
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpirocyclic side chain of a non-fullerene acceptor enables efficient organic solar cells with reduced recombination loss and energetic disorder†
Guangkun Song‡
a,
Yuzhong Huang‡b,
Fangfang Huangb,
Xiangjian Wan b,
Chenxi Lib,
Zhaoyang Yaob,
Yongsheng Chen*b and
Yanhui Hou*a
b,
Chenxi Lib,
Zhaoyang Yaob,
Yongsheng Chen*b and
Yanhui Hou*a
aState Key Laboratory of Separation Membrane and Membrane Processes, Tianjin Municipal Key Laboratory of Advanced Fibers and Energy Storage, School of Materials Science and Engineering, Tiangong University, Tianjin 300387, China. E-mail: houyanhui@tiangong.edu.cn
bState Key Laboratory and Institute of Elemento-Organic Chemistry, The Centre of Nanoscale Science and Technology, Key Laboratory of Functional Polymer Materials, Renewable Energy Conversion and Storage Center (RECAST), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: yschen99@nankai.edu.cn
First published on 25th February 2022
Abstract
Suppressing intramolecular vibration of non-fullerene acceptors (NFAs) by molecular rigidification has been proven to be an effective way to reduce the non-radiative recombination loss and energetic disorder of organic solar cells (OSCs). Thus far, extensive attention has been drawn on rigidifying the fused-ring backbones of NFAs, whereas the highly flexible alkyl side chains are barely concerned. Herein, an effective strategy of side chain rigidification by introducing a spiro-ring is developed for the first time and applied to construct the NFA of Spiro-F. Compared to its counterpart F-2F, the rigid spirocyclic side chain can constrain the vibrational–rotational motion and control the orientation of two highly flexible n-octyl chains effectively. As a result, an optimal molecular packing with enhanced intermolecular actions and lower energetic disorder is achieved by Spiro-F, endowing the OSC based on the as cast blend of PM6:Spiro-F with a significantly improved PCE of 13.56% and much reduced recombination loss compared to that of PM6:F-2F. This work provides a feasible strategy to achieve efficient OSCs through the rigidification of the side chain and could boost the PCEs further if applied to some other efficient systems with smaller bandgaps.
Introduction
Organic solar cells (OSCs) as a green photon-to-current conversion technology have been extensively investigated owing to their excellent merits of light weight, mechanical flexibility, potential low cost etc.1–3 Due to the rapid development of acceptor–donor–acceptor4 (A–D–A) type non-fullerene acceptors (NFAs), especially the emergence of super-star Y6 and its analogs, the current certified efficiencies of more than 18% (ref. 5–10) and 20% (ref. 11) have been reached by single-junction and multi-junction OSCs, respectively. In spite of the rapidly increasing PCE of OSCs, there is still a huge efficiency gap between OSCs and perovskite solar cells or inorganic silicon solar cells due to the much higher energy loss (Eloss) of OSCs in the charge generation process compared to that of the other two counterparts.12,13 Note that the best-performing inorganic solar cell displays a very low Eloss in gallium arsenide (0.32 eV) and crystalline silicon (0.38 eV),14 while the record value for perovskite solar cells is 0.34 eV.15 In sharp contrast, the state-of-the-art OSCs still suffer from a much larger Eloss in the range of 0.6–0.9 eV (ref. 16) than that of not only the inorganic or perovskite counterparts, but also the ideal value of 0.25–0.30 eV predicted by Shockley–Queisser (SQ) theory.17 Therefore, for the sake of further improving PCEs of OSCs, the last but probably most crucial challenge confronted by OSCs should be suppressing Eloss to reach comparable open-circuit voltage (VOC) to that of inorganic solar cells with the similar bandgaps (Eg). For instance, if reducing the Eloss of OSCs towards similar values 0.4 eV or less to that of PSCs etc., over 20% PCEs could be achieved immediately based on our semi empirical model.18 Unfortunately, despite the significant progress having been made thus far,4 it is still unclear in mechanism or lack of effective routes to reduce Eloss greatly in OSCs.Extensive theoretical investigations have indicated that the non-radiative recombination from the lowest-energy charge-transfer (CT) state to ground states (GS) at donor (D)/acceptor (A) interfaces should account for the majority of energy loss in OSCs,19,20 which is tightly related to the intramolecular vibrations of light-harvesting molecules.21–24 Moreover, organic semiconductors generally possess the relatively larger energetic disorder than that of inorganic semiconductors, resulting in a substantial broadening of electronic density of states (DOS)25–27 which is reflected in the absorption of CT states at the D/A interface.17 Note that the enlargement of energetic disorders, and thus the broadening of DOS could invariably lead to the larger recombination loss including not only the non-radiative but also radiative energy loss.28–31 The features of CT states are closely related to the components with small bandgap in the D/A blends,32 indicating that the structure of NFAs is very crucial in reducing energy losses of OSCs due to its dominant role in harvesting low-energy photons. Generally, the state-of-art NFAs usually possess an A–D–A type of molecular architecture, which contributes to not only their superior exciton separation and charge transport, but also the potential to greatly reduce charge generation incurred Eloss.33 Therefore, a lot of structural modifications on NFAs, especially the rigidification of backbones through fused ring strategy to constrain the intramolecular vibrations,24,34–40 have been explored for mitigating the recombination loss and improving PCEs of OSCs successfully. Note that the highly flexible side chains, e.g. alkyl chains, are widely employed in both NFAs and polymeric donors to increase the solubility of materials. However, the resulting severe intramolecular vibrational–rotational motion may cause undesirable molecular packing and more importantly increase the energetic disorders, thus enlarging the recombination loss.41 Although extensive side chains engineering has been conducted thus far, they are mainly focusing on the length or isomerization of aliphatic chains, where the impact of flexibility has not been investigated.42–44 Thereby, unveiling the potentially significant effects of side chain flexibility/rigidification on energy losses of OSCs and more importantly, exploring the effective strategies to reduce energy losses should be highly demanded in the studies of OSCs. Note that the above issue still confronts great challenges in terms of molecular design/synthesis and very few attentions have been drawn on that so far.
In this work, an effective strategy of side chain rigidification by introducing spiro-ring structure is developed for the first time and applied to our previously reported NFA of F-2F45 to construct the target Spiro-F (Fig. 1a). As a result, the spirocyclic side chain on Spiro-F reduces its degree of freedom for vibrational–rotational motion and tunes its orientation, leading to a red-shifted film absorption, further enhanced intermolecular packing and more importantly reduced energetic disorder with respect to those of the control molecule F-2F. When blending with a well-known polymer donor PM6, the OSCs employing Spiro-F as the acceptor achieves an impressive PCE of 13.56% compared with 12.67% of the control molecule F-2F based OSCs, along with an overall ungraded Jsc and FF. What is more important, a significantly suppressed Eloss was achieved for Spiro-F based OSCs with respect to that of F-2F, demonstrating the effectiveness of side chain rigidification as a feasible strategy to reduce Eloss of OSCs.
