
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple single jar “on–off fluorescence” designed system for the determination of mitoxantrone using an eosin Y dye in raw powder, vial, and human biofluids†
Ahmed Abdulhafez Hamad *a,
Ramadan Alia and
Sayed M. Derayeab
*a,
Ramadan Alia and
Sayed M. Derayeab
aDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Al-Azhar University, Assiut Branch, Assiut 71524, Egypt
bDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Minia University, Minia 61519, Egypt
First published on 7th March 2022
Abstract
In this work, a direct, simple, one-pot, and green spectrofluorimetric approach was applied to measure mitoxantrone, a chemotherapeutic agent, through a green validated method. The suggested approach focused on establishing an easy association complex combining mitoxantrone and the eosin Y reagent in a slightly acidic solution. The fluorometric analysis was dependent on off-mitoxantrone action on the emission intensity of the dye (eosin Y) at 544.5 nm (excitation = 301 nm). The devised system has a linear range of 0.07–2.5 μg mL−1 and a detection limit of 0.016 μg mL−1. All system parameters for the formation of mitoxantrone–eosin Y complexes were modulated analytically. Also, the system was reviewed in agreement with ICH criteria. Furthermore, the proposed model was approached to quantify mitoxantrone in its pharmaceutical vial dosage form with high recoveries. Also, the proposed spectroscopic design was efficiently employed to detect the investigated drug in body fluids (blood and urine). Lastly, the designed method was evaluated from a greenness point of view according to eco-scale.
Introduction
Mitoxantrone (MTX; Fig. 1A) is one of the anthracycline oncogenic agents that shows high anticancer activity. MTX is indicated in treating several cancer diseases, such as acute leukaemia, prostate cancer, lymphoma, and breast cancer.1 MTX attacks DNA strands in the human cell, thus obstructing its synthesis and preventing its repair process by disrupting the transcription and translation pathways in the damaged cell.2 Due to its therapeutic importance, MTX was accurately determined in manufactured forms and biofluids. The recommended starting dose of mitoxantrone used as a single agent is 12–14 mg m−2 of body surface area, given as a single intravenous dose, which may be repeated at 21 day intervals.3 Numerous methodologies for analyzing MTX in raw or pharmaceutical formulations have been presented in the literature. HPLC,4–7 an electrophoretic technique,8 chemiluminescence,9 electroanalytical approaches,10–13 and immunoassay14 are among these methodologies. Surprisingly, a few published spectroscopic approaches were found, such as spectrophotometry,15,16 spectrofluorimetry,17 RRS,18 and Raman scattering techniques.19,20 | ||
| Fig. 1 Chemical structures of mitoxantrone (A) and eosin Y (B). | ||
It is noteworthy that HPLC procedures require highly pure organic solvents in a huge quantity, multiple sample pretreatment procedures, and the use of complicated and expensive devices and detectors. Also, the low sensitivity of spectrophotometric tests was an obvious disadvantage. Despite the numerous advantages of spectrofluorimetric methods such as simplicity, selectivity, and high sensitivity, only one method based on quenching the fluorescence of the CdTe quantum dot has been reported to evaluate the MTX.17 Therefore, it is extremely important to design a green, direct, rapid, simple, and sensitive spectrofluorimetric method to analyze the mitoxantrone antineoplastic agent. These purposes can be achieved through the spectrofluorimetric technique by reacting the studied drug with self-fluorescent dyes in a slightly acidic medium. The proposed method was validated entirely concerning ICH criteria.21 It was effectively employed to rapidly analyze MTX in bulk forms, pharmaceutical preparations, and fluids of the human body (urine and blood).
Then again, xanthene-based dyes, for example, erythrosine B and eosin Y, were chemically used to investigate many drugs such as anthelmintic,22,23 and antispasmodic drugs.24 Eosin Y, an acidic dye, is the disodium salt of 2,4,5,7-tetrabromo-fluoresceine (Fig. 1B). In hematoxylin–eosin staining, eosin Y is commonly employed as a counterstain to hematoxylin. Initially, it was used to recognize basic, or eosinophilic, compounds like proteins containing amino acid residues such as arginine and lysine by staining them dark red or pink. Recently, xanthene-based dyes were used for the spectrophotometric and/or spectrofluorimetric determination of proteins25,26 and some pharmaceutical compounds.22–24,27 These methods were based on developing an ion-pair complex between the dye and the compounds with a basic nature. The complex formation was coupled with a quenching effect on the native fluorescence of eosin Y in a linear relationship with the coupled compound. This marked sign can be utilized for the quantitative of the target analyte, which contains a basic center.
MTX contains two secondary amine groups, which could be protonated in an acidic medium and thus could easily react with eosin Y to form an ion-pair complex. The designed approach will be applied to the quantification of MTX based on the “on–off fluorescence system” through the quenching effect of MTX on the inherent emission of the dye upon complex formation. The fluorescence quenching response was directly compatible with the drug concentration at a specific range (0.07–2.5 μg mL−1) and thus provided a very simple and highly sensitive way for drug determination. This method uses inexpensive reagents, and relatively the utilized instrument is commonly available in most quality control laboratories.
Comparable cases (ESI, Table 1†) show fluorescence quenching upon association complex formation with MTX and other compounds (with similar functional groups) and other fluorescence active dyes.
Experimental
Apparatus
All fluorescence tests were conducted using an FS-2 (Korea); SCINCO spectrometer coupled with a light source lamp (150 W Xe-arc). The slit width was fixed at 10 nm for all measurements.A ThermoFisher scientific centrifuge (PICO 21, Germany) was utilized for clinical sample procedures. Also, SONICOR SC-101TH (bath sonicator). The operating medium was corrected using an AD11P pH-meter (Adwa, Romania).
Material and reagents
MTX (purity 99.3%) was provided by the Hikma Specialized Pharmaceutical Company (Badr City, Cairo, Egypt). A Santrone® vial (Batch no. 16010008) labelled to contain 20 mg of MTX per vial was provided by Hikma Specialized Pharmaceutical Co. (Badr City, Cairo, Egypt).Eosin Y (0.02% w/v) was brought from Market Harborough, Leicestershire, United Kingdom, and prepared twice-distilled water.
Solvents and buffer solutions
Acetone, acetonitrile, ethanol, methanol, dimethylformamide, DMSO, 1,2-dioxane, isopropanol, HCl, NaOH, and others were bought from El Nasr Company for Intermediate Chemicals (Cairo, Egypt).A Britton–Robinson controlling solution (pH range 2.0–12.0) consisted of three acids at a concentration of 0.04 M. These acids were boric, phosphoric, and acetic. A suitable volume of this mixture was adjusted to the operating pH value with 0.2 M NaOH contained in another pot.28,30,38 A Teorell–Stenhagen controlling solution with a wide pH range (2.0–12.0).22 A McIlvaine buffer solution (pH 2.2–8.0) is composed of two separate solutions; 0.2 M citric acid and 0.1 M Na2HPO4.29
All used substances were of analytical or pharmaceutical grade and were used in their original state without further purification. All solutions were prepared fresh daily.
Human body fluids
Drug-free human body fluid samples (blood and urine) were drawn from healthy (37 year-old male humans) and frozen until testing. All experiments, including human plasma, were performed in compliance with the instructional guidelines and requirements of the Guide for the Care and Use of Laboratory Animals 8th Edition 2011 (the Guide) and was approved by Commission on the Ethics of Scientific Research, Faculty of Pharmacy, Minia University. In all cases, informed written consent was obtained from each participant.Master solutions
By introducing 10 mg of MTX into a 100 mL marked volumetric flask, dispersing it in a sufficient amount of distilled water, and then filling it to the full volume with distilled water, a standard aqueous solution of MTX was prepared. An operating standard solution was created by dispersing the appropriate volume of the stock solution with twice-distilled water. The solutions were kept in a refrigerator (4 °C) to maintain chemical stability.Practical steps of the system
A fixed aliquot (1.0 mL) of MTX with concentrations ranging from 0.7 to 25.0 μg mL−1 was quantitatively put into 10.0 mL calibrated flasks for the fluorometric assay. To each flask, 1.0 mL of Britton–Robinson controlling solution (pH 4.0) was poured, followed by 1.0 mL of 0.02% w/v eosin Y solution. After 8 minutes, the contents of the flask were finalized with double distilled water and gently shaken. The blank reagent was run parallelly on an equal basis except for the drug solution. The fluorescence-off impact of MTX drug on the eosin Y endogenous emission intensity was recorded using the fluorometric technique at λem of 545 nm (λex = 301 nm), and the resultant value was plotted versus the MTX concentration in μg mL−1. The general derived regression model (y = aC + b) was used to calculate the concentration of the measured sample.Commercial vial analysis
In a 100 mL calibrated marked flask, a precisely measured dose of the Santrone® vial solution containing 10 mg MTX was put, and about 60 mL of twice-distilled water was added. The flask component was well mixed and finalized to 100 mL with water to gain a working solution of 100 μg mL−1 of MTX. Subsequent water dispersion was applied to generate final concentrations of 0.5, 1.0, and 1.5 μg mL−1. In three replicates, the final solution was examined by employing the analytical steps described earlier. By plotting the measured response towards the drug concentration and using the newly obtained regression model, the MTX concentration of each sample solution was calculated.Analysis of MTX in human plasma
A 7.0 mL aliquot of human blood was whirled at 4000 rpm for 10–15 minutes in an anticoagulation tube. The resultant plasma was then transferred to another centrifugation tube in a quantity of 1.0 mL. The standard MTX solution was subsequently added to the tube, resulting in a final MTX concentration of between 0.3 and 2 μg mL−1. The tubes were shaken vigorously, and the protein was extracted by adding an adequate amount of acetonitrile.27,30,38 The tubes were spun for about two minutes before being centrifuged at 4000 rpm for about 10 minutes. Then, 1.0 mL of the translucent supernatant was quantitatively transferred to 5 mL volumetric flasks and monitored as described above. Blank testing was also performed in parallel. A calibration plot in plasma was launched by graphing the recorded response towards the drug concentration in the drug-loaded plasma. Human plasma samples containing varying amounts of MTX within the operating range were treated identically, and their drug concentrations were estimated using the appropriate regression equation.Urine analysis for MTX
A newly drained urine sample from a healthy 35 year-old individual was provided. The urine was screened and doubly filtered, and the supernatant was treated with dignity and respect to plasma. The treated filtrate aliquots were then accurately shifted to a 5 mL standardized volumetric flask, and the evaluation steps were followed.30,35,38Calibration standards' construction and quality control for clinical samples
To design the calibration and quality management standards, human blood and urine were independently spiked with various amounts of MTX standard operating solution. The MTX spiked plasma and urine operating solutions were prepared for the calibration standard (CS) and quality assurance (QA) using the same clear work solution as mentioned for the proposed system procedure. The final plasma and urine DOX concentration ranges were 0.3–2 μg mL−1.The procedure for molar ratio determination
The continuous variation plotting (Job's method)23,31 was mandated in the current designed system to predict the stoichiometry of the MTX–EY reaction under optimal operating conditions. Both the cited drug and eosin Y were introduced as parent solutions at equimolar concentrations (2.8 × 10−4 M). In 10 mL calibrated volumetric flasks, 1.0 mL of the eosin Y parent solution and the drug were prepared in various complementary proportions (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0, 0.1:0.9… 0.9:0.1, 1.0:0).27 The appropriate amount of used buffering system (at the operating pH) was added, and the volume was supplied with twice-distilled water to the mark. The solution's emission intensities were measured at λem = 545 nm (at λex = 301 nm). Concurrently, the preparation and measurement of blanks were performed. The drop in emission intensity (ΔF) towards the mole fraction of the drug was plotted.
