DOI:
10.1039/D1RA09225A
(Paper)
RSC Adv., 2022,
12, 6659-6667
Cellulose-reinforced poly(ethylene-co-vinyl acetate)-supported Ag nanoparticles with excellent catalytic properties: synthesis of thioamides using the Willgerodt–Kindler reaction†
Received
21st December 2021
, Accepted 18th February 2022
First published on 25th February 2022
Abstract
Cellulose, a bio-derived polymer, is widely used in food packaging, dye removal, coatings, and solid-supported catalysis. Heterogeneous catalysts play a critical role in environmental remediation. In this context, the demand for green and cost-effective catalysts has rapidly increased. In this study, cellulose was extracted from rice straw, and a highly active solid-supported catalytic model was developed. First, cellulose was conjugated with poly(ethylene-co-vinyl acetate) (PEVA), and then Ag nanoparticles (AgNPs) were inserted into the cellulose–PEVA composite. The process involved the reduction of AgNPs in the presence of sodium borohydride. The fabricated hybrid catalyst was characterized using Fourier-transform infrared spectroscopy, scanning electron microscopy, energy dispersive X-ray, and powder X-ray diffraction. Thereafter, the obtained hybrid was used as a catalyst for the Willgerodt–Kindler reaction of aromatic aldehydes, amines, and S8 to synthesize thioamides with excellent yields. The developed catalytic system exhibited high stability and recyclability. Moreover, the mechanical properties of the hybrid catalyst were evaluated using tensile strength and impact tests. RGB analysis of digital images was also performed to investigate the primary components of the catalyst.
Introduction
Recently, thioamides have garnered increasing attention in the field of medicinal chemistry owing to their broad biological activity range. Thioamide derivatives, which are critical components of many biologically important entities, exhibit remarkable antioxidant, antimicrobial, anticonvulsant, antithyroid, and anticarcinogenic properties.1–4 Moreover, owing to their unique characteristics, namely their varying bond lengths and bond rotation ability, thioamides are better candidates than amides for peptidomimetics.5–8 Several thioamide synthesis methods have been developed. Among them, the Willgerodt–Kindler reaction, which involves the reaction of an aryl ketone/aldehyde with an amine in the presence of elemental sulfur, is an economical method for thioamide fabrication.9–12 However, the Willgerodt–Kindler reaction presents several disadvantages, including low product yields and long reaction times; moreover, purification of the desired product from the reaction mixture is difficult. Several approaches have been used to overcome these limitations.13–15 The Willgerodt–Kindler reaction has been modified using additives, such as K2S2O8,16 C12H25SH,17 K3PO4,10 TsOH,18 Na2S·9H2O,19 K2CO3,20 and β-CD,21 to improve reaction efficiency. Furthermore, environmentally benign reaction conditions have been developed, such as solvent-less and catalyst-free conditions; in addition, water or glycerol was used as the solvent.22,23 However, these modifications decreased the reaction yield and prolonged the reaction time. In terms of green and sustainable chemistry, the development of a sustainable catalyst that can be recovered and subsequently reused is a priority for Willgerodt–Kindler reaction researchers.
Heterogeneous catalysts are more easily recovered from reaction systems than homogeneous catalysts. Moreover, metal nanoparticles (NPs) play important roles in various organic synthesis reactions. However, the direct use of metal NPs can cause significant human health and environmental problems.24–26 Therefore, heterogeneous metal NPs that are attached to solid supports are required for catalysts with facile recovery and good reusability. Biopolymers, including starch, gelatin, cellulose, chitosan, and sodium alginate, serve as solid supports for heterogeneous catalysts and exhibit remarkable catalytic ability during organic synthesis.27,28 Cellulose is primarily found in plants and also in some animals and bacteria.29,30 Because of the spherical and porous beads of cellulose, its catalytic efficacy can be significantly increased via the insertion of metal ions and NPs into its matrix.31 Moreover, chemically modified hydrophobic cellulose has been increasingly used. The cross-linking properties and large cavity sizes of biopolymers promote the insertion of metals with high binding energies.32 In addition, the primary drawbacks of biopolymers, such as their low water solubility, can be overcome via chemical modification.33
Cellulose has been used to enforce polymer composites because of its high mechanical strength, low density, high surface area, and non-covalent interactions.34–39 In this study, to achieve remarkable thermal stability and high surface area, cellulose was grafted with the PEVA copolymer. The vinyl acetate (VA) content of PEVA ranged between 3% and 40% (w/w), and the mechanical and chemical properties of PEVA depended on its VA content. Thereafter, Ag nanoparticles (AgNPs) were anchored to the cellulose-reinforced PEVA to obtain AgNPs@cellulose–PEVA, which was used as an efficient and sustainable heterogeneous catalyst for the synthesis of thioamides using the Willgerodt–Kindler reaction. The developed catalyst presented high thermal stability and remarkable mechanical strength and was recycled for more than eight cycles without catalytic activity loss.
Experimental section
General information
All the chemicals were purchased from Sigma-Aldrich Co and were used as received without further purification. A PEVA copolymer with a VA content of 12 wt% was used as the composite matrix. Meting points were measured using a SMP30 (BIBBY) melting point meter. Fourier-transform infrared (FTIR) spectra were recorded using an Hyperion 2000 (Bruker Optics) FTIR system. A JEOL instrument operated at 400 and 100 MHz was used to obtain 1H nuclear magnetic resonance (NMR) and 13C NMR spectra. The chemical shifts were measured in parts per million using a deuterated solvent as the internal reference. High-resolution mass spectroscopy (HRMS) was performed using a Xevo G2-XS QTOF (WATERS) mass spectrometer. Scanning electron microscopy (SEM) was performed using a LEO Supra 55 VP (Zeiss) instrument. Powder X-ray diffraction (PXRD) measurements were performed using a Miniflex (Rigaku) diffractometer. Energy-dispersive X-ray spectrometry (EDX) analysis was performed using a JSM-6610-LV (JEOL) instrument. Digital images of the components of the hybrid catalyst were obtained using a smartphone and were utilized for RGB analysis employing the Image J software. Tensile tests were performed using an UTM (INSTRON) tensile tester without an extensometer at 25 °C. The samples used for the tensile tests were 3.20 mm wide and 7.20 mm thick, and their calculated surface area was 23 mm2.
Cellulose isolation from rice straw
Rice straw was cut into small pieces and passed through a 0.5 mm sieve. The obtained mixture was added to distilled water and stirred for 2 d at 50 °C. The obtained solid fraction was washed three times with distilled water, followed by drying. Cellulose was extracted using a literature-reported method, as follows:40 the lignin and hemicellulose fractions of rice straw were removed, followed by alkaline treatment. A 150 mL aqueous solution comprising sodium chlorite (NaClO2; 1.50 g) and acetic acid (CH3COOH; 0.3 mL) was added to the dried rice straw sample (4 g) in a 250 mL round-bottom flask. The mixture was heated at 75 °C for 2 h, and delignification was repeated three times by adding 1.50 g of NaClO2 and 0.3 mL of CH3COOH every hour. The solid residue was extracted and washed with distilled water to neutral pH. Lastly, the dried solid residue was heated to 80–85 °C in 200 mL of a 4.5 wt% potassium hydroxide solution for 2 h, followed by washing four times with distilled water.
Preparation of the AgNPs@cellulose–PEVA hybrid
Preparation of cellulose–PEVA. PEVA (1 g) was dissolved in CHCl3 (10 mL) at 55 °C. Thereafter, cellulose (0.2 g) was added to the solution. The mixture was stirred for 3 h to homogeneity, followed by transfer to a Petri dish and air-drying.
Preparation of AgNPs. A 50 μM citric acid solution (1 mL) was added to a 0.64 mM AgNO3 solution (39 mL), and the mixture was stirred for 30 min to homogeneity. Next, a 25 mM NaBH4 solution (10 mL) was added dropwise to the solution until it turned gray, indicating the formation of AgNPs. Lastly, to prepare the AgNPs@cellulose–PEVA hybrids, the cellulose–PEVA mixture was added to the AgNP dispersion, and the blend was allowed to react for 12 h to ensure the adsorption of AgNPs on the surface of the cellulose–PEVA composite. Thereafter, AgNPs@cellulose–PEVA was separated via centrifugation. Spectroscopy testing confirmed that the AgNPs were dispersed on the surface of the cellulose–PEVA composite.
Catalytic activity of the AgNPs@cellulose–PEVA hybrids for the Willgerodt–Kindler reaction
A mixture of aldehyde (1 mmol), morpholine (1.2 mmol), and elemental S (1.2 mmol) reactants and AgNPs@cellulose–PEVA catalyst (5 mg) in dimethylformamide (DMF; 10 mL) was stirred at 80 °C for 2.5 h. After reaction completion, which was confirmed using thin layer chromatography, the residue was separated by concentrating the organic layer over an evaporator. Lastly, the residue was purified via column chromatography to isolate the final product.
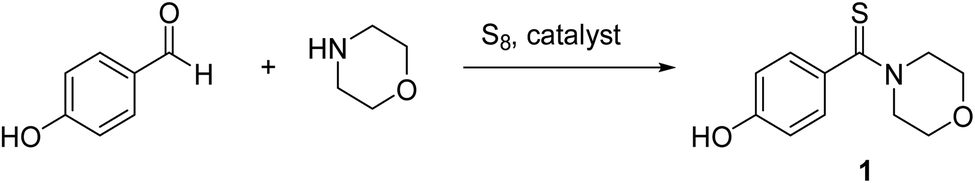
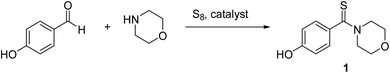
4-Hydroxyphenyl-morpholine-methanethione (1)41. 1H NMR (400 MHz, CDCl3) δ 7.16–7.1 (d, J = 8.3 Hz, 2H Ar–H), 6.71–6.68 (d, J = 8.1 Hz, 2H, Ar–H), 6.01–5.59 (s, 1H –OH), 4.47–4.38 (t, J = 3.8 Hz, 2H –CH2), 3.91–3.84 (t, J = 3.65 Hz, 2H, –CH2), 3.69–3.61 (s, 4H, 2xCH2). 13C NMR (100 MHz, CDCl3), δ 201.4, 157.0, 134.2, 128.1, 115.6, 66.9, 66.6, 52.75, 50.2. HRMS: [M + H]: calculated: 224.0726, found: 224.0746.
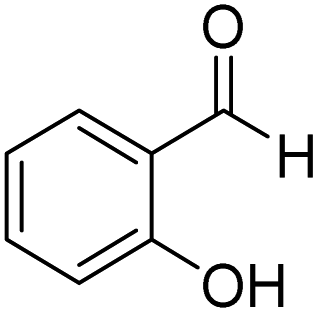
2-Hydroxyphenyl-morpholine-methanethione (2)41. 1H NMR (400 MHz, CDCl3) δ 6.85–6.80 (d, J = 8.5 Hz, 1H Ar–H), 6.49–6.45 (d, J = 1.9, Hz 1H, Ar–H), 6.38–6.35 (d, J = 2.1, Hz 1H, Ar–H), 6.34–6.32 (d, J = 1.8 Hz, 1H, Ar–H) 4.06–4.01 (s, 1H –OH), 3.87–3.70 (m, 6H, –CH2), 3.51–3.43 (dd, J = 14.3 Hz, 1H, –CH2), 2.98–2.86 (d, J = 19.7 Hz, 1H, –CH2). 13C NMR (100 MHz, CDCl3), δ 207.4, 154.4, 131.3, 126.5, 125.9, 66.8, 31.05. HRMS: [M + H]: calculated: 224.0745, found: 224.0746.
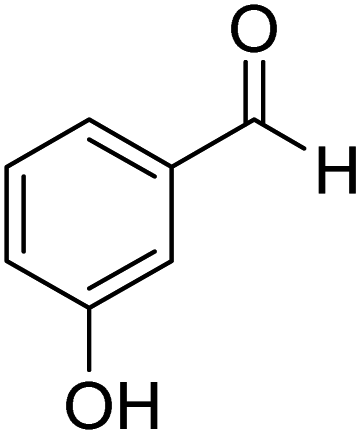
3-Hydroxyphenyl-morpholine-methanethione (3)42. 1H NMR (400 MHz, CDCl3) δ 7.21–7.13 (t, J = 7.7 Hz, 1H–Ar–H), 6.73 (d, J = 7.0 Hz, 2H, Ar–H), 6.04–6.74 (s, 1H, –OH), 4.45–4.37 (t, J = 7.7 Hz, 2H, –CH2), 3.91–3.84 (t, J = 6.7 Hz, 2H, –CH2), 3.61 (m, 4H, 2x–CH2). 13C NMR (100 MHz, CDCl3), δ 200.5, 155.8, 143.2, 130.1, 117.2, 115.9, 112.9, 66.9, 66.6, 52.5, 49.5. HRMS: [M + H]: calculated: 224.0738, found: 224.0745.
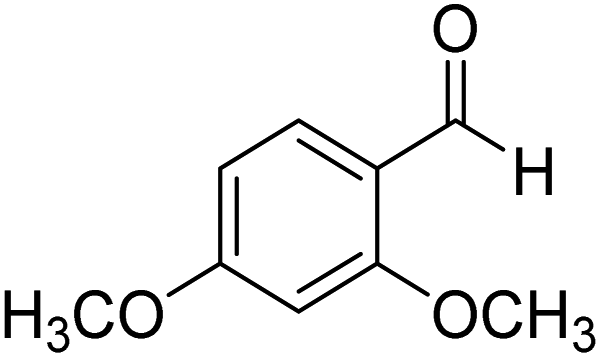
2,4-Dimethoxyphenyl-morpholine-methanethione (4). 1H NMR (400 MHz, CDCl3) δ 7.27–7.24 (d, J = 4.5 Hz, 1H Ar–H), 6.51–6.48 (d, J = 10.5 Hz, 1H, Ar–H), 6.39–6.38 (s, 1H, Ar–H), 4.46–4.41 (dd, J = 9.0 Hz, J = 4.7 Hz, 2H, –CH2), 3.79 (s, 6H, –CH3), 3.70–3.65 (dd, J = 9.0 Hz, J = 4.9 Hz, 1H, –CH2), 3.60–3.54 (dd, J = 8.0 Hz, J = 5.4 Hz, 1H, –CH2), 3.53–3.49 (dd, J = 6.5 Hz, J = 3.0 Hz, 1H, –CH2), 3.48–3.39 (m, 2H –CH2). 13C NMR (100 MHz, CDCl3), δ 199.1, 161.7, 153.8, 130.2, 124.6, 105.3, 98.4, 66.6, 55.7, 51.9, 49.5. HRMS: [M + H]: calculated: 261.1001, found: 261.1007.
4-Hydroxy-3-methoxyphenyl-(morpholinomethanethione) (5). 43 1H NMR (400 MHz, CDCl3) δ 6.95–6.92 (d, J = 2.0 Hz, 1H Ar–H), 6.85–6.82 (s, 1H, Ar–H), 6.76–6.72 (d, J = 2.0 Hz 1H, Ar–H), 5.85–5.76 (s, 1H, –OH), 4.44–4.37 (s, 2H, –CH2), 4.0–3.88 (s, 3H –CH3), 3.88–3.84 (t, J = 3.9 Hz 2H, –CH2), 3.71–3.61 (d, J = 2.9 Hz, 4H, –CH2). 13C NMR (100 MHz, CDCl3), δ 201.2, 146.7, 146.5, 134.5, 119.1, 114.0, 110.4, 66.9, 66.6, 56.1, 53.0, 50.2. HRMS: [M + H]: calculated: 254.0851, found: 254.0832.
4-Dimethylaminophenyl-morpholine-methanethione (6). 42 1H NMR (400 MHz, CDCl3) δ 7.30–7.25 (d, J = 8.4 Hz, 2H Ar–H), 7.63–7.58 (d, J = 8.6 Hz, 2H, Ar–H), 4.54–4.27 (dd, J = 7.3 Hz, 2H, –CH2), 3.91–3.60 (d, J = 18.6 Hz, 6H, –CH2), 2.98–2.96 (s, 6H –CH3). 13C NMR (100 MHz, CDCl3), δ 202.28, 151.31, 129.78, 128.80, 111.15, 66.85, 53.15, 50.13, 40.35. HRMS: [M + H]: calculated: 251.1218, found: 251.1203.
Morpholino(phenyl)-methanethione (7)44. 1H NMR (400 MHz, CDCl3) δ 7.37–7.35 (t, J = 7.66 Hz. 1H Ar–H), 7.35–7.31 (d, J = 5.21 Hz, 2H, Ar–H), 7.29–7.26 (t, J = 7.45 Hz, 1H, Ar–H), 7.26–7.25 (t, J = 7.39 Hz, 1H, Ar–H), 4.47–4.37 (t, J = 4.1 Hz, 2H, –CH2), 3.92–3.82 (t, J = 4.05 Hz, 2H, –CH2), 3.68–3.61 (t, J = 4.0 Hz, 2H –CH2), 3.61–3.54 (t, J = 4.0 Hz, 2H –CH2). 13C NMR (100 MHz, CDCl3), δ 201.1, 142.5, 129.0, 128.7, 126.0, 66.8, 66.6, 52.6, 49.6. HRMS: [M + H]: calculated: 208.0796, found: 208.0797.
4-Fluoro-morpholine-methanethione (8). 1 1H NMR (400 MHz, CDCl3) δ 8.13–8.12 (d, J = 3.2 Hz, 1H Ar–H), 7.30–7.28 (d, J = 5.3 Hz, 1H, Ar–H), 7.15–7.13 (d, J = 8.6 Hz, 1H, Ar–H), 7.07–7.03 (d, J = 8.5 Hz, 1H, Ar–H), 4.43–4.40 (t, J = 4.9 Hz, 1H, –CH2), 3.92–3.83 (d, J = 4.8 Hz, 2H, –CH2), 3.67–3.61 (t, J = 4.4 Hz, 2H –CH2), 3.61–3.56 (t, J = 8.8 Hz, 2H, –CH2), 3.19–3.15 (t, J = 7.7 Hz, 1H, –CH2). 13C NMR (100 MHz, CDCl3), δ 200.11, 170.8, 167.7, 132.9, 128.3, 125.6, 115.7, 66.8, 52.7, 49.8, 44.3. HRMS: [M + H]: calculated: 226.0702, found: 226.0699.
1,3-Phenylenebis(morpholinomethanethione) (9). 22 1H NMR (400 MHz, CDCl3) δ 7.38–7.32 (t, J = 7.0 Hz, 1H, Ar–H), 7.28–7.23 (d, J = 2.9 Hz, 2H, Ar–H), 7.22–7.19 (s, 1H, Ar–H), 4.45–4.34 (t, J = 4.3 Hz, 4H, –CH2), 3.89–3.82 (t, J = 4.8 Hz, 4H, –CH2), 3.67–3.56 (d, J = 15.1 Hz, 8H, –CH2). 13C NMR (100 MHz, CDCl3), δ 199.4, 142.6, 129.0, 128.9, 126.4, 123.9, 66.8, 66.6, 52.7, 49.7. HRMS: [M + H]: calculated: 337.1044, found: 337.1019.
2-Hydroxy-5-nitro-phenyl-(morpholinomethanethione) (10). 1H NMR (400 MHz, CDCl3) δ 11.55 (s, 1H, –OH), 8.09–8.04 (d, J = 11.9 Hz, 1H, Ar–H), 8.0–7.98 (s, 1H, Ar–H), 6.99–6.95 (d, J = 9.1 Hz, 1H, Ar–H), 4.37–4.30 (dd, J = 16.5 Hz, J = 4.5 Hz, 1H, –CH2), 4.21–4.14 (dd, J = 16.7 Hz, J = 4.8 Hz, 1H, –CH2), 3.74–3.69 (t, J = 4.9 Hz, 2H, –CH2), 3.58–3.52 (dd, J = 10.3 Hz, J = 5.2 Hz, 2H, –CH2), 3.45–3.39 (t, J = 4.4 Hz, 2H, –CH2). 13C NMR (100 MHz, CDCl3), δ 192.9, 157.5, 140.0, 130.7, 126.2, 126.3, 66.5, 66.1, 52.3, 49.3. HRMS: [M + H]: calculated: 269.0596, found: 269.05967.
3-Nitrophenyl-morpholine-methanethione (11). 42 1H NMR (400 MHz, CDCl3) δ 8.20–8.18 (s, 1H, Ar–H), 8.15–8.10 (d, J = 9.0 Hz,1H, Ar–H),7.63–7.59 (d, J = 7.6 Hz, 1H, Ar–H), 7.58–7.53 (t, J = 7.8 Hz, 1H, Ar–H), 4.44–4.41 (t, J = 4.7 Hz, 2H, –CH2), 3.91–3.89 (t, J = 4.8 Hz, 2H, –CH2), 3.68–3.65 (t, J = 4.6 Hz, 2H, –CH2), 3.59–3.57 (t, J = 4.3 Hz, 2H, –CH2). 13C NMR (100 MHz, CDCl3), δ 197.3, 148.3, 143.8, 131.8, 130.0, 123.6, 121.1, 66.7, 66.53, 52.8, 49.6. HRMS: [M + H]: calculated: 253.0634, found: 253.0647.
(1H-Imidazole-4-yl) (morpholino)methanethione (12). 1H NMR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 7.67 (s, 1H), 4.17 (t, J = 17.8 Hz, 2H), 3.59 (t, J = 15.6 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 189.9, 142.3, 135.1, 125.1, 67.1, 66.6, 53.0, 51.0. HRMS: [M + H]: calculated: 198.0701, found: 198.0689.
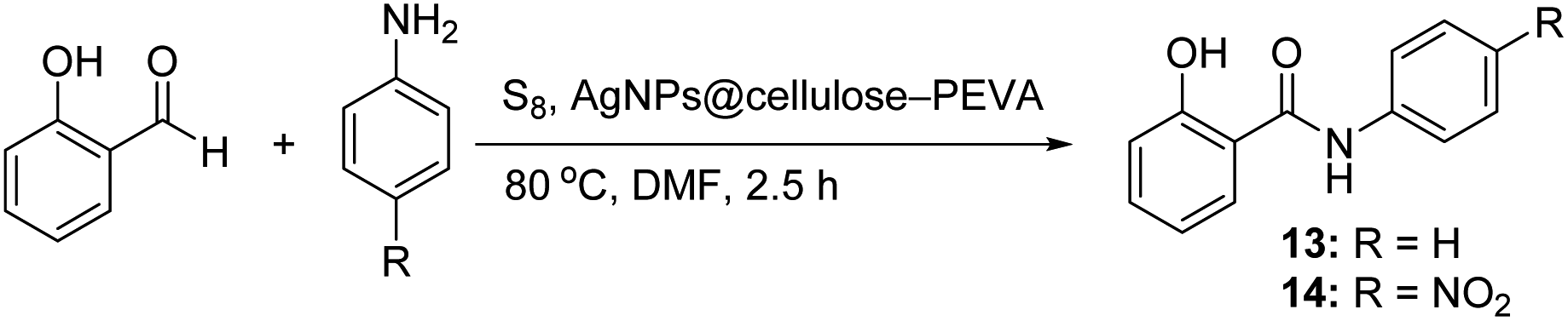
2-Hydroxy-N-phenylbenzothioamide (13). 1H NMR (400 MHz, DMSO-d6) δ 12.11 (s, 1H), 9.89 (s, 1H), 8.35 (t, J = 3.0 Hz, 1H), 8.25 (d, J = 3.1 Hz, 1H), 8.05 (d, J = 7.3 Hz, 1H), 7.46 (t, J = 1.3 Hz, 2H), 7.30 (t, J = 7.0 Hz, 1H), 7.22 (d, J = 1.1 Hz, 1H), 7.19 (dd, J = 3.2, 1.6 Hz, 1H), 7.15 (d, J = 1.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 185.5, 139.1, 137.6, 134.1, 126.8, 124.6, 124.0, 122.7, 121.3, 118.7, 113.0. HRMS: [M + H]: calculated: 230.2552, found 230.2561.
2-Hydroxy-N-(4-nitrophenyl)benzothioamide (14). 1H NMR (400 MHz, CDCl3), δ 8.64 (s, 1H), 8.64 (s, 1H), 8.27 (d, J = 2.6 Hz, 1H), 8.24 (d, J = 2.6 Hz, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.96 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.49 (t, J = 7.1 Hz, 1H), 7.20 (d, J = 5.1 Hz, 1H), 7.18 (d, J = 5.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 172.5, 161.7, 150.4, 136.2, 132.6, 132.2, 131.7, 130.8, 119.7, 117.6, 116.7, 115.6, 113.4. HRMS: [M + H]: calculated: 275.2318, found 275.2350.
Results and discussion
FTIR spectroscopic analysis
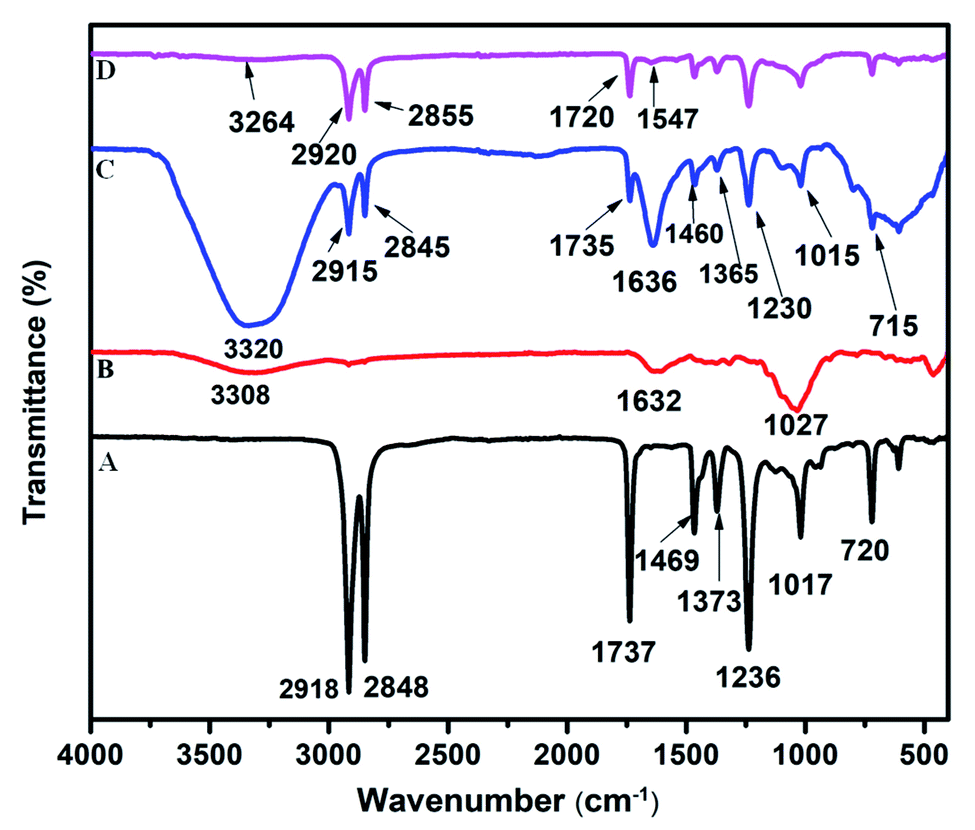
In the FTIR spectrum of PEVA, the characteristic bands of VA emerged at 1736, 1236, and 1017 cm−1, whereas the bands of the ethylene group were observed at 2918, 2848, 1469, 1373, and 720 cm−1 (Fig. 1A). The primary bands at 3308, 1632, and 1027 cm−1 in the FTIR spectrum of cellulose were ascribed to the stretching and bending of the O–H bonds and the stretching of the skeletal C–O–C pyranose ring, respectively (Fig. 1B). The VA bands shifted from 1737, 1236, and 1017 cm−1 in the FTIR spectrum of PEVA to 1735, 1230, and 1015 cm−1, respectively, in the FTIR spectrum of the cellulose–PEVA composite (Fig. 1C). Moreover, the width and intensity of the band at 3600–3000 cm−1, which was attributed to the O–H vibration, increased, indicating that the –OH groups on the cellulose surface and the polar VA groups of PEVA interacted. Upon the addition of AgNPs to the cellulose–PEVA composites, the intensity of the band at 3264 cm−1, which corresponded to the O–H vibration, decreased significantly. Moreover, the bands at 2915 and 2845 cm−1, which were ascribed to the methylene groups, and the band at 1735 cm−1, which was attributed to the vibration of the carbonyl bonds of the acetate groups, shifted to 2920, 2855, and 1720 cm−1, respectively (Fig. 1D). These results indicated that the AgNPs interacted with the cellulose–PEVA composites, and in particular, the –OH groups of cellulose played a critical role in immobilizing the AgNPs and preventing their aggregation via hydrogen bonding.45
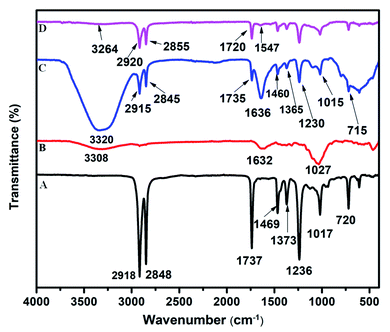 |
| | Fig. 1 Fourier-transform infrared spectra of (A) PEVA, (B) cellulose, (C) cellulose–PEVA composite, and (D) AgNPs@cellulose–PEVA hybrid. | |
PXRD analysis
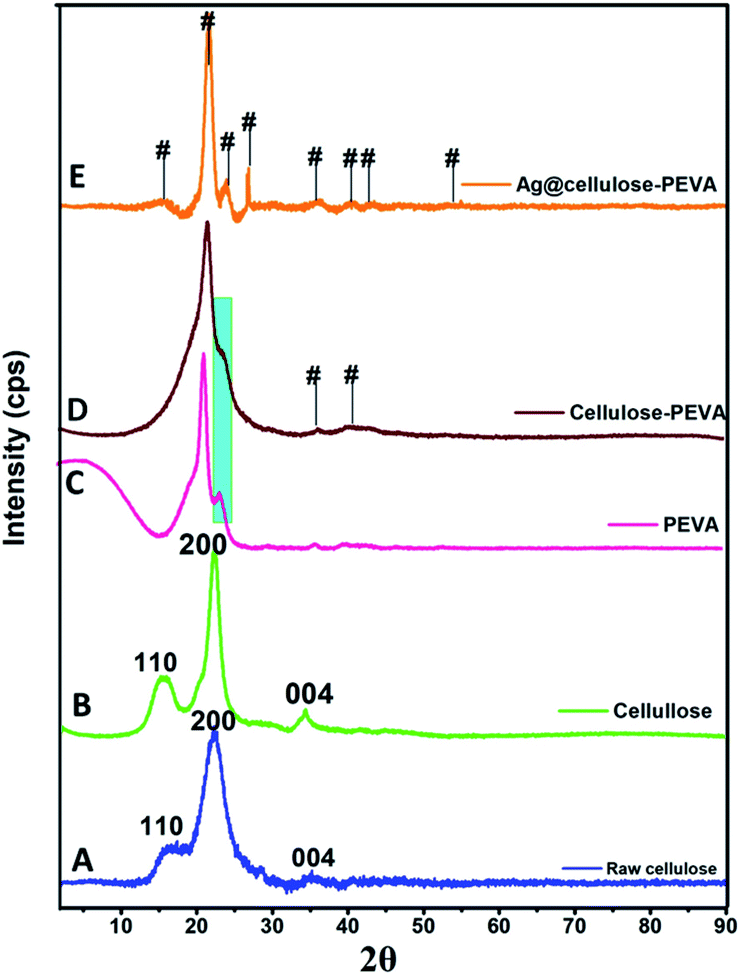
The characteristic peaks at 16.04°, 22.46°, and 34.38°, which corresponded to the (110), (200), and (004) lattice planes of cellulose, respectively, were observed in the PXRD patterns of raw and treated cellulose (Fig. 2A and B, respectively). The primary crystalline peak in the PXRD pattern of PEVA, which was observed at 22.46° and presented an intensity of 100%, confirmed the crystallinity of the polymer. The PXRD patterns of PEVA and the PEVA–cellulose composite are illustrated in Fig. 2C and D, respectively. The characteristic peaks of PEVA at 21.44° and 23.4° were observed in the PXRD pattern of the cellulose–PEVA composite, indicating that cellulose was grafted with PEVA. These peaks were also observed in the PXRD patterns of the AgNPs@cellulose–PEVA hybrid, indicating that the cellulose–PEVA composite served as the support for the hybrid (Fig. 2E). Furthermore, the presence of the peaks at 15.61°, 24.35°, 26.84°, 35.90°, 40.82°, 43.20°, and 54.90° in the PXRD pattern of AgNPs@cellulose–PEVA confirmed the presence of elemental Ag.45 In addition, the presence of these peaks in the PXRD patterns of the freshly prepared and recovered catalysts confirmed that the catalyst was stable and did not undergo structural changes during the reaction. Moreover, the catalyst was reusable and did not lose its catalytic activity.
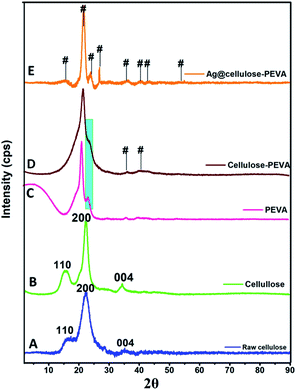 |
| | Fig. 2 Powder X-ray diffraction patterns of (A) raw cellulose, (B) cellulose, (C) PEVA, (D) the cellulose–PEVA composite, and (E) AgNPs@cellulose–PEVA hybrid. Here, # represents the emergence of a new plane. | |
SEM and EDX analyses
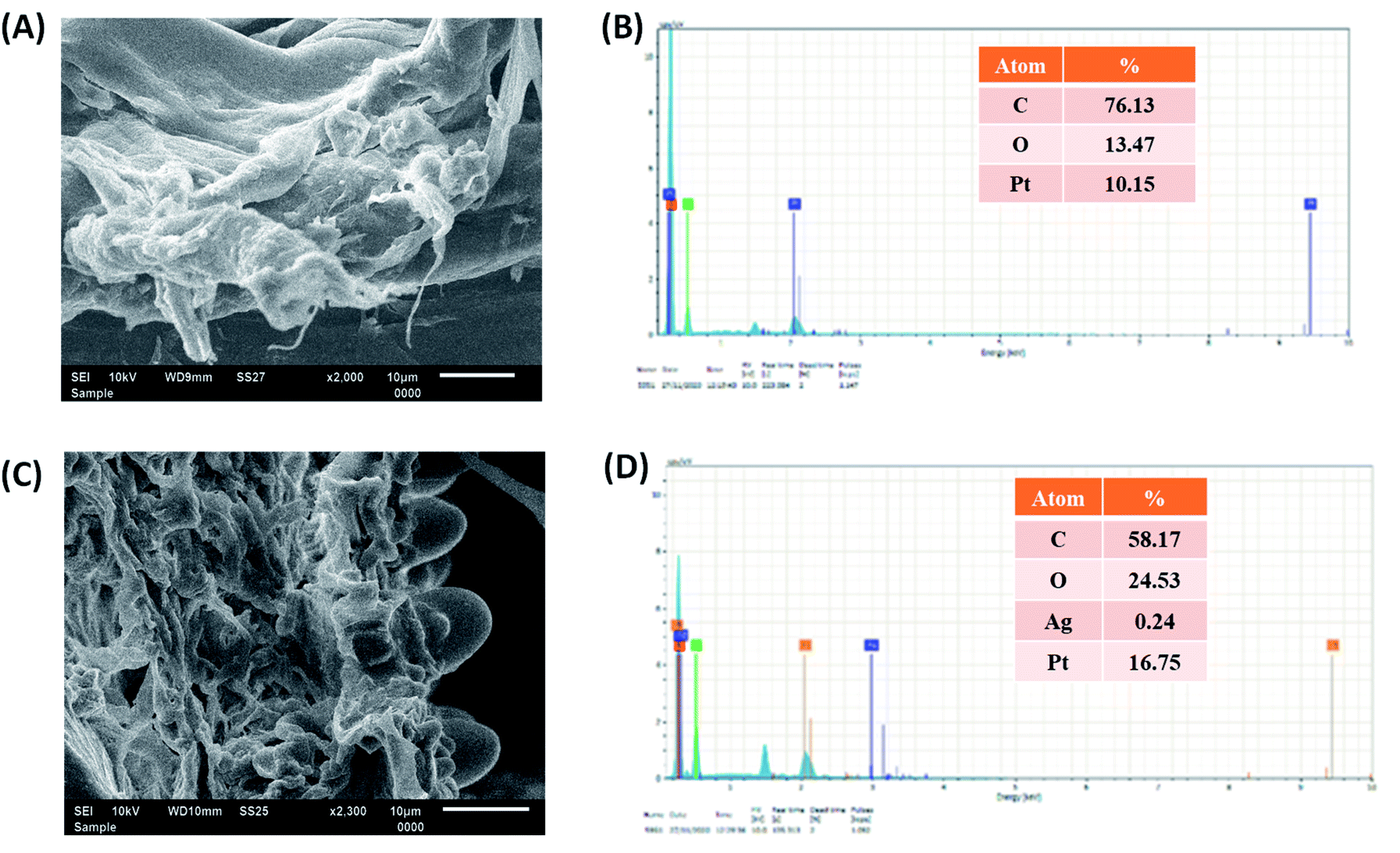
The SEM images revealed the surface morphologies of the cellulose–PEVA composite and AgNPs@cellulose–PEVA hybrid, which varies from particle-to-particle arrangement in a sample. The SEM image of cellulose–PEVA revealed that the composite was denser than the AgNPs@cellulose–PEVA hybrid, confirming the presence of AgNPs on the composite matrix surface (Fig. 3A and C). The SEM image of the AgNPs@cellulose–PEVA hybrid revealed the presence of a crosslinked network in its structure. The mesh-like surface of the cellulose–PEVA composite facilitated the insertion of AgNPs in the crosslinked network and the formation of a heterogeneous catalytic surface for organic reactions. The elemental composition of the cellulose–PEVA composite and AgNPs@cellulose–PEVA hybrid were determined using EDX, and the results are presented in Fig. 3B and D, respectively. The experimental data indicated that the cellulose–PEVA composite comprised C (76.4%), O (13.5%), and Pt (10.2%) and the AgNPs@cellulose–PEVA hybrid contained C (58.2%), O (24.5%), Pt (16.8%), and Ag (0.24%). The presence of Pt in the cellulose–PEVA composite and AgNPs@cellulose–PEVA hybrid was attributed to the Pt coating. Therefore, the EDX results confirmed the presence of Ag in the AgNPs@cellulose–PEVA hybrid.
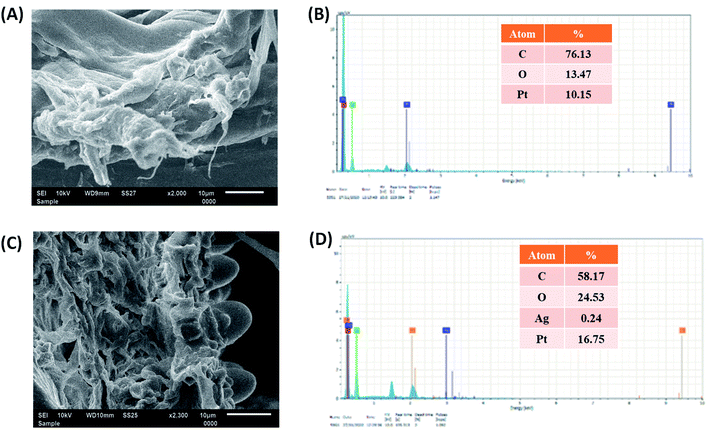 |
| | Fig. 3 Scanning electron microscopy images of the (A) cellulose–poly(ethylene-co-vinyl acetate) (PEVA) composite and (C) AgNPs@cellulose–PEVA hybrid. Energy-dispersive X-ray mappings of the (B) cellulose–PEVA composite and (D) AgNPs@cellulose–PEVA hybrid. | |
RGB analysis
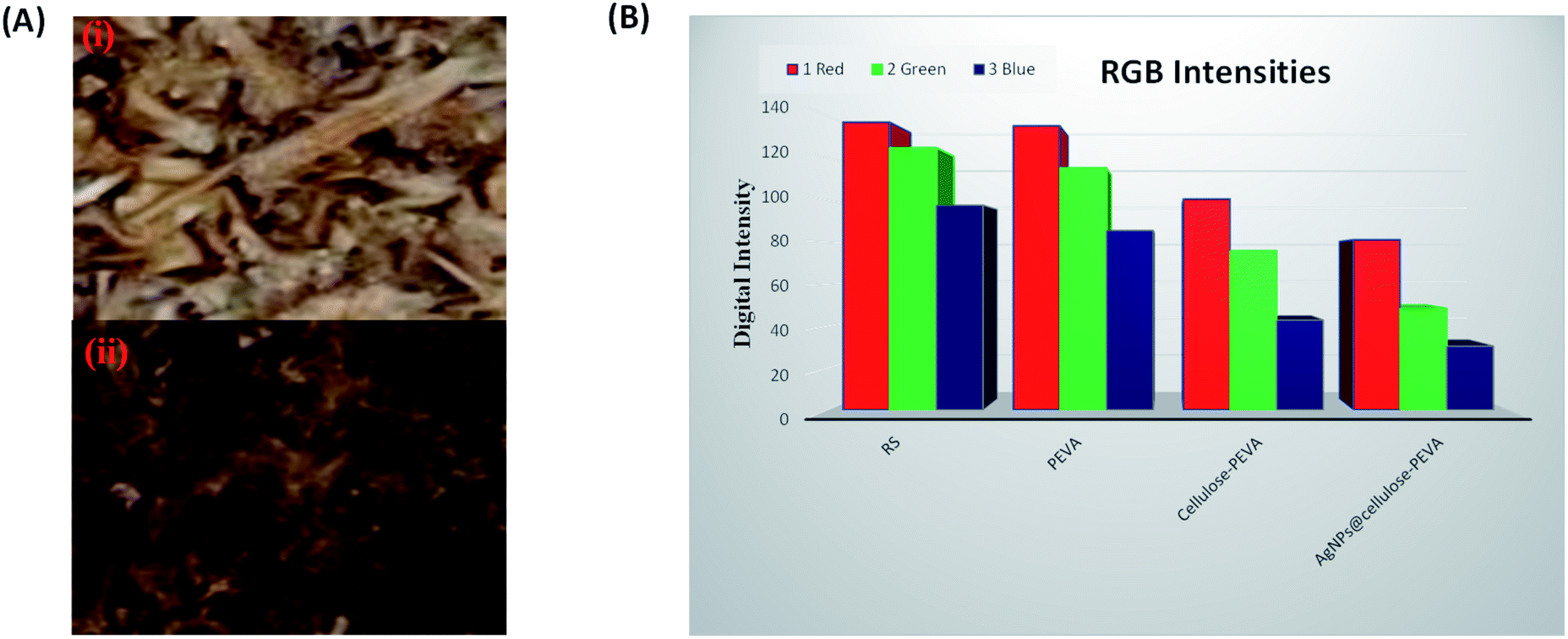
Digital images of the components of the hybrid catalyst were obtained using a smartphone and were used for RGB analysis using the ImageJ software (Fig. 4). The MathWorks software was used to convert the RGB components into three-scale images: red, green, and blue. The digital images revealed the changes in color intensity. In the 8 bit digital images, the intensity of each primary color ranged between 0 and 255. Therefore, 256 values were available for the intensity of each color, where 0 represented black. The remaining 255 values indicated the maximum intensities or pure colors. The images turned dark or bright if the numerical values of the components decreased or increased, respectively. The digital images darkened in the following order: rice straw < PEVA < cellulose–PEVA < AgNPs@cellulose–PEVA (Fig. 4A). The RGB intensities of rice straw, PEVA, cellulose–PEVA, and AgNPs@cellulose–PEVA indicated that the values of the R-, G-, and B-components decreased in the following order: rice straw > PEVA > cellulose–PEVA > AgNPs@cellulose–PEVA (Fig. 4B).
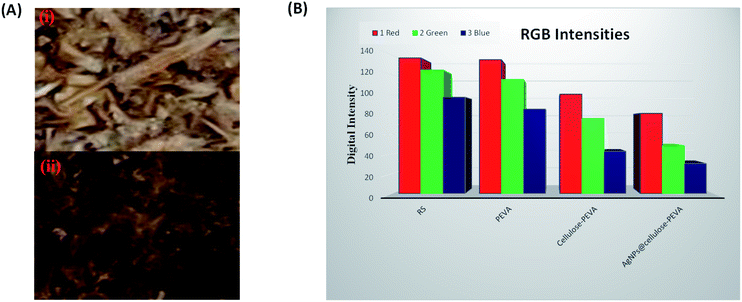 |
| | Fig. 4 (A) Square regions of the digital images selected for RGB analysis of (i) the cellulose–PEVA composite and (ii) the AgNPs@cellulose–PEVA hybrid. (B) Bar graph of the RGB intensities of RS, PEVA, the cellulose–PEVA composite and the AgNPs@cellulose–PEVA hybrid. | |
Mechanical testing
PEVA and the cellulose–PEVA composite were subjected to tensile testing. The strain of PEVA was 68.7% at a stress of 1.6 MPa and the maximum displacement of PEVA was 25.8 mm at a bearing force of 37.8 N (Fig. S2†). The strain and maximum displacement of the cellulose–PEVA composite were 76% at 2.3 MPa and 27.4 mm at a force of 53.0 N, respectively. (Fig. S3†). These results indicated that the cellulose incorporated in the PEVA matrix reinforced the composite and increased its mechanical strength. Furthermore, mechanical strength impact tests were performed in the Izod and Charpy modes. The impact energies of the AgNPs@cellulose–PEVA hybrid in the Izod and Charpy modes were 1.865 and 5.939 kJ m−1, respectively (Table S1†).
Catalytic activity of the AgNPs@cellulose-PEVA nanohybrid for the Willgerodt–Kindler reaction
The catalytic activity of the AgNPs@cellulose–PEVA hybrid was investigated for the synthesis of thioamides. A one-pot multicomponent reaction using 4-hydroxy-benzaldehyde (1 mmol), morpholine (1.2 mmol), and elemental S (1.2 mmol) was selected as the model reaction (Scheme 1). First, the reaction was performed in DMF in the presence of 5 mg of AgNPs@cellulose–PEVA at 110 °C for 6 h, and the yield of the desired product was 71% (Table 1, entry 1). The yields of the reactions performed over cellulose or the cellulose–PEVA composite were lower (Table 1, entries 2 and 3, respectively). Dimethyl sulfoxide (DMSO) has been reported to act as a promoter for the modified Willgerodt–Kindler reaction.46 Therefore, in this study, DMSO was used as the solvent for the reaction. The yield of the reaction in DMSO was lower than that of the reaction in DMF (Table 1, entry 4). In addition, when water was used as a non-toxic solvent, the reaction did not proceed (Table 1, entry 5). Furthermore, the yields of the reactions performed in ethanol and tetrahydrofuran were low (Table 1, entries 6 and 7, respectively). Based on these results, DMF was selected as the optimal solvent for the Willgerodt–Kindler reaction. Next, the reaction performance was monitored by changing the reaction temperature and time. The yield increased with decreasing the reaction temperature and time (Table 1, entries 8–10). Decreasing the amount of catalyst caused a decrease in yield (Table 1, entry 13). The optimal reaction conditions were determined to be 5 mg of catalyst, 80 °C, and 2.5 h (Table 1, entry 11).
 |
| | Scheme 1 Reaction of benzaldehyde, morpholine, and elemental S in the presence of the AgNPs@cellulose–PEVA hybrid as the catalyst. | |
Table 1 Optimization of the reaction conditions for the synthesis of thioamides using the Willgerodt–Kindler reaction
| Entry |
Catalyst |
Solvent |
Catalyst (mg) |
Temp. (°C) |
Time (h) |
Yielda (%) |
| Isolated yield. Here, AgNPs, PEVA, DMF, DMSO, EtOH, and THF denote Ag nanoparticles, poly(ethylene-co-vinyl acetate), dimethylformamide, dimethyl sulfoxide, ethanol, and tetrahydrofuran, respectively. |
| 1 |
AgNPs@cellulose–PEVA |
DMF |
5 |
110 |
6 |
71 |
| 2 |
Cellulose |
DMF |
5 |
110 |
6 |
43 |
| 3 |
Cellulose–PEVA |
DMF |
5 |
110 |
6 |
45 |
| 4 |
AgNPs@cellulose–PEVA |
DMSO |
5 |
110 |
6 |
64 |
| 5 |
AgNPs@cellulose–PEVA |
H2O |
5 |
110 |
6 |
— |
| 6 |
AgNPs@cellulose–PEVA |
EtOH |
5 |
110 |
6 |
37 |
| 7 |
AgNPs@cellulose–PEVA |
THF |
5 |
110 |
6 |
35 |
| 8 |
AgNPs@cellulose–PEVA |
DMF |
5 |
90 |
6 |
75 |
| 9 |
AgNPs@cellulose–PEVA |
DMF |
5 |
80 |
6 |
79 |
| 10 |
AgNPs@cellulose–PEVA |
DMF |
5 |
80 |
4 |
84 |
| 11 |
AgNPs@cellulose–PEVA |
DMF |
5 |
80 |
2.5 |
98 |
| 12 |
AgNPs@cellulose–PEVA |
DMF |
5 |
80 |
2 |
87 |
| 13 |
AgNPs@cellulose–PEVA |
DMF |
4 |
80 |
2.5 |
85 |
Various aromatic aldehydes were used to synthesize thioamides using the Willgerodt–Kindler reaction under the optimal reaction conditions to examine the scope and limitations of the reaction. The results are summarized in Table 2. The yields of the reactions using aromatic aldehydes with electron-withdrawing and electron-donating groups were high, indicating substrate generality. However, the product yields of the reactions using aromatic aldehydes with electron-donating groups were higher than those of the reactions using aldehydes with electron-withdrawing groups.
Table 2 Synthesis of various substituted thioamides using the Willgerodt–Kindler reaction and AgNPs@cellulose–PEVA as the catalyst
To examine the electronic effect of amines, reactions of salicylaldehyde with aniline or p-nitroaniline were performed (Scheme 2). The corresponding amides (13 and 14) were obtained in 90 and 85% yields. These results indicate that the electronic effect of amines affects the efficiency of the reaction, although it was not significant.
 |
| | Scheme 2 The reaction of salicylaldehyde, aromatic amines, and elemental sulfur in the presence of the AgNPs@cellulose–PEVA hybrid as the catalyst. | |
Reusability of the AgNPs@cellulose–PEVA hybrid catalyst
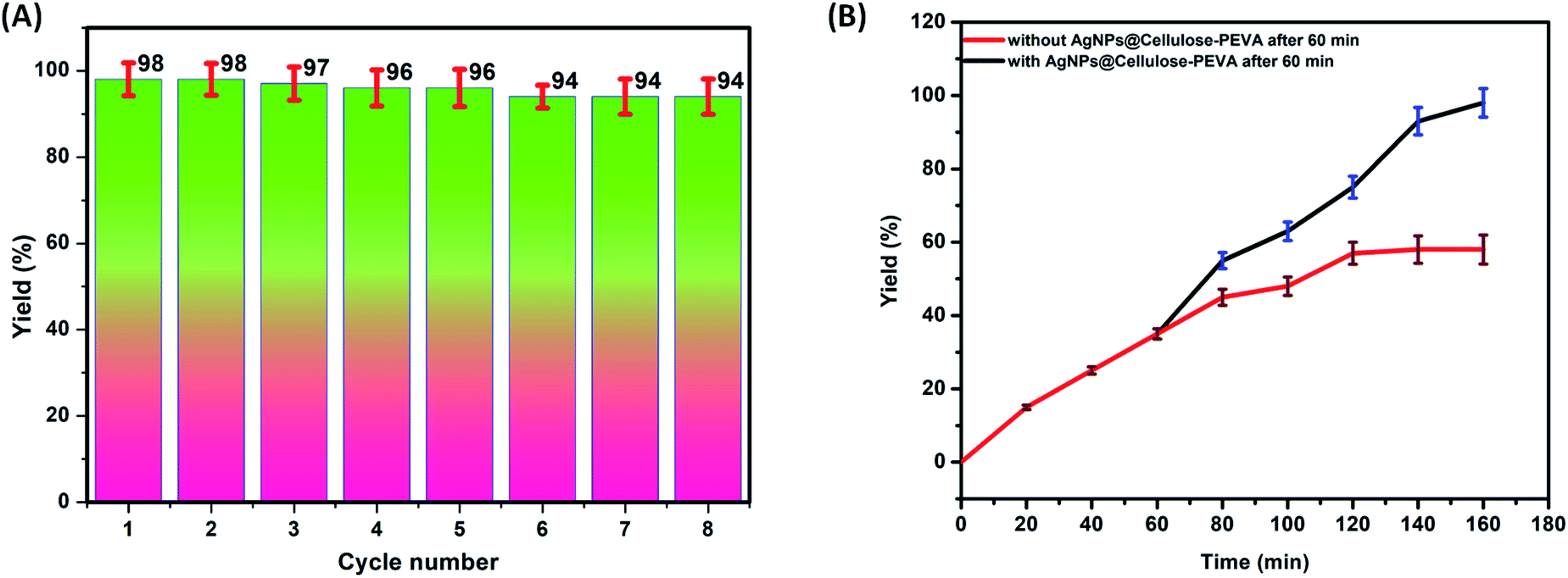
The primary advantage of heterogeneous catalysts is their reusability. Regeneration and reusability are attractive features of the developed catalyst in terms of sustainability. The AgNPs@cellulose–PEVA catalyst was easily separated from the reaction mixtures via centrifugation. Subsequently, the AgNPs@cellulose–PEVA catalyst was washed with methanol and water and dried at approximately 55 °C. Thereafter, the recovered catalyst was used for the next catalytic run. The catalyst was highly stable and could be reused for at least eight runs without significant catalytic activity loss, as illustrated in Fig. 5A. The catalyst recovered after five reaction cycles was subjected to SEM analysis (Fig. S4†). The surface of the recovered catalyst was not significantly different from that of the fresh catalyst, indicating the high recyclability of the AgNPs@cellulose–PEVA catalyst for the synthesis of thioamides. A blank experiment without the AgNPs@cellulose–PEVA catalyst was performed at 80 °C. The efficiencies of the reactions performed with and without catalyst were the same over the first 60 min. However, as the reaction time was increased, the efficiency of the reaction without the AgNPs@cellulose–PEVA catalyst was low; moreover, the reaction was not complete even after 160 min, and the product yield was only 45%. These results indicated that the AgNPs@cellulose–PEVA catalyst was critical for an efficient reaction (Fig. 5B).
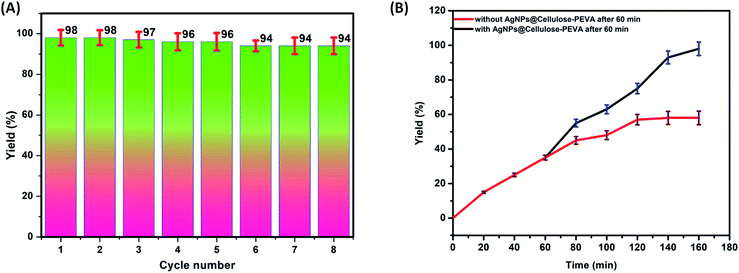 |
| | Fig. 5 (A) Reusability of the AgNPs@cellulose–PEVA catalyst. (B) Reaction yields with or without the AgNPs@cellulose–PEVA catalyst. | |
Proposed mechanism
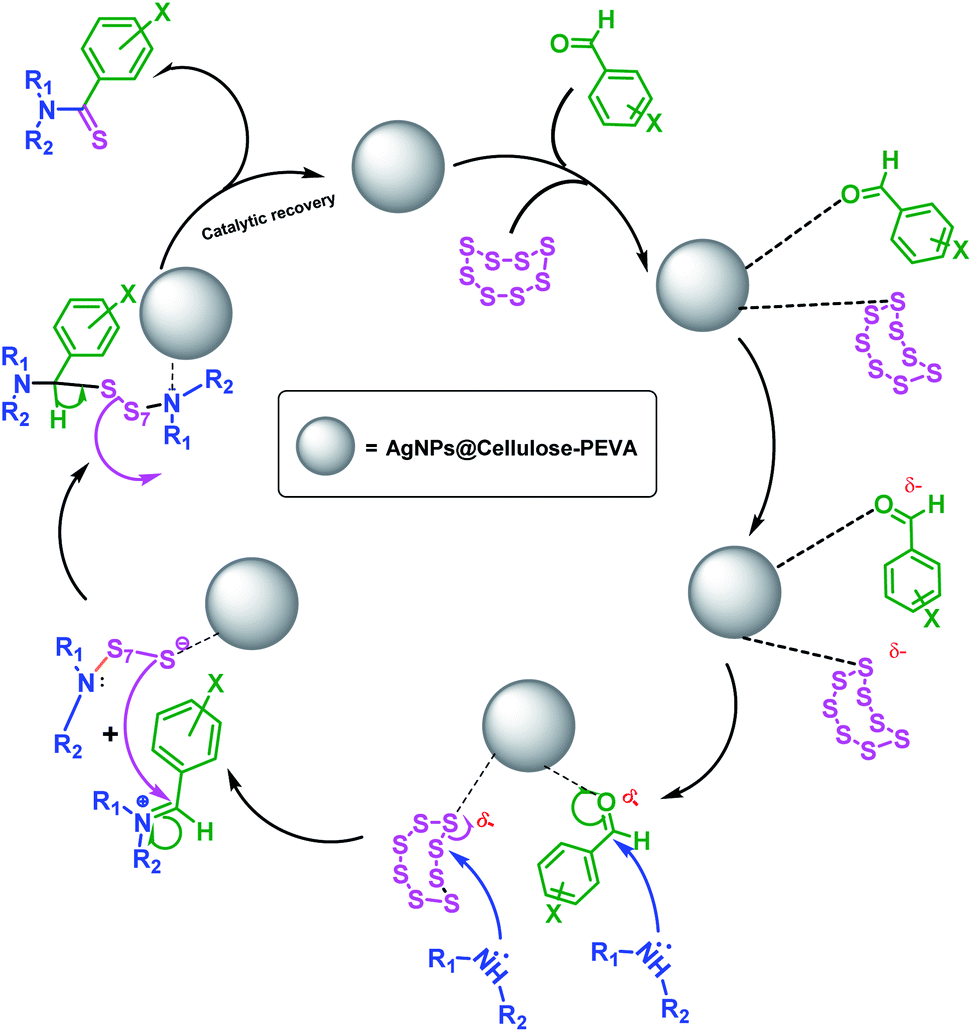
A mechanism for the synthesis of thioamides using the AgNPs@cellulose–PEVA catalyst was proposed, as illustrated in Scheme 3. The AgNPs anchored on the surface of the cellulose–PEVA composite via hydrogen bonding facilitated the nucleophilic attack of morpholine by increasing the electrophilicity of the carbonyl groups through non-covalent interactions.47 The attack of morpholine on S caused the S8 ring to open and induced the formation of polysulfide ions. During the next step, enamine was formed via water removal, and simultaneously, the polysulfide ions acted as a nucleophile, attacking enamine and forming new C–S bonds. Lastly, H+ was eliminated, and the polysulfide moiety formed thioamide and the catalyst.
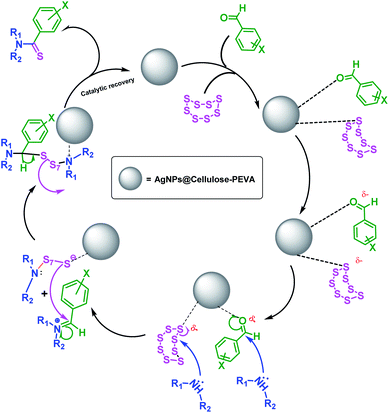 |
| | Scheme 3 Proposed mechanism for the synthesis of thioamides using the Willgerodt–Kindler reaction and the AgNPs@cellulose–PEVA catalyst. | |
Conclusion
A solid support comprising a biomass-derived cellulose–PEVA hybrid embedded with AgNPs was prepared. The developed hybrid catalyst was used for the synthesis of thioamides using the Willgerodt–Kindler reaction. Various thioamides were prepared using aldehydes and the fabricated hybrid catalyst. The AgNPs@cellulose–PEVA hybrid catalyst presented excellent catalytic efficacy for the Willgerodt–Kindler reaction, facilitating the selective formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bonds. The AgNPs@cellulose–PEVA catalyst exhibited high stability, excellent reusability, and sustained heterogeneity in the reaction media. We believe that the fabricated hybrid catalyst can promote the large-scale preparation of thioamides.
S bonds. The AgNPs@cellulose–PEVA catalyst exhibited high stability, excellent reusability, and sustained heterogeneity in the reaction media. We believe that the fabricated hybrid catalyst can promote the large-scale preparation of thioamides.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Anoop Singh is thankful to IIT Ropar for providing a fellowship and the infrastructure necessary to perform his research.
References
- X. Wang, M. Ji, S. Lim and H. Y. Jang, J. Org. Chem., 2014, 79, 7256–7260, DOI:10.1021/jo501378v.
- J. R. Dilworth, S. I. Pascu, P. A. Waghorn, D. Vullo, S. R. Bayly, M. Christlieb, X. Sun and C. T. Supuran, Dalton Trans., 2015, 44, 4859–4873, 10.1039/c4dt03206c.
- Y. Fang, Z. Y. Gu, S. Y. Wang, J. M. Yang and S. J. Ji, J. Org. Chem., 2018, 83, 9364–9369, DOI:10.1021/acs.joc.8b01300.
- X. Chen, E. G. Mietlicki-Baase, T. M. Barrett, L. E. McGrath, K. Koch-Laskowski, J. J. Ferrie, M. R. Hayes and E. J. Petersson, J. Am. Chem. Soc., 2017, 139, 16688–16695, DOI:10.1021/jacs.7b08417.
- S. Batjargal, Y. J. Wang, J. M. Goldberg, R. F. Wissner and E. J. Petersson, J. Am. Chem. Soc., 2012, 134, 9172–9182, DOI:10.1021/ja2113245.
- R. Maini, H. Kimura, R. Takatsuji, T. Katoh, Y. Goto and H. Suga, J. Am. Chem. Soc., 2019, 141, 20004–20008, DOI:10.1021/jacs.9b11097.
- N. Mahanta, D. M. Szantai-Kis, E. J. Petersson and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 142–163, DOI:10.1021/acschembio.8b01022.
- A. Bach, J. N. N. Eildal, N. Stuhr-Hansen, R. Deeskamp, M. Gottschalk, S. W. Pedersen, A. S. Kristensen and K. Strømgaard, J. Med. Chem., 2011, 54, 1333–1346, DOI:10.1021/jm1013924.
- R. Vanjari, T. Guntreddi, S. Kumar and K. N. Singh, ChemComm, 2015, 51, 366–369, 10.1039/c4cc08210a.
- P. Zhang, W. Chen, M. Liu and H. Wu, J. Org. Chem., 2018, 83, 14269–14276, DOI:10.1021/acs.joc.8b01721.
- T. Guntreddi, R. Vanjari and K. N. Singh, Org. Lett., 2014, 16, 3624–3627, DOI:10.1021/ol501482g.
- T. B. Nguyen, M. Q. Tran, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2014, 16, 310–313, DOI:10.1021/ol403345e.
- X. Feng, Y. Song and W. Lin, J. Am. Chem. Soc., 2021, 143, 8184–8192, DOI:10.1021/jacs.1c03561.
- H. Sedighian, M. B. Mamaghani, B. Notash and A. Bazgir, J. Org. Chem., 2021, 86, 2244–2253, DOI:10.1021/acs.joc.0c02391.
- X. K. Qi, L. Guo, L. J. Yao, H. Gao, C. Yang and W. Xia, Org. Lett., 2021, 23, 4473–4477, DOI:10.1021/acs.orglett.1c01412.
- Y. Bian, X. Qu, Y. Chen, J. Li and L. K. Liu, Molecules, 2018, 23, 2225, DOI:10.3390/molecules23092225.
- M. Saito, S. Murakami, T. Nanjo, Y. Kobayashi and Y. Takemoto, J. Am. Chem. Soc., 2020, 142, 8130–8135, DOI:10.1021/jacs.0c03256.
- V. V. Kul’ganek and L. A. Yanovskaya, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1979, 28, 2402–2403, DOI:10.1007/bf00951723.
- K. Okamoto, T. Yamamoto and T. Kanbara, Synlett, 2007, 2687–2690, DOI:10.1055/s-2007-991073.
- H. Xu, H. Deng, Z. Li, H. Xiang and X. Zhou, Eur. J. Org. Chem., 2013, 2013, 7054–7057, DOI:10.1002/ejoc.201301148.
- Y. A. Tayade, A. D. Jangale and D. S. Dalal, ChemistrySelect, 2018, 3, 8895–8900, DOI:10.1002/slct.201801553.
- K. Aghapoor, F. Mohsenzadeh, G. Khanalizadeh and H. R. Darabi, Monatsh. Chem., 2007, 138, 61–65, DOI:10.1007/s00706-006-0560-7.
- B. Mitra, G. C. Pariyar and P. Ghosh, ChemistrySelect, 2019, 4, 5476–5483, DOI:10.1002/slct.201900982.
- Z. Chen, X. Wang, R. Cao, K. B. Idrees, X. Liu, M. C. Wasson and O. K. Farha, ACS Mater. Lett., 2020, 2, 1129–1134, DOI:10.1021/acsmaterialslett.0c00264.
- S. A. Ahmed, M. N. Hasan, D. Bagchi, H. M. Altass, M. Morad, R. S. Jassas, A. M. Hameed, J. Patwari, H. Alessa, A. Alharbi and S. K. Pal, ACS Omega, 2020, 5, 15666–15672, DOI:10.1021/acsomega.0c01898.
- Z. Huang, M. Zhao, C. Wang, S. Wang, L. Dai and L. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 41294–41302, DOI:10.1021/acsami.0c10298.
- L. Wang, H. J. Zhang, X. Liu, Y. Liu, X. Zhu, X. Liu and X. You, ACS Appl. Polym. Mater., 2021, 3, 3197–3205, DOI:10.1021/acsapm.1c00404.
- J. Choi, O. Hasturk, X. Mu, J. K. Sahoo and D. L. Kaplan, Biomacromolecules, 2021, 22, 773–787, DOI:10.1021/acs.biomac.0c01539.
- Q. Liu, Y. Lu, M. Aguedo, N. Jacquet, C. Ouyang, W. He, C. Yan, W. Bai, R. Guo, D. Goffin, J. Song and A. Richel, ACS Sustainable Chem. Eng., 2017, 5, 6183–6191, DOI:10.1021/acssuschemeng.7b01108.
- Y. C. Gorur, M. S. Reid, C. Montanari, P. T. Larsson, P. A. Larsson and L. Wågberg, ACS Appl. Mater. Interfaces, 2021, 13, 32467–32478, DOI:10.1021/acsami.1c06452.
- L. S. S Sobhanadhas, L. Kesavan, M. Lastusaari and P. Fardim, ACS Omega, 2019, 4, 320–330, DOI:10.1021/acsomega.8b03061.
- F. Shen, R. L. Smith, L. Li, L. Yan and X. Qi, ACS Sustainable Chem. Eng., 2017, 5, 2421–2427, DOI:10.1021/acssuschemeng.6b02765.
- Y. Jiang, Z. Wang, X. Liu, Q. Yang, Q. Huang, L. Wang, Y. Dai, C. Qin and S. Wang, ACS Sustainable Chem. Eng., 2020, 8, 17508–17519, DOI:10.1021/acssuschemeng.0c06752.
- W. Liu, H. Du, H. Liu, H. Xie, T. Xu, X. Zhao, Y. Liu, X. Zhang and C. Si, ACS Sustainable Chem. Eng., 2020, 8, 16691–16700, DOI:10.1021/acssuschemeng.0c06561.
- B. Hu, B. Zhang, W. Xie, X. Jiang, J. Liu and Q. Lu, Energy Fuels, 2020, 34, 10384–10440, DOI:10.1021/acs.energyfuels.0c01948.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768, DOI:10.1021/acs.accounts.7b00614.
- G. Wang, J. Xiang, J. Lin, L. Xiang, S. Chen, B. Yan, H. Fan, S. Zhang and X. Shi, ACS Appl. Mater. Interfaces, 2020, 12, 51952–51959, DOI:10.1021/acsami.0c14820.
- D. D. Li, J. W. Zhang and C. Pd. Cai, J. Org. Chem., 2018, 83, 7534–7538, DOI:10.1021/acs.joc.8b00246.
- M. Can, S. Demirci, A. K. Sunol, G. Philippidis and N. Sahiner, ACS Omega, 2020, 5, 15519–15528, DOI:10.1021/acsomega.0c01653.
- N. Kanai, K. Nishimura, S. Umetani, Y. Saito, H. Saito, T. Oyama and I. Kawamura, ACS Agric. Sci. Technol., 2021, 1, 347–354, DOI:10.1021/acsagscitech.1c00041.
- C. K. Khatri, A. S. Mali and G. U. Chaturbhuj, Monatsh. Chem., 2017, 148, 1463–1468, DOI:10.1007/s00706-017-1944-6.
- H. F. Agnimonhan, L. A. Ahoussi, B. Glinma, J. M. Kohoude, F. A. Gbaguidi, S. D. S Kpoviessi, J. Poupaert and G. C. Accrombessi, Org. Chem.: Curr. Res., 2017, 06, 2–6, DOI:10.4172/2161-0401.1000180.
- S. Ray, A. Bhaumik, A. Dutta, R. J. Butcher and C. Mukhopadhyay, Tetrahedron Lett., 2013, 54, 2164–2170, DOI:10.1016/j.tetlet.2013.02.045.
- J. Wei, Y. Li and X. Jiang, Org. Lett., 2016, 18, 340–343, DOI:10.1021/acs.orglett.5b03541.
- W. H. Eisa, A. M. Abdelgawad and O. J. Rojas, ACS Sustainable Chem. Eng., 2018, 6, 3974–3983, DOI:10.1021/acssuschemeng.7b04333.
- T. T. T. Nguyen, L. A. Nguyen, Q. A. Ngo, M. Koleski and T. B. Nguyen, Org. Chem. Front., 2021, 8, 1593–1598, 10.1039/d0qo01654c.
- A. Maleki, H. Movahed and R. Paydar, RSC Adv., 2016, 6, 13657–13665, 10.1039/c5ra21350a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09225a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Doo Ok Jang
*b and
Doo Ok Jang