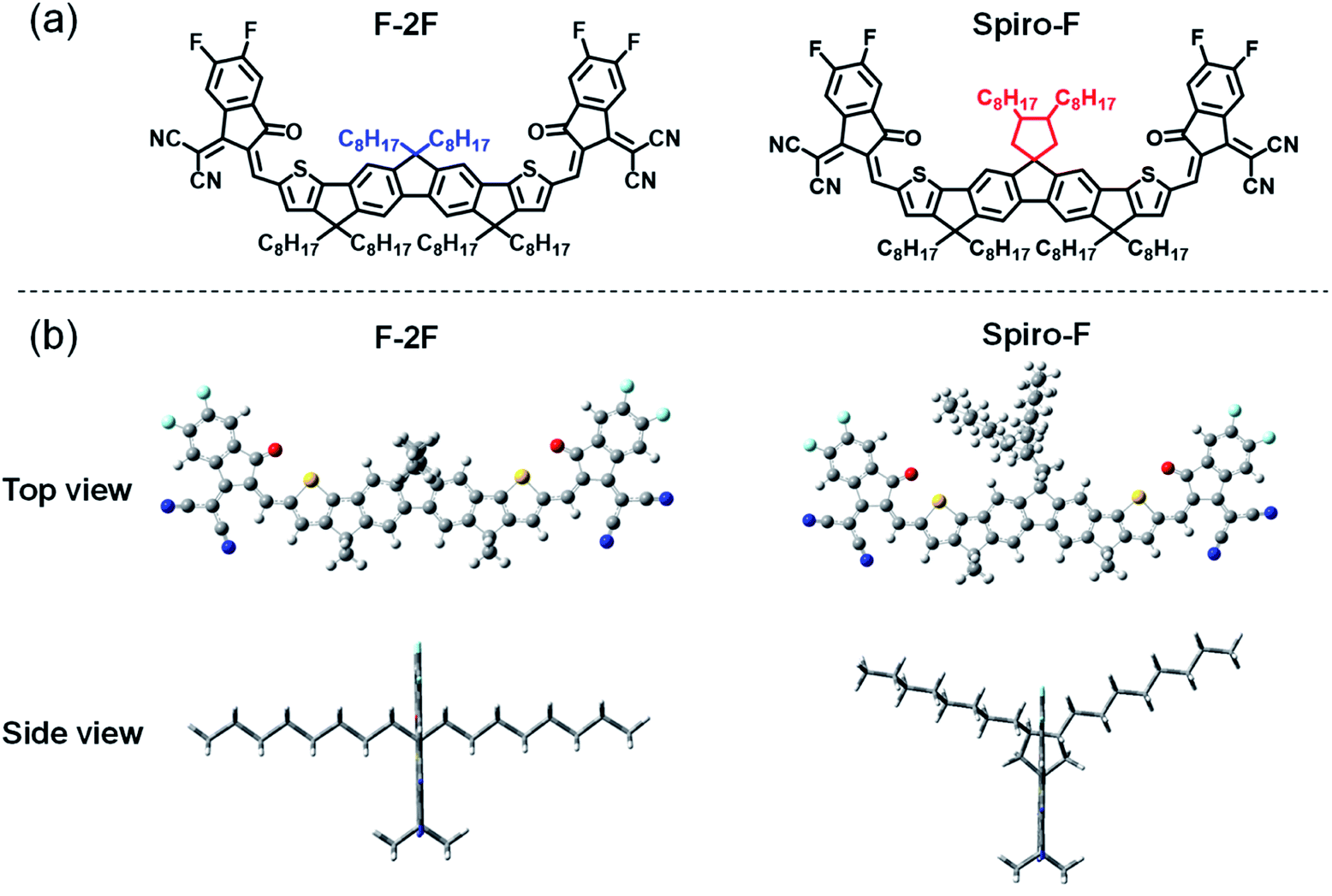 | ||
| Fig. 1 The chemical structures and optimized geometries of (a) F-2F and (b) Spiro-F from top view and side view. | ||
The synthetic route to Spiro-F was shown in Scheme 1 and the detailed procedures and characterizations were presented in the (ESI†). 2,3-Dimethylbuta-1,3-diene (1) was firstly reacted with n-BuLi and t-BuOK at room temperature and following by addition of 1-bromoheptane to afford compound 2 in a good yield (86%). Then compound 2 underwent a borohydride oxidation at the present of B2H6 and H2O2 to generate compound 3, in which the two hydroxy groups were subsequently protected by OTs-groups through reacting with tosyl chloride to yield compound 4 with an excellent yield of 91%. Compound 4 was further dissolved in acetonitrile and reacted with lithium bromide to give 9,10-bis(bromomethyl)octadecane 5 (yield: 74%). Then at the present of KOH, 2,7-dibromo-9H-fluorene (6) was performed a very high yield (92%) alkylation using compound 5 as the alkylating regent to obtain compound 7. Furthermore, compound 7 underwent a halogen–lithium exchange with n-butyllithium at a cryogenic temperature and then transformed to its correlated boric acid ester (8), which was reacted with ethyl 2-bromothiophene-3-carboxylate directly via the Suzuki cross-coupling to afford compound 9 in an overall yield of 83%. Later, compound 9 was hydrolyzed with NaOH as the catalyst, and the hydrolysate was thoroughly acidified with an aqueous hydrochloric acid (1 M) to give compound 10 almost quantitatively. Thereafter, an intramolecular Friedel–Crafts acylation of compound 10 with oxalyl chloride was carried out with the catalysis of acidic AlCl3 to generate the diketone compound 11, which was further transformed into compound 12 by a reduction using hydrazine. Then the alkylation of compound 12 using 1-bromooctane as the alkylating regent was conducted to afford compound 13 with a moderate yield of 67%, which was further converted into its corresponding dialdehyde (14) via the Vilsmeier–Haack reaction. Eventually, the target A–D–A type NFA Spiro-F was afforded with a decent yield of 78% through a condensation reaction between above dialdehyde and 2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile. The chemical structures of key intermediates and target Spiro-F were characterized and confirmed by 1H and 13C NMR spectra (Fig. S8–S18 in ESI†), and high-resolution mass spectrometry (Fig. S18 in ESI†). Note that Spiro-F exhibited good solubility in widely used organic solvents, such as dichloromethane, chloroform and chlorobenzene, in favor of its solution-processing during the device fabrication.
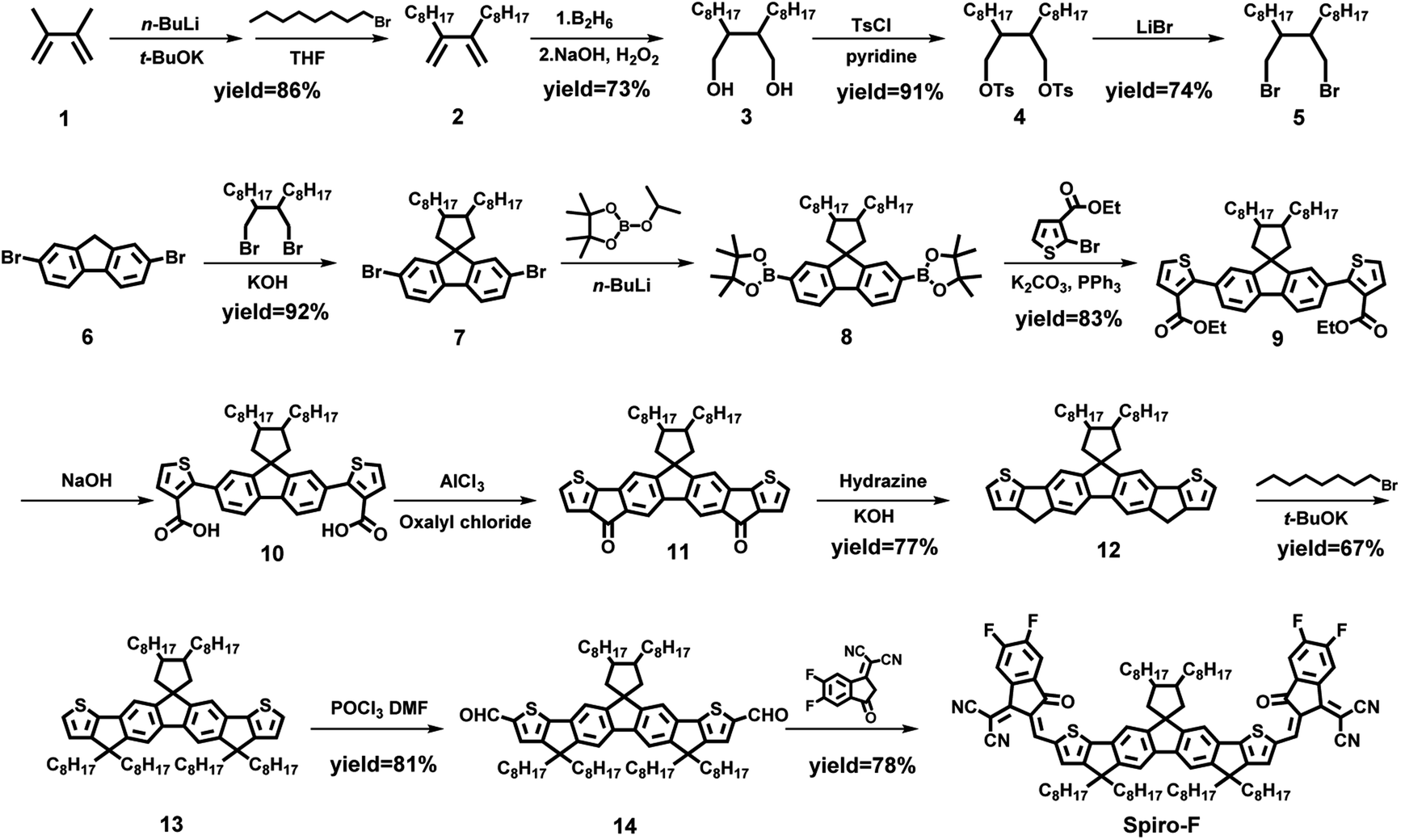 | ||
| Scheme 1 The synthetic route to Spiro-F. | ||
Fig. 1a displayed the chemical structures of F-2F and Spiro-F. Note that both NFAs possess the same fluorene based conjugated backbones, enabling spirocyclic side chain the only difference, so that the side chain effects on energy levels, absorptions, molecular packing, film morphology, even PCEs of OSCs can be clearly investigated below. Density functional theory (DFT) calculations at single molecular level were firstly performed to evaluate the variation of frontier molecular orbital energy levels after introducing the spirocyclic side chain. As it can be expected, both the distributions of frontier molecular orbitals and the corresponding energy levels are not affected by spirocyclic side chain due to the unconjugated property of spiro-rings, thus affording the same highest occupied molecular orbitals (HOMOs) of −5.65 eV and lowest unoccupied molecular orbitals (LUMOs) of −3.45 eV for both F-2F and Spiro-F (Fig. S1†). However, the plane of introduced spiro-ring is perpendicular to the fluorene backbone as observed obviously from the geometry of Spiro-F (Fig. 1b), restricting the orientation of two n-octyl groups. This constraint molecular conformation may result in more ordered and tighter intermolecular packing of Spiro-F than that of F-2F, which should be beneficial to better carrier transportation, lower energetic disorders, smaller recombination loss, and eventually higher PCE of OSCs.
