
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new route for controlling the microstructure and properties of carbon aerogels via sol–gel and impregnation methods
Haixia Yang *a,
Haokun Lia and
Feng Yeb
*a,
Haokun Lia and
Feng Yeb
aSchool of Textile and Materials Engineering, Dalian Polytechnic University, Dalian 116034, P. R. China. E-mail: yanghaixiaedu@163.com
bSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, P. R. China
First published on 24th March 2022
Abstract
A new route to control the microstructure and properties of carbon aerogels via vacuum impregnation is presented. The results show that the enhanced carbon aerogels exhibit uniform pore size distribution (∼50 nm), a high compressive strength of 77.0 MPa and a low thermal conductivity of 0.15–1.62 W m−1 K−1 at 25 to 1600 °C.
Carbon aerogels have been used extensively for thermal insulation applications because of their light weight, low thermal conductivity and excellent heat stability.1,2 However, it is difficult to use carbon aerogels in practical applications because of low strength.3 To improve the mechanical properties of the aerogels, many reinforcements have been used to fabricate carbon aerogel composites, such as ceramic fibres,4 carbon fibres5–7 carbon nanotubes8 and carbon cloth.9 However, the carbon aerogels obtained via carbonization exhibit large shrinkage rates (∼20%), whereas these reinforcements rarely shrink during the process. The different shrinkage rates may lead to a large residual stress, resulting in cracks that will impact the thermal insulation and the mechanical properties.5 To prevent inconsistent contraction, organic PAN fibres reinforced with carbon aerogel composites have been prepared.10 However, organic PAN fibres should be processed into felts, which complicate the preparation process, and the lapped fibres may increase the thermal conductivity. Recently, researchers have improved the strength of porous carbon by combining with silica aerogel.11,12 The silica-modified carbon aerogel showed high strength,12 however its thermal conductivity was also high.
In this study, a simple and effective method to strengthen carbon aerogels uses an organic gel as the skeleton to impregnate RF sol with different catalysts to obtain enhanced carbon aerogels via carbonization. In previous investigations, the gel particle sizes and pore structure of the carbon aerogels could be adjusted by controlling the catalyst concentration.13 As shown in Fig. 1, the gel size increased as the catalyst concentration decreased. An organic aerogel with a larger gel size was used as the skeleton to impregnate the RF sol with a high catalyst concentration. The organic skeleton possesses more hydrophilic groups, which is necessary for the impregnation and bonding. Moreover, the shrinkage rate of the organic aerogel skeleton and of the aerogels within the skeleton is very similar, which can prevent the formation of cracks during carbonization. Additionally, to shorten the drying time and lessen the risk of supercritical drying, freeze drying was used to obtain the organic gel skeleton. Unfortunately, it was difficult to obtain carbon aerogels with a small and uniform pore structure via freeze drying due to the growth of ice crystals during the freeze-drying process. Thus, the impregnated organic gel was dried under atmospheric pressure, and enhanced carbon aerogels were obtained via pyrolysis. The microstructure and mechanical and thermal insulation properties of the carbon aerogels were investigated.
