
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning bimetallic zeolitic imidazolate frameworks (ZIFs) for aqueous catalysis: Co/Zn-ZIF-8 as a cyclic-durable catalyst for hydrogen peroxide oxidative decomposition of organic dyes in water
Osama Abuzalat *a,
Hesham Tantawya,
Mustafa Basunib,
Mohamed H. Alkordib and
Ahmad Barakaa
*a,
Hesham Tantawya,
Mustafa Basunib,
Mohamed H. Alkordib and
Ahmad Barakaa
aDepartment of Chemical Engineering, Military Technical College, Cairo, Egypt. E-mail: osama.abuzalat@mtc.edu.eg
bCenter for Materials Science, Zewail City of Science and Technology, Giza 12578, Egypt
First published on 18th February 2022
Abstract
ZIF-8 is well known hybrid material that is self-assembled from inorganic and organic moieties. It has several potential applications due to its unique structure. One of these potential applications is in advanced oxidation processes (AOP) via a heterogeneous catalysis system. The use of modified ZIF-8/H2O2 for the destruction of the azo dye methyl orange (MO) is presented in this work to explore its efficacy. This work presents the bimetallic Co/Zn-ZIF-8 as an efficient catalyst to promote H2O2 oxidation of the MO dye. Co/Zn-ZIF-8 was synthesized through a hydrothermal process, and the pristine structure was confirmed using XRD, FTIR, and XPS. The Co/Zn-ZIF-8/H2O2 system successfully decolorized MO at the selected pH 6.5. It was found that more than 90% of MO (10 ppm) was degraded within only about 50 minutes. Proposed radical and redox mechanisms are presented for H2O2 decomposition where the redox mechanism is suggested to predominate via a Co(II)/Co(III) redox consecutive cyclic process.
1. Introduction
Zeolitic imidazolate frameworks (ZIFs) represent a particular class of coordination polymers that are built from metal ion nodes and methyl-imidazolate (MIM) linkers. ZIFs are crystalline solids possessing structures analogous to aluminosilicate zeolites with an intrinsic permanent porosity and considerable thermal and chemical stability.1,2 Due to the many desirable characteristics of this class of microporous solids, ZIFs were heavily explored within the last decade for applications such as gas separation, adsorption (gas and liquid phases), sensing3 electronic devices, drug delivery, and catalysis.4,5Indeed, applying ZIFs for heterogeneous catalysis has been systematically investigated and has covered several approaches such as degradation of some dyes/organics (e.g., methylene blue and indigo carmine) via photocatalysis6 catalytic synthesis of some organic derivatives (styrene carbonates),7 hydrogen generation,8 CO2 conversion,9 Knoevenagel condensation,10 Friedel–Crafts acylation,11 trans-esterification,12 and monoglyceride synthesis.13 Although reductive catalytic degradation of some dyes was previously investigated by the ZIF-8/NaBH4 catalysis system,14 or adsorption by ZSM-5 zeolite15 utilization of ZIFs for the counter-redox direction, the oxidative catalytic degradation of organic contaminants (heterogeneous catalysis employing the oxidant H2O2), to best of our knowledge has not previously been reported. This scarcity of in-depth investigations into the catalytic activity of ZIFs for oxidative degradation of organic dyes did not allow for critical evaluation of ZIFs in many environmental applications, one of which is the efficient oxidative remediation of wastewater.
In this work, the known bimetallic Co/Zn-ZIF-8 has been selected for oxidative catalysis degradation primarily due to its significant hydrolytic stability in aqueous solutions, a decisive factor for any material to utilize in aqua-degradation systems.6 Synthesis and characterization of Co/Zn-ZIF-8 are presented, especially detailed XPS analysis. The rate and kinetic-modelling of oxidative catalytic degradation of the stable dye methyl orange by Co/Zn-ZIF-8/H2O2 system are presented as well. To the best of our knowledge, this is the first report presenting the activity of a bimetallic ZIF-8 for degrading a synthetic dye via catalytic H2O2 decomposition.
2. Experimental
2.1 Materials
All chemicals were of analytical grade and have been used without further purification: 2-methylimidazole (2-MIM, 99%, Sigma-Aldrich), triethylamine (TEA, 99%, Alfa Aesar), zinc acetate dihydrate (Zn(CH3CO2)2·2H2O, 98%, Sigma Aldrich), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 98%, Sigma Aldrich), acetone (98%, VWR), and deionized water (DI water).2.2 Co/Zn-ZIF-8 synthesis
The bimetallic Co/Zn-ZIF-8 was synthesized hydrothermally. The molar ratio of cobalt nitrate and zinc acetate was 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 and the molar ratio of metal salts and 2-MIM was 1:1. Typically, Co(NO3)2·6H2O (0.176 g, 0.5 mmol) and Zn(CH3CO2)2·6H2O (0.109 g, 0.5 mmol) were separately dissolved in DI water (10 ml). Another solution was prepared by dissolving 2-MIM (0.082 g, 1 mmol) in a mixture of DI water (10 ml) and TEA (2 ml). Subsequently, the Zn(II) and Co(II) solutions were mixed and magnetically stirred for 15 minutes. The mix-solution was poured into 2-MIM solution under magnetic stirring. A purple suspension was gradually formed which was then heated in an oven (80 °C) for 6 h. Thereafter, the violet solid was partially settled and collected by centrifugation (6000 rpm, 15 min), washed with acetone, water (3 times × 10 ml), and finally dried in an oven at 80 °C overnight.
:0.5 and the molar ratio of metal salts and 2-MIM was 1:1. Typically, Co(NO3)2·6H2O (0.176 g, 0.5 mmol) and Zn(CH3CO2)2·6H2O (0.109 g, 0.5 mmol) were separately dissolved in DI water (10 ml). Another solution was prepared by dissolving 2-MIM (0.082 g, 1 mmol) in a mixture of DI water (10 ml) and TEA (2 ml). Subsequently, the Zn(II) and Co(II) solutions were mixed and magnetically stirred for 15 minutes. The mix-solution was poured into 2-MIM solution under magnetic stirring. A purple suspension was gradually formed which was then heated in an oven (80 °C) for 6 h. Thereafter, the violet solid was partially settled and collected by centrifugation (6000 rpm, 15 min), washed with acetone, water (3 times × 10 ml), and finally dried in an oven at 80 °C overnight.
In addition, two ZIF-8 control samples were also prepared, each was based on a single metal ion, Zn-ZIF-8 and Co-ZIF-8. It was found that while Zn-ZIF-8 was hydro-stable, Co-ZIF-8 readily degraded in hydrogen peroxide solution.