In order to investigate the absorption change of NFAs after substituting by spirocyclic side chain, UV-Vis spectra of F-2F and Spiro-F in diluted chloroform solutions and nest films were recorded in Fig. 2a. Both F-2F and Spiro-F possess almost the identical absorption with the maximum absorption peaks λsolmax at 677 and 678 nm, respectively, which can be expected from the same energy levels and bandgaps predicted by the DFT calculation at single molecular level. Note that the greatly red-shifted maximum absorption peaks λfilmmax can be observed for both F-2F and Spiro-F from solutions to solid films, indicating the strong intermolecular interactions.46,47 What is more interesting, the as cast solid film of Spiro-F displays the λfilmmax at 727 nm, bathochromically shifted by nearly 27 nm comparing to that of 700 nm for F-2F, suggesting the enhanced intermolecular π–π stacking of Spiro-F with a rigid spirocyclic side chain.48 Furthermore, the absorption edge (λonset) of Spiro-F is located at 799 nm, also significantly redshifted with respect to that of 773 nm for F-2F, corresponding to the optical bandgaps of 1.55 and 1.60 eV, respectively. Note that the polymeric donor PM6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49 has been selected as the best to blend with F-2F according to our previous work,45 therefore we further measured the absorption of blend films of PM6:F-2F and PM6:Spiro-F (Fig. 2b). As it can be observed that both NFAs show the complementary absorption with PM6, rendering an efficient light harvesting in a broad range of 500–800 nm.
49 has been selected as the best to blend with F-2F according to our previous work,45 therefore we further measured the absorption of blend films of PM6:F-2F and PM6:Spiro-F (Fig. 2b). As it can be observed that both NFAs show the complementary absorption with PM6, rendering an efficient light harvesting in a broad range of 500–800 nm.
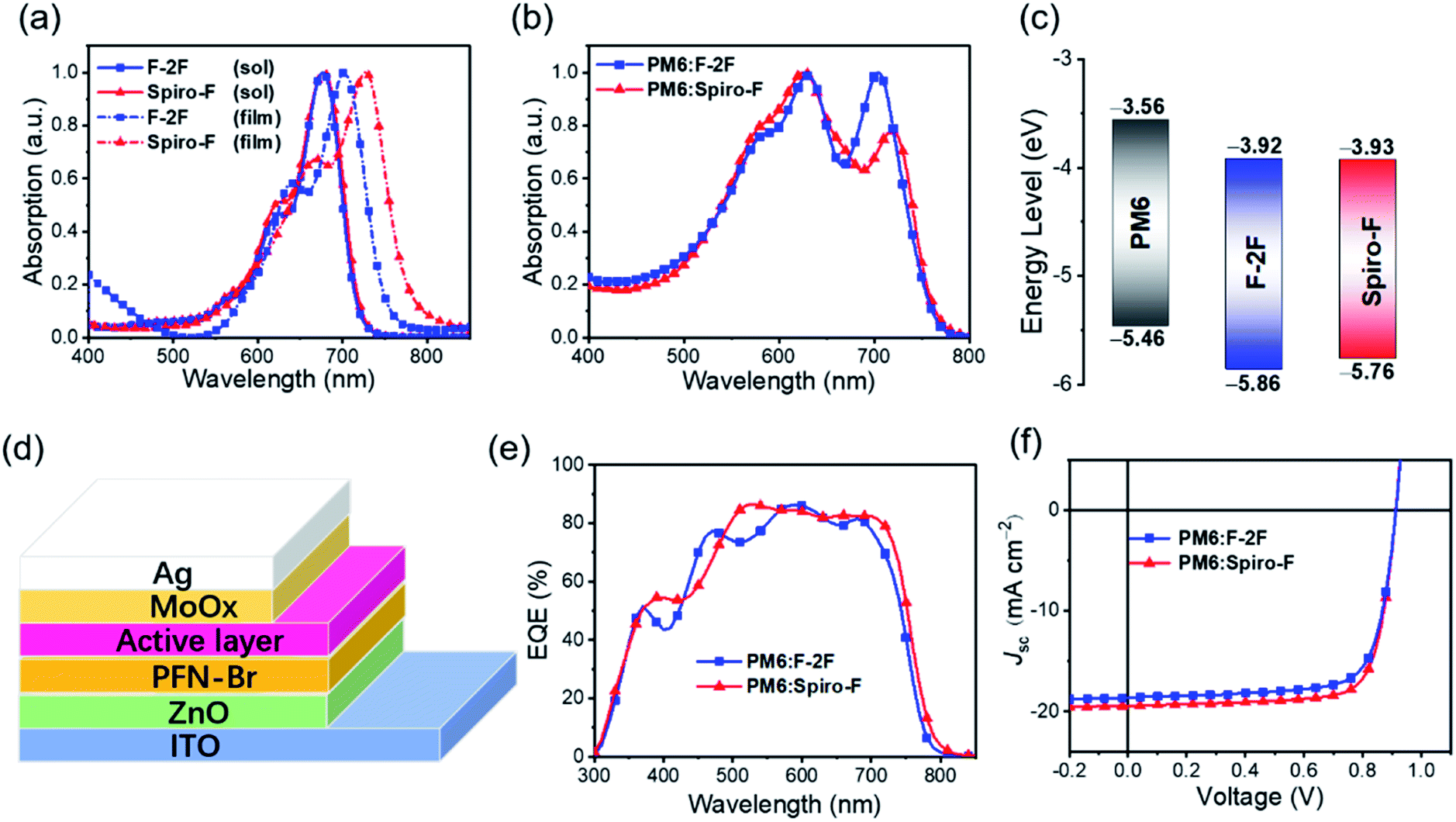 | ||
| Fig. 2 (a) Normalized absorption spectra of F-2F and Spiro-F in diluted solutions of chloroform and neat films. (b) Normalized absorption spectra of PM6:F-2F and PM6:Spiro-F based blend films. (c) Energy level diagram of PM6, F-2F and Spiro-F derived from the electrochemical cyclic voltammetry. (d) The OSC with an architecture of ITO/ZnO/PFN-Br/active layer/MoOx/Ag. (e) EQE curves and (f) current density–voltage (J–V) of the optimized devices. | ||
The experimental energy levels of F-2F and Spiro-F in solid states were investigated by electrochemical cyclic voltammetry (CV) measurements (Fig. S2†). As shown in Fig. 2c, the similar LUMO energy levels of −3.93 eV and −3.92 eV can be observed by Spiro-F and F-2F, respectively. However, the HOMO energy level of Spiro-F can be estimated at −5.76 eV, having a nearly 0.1 eV unshifting comparing to that of −5.86 eV for F-2F. Therefore, the energy gap of Spiro-F could be calculated as 1.83 eV, smaller than that of 1.94 eV for F-2F, which is in good accordance with the redshifted absorption of Spiro-F film. The detailed parameters for optical absorption and energy levels were listed in Table 1 for a clear comparison.