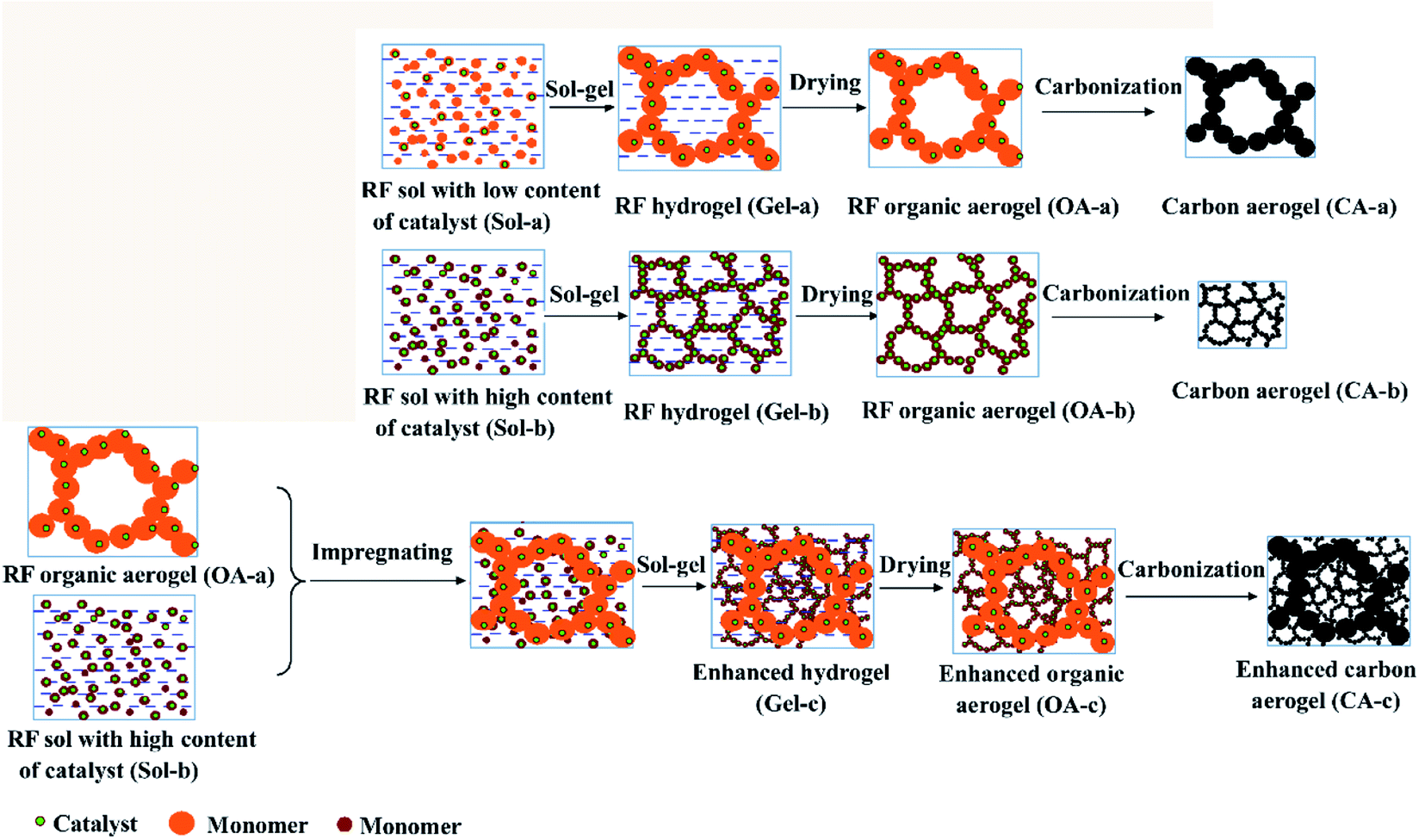 | ||
| Fig. 1 Sketch of the new route to improve the carbon aerogels using an organic network skeleton. | ||
The precursor of carbon aerogel is organic aerogel (OA), which is synthesized by polycondensation of resorcinol (R) and formaldehyde (F), using sodium carbonate (C) as catalyst and deionized water (W) as solvent. Organic aerogel is further carbonized to obtain carbon aerogel (CA). Schematic diagram of sample preparation is shown in Fig. 1. On the one hand, resorcinol dissolved in water, adding formaldehyde and sodium carbonate, thoroughly mixed, to obtain sol. The molar ratios of R, F, C and W are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:0.001:18 respectively, denoting as Sol-a. After a thorough mixing, the Sol-a was sealed into a centrifuge tube and cured at 85 °C for 4 days. The hydrogel was dried in a vacuum freeze dryer to obtain the organic aerogel (OA-a) framework.
:2:0.001:18 respectively, denoting as Sol-a. After a thorough mixing, the Sol-a was sealed into a centrifuge tube and cured at 85 °C for 4 days. The hydrogel was dried in a vacuum freeze dryer to obtain the organic aerogel (OA-a) framework.
On the other hand, resorcinol was dissolved in water by the same process, and formaldehyde and sodium carbonate were added to obtain sol. Where, the molar ratios of R, F, C and W are 1:2:0.01:18 respectively, denoted as Sol-b. Two identical Sol-b were prepared, one of which was impregnated with organic aerogel (OA-a) framework by Sol-b under vacuum condition. Then, the complex was sealed into a centrifuge tube and cured at 85 °C for 4 days. After the 4 day curing period, the water in the hydrogels was exchanged with acetone. Then, the wet gels were dried at room temperature to obtain the organic aerogel (OA-c). As a comparison, another portion of sol b was directly sealed in a centrifuge tube and cured at 85 °C for 4 days, solvent exchanged with acetone, and then dried at room temperature to obtain an organic aerogel (OA-b). The organic aerogel (OA-a, OA-b, and OA-c) were carried out in a tube furnace at 1073 K under nitrogen flow for 2 h, respectively, to obtain carbon aerogels (CA-a, CA-b, and CA-c).
The microstructures were characterized by scanning electron microscopy (SEM, Quanta 200, FEI Co., Oregon, USA) and transmission electron microscopy (TEM, Tecnai G2F30, FEI Co., Oregon, USA). Compressive strength of the specimens was tested at a crosshead speed of 0.5 mm min−1 on specimens (6 mm × 6 mm × 8 mm). The measurements were conducted thrice and the values obtained were averaged. Nitrogen adsorption isotherms were measured by a gas adsorption analyzer (Gemini VII 2390, Micrometics Co., USA) at 77 K in the relative pressure range (p/p0) 0.05–0.99. The carbon aerogels were cut into disk shapes with diameters of 12.6 mm and thicknesses of approximately 2.55 mm, and the thermal diffusivities in the through-plane direction of the samples were determined by the laser flash method using a Netzsch LFA427 apparatus. The thermal conductivities of the carbon aerogels, λ, were calculated by equation: λ = α × ρ × Cp. Where α is the thermal diffusivity, ρ is the density, and Cp is the heat capacity.
The drying shrinkage, density, porosity and compressive strength of the specimens are summarized in Table 1. The drying shrinkages of the CA-a obtained via freeze drying and of the CA-b obtained via ambient drying are 3.4% and 22.6%, respectively. The CA-b has a relatively high density because of the larger drying shrinkage, resulting in its high compressive strength of 113.4 MPa. After impregnating, the drying shrinkage of the CA-c is nearly zero due to the organic skeleton of CA-a, which provides strong support and restrains the shrinkage during drying. By impregnating, the density of the aerogels increased from 0.36 g cm−3 to 0.54 g cm−3, and the corresponding porosity decreased from 86.2% to 79.2%. The compressive strength of the CA-c is 77.0 MPa, which is approximately 4.3 times higher than of the CA-a (14.5 MPa). The increase of compressive strength can be attributed to the fact that the CA-b occupied some pores in the CA-a and was due to the combination them both.
| Specimens | Drying shrinkage of the organic aerogels (%) | Density (g cm−3) | Porosity (%) | Compressive strength (MPa) |
|---|---|---|---|---|
| CA-a | 3.4 | 0.36 | 86.2 | 14.5 ± 1.9 |
| CA-b | 22.6 | 0.96 | 63.0 | 113.4 ± 3.6 |
| CA-c | ∼0 | 0.54 | 79.2 | 77.0 ± 2.9 |
The SEM images and TEM micrographs of the specimens are shown in Fig. 2. The CA-a exhibits a sponge-like microstructure with nanoparticles (10–20 nm) stacked and interconnected into porous network. However, the CA-b rarely has a nanostructure because of its lack of skeleton (with a high catalyst concentration) that collapsed and gathered during drying. After impregnating, the CA-c maintained the nanopore structure, whereas the porosity declined. The TEM micrographs show that most of the larger pores were filled by relatively small nanoparticles, which contributed to the increased mechanical properties.
 | ||
| Fig. 2 SEM images of the carbon aerogels before and after impregnation: (a) CA-a, (b) CA-b, (c) CA-c; TEM micrographs of the carbon aerogels: (d) CA-a, (e) CA-b, (f) CA-c. | ||
The nitrogen adsorption isotherms and pore size distributions of the carbon aerogels are displayed in Fig. 3. As shown in Fig. 3, the CA-b exhibits an adsorption isotherm that suggests a microporous and mesoporous structure, and most of the pores are less than 10 nm. The CA-a and CA-c have an adsorption isotherm of type IV, indicating that it has a well-developed mesoporous structure. By impregnating, the BET surface areas (SBET) decreased from 612 m2 g−1 to 558 m2 g−1, and the adsorption quantity gradually decreased due to the decreased porosity of the composites. The CA-a obtained via freeze drying shows a multimodal pore size distribution (PSD) with peaks at approximately 74 nm and 108 nm. After impregnation, the CA-c has a narrow PSD with the peak centred at approximately 58 nm. Thus, the pore structure of the carbon aerogels is controlled by impregnating the organic sol with different concentrations of catalyst.
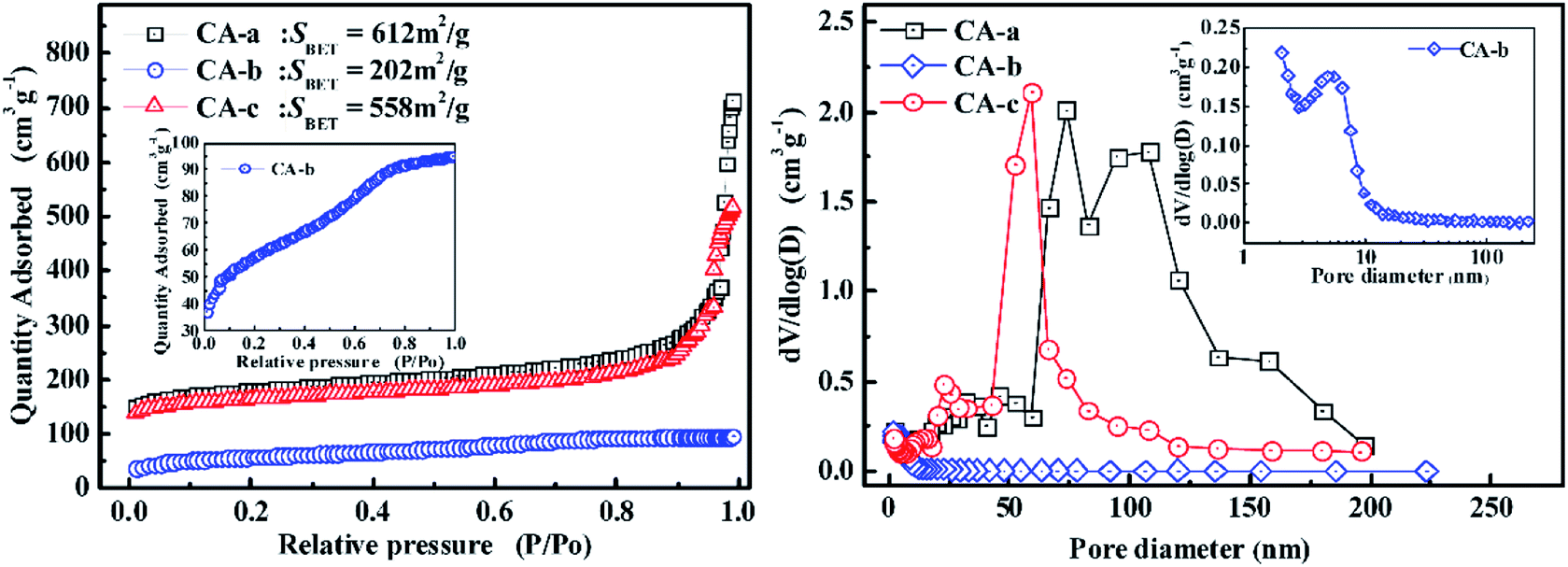 | ||
| Fig. 3 The nitrogen adsorption isotherms and pore size distributions of the carbon aerogels. | ||
The thermal diffusivities and the thermal conductivities of the carbon aerogels are shown in Fig. 4. Literature values of specific heat capacity were used to calculate the thermal conductivity of the carbon aerogels.14 The thermal diffusivities changed insignificantly between 25 and 800 °C, and then they increased as the temperature increased to 1600 °C, which may be due to the microstructural changes in the carbon aerogels. As the temperature increased, the thermal conductivities of the carbon aerogels increased. Because of the higher porosity, the CA-a exhibited a lower thermal conductivity of 0.06–0.80 W m−1 K−1. In contrast, the CA-b with the high density exhibited a high thermal conductivity of 0.59–3.91 W m−1 K−1 from 25 to 1600 °C. After impregnating, the thermal conductivity of the CA-c was 0.14–1.62 W m−1 K−1 from 25 to 1600 °C, more than approximately double that of the CA-a. The increase in the thermal conductivity is primarily attributed to the increase in the solid phase conduction caused by the increase in density and pore wall thickness. Fortunately, the enhanced carbon aerogel CA-c possesses a nanometre pore structure, and the most probable pore size (∼58 nm) is less than the mean free path of the gas molecules (69 nm). Therefore, the composites still possess good thermal insulating properties.(Table 2)
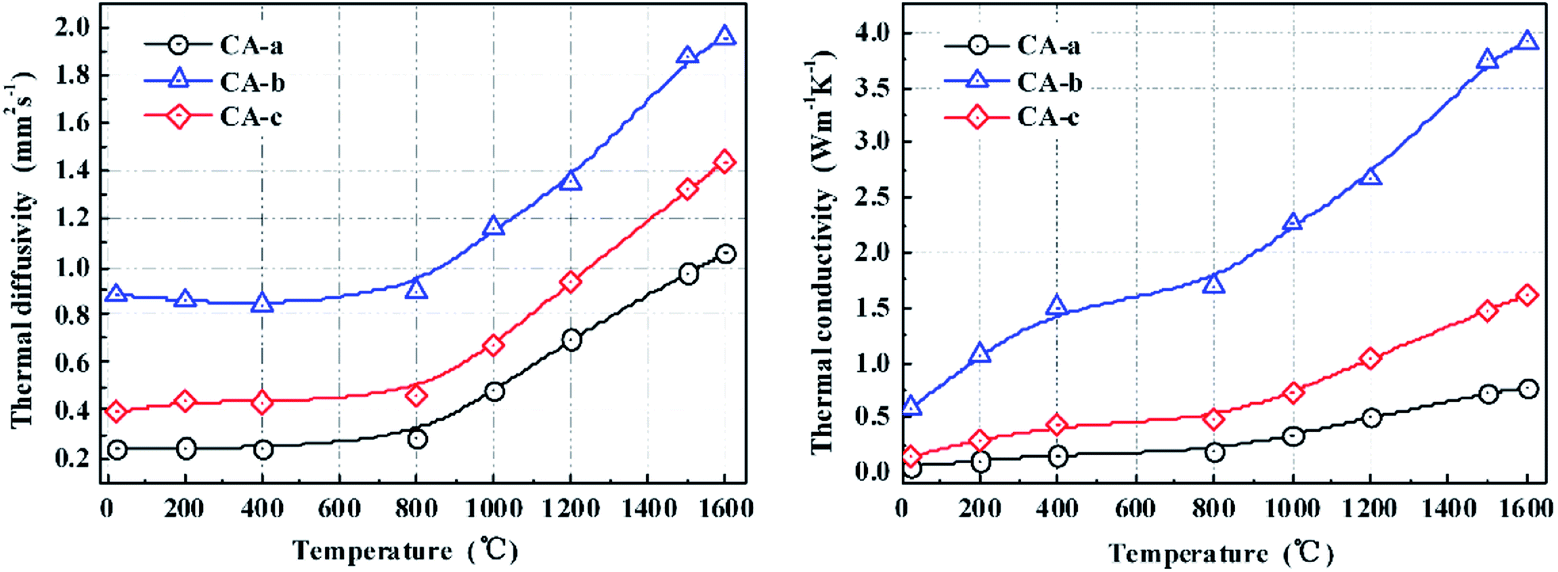 | ||
| Fig. 4 The thermal diffusivity and thermal conductivity of the carbon aerogels. | ||
| Materials | Thermal conductivity (W m−1 K−1) | Compressive strength (MPa) | Ref. |
|---|---|---|---|
| CA-c | 0.14 | 77.0 | In this study |
| Reinforced carbon foam materials | ∼0.25 | 7.43 | 15 |
| Silica-modifies carbon aerogels | 0.118 | 10.0 | 12 |
| SiO2 aerogel/carbon foam composite | ∼0.28 | 12.72 | 11 |
| CBCF/Si–Al aerogels | 0.184 | 5.75 | 16 |
| UCF-carbon aerogels | 0.08 | 5.0 | 17 |
We presented a detailed investigation of the microstructural, mechanical, and thermal insulation properties of carbon aerogels prepared via the sol–gel method. The mechanical properties and thermal conductivity are highly dependent on the densities of the carbon aerogels. Upon impregnation, the carbon aerogels exhibited a uniform pore size distribution (∼58 nm), a low dry shrinkage, and good mechanical and thermal insulation properties. Thus, the carbon aerogels may have potential applications in the field of high-temperature thermal insulation. This route can be more widely used to control the microstructure and mechanical properties of the porous materials.
Author contributions
Haixia Yang: writing – original Draft, investigation, resources. Haixia Yang: writing – review & editing, methodology, funding acquisition. Haokun Li: resources, validation, writing – review & editing. Feng Ye: validation, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financial supported by the Dalian High-level Talent Innovation Support Plan (No. 2019RQ077). We thank the Scientific Research Fund of Liaoning Provincial Education Department (No. J2020104).References
- Y. Hanzawa, H. Hatori, N. Yoshizawa and Y. Yamada, Carbon, 2002, 40, 575–581 Search PubMed.
- A. M. Elkhatat and S. A. Al-Muhtaseb, Adv. Mater., 2011, 23, 2887–2903 Search PubMed.
- Z. Yang, J. Li, X. Xu, S. Pang, C. Hu, P. Guo, S. Tang and H. M. Cheng, J. Mater. Sci. Technol., 2020, 50, 66–74 Search PubMed.
- J. Yang, S. K. Li, Y. M. Luo, L. L. Yan and F. C. Wang, Carbon, 2011, 49, 1542–1549 Search PubMed.
- V. Drach, M. Wiener, G. Reichenauer, H.-P. Ebert and J. Fricke, Int. J. Thermophys., 2007, 28, 1542–1562 Search PubMed.
- C. Liu, J. C. Han, X. H. Zhang, C. Q. Hong and S. Y. Du, Carbon, 2013, 59, 547–554 Search PubMed.
- Y. Zhong, J. Zhang, X. Wu, X. Shen, S. Cui and C. Lu, J. Sol-Gel Sci. Technol., 2017, 84, 129–134 Search PubMed.
- M. A. Worsley, J. H. Satcher and T. F. Baumann, Langmuir, 2008, 24, 9763–9766 Search PubMed.
- X. G. Yang, J. Wei, D. Q. Shi, Y. T. Sun, S. Q. Lv, J. Feng and Y. G. Jiang, Mater. Sci. Eng., R, 2014, 609, 125–130 Search PubMed.
- J. Z. Feng, C. R. Zhang, J. Feng, Y. G. Jiang and N. Zhao, ACS Appl. Mater. Interfaces, 2011, 3, 4796–4803 Search PubMed.
- H. G. Liu, Y. D. Xu, C. L. Tang, Y. Li and N. Chopra, Ceram. Int., 2019, 45, 23393–23398 Search PubMed.
- K. Wu, Q. Zhou, J. X. Cao, Z. Qian, B. Niu and D. H. Long, J. Colloid Interface Sci., 2022, 609, 667–675 Search PubMed.
- A. W. P. Fung, G. A. M. Reynolds, Z. H. Wang, M. S. Dresselhaus, G. Dressehaus and R. W. Pekala, J. Non-Cryst. Solids, 1995, 186, 200–208 Search PubMed.
- M. Wiener, G. Reichenauer, S. Braxmeier, F. Hemberger and H. P. Ebert, Int. J. Thermophys., 2009, 30, 1372–1385 Search PubMed.
- R. Y. Luo, Y. F. Ni, J. S. Li, C. L. Yang and S. B. Wang, Mater. Sci. Eng. A, 2011, 528, 2023–2027 Search PubMed.
- H. M. Li, Y. F. Chen, P. D. Wang, B. S. Xu, Y. B. Ma, W. B. Wen, Y. Z. Yang and D. N. Fang, Ceram. Int., 2018, 44, 3484–3487 Search PubMed.
- P. Guo, J. Li and S. Pang, et al., Carbon, 2021, 183, 525–529 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |