatively charged. When the solution pressure of the metal is less than the osmotic pressure of the ions, the cations migrate from solution and get deposited on the electrode, making it positively charged. When a metallic zinc rod is placed in distilled water, the electrical charge on the zinc rod is always negative.
The description of the electrode–water interface may be assessed from the concept of the oscillatory dipolar behavior of the structure of water at room temperature, which causes charge polarizations on the metal surface of the electrode under consideration. The as-produced electric double layer drives the electrode potential. Particularly, the potential of zero charge (pzc) of the electrode can be assumed for partial charge transfer from the solvating water layer, causing a change in the work function, φ, of the solvated metal electrode in comparison to vacuum. The charge of the electrode (or equivalently the potential) is a useful variable, which is well controlled for the surface in question. In most cases, the electrochemical potentials and the Fermi level of a redox reaction are often considered with electrochemistry. The chemical potential is the work required to add a particle to the system in equilibrium between two phases at constant pressure and temperature. If the added particle is charged, it is considered that it is electrochemical potential. To discuss the function of GME, colloid chemistry provides direct information, which is discussed in the succeeding sections.
3. Metal colloids
Colloidal particles (1–1000 nm) describe huge interfaces, and hence a myriad of reactions and applications has been reported for colloids. This is also the case for metal colloids, which are known as ‘sols’ (suspension of very metal fine particles dispersed uniformly in a medium), where the most interesting cases arise with coinage metal colloids. More than one hundred years ago, it was mentioned by Ostwald that silver sols with a high degree of dispersion exhibit a yellow color with greenish tinge, which has been immensely appreciated recently together with his low dimension chemistry. Interestingly, stable colloidal metal particles under dispersion serve as good “microelectrodes” for many electron transfer reactions although they are not connected to an external potential source as in normal cases.22,23 When the metals are present in finely dispersed form, efficient photochemical reactions become possible with Ag.24 An elegant example is the classical development of photographs involving Ag halides in gelatin. Currently, numerous chemical reactions of Au and Ag colloids in their nanodimensions are being conducted as a function of their size, shape, morphology, and composite with respect to their potential electrochemical reactions. Colloidal gold particles in solution bear a charge, and thus chemical control of their nanocrystal shape, composition, and charge-carrier density further broadens their potential properties and applications. The reactions of colloidal metal particles in a dispersion involve the accumulation of electrons and positive holes, which leads to changes in their Fermi level, affecting their redox properties. Again, the oxidation of water by hot positive holes from a gold microelectrode occurred at positive potential, which has now become a regular and popular area of research.25 A discussion on metal semiconductor interfaces has been omitted for brevity.A metal nanoparticle is an assembly of many metal atoms with the approximate size of 1–100 nm with a surface positive (δ+) charge. The most stable type of assembly is spherical assemblies due to their minimum surface energy. At the center of a spherical assembly, there is one central atom, which is coordinatively saturated (typically surrounded by other atoms), and the peripheral atoms are coordinatively unsaturated because of the dispersion medium. Thus, there is a lack of electron density in the surface atoms, which makes the surface of the assembly positively charged. To maintain electroneutrality, negative ions from the dispersing phase are dragged onto the surface of the nanoassembly. An elegant example is the self-assembled monolayers of alkane thiols on gold surfaces. Thus, the metal nanoparticle assemblies acquire a negative charge unless otherwise indicated,26 and the electrostatic repulsion between the negatively charged particles stabilizes (electrostatic stabilization) the metal nanoparticles in the dispersion. Small particles of metal and metalloid may be obtained easily as a stable colloid via the reduction of the respective compounds via the ‘bottom up’ method.27 The simple photochemical method is another reproducible technique.28 Eventually, metal colloids, i.e., MNPs, often behave as a microelectrode without a connection to the battery. These MNPs can then transfer electrons for a chemical reaction with or without illumination. The optical property of the surfaces of coinage metals, i.e., plasmon absorption in the visible range is unique, rich and distinct. This happens due to the collective surface oscillations of electron gas. However, although there is a discrete excitation for Ag, in the case of Au and Cu, a discrete but superimposed excitation occurs. Fluorescence may be another surface property of these MNPs.29 The stepwise enlargement of AuNP seeds to any desired size has been shown in a controlled fashion using Au(CN)−.30 An excellent depiction is the stable gold nanoparticle dispersion demonstrated by Michael Faraday. In this context, it is relevant to mention that molecular colloids always become a selective adsorbent of their own ions, in general similar to colloidal AgCl, which attracts either Ag(I) or Cl− ions to achieve electrostatic stability and remain dispersed in a polar solvent (Fig. 2) such as water, and are known as hydrosols.
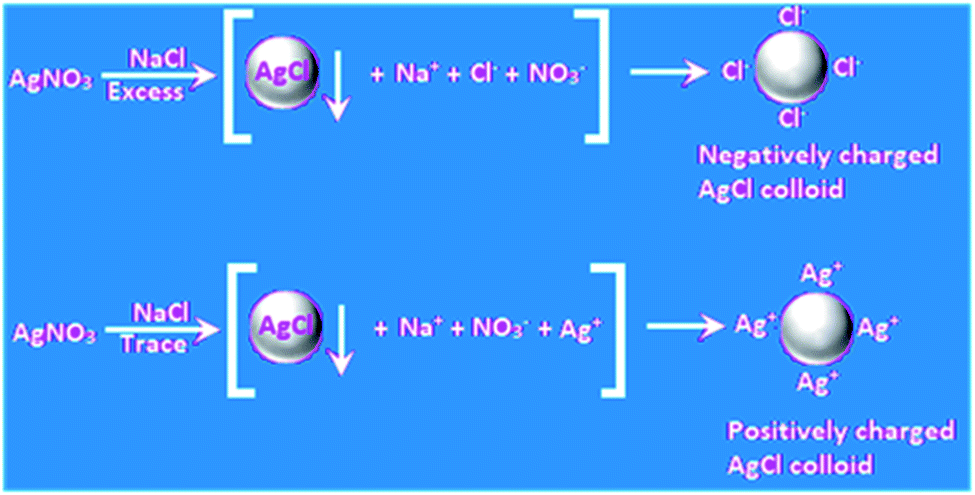 | ||
| Fig. 2 Formation of AgCl colloids with surface charges. | ||
A remarkable paper appeared in the literature, where AuNPs were stabilized by long chain thiols or amines,31 and consequently the surface of the AuNPs behave as a non-polar assembly and is dispersed in non-polar solvents (steric stabilization). Subsequently, the organic solvent (dispersing phase, non-polar solvent) may easily be removed by evaporation from the organosol and nanoparticle, resulting in the formation of a powder.26 The binding propensity of ligands is commonly described in the literature. However, the effect of nucleophiles is rarely discussed, which becomes interesting to consider for coinage metals.
4. Chemistry of coinage revisited in aqueous solution
4.1. Coinage metals in aqueous medium and effect of nucleophiles
The functioning of coinage metals in water has been described with little experimental detail. Analytical grade Cu, Ag and Au wires were used after thorough cleaning, and the stock solutions of CuSO4, AgNO3 and HAuCl4 in distilled water were reserved for further experimentations. The reagent solutions were obtained by employing the usual reported procedure. At room temperature (25 °C), nucleophiles such as I−, SCN−, and CN− were employed individually for the dissolution of the coinage metal wires in water under sonication and the observations are reported in Table 1 and (Fig. 3).32 Sonication prompted the dissolution process, resulting in the formation of a coinage metal hydrosol. Explicitly, the hydrosols of Au, Ag and Cu exhibited λmax values at about 525, 410 and 400 nm, respectively. The aggregation of the small hydrosol shifted the λmax values to the red region (bathochromic shift). On the contrary, a hypsochromic shift (blue shift) was observed when the hydrosols were simply nanoparticle dispersions in aqueous medium. Upon nucleophilic attack, the stable nanoparticle dispersions of the coinage metals dissolved in water, which was beneficial to study dissolution reactions. Parallel reactions with Au, Ag and Cu wires were also carried out in the presence of different nucleophiles to study the dissolution of the respective metals in aqueous solution (Fig. 3). The dissolution of stable dispersions of nanoparticles was monitored spectrophotometrically at the respective λmax values. However, the dissolution reactions of the metal wires were determined gravimetrically. The rate of dissolution of the wires followed the order of Cu > Au > Ag. Three common nucleophiles, CN−, SCN−and I−, were employed, and upon the addition of the nucleophiles, a decrease in E1/2 values was observed, as experimentally evidenced by cyclic voltammetry. The nucleophiles increased the Fermi level of the metals. This increase in Fermi level makes the metal surface vulnerable to oxidation, as evidenced by the measured E1/2 values. As expected, the hydrosols dissolved promptly, leaving behind the metal wires, and the color-bleaching reactions of the sol systems were always faster than the dissolution of wires due to their surface area. After the dissolution of the metals the presence of Au(III), Ag(I) and Cu(II) in aqueous solutions were individually tested with NaBH4 solution, and in each case, stable MNP dispersions, i.e., M(0) in solution, were confirmed spectrophotometrically. The ease of the construction of an electrochemical cell involving the I2/I− system helped to study Au(III)/Au(0) elaborately. It is very important to note that the I2/I−-assisted dissolution of Au is facile, which is much faster than the rate of the CN−-assisted dissolution of Au(0) in air. Thus, the latter is an inferior agent for the oxidation of Au(0) (Fig. 3).32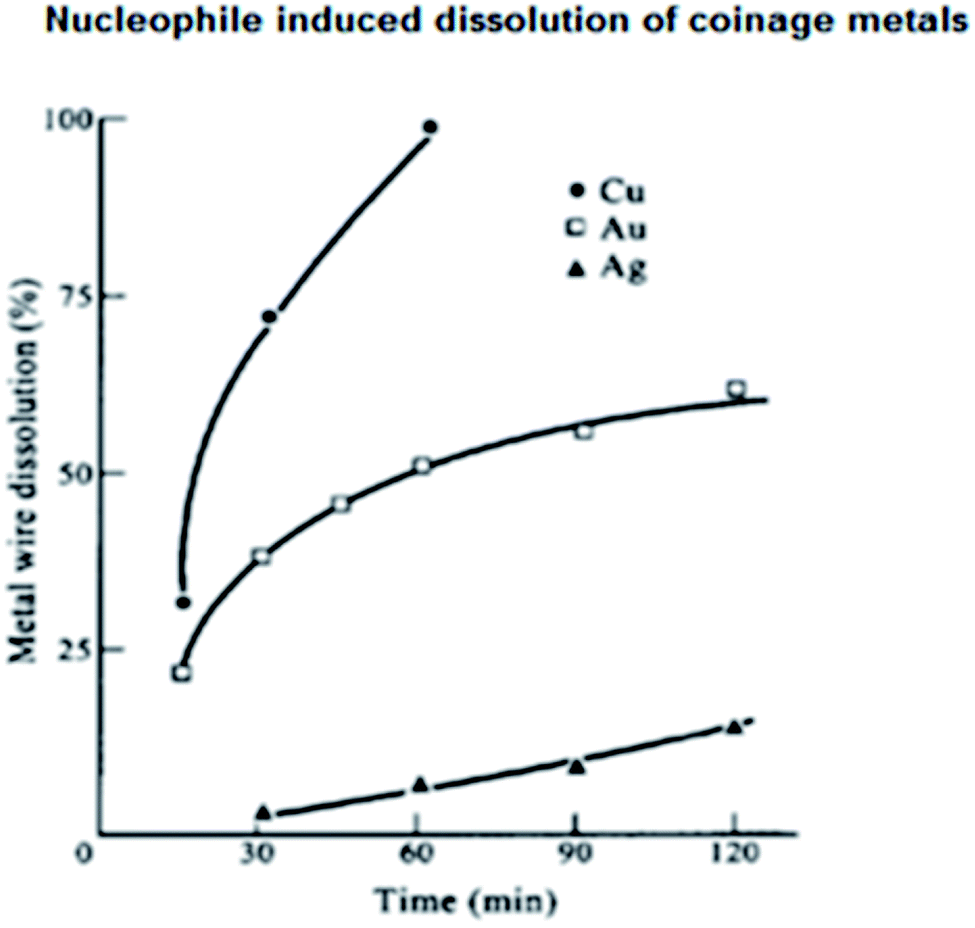 | ||
| Fig. 3 Dissolution of Cu, Ag and Au wires in I2/I−or I2/SCN− solution under sonication at 25 °C: ● for Cu; ▲ for Ag; and □ for Au. Conditions: [I2] = 10−2 mol dm−3, [I−] = [SCN−] = 0.1 mol dm−3. Reproduced with permission from Elsevier (ref. 32). | ||
The probable redox reactions at the gold surface for its dissolution by the I2/I− system are as follows:
| 2Au + 2I− + I2 = 2AuI + 2I− | (4) |
| 2AuI + 2I2 + 2I− = 2[AuI4]− | (5) |
The presence of I−, a nucleophile, is essential for the oxidation of Au(0), and molecular I2 alone did not dissolve Au(0) in solution but dissolved Ag(0) easily.32 It is pertinent to mention that the ‘repetition of property’ was the mother of the invention behind the idea of framing the periodic table. Among three elements, even for the coinage metals, Cu, Ag, and Au, Mendeleev indicated the gradation of their periodic property. The harmony, correlation or gradation of properties always helps in formulating a neat conclusion. The farsighted Mendeleev did that, where currently, one similar elegant topic has emerged, i.e., redox reactions, which are related to the redox potential value. Thus, the gradation of the redox potential values of the coinage metals in the periodic table has far-reaching revelation, which was corroborated by Mendeleev. In addition, the pattern of the accommodation of the elements in periods and groups is the strength of the periodic table. Metallic gold dissolves in water at room temperature (25 °C) in the presence of nucleophiles such as I−, SCN−, and CN−. All these nucleophiles also perform well for the dissolution of other metals, which is a general observation.