To evaluate the photovoltaic properties of the two acceptors, bulk heterojunction OSCs with a device structure of ITO/ZnO/PFN-Br/active layer/MoOx/Ag (Fig. 2d) were fabricated. In the device optimization, polymer PM6 was used as the donor, in considering its well complementary absorption and matched energy levels with both F-2F and Spiro-F. The detailed data of device optimization were listed in Tables S1 and S2 in ESI.† Fig. 2e displayed the external quantum efficiencies (EQEs) of F-2F and Spiro-F based OSCs. Among them, over 80% EQE can be achieved in a broad range of 490–725 nm for Spiro-F based OSCs, indicating the efficient photon harvesting, exciton dissociation, charge transport and collection. Therefore, the Spiro-F based OSCs afford a much larger integrated Jsc of 18.89 mA cm−2 than that of 17.96 mA cm−2 for F-2F, in good accordance with the redshifted absorption of Spiro-F.
Fig. 2f recorded the current density–voltage (J–V) curves of OSCs based on F-2F and Spiro-F. For a clear comparison, the related photovoltaic parameters of OSCs were summarized in Table 2. The OSC based on PM6:F-2F blend exhibits a good PCE of 12.67% with a VOC of 0.913 V, short-circuit current density (Jsc) of 18.63 mA cm−2 and fill factor (FF) of 74.49%, which is comparable to that reported in our previous work.45 After replacing the two n-octyl groups on fluorene of F-2F with a spirocyclic alkyl chains, it is worth noting that the OSC based on PM6:Spiro-F blend exhibits an impressive PCE of 13.56% with a comparable VOC of 0.912 V. However, both upgraded Jsc of 19.45 mA cm−2 and FF of 76.35% with respect to that of PM6:F-2F based OSCs. In comparison with the OSCs based on F-2F, the enlarged Jsc of Spiro-F should be mainly ascribed to the redshifted absorption, which agrees well with the integrated Jsc from the EQE spectrum. The improved FF of Spiro-F based device should be attributed to the spirocyclic alkyl chains, which can result in the tighter intermolecular packing, improved and well-balanced charge mobilities discussed below. The greatly improved PCE of Spiro-F based OSCs indicates the effectiveness of introducing rigid spirocyclic alkyl chains to boost the efficiency of OSCs.
| Devices | VOC [V] | Jsc [mA cm−2] | FF [%] | PCE [%] |
|---|---|---|---|---|
| a The values in parenthesis are average parameters obtained from 10 devices. | ||||
| PM6:F-2F | 0.913 (0.913 ± 0.02)a | 18.63 (18.27 ± 0.29) | 74.49 (75.30 ± 0.63) | 12.67 (12.55 ± 0.19) |
| PM6:Spiro-F | 0.912 (0.909 ± 0.03) | 19.45 (19.37 ± 0.28) | 76.35 (75.69 ± 0.69) | 13.56 (13.31 ± 0.19) |
In order to reveal the origin of improved Jsc and FF for Spiro-F based OSCs, the exciton dissociation efficiencies (Pdiss) were firstly evaluated by measuring the photocurrent densities (Jph) versus effective voltages (Veff) plots for the optimized devices (Fig. 3a). The Jph is obtained from the formula of Jph = JL − JD, where JL and JD are the current densities under illumination and in the dark, respectively. Veff is defined as V0 − V, where V0 is the voltage when Jph is zero and V is the applied voltage.50 It is supposed that all photogenerated excitons would be dissociated into free carriers at Veff = 2 V where the photocurrent reaches saturation condition (Jsat). Therefore, the Jph/Jsat ratio can be used to characterize the exciton dissociation and charge collection efficiency. Under short circuit conditions, the Jph/Jsat values for F-2F and Spiro-F based OSCs are 97% and 96%, respectively, indicating the highly efficient exciton dissociation in both devices. Furthermore, the dependence of Jsc on the light intensity (P) has been performed to investigate the bimolecular recombination of OSCs51 (Fig. 3b). The relationship between Jsc and P follows the equation of Jsc ∝ Pα, and the bimolecular recombination can be regarded as negligible when α is close to unit.51 As shown in Fig. 3b, the α values of the optimized PM6:F-2F and PM6:Spiro-F devices are 0.961 and 0.968, respectively. The indicative negligible charge recombination will be in favor of the high VOC and FFs for both devices. Thereafter, Fig. 3c recorded the VOC dependence of light intensity (P) for the optimized OSCs to evaluate the trap-assist recombination. The slopes (S) of VOC versus the natural logarithm of P were calculated to be 1.40 and 1.26 kT/q for PM6:F-2F and PM6:Spiro-F based OSCs, respectively. The smaller slope for PM6:Spiro-F based OSCs indicates the lower trap-assisted recombination loss,52 which can be further confirmed by its lower energetic disorders below.