2.3 Co/Zn-ZIF-8 characterization
The chemical structure of Co/Zn-ZIF-8 was explored via FTIR analysis (KBr pellet method using Jasco FT/IR 4100). For its crystalline nature, powder X-ray diffraction (PXRD, Shimadzu XD-l) was applied. SEM/EDX analysis (Zeiss EVO-10 microscopy) was utilized to study the surface morphology and to acquire more information about elemental composition. The X-ray photoelectron spectrum of Co/Zn-ZIF-8 (XPS-Thermo Fisher Scientific, USA) was acquired with a monochromatic Al-Kα (1486.7 eV). BET analysis (N2 adsorption, NOVA Station A) was also performed to determine surface area.2.4 MO degradation
Three MO solutions (250 ml each with adjusted initial pH at 6.5) of different initial concentrations, 2, 5 and 10 ppm, were used to examine MO degradation rate by adding Co/Zn-ZIF-8 (0.05 g) and H2O2 (1 ml (45% H2O2, 0.5 mM)) for each. With no agitation or shacking, the three samples were left in dark at ambient temperature (≈27 °C). The darkness condition was applied to avoid the probable photocatalysis that may happen due to lab stray lights. Periodically, every 10 minutes, about 3 ml from each sample were withdrawn to measure the absorbance of MO remnants using UV-Vis spectrophotometer (Jasco Model 7850 at λ = 591 nm). The dosage effect was also considered by using different amounts of Co/Zn-ZIF-8, 0.01, 0.025, 0.05, and 0.1 g, keeping other conditions the same as previous.It is important to mention that the selection of pH 6.5, near neutral, was decided to avoid probable metal ion leaching from Co/Zn-ZIF-8 structure by acidity and to avoid possible hydrolysis by alkalinity.16 In addition, such selection is appropriate to circumvent Co/Zn-ZIF-8 surface interaction with acidity or alkalinity, because of the probable buffering MIM moieties, which might affect H2O2-produced oxidative species.17,18 In addition, the decomposition rate of H2O2 by Co/Zn-ZIF-8 was monitored and recorded by the traditional triiodide-Ghormley method.19,20
3. Results and discussion
3.1 Co/Zn-ZIF-8 characterization before and after MO degradation
Fig. 1(A) shows the SEM image of Co/Zn-ZIF-8 crystals before and after 5 cycles of catalysis. The crystals morphology can be sought as of dodecahedron type of similar average sizes, about 0.4 μm. This shape is conventional to most of the ZIF-8 samples.3 Some deterioration in morphology has been noticed after 5 cycles. This may be due to the harsh condition by using H2O2.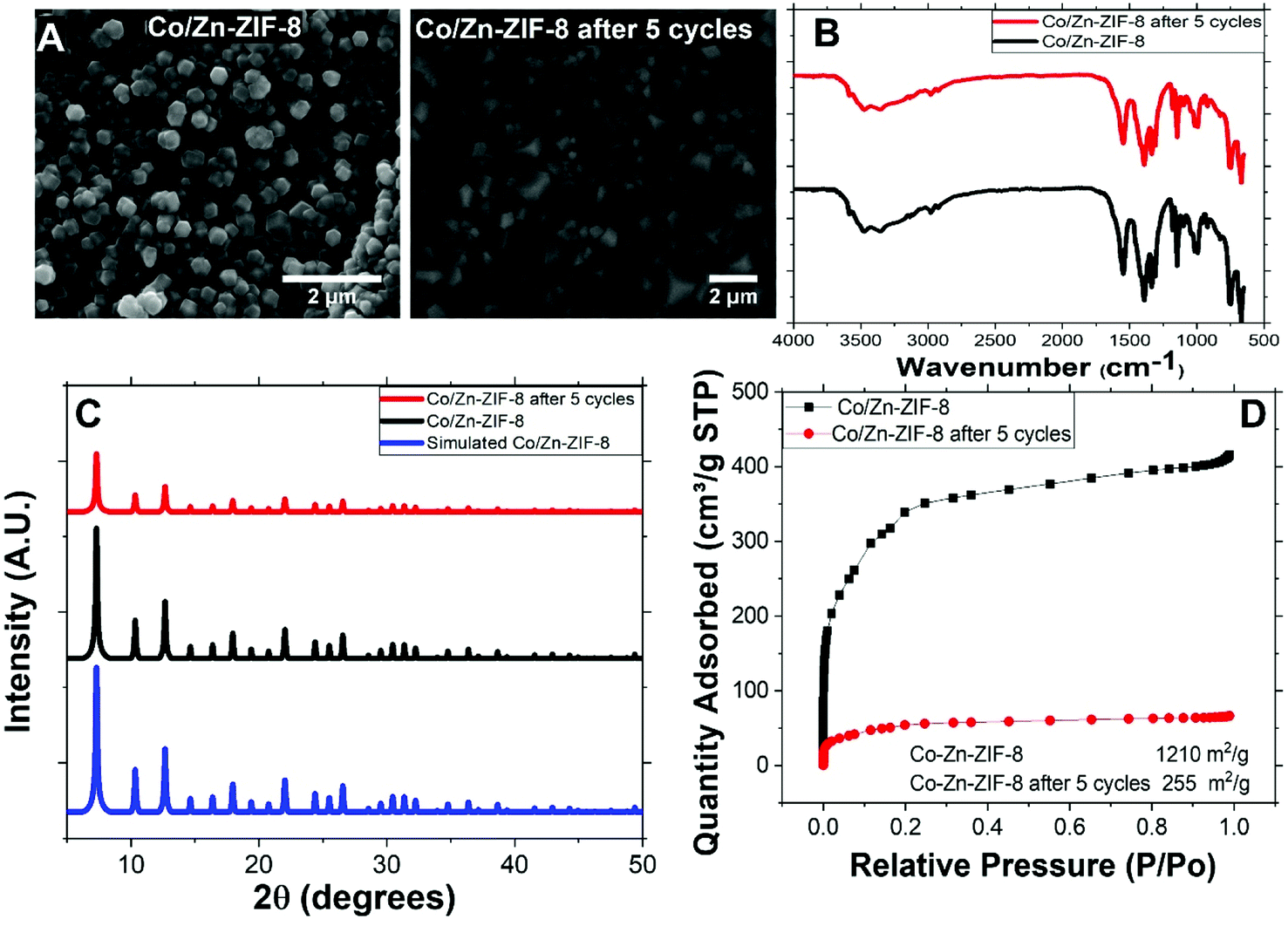 | ||
| Fig. 1 (A) SEM of Co/Zn-ZIF-8, (B) FTIR of BE-S and AF-S, (C) XRD patterns of BE-S and AF-S, and (D) N2 adsorption isotherm and BET surface area of BE-S and AF-S. | ||
Fig. 1(B) shows the FTIR spectra of two Co/Zn-ZIF-8 samples: the before degradation sample (BE-S) and the after 5 degradation-cycles sample (AF-S). For both spectra, the vibrations recorded in the range 760–1422 cm−1 are attributed to the 2-MIM ring. Also, for both spectra, the characteristic band at 1585 cm−1 arose from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration while the peaks at 2800–3100 cm−1 correspond to the aliphatic and aromatic stretching C–H of 2-MIM, respectively. The broad peak around 3400 cm−1 could be assigned to adsorbed moisture from the atmosphere into the ZIF crystals, with spikes that can be associated with the N–H stretching vibration of the residual 2-MIM. The very similarity of these spectra indicates the chemical stability of Co/Zn-ZIF-8 against H2O2, especially the organic part.
N stretching vibration while the peaks at 2800–3100 cm−1 correspond to the aliphatic and aromatic stretching C–H of 2-MIM, respectively. The broad peak around 3400 cm−1 could be assigned to adsorbed moisture from the atmosphere into the ZIF crystals, with spikes that can be associated with the N–H stretching vibration of the residual 2-MIM. The very similarity of these spectra indicates the chemical stability of Co/Zn-ZIF-8 against H2O2, especially the organic part.
XRD patterns of BE-S, AF-S, and the simulation are shown in Fig. 1(C). These patterns are almost identical which verifies the successful preparation of Co/Zn-ZIF-8 as a ZIF-8 material with a body-centred cubic crystal lattice structure.21 Specifically, the similarity of BE-S and AF-S patterns validates its crystalline robustness against H2O2. Hence, it can be suggested that the incorporation of cobalt does not alter the topology of the framework structure and the original topology was retained due to: (i) the similarity of the ionic size of cobalt and zinc (Co2+ 0.79 Å and Zn2+ 0.74 Å) and (ii) the similarity of coordination number for the Co(II) and Zn(II) ions in this framework structure.22
Fig. 1(D) presents the N2-adsorption isotherms of BE-S and AF-S and the accordingly determined BET surface areas are 1210 and 255 m2 g−1 respectively. The obvious reduction in the surface area could be referred to as the adsorption of degradation products of MO which does not affect the successive degradation cycles as will be shown later.
Fig. 2 presents the XPS survey spectra of Co/Zn-ZIF-8 samples, BE-S (A) and AF-S (B). The presence of the principal Co 2p, Zn 2p, N 1s, C 1s, and O 1s core levels before and after degradation with relatively similar abundances proposes the significant chemical stability of Co/Zn-ZIF-8 during H2O2 decomposition process. Table 1 indicates that pristine Co/Zn-ZIF-8 suffered a slight degradation due to the 5-cycles of degradation where a significant increase in oxygen abundance was recorded. Moreover, the observed similarities of atomic ratios of Co/Zn and C/N before and after H2O2 treatment confirms the successive H2O2-decomposition without any significant change in the chemical composition of Co/Zn-ZIF-8.
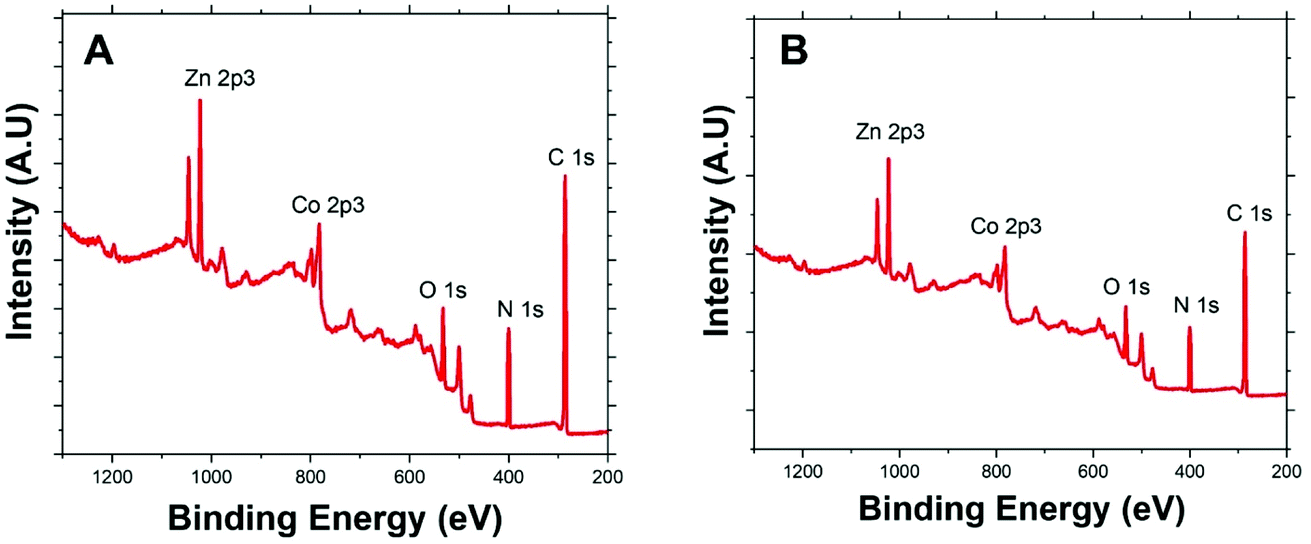 | ||
| Fig. 2 XPS survey spectra of Co/Zn-ZIF-8: (A) BE-S and (B) AF-S. | ||
| Sample | Co | Zn | N | C | O | |
|---|---|---|---|---|---|---|
| BE-S | Atomic wt% | 3.1% | 3.7% | 28.6% | 54.4% | 10.3% |
| Relative abundance to Co ions | 1.0 | 1.2 | 9.3 | 17.7 | 3.3 | |
| AF-S | Atomic wt% | 2.9% | 3.3% | 27.1% | 52.0% | 14.7% |
| Relative abundance to Co ions | 1.0 | 1.1 | 9.2 | 17.6 | 5.0 |
Further, in-depth analysis concerning the XPS results was carried out to investigate the possible effects of H2O2 treatment on Co/Zn-ZIF-8 structure and to assist the understanding of the decomposition mechanism.
Fig. 3(A and B) presents Co 2p high-resolution XPS spectra of BE-S and AF-S. Two main peaks are present for both samples. These peaks are indexed to a spin–orbit splitting of Co 2p3/2 and 2p1/2 at 781.16 and 797.08 eV respectively for BE-S and 781.28 and 797.27 eV for AF-S respectively. The two peaks of BE-S are very similar to those of AF-S which indicate the chemical stability of Co/Zn-ZIF-8. The splitting energies of the 2p doublet (Δ = 2p1/2 − 2p3/2) for both samples were found to be close and equal to about 16 eV, which is quite similar to the literature.23,24
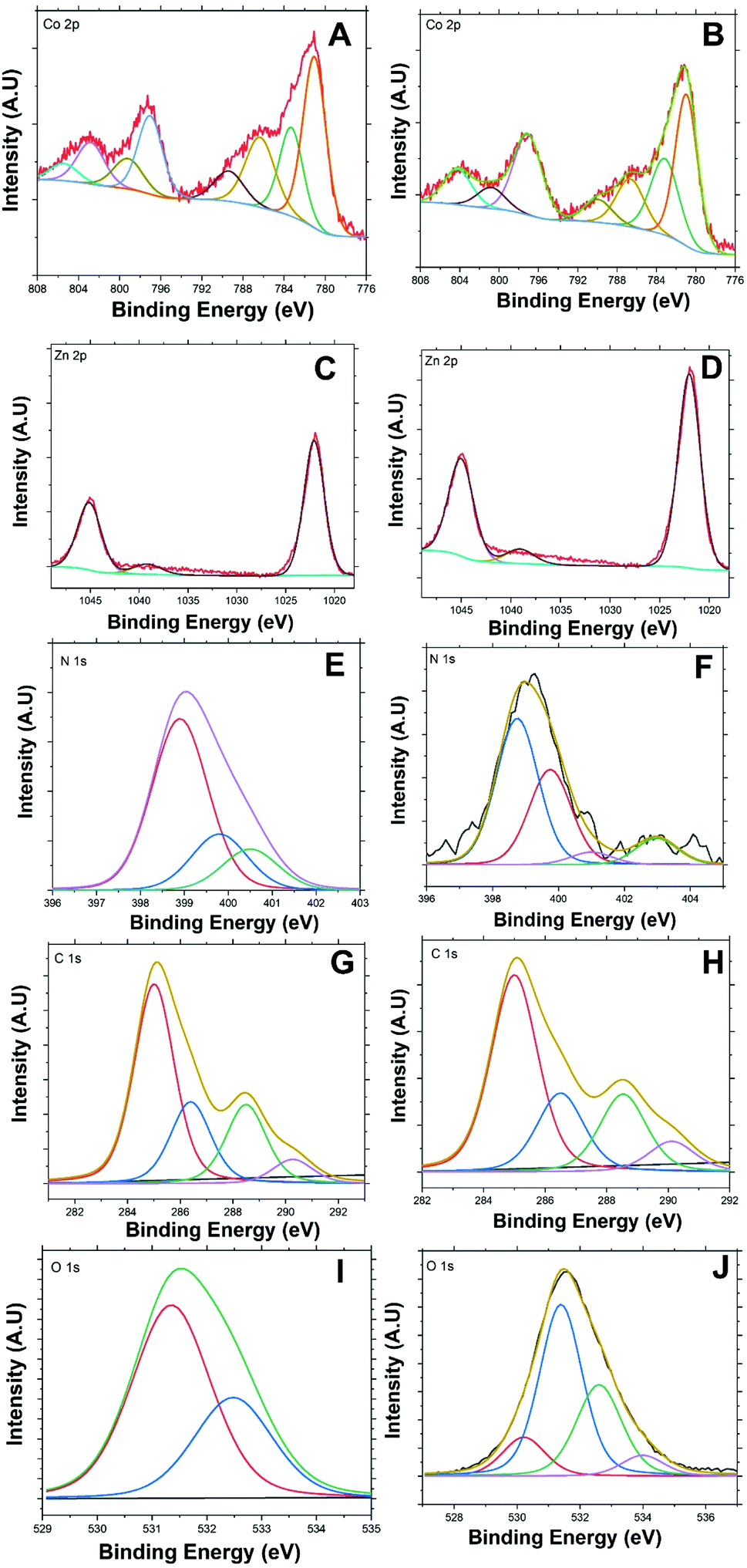 | ||
| Fig. 3 XPS detailed line scans of the Co/Zn-ZIF-8 for BE-S (left column) and AF-S (right column): (A and B) Co 2p peak, (C and D) Zn 2p peak, (E and F) N 1s peak, (G and H) C 1s peak, and (I and J) O 1s peak. | ||
Deconvoluting cobalt Co 2p3/2 and Co 2p1/2 spectra present two main peaks under each one along with their significant corresponding satellites peaks. The fitted peak at 780.96 eV (BE-S) is very similar to the peak at 781.04 eV (AF-S) and the fitted peak at 783.16 eV (BE-S) is also very similar to the peak at 783.3 eV (AF-S). These peaks should be attributed mainly to Co 2p3/2 of Co2+/Co3+ mixed valency.24–29 Further, the observed peaks around 797.03 eV (BE-S) and 797.1 eV (AF-S) and 799.19 (BE-S) and 800.82 eV (AF-S) could be also attributed to Co 2p1/2 of Co2+/Co3+ mixed structure.30–33 Accordingly, both oxidation states, Co2+ and Co3+, are always present within Co/Zn-ZIF-8 structure before and after H2O2 treatment which also indicates the reasonable stability of the chemical structure of the pristine sample.
Co 2p3/2 satellite peaks are observed at 786.33 (BE-S) and 786.66 eV (AF-S) and 789.36 (BE-S) and (AF-S) 789.95 eV.26,34,35 Also, Co 2p1/2 satellite peaks are observed at 805.1 (BE-S) and 804.69 eV (AF-S) in addition to another one at 802.82 (AF-S) which also supports the possibility of having a stable Co2+/Co3+ mixed structure before and after H2O2 treatment.27,36–38 The disappearance of the satellite peaks at 802.82 eV after H2O2 treatment may indicate the possibility of having a slight difference within the electronic environment and charge transfer around Co2+/Co3+ after H2O2 decomposition.39–41
Recalling that cobalt precursor was divalent, however, it seems that some Co2+ ions have been oxidized to Co3+ during synthesis.34,42,43 This depiction confirms that cobalt ions should have a different coordination environment,44–46 which subsequently allows consecutive redox electron transfer.45,47,48 In addition, the presence of a mixed metal system, such as the present case (Zn/Co), rises the metal-heterogeneity mode of the mixed valence system that should enhance the redox activity a character that is essential for catalytic reactions.49,50
Fig. 3(C and D) presents the 2p3/2 and 2p1/2 lines for Zn2+ for both BE-S and AF-S which are assigned at 1022.02 and 1045.03 eV for BE-S and 1022.09 and 1045.1 eV for BE-S and AF-S, respectively. The binding-energy difference between these lines is about 23.0 eV and they are free from multiple splitting and other complicating effects.51–53 A distinct weak peak located at 1039.09 eV (BE-S) and 1039.2 eV (AF-S) is observed, which may be due to the modulation of binding energies arising from some ligand dissimilarities of Zn2+ ions.53–55 The existence of some Co3+ nods, in addition to Zn2+ and Co2+ nods, throughout Co/Zn-ZIF-8 structure must be balanced with the negative anion (COO−) available within the reaction medium. This phenomenon may induce some local dipole moments at Co3+ sites influencing spots of higher surface energies.53,56,57
Fig. 3(E and F) presents N 1s core-level spectra of the Co/Zn-ZIF-8 for both BE-S and AF-S. They are resolved into multiple peaks for different nitrogen environments. The de-convolutions were decomposed into three (BE-S) and four (AF-S) peaks respectively. The most intense N 1s peak at 398.8 eV (BE-S) and 398.91 eV (AF-S) could be attributed to the pyridinic nitrogen atoms.58–62 Because of the little difference between the binding energy of the metal-coordinated N and pyridinic N,59,63 this peak indicates the successful coordination through nitrogen-sp2 bonding with metal nods.59,63,64 In addition, it also designates that most reacted methylimidazole should exist as methyl imidazolate.65,66
Here are some important observations to comment on which reveals some side effect of H2O2 on Co/Zn-ZIF-8 structure. The pyrrolic peak of nitrogen is observed at 399.75 eV (BE-S) and at 399.83 eV (AF-S).58,59,61,62 The peak intensity increased after H2O2 treatment which could imply some partial consumption of the pyridinic species. Also, the intensity of N-graphitic58,59,62,67 protonated pyrrolic68 peak at 400.5 eV (BE-S) has been diminished, at 401.11 eV (AF-S), due to repetitive oxidation. On the other hand, a weak peak around 402.95 eV representing the oxidized N (pyridine-N-oxide) species67–69 appears for (AF-S). This new peak is of higher energy concerning N-pyridinic due to relatively stronger electron localization associated with poorer conjugation at sp3-bonded sites. Hence, lower charge mobility is expected to lead to only a minor decline in degradation performance.70,71 These results propose a minor oxidative deterioration of the Co/Zn-ZIF-8 after five successive oxidation cycles with a survivable and stable structure.
Fig. 3(G and H) presents the de-convoluted C 1s spectra. Each consisted of four main peaks for both (BE-S) and (AF-S). These peaks are observed at 285.13, 286.57, 288.44, and 289.97 eV for BE-S and at 284.90, 286.26, 288.46, and 290.11 eV for AF-S which can be assigned to C–C/CC,61,72,73 CN,74–76 C–N61,76,77 and CO,78–80 respectively.
The presence CO species, even with low abundance, confirms the possibility of incorporation of acetate anion within the structure as it was mentioned previously. It is obvious that there are no significant changes within the de-convoluted C 1s due to H2O2 treatment, and this suggests that Co/Zn-ZIF-8 chemical structure withstands H2O2 oxidative medium to an acceptable extent. However, the cautious analysis showed a slight relative decrease in CN abundance with respect to that of C–N after H2O2 treatment. This observation agrees with the decrease of pyridinic-N with respect to pyrrolic-N after H2O2 treatment. Furthermore, CO peak showed a diminutive increase after H2O2 treatment, at 290.11 eV, which was expected due to possible minor oxidation of trace pyridinic units to form pyridone species integrated within Co/Zn-ZIF-8 structure.81,82
Fig. 3(I and J) presents the de-convoluted O 1s spectra. Although the relative abundance of oxygen is low with respect to other elemental compositions, specifically C and N, the O 1s core-level de-convoluted seems to be very interesting. The O 1s core-level spectra of BE-S and AF-S have similarities and have a difference. There are two main observable peaks at 531.35 eV (BE-S) and at 531.39 eV (AF-S) and at 532.48 eV (BE-S) and at 532.61 eV (AF-S) which shows similarities and could be assigned for (CO) acetate/quinone/pyridone83–85 and (O–C) acetate,83,84,86–88 respectively. In addition, the possibility of having trapped or intercalated hydroxyl anions within the structure could be considered.60,89,90 The difference is that there are two more peaks observed for AF-S, which appear at 530.1 and 533.9 eV. These peaks could be attributed to some adsorbed/intercalated oxygen89,91 and N–O species.88,92 Hence it should be assumed that, during H2O2 decomposition process, there is a likelihood of oxidation of some pyridinic nitrogen leading to the formation of the corresponding N-oxides.93–95 This hypothesis supports the previous discussion about N 1s peak considering oxidized N (pyridine-N-oxide).
3.2 Decomposition of H2O2 and degradation of MO
Fig. 4(A) shows the dark-condition decomposition rate of H2O2 (C0 = 0.5 mM) by Co/Zn-ZIF-8. A gradual yet rapid decrease of H2O2 concentration is quite observable which signifies the likely catalytic affinity of Co/Zn-ZIF-8 towards H2O2 decomposition. It is important to mention that without Co/Zn-ZIF-8, the dark-condition decomposition rate of H2O2 is limited. The heterogeneous catalytic decomposition of H2O2 usually obeys a pseudo-first-order kinetic model with respect to H2O2 regarding in-excess catalyst.96 The known integrated form of decomposition rate equation is given as follow:where kdec is the H2O2 decomposition rate constant, t is the reaction time, [H2O2]0 and [H2O2] are the initial concentration and the concentration of H2O2 at any time t. Fig. 4(B) shows the pseudo-first-order plot of H2O2 decomposition by Co/Zn-ZIF-8. Linear fitting (R2 = 0.976) revealed the decomposition rate constant, kdec = 0.065 min−1. In comparison to some studied catalysts of different types, Table 2, Co/Zn-ZIF-8/H2O2 system is competitive. To elucidate the catalytic influence of Co/Zn-ZIF-8 towards H2O2, the turnover number (TON) and turnover frequency (TOF) were calculated and found to be 2350 and 2820 h−1 respectively.
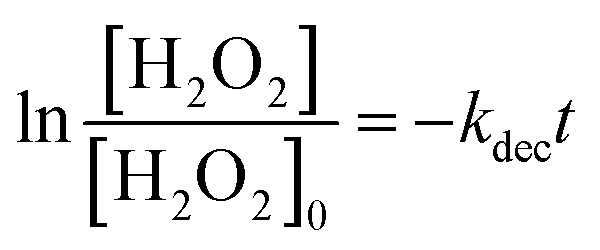
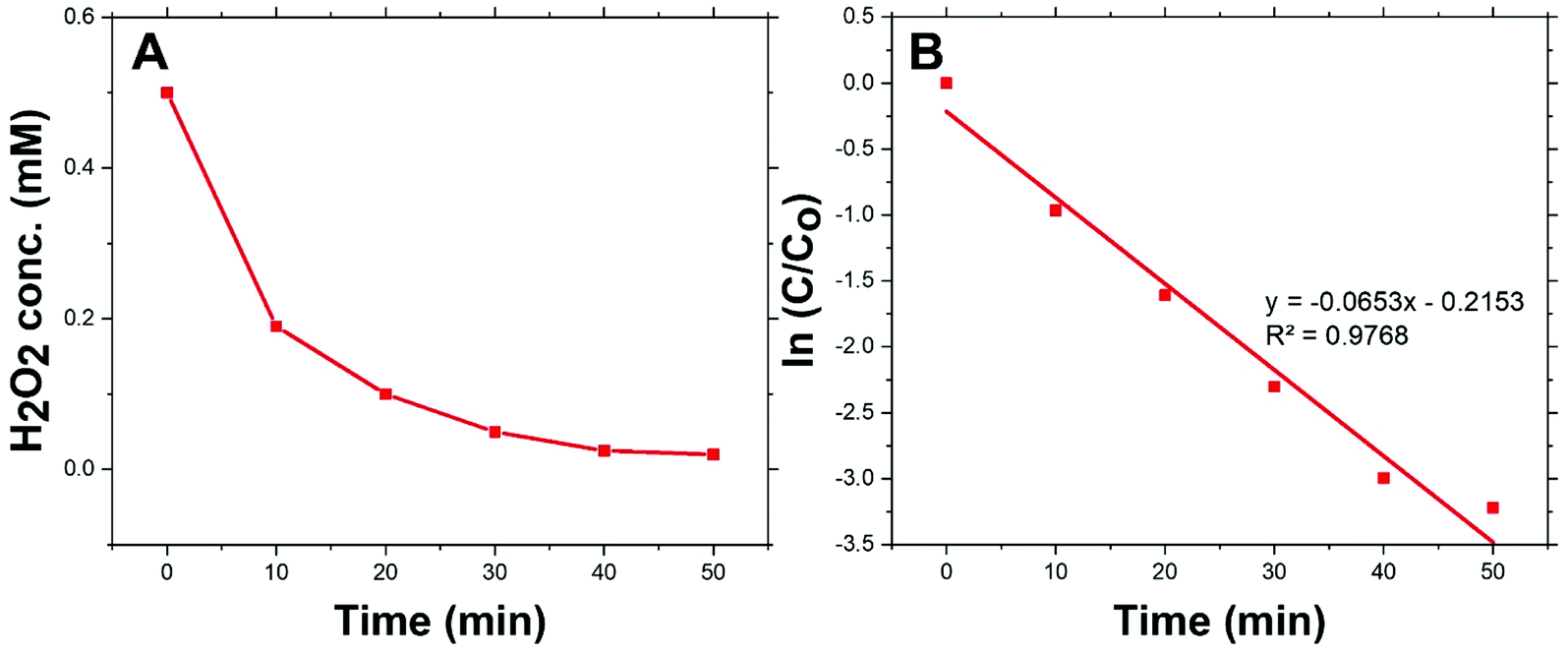 | ||
| Fig. 4 (A) Decomposition of H2O2 over 0.05 g Co/Zn-ZIF-8 catalyst, and (B) pseudo-first-order kinetics of H2O2 decomposition. | ||
3.3 Degradation of MO
Fig. 5(A, B, and C) shows the degradation rates of MO, with initial concentrations 2, 5, and 10 ppm respectively, compared to degradation rates under conditions other than catalyst with H2O2. For all applied initial concentrations, degradation rates for catalyst with H2O2 are remarkably faster than: catalyst only, H2O2 only, and under vis. light solely. This designates Co/Zn-ZIF-8/H2O2 system as a successful system for MO degradation. Fig. 5(D) shows the effect of catalyst dose on MO degradation rate. It is obvious that as dose increases, degradation rate increases, and this is expected due to the increase of available catalyst's active sites.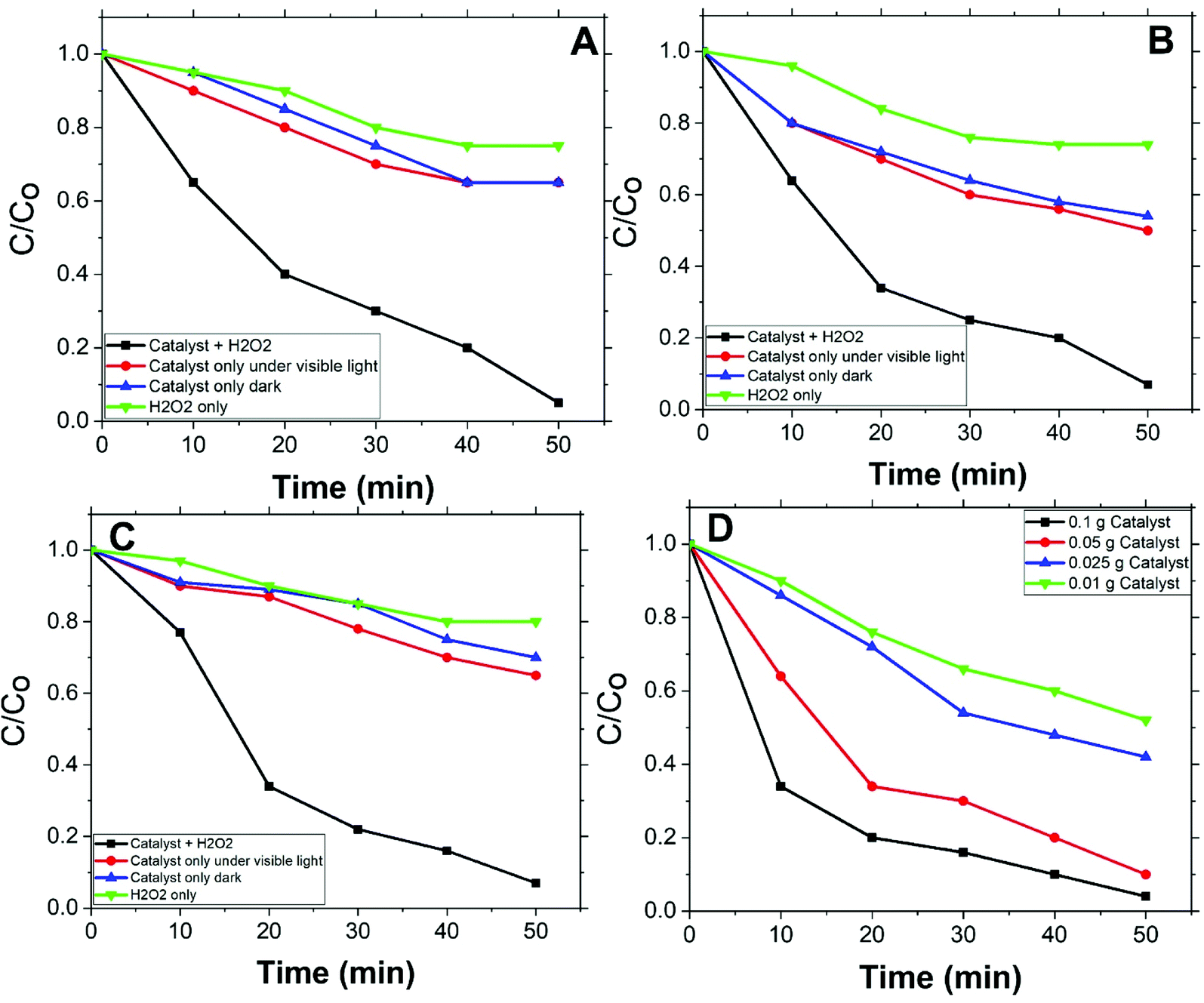 | ||
| Fig. 5 MO degradation at different initial concentrations (0.05 g catalyst, 0.5 mM H2O2), (A) 2 ppm, (B) 5 ppm, (C) 10 ppm, and (D) degradation of 5 ppm MO at different catalyst does (0.01, 0.025, 0.05, 0.10 g). | ||
Degradation rates of the different initial concentrations of MO were modelled applying a pseudo-first-order equation. This equation is similar to the previous one and is as follows:

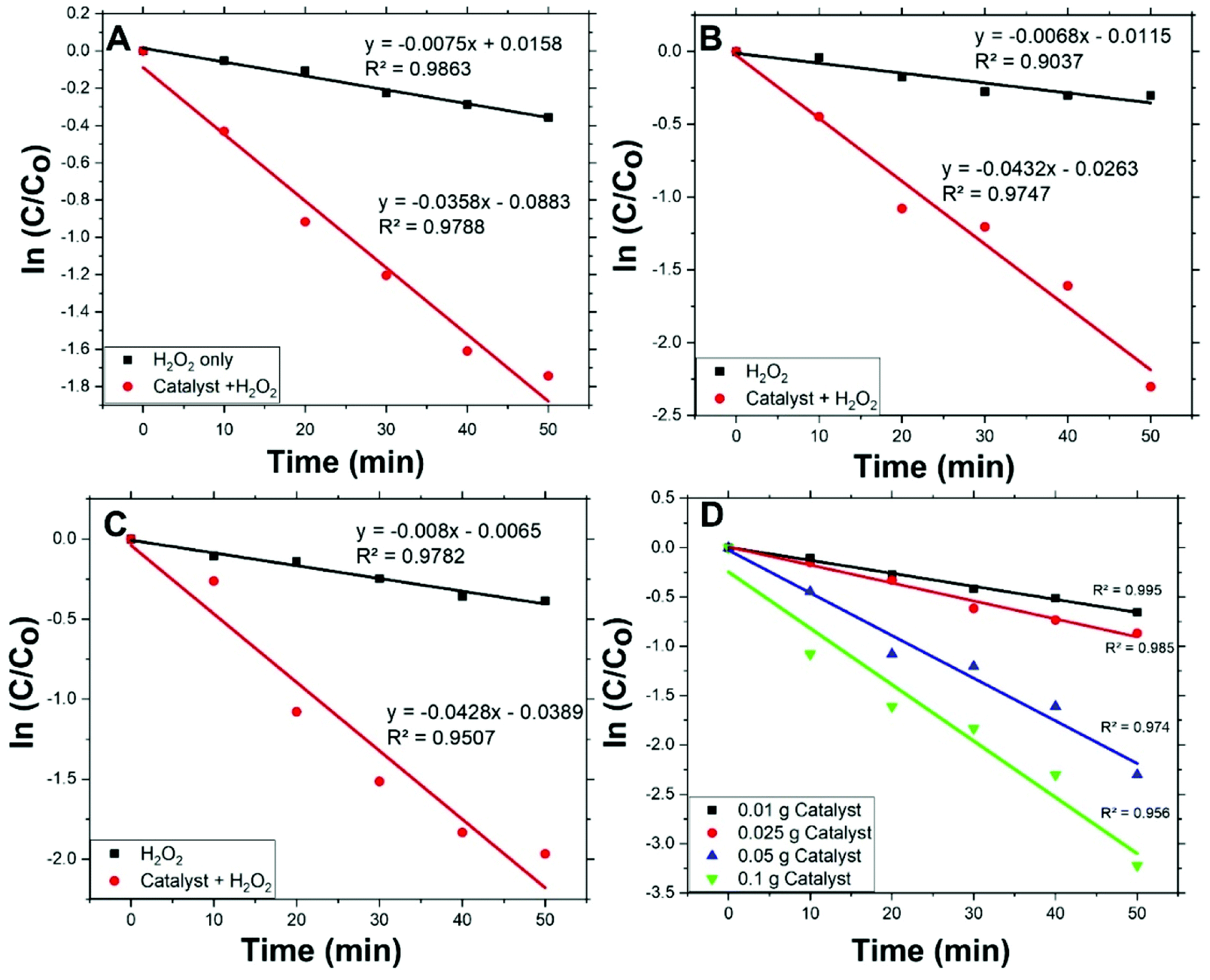 | ||
| Fig. 6 Fitting of pseudo-first-order equation for MO degradation at different initial concentrations (A) 2 ppm, (B) 5 ppm, (C) 10 ppm, and (D) different dose of Co/Zn-ZIF-8 (5 ppm). | ||
Values of rate constants, kdeg, are given in Table 3. Form the table, first: MO degradation by Co/Zn-ZIF-8/H2O2 system is considerably more effective than by H2O2 solely with an average rate constant about 5 times that of without catalyst, second: kdeg values are similar to an acceptable extent, i.e. kdeg does not change with MO initial concentration, and hence one effective decomposition/degradation mechanism can be suggested, and third: kdeg values for Co/Zn-ZIF-8/H2O2 system are of the same order of kdec, where kdeg/kdec ≈ 0.65. This ratio means that the suitable in-excess amount of degrading radicals (not too much or less) should be always sufficient for continual degradation of the applied dye concentration. The adjustment of this ratio influences the efficiency and economy of the catalytic decomposition process of dyes and should be taken into consideration, i.e., adjusting applied conditions to have kdec value properly higher than kdeg.
| [MO]0 (ppm) | kdeg (min−1) Co/Zn-ZIF-8/H2O2 | R2 | kdeg (min−1) H2O2 | R2 |
|---|---|---|---|---|
| 2 | 0.035 | 0.978 | 0.007 | 0.986 |
| 5 | 0.043 | 0.974 | 0.006 | 0.903 |
| 10 | 0.042 | 0.950 | 0.008 | 0.978 |
It is important to also mention that in an investigatory preliminary experiment, mixing of MO solution and Co/Zn-ZIF-8 did not present any significant decrease in MO concentration, for about three hours, which indicates no degradation without H2O2 and even no observable adsorption. Hence kdeg can be sought catalysis-intrinsic behaviour rather than apparent and that a pure chemical process is responsible for degradation.
Another imperative observation, it has been elucidated previously that the decomposition rate of H2O2 without Co/Zn-ZIF-8 is almost neglected. However, MO degradation rate using H2O2 without Co/Zn-ZIF-8 is measurable as given in Table 3, kdeg = 0.007 min−1 on average. This suggests the importance of radical consumption, due to dye degradation, to draw further H2O2 decomposition.
Fig. 6(D) shows pseudo-first-order modelling of MO degradation for the different applied amounts of Co/Zn-ZIF-8 and Table 4 presents the corresponding kdeg values according to fitting. From the figure and the table, the amount of Co/Zn-ZIF-8 causes a steady increase of kdeg value. This signifies the inherent fervent of Co/Zn-ZIF-8 towards H2O2 decomposition. In addition, though, R2 value constantly decreases with applied catalyst amount which points to a deviation from pseudo-first-order modelling obedience, and this may indicate the contribution of more than one mechanism as Co/Zn-ZIF-8 amount increases and/or the need to control the dose of Co/Zn-ZIF-8 with respect to applied H2O2 concentration. Table 5 gives a comparison for degradation rate constants given by Co/Zn-ZIF-8/H2O2 and some other selected catalysts/H2O2 systems which indicate Co/Zn-ZIF-8 as a useful catalyst.
| Co/Zn-ZIF-8 (g) | kdeg (min−1) Co/Zn-ZIF-8/H2O2 | R2 |
|---|---|---|
| 0.010 | 0.013 | 0.995 |
| 0.025 | 0.018 | 0.985 |
| 0.050 | 0.043 | 0.974 |
| 0.100 | 0.057 | 0.956 |
4. Radical scavenging experiments and reusability
In general, ˙OH and HO2˙ radicals are known as the key active type in the catalytic oxidation process. ˙OH, radical is a strong oxidant for many organic molecules. 2-Propanol and 1,4-benzoquinone (BQ) are known as ˙OH and HO2˙ scavengers respectively.103–105 These scavengers were introduced into the reaction medium to capture ˙OH and HO2˙ during the MO degradation process. The results are illustrated in Fig. 7 and point out that the addition of scavengers causes a sharp decrease in the degradation of MO from 96% to 3.5, and 6% in the presence of 2-propanol, and BQ respectively. According to the above results, it is furthermore established that the degradation of MO is mainly dependent on the availability of ˙OH and HO2˙ radical species.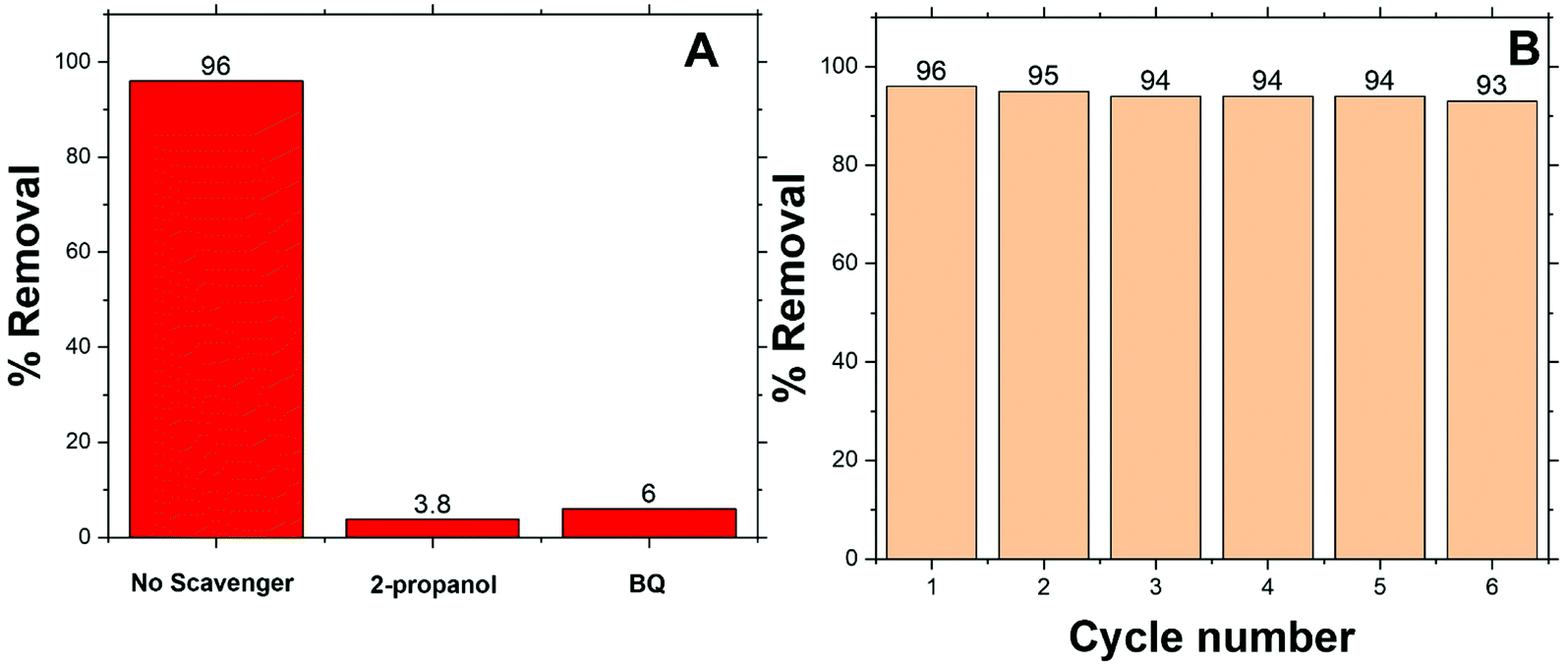 | ||
| Fig. 7 (A) Quenching experiment of the active species during catalytic degradation of MO (5 ppm) over Co/Zn-ZIF-8, 0.5 mM H2O2, with the addition of methanol, ethanol and 2-propanol as scavengers for ˙OH, (B) 6-cycle reuse of Co/Zn-ZIF-8, 0.05 mM H2O2 in the degradation of MB (initial pH 6.5, 5 ppm). | ||
The reusability of a catalyst is important to consider of any heterogeneously catalyzed reaction. However, only limited experiments are there considering the reusability of powder photocatalysts in the powder form. This may be due to the difficulty in the separation of the catalyst powder after degradation processes. In the present case, it is simple to separate Co/Zn-ZIF-8 catalyst from the degradation solutions either by centrifugation or even by simple filtration. Regeneration of the catalyst was done after each dye-removal experiment, by centrifugation at 6000 rpm for 30 min, washing with water, drying at 120 °C, for 2 hours. The catalytic activity of Co/Zn-ZIF-8 remains efficient even up to 6 consecutive experiments, as depicted in Fig. 7(B), under the selected applied conditions. The remarkable stability of Co/Zn-ZIF-8 in water, as shown in the XRD patterns, sustains the chance for its reusability.
5. Proposed mechanisms of H2O2 decomposition
For the present work, it is important to mention the following: (i) Zn-based ZIF-8 has also been synthesized and has been applied for decomposing H2O2, as a supporting auxiliary experiment, however, no measurable H2O2-decomposition was recorded and accordingly, it can be concluded that the sole presence of Zn(II) in the nodes does not intrinsically contribute in H2O2 catalytic decomposition by either radical mechanism or redox mechanism, (ii) the quenching experiments revealed that ˙OH is the main formed radical upon H2O2 decomposition and hence being responsible for dye-degradation, and (iii) no significant change of pH value was recorded.Hence, the proposal of H2O2-decomposition over Co/Zn-ZIF-8 is based on the following suppositions: (i) nodes of Co/Zn-ZIF-8 are sought as nitrides moieties, zinc nitride and cobalt nitride, (ii) these metal nitrides are very similar to metal oxides, because of the very similarity of electronegativity of nitrogen and oxygen, (iii) cobalt-nods are the active sites at which H2O2-decomposition takes place (due to possible redox), and (iv) hydrogen bonding of H2O2 with nitrogen (replacing some hydrogen-bonded H2O) can cause accumulation of H2O2 onto catalyst surface and then a stronger interaction can cause adsorption of H2O2 with zinc nitride and cobalt nitride moieties (similar to oxides) by the interaction of its O atoms with the surface metal cations leading to a strong type of interaction.96
Fig. 8 shows the EPR patterns (range: 310–370 mT) for BE-S and AF-S. Comparison of AF-S pattern with respect to that of BE-S, confirms the presence of free radicals after H2O2 treatment as it shows six characteristic peaks corresponding to the Zn2+ and Co2+ catalytic centers within Co/Zn-ZIF-8 to produce free radicals via electron transfer upon treating the Co/Zn-ZIF-8 with hydrogen peroxide. In some previous works, EPR/ESR measurements revealed the presence of ˙OH and HO2˙ species on the surface of different oxides catalysts. These species are normally short-lived and reactive, however, they become long-lived because of the enhanced stabilization due to their adsorption onto the oxide surface.106
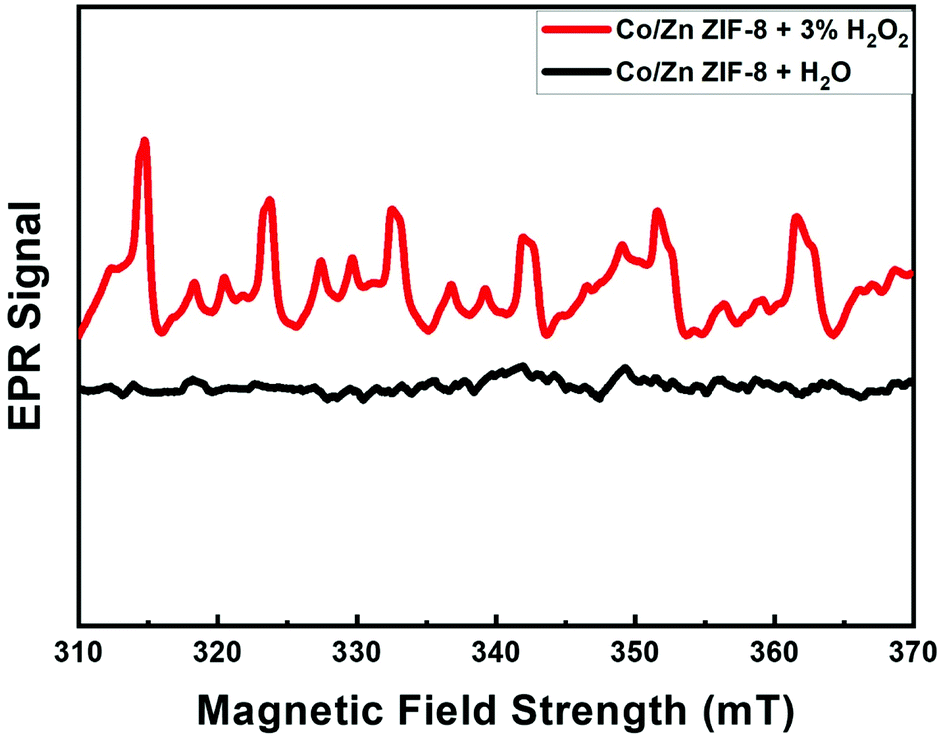 | ||
| Fig. 8 EPR signal of Co/Zn-ZIF-8. | ||
Decomposition of hydrogen peroxide on a solid surface is a spontaneous process at temperatures that range from room temperature to 286 °C. In addition, it was revealed that energy barrier for reactions catalyzed by surface sites where the metal atoms are of under-coordination should be lower than the energy barrier for equivalent reactions catalyzed by non-defected surfaces.106 Two possible mechanisms can be given as follows:
(1) The first (radical mechanism) is based on the complexation of H2O2 with catalyst surface where H2O2 coordinates with surface-metal followed by a series of surface reactions leading, eventually, to the decomposition of H2O2 into the oxidizing radicals, ˙OH and HO2˙:107,108
(I) Being in an aqueous medium, adsorption of H2O preponderates the ZIF-surface. Continual coordination-adsorption of H2O2 molecules onto ZIF-surface occurs via replacing some H2O molecules where coordinated H2O2 suffers decomposition. This repeated adsorption step grantees the continuous-controlled consumption of H2O2:
| [H2Oads–Co(II)]s + H2O2 ↔ [H2O2ads–Co(II)]s + H2O |
(II) Decompositions of H2O2 and desorption of radicals:
| [H2O2ads–Co(II)]s ↔ [HO˙–Co(II)]s + ˙OH (homolytic) |
| [H2O2ads–Co(II)]s ↔ [HO2˙–Co(II)]s + ˙H (heterolytic) |
| [HO2˙ads–Co(II)]s ↔ –Co(II) + HO2˙ |
In this mechanism, Co(III) nodes may contribute to the decomposition process likewise Co(II).
(2) The second (redox mechanism) is analogous to the Haber–Weiss mechanism albeit with a metal ion belongs to solid surface:109
| [H2Oads–Co(II)]s + H2O2 ↔ [H2O2ads–Co(II)]s + H2O | (1) |
| [H2O2ads–Co(II)]s + H2O → [H2Oads–Co(III)]s + ˙OH + OH− | (2) |
| [H2Oads–Co(III)]s + H2O2 → [H2O2ads–Co(III)]s + H2O | (3) |
| [H2O2ads–Co(III)]s + H2O → [H2Oads–Co(II)]s + HO2˙ + H+ | (4) |
Monitoring of pH during decomposition did not give a significant change which suggests similar rates of both the eqn (2) and (4) where a balance occurs for the formation of OH− and H+. Hence, the catalytic overall decomposition reaction, via Co(II)/Co(III) redox consecutive cyclic process, should be:
| 2H2O2 → +HO2˙ + ˙OH + H2O | (5) |
Herein, Co(III) nodes essentially contribute to the decomposition process by forming a redox pair with Co(II), i.e. Co(II)/Co(III) redox system.
Both stability of pH and the previously discussed XPS analysis, which revealed that cobalt ions of different coordination environments and different oxidation states permit the consecutive redox reaction suggesting a redox mechanism to predominate the decomposition process (Fig. 9).
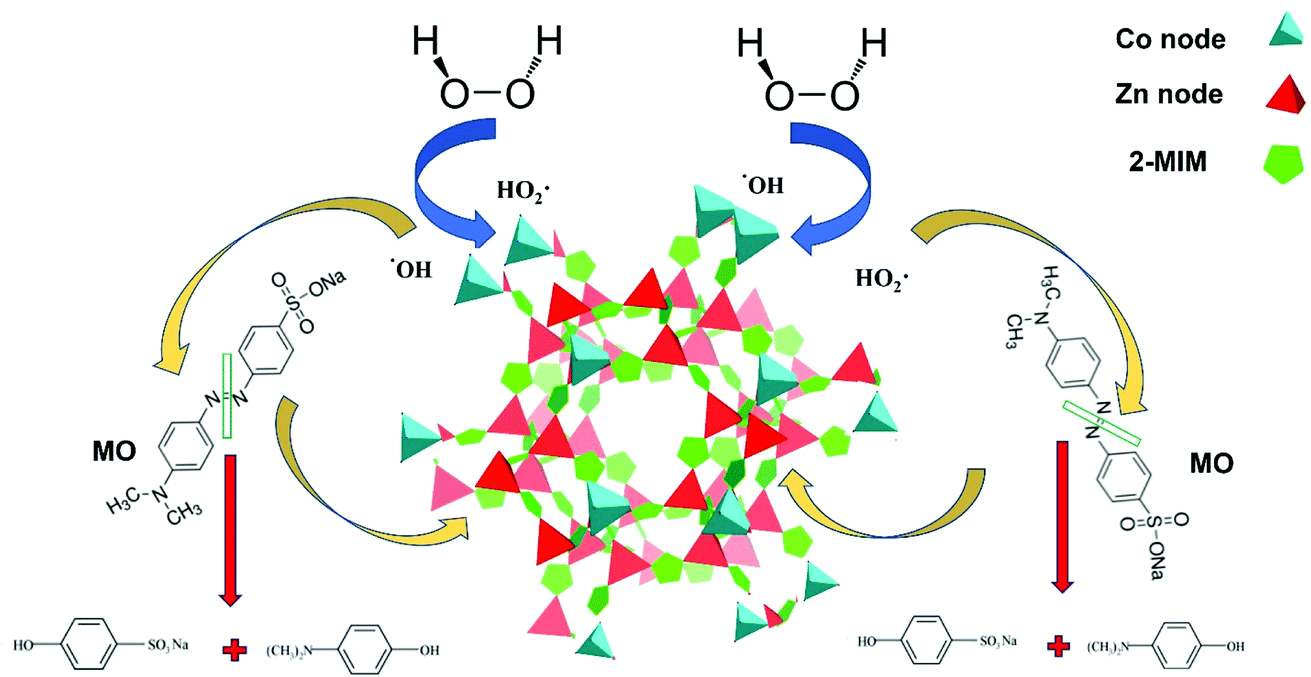 | ||
| Fig. 9 Schematic illustration of the proposed redox mechanism of H2O2 decomposition by Co/Zn-ZIF-8 catalyst and the subsequent MO degradation. | ||
6. Conclusions
The successful synthesis of Co/Zn-ZIF-8 in an aqueous medium points to the importance of zinc nodes to stabilize the material against hydro-chemical stress. Detailed study of XPS patterns of a hybrid material before and after catalysis is a strong tool to disclose some verities about the changes that happened within the material structure and the function(s) of its compositions. For Co/Zn-ZIF-8, XPS detailed analysis made known the importance of the mixed oxidation of cobalt for H2O2 redox decomposition. For Co/Zn-ZIF-8, H2O2-decomposition rate constant, kdec, and the average MO-degradation rate constant, kdeg, were found of the same order, where kdeg/kdec ≈ 0.65. Hence, regulating degradation conditions, specifically H2O2 and dye concentration and catalyst amount, is important for the appropriate decomposition/degradation process.Conflicts of interest
There are no conflicts to declare.References
- J. K. Zareba, M. Nyk and M. Samoć, Cryst. Growth Des., 2016, 16, 6419–6425 CrossRef CAS
.
- K. Zhou, B. Mousavi, Z. Luo, S. Phatanasri, S. Chaemchuen and F. Verpoort, J. Mater. Chem. A, 2017, 5, 952–957 RSC
- O. Abuzalat, D. Wong, S. S. Park and S. Kim, Nanoscale, 2020, 12, 13523–13530 RSC
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2014, 2, 16811–16831 RSC
- S.-G. Jeon, J.-W. Ko and W.-B. Ko, Catalysts, 2021, 11, 1022 CrossRef CAS
- T. N. Nguyen, H. P. Nguyen, T.-H. Kim and S. W. Lee, Korean J. Mater. Res., 2018, 28, 68–74 CrossRef CAS
- M. Zhu, D. Srinivas, S. Bhogeswararao, P. Ratnasamy and M. A. Carreon, Catal. Commun., 2013, 32, 36–40 CrossRef CAS
- P.-Z. Li, K. Aranishi and Q. Xu, Chem. Commun., 2012, 48, 3173–3175 RSC
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS
- Y. Guan, J. Shi, M. Xia, J. Zhang, Z. Pang, A. Marchetti, X. Wang, J. Cai and X. Kong, Appl. Surf. Sci., 2017, 423, 349–353 CrossRef CAS
- L. T. Nguyen, K. L. Ky and T. Nam, Chin. J. Catal., 2012, 33, 688–696 CrossRef CAS
- C. Chizallet, S. Lazare, D. Bazer-Bachi, F. Bonnier, V. Lecocq, E. Soyer, A.-A. Quoineaud and N. Bats, J. Am. Chem. Soc., 2010, 132, 12365–12377 CrossRef CAS PubMed
- L. H. Wee, T. Lescouet, J. Ethiraj, F. Bonino, R. Vidruk, E. Garrier, D. Packet, S. Bordiga, D. Farrusseng and M. Herskowitz, ChemCatChem, 2013, 5, 3562–3566 CrossRef CAS
- S. Chirra, L.-F. Wang, H. Aggarwal, M.-F. Tsai, S. S. Soorian, S. Siliveri, S. Goskula, S. R. Gujjula and V. Narayanan, Mater. Today Commun., 2021, 26, 101993 CrossRef CAS
- S. Radoor, J. Karayil, A. Jayakumar, J. Parameswaranpillai and S. Siengchin, Colloids Surf., A, 2021, 611, 125852 CrossRef CAS
- J. Wei, D. Zhang, L. Zhang, H. Ouyang and Z. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 35597–35603 CrossRef CAS PubMed
- H. Hatem, M. S. El-Geundi, H. Tantawy and A. Baraka, J. Solid State Chem., 2020, 286, 121271 CrossRef CAS
- M. Mohsen, I. Naeem, M. Awaad, H. Tantawy and A. Baraka, J. Solid State Chem., 2020, 289, 121493 CrossRef CAS
- C. Hochanadel, J. Phys. Chem., 1952, 56, 587–594 CrossRef CAS
- Z. Liu, Q. Shen, C. Zhou, L. Fang, M. Yang and T. Xia, Catalysts, 2018, 8, 445 CrossRef
- O. Karagiaridi, M. B. Lalonde, W. Bury, A. A. Sarjeant, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 18790–18796 CrossRef CAS PubMed
- N. Nagarjun and A. Dhakshinamoorthy, New J. Chem., 2019, 43, 18702–18712 RSC
- T. Ivanova, A. Naumkin, A. Sidorov, I. Eremenko and M. Kiskin, J. Electron Spectrosc. Relat. Phenom., 2007, 156, 200–203 CrossRef
- M. Fantauzzi, F. Secci, M. S. Angotzi, C. Passiu, C. Cannas and A. Rossi, RSC Adv., 2019, 9, 19171–19179 RSC
- P. Shelke, Y. Khollam, K. Patil, S. Gunjal, S. Jadkar, M. Takwale and K. Mohite, J. Nano- Electron. Phys., 2011, 3, 486 Search PubMed
- C. Vaz, D. Prabhakaran, E. Altman and V. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155457 CrossRef
- Q. Duan and H. Chen, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 207(1), 012020 Search PubMed
- D. G. Brown and U. Weser, Z. Naturforsch., B: J. Chem. Sci., 1979, 34, 1468–1470 CrossRef
- N. Alotaibi, H. H. Hammud, R. K. Karnati, S. G. Hussain, J. Mazher and T. Prakasam, RSC Adv., 2020, 10, 17660–17672 RSC
- Y. Yang, Y. Tu, P. Zhu, L. Zhang, T. Li, H. Zheng, R. Sun and C. Wong, Sustainable Energy Fuels, 2018, 2, 2345–2357 RSC
- C. Li, Q. Yang, M. Shen, J. Ma and B. Hu, Energy Storage Mater., 2018, 14, 82–89 CrossRef
- Z. Jiang, Z. Li, Z. Qin, H. Sun, X. Jiao and D. Chen, Nanoscale, 2013, 5, 11770–11775 RSC
- A. Gabe, J. García-Aguilar, Á. Berenguer-Murcia, E. Morallón and D. Cazorla-Amorós, Appl. Catal., B, 2017, 217, 303–312 CrossRef CAS
- N. Osenciat, D. Bérardan, D. Dragoe, B. Leridon, S. Holé, A. K. Meena, S. Franger and N. Dragoe, J. Am. Ceram. Soc., 2019, 102, 6156–6162 CrossRef CAS
- B. J. Tufts, I. L. Abrahams, C. E. Caley, S. R. Lunt, G. M. Miskelly, M. J. Sailor, P. G. Santangelo, N. S. Lewis, A. L. Roe and K. O. Hodgson, J. Am. Chem. Soc., 1990, 112, 5123–5136 CrossRef CAS
- T. J. Chuang, C. R. Brundle and D. W. Rice, Surf. Sci., 1976, 59, 413–429 CrossRef CAS
- E. Meza, J. Ortiz, D. Ruíz-León, J. F. Marco and J. L. Gautier, Mater. Lett., 2012, 70, 189–192 CrossRef CAS
- Y. Duan, T. Hu, L. Yang, J. Gao, S. Guo, M. Hou and X. Ye, J. Alloys Compd., 2019, 771, 156–161 CrossRef CAS
- M. Bravo Sanchez, J. A. Huerta-Ruelas, D. Cabrera-German and A. Herrera-Gomez, Surf. Interface Anal., 2017, 49, 253–260 CrossRef CAS
- W. Xi, G. Yan, Z. Lang, Y. Ma, H. Tan, H. Zhu, Y. Wang and Y. Li, Small, 2018, 14, 1802204 CrossRef PubMed
- J. Janas, J. Gurgul, R. P. Socha, T. Shishido, M. Che and S. Dzwigaj, Appl. Catal., B, 2009, 91, 113–122 CrossRef CAS
- T. Zhou, Z. Cao, H. Wang, Z. Gao, L. Li, H. Ma and Y. Zhao, RSC Adv., 2017, 7, 22818–22824 RSC
- Q. Zhang, H. Chen, X. Han, J. Cai, Y. Yang, M. Liu and K. Zhang, ChemSusChem, 2016, 9, 186–196 CrossRef CAS PubMed
- P. Srinivasan, A. J. Kulandaisamy, G. K. Mani, K. J. Babu, K. Tsuchiya and J. B. B. Rayappan, RSC Adv., 2019, 9, 30226–30239 RSC
- P. W. Menezes, A. Indra, A. Bergmann, P. Chernev, C. Walter, H. Dau, P. Strasser and M. Driess, J. Mater. Chem. A, 2016, 4, 10014–10022 RSC
- W. Guo, Z. Liang, J. Zhao, B. Zhu, K. Cai, R. Zou and Q. Xu, Small Methods, 2018, 2, 1800204 CrossRef
- A. V. Piskunov, K. I. Pashanova, A. S. Bogomyakov, I. V. Smolyaninov, A. G. Starikov and G. K. Fukin, Dalton Trans., 2018, 47, 15049–15060 RSC
- Y. K. Penke, G. Anantharaman, J. Ramkumar and K. K. Kar, ACS Appl. Mater. Interfaces, 2017, 9, 11587–11598 CrossRef CAS PubMed
- L. Huang, D. Chen, G. Luo, Y. R. Lu, C. Chen, Y. Zou, C. L. Dong, Y. Li and S. Wang, Adv. Mater., 2019, 31, 1901439 CrossRef PubMed
- S. Dou, C.-L. Dong, Z. Hu, Y.-C. Huang, J.-l. Chen, L. Tao, D. Yan, D. Chen, S. Shen, S. Chou and S. Wang, Adv. Funct. Mater., 2017, 27, 1702546 CrossRef
- M. C. Biesinger, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS
- M. Vijayaraj and C. S. Gopinath, J. Catal., 2006, 241, 83–95 CrossRef CAS
- A. K. Yadav, R. Dey, R. Bhunia, S. Hussain, S. N. Jha, D. Bhattacharyya, R. Bhar and A. K. Pal, J. Polym. Res., 2016, 23, 1–10 CrossRef CAS
- S. Sampath, M. Shestakova, P. Maydannik, T. Ivanova, T. Homola, A. Bryukvin, M. Sillanpää, R. Nagumothu and V. Alagan, RSC Adv., 2016, 6, 25173–25178 RSC
- J. P. Singh, S. H. Kim, S. O. Won, W. C. Lim, I.-J. Lee and K. H. Chae, CrystEngComm, 2016, 18, 2701–2711 RSC
- C. Chen, Z. Cao, X. Zhang, Y. Li, L. Yu and X. Jiang, Chin. J. Chem., 2020, 38, 1045–1051 CrossRef CAS
- Z. Cao, X. Deng, C. Chen, Y. Liu, L. Yu and X. Jiang, React. Chem. Eng., 2021, 6, 454–458 RSC
- Y. Wang, Q. Du, H. Zhao, S. Hou, Y. Shen, H. Li, X. Kong, W. Sun, B. Zhang and S. Li, Nanoscale, 2018, 10, 17958–17964 RSC
- H. Chen, X. Wu, R. Zhao, Z. Zheng, Q. Yuan, Z. Dong and W. Gan, Microchim. Acta, 2019, 186, 1–9 CrossRef PubMed
- S. Bibi, E. Pervaiz, M. Yang and O. Rabi, Catalysts, 2021, 11, 304 CrossRef CAS
- L. Xu, F. Meng, X. Wei, C. Lin, L. Zheng and J. Liu, Sci. China: Technol. Sci., 2020, 63, 1730–1738 CrossRef CAS
- S. Zhuang, E. S. Lee, L. Lei, B. B. Nunna, L. Kuang and W. Zhang, Int. J. Energy Res., 2016, 40, 2136–2149 CrossRef CAS
- Y. Zhu, K. Miyake, Y. Shu, A. Gabe, Y. Hirota, Y. Uchida, S. Tanaka, E. Morallón, D. Cazorla-Amorós and N. Nishiyama, Catal. Sci. Technol., 2019, 9, 578–582 RSC
- Y. Jiang, Y. Lu, X. Wang, Y. Bao, W. Chen and L. Niu, Nanoscale, 2014, 6, 15066–15072 RSC
- M. Mohsen, I. Naeem, M. I. Awaad, H. R. Tantawy and A. Baraka, J. Solid State Chem., 2020, 289, 121493 CrossRef CAS
- C. S. Budi, J. R. Deka, W.-C. Hsu, D. Saikia, K.-T. Chen, H.-M. Kao and Y.-C. Yang, J. Hazard. Mater., 2021, 407, 124392 CrossRef CAS PubMed
- A. Vesel, R. Zaplotnik, G. Primc and M. Mozetič, Nanomaterials, 2020, 10(11), 2286 CrossRef CAS PubMed
- W. Kiciński and S. Dyjak, Molecules, 2021, 26, 668 CrossRef PubMed
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L.-J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755–11759 CrossRef CAS PubMed
- X. L. Wei, M. Fahlman and A. J. Epstein, Macromolecules, 1999, 32, 3114–3117 CrossRef CAS
- H. R. Tantawy, B.-A. F. Kengne, D. N. McIlroy, T. Nguyen, D. Heo, Y. Qiang and D. E. Aston, J. Appl. Phys., 2015, 118, 175501 CrossRef
- H. S. OnáChan, L. MingáGan, C. HaráChew and S. HowáSeow, J. Mater. Chem., 1993, 3, 1109–1115 RSC
- H. Lee, Y. N. Choi, S. B. Choi, J. H. Seo, J. Kim, I. H. Cho, S. Gang and C. H. Jeon, J. Phys. Chem. C, 2014, 118, 5691–5699 CrossRef CAS
- Y. Wu, X. Song, S. Xu, J. Zhang, Y. Zhu, L. Gao and G. Xiao, Catal. Lett., 2019, 149, 2575–2585 CrossRef CAS
- A. Indra, P. W. Menezes, K. Kailasam, D. Hollmann, M. Schröder, A. Thomas, A. Brückner and M. Driess, Chem. Commun., 2016, 52, 104–107 RSC
- H. Liu, Z. Li, L. Zhang, H. Ruan and R. Hu, Nanoscale Res. Lett., 2019, 14, 1–10 CrossRef PubMed
- B. Yin, X. Cao, A. Pan, Z. Luo, S. Dinesh, J. Lin, Y. Tang, S. Liang and G. Cao, Adv. Sci., 2018, 5, 1800829 CrossRef PubMed
- S. Dorey, F. Gaston, S. R. Marque, B. Bortolotti and N. Dupuy, Appl. Surf. Sci., 2018, 427, 966–972 CrossRef CAS
- L. Ma, X. Zhang, M. Ikram, M. Ullah, H. Wu and K. Shi, Chem. Eng. J., 2020, 395, 125216 CrossRef CAS
- B. Ding, J. Wang, Z. Chang, G. Xu, X. Hao, L. Shen and X. Zhang, ChemElectroChem, 2016, 3(4), 668–674 CrossRef CAS
- J. P. Sousa, M. F. Pereira and J. L. Figueiredo, Fuel Process. Technol., 2013, 106, 727–733 CrossRef CAS
- H. Sepehrmansourie, M. Zarei, M. A. Zolfigol, S. Babaee and S. Rostamnia, Sci. Rep., 2021, 11, 1–15 CrossRef PubMed
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CrossRef CAS
- Z. Xing, Z. Ju, Y. Zhao, J. Wan, Y. Zhu, Y. Qiang and Y. Qian, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed
- M. P. Woods, E. J. Biddinger, P. H. Matter, B. Mirkelamoglu and U. S. Ozkan, Catal. Lett., 2010, 136, 1–8 CrossRef CAS
- M. González-Torres, M. G. Olayo, G. J. Cruz, L. M. Gómez, V. Sánchez-Mendieta and F. González-Salgado, Adv. Chem., 2014, 2014, 1–8 CrossRef
- B. Huang, G. Liu, P. Wang, X. Zhao and H. Xu, Processes, 2019, 7, 167 CrossRef CAS
- J. L. Hueso, J. P. Espinós, A. Caballero, J. Cotrino and A. R. González-Elipe, Carbon, 2007, 45, 89–96 CrossRef CAS
- Y.-C. Cheng, S.-P. Chang, S.-J. Chang, T.-H. Cheng, Y.-L. Tsai, Y.-Z. Chiou and L. Lu, ECS J. Solid State Sci. Technol., 2019, 8, Q3034 CrossRef CAS
- F. Gu, W. Liu, Y. Song, L.-M. Liu and Y. Zhu, Ionics, 2020, 26, 5897–5906 CrossRef CAS
- M. Scardamaglia, T. Susi, C. Struzzi, R. Snyders, G. Di Santo, L. Petaccia and C. Bittencourt, Sci. Rep., 2017, 7, 7960 CrossRef PubMed
- S. Shahriar, V. Castaneda, M. Martinez, A. K. Mishra, T. Akter, K. Schutt, J. A. Boscoboinik and D. Hodges, J. Renewable Sustainable Energy, 2019, 11, 053504 CrossRef
- G. Gajeles, S. M. Kim, J.-C. Yoo, K.-K. Lee and S. H. Lee, RSC Adv., 2020, 10, 9165–9171 RSC
- F. F. Bamoharram, M. M. Heravi, M. Roshani and N. Tavakoli, J. Mol. Catal. A: Chem., 2006, 252, 219–225 CrossRef CAS
- W. Xie, Y. Zheng, S. Zhao, J. Yang, Y. Liu and P. Wu, Catal. Today, 2010, 157, 114–118 CrossRef CAS
- C. M. Lousada, M. Yang, K. Nilsson and M. Jonsson, J. Mol. Catal. A: Chem., 2013, 379, 178–184 CrossRef CAS
- A. Maharjan, P. K. Dikshit, A. Gupta and B. S. Kim, J. Chem. Technol. Biotechnol., 2020, 95, 2495–2508 CrossRef CAS
- C. Chen, X. Zhang, H. Cao, F. Wang, L. Yu and Q. Xu, Adv. Synth. Catal., 2019, 361, 603–610 CrossRef CAS
- H. Sun and Z. Zhang, J. Chem. Eng. Jpn., 2017, 50, 26–30 CrossRef CAS
- M. Shaban, M. R. Abukhadra, S. S. Ibrahim and M. G. Shahien, Appl. Water Sci., 2017, 7, 4743–4756 CrossRef CAS
- S. D. Ovhal, C. S. Rodrigues and L. M. Madeira, J. Chem. Technol. Biotechnol., 2021, 96, 349–359 CrossRef CAS
- I. A. Salem, H. A. El-Ghamry and M. A. El-Ghobashy, Beni Suef Univ. J. Basic Appl. Sci., 2014, 3, 186–192 Search PubMed
- O. Abuzalat, H. Tantawy, R. Abdlaty, M. Elfiky and A. Baraka, Dalton Trans., 2021, 50(24), 8600–8611 RSC
- X. Wang, M. Brigante, W. Dong, Z. Wu and G. Mailhot, Chemosphere, 2020, 258, 127268 CrossRef CAS PubMed
- M. Sadeghi, S. Farhadi and A. Zabardasti, RSC Adv., 2020, 10, 10082–10096 RSC
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, J. Phys. Chem. C, 2012, 116, 9533–9543 CrossRef CAS
- X.-j. Yang, P.-f. Tian, H.-l. Wang, J. Xu and Y.-f. Han, J. Catal., 2016, 336, 126–132 CrossRef CAS
- M. Sadeghi, S. Farhadi and A. Zabardasti, RSC Adv., 2020, 10, 44034–44049 RSC
- K. Rusevova, F.-D. Kopinke and A. Georgi, J. Hazard. Mater., 2012, 241, 433–440 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2022 |