The important effect of ions on noble metal surfaces has not been addressed in a comprehensive way.33 The chemical adsorption of nucleophilic molecules/ions or deposition of another metal has been studied in detail. Here, the resulting changes in the chemical, photochemical, and optical properties are discussed. Metal particles carrying excess electrons initiate electrochemical reactions such as the reduction of water or the deposition of metals. The dependence of the standard redox potential on particle size is also discussed.34–39 Both galvanic replacement and exchange reactions have been explained straightforwardly40,41 based on thermodynamics for up- or down-scaling and tuning metal particles, which are related to the structure–property relationship. The electronic structure of MNP is important to infer surface phenomena of all types, i.e., adsorption processes, which determine electrochemistry, spectroscopy and catalysis. Adsorption occurs in the case of catalysis of a single atom. Furthermore, the environment surrounding the catalyst particle has different effects. Ion exchangers and solubilizers fall in the category of adsorption, producing better product yields.
4.2. Demonstration of down scaling and nucleophile-induced dissolution of coinage metals in water
The above-mentioned nucleophiles also work well for the dissolution of other metals.42 The concentration of nucleophile used is in the range of 10−3–10−1 mol dm−3 to perform dissolution reactions. Finely divided metals with an increased surface area, that is, with a low aggregation number of atoms (n), dissolve quickly and produce a “microelectrode”.43 The dissolution of coinage metals becomes eye catching because of their colours in the nanoscale under dispersion. However, the dispersion of stable nanoparticles in water or in dilute (10−3 mol dm−3) aqueous surfactant solutions with a metal concentration in the range of 10−3–10−4 mol dm−3 effectively stabilizes the metals in their nanodimensions (1–100 nm). Stable dispersions of these metal nanoparticles exhibit a rich plasmon band in the visible range.44 Then, it is easy to follow the color bleaching (disappearance of yellow colour of Cu and Ag, and pink colour of Au) of nanoparticle dispersions as their dissolution (oxidation) proceeds32 in air. Absorption spectrophotometric measurements revealed that the maximum absorption appears at ∼400 nm for Cu and Ag and ∼520 nm for Au NPs in air.32The reproducible demonstration of experimental details for the dissolution reaction of the cheaper silver nanoparticles (as a representative) in aq. NaBH4 (E0 = −1.24 V) solution is versatile,45 reversible and cost effective. A similar concentration of NaBH4 was used throughout the experiment, which produced intense yellow coloration19,45 with aq. AgNO3 (10−3 mol dm−3) in the presence of NaBH4 (10−2 mol dm−3) in air. The redox reaction can be written as follows:
| 2AgNO3 + 2NaBH4 + 6H2O → 2Ag(0) + 2NaNO3 + 2H3BO3 + 7H2 | (6) |
Evolution of the plasmon band of silver particles needs careful manipulation/introduction of NaBH4 solution. The dropwise addition of NaBH4, i.e., addition in portions, produces black Ag(0) particles, whereas the introduction of NaBH4 solution all at once produces a yellow solution. In this reaction, the adsorption of BH−4 ion on the Ag nanoparticle surface makes it negatively charged, which become susceptible to oxidation in the presence of air/oxygen (the reduction potential of O2/OH− should reach ≤+0.40 V). The oxidation of Ag(0) can be written as
| 4Ag + O2 + 2H2O → 4Ag+ + 4OH−, | (7) |
The homogenization of compounds in surfactant solution not only compartmentalizes the reaction products but also takes care of the cleansing action. Both these abilities are demonstrated in the following paragraph for the redox reaction of silver(I) with the appearance of its plasmon band.
4.3. Reversible redox reaction of silver nanoparticles in aqueous surfactant solution
Again, in this section, the moderately cheaper silver nanoparticles under dispersion are considered. Upon shaking the yellow solution of AgNPs in air, the surface of the dispersed AgNPs gets oxidized. Thus, the yellow colour is successively bleached due to the oxidation of the AgNPs in the surfactant solution, and finally their plasmon band disappears completely due to their quantitative oxidation, i.e., Ag microelectrode dissolution at air/water interface. On leaving the clear solution to stand, NaBH4 (already introduced in excess in the solution) again reduces the oxidized Ag(I) and brings back the yellow colour of the AgNPs, as presented in Fig. 4.45 Thus, the reversible oxidation and reduction proceed for the AgNPs in the solution as long as the nucleophile BH4−, i.e., reductant remains in the aq. medium. The surface of the particles upon oxidation by O2 in the air, gets covered with a silver oxide layer. To refresh the silver surface, ionic or non-ionic surfactant (10−3 mol dm−3) solution is very effective (Fig. 4), which removes Ag2O.45 Common surfactants solubilize the silver oxide layer on the Ag nanoparticle surface and oxidation progresses steadily.45,46 However, BH4−, being more diffuse, readily supports the dissolution of the AgNPs. Thus, BH4− successively produces size-controlled AgNPs in steps from the dissolution of the oxide layer. | ||
| Fig. 4 (a) Absorption spectra of silver plasmon resonance during bleaching by purging O2 (−) and reappearance in CTAB media (…). Curves 1–4 represent successive stages of bleaching starting from stable sol (1) to completely bleached sol (4) and curves 5–7 represent the successive stages of the reappearance of the sol, where 14 represents it complete conversion. (b) Mechanism of the reversible formation and dissolution of a silver nanoparticle. Reproduced with permission from the American Chemical Society (ref. 45). | ||
An idea that has provided a unique way to demonstrate the size-dependent E0 values of small metal particles from the reduction reaction of different dyes experimentally is described here. The introduction of different indicator dye solutions (10−5 mol dm−3) of variable E0 values in an aqueous reaction mixture of AgNO3 solution (10−3 mol dm−3), surfactant (2 mol dm−3) and NaBH4 (10−2 mol dm−3) leads to the reduction of the dye by the evolved GME (grown microelectrode) of AgNPs. The chosen dye molecules (λmax is widely different from the plasmon λmax of AgNP solution) get reduced at different intervals by GMEs, which clearly indicates that the grown GME of AgNP acts as a catalyst for dye reduction. The dye reduction is followed spectrophotometrically at the respective λmax values of the dye solution. It is interesting to mention that with time, the GME grows successively but its size remains the same for the entire period of a particular dye reduction. Subsequently, the growth of the GME progresses. Surfactants aid the catalytic property of the particles by controlling their growth by inhibiting the adsorption of reactants/products on their surface. Various dye solutions can be reduced with the in situ-produced AgNPs (Fig. 5), as previously reported.20 Furthermore, this has been shown to be a general feature for other metal GMEs. The above discussion is the manifestation of the size-dependent reduction potential value of AgNPs. The E0 value at the initial point of dye reduction becomes the reference E0 for the size-dependent E0 value of the GME. The size of the GME remains the same once a particular dye is quantitatively reduced, which subsequently starts to increase. Gelatine (aq.1% solution), Triton X-100 (TX-100, alkylphenol-polyoxyethylene ether), β-cyclodextrin (β-CD), etc. have been found to stabilize metal nanoparticles in aqueous solution.47,48 Thus, the size tuning of coinage MNPs in polymer, surfactant, etc. can be generalized in aq. solution.
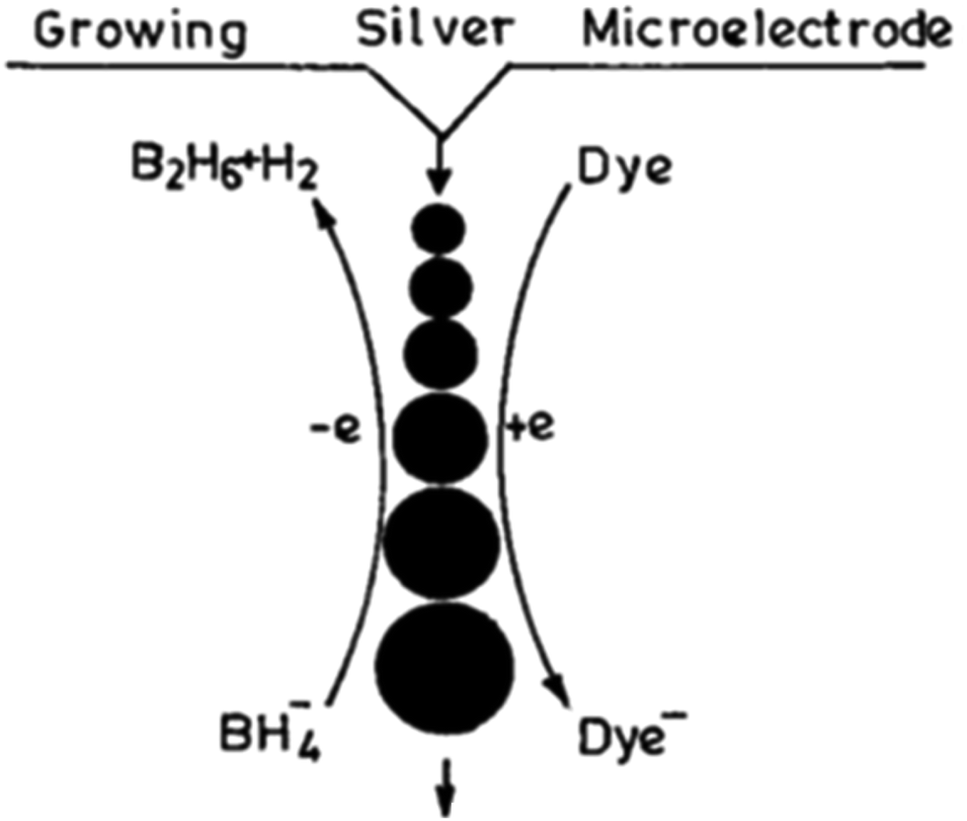 | ||
| Fig. 5 Scheme of growing silver particle-catalyzed redox reaction. Reproduced with permission from the American Chemical Society (ref. 20). | ||
In the same context, a reproducible photochemical technique has been reported for the evolution of highly stable Au nanoparticles in TX-100 medium. The evolved Au nanoparticles have application for the determination of cyanide ions, avoiding the aggregation of the Au particles in solution.28
4.4. Consequence of Fermi level shift of metal nanoparticles upon electron injection by nucleophiles
In the present discussion, electron donation by nucleophiles is hypothesized to demonstrate the shift in Fermi level (Fig. 6)43 as an indication of colloidal particles of Au and Ag. A study also validated the changes in their optical absorption, spectroscopy and catalysis. It has been authenticated that excess electron deposition by nucleophiles on the particles leads to a blue-shift (filling of the conduction band states at the Fermi level) in their plasmon absorption band. On the contrary, the plasmon absorption band shifts to longer wavelengths with electrophiles. The shift in the plasmon band shift is in good agreement with theory.49–51 It is pertinent to mention that the size of coinage metal NPs and/or their aggregation in solution shifts the λmax values of NP dispersions, which is different from the above-mentioned shifts in Fermi level.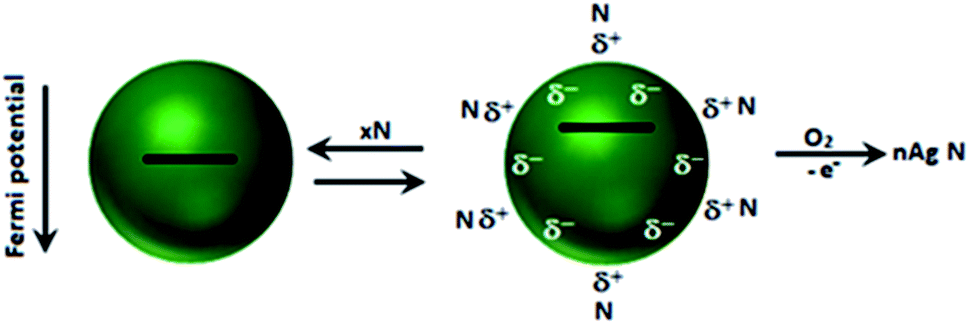 | ||
| Fig. 6 Adsorption of nucleophile N: schematic description of the adsorption/desorption equilibrium and accompanying shift in the Fermi potential. Reproduced with permission from the American Chemical Society (ref. 43). | ||
4.5. Typical role of cyanide in coinage metal chemistry
Generally, ligands are electron donors or Lewis bases, where when written in order of their field strengths, the spectrochemical series is obtained. Consequently, the cyanide ion (CN−) acts as a typical ligand in coordination chemistry and in redox reactions. The redox processes become interesting in the case of coinage metals. Henglein reported that cyanide being a strong nucleophile, shifts the Fermi level of the metals noticeably and can be studied easily for coinage metals using a visible spectrophotometer27 and at that point, H2O is decomposed upon UV light irradiation.52 The reactivity of the metal particles increases, creating (i) successive subdivision of the metal particles and (ii) unsaturated surface atoms of small metal particles with a nucleophile. In the former case, metal colloids are evolved under dispersion. Thus, the seed idea of colloid science,12 i.e., colloid-to-molecule transition, was described by Henglein.51,53 Finally, one AgNP in a solution but under dispersion may be considered as a metal microelectrode in equilibrium with one Ag(I) ion. The nuclearity-dependent potential changes were explained about 30 years ago.54Cyanide, a pseudo halide, acts as a strong ligand, reducing agent, masking agent, and strong complexing agent, and this is considered a nucleophile (Fig. 7). However, the nucleophilic property of cyanide has not been addressed in a comprehensive way for the extraction processes of gold and silver. It is known that cyanide shifts the Fermi level of metals to a great extent, making it negative. Thus, the metal becomes vulnerable to oxidation, and therefore in air, O2 can oxidize the metal in the presence of cyanide. A series of ligands, based on their strength of interaction with metal ions, is described in the spectrochemical series, where the position of the cyanide ion validates its strong ligand field strength.42 It is important to mention that the strong nucleophilic cyanide ion has been well exploited in redox processes. As a nucleophile, the Fermi level of small gold particles shifts towards the (negative) conduction band by the cyanide ion. Consequently, gold becomes vulnerable to oxidation. Gold can be dissolved in water when gold particles are stirred in an aqueous solution of iodide, cyanide, etc. in air. A demonstrable experiment becomes eye catching if some surfactant molecules are added to alkali cyanide in aqueous solution. This is more easily observed with small metal particles/metal nanoparticles under dispersion. Under these conditions, the reduction potential values of the metal become negative with a low aggregation number of metal atoms. Surfactants and polymers stabilize metal nanoparticles. The surfactant refreshes the gold surface by removing the oxidized gold from the surface of the metal nanoparticle and dissolves the oxidized layer from gold particles. Actually, gold dissolves in water without any acid! This type of dissolution has also been demonstrated for silver, and interestingly the reversible formation of silver nanoparticles (AgNPs) and their dissolution are perceptible even with the naked eye.45 Moreover, this information provides a clue to understand size-selective catalysis and size-tunable catalyst formation.
 | ||
| Fig. 7 Schematic representation of the potential action of cyanide. | ||
5. Core–shell metal particles of Au and Ag
According to the above understanding, monometallic Au and Ag nanoparticles and bi-metallic core–shell particles from silver and gold are obtained in a stepwise fashion. Because of the comparable lattice spacing (∼0.408 nm) of Ag and Au, their integration occurs easily to produce alloy nanoparticles. Even transient laser irradiation results in the formation of Ag(0)–Au(0) alloys if they are present together in a reaction medium.47 However, from a stepwise reaction, the core–shell architecture evolves.48 Similar manipulation has been elaborated from the galvanic replacement reaction to obtain a ‘hollow core–shell’ structure over time.28,55 In catalysis, when larger gold particles are needed, a gold-coated base metal can be used. The chemical and physical properties of the nanoparticles can be tuned by using the core–shell structure. The core–shell structure is generally preferred due to its extraordinary physicochemical properties, which are very different from that of its constituent metals.56–58 To obtain well-defined surface plasmon absorption bands (around 400 nm for Ag and 520 nm for Au), Aucore–Agshell nanoparticles have received great attention.59 Also core–shell nanoparticles have been proven to be a promising substrate for SERS studies because this architecture allows the tuning of localized surface plasmon resonance (LSPR). This helps to achieve favourable SERS signals by varying the size of the core and thickness of the shell in association with the electromagnetic field effect (EM).60,61The formation of the Aucore–Agshell structure may be explained by the different reduction potential values of Au and Ag (Au > Ag). Commonly, the nobler metal Au becomes the core (Au(III) is reduced preferentially) in the 1st step and the less noble metal Ag always remains as the shell (as Ag(I) is reduced later). In this case, Aucore–Agshell bimetallic particles evolve followed by the reduction of the added AgNO3 in the 2nd step. The reduction of AgNO3 was carried out using preformed Au nanoparticles (seed). Freshly introduced silver ions were skillfully reduced under UV (∼365 nm) light irradiation or by a weaker reducing agent (E0 = +0.35 V) such as ascorbic acid (10−3 mol dm−3). Then, the Au(0) surface was covered autocatalytically by the silver shell. For the reduction of AgNO3, the reducing agent should be less diffusive and weaker than BH−4, otherwise the adsorbed Ag+ ions are desorbed from the Au(0) surface and a mixture of Ag(0) and Au(0) particles are dispersed in the solution. However, employing ascorbic acid or UV exposure (∼365 nm), Aucore–Agshell particles are obtained. Alternatively, upon the addition of excess of Au(III) ions to a solution containing preformed Ag(0), Au(III) is reduced, and in turn Ag(0) is oxidized to Ag(I) because of the higher reduction potential of Au(III)/Au(0) (E0, +1.50 V) than the Ag(I)/Ag(0) (E0, +0.79 V) couple. Consequently, hollowcore–Aushell evolves. An interesting structural feature has been observed that if the added Au(III) ions are insufficient compared to preformed Ag(0), then the inverted Agcore–Aushell is formed (Fig. 8) but with a smaller Agcore.40 In this case, galvanic replacement (GR) of simultaneous oxidation and in situ reduction reactions have attracted interest in nanoscience for the size and shape tuning of various metal nanoparticles (MNPs).
 | ||
| Fig. 8 HRTEM images of (A) Aucore–Agshell and (B) Agcore–Aushell bimetallic nanoparticles. Reproduced with permission from the American Chemical Society (ref. 63). | ||
A suitable stabilizer combined with a weak reducing agent provides more interesting features, where kinetic control of the growth process of Ag and Au MNPs become perceptible. Kinetic control was observed by using suitable photons.41,62,63 Silver particles grow autocatalytically onto the preformed Au seed particles (λmax 523 nm) with a 1:2 molar ratio of Au and Ag(I). This bimetallic particle formation is driven by thermodynamics as argued from the standard reduction potential values of the two participating Au and Ag couples. Then, the λmax of Au(0) shifts from 523 nm to the more red region of ∼450 nm depending on the Ag(0) shell thickness on the Au(0) core. The result is presented in Fig. 9.63
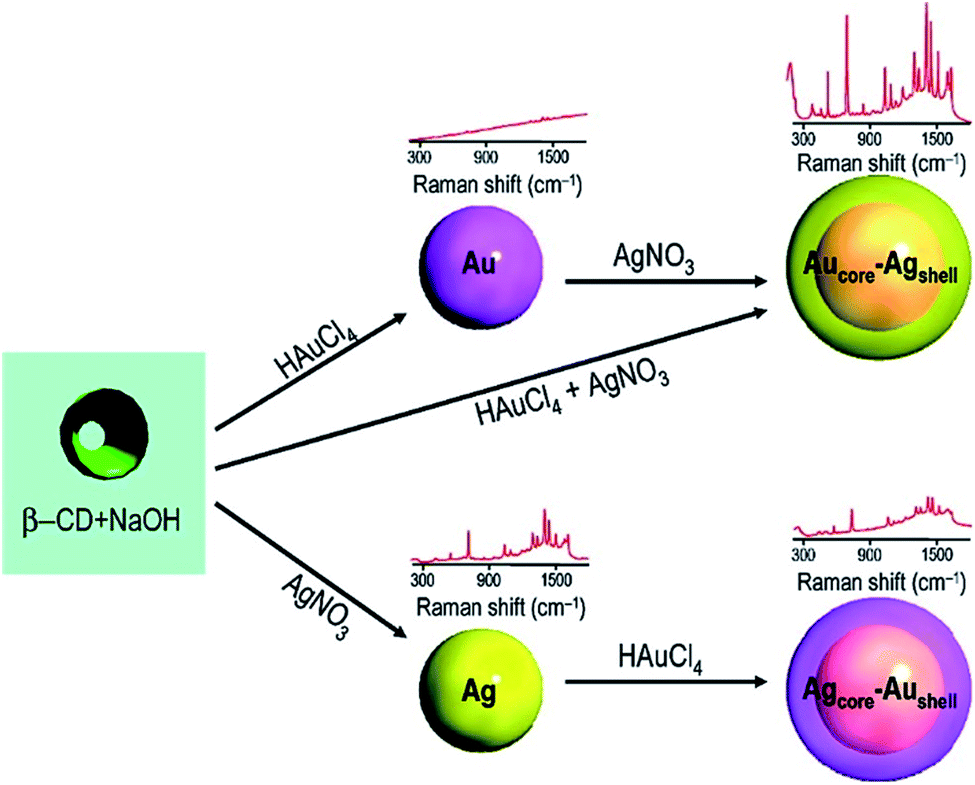 | ||
| Fig. 9 Schematic representation of the formation of mono- and bimetallic nanoparticles and their corresponding SERS spectra. Reproduced with permission from the American Chemical Society (ref. 63). | ||
Classically, the careful but preferential cyanide-assisted dissolution of the Ag shell of Aucore–Agshell particles resulted in the appearance of the original Au(0) plasmon peak at 523 nm. This experiment unequivocally established the nobility of gold (E0, +1.50 V) in comparison to silver (E0, +0.79 V). More importantly, once again this unique experiment well justified the different periodic properties of the participating coinage metals in terms of nobility. Recently, water-soluble noble metal NPs were evolved in aqueous solution.64
Although it is convenient to evolve normal Aucore–Agshell particles under normal conditions with preformed Au nanoparticles, the reverse case of bimetallic particle formation has become possible.63 Again, selective gold dissolution by cyanide has been experimentally explored using fluorescence probes.65 The description of Au and Ag metal dissolution at the air/water interface with nucleophiles such as cyanide and BH4− has become a general methodology, as presented in the preceding section with a variety of nucleophiles. The normal course of the reduction of Au(III) and Ag(I) mixtures in aqueous solution may lead to the formation of bimetallic Au–Ag alloy nanoparticles. Both Au and Ag have comparable lattice parameters (4.078 and 4.08 Å, respectively), which endorse the facile production of their alloyed bimetallic structures. However, the formation of Au–Ag alloys depends on the Au(III):Ag(I) ratio and the tendency of the reductant. Therefore, the concentration and kinetics control the reduction, which can lead to the formation of different types of bimetallic particles. For fast kinetics, Aucore–Agshell structures evolve.23 In this case, the higher reduction potential of +1.50 V of the Au(III) couple evolves the nobler Au(0) core in the 1st instance, and then Ag(I) is reduced onto the shell. During the course of slow reduction kinetics, Au(0) core becomes the adsorbent for the incoming Ag(I) ion, which becomes an adsorbate. The formation of Au(0)core–Ag(I)shell is authenticated from the red (bathochromic) shift in the plasmon resonance of the preformed Au(0) particles after the adsorption of Ag(I). Subsequently, the evolved Au(0)core–Ag(I)shell particles are reduced to produce normal Aucore–Agshell structures. Under radiolysis condition, if the mono-anionic Au(CN)2− and Ag(CN)2− complexes are employed in solution in presence of CN− ions, Au(0)core–Ag(I)shell is evolved, which is formed for chemical reduction. Subsequently, it is observed from the dynamics of the reduction reaction that the interesting electron relay takes place from the Au(0) core to the surface-adsorbed Ag(I) ion. Thus, this electron relay authenticates the oxidation of the Au core and reduction of the adsorbate, Ag(I). This process authenticates the reversal of the nobility of Au(0), i.e., gold becomes less noble than Ag.66 Furthermore, the kinetics of the reduction of Au(III) and Ag(I) through radiolysis revealed that slow kinetics of the process evolves Aucore–Agshell bimetallic particles and fast kinetic supports Au–Ag alloying.23 Similar redox chemistry results for the evolution of normal and inverted core–shell bimetallic particles by controlled kinetics were reported by Pande et al.63
6. Au–Ag alloy nanoparticles (NPs)
Small alloy particles of Au and Ag were discovered long ago, as discussed in the preceding section.67 This is ascribed to their similarities in lattice parameter and corresponding plasmon band shift.68 Alloy formation and the corresponding electronic spectral information have been explicitly explained based on the similarity of plasma frequencies of the participating Au and Ag atoms. The plasmon absorption of these two noble metals as a hybrid resonance results from the cooperative behavior of both their d-band and conduction electrons. In another report, the formation of non-homogeneous but monodisperse Aucore–Agshell was reported via the co-reduction of gold and silver salts.69 The growth process resulted in the replacement of the surface Ag atom by Au. Thus, the formation of Au–Ag alloys and their interparticle mixing are prompted by possible galvanic replacement reaction and place exchange reactions.7. Gold is unique and worthy of importance
Surface plasmons are coherent oscillations of conduction electrons on a metal surface excited by electromagnetic radiation at the metal–dielectric interface. Plasmonics deals with the study of light–matter interaction, where the wavelength of light is significantly larger than the size of the metal particles. Plasmon excitation enables tunable light absorption and photon confinement at the nanoscale by coupling a photon with the collective oscillations of the conduction electrons in the metal. Surface plasmons exist as either propagating surface plasmon polaritons (SPPs) on planar interfaces or localized surface plasmon resonances (LSPRs), which are confined to the surface of a particle. Surface plasmons are readily damped; thus, after a short time, the plasmon will start to decay radiatively into a reemitted photon or non-radiatively via intraband or interband transitions, forming energetic or “hot” electrons. Metal nanocrystals have large absorption cross-sections and can efficiently enhance and trap light, and therefore plasmon excitation has been widely viewed as an efficient mechanism for generating non-thermal ‘hot’ carriers. Hot carriers generated from non-radiative plasmon decay offer new opportunities for harnessing absorption loss. The hot electrons thermalize further through electron–electron (e–e) and electron–phonon (e–p) coupling and the energy is transferred as heat. Further, non-radiative plasmon decay results in the formation of a pair of excited ‘hot’ carriers, which can be injected from the metal into an adjacent semiconductor via internal photoemission (IPE) to yield a photocurrent. In the sequential processes, a photon excites the LSPR of the nanoparticle, which subsequently decays to yield an excited electron–hole pair. The electron–hole pairs are distributed over a range of energies, some of which are high enough to allow electrons to tunnel into the vacant states of nearby semiconductors. The photogeneration of hot electrons in a metallic structure and their injection into an adjacent semiconductor are a crucial mechanism, given that they allow electrons to be generated in the conduction band of the semiconductor with incident photon energies larger than the energy barrier between the two media, which is in general much smaller than the bandgap of the semiconductor (1.1 eV for a gold–titania interface). The hot electrons can be captured before thermalization by an adjacent semiconductor, providing a novel photoelectrical energy conversion scheme for photovoltaics or for driving chemical reactions. In this case, carriers excited with photon energies lower than the semiconductor bandgap can be captured, circumventing bandgap limitations and opening pathways for additional energy harvesting. Hot carriers can be harnessed for applications ranging from chemical catalysis to photothermal heating, photovoltaics, and photodetection. Although much effort has been devoted to mitigating plasmon nonradiative decay, recent research has uncovered exciting opportunities for harnessing this process, such as in photothermal heat generation, photovoltaic devices, photocatalysis, driving material phase transitions, photon energy conversion, and photodetection.Small particles (1–100 nm) of gold display a spectacular colored nanoparticle effect in glass. This effect and the stability of small gold particles against air oxidation was discussed in the Bakerian Lecture delivered by Michael Faraday.70 He thought that it is possible to prepare Au particles much smaller than that of the wavelength of visible light and a variety of colors may be observed from the solution due to the particle size variation under dispersion. G. Mie rationalized the red color of AuNPs by solving Maxwell's equation for particles of sizes equal to the wavelength of light.71 Now, we know that the nobility of AuNPs and their color is due to the collective oscillation of conducting electrons, which resonate (surface plasmon resonance, SPR) with the incident photon frequency and distinctly appear in the visible range. This makes AuNPs unique, even for applications.72 Again, the SPR is dependent on the size, shape, interparticle distance, and dielectric properties of the medium in relation to the local environment of AuNPs.29,73
Relativistic effects explain the distinctive properties of gold. The relativistic effects of heavier elements have long been recognized. The unusual large relativistic effect of gold is now well-established fact. Explicitly, the 6s-orbital contraction (decrease in 6s orbital distance from the nucleus), and simultaneously 5d-orbital expansion (increase in distance from the nucleus) for gold atoms are supported by different experimental facts and figures. The mass of an electron (travelling with half the speed of light) in the orbital increases for gold. Thus, the removal of an electron becomes difficult, and consequently blue light is absorbed, resulting in the appearance of the complimentary yellow color. The yellow color of gold corresponds to an energy difference of 2.3 eV (absorption energy). Cu has strong absorption at a slightly lower energy of 2.15 eV, which corresponds to its strong orange color. Similarly, Ag has an energy difference of 4.0 eV and it has high reflectivity evenly across the visible spectrum, which results in a pure white color. In general, all these effects are the result of interband transition. The relativistic effect, which raises the 5d-orbital and causes lowering of the 6s-orbital, also explains the color of metals. This effect is more prominent in Au than in Ag, which is also explained practically by its color. Thus, nonrelativistic gold would be white. The most popular plasmonic nanomaterials come from Cu, Ag and Au, which can directly convert incident photons (light) into electricity via the generation of hot electrons. When light is absorbed by plasmonic nanomaterials, they undergo two types of decay, as follows: (i) radiative decay and (ii) non-radiative decay. Ten years ago, researchers neglected this non-radiative decay process, and also tried to eliminate this process to maintain the optical performance of plasmonic devices. However, in 2004, Tian et al. made a breakthrough discovery that has been intensively investigated recently.16 Plasmonic metals experience surface plasmon resonance decays and generate hot carriers (hot electrons and hot holes) via localized surface plasmon resonance (LSPR), i.e., collective oscillations of the free electrons in plasmonic nanostructured metals. This happens when the resonant frequency of the electrons exactly matches the incident light.74 Consequently, they transfer their energy to the electrons in the metal and generate hot electrons. Hot electrons are produced in metal nanostructures due to (i) intraband excitation from the occupied s-band to the empty s-band or through (ii) interband transitions involving the d-band to the empty s-band. Interband transitions from other bands to the unoccupied conduction band states are also evident. Therefore, when silver is illuminated with visible light, hot electrons can only be produced by intraband excitation. Alternatively, gold and copper are capable of interband excitation in the visible region and hot electrons with a relatively lower energy are generated than that via intraband excitation in Ag.75,76 When the metal is kept in contact with a semiconductor, it will transfer its electron to the semiconductor. However, the electron transfer may be indirect transfer or direct transfer. In the case of indirect transfer, hot electron injection has to compete with e−–e−scattering. Also, because a very small fraction of hot electrons can be transferred, in this pathway, a very low efficiency is observed, i.e., <2%. However, in the direct pathway, there is no such e−–e−scattering, and thus the energy loss will be less, increasing its efficiency. In this case, when the metal and semiconductor remain close, it results in the formation of an empty hybridized orbital, and the generated hot electrons are directly placed in the empty hybridized orbital.74 However, to transfer the electron, it needs to overcome certain conditions. When the metal (Au) and semiconductor (TiO2) are kept in contact with each other, their different work function (φ) leads to the formation of a space charge region. Due to the effect of this space charge region, the band edges of the semiconductor will also shift, causing band bending in the semiconductor. If the work function of the metal (φm) is greater than that of the n-type semiconductor (φs), a Schottky barrier (φSB) will form at the metal–semiconductor interface, which will suppress the recombination of hot carriers. In that case, the electron will transfer from the semiconductor to metal until the Fermi levels of the metal (Ef,m) and semiconductor (Ef,s) reach the same level and the bands will bend upward towards the interface. Also, high energetic hot carriers can overcome this Schottky barrier. However, if φm < φs, then there will be no Schottky barrier. Electron transfer will take place from the metal to semiconductor. In that case, the contact between the metal and semiconductor is regarded as ohmic contact and the bands will bend downward towards the interface.77 To allow this process to continue, there should be some electron donor in the solution, which will donate electrons continuously to the Au nanoparticles. Firstly, Tian et al. tried I− and found that the incident photon to current conversion efficiency (IPCE) was very low, i.e., 1%.16 The corrosive property of I−/I3− towards the metal (Au) was the major problem associated with this electron donor. Then, the same group optimized this problem and tried other electron donors such as I−, F−, Cl−, and Fe2+ and they solved this problem by using the Fe2+/Fe3+ redox couple.17 In this case, Fe2+ acts as an electron donor and Fe3+ acts as an electron acceptor, as shown in the schematic in Fig. 10. However, this redox couple can work only when the potential of the redox couple is more negative than that of the hole and more positive than that of the conduction band of the semiconductor (TiO2), otherwise the metal (Au) nanoparticle cannot accept electrons from the donor and acceptor also cannot accept back transfer electrons from Au via TiO2. Fe2+ is considered to be the best donor because it shows the largest photocurrent of about 46 μA cm−2 and photovoltage of about 0.68 V. In this whole process, the most important thing is that the whole mechanism proceeds without the use of an external electric field. The hot-electron production efficiency in a nanocrystal strongly depends on its size, shape, material and local environment.78 Hot electrons, which are generated in semiconductor-plasmonic nanostructure composites, are ubiquitously used for the advancement of solar energy conversion due to their superlative ability to tune the wavelength over a wide range of the electromagnetic spectrum by controlling the shape of the nanoparticles. They operate across the visible-to-near-infrared (NIR) region,79 showing a wide range of applications such as photodetectors, photocatalysts, photovoltaics, chemical sensors, and biosensors.74 Plasmonic hot electrons are imperishable but have a very short time frame for their generation, emission and transfer, limiting their applications. Similarly, large relativistic effects lead to an increased ionization potential of ca. 2.3 eV, large work function, φ value (work necessary to extract an electron), negatively charged gold cluster, unusual lattice geometries of AuCl, AuBr and AuI with short Au–Au distances, helical gold nanowires.73 Furthermore, gold has the highest electronegativity (Pauling scale) and softest acid character among the metals in the periodic table. Therefore, the formation of Au− is understandable. All the other metals in the periodic table exhibit harder acidity than gold, and interestingly the hardness of the metal increases in proportion to the distance from the position of the gold.29 On the contrary, silver takes high energy UV photon, as already mentioned, to remove an electron and low energy visible photons are reflected, which give silver the look of a reflecting mirror. In conclusion, it is worth mentioning that a superimposed collective and discrete excitation occurs for Au and Cu, whereas in the case of Ag, discrete electronic oscillation occurs due to its interband transition at a shorter wavelength.
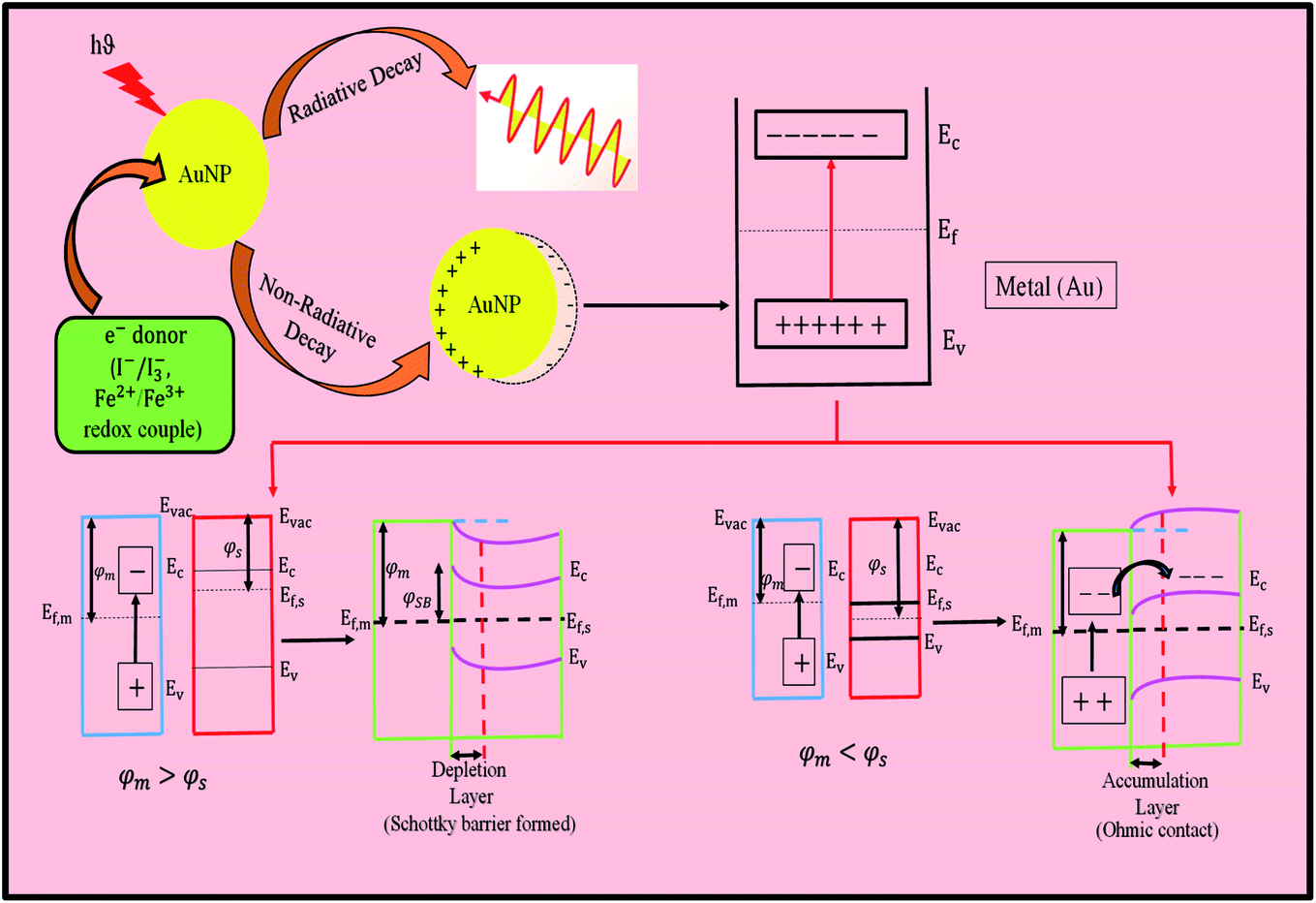 | ||
| Fig. 10 Schematic diagram showing the mechanism of the generation of hot electrons, where Ec, conduction band minimum; Ev, valence band maximum; and Evac, vacuum energy. | ||
Bacteria are very helpful and vital to the ecosystem. They are found in various sizes. However, a great challenge associated with them is their adherence to the surface of materials, initiating the process of colonization, namely the development of a biofilm. This has an adverse effect on human health and industrial applications. These challenges have encouraged researchers to explore solutions, where emphasis has been placed on the development of antibacterial metals. The state-of-the-art of coinage metals Cu, Ag, and Au are capable of being used as antibacterial agents.80 Based on this fact, the size, shape and surface chemistry of MNPs have been proven to affect their biological activity. Moreover, the dimensions of Ag nanoparticles play a vital role in their activity. Smaller particles impart a great antibacterial effect. Sharma et al. revealed that the antibacterial activity of Ag nanoparticles is size dependent.81 AgNPs with a size of 25 nm showed the highest antibacterial activity among the other sizes tested up to 450 nm. Their antibacterial performance was very more prominent at a very low concentration of silver (as low as 1.69 μg mL−1 Ag). In contrast, the low toxicity of gold to bacteria prevents it from being used as an antibacterial agent. However, bimetallic Au@Ag nanostructures can reverse this. It was observed that Au@Ag core–shell nanoparticles exhibited enhanced antibacterial activity because of the Ag shell around the Au core.82
8. Catalysis with metal particles
The word ‘catalysis’ was 1st used by Berzelius, which eventually acquired significance in the development of physical chemistry. The 1909 Nobel Prize in Chemistry was awarded to Ostwald for his contribution, which rightly steered the world of catalysis for innumerable practical applications. The grinding-mixing protocol (GMP) is a strategy to achieve size-dependent catalysis. Then, size-, shape- and facet-selective catalysis have enriched the subject for a better understanding of catalytic reactions. The modification of the catalyst surface is now explained with the change in work function (φ) value, which can currently be used to explain the improved product yield of a catalytic reaction in relation to the facet selection of a catalytic crystal.8.1. Catalysis with gold particles: work function (φ), size-, shape- and facet-selective catalysis
In the groundbreaking report on Au nanocatalytic reactions, Haruta described the oxidation of CO.83 Much emphasis was given to restructuring the crystal structure of the catalyst to determine the energetics of the surfaces.84 Recently, real time work function measurements were carried out for heterogeneous catalysis. Direct real-space information about the dynamics of catalyzed reactions has been elaborated to understand the work function-related changes of a catalytic reaction. Hence, in situ SEM studies have provided reaction fronts and the spillover of activated species.85 Several attempts have been made to rationalize the particle size effect in catalytic reactions. In particular, Au-catalyzed CO oxidation has been studies but the role of the altered electronic factors of the supported AuNP surfaces has been largely ignored. According to the ‘electron theory of catalysis’ or the ‘rigid band model’, the re-distribution and concentration of electrons on the catalyst surface/interface govern the catalytic processes, which is now being considered.Thermodynamic equilibrium is not achieved while catalysis proceeds in an open system given that heat and matter are exchanged with the surroundings. Therefore, the complex dynamics of catalysis should be studied on a real-time basis. In situ electron microscopy has paved the way for investigations of the surface changes of a catalyst for adsorption–desorption processes during a reaction.85–87 Reactions between the adsorbed species in the grain boundary region have been detected with respect to the contrast variations in SEM images, which has been related qualitatively to work function (φ) changes.
The changeable property of nanomaterials with large exposed surfaces has been well described in catalysis. In heterogeneous catalysis, generally polycrystalline catalyst particles are employed when the surface area of the catalyst particles, i.e., radius to charge ratio of the catalyst, becomes an important issue in terms of product yield and faster kinetics. Amongst the coinage metals, bulk gold does not support catalysis very well. There is no energy-permitted vacant orbital available to establish efficient substrate–gold interaction, a necessary condition for catalysis. However, a simple dispersion of gold nanoparticles (AuNPs) has been shown to perform well for the 4-nitrophenol reduction reaction.88 Furthermore, the importance of the size variation of AuNPs has been critically illustrated, which led to size regime-dependent catalysis.89 Eventually, the 4-nitrophenol reaction has become the ‘bench-mark model reaction’90 and widely used to test the catalysis of variety of MNPs.91 In most heterogeneous catalytic reactions, the surface area of the catalyst particles is increased by the ‘grinding mixing protocol’, resulting in an intimate reaction for efficient catalysis.92 Then, in catalysis, shape selectivity has been reported, where the reaction involves high-energy surfaces.93
In the case of facet-selective catalysis,94 catalyst particles with a selective facet catalyze a particular reaction87 and other facets may remain innocent or totally dormant. An analogous conclusion may be obtained from the work function (φ) value of a catalyst, which is also surface morphology or impurity driven. The origin of the catalytic activity of supported AuNPs is still far from being understood. The genesis of the high activity of supported Au catalysts is ascribed to the presence of negatively charged top Au atoms. An atom with low coordinated Au, in other words, high coordination vacancy or “crown jewel” concept, was introduced by Toshima et al.95 This idea has generated many reports on low coordinated Au catalysis. Recently, C–H oxidation reaction by Au nanoislands has been advocated, where the stabilization of the spin state, hybridization and charge-polarization are considered for a successful reaction.96
The exhibition of new physical and chemical properties with thiolate-protected gold clusters has received significant attention as a typical functional nanomaterial with limited active sites in photocatalysis.97 Recently, it has been rationalized that miniaturization of the co-catalyst plays a role in increasing the number of surface gold atoms, which is related to an increase in photocatalytic activity. However, the role of the co-catalyst is complicated and not well understood. Presumably, the gold cluster seizes a photoelectron, preventing electron–hole recombination and/or transfer of the seized electron to the gold surface (active site). Thus, confinement of the cluster on the catalyst surface and electron confinement on the co-catalyst site come into play. The catalytic activity is believed to have evolved from the changes in the number of surface gold atoms (co-catalyst) that react with hydrogen. In this case, the photoelectron reduces H+ of water to H2; in turn, OH− produces O2, justifying water splitting by confined gold clusters.
Based on the understanding of the facet-selective affinity of molecules and ions, the quantitative separation of Au nanorods (AuNRs) with a high aspect ratio was reported.87 Tetraazaanthracene derivatives from the oxidation reaction on selected Au(111) and Ag(111) facets via covalent C–N bond formation were discussed. This thermal oxidative cyclodehydrogenation reaction was supported by ab initio density functional theory calculations. The detailed mechanistic analysis was consistent with the experimental reaction conditions.98 Facet-dependent work function values are available for metals. A comparative account of the work function values of metals showed that they are crystal structure dependent. Alternatively, examples of size-, shape-, and then facet-selective catalysis are the progressive developments in the field of catalysis. However, facet-selective catalysis could not be related with the work function values of metals, and types of catalysis were varied.99 Partial charge transfer has been shown from the solvating water layer to the Pt electrode, leading to a dipolar polarization distribution along the interface, which is normal. The creation of an interface dipole is essential to understand the work function change from Pt(111) in vacuum to the Pt electrode covered by a film of an aqueous electrolyte. The partial charge transfer at the point of zero charge is a feature that has been typically neglected in traditional electrochemistry, which rather focused on the arrangement of point charges and the orientation of solvent molecules from theoretical calculation. It was shown in the calculation that the redox potential of the catalyst contributes to the driving force of the reaction, but the electrode regenerates the active form of the catalyst. From a chemical reaction, an exclusive concave hexagonal molecule with a high index facet {hkl} was successfully prepared via the Cu(II)-assisted etching of hexagonal (NH4)2Cu(MoO4)2(ACM) for the first time. The higher reactivity of the {001} crystal plane than that of the {010} plane was observed after the etching process. It was shown that the high index facet-exposed concave hexagonal ACM serves as a better catalyst for the photodegradation of dyes than the other microstructures enclosed by low index facets.94 Thus, the energetics of the catalyst surface has become very important.
Chemisorbed O2 binding weakens due to the work function of the Pt catalyst. It has been shown that activation energy of oxygen desorption depends linearly on the potential and work function of the catalyst. Experimental correlation between surface structure and catalytic behavior is difficult to generalize. It is even more difficult to identify the low-coordinated active sites of a catalyst for the preferred anchoring of different substrates.100 Again, a catalyst with an exposed surface is very selective sometimes in relation to a particular reaction, facet, interface, coordination site, reagent, etc.101 The active sites change during the reaction process, further complicating the situation, which may be explained from real-time monitoring of the work function (φ).
Here, the synthesis of highly fluorescent water dispersible silver clusters is presented. The clusters on the Au(I) surface exhibit strong fluorescence due to synergism/cooperativity. Moreover, the green synthesis and reversible dispersion of stable silver cluster in water and common organic solvents make them useful.15 Thus, fluorescing Ag clusters on the surface of Au present a platform to enhance the fluorescence of Ag and have potential for sensing activity. The Fermi level does not include the work required to remove an electron from wherever it came. A precise understanding of the Fermi level provides information about the electronic band structure, voltage and flow of charge.
The binding energy shift of Ag is mainly attributed to the electron transfer from metallic Ag to ZnO crystals (i.e., formation of monovalent Ag). The Fermi levels of two components become balanced when the metal particles come into contact with a semiconductor, such as Au/ZnO and Ag/ZnO heterostructures.102 Accordingly, when Ag (work function = 4.26 eV) is attached to ZnO (work function = 5.3 eV), some of the electrons are transferred from Ag to ZnO at the interfaces of the Ag/ZnO core–shell heterostructures, resulting in higher valence Ag. The binding energy of monovalent Ag is lower than that of zero-valent Ag; therefore, the shift to lower binding energies for Ag 3d5/2 and Ag 3d3/2 further verifies the formation of Ag/ZnO heterostructure composites.
It was described by theory and experiment by Giovannetti et al. that graphene retains its unique capacitance property with Au.103 Graphene upon doping onto Au(111) facets weakly interacts with Au and its Fermi level shifts. Here, this weak interaction results in a high contact resistance. However, the φ value of graphene changes with Au, similar to that with other metals. This result becomes important for fabricating high-performance graphene-based electronic devices. The authors indicated that metals may be divided into two groups and this classification becomes important for the fabrication of devices. This was supported by Song et al.104 It has already been indicated that defects, kinks, stairs, impurities, etc. drive catalytic reactions easily.93 The reasons for this are not well understood in the case of low coordinated/under coordinated catalyst particles. However, the importance of heterojunctions is understood but their manipulation is done in a ‘hit-or-miss’ way.
The work function (φ) of a metal is defined as the minimum energy needed to remove one electron from the metal, which depends on the structure, morphology and chemical composition of the metal surface. For example, different crystallographic surfaces of the same metal exhibit different work function values. Thus, the chemical modification of a surface can also induce altered φ values. Therefore, the metal surface becomes susceptible to oxidation in the presence of suitable nucleophiles. It is worth mentioning that in the groups of alkali and alkaline earth metals in the periodic table, the value of the work function decreases progressively from the top to bottom. This trend in the change of work function is reverse for the groups of transition metals. Similarly, E0 changes for most metals. The work function value is anomalous for the coinage metals, as follows: Au > Cu > Ag, which is explained by the relativistic effect. Electrochemistry has much in common with surface science in relation to the solid surface in contact with another phase. As mentioned already, for electrons in a metal, the work function, φ, is defined as the minimum work required to transfer an electron from the inside of the metal to the outside. Electrons may go from the inside to the surface of the metal (work is done against the surface dipole potential). Therefore, φ is a surface term, and hence it is different for different surfaces of a single crystal. It is important to note that the work function, φ, depends on the surface structure at the single atomic level, and consequently must be defined for each crystal face. Therefore, facet selectivity appears in catalysis. The work function is the negative value of the Fermi level, provided that the reference point for the Fermi level is chosen just outside the surface of the metal. The standard reduction potential (E0 in V) is a thermodynamic property of the elements. This value (referred with reference to the standard H2 electrode) are displayed in textbooks, which is related to the Gibbs free energy change. Eventually, the E0 values sequentially designate a group property of the elements, as presented in the periodic table. Similarly, the group properties of the elements are described by the work function (φ, eV) values. Both values are useful for catalytic reactions to further explain the chemical reactions. Furthermore, the work function values are not only facet dependent but also impurity driven, which makes a particular metal selective for a particular reaction. Thus, the controllable work function results in a dramatic change in catalytic activity and selectivity.105 The work function, φ, of metals varies from 2.3 eV for potassium to 5.3 eV for gold. A metal such as gold, which has higher work function, φ, requires more energy to transfer an electron. The valence bands are filled with electrons up to the Fermi energy. The energy difference between Fermi energy and vacuum level corresponds to the work function (φ). One of the key electronic properties of a metal that is affected by adsorption is its work function, φ.106 The work function, φ, and its related quantity, the potential of zero charge, are significant in understanding several surface and electrochemical phenomena, namely, catalytic activity, redox kinetics, and electric double layer dynamics. The work function (φ) of metals is reduced by ∼4% upon water adsorption, which has been observed to be a function of surface coverage.107–110 Thus, the surface roughness of the metal in the case of nanostructured materials should be considered. The presence of a minute amount of contamination (less than a monolayer of atoms or molecules) or the occurrence of surface reactions (oxidation or similar) can change the work function substantially. Changes in the order of 1 eV are common for metals and semiconductors, depending on the surface conditions. These changes are the result of the formation of electric dipoles at the surface, which changes the energy required for the electron to leave the sample. Due to the sensitivity of the work function to chemical changes, its measurement can give valuable insight into the condition of a given surface. The results justify metal–semiconductor band bending as an interfacial activity. The work function also has a significant influence on the band line-up at semiconductor interfaces. Thus, it is a major module to study electrochemistry and catalysis. The work function of a metal surface is now known to be affected by surface adsorbates, as exemplified by surfactant-modified particles.111 The removal of heavy metals and preparation of a new generation of catalysts are never-ending demands to achieve higher product yields. This is achieved reproducibly through adsolubilization111 and exploitation of commercial ion exchange resins.112 The former is a process in which adsorption and solubilization occur concurrently.111 However, the latter performs extremely well for the synthesis of mono- bi- and multi-metallic nanoparticle catalysts.88,112 In both processes, ionic surfactants are used. More reports on solid heterogeneous catalysis are warranted to quantitatively relate catalysis and the work function of metals.
9. Graphene with gold and silver
The interparticle coupling effect on the surface plasmon resonance of gold nanoparticles has been described in the literature.72 The SERS (surface-enhanced Raman scattering) analysis of a graphene–Ag nanowire hybrid revealed that the plasmon-driven electron source is available due to strong plasmon-exciton coupling. As plasmon-to-electron conversion readily occurs, the evolved electrons then propagate in chemical reactions due to strong plasmon–exciton coupling. However, experiments have categorized that electron storage is not possible in graphene–Ag nanowire hybrids and monolayer graphene or even single Ag nanowire. Therefore, all these systems cannot drive chemical reactions with stored electrons. The latter two systems cannot compete for plasmon-to-electron conversion, whereas graphene–Ag nanowire hybrids can and drive a chemical reaction favorably, as already mentioned.103 However, the work function change of Ag and Au surfaces has not been investigated.111 All the above-mentioned heterogeneous materials help to study chemical reactions because of the easier separation of the used materials, their higher shelf-life, easier handling process, easier recovery/reuse, and in many cases higher efficiency. In this case, the solid surface sometimes becomes a promoter, as is often observed in the case of adsolubilization.10910. Surface-enhanced Raman scattering (SERS) involving Au and Ag nanoparticles (NPs)
The most remarkable discovery was made from an electrolytic cell fitted with a silver electrode. In 1974, the huge enhancement in the Raman signal of an organic molecule, pyridine, using a bulk Ag electrode was reported.113 Subsequently, the manipulation of gold and silver particles has found innumerable applications in surface-enhanced Raman scattering (SERS) studies.114,115 Recently, copper nanoparticles have been exploited as an SERS substrate.116 Currently, SERS is considered the most sensitive and selective analytical detection method. The Raman signal enhancement (million times) is explained by (i) the charge transfer mechanism, CT117 and (ii) electromagnetic mechanism, EM.118 In the initial stage, gold and silver sol systems were employed for SERS studies and extended to different analyses on Au and Ag micro-electrode systems.119 For the first time, Moskovits noted the SERS intensities arising from localized surface plasmons in nanostructured metals.120 Furthermore, individual particles with Au or Ag cores have been found to enhance the Raman signal of the CN− ion despite the platinum metal coating on Au and Ag particles. The vibrational spectral profiles of CN− have been compared with electrochemical findings.121 Gold or silver was employed for SERS studies on a shell-protected Fe3O4 core. The shell thickness was fine tuned for the study and the effect of the magnetic core was investigated. Temperature-dependent magnetic studies were also conducted to relate the SERS enhancement.122 Pande et al. elegantly discussed the formation of normal and inverted core–shell particles of Au and Ag.63 The morphologies were not only been confirmed but also compared and the superiority of Ag over Au for SERS activities63 was categorically described, as shown in Fig. 9. This enhancement has also been extended to transition metal nanoparticles with well-known chelating ligands.123Fe3O4 nanoparticles with a size approximately of 13 nm have been prepared successfully in aqueous micellar medium. Thus, Fe3O4 nanoparticles are protected from surface poisoning, a new route to develop a shell with noble metals such as gold or silver. The shell thickness of the core–shell particles becomes tunable through the adjustment of the ratio of their constituents. Thus, this route yields well-defined core–shell structures with a size in the range of 18 to 30 nm with varying proportions of Fe3O4 to the noble metal precursor salts. These magnetic nanoparticles were characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), Fourier-transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), Raman and temperature-dependent magnetic studies for the fruitful application of SERS studies on magnetic hybrid materials.123 Here, the magnetic enhancement, i.e., field effect, was observed due to the enhancement of the SERS signal intensity. The interaction between Au–TiO2 can be easily visualized from the spectral broadening of AuNPs. The direct conversion of plasmons to electrons has gained wide scientific interest and has been capitalized, as discussed earlier. The interaction with a suitable electron donor generates hot electrons and is not discussed in SERS given that it involves a semiconductor–Au composite.
11. Fluorescence and metal clusters of Au and Ag
It is known that with a decrease in the nanoparticle size, the intrinsic size effect predominates, resulting in damping of the LSPR (localized surface plasmon resonance band) effect.14,15,124,125 The intrinsic size effect is a microstructural effect and the extrinsic size effect is a dimensional effect. The spectral properties of SPR have been extensively studied for different noble nanoparticle systems and compared with the Mie theory. For very large nanoparticles (diameter D ≥ 20 nm), the resonant peak energy, ER, experiences a red shift with an increase in sizes due to retardation effects and the increasing contribution from multipolar terms. In addition, for nanoparticles larger than 50 nm, radiative damping of the collective electronic excitation significantly broadens the line width of the SPR band. For smaller nanoparticles (D ≤ 20 nm), these extrinsic size-effects become negligible and intrinsic size-effects exist. Size-dependent modifications of the dielectric constant with respect to the bulk values have been critically considered. However, the disparities in nanoparticle size, shape, and local environment are complicated issues to explain the width of the SPR band.126 The fluorescence of Au/Ag nanoparticles at the sub-nanometer level is generated due to the interband transition and the consequence of metal–ligand interaction, which is now an established fact.127 Ultra-small MNPs have discrete energy levels and interact with incident light via electronic transitions between the discrete energy levels, leading to strong light absorption and emission. The size of MNPs should normally be less than 2 nm for them to be luminescent. Some recent studies reported that fluorescence can be detected from much larger silver nanoparticles due to strong ligand participation. The fluorescence emission wavelength and quantum yields of MNPs are strongly dependent on the surface ligands used for nanoparticle stabilization. Again, the fluorescence of nanoparticles has an inverse relationship with the size regime and damping with larger particles. The damping of fluorescence relates to quantum confinement, interband transition, etc.128 Here, in conclusion, it can be stated that fluorescence originates from the quantum states at the inorganic–organic interface from the metal core or mixed ligand moieties. In some cases, very strong fluorescence becomes very similar to ligand-related states.129 Gold is now being exploited in heterogeneous or homogeneous catalysis, as mentioned in the preceding sections. Thus, many more new discoveries are expected with gold in the nanoscale.12. Future applications of Au and Ag nanoparticles (NPs)
In the field of sensors, Au and Ag NP clusters and their heterostructures with carbon dots (CDs) and graphene generate many nanotechnological applications. Thus, considering all these nanostructured materials, the fabrication of new devices has become desirable for bioscience and therapeutic usages.112 It is important to mention that the majority publications on MNPs are focused on AuNPs/clusters.130 Also, the competing emission applications of the behaviour of molecules and Au–Ag NP systems are sometimes compared not from a sensitive analytical detection point of view but selectivity.Furthermore, plasmon-induced hot electron generation has been unambiguously proven without any bias voltage,16–19 resulting in energy storage, with continuous research on photocatalysis, photoelectrochemistry, and even the clean stereo-selective synthesis of drug molecules.16–19,131
13. Conclusions
In conclusion, the manipulation of coinage metals, predominantly silver and gold colloids, individually or co-jointly, have been documented from bulk material to cluster formulations. According to the discussion herein, the representative noble (bulk) metal Au can be made less noble upon subdivision. A lower reduction potential value (<+1.50 V) is observed for smaller Au particles. Electrochemistry with added nucleophiles and properties of smart material tuning even for hot electron generation has been considered quantitatively for energy production and product formulation. It was shown that control of the electrode potential and consideration and alteration of the work function value and facet-selectivity of AuNPs compared to other MNPs can pave the way for their catalytic and spectroscopic applications. Most industries rely on catalysis and the importance of supports for catalyst particles especially for gold, despite its natural inactivity, has been deliberated. The size, shape, facet, and coordinatively unsaturated surface atoms for the design of a new generation of catalysts were presented, which can further inspire future scientists. Then, the present status of SERS, the most sensitive technique, for the detection of analytes down to the single molecular level was discussed, together with the plasmonic, CT and EM mechanisms. Furthermore, the emissive behaviour of gold with silver clusters and their synergistic propensity have recently been capitalized for detection and cell imaging applications.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors are thankful to Dr Teresa Aditya of Penn. State Univ. for helpful corrections and gratefully acknowledge the facilities received from the University of Johannesburg, SA and Indian Institute of Technology Kharagpur, India.References
- F. Selmi, Studi sulla dimulsione di cloruro d'argento, Nuovi Annali delle Scienze Naturali, 1845 Search PubMed.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- T. Graham, Philos. Trans. R. Soc. London, 1861, 151, 183–224 CrossRef.
- R. Zsigmondy, J. Am. Chem. Soc., 1909, 31, 951–952 CrossRef.
- Y. Xin, K. Yu, L. Zhang, Y. Yang, H. Yuan, H. Li, L. Wang and J. Zeng, Adv. Mater., 2021, 33, 2008145 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- T. Hou, H. Peng, Y. Xin, S. Wang, W. Zhu, L. Chen, Y. Yao, W. Zhang, S. Liang and L. Wang, ACS Catal., 2020, 10, 5502–5510 CrossRef CAS.
- S. Kim, J. M. Kim, J. E. Park and J. M. Nam, Adv. Mater., 2018, 30, 1704528 CrossRef PubMed.
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC.
- N. N. Greenwood and A. Earnshaw, Chemistry of the element, Elsevier Sci. &Tech. Butterworth-Heinemann Ltd, Oxford, UK, 2nd edn, 1997 Search PubMed.
- W. Ostwald, Die Welt der vernachlässigten Dimensionen, Verlag von Theodor Steinkopff: Dresden und Leipzig, 1915 Search PubMed.
- A. I. Vogel, A textbook of qualitative chemical analysis, Longmans, Green and Co., London, New York, Toronto, 1937 Search PubMed.
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC.
- L. A. Peyser, A. E. Vinson, A. P. Bartko and R. M. Dickson, Science, 2001, 291, 103–106 CrossRef CAS PubMed.
- M. Ganguly, J. Pal, S. Das, C. Mondal, A. Pal, Y. Negishi and T. Pal, Langmuir, 2013, 29, 10945–10958 CrossRef CAS PubMed.
- Y. Tian and T. Tatsuma, Chem. Commun., 2004, 10, 1810–1811 RSC.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- J. Li, Y. Long, Y. Liu, L. Zhang, Q. Wang, X. Wang, S. Song and H. Zhang, Angew. Chem., 2020, 59, 1103–1107 CrossRef CAS PubMed.
- K. Kitano, Y. Sakakibara, M. Kago, T. Doe, M. Ueda, T. Ryowa, M. Izumi, H. Nishi, T. Tatsuma and Y. Arakawa, Appl. Phys. Lett., 2021, 118, 063505–063506 CrossRef CAS.
- N. R. Jana, T. K. Sau and T. Pal, J. Phys. Chem. B, 1999, 103, 115–121 CrossRef CAS.
- G. Inzelt, J. Chem. Educ., 1996, 73, A294–A296 CrossRef CAS.
- A. Henglein, Topics in Current Chemistry,Electrochemistry II, 1988, 143, 113–180 CAS.
- M. Treguer, C. de Cointet, H. Remita, J. Khatouri, M. Mostafavi, J. Amblard, J. Belloni and R. de Keyzer, J. Phys. Chem. B, 1998, 102, 4310–4321 CrossRef CAS.
- T. Linnert, P. Mulvaney and A. Henglein, Ber. Bunsenges. Phys. Chem., 1991, 95, 838–841 CrossRef CAS.
- C. Loo, A. Lowery, N. Halas, J. West and R. Drezek, Nano Lett., 2005, 5, 709–711 CrossRef CAS PubMed.
- S. Nath, S. Jana, M. Pradhan and T. Pal, J. Colloid Interface Sci., 2010, 341, 333–352 CrossRef CAS PubMed.
- A. Henglein, Isr. J. Chem., 1993, 33, 77–88 CrossRef CAS.
- A. Pal and M. Bandyopadhyay, Indian J. Chem. Technol., 2000, 7, 75–78 CAS.
- G. Wulfsberg, Foundation of inorganic chemistry, University Science Books, CA, USA, Indian edn, 2002, p. 202 Search PubMed.
- A. Henglein and D. Meisel, Langmuir, 1998, 14, 7392–7396 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, Chem. Commun., 1994, 801–802 RSC.
- T. Pal, N. R. Jana and T. K. Sau, Corros. Sci., 1997, 39, 981–986 CrossRef CAS.
- O. M. Magnussen, B. M. Ocko, R. R. Adzic and J. X. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 5510–5513 CrossRef CAS PubMed.
- O. S. Ivanova and F. P. Zamborini, J. Am. Chem. Soc., 2010, 132, 70–72 CrossRef CAS PubMed.
- M. Mostafavi, N. Keghouche, M. O. Delcourt and J. Belloni, Chem. Phys. Lett., 1990, 167, 193–197 CrossRef CAS.
- J. Belloni, M. Mostafavi, J. L. Marignier and J. Amblard, J. Imaging Sci., 1991, 35, 68–74 CAS.
- O. Platzer, J. Amblard, J. L. Marignier and J. Belloni, J. Phys. Chem., 1992, 96, 2334–2340 CrossRef CAS.
- J. Amblard, O. Platzer, J. Ridard and J. Belloni, J. Phys. Chem., 1992, 96, 2341–2344 CrossRef CAS.
- A. Henglein, P. Mulvaney and T. Linnert, Faraday Discuss., 1991, 92, 31–44 RSC.
- K. Mallik, M. Mandal, N. Pradhan and T. Pal, Nano Lett., 2001, 1, 319–322 CrossRef CAS.
- Y. Sun, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- T. Pal, A. Ganguly and D. S. Maity, Anal. Chem., 1986, 58, 1564–1566 CrossRef CAS.
- A. Henglein, J. Phys. Chem., 1993, 97, 5457–5471 CrossRef CAS.
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881–3891 RSC.
- T. Pal, T. K. Sau and N. R. Jana, Langmuir, 1997, 13, 1481–1485 CrossRef CAS.
- T. Pal and P. K. Das, Analyst, 1988, 113, 1601–1603 RSC.
- T. Pal, J. Chem. Educ., 1994, 71, 679–681 CrossRef CAS.
- T. Pal, Inorg. Chim. Acta, 1983, 79, 283–284 CrossRef.
- W. T. Doyle, Phys. Rev., 1958, 111, 1067–1072 CrossRef CAS.
- U. Kreibig, J. Phys. F: Met. Phys., 1974, 4, 999–1014 CrossRef CAS.
- A. Henglein, P. Mulvaney, T. Linnert and A. Holzwarth, J. Phys. Chem., 1992, 96, 2411–2414 CrossRef CAS.
- J. L. Marignier, J. Belloni, M. O. Delcourt and J. P. Chevalier, Nature, 1985, 317, 344–345 CrossRef CAS.
- M. Michaelis and A. Henglein, J. Phys. Chem., 1992, 96, 4719–4724 CrossRef CAS.
- A. Henglein, Ber. Bunsenges. Phys. Chem., 1990, 94, 600–603 CrossRef CAS.
- J. H. Hodak, A. Henglein, M. Giersig and G. V. Hartland, J. Phys. Chem. B, 2000, 104, 11708–11718 CrossRef CAS.
- J. Zhu, Y. Wang, L. Huang and Y. Lu, Phys. Lett. A, 2004, 323, 455–459 CrossRef CAS.
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- L. M. Liz-Marzán, Langmuir, 2005, 22, 32–41 CrossRef PubMed.
- J. C. Hernández-Garrido, M. S. Moreno, C. Ducati, L. A. Pérez, P. A. Midgley and E. A. Coronado, Nanoscale, 2014, 6, 12696–12702 RSC.
- J. B. Jackson, S. L. Westcott, L. R. Hirsch, J. L. West and N. J. Halas, Appl. Phys. Lett., 2003, 82, 257–259 CrossRef CAS.
- L. Lu, G. Burkey, I. Halaciuga and D. V. Goia, J. Colloid Interface Sci., 2013, 392, 90–95 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- S. Pande, S. K. Ghosh, S. Praharaj, S. Panigrahi, S. Basu, S. Jana, A. Pal, T. Tsukuda and T. Pal, J. Phys. Chem. C, 2007, 111, 10806–10813 CrossRef CAS.
- M. Perez-Lloret, A. Fraix, S. Petralia, S. Conoci, V. Tafani, G. Cutrone, A. Vargas-Berenguel, R. Gref and S. Sortino, Chem.–Eur. J., 2019, 25, 14638–14643 CrossRef CAS PubMed.
- O. V. Makarova, A. E. Ostafin, H. Miyoshi, J. R. Norris and D. Meisel, J. Phys. Chem. B, 1999, 103, 9080–9084 CrossRef CAS.
- C. de Cointet, J. Khatouri, M. Mostafavi and J. Belloni, J. Phys. Chem. B, 1997, 101, 3517–3522 CrossRef CAS.
- G. C. Papavassiliou, J. Phys. F: Met. Phys., 1976, 6, L103–L105 CrossRef CAS.
- S. Link, Z. L. Wang and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 3529–3533 CrossRef CAS.
- D. Rioux and M. Meunier, J. Phys. Chem. C, 2015, 119, 13160–13168 CrossRef CAS.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- P. Schwerdtfeger, Heteroat. Chem., 2002, 13, 578–584 CrossRef CAS.
- H. Tang, C. J. Chen, Z. Huang, J. Bright, G. Meng, R. S. Liu and N. Wu, J. Chem. Phys., 2020, 152, 220901 CrossRef CAS PubMed.
- R. Sundararaman, P. Narang, A. S. Jermyn, W. A. Goddard and H. A. Atwater, Nat. Commun., 2014, 5, 5788 CrossRef CAS PubMed.
- A. M. Brown, R. Sundararaman, P. Narang, W. A. Goddard and H. A. Atwater, ACS Nano, 2016, 10, 957–966 CrossRef CAS PubMed.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- L. V. Besteiro, X. T. Kong, Z. Wang, G. Hartland and A. O. Govorov, ACS Photonics, 2017, 4, 2759–2781 CrossRef CAS.
- A. Furube and S. Hashimoto, NPG Asia Mater., 2017, 9, e454 CrossRef CAS.
- S. Y. Tee and E. Ye, Mater. Adv., 2021, 2, 1507–1529 RSC.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. Nevěčná and R. Zbořil, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef PubMed.
- P. Nalawade, P. Mukherjee and S. Kapoor, J. Nanostruct. Chem., 2014, 4, 113 CrossRef.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- H. Yoshida, Y. Kuwauchi, J. R. Jinschek, K. Sun, S. Tanaka, M. Kohyama, S. Shimada, M. Haruta and S. Takeda, Science, 2012, 335, 317–319 CrossRef CAS PubMed.
- C. Barroo, Z. J. Wang, R. Schlögl and M. G. Willinger, Nat. Catal., 2020, 3, 30–39 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Commun., 2001, 617–618 RSC.
- B. P. Khanal and E. R. Zubarev, J. Am. Chem. Soc., 2008, 130, 12634–12635 CrossRef CAS PubMed.
- N. Pradhan, A. Pal and T. Pal, Langmuir, 2001, 17, 1800–1802 CrossRef CAS.
- T. K. Sau, A. Pal and T. Pal, J. Phys. Chem. B, 2001, 105, 9266–9272 CrossRef CAS.
- S. Gu, S. Wunder, Y. Lu, M. Ballauff, R. Fenger, K. Rademann, B. Jaquet and A. Zaccone, J. Phys. Chem. C, 2014, 118, 18618–18625 CrossRef CAS.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC.
- S. Sarkar, S. Dutta, C. Ray, B. Dutta, J. Chowdhury and T. Pal, CrystEngComm, 2015, 17, 8119–8129 RSC.
- A. K. Sinha, M. Pradhan, S. Sarkar and T. Pal, Environ. Sci. Technol., 2013, 47, 2339–2345 CrossRef CAS PubMed.
- J. Pal, M. Ganguly, C. Mondal, Y. Negishi and T. Pal, Nanoscale, 2015, 7, 708–719 RSC.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49–52 CrossRef PubMed.
- Z. Lin, H. Hirao, C. Sun and X. Zhang, Phys. Chem. Chem. Phys., 2020, 22, 14458–14464 RSC.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS.
- I. Piskun, R. Blackwell, J. Jornet-Somoza, F. Zhao, A. Rubio, S. G. Louie and F. R. Fischer, J. Am. Chem. Soc., 2020, 142, 3696–3700 CrossRef CAS PubMed.
- S. Sakong and A. Gross, J. Chem. Phys., 2018, 149, 084705–084711 CrossRef PubMed.
- C. Mohr, H. Hofmeister, J. Radnik and P. Claus, J. Am. Chem. Soc., 2003, 125, 1905–1911 CrossRef CAS PubMed.
- T. Aditya, J. Jana, N. K. Singh, A. Pal and T. Pal, ACS Omega, 2017, 2, 1968–1984 CrossRef CAS PubMed.
- H. R. Liu, G. X. Shao, J. F. Zhao, Z. X. Zhang, Y. Zhang, J. Liang, X. G. Liu, H. S. Jia and B. S. Xu, J. Phys. Chem. C, 2012, 116, 16182–16190 CrossRef CAS.
- G. Giovannetti, P. A. Khomyakov, G. Brocks, V. M. Karpan, J. van den Brink and P. J. Kelly, Phys. Rev. Lett., 2008, 101, 026803–026804 CrossRef CAS PubMed.
- S. M. Song, J. K. Park, O. J. Sul and B. J. Cho, Nano Lett., 2012, 12, 3887–3892 CrossRef CAS PubMed.
- C. G. Vayenas, S. Bebelis and S. Neophytides, J. Phys. Chem., 1988, 92, 5083–5085 CrossRef CAS.
- H. D. Hagstrum, Phys. Rev., 1956, 104, 1516 CrossRef CAS.
- J. Kaur and R. Kant, J. Phys. Chem. C, 2017, 121, 13059–13069 CrossRef CAS.
- J. M. Heras and L. Viscido, Appl. Surf. Sci., 1980, 4, 238–241 CrossRef CAS.
- Q. Ding, Y. Shi, M. Chen, H. Li, X. Yang, Y. Qu, W. Liang and M. Sun, Sci. Rep., 2016, 6, 32724 CrossRef CAS PubMed.
- H. D. Hagstrum and Y. Takeishi, Phys. Rev., 1965, 137, 304–314 CrossRef CAS.
- S. Biswas, A. Pal and T. Pal, RSC Adv., 2020, 10, 35449–35472 RSC.
- S. Praharaj, S. Nath, S. K. Ghosh, S. Kundu and T. Pal, Langmuir, 2004, 20, 9889 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- H. Turan, B. Calis, A. N. Dizaji, S. Tarhan, H. Mazlumoglu, F. Aysin, A. Yilmaz and M. Yilmaz, Nanotechnology, 2021, 32, 315702 CrossRef CAS PubMed.
- M. T. Yaraki and Y. N. Tan, Chem.–Asian J., 2020, 15, 3180–3208 CrossRef CAS PubMed.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- M. Moskovits, J. Chem. Phys., 1978, 69, 4159 CrossRef CAS.
- E. Hesse and J. A. Creighton, Chem. Phys. Lett., 1999, 303, 101–106 CrossRef CAS.
- M. Mandal, S. Kundu, S. K. Ghosh, S. Panigrahi, T. K. Sau, S. M. Yusuf and T. Pal, J. Colloid Interface Sci., 2005, 286, 187–194 CrossRef CAS PubMed.
- S. Sarkar, A. K. Sinha, M. Pradhan, M. Basu, Y. Negishi and T. Pal, J. Phys. Chem. C, 2011, 115, 1659–1673 CrossRef CAS.
- J. Piella, N. G. Bastus and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- J. Wang, L. Jiang, F. Liu, M. Jia, M. Liu, J. Li and Y. Lai, Chem. Eng. J., 2021, 407, 127195 CrossRef CAS.
- S. Berciaud, L. Cognet, P. Tamarat and B. Lounis, Nano Lett., 2005, 5, 515–518 CrossRef CAS PubMed.
- Y. Huang, L. Fuksman and J. Zheng, Dalton Trans., 2018, 47, 6267–6273 RSC.
- H. Baida, P. Billaud, S. Marhaba, D. Christofilos, E. Cottancin, A. Crut, J. Lermé, P. Maioli, M. Pellarin, M. Broyer, N. Del Fatti, F. Vallée, A. Sánchez-Iglesias, I. Pastoriza-Santos and L. M. Liz-Marzán, Nano Lett., 2009, 9, 3463–3469 CrossRef CAS PubMed.
- B. A. Ashenfelter, A. Desireddy, S. H. Yau, T. Goodson and T. P. Bigioni, J. Phys. Chem. C, 2015, 119, 20728–20734 CrossRef CAS.
- Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2015, 44, 8636–8663 RSC.
- Z. Lin, X. Wang, J. Liu, Z. Tian, L. Dai, B. He, C. Han, Y. Wu, Z. Zeng and Z. Hu, Nanoscale, 2015, 7, 4114–4123 RSC.
Footnote |
| † Present address: Institute of Analytical and Environmental Sciences, National Tsing Hua University, Hsinchu 30013, Taiwan. |
| This journal is © The Royal Society of Chemistry 2022 |