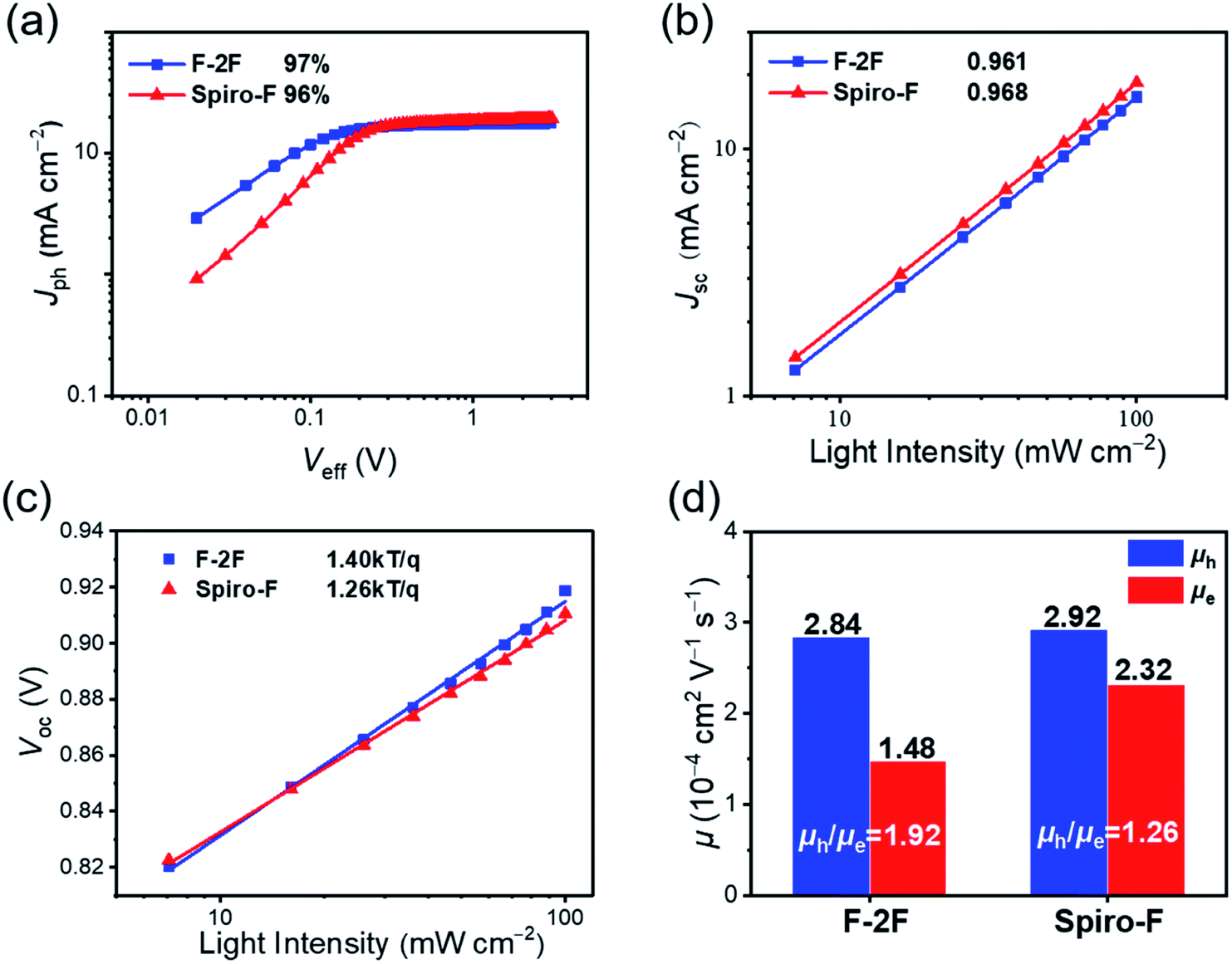 | ||
| Fig. 3 (a) Jph versus Veff plots. (b) Light intensity (P) dependence of Jsc and (c) light intensity (P) dependence of VOC plots for the optimized PM6:F-2F and PM6:Spiro-F based OSCs. (d) The hole (μh) and electron (μe) mobilities of PM6:F-2F and PM6:Spiro-F devices. | ||
The dynamics of free charge were further investigated by employing transient photocurrent (TPC) and transient photovoltage (TPV) measurements in Fig. S3,† respectively, to unveil the carrier transport chrematistics in PM6:F-2F and PM6:Spiro-F blends. Among them, TPCs were measured at the short circuit condition and can reveal the charge extraction process of OSCs.53 As displayed in Fig. S3a,† the PM6:Spiro-F device demonstrates a faster charge extraction (0.99 μs) comparing to that of PM6:F-2F device (1.03 μs), which is beneficial for the charge transport in blend films45 and also in good agreement with the improved charge mobility of PM6:Spiro-F device discussed below. On the other hand, the TPV measurement could disclose the charge recombination kinetics in OSCs.54 As shown in Fig. S3b,† the PM6:Spiro-F device possesses a longer carrier lifetime of 52 μs than that of 38 μs for PM6:F-2F device, indicating a suppressed charge recombination in PM6:Spiro-F based OSCs.55 Moreover, the charge carrier transport ability including the electron mobility (μe) and hole mobility (μh) of the blended films were estimated by the space-charge limited current (SCLC) method56 (Fig. S4 and S5†). As shown in Fig. S4,† the electron mobilities of the neat films for F-2F and Spiro-F are 1.51 and 2.25 cm2 V−1 s−1, respectively. The slightly increased electron mobility of Spiro-F should be attributed to its rigid spirocyclic side chain. As displayed in Fig. 3d, the PM6:F-2F based device exhibits a hole and electron mobility of 2.84 × 10−4 and 1.48 × 10−4 cm2 V−1 s−1, respectively, rendering a μh/μe ratio of 1.92. Note that for the PM6:Spiro-F based device, a similar hole mobility of 2.92 × 10−4 cm2 V−1 s−1, but much larger electron mobility of 2.32 × 10−4 cm2 V−1 s−1 can be achieved with respect to that of PM6:F-2F, resulting in a more balanced μh/μe ratio of 1.26. Apparently, the higher and more balanced charge mobilities for the PM6:Spiro-F blend may be caused by the more ordered molecular packing of Spiro-F with a rigid spirocyclic alkyl side chain, and should account for the improved FF of Spiro-F based device. Note that all the parameters discussed above have been summarized in Table S3† for a clear presentation.
The introduction of rigid spirocyclic alkyl chain on Spiro-F will inevitably result in the variation of microscopic morphology of the active layer, which should play a dominant role in the improved Jsc, FF and thus PCE for Spiro-F based OSCs discussed above. Therefore, it is crucial to unveil the morphology change of active layers from PM6:F-2F to PM6:Spiro-F at a level of nanoscale. Fig. 4a and b exhibited the images of atomic force microscope (AFM) for PM6:F-2F and PM6:Spiro-F blends, respectively. The relatively smooth surface can be observed by both PM6:F-2F and PM6:Spiro-F blends with a root-mean-square (RMS) roughness of 1.57 and 1.32 nm, respectively, due to the good miscibility of PM6 with both F-2F and Spiro-F. The smooth surface morphologies in both blended films will be beneficial for the effective contact between active layer and electrode, and thus efficient interfacial charge transfer.