:1.0, 0.1:0.9… 0.9:0.1, 1.0:0).27 The appropriate amount of used buffering system (at the operating pH) was added, and the volume was supplied with twice-distilled water to the mark. The solution's emission intensities were measured at λem = 545 nm (at λex = 301 nm). Concurrently, the preparation and measurement of blanks were performed. The drop in emission intensity (ΔF) towards the mole fraction of the drug was plotted.
Results and discussion
The system spectra
In the developed estimation system, eosin Y (as a xanthene dye example) was tapped to quantify the suggested drug by suppressing its inherent emission intensity. Because the eosin Y reagent has an intrinsic emission at 544.5 nm (excitation at 301 nm) that can be quantitatively suppressed by the action of MTX (Fig. 2), the drug can also be quantified using the fluorometric response. Furthermore, for confirmation and comparison purposes, the pure spectra for MTX were provided (ESI, 1.1†). Also, full spectra (excitation and emission) of the fully optimized MTX–eosinY system conditions at different MTX concentrations within the working range were present in (ESI, 1.2†).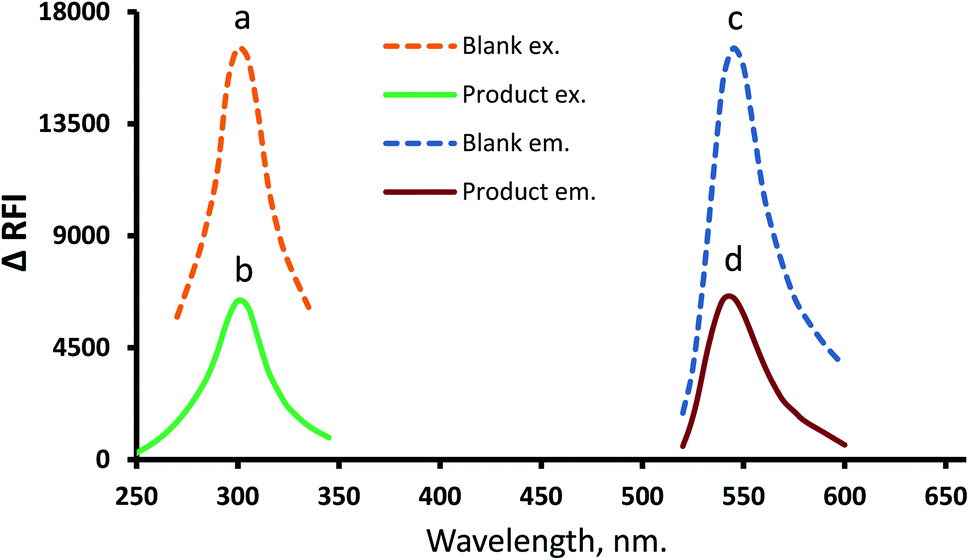 | ||
| Fig. 2 Full spectra (excitation and emission) of the formed association complex MTX–EY (b and d) and reagent blank (a and c). | ||
Control of the experimental conditions
The reaction parameters that might be predicted to have an impact on the system response (a quenching of the emission intensity of eosin Y reagent) were reviewed and corrected to get the best results for this recommended spectroscopic application.Effects of pH and buffer volume
In the current spectroscopic approach, the effect of pH on the binary complex formed between MTX and eosin Y was examined in the 2.0–6.0 pH range. The pH of the sample was observed to have a significant impact on the development of the MTX–eosin Y complex. For the quenching mechanism, the highest ΔRFI values were recorded in the pH range of 3.5–4.5. Outside of this range, decreasing or increasing the pH values resulted in a significant drop in the values of ΔRFI (Fig. 3). | ||
| Fig. 3 Effect of pH on the fluorescence quenching effect of the association complex formation between MTX (1.0 μg mL−1) and EY. | ||
In conclusion, the suitable pH value in the current investigation was 4.0. Using quantities ranging from 0.2 to 2.2 mL of Britton–Robinson buffer solution, the binary complex formed from MTX and eosin Y was examined using the designed spectroscopic approach (pH 4). The buffer volume had a considerable effect on the development of the MTX–eosin Y complex. The strongest responses (maximum decrease in the dye emission) were obtained using a buffer volume of 1.2–1.6 mL for the current spectroscopic approach. Lower or higher volumes than those in this range resulted in lower response values. Low buffer volume may be inadequate to keep the pH, but large buffer volume increases the ionic strength of the solution, causing the positive component of the buffer to compete with drug cations for coupling with the anionic dye, hindering the complex formation process. As a result, the best volume used for this investigation was 1.4 mL (ESI, 2.1†). Full excitation and emission spectra at different pH values and buffer volumes for the formed association complex between MTX (1.0 μg mL−1) and eosin Y were shown in (ESI, 2.2†).
The impact of buffer types
To get the best response for the applied procedure, different buffer types (pH 4) in 1.4 mL volumes were tested. The studies revealed that all of the buffer solutions utilized (Toerell–Stanhagen, Britton–Robinson, acetate, and McIlvaine) produced results that were close to each other, with the Britton–Robinson buffer producing somewhat higher values and the most robust responses. As a result, it was the buffer of choice (ESI, 3†).The impact of eosin Y (0.02% w/v) volume
Various volumes of 0.02% w/v eosin Y reagent were tested to obtain the best response from the designed system. It was observed that 1.0 mL of (0.02% w/v) eosin Y solution was the most efficient volume that displayed the topmost fluorescence quenching effect (Fig. 4). Because of the incomplete reaction, the low response was detected if a low concentration of eosin Y was utilized, as seen in Fig. 4. Higher eosin concentrations, on the contrary, resulted in lower responses, which might be due to eosin Y self-aggregation. Excitation and emission spectra at different volumes of EY for the formed association complex between MTX (1.0 μg mL−1) and eosin Y (0.02% w/v) were present in (ESI, 2.3†).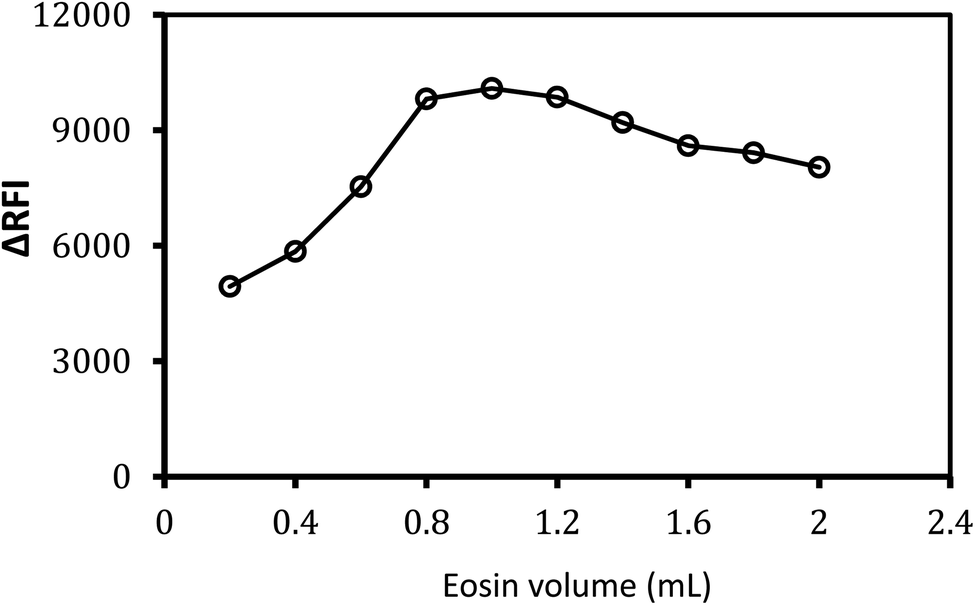 | ||
| Fig. 4 Effect of eosin Y (0.02% w/v) volume on the fluorescence quenching of the formed association complex with MTX (1.0 μg mL−1). | ||
Reaction completion and stability times
At ambient temperature, the MTX–eosin Y formed shortly after the reactant solutions collided with each other, and it was completed in a short period of time (6 min). Furthermore, the developed complex observable response (fluorescence quenching effect) remains considerably constant for more than 60 minutes (ESI, 4 and 5†).Dispersing solvent effect
Many dispersing solvents were investigated, including ethyl alcohol, methyl alcohol, 2-propanol, acetone, dioxane, and distilled water (ESI, 6.1†). For the designed spectroscopic approach, the greatest fluorescence quenching values were gained when distilled water (the best green solvent38) was the dispersion medium. The low response obtained with organic solvents in the present study may be due to the destructive effect of these solvents on the formed complex. These solvents may cause denaturation of the complex system and therefore change the fluorescence value. Moreover, alcohols with short chains, such as ethanol and methanol, are easily dissolved in the aqueous fraction and consequently break down the complex formation process by altering solvent characteristics. Alcohol disrupts the complex system, and at higher concentrations, the system may suffer intensive disruption.32 Fortunately, the highest response was achieved when using distilled water as the final diluting solvent. Also, this may be due to the higher polarity index (9) and high dielectric constant (80.2) of water than other solvents used30,33 (ESI, Table 2†).Furthermore, with all constituents of the system dissolved in water, all solutions are likely to be completely miscible and the system freely formed. In contrast, the presence of other organic solvents (having different dielectric constants) may be of low miscibility, which would hinder the system's formation. A full excitation and emission spectra showing the effect of different diluting solvents on the formed association complex between MTX (1.0 μg mL−1) and eosin Y present in (ESI, 6.2†).
Mechanism and pathway of complex formation
The peak inherent fluorescence of xanthene-based dyes (erythrosine, fluorescein, and eosin) has been figured out to be suppressed following complexation with many nitrogen-containing drugs.22,23 The designed spectroscopic approach in this research relies on an ion-pair association complex in a moderately acidic medium between MTX (which has two basic centers) and eosin Y reagent to produce a binary complex (Scheme 1). | ||
| Scheme 1 The proposed pathway and suggested ion association mechanism between MTX and EY dye. | ||
According to the solution pH, the eosin Y molecule can appear in one of the following forms:

FT-IR confirmation
FTIR spectroscopy was used to verify the presence of functional groups in both the pure drug sample and the EY-formed product. MTX FTIR spectra revealed the predicted band of amino groups, the characteristic features that were discovered to be lost in the product IR spectrum, confirming the birth of a new product. Thus, the absence of many MTX spectroscopic features and new functionalities in the formed complex confirm the formation of a new product. It was concluded that the amino groups in the MTX molecule were either absent or had been included in an additional bond with EY because the –NH peak in the MTX–EY complex was no longer apparent (ESI, 7†).The reaction stoichiometry
Then, Job's plotting procedure was executed to calculate the molar ratio of the developed MTX–EY ion-pair complex. To do this, equimolar solutions (2.8 × 10−4 M) of both the drug and the dye were coupled in various molar ratios while maintaining the total molarity constant. The analytical steps were undergone exactly as detailed in the experimental section. Job's plots were designed to show the correlation between the acquired response and the matching mole fraction of the drug. The plots attained a maximum value at a mole fraction of roughly 0.66 for the employed approach, as proven in ESI, 8.† This value indicated the development of a drug:dye combination in the quota of 1:2. This ratio is indicative of the existence of two secondary amino groups in the drug molecule that may ion-pair with two dye molecules.
Application of Stern–Volmer formula
The Stern–Volmer plot23 was applied to study the quencher suppression mechanism on the inherent fluorescence of the utilized xanthene based dye (EY). Plotting the fluorescence strengths of the dye alone (EY) represented as (f0) and complexed with the suppressor molecule (2MTX–EY) expressed as (f) against the quencher (MTX) concentration represented as [Q] resulted in the Stern–Volmer graph. The plot gave a linear relationship that matched eqn (1).22| f0/f = 1 + ksv[Q] = 1 + kqτo[Q] | (1) |
The constructed plot resulted in a straight line with an intercept and slope called the Stern–Volmer constant (ksv), which in turn was the outcome of multiplying the natural radiation lifetime (τo) by the quenching rate constant kq (Ksv = kqτo). Because EY has a reported fluorescence lifetime value of 1.21 ns,36 and following the previous data, the calculated value of the quenching rate constant was 4.81 × 1014 L mol−1 s−1, which exceeds the highest documented Kq value for collisional quenching (2 × 1010 L mol−1 s−1).37 Thus, the proposed suppressing process for the MTX–EY interaction was considered a single static process.
The binding constant and free energy
The reaction of the cited drug with the EY dye had a binding constant (Kd), which can be computed using the following equation formula (2) similarly as applied in the estimation of the benzimidazole drug.23|
log(f0 − f/f) = logkd + nlog[Q]
| (2) |
:1) drug:dye quota. This was additionally confirmed by the MTX drug mole number (n), which was noted to be approximately two (1.94).
The plots (ESI, 9†) revealed that the results exhibited good linearity, which also verified that the quenching mechanism and the stoichiometry of the reaction between MTX and eosin Y follow the static mode.
The calculated binding constant can be applied to obtain Gibb's free energy value (ΔG°), from the formula: ΔG° = −2.303RTlogKd, where R stands for gas constant (8.314 J K−1 mol−1), T for the absolute temperature (in Kelvin), and Kd for the binding constant.27 The resulting Gibb's free energy value was about −28.3 kJ mol−1 for the designed reaction. This large negative free energy value indicates that the response exhibits a high degree of immediacy and practicality at ambient temperature.22,27
Methods validation
Based on the guidelines of ICH,21 the designed spectroscopic system was validated. The included parameters for validation were linearity, range, precision, accuracy, robustness, the quantification limit (LOQ), and the detection limit (LOD).Linearity and range
A series of standard MTX solutions were quantified with the designed spectrofluorimetric system. The calibration graph for the constructed spectroscopic method was constructed by plotting the ΔFI values towards the concentrations of MTX in μg mL−1. The response of the designed method was linear over the concentration ranged from 0.07 to 2.5 μg mL−1. In addition, the linear decline analysis was applied to the prescribed method, and the analytical parameters were calculated and listed in Table 1. Furthermore, a comparison between the sensitivity of the current fluorescence method other techniques for the determination of MTX was present in ESI, Table 3.†| Parameter | Value |
|---|---|
| Linear range (μg mL−1) | 0.07–2.5 |
| Slope | 3.413 |
| SD of slope (Sb) | 0.013 |
| Intercept | 6056.7 |
| SD of intercept (Sa) | 16.97 |
| Correlation coefficient (r) | 0.9999 |
| Determination coefficient (r2) | 0.9998 |
| SD of residuals (Sy/x) | 32.91 |
| Limit of quantitation (μg mL−1) | 0.049 |
| Limit of detection (μg mL−1) | 0.016 |
Limits of quantification and detection
The following equations were applied for the estimation of the quantitation limit (LOQ) and detection limit (LOD);| LOQ = 10SDi/P and LOD = 3.3SDi/P |
Accuracy and precision
At three concentration levels (0.1, 1.0, and 2.0 μg mL−1), the accuracy of the used spectroscopic approach was examined. The determining criteria for evaluating accuracy are the recovery percentage and relative error. The data in Table 2 shows the high accuracy of the designed method.| No. | Concentration (μg mL−1) | Recovery% ± SD | Er% | RSD% |
|---|---|---|---|---|
| a The value is the mean of three replicate measurements, SD is the standard deviation, RSD is the relative standard deviation, and Er% is the relative error percentage. | ||||
| 1 | 0.1 | 99.30 ± 1.79 | −0.70 | 1.80 |
| 2 | 1.0 | 100.12 ± 0.59 | 0.12 | 0.58 |
| 3 | 2.0 | 100.18 ± 0.35 | 0.18 | 0.35 |
Similarly, the proposed system was employed to assess the inter-day and intra-day precision of the drug sample solutions at three concentration levels (0.1, 1.0, and 2.0 μg mL−1). The RSD value was utilized as the parameter in this example to assess the accuracy of the provided spectroscopic technique. As Table 3 shows, the RSD values did not reach 2%, which indicates the high precision (at both levels) of the proposed approach.
| Precision level | Concentration (μg mL−1) | Recovery% | RSD% |
|---|---|---|---|
| a The value is the mean of three replicate measurements, SD is the standard deviation, and RSD is the relative standard deviation. | |||
| Inter-day | 0.1 | 99.62 ± 1.93 | 1.96 |
| 1.0 | 100.44 ± 0.71 | 0.71 | |
| 2.0 | 100.34 ± 0.41 | 0.41 | |
|
|||
| Intra-day | 0.1 | 96.18 ± 1.63 | 1.70 |
| 1.0 | 100.39 ± 0.76 | 0.76 | |
| 2.0 | 100.02 ± 0.49 | 0.49 | |
Robustness
The proposed spectroscopic system was validated from the robustness point of view by testing its performance under slight variations in system parameters such as pH (4.0 ± 0.2), eosin Y solution volume (1.0 ± 0.2 mL), and reaction rate. The parameters for measuring the approach robustness were the percent recovery and standard deviation values. The slight alterations were realized to have no significant influence on fluorescence quenching readings, demonstrating the robustness of the proposed spectrofluorimetric application (Table 4).| Parameter | Value | Recovery% ± SD |
|---|---|---|
| a The value is the mean of three measurements, SD is the standard deviation, and RSD is the relative standard deviation. | ||
| pH | 3.8 | 98.68 ± 0.92 |
| 4.2 | 101.15 ± 0.55 | |
| Buffer volume (mL) | 1.2 | 101.33 ± 1.00 |
| 1.6 | 99.48 ± 0.32 | |
| EY volume (mL) | 0.8 | 96.87 ± 1.63 |
| 1.2 | 100.64 ± 1.31 | |
| Time (min) | 8 | 96.92 ± 1.67 |
| 12 | 101.84 ± 1.03 | |
Application to the pharmaceutical form
The recommended spectroscopic methodology was employed to assess MTX in its pharmaceutical prescription form (Santrone® vial). The reported spectrofluorimetric technique38 was also used to examine the same dosage form. Using two statistical tests (t-test and F-test), the percentage recovery values of the reported spectrophotometric technique were statistically rated against those obtained from the developed system. Because the obtained t- and F-values at the 95 percent confidence level were less than the theoretical values, there was no discernible difference in accuracy and precision between the proposed spectroscopic system and the documented method for MTX analysis, as shown in Table 5.Furthermore, the designed system outperformed other documented methods in terms of sensitivity, simplicity, time-saving, employment of an “eco-friendly” solvent (water), and detection limits. Additionally, the high percentage of recovery and absence of interference from prescription packaging additions make the described system suitable for testing MTX-containing dosage forms and assays in QC laboratories.
Human blood and urine analysis
The excellent sensitivity (70–2500 ng mL−1) of the proposed method allowed it to be used in a clinical study for MTX in human blood and urine (Cmax = 318 ng mL−1).39 To make the calibration graphs, plasma and urine samples were created by diluting the operating drug solutions with the supernatant of the relevant biological fluid. The suggested spectroscopic system was used to detect the final MTX concentrations in the clinical samples, which ranged from 0.3 to 2.0 μg mL−1. With calibration curves being executed utilizing the collected data and the equations derived from linear regression analysis (ESI, 10 and 11†). The drug analysis linear ranges for blood and urine were 0.3–2.0 μg mL−1. Three replicated samples of plasma or urine were supplemented with varying doses of MTX standard solution before being evaluated according to the methodology. The suggested method recovery ranges were nearly 100% with a very low RSD (Table 6), demonstrating the method's feasibility for MTX measurement in human blood and urine.| Matrix | Conc. (μg mL−1) | Recovery% ± SD | Er% | RSD% |
|---|---|---|---|---|
| a The value is the mean of three measurements, SD is the standard deviation, RSD is the relative standard deviation, and Er is the relative error. | ||||
| Plasma | 0.5 | 99.78 ± 1.75 | −0.22 | 1.75 |
| 1.0 | 100.21 ± 1.54 | 0.21 | 1.54 | |
| 1.5 | 101.87 ± 0.66 | 1.87 | 0.65 | |
|
||||
| Urine | 0.5 | 100.03 ± 1.79 | 0.03 | 1.79 |
| 1.0 | 100.28 ± 0.97 | 0.28 | 0.97 | |
| 1.5 | 100.99 ± 0.54 | 0.99 | 0.54 | |
System greenness evaluation
Analysts bear a significant level of authority for shielding both the environment and people from harsh chemicals and organic waste generated as a result of their implementation in chemical and pharmaceutical activities.30 The creation and upgrading of green chemistry must be conducted regularly. Recent considerations, such as the analytical eco scale score40 and the Environmental Quality Methods Index marking,41 have been used to assess an analytical method's “ecological value”. We used the eco-scale to determine the greenness of the proposed system. An analytical eco-scale assessment result is a number signifying a penalty point issued that was deducted from 100, a result acquired for “ultimate green analysis”. These points represent the dangers encountered throughout the research method. The greater the score (represented by a high number), the greener the analysis.30 Because the developed technique contained no extraction process and no heating required, the energy consumed was less than 0.1 kW h per sample. Furthermore, because of the use of an aqueous medium throughout the procedure, the eco-scale score for the created procedure was high (95), as shown in Table 7. This means that our technique was environmentally sustainable.| Item | Parameter | Word sign | PP score |
|---|---|---|---|
| a LSH is an abbreviation for the less severe hazard, and TPPs for the total penalty points. | |||
| Technique | Fluorimetry | 0 | |
| Reagent | Eosin Y | LSH | 1 |
| Amount of reagent | >10 mL | 1 | |
| Solvent(s) | Water | Green solvent | 0 |
| Heating | — | 0 | |
| Temperature | 25 °C | 0 | |
| Cooling | — | 0 | |
| pH | 4.0 | 0 | |
| Energy (kWh per sample) | 1.0> | 0 | |
| Waste | 1–10 (mL) | 3 | |
| Occupational hazards | 0 | ||
| (TPPs) | 5 | ||
| Eco-scale total score | = 100 − TPP | 95 | |
Conclusion
In this work, a new, selective, sensitive, and accurate spectrofluorimetric method was described for MTX assay. An electrostatic attraction in a slightly acidic medium was used to quantify MTX in the range of 0.07–2.5 μg mL−1. Furthermore, eosin Y is a less hazardous regent. Besides environmental safety, the most significant benefit of the procedure was its simplicity since the sample was recovered with water, and the generated ion pair was readily quantified in the aqueous solution. The present system is based on forming an ion pair-associated complex between the two secondary amino groups of MTX and the protonated hydroxyl group of eosin Y. The method was characterized by simplicity and rapidity, which were explained by the freeing of the extraction process, the lack of tedious steps, shortened analytical time, and reduced cost. Moreover, water is the reaction solvent, which is the most significant green solvent. The suggested techniques are sensitive and ecologically safe. Volatile solvents were not used in this study. Also, the designed system was successfully executed to detect an investigated drug in its dosage form, blood, and urine without any interference from the matrices at a range of 0.3 to 2.0 μg mL−1. In terms of the method's greenness, the method was evaluated following the eco-scale and offered a high greenness value. Therefore, this method can be considered for quality control analysis of this drug in research laboratories, clinical studies, and pharmaceutical industries.Conflicts of interest
The authors declare that there is no conflict of interest. They also state that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.References
- S. Agarwal, D. K. Jangir and R. Mehrotra, J. Photochem. Photobiol., B, 2013, 120, 177–182 CrossRef CAS PubMed.
- A. Buczkowski, P. Tokarz, A. Stepniak, J. Lewkowski, A. Rodacka and B. Palecz, J. Mol. Liq., 2019, 290, 111190 CrossRef CAS.
- C. K. Lee, V. J. Gebski, A. S. Coates, A.-S. Veillard, V. Harvey, M. H. Tattersall, M. J. Byrne, B. Brigham, J. Forbes and R. J. Simes, SpringerPlus, 2013, 2, 1–10 CrossRef PubMed.
- G. Micelli, A. Lozupone, M. Quaranta, A. Donadeo, M. Coviello and V. Lorusso, Biomed. Chromatogr., 1992, 6, 168–171 CrossRef CAS PubMed.
- J. L. Johnson, A. Ahmad, S. Khan, Y.-F. Wang, A. W. Abu-Qare, J. E. Ayoub, A. Zhang and I. Ahmad, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 799, 149–155 CrossRef CAS PubMed.
- G. An and M. E. Morris, J. Pharm. Biomed. Anal., 2010, 51, 750–753 CrossRef CAS PubMed.
- R. J. van de Nesse, R. J. van der Wegen, C. Gooijer, A. T. Udo and N. H. Velthorst, Anal. Chim. Acta, 1995, 309, 135–144 CrossRef CAS.
- S. Han and H. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2901–2904 CrossRef CAS PubMed.
- H. Yao, M. Zhang, W. Zeng, X. Zeng and Z. Zhang, Spectrochim. Acta, Part A, 2014, 117, 645–650 CrossRef CAS PubMed.
- A. O. Brett, T. Macedo, D. Raimundo, M. Marques and S. Serrano, Anal. Chim. Acta, 1999, 385, 401–408 CrossRef.
- S. Wang, T. Peng and C. F. Yang, Biophys. Chem., 2003, 104, 239–248 CrossRef CAS PubMed.
- S. Golabi and V. Hassan-Zadeh, Talanta, 1996, 43, 397–406 CrossRef CAS PubMed.
- Y. Mao, J. Hu, Q. Li and P. Xue, Analyst, 2000, 125, 2299–2302 RSC.
- S. U. Flavell and D. J. Flavell, J. Immunol. Methods, 1988, 115, 179–185 CrossRef CAS PubMed.
- M. Enache and E. Volanschi, J. Pharm. Sci., 2011, 100, 558–565 CrossRef CAS PubMed.
- Z. Zhou, C. Wu, L. Zhang, L. Li and X. He, Chin. J. Pharm. Anal., 1997, 17, 403–405 CAS.
- A. Mohammadinejad, Z. Es' haghi, K. Abnous and S. A. Mohajeri, J. Lumin., 2017, 190, 254–260 CrossRef CAS.
- F. Wang, Z. Liu and S. Liu, Chin. J. Chem., 2005, 63, 1991–1998 CAS.
- C. McLaughlin, D. MacMillan, C. McCardle and W. E. Smith, Anal. Chem., 2002, 74, 3160–3167 CrossRef CAS PubMed.
- G. Breuzard, J.-F. Angiboust, P. Jeannesson, M. Manfait and J.-M. Millot, Biochem. Biophys. Res. Commun., 2004, 320, 615–621 CrossRef CAS PubMed.
- M. E. Swartz and I. S. Krull, Analytical Method Development and Validation, CRC press, 2018 Search PubMed.
- S. M. Derayea, A. A. Hamad, R. Ali and H. R. H. Ali, Microchem. J., 2019, 149, 104024 CrossRef CAS.
- A. A. Hamad, R. Ali, H. R. H. Ali, D. M. Nagy and S. M. Derayea, RSC Adv., 2018, 8, 5373–5381 RSC.
- S. M. Derayea, Anal. Methods, 2014, 6, 2270–2275 RSC.
- H.-Y. Hong, G.-S. Yoo and J.-K. Choi, 1999.
- M. E. Selsted and H. W. Becker III, Anal. Biochem., 1986, 155, 270–274 CrossRef CAS PubMed.
- S. M. Derayea, A. A. Hamad, D. M. Nagy, D. A. Nour-Eldeen, H. R. H. Ali and R. Ali, J. Mol. Liq., 2018, 272, 337–343 CrossRef CAS.
- H. T. S. Britton and R. A. Robinson, J. Chem. Soc., 1931, 1456–1462 RSC.
- T. McIlvaine, J. Biol. Chem., 1921, 49, 183–186 CrossRef CAS.
- S. M. Derayea, R. Ali and A. A. Hamad, Arabian J. Chem., 2020, 13, 8026–8038 CrossRef CAS.
- C. Y. Huang, in Methods in Enzymology, Elsevier, 1982, vol. 87, pp. 509–525 Search PubMed.
- S. Shiao, V. Chhabra, A. Patist, M. Free, P. Huibers, A. Gregory, S. Patel and D. Shah, Adv. Colloid Interface Sci., 1998, 74, 1–29 CrossRef CAS.
- L. B. Kier, J. Pharm. Sci., 1981, 70, 930–933 CrossRef CAS PubMed.
- L. Yu, Z. Liu, X. Hu, L. Kong and S. Liu, Microchim. Acta, 2010, 169, 375–382 CrossRef CAS.
- C. Li, S. Liu, Z. Liu and X. Hu, J. Fluoresc., 2011, 21, 723–732 CrossRef CAS PubMed.
- X.-F. Zhang, J. Zhang and L. Liu, J. Fluoresc., 2014, 24, 819–826 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- A. A. Hamad, R. Ali and S. M. Derayea, Luminescence, 2021, 36, 443–453 CrossRef CAS PubMed.
- D. R. Liston and M. Davis, Clin. Cancer Res., 2017, 23, 3489–3498 CrossRef CAS PubMed.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
- M. De La Guardia and S. Armenta, Green Analytical Chemistry: Theory and Practice, Elsevier, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00120a |
| This journal is © The Royal Society of Chemistry 2022 |