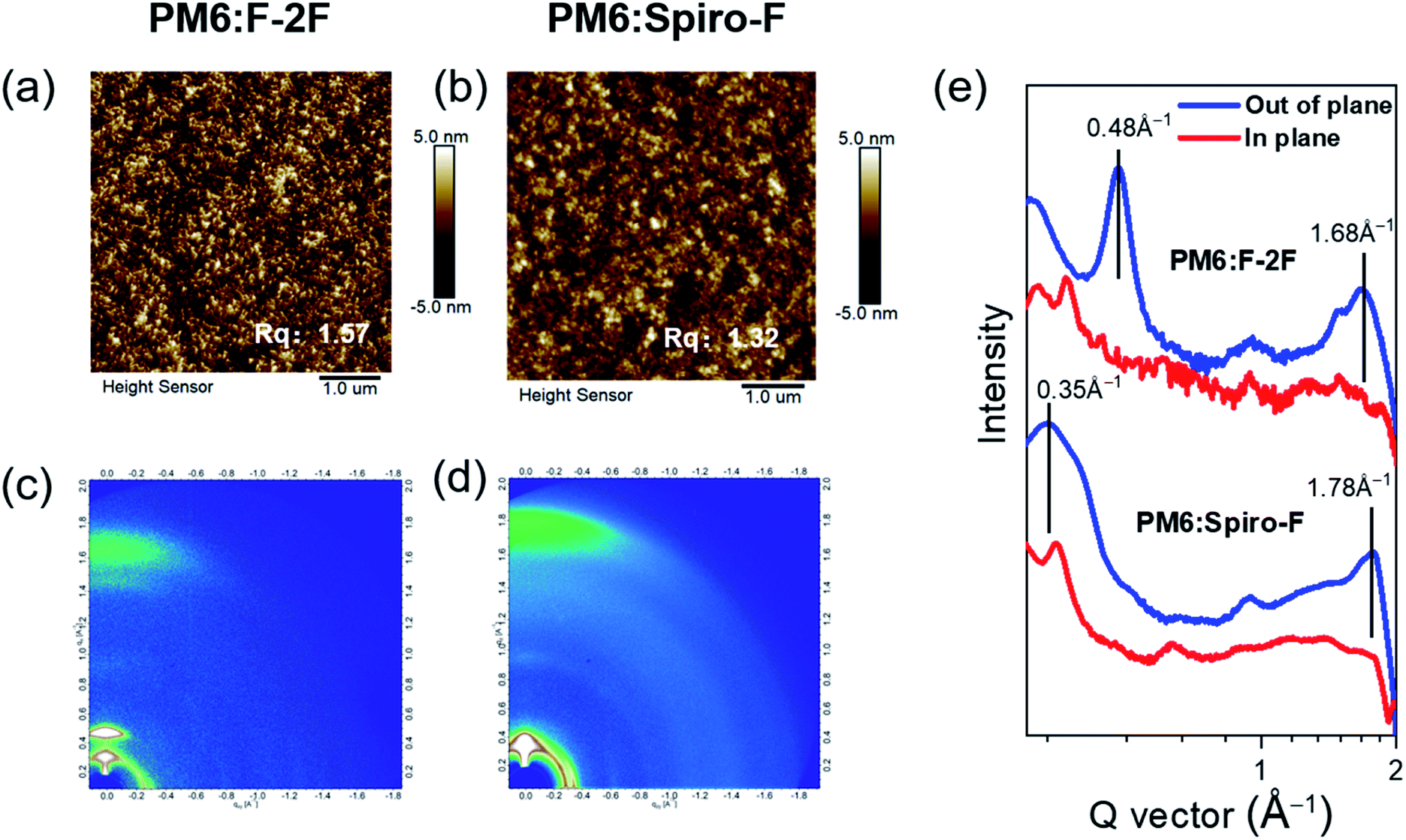 | ||
| Fig. 4 AFM images for (a) PM6:F-2F blend film and (b) PM6:Spiro-F blend film. Two dimensional GIWAXS patterns for (c) PM6:F-2F blend and (d) PM6:Spiro-F blend. (e) In-plane (red lines) and out-of-plane (blue lines) line cuts of the corresponding GIWAXS patterns. | ||
The introduction of a rigid spirocyclic alkyl side chain on central fluorene unit of Spiro-F should affect the intermolecular packing markedly, thereby the grazing incidence wide-angle X-ray diffraction (GIWAXS) was further carried out to study the molecular packing and orientation in blended films of PM6:F-2F and PM6:Spiro-F. Fig. 4c and d displayed the two dimensional GIWAXS patterns for PM6:F-2F and PM6:Spiro-F blends, respectively. The derived in-plane and out-of-plane line cuts were exhibited in Fig. 4e and the detailed parameters were summarized in Table 3. Note that both blended films possess the preferential face-on orientation as evidenced by the strong (010) π–π stacking peaks at ∼1.7 Å−1 in the out-of-plane direction. When taking a more in-depth observation, we found that the PM6:Spiro-F film demonstrates a (010) π–π stacking peak at 1.78 Å−1, corresponding to a distance of 3.52 Å. This is smaller than that of 3.75 Å for PM6:F-2F film indicated by the (010) π–π stacking peak at 1.68 Å−1. Moreover, the crystal coherence length (CCL) in the (010) region for PM6:Spiro-F blend can be estimated as 32.9 Å, much larger than that of 20.8 Å for PM6:F-2F blend. The enhanced CCL of PM6:Spiro-F indicates the more ordered molecular packing,39 in good accordance with the improved hole/electron mobilities comparing to that of PM6:F-2F. Generally, the rigid spirocyclic alkyl side chain in Spiro-F constraints the vibrational–rotational motion and controls the orientation of two highly flexible n-octyl chains, in favour of the preferred face-on orientation, ordered and enhanced molecular π–π stacking and increased CCL. The above superior characteristics of Spiro-F facilitate the efficient charge transport and suppressed charge carrier recombination, accounting for the better photovoltaic performance of PM6:Spiro-F based OSCs comparing to that of PM6:F-2F based one.
| Blends | qa (010, Å−1) | db (010, Å) | FWHMc (010, Å−1) | CCLd (010, Å) | q (100, Å−1) | d (100, Å) |
|---|---|---|---|---|---|---|
| a The location of diffraction peaks.b The π–π stacking distance.c The full width at half maxima of peaks.d The crystal coherence lengths calculated from the Scherrer equation: CCL = 2πk/FWHM.57 | ||||||
| PM6:F-2F | 1.68 | 3.75 | 0.27 | 20.8 | 0.48 | 13.09 |
| PM6:Spiro-F | 1.78 | 3.52 | 0.17 | 32.9 | 0.33 | 19.04 |
As we discussed above, the rigid spirocyclic alkyl side chain on Spiro-F could facilitate more ordered and enhanced molecular π–π stacking, thus suppress the recombination loss in OSCs. Therefore, we analysed the energy losses of OSCs based on both PM6:F-2F and PM6:Spiro-F blends to disclose the inner relationship of structure-energy loss. The total Eloss in OSCs can be categorized into three contributions (ΔEloss = ΔE1 + ΔE2 + ΔE3) based on the detailed balance theory58 (Fig. 5a): (1) ΔE1 (= Eg − qVSQOC) is the energy offset between the bandgap edge (Eg) and qVOC in the Shockley–Queisser (SQ) limit (qVSQOC); (2) ΔE2 (= qVSQOC − qVradOC) is the energy offset between the qVSQOC and qVOC in the radiative limit (qVradOC); (3) ΔE3 (= qVradOC – qVOC) is the energy offset between the qVradOC and qVOC in a realistic OPV devices. As shown in Fig. S6,† the Eg can be accurately determined by the crossing point of normalized absorption and photoluminescence (PL) spectra, being 1.715 and 1.656 eV for PM6:F-2F and PM6:Spiro-F blends, respectively. Thereby the total Eloss can be calculated as 0.802 and 0.744 eV for PM6:F-2F and PM6:Spiro-F based OSCs, respectively, demonstrating a 0.058 eV reduction after the spiro cyclization of alkyl chains. Among them, the ΔE1 is the inevitable loss in any types of solar cells58 and can be calculated to be the nearly identical values of 0.284 and 0.280 eV for PM6:F-2F and PM6:Spiro-F based OSCs, respectively. The ΔE2 can be regarded as the additional radiative recombination loss from absorption below the bandgap,59 which are 0.145 and 0.111 eV for PM6:F-2F and PM6:Spiro-F based OSCs, respectively. The ΔE3 is non-radiative energy loss, which usually contributes to the largest part of Eloss. Herein, the ΔE3 for PM6:Spiro-F based OSCs is 0.353 eV, smaller than that of 0.373 eV for PM6:F-2F based ones. The detailed calculation of three parts energy losses above were described in ESI.† Moreover, the reduced ΔE3 of PM6:Spiro-F based OSCs is also confirmed by the much higher external electroluminescence quantum efficiency (EQEEL) of devices in Fig. 5b, based on the equation: ΔE3 = −kBTln(EQEEL),60 where kB is Boltzmann's constant and T is absolute temperature. As shown in Fig. S7,† we have measured the PL and PL quenching yields (PLQY) of F-2F and Spiro-F films. The PLQY of F-2F and Spiro-F films are 0.87% and 1.07%, respectively. The increased PLQY for Spiro-F film with respect to that of F-2F could reduce the non-radiative decay, which should be in favor of the smaller ΔE3 for corresponding OSCs.20 Note that low photon energy losses or recombination losses achieved by the PM6:Spiro-F device is mainly a combined result of both reduced ΔE2 and ΔE3 (Table 4).
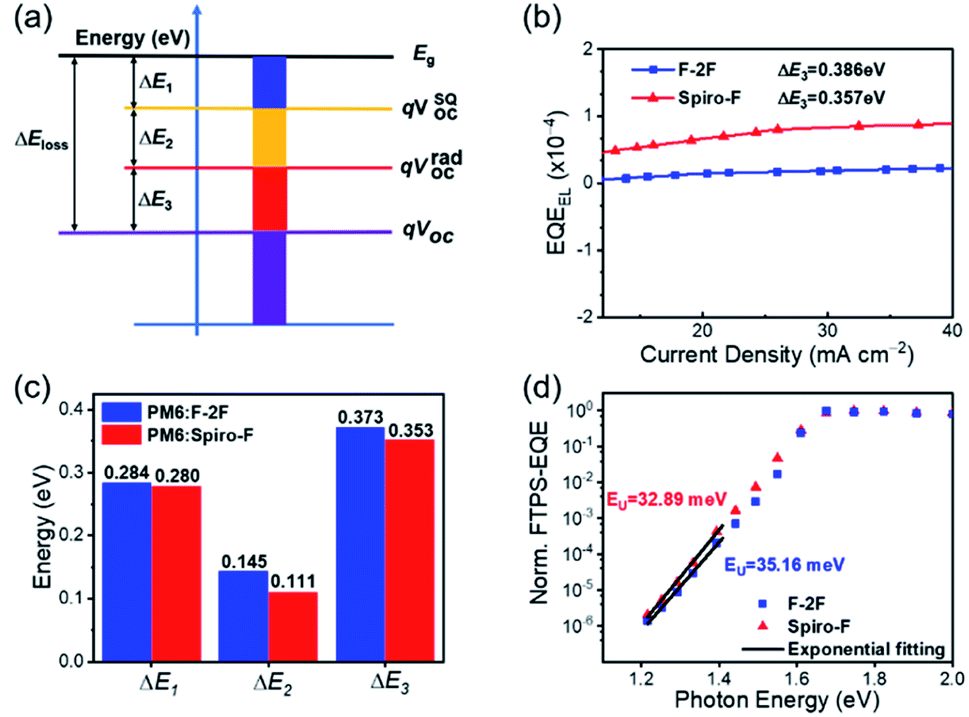 | ||
| Fig. 5 (a) The total energy losses analysis in OSCs. (b) EQEEL of the devices based on the as-cast PM6:F-2F and PM6:Spiro-F blended films. (c) Radiative and non-radiative energy losses in the OSCs studied here. (d) FTPS-EQE of the PM6:F-2F and PM6:Spiro-F based devices at the absorption onset. | ||
As discussed in recent dedicated studies,29,30,61,62 the absorption edge broadening effects63 (reflecting the ΔE2) and the non-radiative recombination loss (ΔE3) are closely correlated to the energetic disorder which is determined by the microscopic properties of blended films. For an organic semiconductor, the relatively larger energetic disorder than that of the inorganic semiconductor is generally observed, accounting for a substantial broadening of the electronic density of states (DOS) of light-harvesting materials.25–27 The broadening of the DOS invariably thrusts tail states further into the bandgap, resulting in the large recombination losses in OSCs.28 In the range of low photon energy, the optical absorption coefficient (α(E)) of disordered organic semiconductors obeys the Urbach rule: α(E) =α0e(E−Eg)/EU, where α0 represents the optical absorption coefficient at the band edge, E is the photon energy, EU is the Urbach energy,64 which is usually described as the width of the tail of the DOS and a measure of energetic disorder.33 Note that a smaller Urbach energy generally suggests a smaller degree of energy disorder. As shown in Fig. 5d, the blend films of PM6:Spiro-F showed a smaller Urbach energy of 32.89 meV than that of 35.16 meV for PM6:F-2F, which could be also suggested by the slightly sharper absorption onset (Fig. 2b) and enlarged CCL (Table 3) relative to those of PM6:F-2F blend.39 Obviously, the ordered molecular stacking in PM6:Spiro-F blend caused by the introduction of rigid spirocyclic alkyl chain, should account for the smaller energetic disorder and thus migrate the recombination loss of OSCs.
Conclusions
To summarize, rigidifying backbones of NFAs through the constraint of vibrational–rotational motion of highly flexible aliphatic side chains could lead a smaller energy loss. Based on a typical NFA of F-2F, a rigid spirocyclic alkyl side chain is applied in NFAs for the first time to replace the two highly flexible n-octyl chains on fluorene unit, affording an optimal molecular packing with enhanced intermolecular actions and lower energetic disorders for Spiro-F. As a result, the OSC based on as cast PM6:Spiro-F blended film achieves a significantly higher PCE of 13.56% and much reduced recombination loss comparing to that of the control device of F-2F. This work demonstrated an effective strategy to reduce the energy loss and even improve the PCE of OSCs through the rigid spirocyclic alkyl side chain. Moreover, we believe this strategy could be applied for other efficient systems with much smaller bandgaps, though the molecule design might be challenging.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support from NSFC (21935007, 52025033, 51873089) and MOST (2019YFA0705900) of China, Tianjin city (20JCZDJC00740) and 111 Project (B12015).Notes and references
- J. S. Wu, S. W. Cheng, Y. J. Cheng and C. S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- Z. Li, G. He, X. Wan, Y. Liu, J. Zhou, G. Long, Y. Zuo, M. Zhang and Y. Chen, Adv. Energy Mater., 2012, 2, 74–77 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, e2102420 CrossRef PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y. Cai, Y. Li, R. Wang, H. Wu, Z. Chen, J. Zhang, Z. Ma, X. Hao, Y. Zhao, C. Zhang, F. Huang and Y. Sun, Adv. Mater., 2021, 33, e2101733 CrossRef PubMed.
- S. Bao, H. Yang, H. Fan, J. Zhang, Z. Wei, C. Cui and Y. Li, Adv. Mater., 2021, 33, e2105301 CrossRef PubMed.
- J. Zhang, F. Bai, I. Angunawela, X. Xu, S. Luo, C. Li, G. Chai, H. Yu, Y. Chen, H. Hu, Z. Ma, H. Ade and H. Yan, Adv. Energy Mater., 2021, 11, 2102596 CrossRef CAS.
- Y. Pan, X. Zheng, J. Guo, Z. Chen, S. Li, C. He, S. Ye, X. Xia, S. Wang, X. Lu, H. Zhu, J. Min, L. Zuo, M. Shi and H. Chen, Adv. Funct. Mater., 2021 DOI:10.1002/adfm.202108614.
- Z. Zheng, J. Wang, P. Bi, J. Ren, Y. Wang, Y. Yang, X. Liu, S. Zhang and J. Hou, Joule, 2021, 6, 171–184 CrossRef.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS.
- K. Yoshikawa, H. Kawasaki, W. Yoshida, T. Irie, K. Konishi, K. Nakano, T. Uto, D. Adachi, M. Kanematsu, H. Uzu and K. Yamamoto, Nat. Energy, 2017, 2, 17032 CrossRef CAS.
- Z. Liu, L. Krückemeier, B. Krogmeier, B. Klingebiel, J. A. Márquez, S. Levcenko, S. Öz, S. Mathur, U. Rau, T. Unold and T. Kirchartz, ACS Energy Lett., 2018, 4, 110–117 CrossRef.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- M. Gruber, J. Wagner, K. Klein, U. Hörmann, A. Opitz, M. Stutzmann and W. Brütting, Adv. Energy Mater., 2012, 2, 1100–1108 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H. L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- J. Benduhn, K. Tvingstedt, F. Piersimoni, S. Ullbrich, Y. Fan, M. Tropiano, K. A. McGarry, O. Zeika, M. K. Riede, C. J. Douglas, S. Barlow, S. R. Marder, D. Neher, D. Spoltore and K. Vandewal, Nat. Energy, 2017, 2, 17053 CrossRef CAS.
- X.-K. Chen, D. Qian, Y. Wang, T. Kirchartz, W. Tress, H. Yao, J. Yuan, M. Hülsbeck, M. Zhang, Y. Zou, Y. Sun, Y. Li, J. Hou, O. Inganäs, V. Coropceanu, J.-L. Bredas and F. Gao, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- K. Vandewal, J. Benduhn, K. S. Schellhammer, T. Vangerven, J. E. Ruckert, F. Piersimoni, R. Scholz, O. Zeika, Y. Fan, S. Barlow, D. Neher, S. R. Marder, J. Manca, D. Spoltore, G. Cuniberti and F. Ortmann, J. Am. Chem. Soc., 2017, 139, 1699–1704 CrossRef CAS PubMed.
- Q. Liu, S. Smeets, S. Mertens, Y. Xia, A. Valencia, J. D'Haen, W. Maes and K. Vandewal, Joule, 2021, 5, 2365–2379 CrossRef CAS.
- M. Panhans, S. Hutsch, J. Benduhn, K. S. Schellhammer, V. C. Nikolis, T. Vangerven, K. Vandewal and F. Ortmann, Nat. Commun., 2020, 11, 1488 CrossRef CAS PubMed.
- X. K. Chen and J. L. Brédas, Adv. Energy Mater., 2017, 8, 1702227 CrossRef.
- S. D. Collins, C. M. Proctor, N. A. Ran and T.-Q. Nguyen, Adv. Energy Mater., 2016, 6, 1501721 CrossRef.
- G. Garcia-Belmonte, P. P. Boix, J. Bisquert, M. Lenes, H. J. Bolink, A. La Rosa, S. Filippone and N. Martín, J. Phys. Chem. Lett., 2010, 1, 2566–2571 CrossRef CAS.
- N. I. Craciun, J. Wildeman and P. W. Blom, Phys. Rev. Lett., 2008, 100, 056601 CrossRef CAS PubMed.
- G. Garcia-Belmonte and J. Bisquert, Appl. Phys. Lett., 2010, 96, 113301 CrossRef.
- M. Kuik, L. J. Koster, G. A. Wetzelaer and P. W. Blom, Phys. Rev. Lett., 2011, 107, 256805 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- S. Xie, Y. Xia, Z. Zheng, X. Zhang, J. Yuan, H. Zhou and Y. Zhang, Adv. Funct. Mater., 2018, 28, 1705659 CrossRef.
- L. Zuo, S. B. Jo, Y. Li, Y. Meng, R. J. Stoddard, Y. Liu, F. Lin, X. Shi, F. Liu, H. W. Hillhouse, D. S. Ginger, H. Chen and A. K. Jen, Nat. Nanotechnol., 2022, 17, 53–60 CrossRef CAS PubMed.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- W. Gao, Q. An, C. Zhong, Z. Luo, R. Ming, M. Zhang, Y. Zou, F. Liu, F. Zhang and C. Yang, Chem. Sci., 2018, 9, 8142–8149 RSC.
- Q. An, W. Gao, F. Zhang, J. Wang, M. Zhang, K. Wu, X. Ma, Z. Hu, C. Jiao and C. Yang, J. Mater. Chem. A, 2018, 6, 2468–2475 RSC.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- S. Dai, F. Zhao, Q. Zhang, T. K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed.
- G. Chai, J. Zhang, M. Pan, Z. Wang, J. Yu, J. Liang, H. Yu, Y. Chen, A. Shang, X. Liu, F. Bai, R. Ma, Y. Chang, S. Luo, A. Zeng, H. Zhou, K. Chen, F. Gao, H. Ade and H. Yan, ACS Energy Lett., 2020, 5, 3415–3425 CrossRef CAS.
- Z. Zhang, Y. Li, G. Cai, Y. Zhang, X. Lu and Y. Lin, J. Am. Chem. Soc., 2020, 142, 18741–18745 CrossRef CAS PubMed.
- F. Lin, K. Jiang, W. Kaminsky, Z. Zhu and A. K. Jen, J. Am. Chem. Soc., 2020, 142, 15246–15251 CrossRef CAS PubMed.
- D. Luo, X. Lai, N. Zheng, C. Duan, Z. Wang, K. Wang and A. K. K. Kyaw, Chem. Eng. J., 2021, 420, 129768 CrossRef CAS.
- K. Jiang, Q. Wei, J. Y. L. Lai, Z. Peng, H. K. Kim, J. Yuan, L. Ye, H. Ade, Y. Zou and H. Yan, Joule, 2019, 3, 3020–3033 CrossRef CAS.
- S. Dong, T. Jia, K. Zhang, J. Jing and F. Huang, Joule, 2020, 4, 2004–2016 CrossRef CAS.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- X. Ke, L. Meng, X. Wan, M. Li, Y. Sun, Z. Guo, S. Wu, H. Zhang, C. Li and Y. Chen, J. Mater. Chem. A, 2020, 8, 9726–9732 RSC.
- X. Li, S. Luo, H. Sun, H. H.-Y. Sung, H. Yu, T. Liu, Y. Xiao, F. Bai, M. Pan, X. Lu, I. D. Williams, X. Guo, Y. Li and H. Yan, Energy Environ. Sci., 2021, 14, 4555–4563 RSC.
- D. Luo, L. Li, Y. Shi, J. Zhang, K. Wang, X. Guo and A. K. K. Kyaw, J. Mater. Chem. A, 2021, 9, 14948–14957 RSC.
- R. Steyrleuthner, M. Schubert, I. Howard, B. Klaumunzer, K. Schilling, Z. Chen, P. Saalfrank, F. Laquai, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2012, 134, 18303–18317 CrossRef CAS PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- X. Gong, M. Tong, F. G. Brunetti, J. Seo, Y. Sun, D. Moses, F. Wudl and A. J. Heeger, Adv. Mater., 2011, 23, 2272–2277 CrossRef CAS PubMed.
- A. K. Kyaw, D. H. Wang, D. Wynands, J. Zhang, T. Q. Nguyen, G. C. Bazan and A. J. Heeger, Nano Lett., 2013, 13, 3796–3801 CrossRef CAS PubMed.
- M. M. Mandoc, F. B. Kooistra, J. C. Hummelen, B. de Boer and P. W. M. Blom, Appl. Phys. Lett., 2007, 91, 263505 CrossRef.
- C. G. Shuttle, B. O’Regan, A. M. Ballantyne, J. Nelson, D. D. C. Bradley, J. de Mello and J. R. Durrant, Appl. Phys. Lett., 2008, 92, 093311 CrossRef.
- D. Hu, Q. Yang, H. Chen, F. Wobben, V. M. Le Corre, R. Singh, T. Liu, R. Ma, H. Tang, L. J. A. Koster, T. Duan, H. Yan, Z. Kan, Z. Xiao and S. Lu, Energy Environ. Sci., 2020, 13, 2134–2141 RSC.
- Y. Shen, A. R. Hosseini, M. H. Wong and G. G. Malliaras, ChemPhysChem, 2004, 5, 16–25 CrossRef CAS PubMed.
- D. M. Smilgies, J. Appl. Crystallogr., 2009, 42, 1030–1034 CrossRef CAS PubMed.
- U. Rau, B. Blank, T. C. M. Müller and T. Kirchartz, Phys. Rev. Appl., 2017, 7, 044016 CrossRef.
- S. Chen, Y. Wang, L. Zhang, J. Zhao, Y. Chen, D. Zhu, H. Yao, G. Zhang, W. Ma, R. H. Friend, P. C. Y. Chow, F. Gao and H. Yan, Adv. Mater., 2018, 30, e1804215 CrossRef PubMed.
- U. Rau, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 085303 CrossRef.
- T. Kirchartz and J. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 165201 CrossRef.
- T. Kirchartz, B. E. Pieters, J. Kirkpatrick, U. Rau and J. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115209 CrossRef.
- J. Yao, T. Kirchartz, M. S. Vezie, M. A. Faist, W. Gong, Z. He, H. Wu, J. Troughton, T. Watson, D. Bryant and J. Nelson, Phys. Rev. Appl., 2015, 4, 014020 CrossRef.
- F. Urbach, Phys. Rev., 1953, 92, 1324 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00253a |
| ‡ Guangkun Song and Yuzhong Huang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |