
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into ZnO-based doped porous nanocrystal frameworks
Buzuayehu Abebe
 * and
H. C. Ananda Murthy
*
* and
H. C. Ananda Murthy
*
Adama Science and Technology University, Department of Applied Chemistry, 1888, Adama, Ethiopia. E-mail: buzea8@gmail.com; anandkps350@gmail.com
First published on 16th February 2022
Abstract
Colloidal nanocrystals play a vital role in several applications. The doping of cations in the nanocrystal matrix enhances the optical, electrical, and magnetic properties. The number and well-defined distribution of the dopant are crucial to protect the nanocrystal from clustering. The XRD, XPS, and XAS instruments reveal the change in the lattice parameters, chemical states, and local coordination environment information. In addition of detecting the position and distribution of the dopant, the 4D-STEM detector mode gathers all types of real-space atomic-resolution images by collecting all diffraction datasets from each electron probe with high-speed and efficient detection. Dopant–host ligand type, reactions conditions, and reaction time optimization during synthesis are critical for the host and dopant reactivity balance. Pearson's hard/soft acids/bases theory would be a base for balancing the solubility of the dopant–host in the given solvents/surfactant. In addition, tuning the colloidal nanocrystals to secondary structures, which enhances the mass-/ions transport, can contribute a combination of properties that do not exist in the original constituents.
 Buzuayehu Abebe | Dr Buzuayehu Abebe is currently working at Adama Science and Technology University Adama, Ethiopia, East Africa. He is doing his research on the synthesis of nanoscale materials, specifically nanocomposite materials for photocatalysis and antimicrobial application. Buzuayehu Abebe has published more than 20 research articles in Web of Science indexed journals and presented many papers in national as well as international conferences. He is also a review editor of Frontiers in Catalysis journal. |
 H. C. Ananda Murthy | Dr H. C. Ananda Murthy has been a sincere, committed, and dedicated faculty member at various prestigious universities in India, Tanzania, and Ethiopia for the last 24 years. He is currently working as Associate Professor, Department of Applied Chemistry, Adama Science and Technology University, Adama, Ethiopia, East Africa. Prof. Ananda has authored a number of books, compendia, book chapters, published more than 100 research articles in journals of international repute, and has presented many papers in national as well as international conferences. He has been a guest editor for Journal of Nanomaterials and Journal of Renewable Materials. He is also a review editor of Frontiers in Catalysis journal and editorial board member of Annals of Applied Science journal. He has 4 patents to his credit. He has delivered many invited talks at various platforms. He has taught various chemistry courses at the UG, PG, and PhD level of the universities, and supervised 7 MSc and 1 PhD students. He is currently guiding 2 MSc and 7 PhD students (1 awarded). He has successfully completed 2 projects sanctioned by Adama Science and Technology University, Ethiopia and National Innovation Foundation of India (NIFI). He is currently associated with research projects related to the green synthesis of metal and metal oxide nanoparticles/nanocomposites for multifunctional applications. |
1. Introduction
Nanotechnology is a progressive area of science used for manufacturing nanoscale materials.1 Among different nanomaterials, semiconductor metal oxides are highly stable, non-toxic, inexpensive, and have superior catalytic properties. ZnO, a wide bandgap semiconductor (3.37 eV), has all the abovementioned properties; however, it cannot degrade pollutants under visible light.2 The photocatalytic properties of the materials could be further enhanced by doping noble metals as an electron reservoir.3 Industrially, the incorporation of impurities in the host matrix for improved device functionality is expensive.4,5The unique properties of nanoscale materials (NMs) such as electronic density, charge distribution, and lattice distortion have great impacts on their applications.6 The effective doping of metal or metal ions, which creates either an n-type or p-type dopant, is one of the main ways to tune the optical, magnetic, and optical properties. Besides, the incorporation of impurities also increases the surface-to-volume ratios, creates crystal defects, and traps charge carriers.7–9 However, the dopant should substitute the host atoms effectively so that decent dopant–host balance reactivity occurs. The selection of suitable host–dopant type, synthetic approach, solvent, and surfactant are crucial parameters for balancing the reactivity. For the successful incorporation of the dopant in the host lattice, the host–dopant type is essential. According to Pearson's hard/soft acids/bases (HSAB) theory, for the successful diffusion of the dopant ions into the host lattice, the hardness of the dopant should be less than that of the host in a hard base solvent such as water.10,11
The doping process may be under the control of either kinetic or thermodynamic equilibrium.12–14 Compared to the former, the latter synthetic equilibrium process creates stable and efficiently doped nanocomposites.15 Several synthetic approaches such as single-source precursors, nucleation-doping, growth-doping, and cation diffusion follow either the thermodynamic or kinetic equilibrium process. Nowadays, the cation diffusion technique is a progressive approach.16
The properties of the doped materials can be understood using advanced analytical techniques. The XRD pattern confirms the formation of a local heterojunction and interstitial or/and substitutional incorporation depending on the angle shift. The optical properties of the doped materials were understood from the DRS/UV-vis and PL techniques. Soft Lewis acid dopants (such as Ag and Cu) improve the light absorption efficiency and also condense the bandgap by creating an interband between the valence band (VB) and conduction band (CB) of the host.17 Intensity reduction in the PL spectra is an indication of the electron–hole recombination diminishing properties of the materials,18 which enhances its application, especially in photocatalysis. XPS and XAS techniques give the composition, chemical state, local coordination environment, and oxidation states of the doped material.19,20 In addition, a radical instruments such as ADF-STEM and EELS-STEM detect the atomic level incorporated dopants based on the contrasts.21,22
In addition, tuning the material to have a secondary structure during synthesis (ordered porous crystalline frameworks) boosts the mass-/ion-transports of the materials toward precise applications such as energy devices.10,23 For the development of decent ordered porous crystalline frameworks, selecting a suitable solvent, reaction temperature, nanocrystal tethering, and pore generating architecture-directing agent (ADA) is vital. Herein, this review also provides awareness about the dopant–host reactivity balance, characterization techniques for the doped materials, and porous crystalline frameworks. The paper also gives detailed insight into Cu- and Ag-doped and co-doped ZnO-based materials using crucial analytical techniques. Besides, the photocatalytic and antibacterial activities of the doped materials, and the mechanisms involved in these activities have also been discussed.
2. Dopant–host reactivity balances
The host–dopant reactivity balance is highly dependent on sufficient time for effective dopant trapping, surface morphology, shape, dopant host ligand type, and reaction conditions.16 The HSAB theory helps in tuning the solubility balance by the careful selection of metal ions to the ligands/solvents affinity.24 According to the HSAB theory, hard acids favor hard bases, while soft acids favor soft bases to bind and yield ionic and covalent complexes, respectively. The HSAB absolute hardness (η) of some cations and ligands are given in Table 1. Besides, Fig. 1 also groups the soft, borderline, and hard acids/bases and molecular recognition based on the HSAB theory. Hard/borderline Lewis acids such as Zn(II) ion are easily exchanged with soft acids such as Cu(I) and Ag(I) ion when hard base solvents (alcohols and water) are used. On the other hand, soft acids cations are spontaneously exchanged with harder acid cations if soft bases are used.10,11,25 In the presence of a hard base (water as a solvent) and poly(vinyl alcohol) surfactant, the non-inclusion of the manganese hard Lewis acid in the ZnO lattice was verified in our recent study.26 In its place, the local heterojunction was formed between manganese oxide and zinc oxide after the oxidation of the adsorbed manganese ion on the surface of zinc oxide at 500 °C.| Lewis acid | η | Lewis base | η |
|---|---|---|---|
| Cu(I) | 6.3 | Aniline (C6H5NH2) | 4.4 |
| Pd(II) | 6.8 | Benzenethiol (C6H5SH) | 4.6 |
| Ag(I) | 7.0 | Phenolate (C6H5OH) | 4.8 |
| Fe(II) | 7.2 | Pyridine (C5H5N) | 5.0 |
| Hg(II) | 7.7 | Acetone (CH3COCH3) | 5.6 |
| Sn(II) | 7.9 | Acetaldehyde (CH3CHO) | 5.7 |
| Pt(II) | 8.0 | Dimethylformamide (DMF) | 5.8 |
| Co(II) | 8.2 | Trimethylphosphine ((CH3)3P) | 5.9 |
| Cu(II) | 8.3 | Phosphine (PH3) | 6.0 |
| Au(III) | 8.4 | Dimethyl sulfide ((CH3)2S) | 6.0 |
| Pb(II) | 8.5 | Formaldehyde (CH2O) | 6.2 |
| Co(III) | 8.9 | Formamide (HCONH2) | 6.2 |
| Mn(II) | 9.0 | Trimethylamine ((CH3)3N) | 6.3 |
| Ge(II) | 9.1 | Methyl formate (HCO2CH3) | 6.4 |
| Cd(II) | 10.3 | Acetonitrile (CH3CN) | 7.5 |
| Zn(II) | 10.9 | Chloromethane (CH3Cl) | 7.5 |
| Fe(III) | 12.1 | Dimethyl ether ((CH3)2O) | 8.0 |
| In(III) | 13.0 | Ammonia (NH3) | 8.2 |
| Ga(III) | 17.0 | Fluoromethane (CH3F) | 9.4 |
| Al(III) | 45.8 | Water (H2O) | 9.5 |
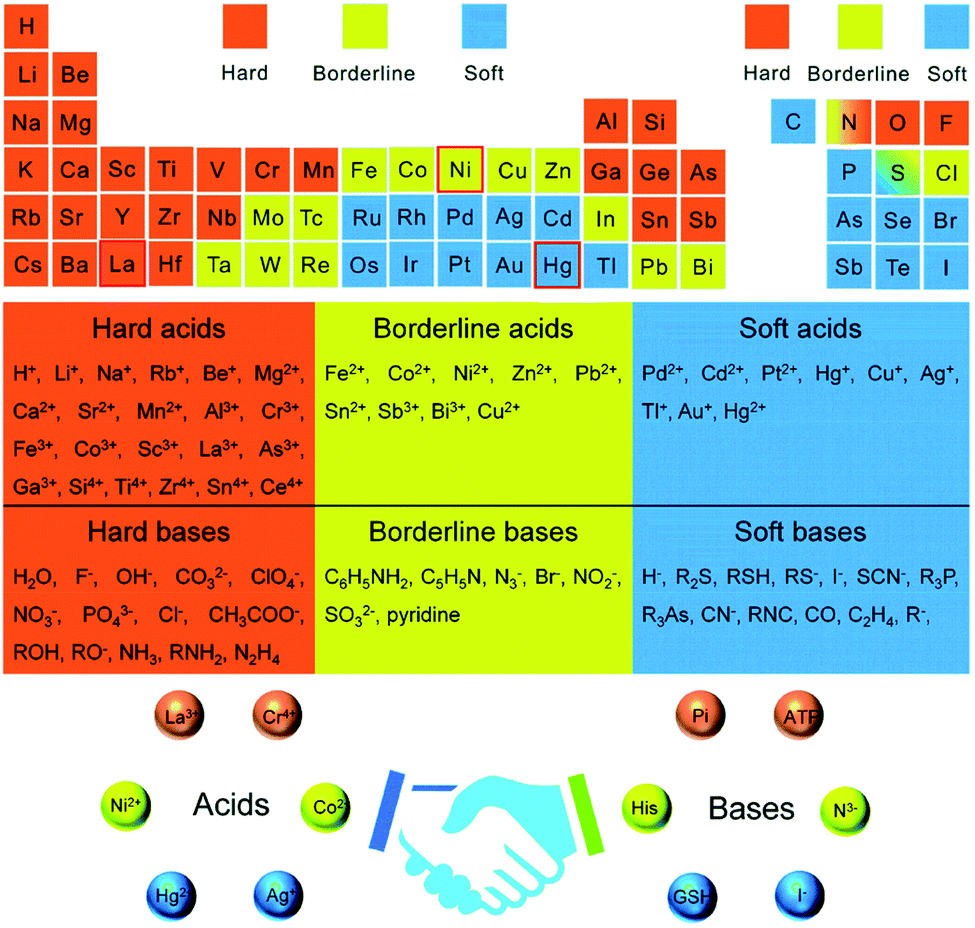 | ||
| Fig. 1 Periodic element table for grouping hard, soft, and borderline acids/bases and molecular recognition based on the hard–soft–acid–base theory.24 | ||
As reported by Buonsanti and Milliron,16 the common approaches used for balancing the reactivity of the dopant and host precursors are: (i) single-source precursors, (ii) nucleation-doping, (ii) the growth-doping, (iii) tuning of ligand–metal bond strength, and (vi) cation diffusion (Scheme 1). A single-source precursor is effective in controlling the final stoichiometry of the constituents and easily forms a direct chemical bond between the host matrix and the dopant. Still, stronger attraction among the elements composing the cluster is a requirement compared to the ligands and the cluster. If the attraction among the elements is not strong enough and, if the other attraction dominates, a complex decomposition takes place and diminishes the dopant concentration. Using chimie douce single-source molecular precursors approach, mixing of similar zinc- and copper-ketoacidooximates precursors, Pashchanka et al. synthesized a ‘green body’ nanorod arrays morphology of Cu(II)-doped ZnO.27 In this study, chemical processing techniques (dichloromethane (CH2Cl2)) were used to remove the polycarbonate film template-forming agent.
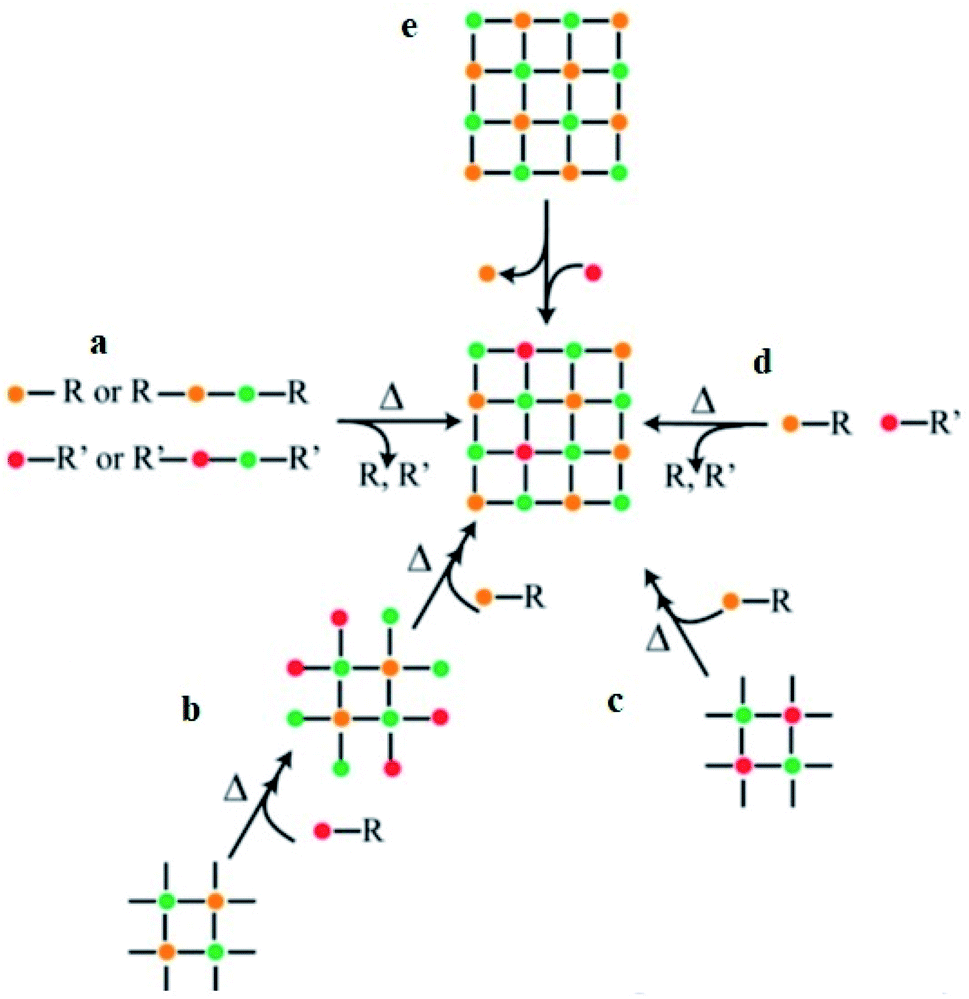 | ||
| Scheme 1 General synthetic approaches for colloidal nanocrystals' doping. The single-source precursors (a), nucleation-doping approaches (b), the growth-doping (c), the tuning of ligand–metal bond strength (d), and cation diffusion (e).16 | ||
During the nucleation-doping method, in which the dopant and the host precursors are mixed once, their reactivity is regulated so that the dopant nucleates first, followed by its overcoating by the host shell. On the other hand, in the growth-doping method, the dopant precursors are added to the growing host shell by controlling the reaction conditions/temperature to reach the required size so that the dopant is encapsulated by the overgrowing host matrix shell and confines the dopant in the core. In general, the growth process of the host materials is controlled by decreasing the temperature of the reaction. In addition, using a reactive host precursor, and decreasing the overcoating temperature were reported in several studies,15,28,29 which also prevents the diffusion of the dopant from the core at high temperatures.
From the concept of HSAB theory, appropriate ligand–metal bond strength classification is also used to balance the host and dopant reactivity. For successful dopant incorporation into the zinc oxide semiconductor, soft Lewis acid dopant cation (low positive charge and large size) such as Cu and Ag are chosen. The softness and hardness of the coordinating ligands and the size of the cations also affect the dopant–host reactivity. If the dopant cation is a harder Lewis acid compared to the host cation, more reactive dopant coordinating ligands are required for successful incorporation.30 In addition, if a comparable size of the host and dopant cations is used, the reactivity becomes less for dopant integration.31 Besides, similar coordinating ligands can also be used if the host and dopant cations have similar reactivity.32,33 Similarities in reactivity and ionic radii were also reported34 for improved dopant concentration insertion.
The final approach described by Buonsanti and Milliron16 that was used for the incorporation of both cationic and anionic dopants was ion diffusion. This approach allows ions of the dopant to be exchanged within a few seconds in the pre-shaped host matrix. The ion diffusion approach exchanges the cation with remarkable speeds under kinetic control at low temperatures. The host–dopant cations were exchanged without affecting the crystal framework.35 The (a) association and dissociation process of the crystals, which is defined in terms of lattice energy (lattice enthalpy) and surface energy, and (b) the solvation and desolvation process of the ions leads to a successful cation-exchange procedure.35 The association and dissociation process of the crystal measures the strength of the chemical bonding, while the solvation and desolvation process of the ions depends on the number of ions solvated from the host precursor and ions desolvated from the dopant precursor. If the number of ions solvated is greater than the ions desolvated, the reaction is favored by increasing the overall entropy of the system and such a reaction does not take place unless a careful choice of the solvents and soft Lewis bases/ligands is used.10,36,37
In addition to the thermodynamic factors, the cation-exchange tactic is also dependent on the kinetic factor of the reaction process such as ions diffusivity or/and activation energy barriers.38 Actually, the cation-exchange process is also reported to occur during the growth of the host matrix.4 The diffusion of the dopant into the host cell is also dependent on the dopant concentrations, the type of the coordinating ligands used, and temperature.15,35 The process of dopant adsorption, diffusion, and ejection in the host lattice is similarly dependent on the optimization of the critical temperature of the overall synthetic process.15
2.1. Silver-doped ZnO nanocrystals
Silver ion has a larger ionic size (0.126 nm) compared to zinc ion (0.074 nm); thus, the doping of silver into the ZnO lattice creates a doped band, which shifts the Fermi level toward the VB (deep acceptor level) and induces a p-type property.39,40 Besides, the interstitial and substitutional incorporation of Ag into the ZnO lattice causes quantifiable higher and lower angle shifts, respectively.41–46 However, the segregation or formation of a local heterojunction47 between Ag and ZnO does not result in any peak shift in XRD.48,49 In fact, the formation of a local heterojunction, and interstitial and substitutional defects were dependent on the synthetic approach, cation hardness, and surfactant or/and solvent type. To indicate this, Yıldırım et al.42 showed an XRD pattern lower degree shift due to the substitution of Ag+ into the ZnO lattice and a higher degree shift was shown by Modwi et al.46 due to the interstitial sites' doping. The formation of deep-level acceptor and stability improvement were proved by density functional theory calculations within the generalized gradient approximation analysis.50The detailed procedure for the synthesis of tri-ethanolamine (C6H15NO3) surfactant-assisted Ag-doped ZnO nanocomposites (sol–gel-based synthetic approach) was proposed in the study by Sagadevan et al.51 The composite was synthesized by the dropwise addition of AgNO3 in an aqueous solution of Zn(CH3COO)2·2H2O, possibly following the growth-doping type dopant–host reactivity balance approach. The addition of excess NH4OH solution results in the precipitation of the acetate ion to ammonium acetate (NH4CH3COO) and the development of metal hydroxide colloidal particles (Zn(OH)2 and AgOH). Ammonium acetate was removed by washing with deionized water, and then it was washed with acetone to remove water. Lastly, the final product was calcined at 300 °C to oxidize the metal hydroxides (removal of water of crystallization) to stable metal oxide or/and doped metal oxide nanocomposites.
Using the hydrothermal approach, Jin et al.43 proposed the substitution of Ag in the ZnO lattice. The experiment was conducted by the dropwise addition of ammonia in the mixture of zinc and silver solution. With the continuous addition of ammonia, first, a milky colored zinc hydroxide (Zn(OH)2), then the Zn complex (Zn(OH)42−, and finally, a clear solution was formed when the molar ratio of zinc ion and ammonia base reached 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. In this process, the final solutions, which contain ZnAg(OH)2 and ZnAg(OH)42− , were allowed to oxidize in an autoclave to form Ag-doped ZnO (ZnAgO). A related solvothermal synthetic approach was also reported in Zheng et al.’s study by the dropwise addition of NaOH in silver and zinc solution mixture.52
:4. In this process, the final solutions, which contain ZnAg(OH)2 and ZnAg(OH)42− , were allowed to oxidize in an autoclave to form Ag-doped ZnO (ZnAgO). A related solvothermal synthetic approach was also reported in Zheng et al.’s study by the dropwise addition of NaOH in silver and zinc solution mixture.52
Using digital mechanoelectrospinning direct-writing for polyethylene oxide template deposition, a hydrothermal method for ZnO nanoarray growth and photoreduction for doping silver approaches were reported; aligned hierarchical Ag-deposited ZnO nanoheterostructure arrays with high nitrogen dioxide gas sensing performance were synthesized by Yin et al.53 The formation of more than zero-dimensional nanomaterials was stated to enhance the surface area and gas sensing properties of the materials; a related surface area improvement interpretation was also given in Singhet al.’s work.39 From the results,53 a hexagonal wurtzite ZnO and face-centered cubic Ag structure with d spacing values of 0.26 nm and 0.23 nm, respectively, were obtained using the HRTEM image analysis, with no ZnO lattice distortion. Besides, the deposition of Ag NPs on the surface of the ZnO nanorods was further confirmed from a highly sensitive HAADF-STEM image.
The non-incorporation of Ag in the ZnO lattice was also confirmed by Ansari et al.48 (Fig. 2(A)) and Alharthi et al.49 In Ansari et al.’s work, the Ag-anchored ZnO material was synthesized by a biogenic approach using an electrochemically active biofilm.48 The deposition of Ag on the surface of ZnO was also indirectly understood from DRS analysis (Fig. 2(C)), which shows Ag localized surface plasmon resonance (LSPR) band characteristics in the wavelength range of 400–550 nm. The LSPR features of the materials occurred when frequency matching between the conduction bands confined the electron oscillation and the incident light exits.54 This LSPR effect is also reported in other works55–57 and is attributed to the intense light absorption and scattering reflection behavior of the doped Ag clusters. Besides, with an increase in the amount of AgNO3, higher energy plasmon peak shift and intensity heightening were noticed due to the presence of silver on the surface and increase in the cluster size, respectively.56
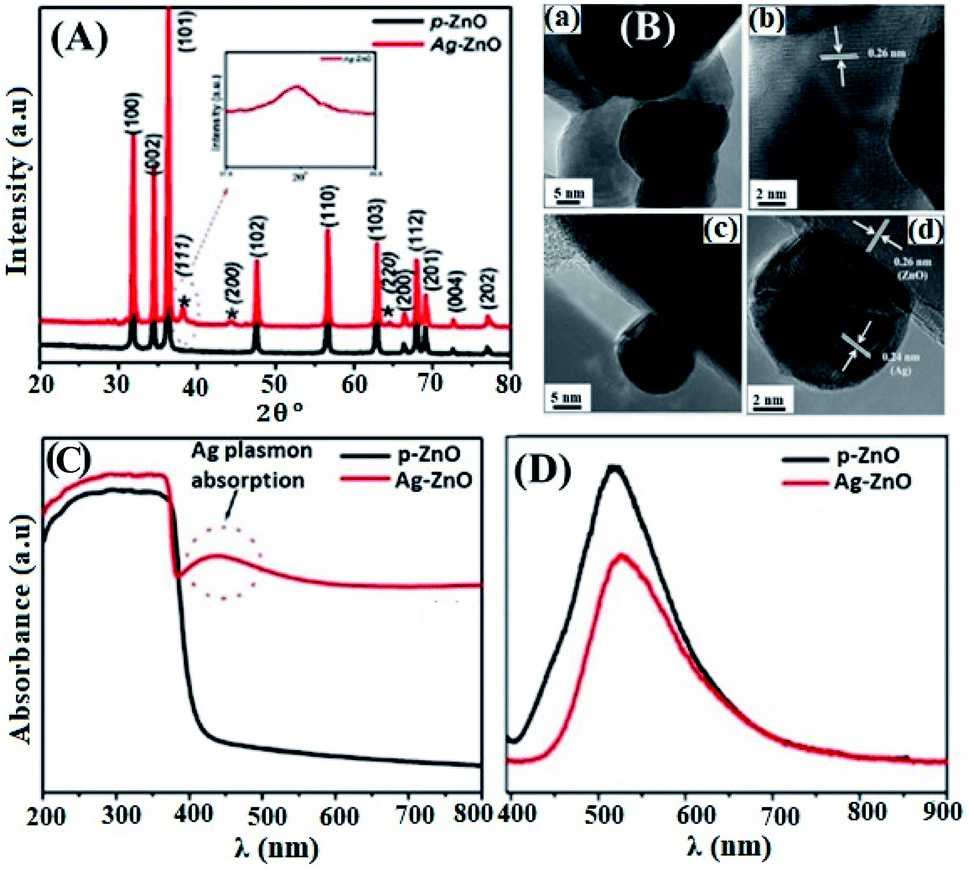 | ||
| Fig. 2 (A) XRD patterns of pure-ZnO and Ag–ZnO. The peaks marked with (*) represent the signals of Ag and the remaining peaks represent the signals from pure-ZnO. The inset shows the broadening of the Ag peak, (B) HR-TEM images of pure-ZnO (a and b), and Ag–ZnO (c and d), (C) UV-vis diffuse absorption spectra of pure-ZnO and Ag–ZnO, (D) photoluminescence spectra of pure-ZnO and Ag–ZnO.48 | ||
Besides, Ziashahabi et al.57 reported the lower wavelength plasmonic peak shift on aged Ag/ZnO samples due to silver oxidation, which was not observed on the freshly synthesized Ag/ZnO sample. The HR-TEM images (Fig. 2(B)) and XPS analysis also confirmed the presence of strong interaction between Ag and ZnO in Ansari et al.’s work, and a reliable clarification was also reported.58 The shift in the binding energy of Ag 3d5/2 for the doped composite toward a lower binding energy and 6 eV splitting difference between Ag 3d5/2 and Ag 3d3/2 (high-resolution scans) confirm the existence of interaction between Ag and ZnO, which leads to the Fermi level tuning and the reduction of Ag+ to Ag metal, respectively. Comparable XPS binding energy shift and splitting values were also reported in the literature.42,43,49,56,58–60 The lower energy shift is ascribed to the binding energy difference between Ag(0) and Ag(I), in which Ag(I) has much greater binding energy than Ag(I).
Besides, in Ansari et al.’s work, the formation of local contact/composite between Ag and ZnO, which enhances electron transfer without recombination, was also further confirmed via PL analysis (Fig. 2(D)); the smaller the peak intensity, the lower the electron–hole recombination activity.18 Similar explanations were also given in other studies and reported to be due to the occurrence of metal–semiconductor Schottky contact.3,40,49 This electron transfer without recombination was described by the band energy difference between Ag (more positive band energy) and ZnO (Fig. 3), which leads to continuous electron transfer from ZnO to Ag until their Fermi level values become equivalent.3,42 The electron–hole separation is dependent on the Ag disparity, the concentration of the Ag–ZnO interface, and vacancy defects.52 As seen in Fig. 3, during the irradiation of the doped photocatalyst in the presence of vacancies, the electron transfer follows either path I (toward doped Ag) or path II (toward the created ZnO vacancy). The method used to synthesize Ag-doped ZnO material, reagents, substrate or/and surfactants used, and the morphology of the synthesized material is also given in Table 2.
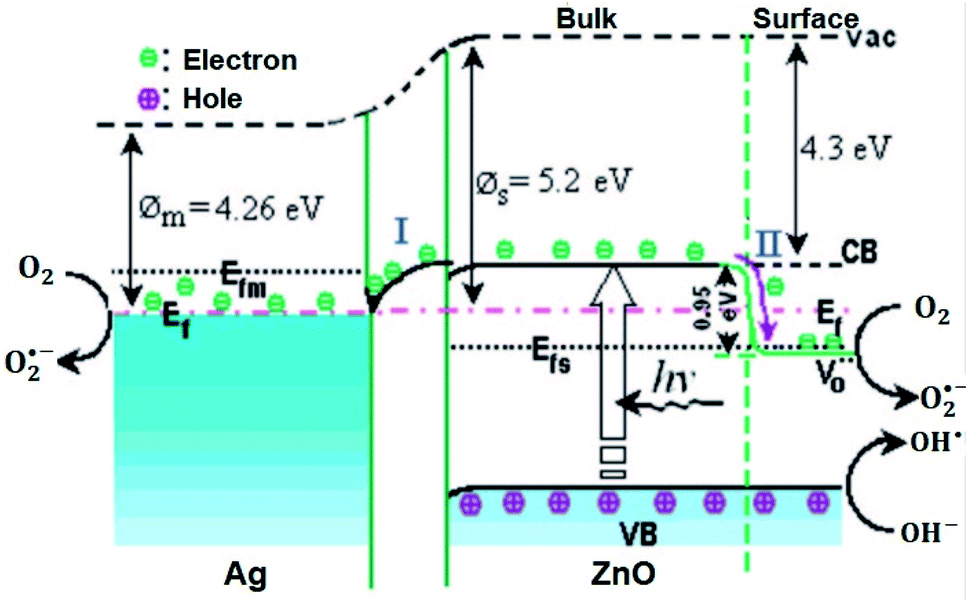 | ||
Fig. 3 Photogenerated electron transfer in Ag/ZnO nanocatalyst during the catalytic process. EF: Fermi level; 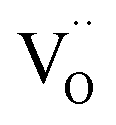 : ø: work function; oxygen vacancy; VB: valence band; CB: conduction band; m: metal; vac: vacuum level; and s: semiconductor.52 : ø: work function; oxygen vacancy; VB: valence band; CB: conduction band; m: metal; vac: vacuum level; and s: semiconductor.52 | ||
| Approach | Reagents | Morphology | Ref. | ||
|---|---|---|---|---|---|
| Precursor | Solvent | Surfactant/substrate/other | |||
| a Acetate (CH3COO). | |||||
| Hydrothermal and seed-mediated growth | ZnCl2 and AgNO3 | Ethanol | Cetyl trimethylammonium bromide triethyl- amine | Hybrid core–shell nanorods | 3 |
| 3-Aminopropyl-trimethoxysilane | |||||
| Precipitation | Zn(aca)2·2H2O | Water, ethylene glycol | Polyvinylpyrrolidone | — | 42 |
| C2H3AgO2 | |||||
| Hydrothermal | Zn(aca)2·2H2O | Water | Flower-shaped structures | 43 | |
| AgNO3 | |||||
| Electrospinning | Zn(NO3)2·6H2O | Ethanol | Polyvinylpyrrolidone | Nanofiber | 44 |
| AgNO3 | |||||
| Biogenic synthesis | Nano ZnO | Water | — | Spherical | 48 |
| AgNO3 | |||||
| Deposition–precipitation solvothermal method coprecipitation | Zn(aca)2·2H2O | Ethanol | — | Porous | 52 |
| C2H3AgO2 | |||||
| Photodeposition | Zn(aca)2·2H2O | Water and ethyl alcohol | Polyethylene oxide | Film, ribbon, and nanowires | 53 |
| AgNO3 | |||||
| Combustion | Zn(NO3)2·6H2O | Polyethylene glycol fuel | THF and thermoplastic polyurethane | Agglomerated spherical NPs | 56 |
| AgNO3 | |||||
| Chemical reduction | Zinc rods AgNO3 | Water | Trisodium citrate (reducing agent) | Spherical | 57 |
| Polyacrylamide gel | Zn(NO3)2 | Water | Acrylamide and N,N′-methylene-bisacrylamide | Quasispherical | 58 |
| AgNO3 | |||||
| Facile surfactant-free | Zn(NO3)2·6H2O | Ethanol | Ethylene glycol (reducing agent) | Nanorods | 60 |
| AgNO3 | |||||
2.2. Copper-doped ZnO nanocrystals
Zinc and copper have similar electronic configurations and comparable atomic radii but different structures, which limits the stability of the doped copper toward dissolution. Doping of copper into the ZnO host improves the optical, electrical, and magnetic properties, which is not observed in isolated constituents. A higher bandgap semiconductor metal oxide such as zinc oxide, ZnO, has outstanding thermal, optical, and electrical properties. ZnO also has higher light spectrum absorption efficiency and lower production cost.61 However, the number and exact position of the dopant should be achieved to prevent the clustering of dopant atoms that have a damaging effect.62 The oxidation states of copper (Cu, Cu(I), or Cu(II)) in the ZnO matrix is contentious and seems to be dependent on various parameters such as dopant concentration, temperature, and type of synthetic approach.19,27 The general host–dopant molecular precursor's decomposition and reaction process for nitrate precursor (as an example) are given in eqn (1) for ZnO and eqn (2) for copper-doped ZnO, where x is the doping amount and δ is the spillover valence of oxygen.63| 9[Zn(NO3)2·6H2O] + 10[NH2CH2COOH] (fuel) → 9ZnO + 20CO2 + 14N2 + 79H2O | (1) |
| xCu(NO3)2·3H2O + (1 − x)Zn(NO3)2·6H2O + oxidant → CuxZn(1−x)O(2−δ) + gaseous products | (2) |
The solution combustion process enables the final products of the nanomaterials with precise stoichiometric ratio, pronounced disparity, and uniform composition.64 Fig. 4 shows the solution combustion synthetic process accompanied by the heating of the fuel and oxidant to its ignition temperature. When a mixture of the metal nitrate precursors (which acts as oxidizer) and the urea fuel (as reducer) is heated to its ignition temperature, the metal ion–fuel complex starts to undergo combustion and form oxides. The combustion process is a self-propagation process, which could be controlled only by adjusting the ratio of reducing and oxidizing valances. Further calcination and reduction may help to stabilize the structure and reduce the metal as needed.
 | ||
| Fig. 4 Schematic diagram of the synthetic process of catalysts by the solution combustion approach.64 | ||
Gupta et al. synthesized highly porous doped materials using a simple combustion approach in the presence of glycine as a fuel.63 The combustion method is one of the methods that create pores/voids within the nanoparticles by releasing gas due to the exothermic reactions.65 The fuel/reducer has a prominent role in facilitating the combustion process by creating an exothermic reaction and change in the morphology of the NPs. However, the porous materials synthesized by this approach exist in a disordered form rather than an ordered form, which can be synthesized by choosing appropriate nanocrystal tethering and pore-generating domain surfactants.
Nowadays, several approaches such as sol–gel, Pechini, coprecipitation, solid-state reaction, hydrothermal, solvothermal, successive ionic layer adsorption and reaction, chemical bath deposition, plasma-enhanced chemical vapor deposition, spray pyrolysis, pulsed laser deposition, ball-milling, and sonochemical method are being employed for synthesizing transition metal-doped ZnO materials.66 In the sol–gel and Pechini approaches, the pH of the solution, aging, and annealing temperature are the major parameters affecting the particle size, the nucleation and growth units, and the morphology of the NMs.67 The coprecipitation technique is applied by the dropwise addition of the precipitating agent in the precursor solution, and calcinating the product at high temperature. The coprecipitation method is suitable for the atomic-level mixing of the host and dopant at low temperature; however, it is difficult to control the nanocrystals' (NCs) geometry.66 Both the solid-state reaction and coprecipitation technique present a decent stoichiometry of the product, in which the former needs higher temperature than the latter. The solid-state reaction follows the homogeneous mixing of oxides and calcination at high temperatures. The synthesis of NMs by the hydrothermal and solvothermal techniques employs an autoclave with stepwise heating and cooling in the temperature range between 100 and 300 °C and then storing for several days. The solvothermal method uses a different solvent than that in the hydrothermal method, which uses only water as a solvent. The spray pyrolysis and pulsed laser deposition methods are used to deposit thin films. Ball-milling is a simple process but needs high annealing temperature and consumes more time.68
Various concentrations of Cu(II) ion-doped ZnO films were synthesized by sputter chamber deposition techniques with the help of sapphire as the substrate.19 From the XRD pattern, a contraction of the lattice due to the smaller size of Cu(I) and Cu(II) compared to the Zn(II) ion was noticed. Increasing the dopant concentration showed decreasing intensity and increasing FWHM values, which corroborates the reduction of both the crystallite size and crystallinity of the composite. Similarly, the observed shift in the peak position toward a higher 2θ value, decreasing peak intensity, and peak broadening were also reported in various works69–73 and explained to be due to the contraction of the c lattice due to the doping of Cu atoms into the ZnO lattice. No impurity peak conforming to copper was found, which indicates the absence of structural distortion in the ZnO lattice, although the XRD technique is not sensitive to small doped particles.
Transmission electron microscopy (TEM) can directly observe the doped nanocrystal; however, it cannot give the correct oxidation state. Based on the HTEM image analysis, Pashchanka et al. interpreted the effective doping of copper ions into the ZnO lattice.27 The experimentally obtained lattice fringe for Cu-doped ZnO (0.254 nm) is smaller compared to the normal ZnO interplanar spacing,74 which confirms the substitution of copper ion (which has an ionic radius of 0.057 nm) with zinc ions (which has an ionic radius of 0.060 nm).
An element-specific sensitive technique such as XAS (containing XANES and EXAFS) reveals the detailed information about the dopant oxidation state. Liu et al. reported that the XAS technique can be effectively used to study the local order, electronic environment, position, and distribution of doped copper.19 As indicated, the edge change is due to the incorporation of Cu at the Zn site, which causes minor disorder but does not affect the ZnO structure. From the background-subtracted and normalized Cu K edge XANES spectra, the detected pre-edge peaks, ca. 8977.5 eV and 8981 eV show the presence of Cu(II) and trace amount of Cu(I) oxidation state, respectively. Related XAS interpretation was also given on the bullet-like morphologies of Cu-doped ZnO crystal synthesized by the sol–gel route.75 The shoulder at 9000.5 eV in all the doped films is due to the substitution of Cu in the Zn lattice. Also, the likenesses of the oscillations and magnitude of the Zn K-edge k3-weighted EXAFS spectra for both bare ZnO and doped films show the substitution of Zn by Cu without distortion. The similarities between Zn and Cu K-edge in EXAFS analysis show the development of ZnO-like wurtzite structure around the Cu atom with Cu(II) oxidation state.
Agarwal et al.17 synthesized copper-doped ZnO by neutral beam sputtering techniques and reported the absence of any structural distortion at low dopant concentration. However, increasing the dopant concentration results in the development of the copper peaks on the XRD pattern. From the optical properties obtained by UV-vis spectral analysis, the bandgap energy of Cu-doped ZnO (15%) was obtained to be smaller (2.8 eV) than the ZnO bandgap energy (3.4 eV). This is probably due to the interband transition within the Zn 4s and Cu 3d bands. As is well interpreted on the surface plasmon resonance analysis, at a lower concentration, the doped material reveals the host behavior. On increasing above the optimum value, the material can exhibit dual behavior. With the help of XANES spectra, the substitution of empty ZnO d states by Cu(II) with trace amounts of Cu(I) was confirmed. A consistent XAS-based XANES Cu(I) substitution result was interpreted in another work.72 In addition to XANES, the substitution of cuprous state (Cu(I)) in the ZnO lattice was confirmed by XPS analysis.76 The sealed microspheres/semispherical shell morphology of the Cu-doped ZnO composite was synthesized by chemical vapor deposition techniques.
In addition to XAS, XPS analysis was also used to understand the doped copper oxidation states.20 Herein, the Cu-doped ZnO material was synthesized by radiofrequency magnetron sputtering technique from ZnO and CuO powders. XPS analysis revealed the presence of both Cu(II) and Cu(I) oxidation states with Cu(I) in the interstitial position. Besides, increasing the dopant concentration results in a reduction in the amount of Cu(II) oxidation state and increasing the Cu(I) oxidation state. The ZnO and CuO powders were also used to synthesize Cu-doped ZnO films deposited using pulsed laser deposition technique under an oxygen partial pressure of 10−3 and 10−5 torr.77 The composition, chemical state, and oxidation states of doped copper were analyzed by XPS analysis. In the oxygen-deficient sample (10−3 torr partial pressure), the Cu(I) state is dominant, while the Cu(II) state is in the oxygen-rich sample (10−5 torr partial pressure). The method used to synthesize Cu-doped ZnO material, reagent, substrate, or surfactants, and the dopped Cu oxidation state are given in Table 3.
| Approach | Reagents | Copper OSb | Ref. | ||
|---|---|---|---|---|---|
| Precursor | Solvent | Surfactant/substrate | |||
| a Acetate (CH3COO)b Oxidation state. | |||||
| Co-sputtering | Cu and ZnO powder | — | Si and quartz substrate | +II major | 17 |
| +I trace | |||||
| Sputter chamber deposition | Cu and Zn powder | — | c-Plane sapphire substrate | +II major | 19 |
| +I trace | |||||
| Sputtering | ZnO and CuO powders | — | Quartz substrate | +II, +I | 20 |
| Chimie douce single source molecular precursors | Zinc and copper ketoacidooximates | H2O | Porous polycarbonate film | +II | 27 |
| CH2Cl2 (to remove the polymeric template) | |||||
| Sol–gel | Zn(aca)2·2H2O | Methanol | Polyvinyl alcohol | — | 69 |
| CuCl2·2H2O | |||||
| Co-sputtering | Cu and Zn powder | — | Pt(111)/Ti/SiO2/Si(100) substrate | +II, +I | 72 |
| Facile soft-chemical | Zn(ac)2·2H2O | Diethylene glycol | — | — | 73 |
| Cu(ac)2·xH2O | Polyethylene glycol | ||||
| Sol–gel | Zn(NO3)2·6H2O | H2O | — | +II | 75 |
| Cu(NO3)2·6H2O | |||||
| Chemical vapor deposition | Zn and CuCl2·2H2O powders | — | n-Type Si(100) substrates | +I | 76 |
| Pulsed laser deposition | ZnO and CuO powders | — | Pt (200 nm)/Ti (45 nm)/Si (001) substrates | +II, +I | 77 |
2.3. Bi-metallic/co-doped ZnO
The precise position and distribution of the dopant can create shallow defect levels in the host matrix and tune the properties of the materials toward a specific application. Recently, two kinds of element doping into the semiconductor lattice have attracted substantial interest as it could result in unusual characteristics compared with one element doping.18 p-type doping and n-type doping were found to be difficult for low VB maximum and high CB minimum energy, respectively.78 Thus, concurrently integrating the n-type and p-type dopants can be a feasible solution for the aforementioned problems. Also, co-doping increases the activation rate, dopant solubility, and carrier mobility. The Cu–Bi codoped ZnO nanospheres were synthesized by sol–gel-assisted hydrothermal method.79 From the XRD pattern, the doping of Bi and Cu separately showed a lower angle and higher angle diffraction peak shift owing to the respective higher and small size, compared to Zn size. This is attributed to the expansion and contraction of the ZnO lattice during Bi and Cu substitution, respectively. The high-resolution scan XPS analysis confirms, the Bi3+ to be the major dopant chemical state. Besides, the main peak for the O 1s XPS spectrum of Cu–Bi codoped ZnO is divided into five different peaks. Among these five peaks, the peak obtained at the binding energy of 530.54 eV and 532.18 eV fit with Cu–O and Bi–O, respectively. The photocatalytic activity of Cu–Bi codoped ZnO is greater than ZnO, which is ascribed to the bandgap reduction for codoped materials, compared to Cu-, Bi-doped ZnO, and bare ZnO.Spherical Cu and Ag NPs anchored on the surface of the ZnO materials synthesized by the green method showed UV-vis absorption spectra intensity reduction and greater photocatalytic activity.80 This local heterojunction between Ag/Cu NPs and ZnO was further confirmed by XPS analysis. The optical properties' adjustment, attributed to the characteristics of Co+2 d–d transitions in the Fe–Co codoped ZnO, were also clearly shown by Beltrán et al.81 The presence of Fe and Co resulted in more charge transfer, which improved the ferromagnetic properties. The improvement in the optical and ferromagnetism properties ascribed to codoping (Co–Mn codoped ZnO) was also reported in Pragna et al.’s study.82 The Ni–Cu codoped ZnO spherical materials synthesized by the wet hydrolysis approach were shown to have pronounced PL spectra intensity reduction than bare and Ni- and Cu-doped ZnO counterparts.83 The codoped material showed complete quenching of the UV emission and growth of broad two visible emission bands at 380 nm and 460 nm, which is attributed to the Ni and Co dopant, respectively, indicating their ability to serve as UV photodetectors.
The use of codoped ZnO as a photodetector was also studied in depth by GuruSampath et al.84 The heterostructured ZnO/Ga–Ag codoped ZnO nanorod material was synthesized by the two-step process, sol–gel-assisted hydrothermal approach. A slight lower angle shift was observed in the XRD pattern of the codoped material, which makes it confusing to interpret the effect based on the ionic radii because Ga(III) has a smaller ionic radius (0.62 Å) than Zn(II) (0.74 Å) and Ag(I) has a greater radius (1.02 Å) than zinc ion. The incorporation of bimetallic dopant showed a superior (nearly 20 times) PL emission intensity drop, which increases additional CB electrons and also reduces the material bandgap. The fabricated codoped heterostructured nanorod UV-photodetector showed two times greater photoresponse compared to bare ZnO at 13.5 mW cm−2 UV power density.
In addition to transition metal doping, nonmetal doping participates in band narrowing and amends the materials property toward visible light absorption. Transition metal ion (Cu) as a p-type and non-metal ion (N) as n-type codoped ZnO porous material were synthesized by Gupta et al.63 using the combustion and hydrothermal methods. The materials were synthesized by keeping the amount of N constant and increasing the amount of copper. From the XRD pattern analysis, increasing the amount of copper showed lattice contraction, which is attributed to the substitution of smaller Cu ionic radii with greater Zn ionic radius. In this study, the absorption of light in the visible region and bandgap narrowing were seen for N–Cu codoped ZnO than Cu-, N-doped, and bare ZnO. The incorporation of both Cu (Cu(I) and C(II)) and N in the ZnO lattice were also confirmed by the XPS analysis. Herein, the creation of a sub-energy level band below the host CB and above the VB was suggested due to the transition metal and nonmetal doping, respectively. The created sub-energy level acts as a photogenerated electron–hole sink, thus prolonging electron–hole recombination.
Besides, Ce–Ag codoped-ZnO material prepared by the solvothermal method also showed a superior surface area and optical property improvement.18 Compared to bare ZnO, the codoped-ZnO material showed approximately five times lesser average crystallite size. The greater the surface area, the higher the sorption properties and the greater the photon-assisted degradation. The redshift in the optical spectra occurred due to a new energy level in the DRS spectra, which is attributed to the existence of noble contact between the host and the dopant (Ag–ZnO–Ce). Besides, the lower PL intensity for the codoped sample compared to the bare ZnO also shows the lessening of electron–hole recombination. The smaller the recombination, the higher the Naphthol Blue Black dye degradation in aqueous solution under solar light irradiation. The photo-generated electron transfer from the ZnO CB to both the doped Ce and Ag metals was also reported by reducing the electron–hole recombination synergistically.
In addition to the magnetic and optical properties, the electrical properties of ZnO were also boosted by the doping of Cu in the ZnO lattice and further decorating it with different percentages of Ag (Ag@Cu–ZnO). Small peaks ascribed to Cu and Ag and higher angle shift owing to interstitial site impurity incorporation were detected in the XRD pattern. The increase in the average crystallite size with increasing Ag amount was attributed to the replacement of greater size Ag ion (1.26 Å) with smaller size Zn ion (0.74 Å). In this study, the incorporation of Cu into the ZnO lattice was understood from the FTIR band shift. The method used to synthesize Ag–Cu codoped ZnO material, reagents, substrate/surfactants, and the morphology of the synthesized material are also given in Table 4.
| Approach | Reagents | Morphology | Ref. | |||
|---|---|---|---|---|---|---|
| Host precursor | Dopant precursor | Solvent | Surfactant/substrate | |||
| a Acetate (CH3COO). | ||||||
| Solvothermal | Zn(NO3)2·6H2O | Ce(NH4)2(SO4)4·2H2O | Water | Oxalic acid dihydrate | Hexagonal | 18 |
| AgNO3 | ||||||
| Sol–gel | Zn(aca)2·2H2O | Cu(NO3)2·5H2O AgNO3 | Methanol | Tartaric acid | Quasi-spherical (Cu–ZnO), spherical (Ag@Cu–ZnO) | 46 |
| Combustion | ||||||
| Hydrothermal | Zn(NO3)2·6H2O | Cu(NO3)2·3H2O triethanolamine | Water | Glycine (fuel) | Porous spherical | 63 |
| Sol–gel aided hydrothermal | Zn(aca)2·2H2O | Bi(NO3)3·5H2O | Ethyl alcohol | Cotton fabrics | Spherical | 79 |
| Zn(NO3)2·6H2O | Cu(NO3)2·3H2O | |||||
| Green (Acacia caesia) | ZnO powder | AgNO3, and CuNO3 | Water | Flower extract | Spherical Ag and Cu anchored on ZnO | 80 |
| Sol–gel | Zn(NO3)2·6H2O | Co(NO3)2·6H2O | Ethylene glycol | Citric acid | — | 81 |
| Fe(NO3)3·9H2O | ||||||
| Combustion | Zn(NO3)2·6H2O | Co(NO3)2·6H2O Mn(aca)2·4H2O | Water | Polyethylene glycol | Spherical | 82 |
| Hydrolysis | Zn(aca)2·2H2O | Nickel nitrate copper chloride | Water | Poly(vinyl alcohol) | Spherical | 83 |
| Sol–gel-aided hydrothermal | Zn(aca)2·2H2O | Ga(NO3)2·H2O | 2-Methoxyethanol | Monoethanolamine (stabilizer) | Hexagonal nanorods | 84 |
| ZnCl2 | AgNO3 | |||||
| One-pot hydrothermal | Zn(aca)2·2H2O | HAuCl4 | Water | — | Nanorods | 85 |
| Zn(NO3)2·6H2O | AgNO3 | |||||
3. Characterization techniques
A combination of characterization techniques should be applied for understanding the efficient doping of the dopant into the host lattice. The optical, compositional, crystalline phase change analyses are common to study the dopant characteristics. X-ray diffraction (XRD), UV-vis/DRS, and inductively coupled plasma-atomic emission spectroscopy or mass spectrometry (ICP-AES/MS) are normally used for understanding the change in the lattice parameters, bandgap modifications, and atomic percentage, respectively. The changes in the lattice parameters, such as contraction/expansion, which occurred due to the difference in the size of the host and the dopant, further creates a change in the electronic structure of the host materials. However, using the XRD pattern, the visualization of the evenly distributed doped materials is impossible. Besides, characterization techniques such as electron paramagnetic resonance spectroscopy (EPR), X-ray photoelectron spectroscopy (XPS), photoluminescence spectroscopy (PLS), and X-ray absorption (XAS), including X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure spectroscopy, are used for understanding the local coordination environment (chemical states), optical properties, and magnetic properties of the dopant.86,87 The near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) indicates the dopant substitution and bond distance between the dopant and the host, respectively.19 Since all elements have characteristics of core binding energy, EXAFS is used for analyzing the local structure of all the elements. In addition, the development of the atomic-resolution characterization technique helps in the precise imaging of dopant atoms' location and distribution.Advanced hyphenated instruments such as annular dark-field scanning transmission electron microscopy (ADF-STEM)22 and EELS-STEM62 provide the actual three-dimensional location and distribution, as well as element-specific analysis of the isolated impurities in the host matrix. ADF-STEM is an advanced instrument, which works by scanning the surface of the sample by focused electron beam and collecting the scattered electron by the annular dark-field detector. Higher atomic number impurities, which have different contrast compared to the host, can be detected as an image, which is difficult for small atomic number elements. Based on the characteristic core-edge electron-energy-loss signal, the single-atom sensitive EELS technique is also used to detect the dopant.22 Postica et al. used the high angle annular dark-field (HAADF)-STEM imaging technique for exact Ag-doped size measurement.21
3.1. STEM with different detectors
Controlling the position and distribution of doped light and heavy metal elements improves the device application potential. Besides, noticing the dopant-induced defects (point or/and extended) and heterogeneities89 gives information for tailoring the properties of the material. The 0D defects (dopants and vacancies), 1D defects (inclusion and dislocations), grain boundaries, and van der Waals gaps are the main dopant-induced structural disorders.90 This position, distribution, and disorder in materials, which affect the characteristics of the materials, can be identified by STEM analytical technique with atomic resolution.91 The working principle of the STEM technique is based on the idea of the angular selection of the scattered signal, in which the detector collects only the electrons scattered to large angles and avoids the Bragg reflections.92 As shown in Fig. 5(a), the various lenses assist the magnetic fields and the aberration corrector passes the beam to the EELS and other various modes of STEM detectors without affecting its energy. Fig. 5(b) show the electron beam monochromation, in which the transmitted beam is dispersed on the prism and detected on the EELS detector.88 The STEM technique detects both the small atomic number and large atomic number elements by annular dark-field (ADF-STEM) as bright spots and annular bright-field (ABF-STEM) as dark spots imaging, respectively. The BF-STEM has small-, middle-, and large-angle categories, in which the middle angle bright-field STEM (MABF-STEM) is used for detecting low atomic number elements with high accuracy.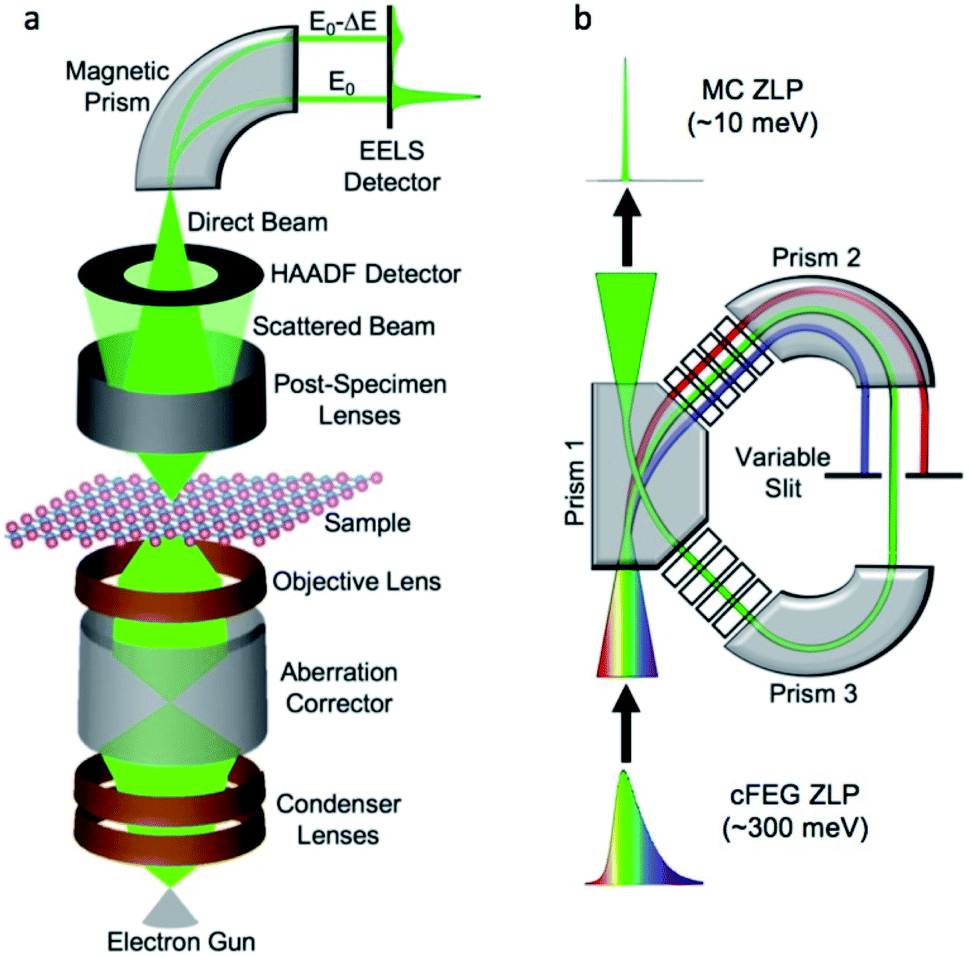 | ||
| Fig. 5 Electron energy loss spectroscopy and monochromation: (a) schematic of electron energy-loss spectroscopy (EELS) experiment in a scanning transmission electron microscope (STEM), (b) schematic of monochromation of an electron beam (occurring between the electron gun and the condenser lenses).88 | ||
Bazioti et al. used a STEM technique to validate the role of nitrogen doping in the evolution of point (vacancies and interstitials) and extended (stacking faults) defects.93 Multiple detectors (ABF, ADF, and HAADF) were used to simultaneously detect the chemical information and atomic structure of the material. The common HAADF detector correlated to the Z-contrast detects the Z-contrast image scattered from the doped heavy metals. The ADF detector gives information based on the strong diffraction contrast by selecting the angular range of the scattered electrons. Here, in the ADF detector, the existence of an extended defect is easily sensed. The BF detector can be used for the detection of light and heavy elements simultaneously by aligning the detector with all/part of the unscattered probe. The ABF detector operates by removing the circle from the center of the detector. The electronic and compositional information is collected using the EELS spectroscopic technique.93,94 Currently, the 4D-STEM modes of imaging detector, commonly known as scanning electron nanodiffraction, is an advanced technology used for crystal orientation mapping of any dopant. It gathers all types of real-space atomic-resolution images by collecting all diffraction datasets from each electron probe with high-speed and efficient detection.95
In Tang et al.’s study, the in situ substitution and distribution of niobium on the surface of transition metal dichalcogenides for the modification of the electronic structure was clearly visualized in the HAADF-STEM images.96 Strontium dopant-induced tetragonal phase to cubic phase structural transition on barium titanate was confirmed from powder XRD analysis.97 This solid result obtained from the XRD pattern was further confirmed from HAADF-STEM, in which the phase change results from the substantial compositional and microstructural inhomogeneities. Wen et al. detected both the low Z-C atoms and high Z-Mo atoms simultaneously by the ADF-STEM techniques. Remarkably, the 2D pixelated electron detector was used here to capture the low Z-number atoms convergent beam electron diffraction patterns in a 4D STEM geometry. From the ADF (LAADF), BF, and ABF STEM modes, the atomic sites in bright contrast to MoS2 and WS2 and the holes in dark contrast due to the carbon-based contamination, which result from either organic solvent residue or microscope vacuum, were clearly identified.98
The effect of transition metal dopant-induced oxygen vacancies on the photocatalytic activities was reported by Kim et al.99 Cr doping in the ZnO matrix showed a significant positive effect on the photocatalytic degradation of 4-chlorophenol and formic acid contaminates, whereas a negative result was seen for Co doping. As understood from the multimodal characterization techniques (STEM-EDX and E STEM-EELS), the occurrence of Cr+3, which has a charge mismatch with ZnO, induces higher vacancies in the ZnO crystal. The charge mismatch between two transition metal ions boosts the charge transfer properties. The doping of bismuth (Bi) in the tin oxide (SnOx) for fascinating catalytic properties was also reported.101 The Bi dopant position and distribution in Bi–SnOx synthesized from SnCl2 and BiI3 precursors were investigated by HAADF-STEM and EDX-STEM techniques.
Crystallographic defects detection based on ADF-STEM one-class support vector machine were proposed by Guo et al.102 ZrO2 and bilayer (MoW)Te2 grown from bulk crystal were used for their experimental detection of point, line (in 2D), and boundary (in 3D) defects. HAADF-STEM image-based dopant-segregation induced atomic-scale fractures arising within the Al2O3 ceramic grain boundary core were also confirmed by Kondo et al.103 Loche et al. optimized the maximum (7.5%) lanthanum doping in the CeO2 matrix, which possesses an outstanding oxygen storage capacity. In this study, the dopant-induced oxygen vacancy stabilization, shape, and size of the NPs were optimized using a surfactant-mediated hydro-solvothermal synthetic approach. Fig. 6 shows the conventional TEM, HRTEM, aberration-corrected HAADF-STEM, and EDS-STEM image of La-doped CeO2. From these characterization techniques, the cubic shape and d-spacing confirm the material authenticity, homogeneous distribution of La, and amounts of La.100
 | ||
| Fig. 6 CTEM, AC-HRTEM, HAADF-AC-STEM, and AC-STEM-EDS mapping of the nominal 7.5 mol% La-doped CeO2 nanocubes: (A) CTEM image showing clusters of La-doped CeO2 nanocubes, (B) AC-HRTEM image showing a single crystalline La-doped CeO2 nanocube projected along the [001] zone axis, with the d-spacings for the (200) and (220) lattice planes being shown. Inset shows the corresponding 2D-FFT. (C) HAADF-AC-STEM image of the same nanocubes, showing the scan region imaged for EDS mapping, (D and E) EDS maps showing the distribution of Ce and La throughout the nanocubes, respectively. The EDS overall quantification indicates a molar percentage of Ce and La of 94.5% and 5.5%, respectively.100 | ||
4. Doped colloidal nanocrystal frameworks
In addition to doping, tuning the NMs into a subordinate structure to form colloidal nanoparticle clusters (CNCs) and then assembling to the colloidal nanocrystal frameworks (CNFs) allows the material to have a combination of properties, which did not exist in the original constituents. The secondary structure, which is organized by optimizing the surfactants, solvents, and reaction temperatures, also regulates the geometry and consequently becomes functional for chosen applications. Thus, a CNCs is an inorganic core, which is shaped following the nucleation and growth steps, stabilized by surfactants molecules in the solution.104 Similarly, CNFs are ordered arrangements of porous CNCs material containing both matter and void space.105,106 The CNFs may have (1) microporous pore size distribution such as MOFs and zeolites,107,108 (2) mesoporous,23,109 and macroporous distribution, which employs hard or soft templates to form the framework.During CNFs growth, first, the molecular precursors were decomposed in the presence of template-forming surfactants by stepwise heating at moderate temperatures. The direct calcination of the colloidal particles synthesized without the template-forming surfactant provides disordered nanoparticles, while it provides ordered nanoparticles if the surfactant is used. Subsequent room and different temperature aging (inorganic condensation/encapsulation), evaporation, and crystallization yield the ordered mesoporous inorganic framework. After thermal or chemical processing (chemicals such as CH2Cl2 (ref. 27)), the self-supporting properties of the CNFs are understood from an instrument such as SEM and grazing incidence small-angle X-ray scattering (GISAXS) analysis.105 Although, the surfactants stabilize the nanoparticle during synthesis, however, they protect the target NPs from the light source, which diminishes the material applications such as catalysis and bio-separation. Thus, post-assembly of the CNFs, calcination/chemical processing treatment is used to form well-structured and clean porous framework architecture stabilized by thermal fusion (Fig. 7).5,110 However, high-temperature treatment for purifying the CNFs assists the framework crosslinking or/and aggregation/agglomeration process, which increases the crystallite sizes.
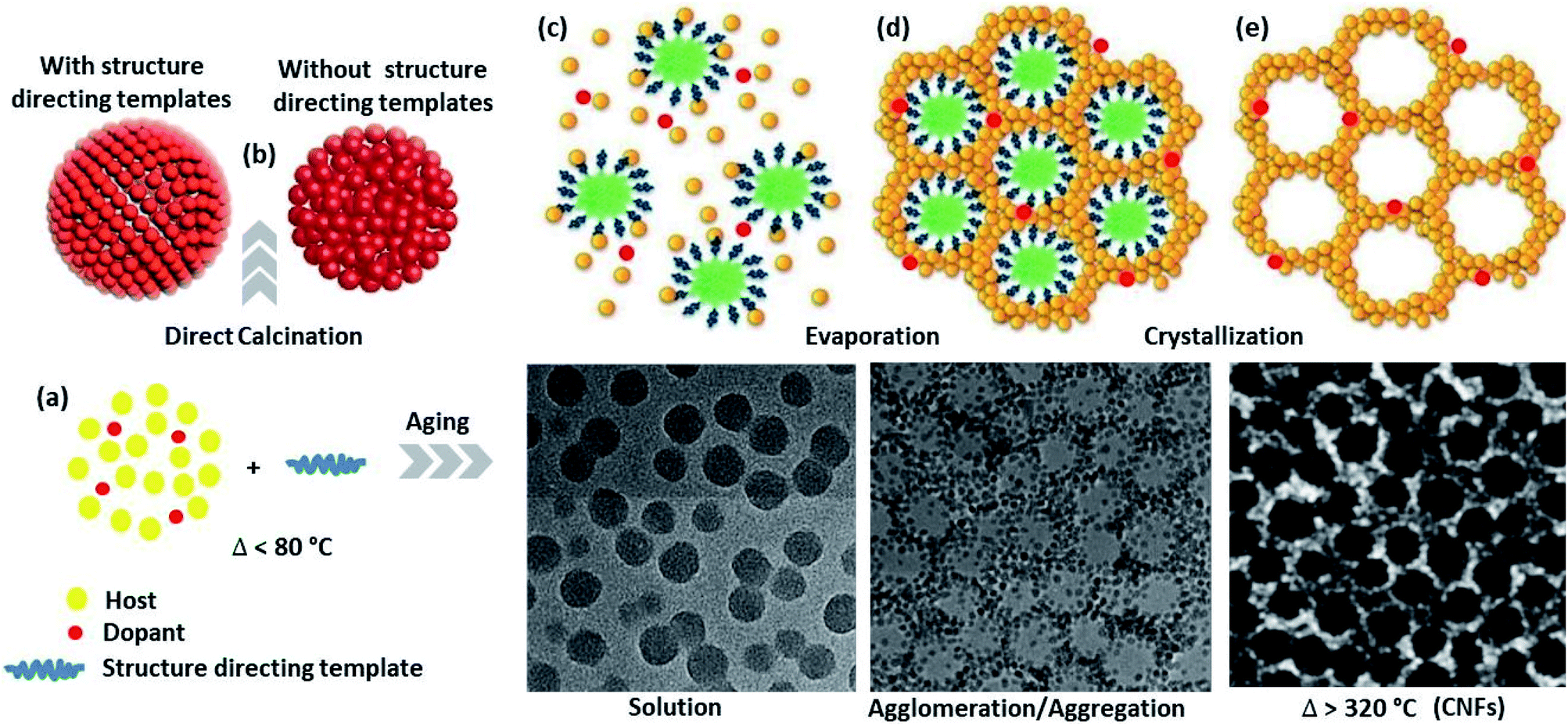 | ||
| Fig. 7 Colloidal nanocrystal frameworks formation steps: (a) nanocrystal formation by the decomposition of molecular precursors, (b) steps show an ordered nanocrystal formation in the presence of surfactant and disordered nanocrystal formation in the absence of surfactant, (c–e) steps show the condensation and evaporation upon heating at low temperature and crystallization upon calcination at higher temperature to produce an ordered porous framework. | ||
The frameworks can be synthesized either from molecular precursors or from the pre-formed CNCs, using the block copolymer architecture-directing agent (ADA) as nanocrystal tethering and a pore-generating domain.111 The chimie douce single molecular precursor's approach using zinc and copper ketoacidooximates precursor was applied by Pashchanka et al. to dope Cu ion into the ZnO host matrix.27 From different spectroscopic and microscopic examinations, the oxidation state of copper was found to be Cu(II).
Advanced methods such as sol–gel and solvo-/hydrothermal were reported112 to control the size, shape, dispersity, and geometry of the framework. During the synthesis, the interaction between the CNCs and ADA needs to be electrostatic rather than van der Waals or hydrogen bonding, which can resist thermal processing.113–115 The thermal treatment applied to transfer the precursor to active atomic or molecular species may go to up to 320 °C, and be grown to the NMs, then assembled into cluster. The layer-by-layer (LBL), liquid–liquid interface (LLI), and evaporation-induced self-assembly (EISA) are some of the approaches used to assemble the pre-formed NPs. Frequently, the EISA that follows the evaporation of the solvent in the presence of block copolymer utilizes crystalline NPs instead of molecular precursors.116,117 The LBL approach is the process of adsorption of the NPs on the surface of submicrometer beads.118 The LLI is formed at the interface of two immiscible liquids.119
The size can be controlled by changing the concentration of the ligands or precursors and stopping the growth at different steps, while the shape can be controlled by the selective adhesion of ligands to specific crystalline facets.104 Compared to ligand-coated NCs copolymer ADA interaction, the ligand-stripped NCs are suggested to avoid the stability issue.120 From a surface chemistry perspective, copolymers such as poly(4-vinyl pyridine) (P4VP) are promising toward organic–organic interaction,121 while the surface charge on the NCs and the type of the copolymer is reported104,106 to be the most important consideration for copolymer selection toward organic–inorganic interaction. For instance, an ordered mesoporous structure was synthesized by Buonsanti et al. using poly(N,N-dimethyl acrylamide) and porogenic polystyrene block copolymer structure-directing agents.114 During synthesis, different block copolymer ratios and molecular weights were taken to optimize the mesoporosity. The group used the reversible addition–fragmentation chain transfer polymerization ligand-stripped NCs approach.
The residual surface charge on molecular precursors (metal cation or/and hydroxyl-terminated metal oxide NCs) can be generated by solvothermal or sol–gel methods.106 Thermally heating the assembled composite led to the formation of hexagonal phase-segregated morphology and then well-ordered CNFs on further thermal processing.113 In this regard, the distinctive recyclability/stability and rate performance of metal oxide semiconductor-based CNFs make them a suitable choice.114
5. Applications
5.1. Bacterial activity
Nowadays, several conventional antibiotic resistant-bacteria have been emerging, leading to enhanced infections in human beings. Thus, searching for safe and effective antimicrobial agents for therapeutic and non-therapeutic purposes has been continuously encouraged. In recent decades, nanomaterials such as metal oxides have shown novel bactericidal potential. The antibacterial action is believed to have taken place when the negatively charged bacterial cells electrostatically interact with the positively charged metal oxides NPs.122 The cell wall of Gram-negative (GN) bacteria is complex and has a higher negative potential due to the presence of the lipopolysaccharide layer, compared to the Gram-positive (GP) bacteria.123 This complex structure and negatively charged behavior of the GN bacteria shield the negatively charged reactive oxygen species (ROS) and avoid their absorption.124The bactericidal mechanism of NPs is limited, although the direct and indirect interactions were reported as the main mechanisms.125 The direct mechanism occurs by the direct interaction of the positively charged NPs with negatively charged microorganisms.126 The indirect mechanism occurs through the interaction of the NPs with the intercellular space.127 The direct and indirect mechanism can damage the bacterial cell through the generation of ROS, by the release of ions, and NPs interaction with the cell membrane.128–130 The ROS generation is dependent on the interaction of the NPs with light that results in the generation of electrons and holes. The photo-induced electrons and holes further react with oxygen and water to form a highly oxidizing agent, hydroxyl radical (OH˙). Photocatalytic ROS generation under visible light irradiation for the inactivation of E. coli by Ag-doped ZnO has been reported in different works.132,133
The three important features that play a vital role in the generation of ROS are active redox cycling, the presence of pro-oxidant functional groups on the surface of NPs, and cell–particle interactions.134 The change in the electronic properties and reduction in the particle size produce reactive groups on the surface of the NPs. These reactive sites are the center of interaction between molecular oxygen and electron donor/acceptor active sites, which result in the formation of 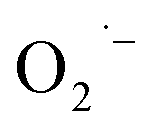 . The generated
. The generated 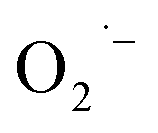 damage the iron–sulfur (Fe–S) clusters in the electron transport chain, releasing more ferrous ions, and these ferrous ions are oxidized by the Fenton reaction for generating moreOH˙.135,136 Antibacterial activity through the release of Ag+ ions from Ag–SiO2 NCs was proposed by Cui et al. As seen in Fig. 8, the electrostatic force of interaction between the positively charged NCs and negatively charged bacterial cells led to the release of Ag+ ions and transported them to the cytoplasm. The direct interaction of Ag+ with the mitochondria results in the generation of ROS by the Fenton reaction and inhibits DNA replication. After the interaction of the NCs and bacterial cells, the disruption of the membrane/cell wall and change in the bacterial cell morphology from cylindrical to spherical shape was also observed in the TEM analysis.131
damage the iron–sulfur (Fe–S) clusters in the electron transport chain, releasing more ferrous ions, and these ferrous ions are oxidized by the Fenton reaction for generating moreOH˙.135,136 Antibacterial activity through the release of Ag+ ions from Ag–SiO2 NCs was proposed by Cui et al. As seen in Fig. 8, the electrostatic force of interaction between the positively charged NCs and negatively charged bacterial cells led to the release of Ag+ ions and transported them to the cytoplasm. The direct interaction of Ag+ with the mitochondria results in the generation of ROS by the Fenton reaction and inhibits DNA replication. After the interaction of the NCs and bacterial cells, the disruption of the membrane/cell wall and change in the bacterial cell morphology from cylindrical to spherical shape was also observed in the TEM analysis.131
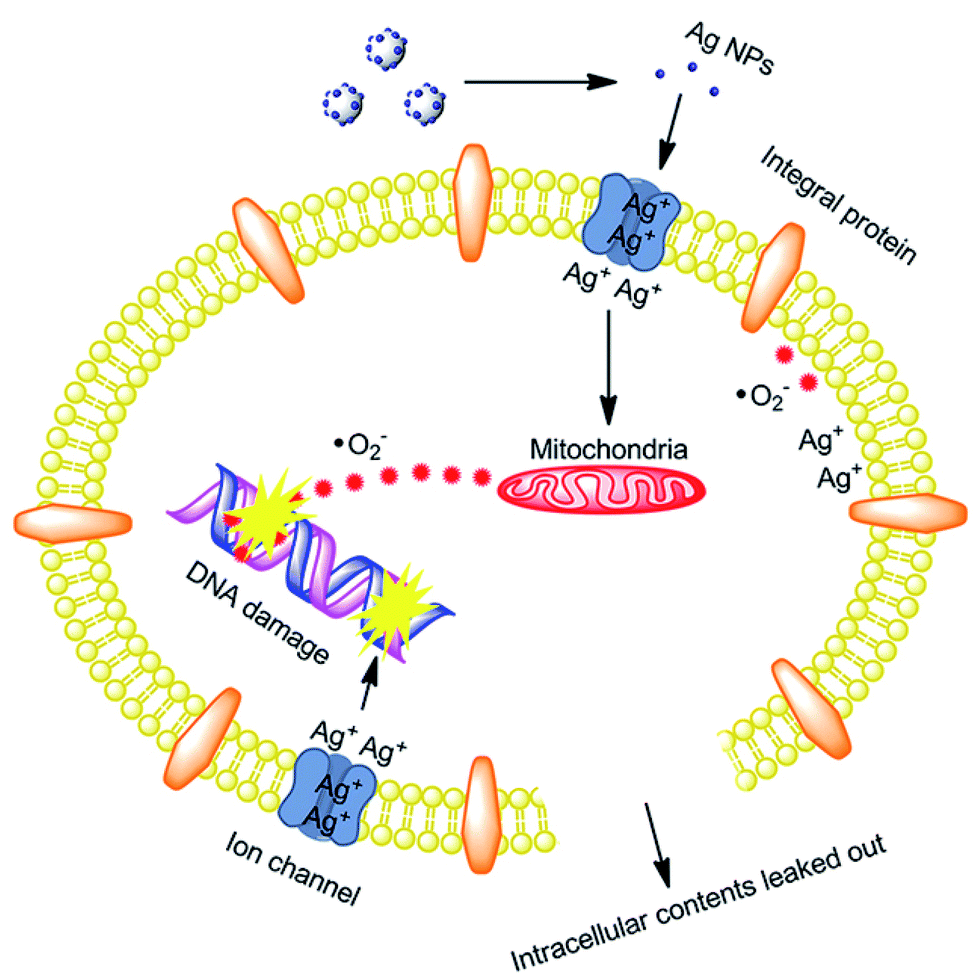 | ||
| Fig. 8 Schematic of the antibacterial mechanism of the SiO2–Ag composites.131 | ||
ZnO NPs have good antibacterial performance against both GP and GN bacteria. In addition, the antibacterial activities of copper and silver were also reported,137 although the bactericidal activities of Ag NPs would be diminished as a result of its oxidation and aggregation behavior.131 In Wang et al.’s study138 significant antibacterial activities of Ag-doped ZnO were reported, compared to the single ZnO NPs, which results from the synergistic effect of both Ag and ZnO. In this work, since no visible/UV light was applied during the antibacterial study, bacterial damage by the release of Zn+2/Ag+ ions or/and Ag/ZnO NPs interaction with cell membrane was proposed to be the probable mechanism for bacterial death. Similar synergistic effect antibacterial activities owing to the band structure modification through dopant inclusion were also proposed in Zare et al.’s139 studies. The disruption of the bacterial cell membrane at a cellular level and the inhibition of DNA/proteins at a molecular level by either Zn2+/Ag+ ions or ROS formation were proposed in Matai et al.’s140 study. Herein, cell membrane shrinkage due to disruption, followed by the leakage of intracellular materials, was clearly supported by FE-SEM, AFM, and TEM analysis. Hydrothermally synthesized Cu-doped ZnO by Khalid et al.141 also verified the synergistic antibacterial effect, especially on GP bacteria (results of structural differences among GP and GN bacteria) with a superior zone of inhibition. ROS generation by the Fenton-type reaction following the release of ions (Zn2+, Cu, Cu+, and Cu2+) is a probable mechanism for the bacterial inactivation/death in this study.
5.2. Photocatalytic activity
The rapid photo-induced electron–hole recombination and narrow light-absorption range are believed to diminish the photocatalytic efficiency of ZnO.142 The doping of metal reduces the electron–hole recombination by forming a Schottky junction, also improves the crystalline transformation, visible light absorption, and materials' surface area-to-volume ratio.143 Such doping also affects the optical, electrical, and magnetic properties. Yang et al. synthesized Ag-doped ZnO in the presence of higher surface area zeolitic imidazolate framework-8. The doped Ag metal acts as an electron capture site from the CB of ZnO and helps to improve the electron–hole separation, and this in turn enhances the photocatalytic degradation of rhodamine B dye.144 The electron capturing properties of Ag in Ag–ZnO resulted due to the establishment of the Ag/ZnO Fermi level between the CB and Fermi level of pure ZnO, which is known as a Schottky junction (more negative Fermi level potential for Ag–ZnO than bare ZnO).145,146The doping of copper also creates an electron acceptor level and modifies the ZnO crystal band structure, and it further enhances the visible light absorption efficiency of the material. Using hydrated zinc and copper acetate precursors in the presence of citrate and the dropwise addition of NaOH, Ma et al. synthesized porous Cu-doped ZnO NCs for MB dye degradation. The improved degradation behavior of Cu-doped ZnO than bare ZnO was reasonably explained as the occurrence of interfacial charge transfer from ZnO to Cu dopant (Fig. 9(c) and (d)). For single ZnO, after the excitation of the photo-induced electron to the CB, it directly returns to the VB and recombines with a hole (Fig. 9(c)). However, the excited electron on the Cu–ZnO nanocomposite would be accepted by Cu+2 and then reduced to Cu+, which further extends the life span of the hole in the VB (Fig. 9(d)).142 As a result, the reduction of oxygen by electron and oxidation of water by the hole occurred.
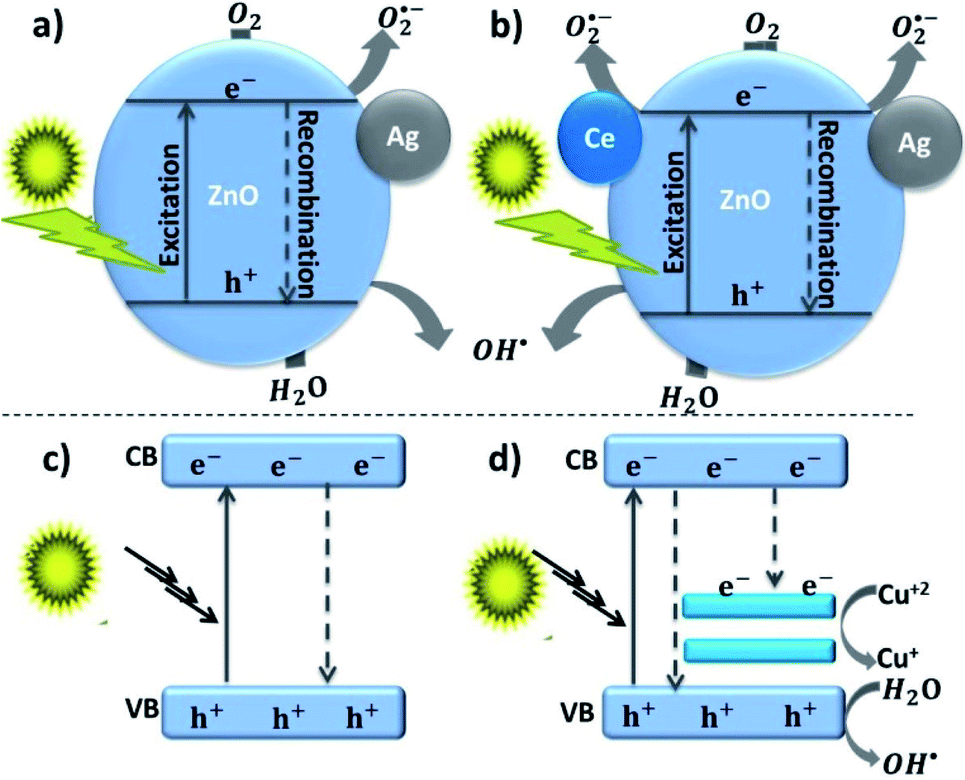 | ||
| Fig. 9 (a and b) Photocatalytic naphthol blue-black dye degradation mechanism of Ce–Ag co-doped ZnO,18 (c and d) photocatalytic methylene blue dye degradation mechanism of Cu-doped ZnO hierarchical nanostructures.142 | ||
Thus, an intermediate oxidizing agent and hydroxyl radical are generated.
To understand the detailed photocatalytic dye degradation mechanism, testing the existence of ROS and a free hole is important. Karthik et al.147 used isopropyl alcohol and 1,4-benzoquinone as a 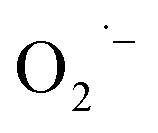 and OH˙ trapper, and ammonium oxalate as a hole trapper on the surface of Cu-doped ZnO NCs. In the presence of a hole trapper, greater than 85% dye degradation took place, whereas only about <20% degradation took place in the presence of ROS trapper. Hence, the ROS is the main degradation species of the dyes (MB, indigo carmine, and rhodamine B) compared to the hole under UV light irradiation. Here, in this work, the 3% and 5% dopant concentrations showed better degradation activity compared to the 7% and 9% doped NCs. The reason for the diminishing photocatalytic activities with an increase in the dopant amount is the development of CuO that covers the ZnO surface, which significantly shields the UV absorption capacity of the material. The development of CuO also acts as a photo-induced electron–hole recombination center. This shielding and recombination center properties of the developed CuO (doping above 5%) were also explained by Kuriakose et al.148 The improvement in the degradation of the MB dye with increasing Cu percentage up to 5% was reported by Okeke et al. Herein, the degradation on the doped nanocomposite resulted from the occurrence of ZnO bandgap shrinkage and developments of different types of defects in the ZnO matrix during Cu doping.149
and OH˙ trapper, and ammonium oxalate as a hole trapper on the surface of Cu-doped ZnO NCs. In the presence of a hole trapper, greater than 85% dye degradation took place, whereas only about <20% degradation took place in the presence of ROS trapper. Hence, the ROS is the main degradation species of the dyes (MB, indigo carmine, and rhodamine B) compared to the hole under UV light irradiation. Here, in this work, the 3% and 5% dopant concentrations showed better degradation activity compared to the 7% and 9% doped NCs. The reason for the diminishing photocatalytic activities with an increase in the dopant amount is the development of CuO that covers the ZnO surface, which significantly shields the UV absorption capacity of the material. The development of CuO also acts as a photo-induced electron–hole recombination center. This shielding and recombination center properties of the developed CuO (doping above 5%) were also explained by Kuriakose et al.148 The improvement in the degradation of the MB dye with increasing Cu percentage up to 5% was reported by Okeke et al. Herein, the degradation on the doped nanocomposite resulted from the occurrence of ZnO bandgap shrinkage and developments of different types of defects in the ZnO matrix during Cu doping.149
Simultaneously doping two metals generates special characteristics and results in a higher photodegradation activity.18 Subash et al. synthesized Ce and Ag co-doped ZnO superstructures for the photocatalytic degradation of naphthol blue-black dye under solar light irradiation. Both the adsorption and photocatalytic activities of Ag and Ce co-doped ZnO NCs are higher than those of bare ZnO NPs, Ce–ZnO, and Ag–ZnO NCs. This indicates that co-doping results in increased surface active sites by increasing the overall surface area and photocatalytic activity resulting from the interfacial charge transfer from ZnO to Ce and Ag dopant as shown in Fig. 9(a) and (b).18 MB dye degradation and bactericidal activities' improvement as a result of synergistic effect (Sn/Cu co-doped ZnO), interfacial charge transfer, and creation of extrinsic defects were also reported by Shanmugam et al.150 The five cycle recyclability test also proved the stability of the Sn/Cu-co-doped ZnO materials with an insignificant potential reduction in the 5th cycle. These Sn/Cu-co-doped ZnO materials synthesized by microwave-assisted coprecipitation approach also showed superior antibacterial activity against GN and GP bacteria. In addition to antibacterial and photocatalytic applications, tuning the materials to have an interconnected porous framework improves the mass- or ion-transport properties in several fields such as adsorption,151 capacitors/storage,152 electrocatalysis,153 windows,154 and sensors.155
6. Conclusions and future outlook
Nanotechnology is a fundamental field for producing advanced materials with interesting properties and unusual application potentials. Nanocrystals synthesized by common approaches have had essential applications for several years. Precise inclusion and good distribution of the impurities in the nanocrystal semiconductor materials further advance the material properties toward a particular application. For the precise inclusion and distribution of the dopant, controlling several conditions such as host–dopant and surfactant hardness, reaction time, and synthetic approach are significant. Common instruments (XRD, TEM, XAS, and XPS) can give information about the properties of the doped materials. In addition, advanced imaging modes of the STEM technique have been valued in direct atomic level defect and visualization of heterointerfaces and their impact on the properties of materials. Thus, the detection of the defects position, distribution, composition, and level of defects has received attention and should also be future viewpoints, especially in the case of in situ STEM control. In addition, forming a secondary structure, a colloidal nanocrystals framework can enhance the mass-/ion transport properties. Thus, tuning the crucial parameters under the control of advanced instruments and modifying the nanocrystals to the secondary structure should be the nonstop evolution for the future.Abbreviations
| HSAB | Pearson's hard/soft acids/bases |
| NMs | Nanoscale materials |
| ADF-STEM | Annular dark-field scanning transmission electron microscopy |
| EELS-STEM | Electron energy loss spectroscopy-scanning transmission electron microscope |
| XRD | X-ray diffraction |
| XPS | X-ray photoelectron spectroscopy |
| XAS | X-ray absorption |
| EXAFS | X-ray absorption fine structure |
| EXAFS | Extended X-ray absorption fine structure |
| ADA | Architecture-directing agent |
| CNCs | Colloidal nanoparticle clusters |
| CNFs | Colloidal nanocrystal frameworks; |
| ROS | Reactive oxygen species; |
| GP | Gram-positive |
| GN | Gram-negative |
| CB | Conduction band |
| VB | Valence band |
| MB | Methylene blue |
Author contributions
This review manuscript is written by Buzuayehu Abebe. The write-up improvement was done by H. C. Ananda Murthy. Both authors contributed to the study's conception and design.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Adama Science and Technology University.References
- T. A. Saleh, Environ. Technol. Innovation, 2020, 20, 101067 CrossRef CAS.
- R. Ebrahimi, K. Hossienzadeh, A. Maleki, R. Ghanbari, R. Rezaee, M. Safari, B. Shahmoradi, H. Daraei, A. Jafari, K. Yetilmezsoy and S. H. Puttaiah, J. Environ. Health Sci. Eng., 2019, 17, 479–492 CrossRef CAS PubMed.
- V. P. Dinesh, P. Biji, A. Ashok, S. K. Dhara, M. Kamruddin, A. K. Tyagi and B. Raj, RSC Adv., 2014, 4, 58930–58940 RSC.
- J. Zhang, Q. Di, J. Liu, B. Bai, J. Liu, M. Xu and J. Liu, J. Phys. Chem. Lett., 2017, 8, 4943–4953 CrossRef CAS PubMed.
- Z. Lu and Y. Yin, Chem. Soc. Rev., 2012, 41, 6874 RSC.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583 RSC.
- Y. Jun, J. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414–3439 CrossRef CAS PubMed.
- R. Buonsanti, V. Grillo, E. Carlino, C. Giannini, T. Kipp, R. Cingolani and P. D. Cozzoli, J. Am. Chem. Soc., 2008, 130, 11223–11233 CrossRef CAS PubMed.
- M. M. Rahman, H. B. Balkhoyor and A. M. Asiri, New J. Chem., 2019, 43, 18848–18859 RSC.
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef CAS PubMed.
- J. Gui, M. Ji, J. Liu, M. Xu, J. Zhang and H. Zhu, Angew. Chem., 2015, 127, 3754–3758 CrossRef.
- G. M. Dalpian and J. R. Chelikowsky, Phys. Rev. Lett., 2006, 96, 226802 CrossRef PubMed.
- F. V. Mikulec, M. Kuno, M. Bennati, D. A. Hall, R. G. Griffin and M. G. Bawendi, J. Am. Chem. Soc., 2000, 122, 2532–2540 CrossRef CAS.
- S. C. Erwin, L. Zu, M. I. Haftel, A. L. Efros, T. A. Kennedy and D. J. Norris, Nature, 2005, 436, 91–94 CrossRef CAS PubMed.
- D. Chen, R. Viswanatha, G. L. Ong, R. Xie, M. Balasubramaninan and X. Peng, J. Am. Chem. Soc., 2009, 131, 9333–9339 CrossRef CAS PubMed.
- R. Buonsanti and D. J. Milliron, Chem. Mater., 2013, 25, 1305–1317 CrossRef CAS.
- D. C. Agarwal, U. B. Singh, S. Gupta, R. Singhal, P. K. Kulriya, F. Singh, A. Tripathi, J. Singh, U. S. Joshi and D. K. Avasthi, Sci. Rep., 2019, 9, 6675 CrossRef CAS PubMed.
- B. Subash, B. Krishnakumar, R. Velmurugan, M. Swaminathan and M. Shanthi, Catal. Sci. Technol., 2012, 2, 2319 RSC.
- H. Liu, F. Zeng, S. Gao, G. Wang, C. Song and F. Pan, Phys. Chem. Chem. Phys., 2013, 15, 13153–13161 RSC.
- N. Ali, A. R. Vijaya, Z. A. Khan, K. Tarafder, A. Kumar, M. K. Wadhwa, B. Singh and S. Ghosh, Sci. Rep., 2019, 9, 20039 CrossRef CAS PubMed.
- V. Postica, A. Vahl, D. Santos-Carballal, T. Dankwort, L. Kienle, M. Hoppe, A. Cadi-Essadek, N. H. de Leeuw, M.-I. I. Terasa, R. Adelung, F. Faupel and O. Lupan, ACS Appl. Mater. Interfaces, 2019, 11, 31452–31466 CrossRef CAS PubMed.
- A. A. Gunawan, A. Wills, A. Mkhoyan, M. G. Thomas and D. J. Norris, Microsc. Microanal., 2012, 18, 298–299 CrossRef.
- Y. Deng, J. Wei, Z. Sun and D. Zhao, Chem. Soc. Rev., 2013, 42, 4054–4070 RSC.
- H. Hu, H. He, J. Zhang, X. Hou and P. Wu, Nanoscale, 2018, 10, 5035–5046 RSC.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- B. Abebe, E. A. Zereffa and H. C. A. Murthy, ACS Omega, 2021, 6, 954–964 CrossRef CAS PubMed.
- M. Pashchanka, R. C. Hoffmann, A. Gurlo, J. C. Swarbrick, J. Khanderi, J. Engstler, A. Issanin and J. J. Schneider, Dalton Trans., 2011, 40, 4307 RSC.
- N. Pradhan, D. Goorskey, J. Thessing and X. Peng, J. Am. Chem. Soc., 2005, 127, 17586–17587 CrossRef CAS PubMed.
- S. K. Panda, S. G. Hickey, H. V. Demir and A. Eychmüller, Angew. Chem., Int. Ed., 2011, 50, 4432–4436 CrossRef CAS PubMed.
- S. Sarkar, N. S. Karan and N. Pradhan, Angew. Chem., 2011, 123, 6189–6193 CrossRef.
- Y. Yang, Y. Jin, H. He, Q. Wang, Y. Tu, H. Lu and Z. Ye, J. Am. Chem. Soc., 2010, 132, 13381–13394 CrossRef CAS PubMed.
- R. A. Gilstrap, C. J. Capozzi, C. G. Carson, R. A. Gerhardt and C. J. Summers, Adv. Mater., 2008, 20, 4163–4166 CAS.
- S. Il Choi, K. M. Nam, B. K. Park, W. S. Seo and J. T. Park, Chem. Mater., 2008, 20, 2609–2611 CrossRef.
- I. Bilecka, L. Luo, I. Djerdj, M. D. Rossell, M. Jagodič, Z. Jagličić, Y. Masubuchi, S. Kikkawa and M. Niederberger, J. Phys. Chem. C, 2011, 115, 1484–1495 CrossRef CAS.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- G. D. Moon, S. Ko, Y. Xia and U. Jeong, ACS Nano, 2010, 4, 2307–2319 CrossRef CAS PubMed.
- S. E. Wark, C.-H. Hsia and D. H. Son, J. Am. Chem. Soc., 2008, 130, 9550–9555 CrossRef CAS PubMed.
- B. J. Beberwyck, Y. Surendranath and A. P. Alivisatos, J. Phys. Chem. C, 2013, 117, 19759–19770 CrossRef CAS.
- S. K. Singh, D. Dutta, A. Dhar, S. Das, M. C. Paul and T. K. Gangopadhyay, Phys. Status Solidi A, 2019, 216, 1900141 CrossRef.
- S. M. Hosseini, I. A. Sarsari, P. Kameli and H. Salamati, J. Alloys Compd., 2015, 640, 408–415 CrossRef CAS.
- C. Karunakaran, V. Rajeswari and P. Gomathisankar, J. Alloys Compd., 2010, 508, 587–591 CrossRef CAS.
- Ö. A. Yıldırım, H. E. Unalan and C. Durucan, J. Am. Ceram. Soc., 2013, 96, 766–773 CrossRef.
- Y. Jin, Q. Cui, K. Wang, J. Hao, Q. Wang and J. Zhang, J. Appl. Phys., 2011, 109, 053521 CrossRef.
- Y. Ning, Z. Zhang, F. Teng and X. Fang, Small, 2018, 14, 1703754 CrossRef PubMed.
- S. Kaenphakdee, S. Yodyingyong, J. Leelawattanachai, W. Triampo, N. Sanpo, J. Jitputti and D. Triampo, Mater. Sci. Forum, 2020, 1007, 143–147 Search PubMed.
- A. Modwi, K. K. Taha, L. Khezami, M. Boudina, M. Khairy, O. K. Al-Duaij and S. Talab, Z. Phys. Chem., 2021, 235, 745–767 CrossRef.
- Y. Liu, Q. Zhang, M. Xu, H. Yuan, Y. Chen, J. J. Zhang, K. Luo, J. J. Zhang and B. You, Appl. Surf. Sci., 2019, 476, 632–640 CrossRef CAS.
- S. A. Ansari, M. M. Khan, M. O. Ansari, J. Lee and M. H. Cho, J. Phys. Chem. C, 2013, 117, 27023–27030 CrossRef CAS.
- F. A. Alharthi, A. A. Alghamdi, N. Al-Zaqri, H. S. Alanazi, A. A. Alsyahi, A. El Marghany and N. Ahmad, Sci. Rep., 2020, 10, 20229 CrossRef CAS PubMed.
- S. Masoumi, E. Nadimi and F. Hossein-Babaei, Phys. Chem. Chem. Phys., 2018, 20, 14688–14693 RSC.
- S. Sagadevan, K. Pal, Z. Z. Chowdhury and M. E. Hoque, J. Sol-Gel Sci. Technol., 2017, 83, 394–404 CrossRef CAS.
- Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei and J. Zhu, J. Phys. Chem. C, 2008, 112, 10773–10777 CrossRef CAS.
- Z. Yin, X. Wang, F. Sun, X. Tong, C. Zhu, Q. Lv, D. Ye, S. Wang, W. Luo and Y. Huang, Sci. Rep., 2017, 7, 12206 CrossRef PubMed.
- S. T. Kochuveedu, Y. H. Jang and D. H. Kim, Chem. Soc. Rev., 2013, 42, 8467 RSC.
- Y. Wei, J. Kong, L. Yang, L. Ke, H. R. Tan, H. Liu, Y. Huang, X. W. Sun, X. Lu and H. Du, J. Mater. Chem. A, 2013, 1, 5045–5052 RSC.
- R. J. V. Michael, B. Sambandam, T. Muthukumar, M. J. Umapathy and P. T. Manoharan, Phys. Chem. Chem. Phys., 2014, 16, 8541 RSC.
- A. Ziashahabi, M. Prato, Z. Dang, R. Poursalehi and N. Naseri, Sci. Rep., 2019, 9, 11839 CrossRef PubMed.
- Q. Zhang, G. Xie, M. Xu, Y. Su, H. Tai, H. Du and Y. Jiang, Sens. Actuators, B, 2018, 259, 269–281 CrossRef CAS.
- O. Lupan, V. Cretu, V. Postica, M. Ahmadi, B. R. Cuenya, L. Chow, I. Tiginyanu, B. Viana, T. Pauporté and R. Adelung, Sens. Actuators, B, 2016, 223, 893–903 CrossRef CAS.
- J. Ding, J. Zhu, P. Yao, J. Li, H. Bi and X. Wang, Ind. Eng. Chem. Res., 2015, 54, 8947–8953 CrossRef CAS.
- B. Abebe, H. A. Murthy and E. Amare, Environ. Nanotechnol. Monit. Manag., 2020, 14, 100336 Search PubMed.
- M. D. Rossell, Q. M. Ramasse, S. D. Findlay, F. Rechberger, R. Erni and M. Niederberger, ACS Nano, 2012, 6, 7077–7083 CrossRef CAS PubMed.
- R. Gupta, N. KrishnaRao Eswar, J. M. Modak and G. Madras, RSC Adv., 2016, 6, 85675–85687 RSC.
- Y. Gao, F. Meng, X. Li, J. Z. Wen and Z. Li, Catal. Sci. Technol., 2016, 6, 7800–7811 RSC.
- K. Nagaveni, G. Sivalingam, M. Hegde and G. Madras, Appl. Catal., B, 2004, 48, 83–93 CrossRef CAS.
- P. Singh, R. Kumar and R. K. Singh, Ind. Eng. Chem. Res., 2019, 58, 17130–17163 CrossRef CAS.
- S. S. Alias, A. B. Ismail and A. A. Mohamad, J. Alloys Compd., 2010, 499, 231–237 CrossRef CAS.
- O. M. Lemine, T. Almusidi, M. B. Kanoun, S. Goumri-Said, M. Alshammari, N. Abdel All, A. Z. Alanzi, F. S. Alghamdi and A. Alyamani, J. Mater. Sci.: Mater. Electron., 2019, 30, 19833–19840 CrossRef CAS.
- A. Mahmoud, M. Echabaane, K. Omri, J. Boudon, L. Saviot, N. Millot and R. Ben Chaabane, Molecules, 2021, 26, 929 CrossRef CAS PubMed.
- Y.-C. Chang, P.-S. Lin, F.-K. Liu, J.-Y. Guo and C.-M. Chen, J. Alloys Compd., 2016, 688, 242–251 CrossRef CAS.
- C. Xu, K. Yang, L. Huang and H. Wang, J. Chem. Phys., 2009, 130, 124711 CrossRef PubMed.
- H. Liu, Y. Wang, J. Wu, G. Zhang and Y. Yan, Phys. Chem. Chem. Phys., 2015, 17, 9098–9105 RSC.
- S. Chomean, S. Ingkananth, M. Kiatchaipar and C. Kaset, Anal. Chim. Acta, 2017, 1180, 338884 CrossRef PubMed.
- S. Thakur and S. K. Mandal, Mater. Adv., 2021, 2, 511–524 RSC.
- R. Bhardwaj, A. Bharti, J. P. Singh, K. H. Chae and N. Goyal, Nanoscale Adv., 2020, 2, 4450–4463 RSC.
- D. H. Xu and W. Z. Shen, J. Phys. Chem. C, 2012, 116, 13368–13373 CrossRef CAS.
- X. Luo, L. T. Tseng, W. T. Lee, T. T. Tan, N. N. Bao, R. Liu, J. Ding, S. Li, V. Lauter and J. B. Yi, Sci. Rep., 2017, 7, 6341 CrossRef CAS PubMed.
- J. Zhang, K. Tse, M. Wong, Y. Zhang and J. Zhu, Front. Phys., 2016, 11, 117405 CrossRef.
- L. Cao, L. Wang, L. Xu, Y. Shen, M. Xie and H. Hao, RSC Adv., 2021, 11, 29416–29425 RSC.
- G. Manjari, S. Saran, S. Radhakrishanan, P. Rameshkumar, A. Pandikumar and S. P. Devipriya, J. Environ. Manage., 2020, 262, 110282 CrossRef CAS PubMed.
- J. J. Beltrán, J. A. Osorio, C. A. Barrero, C. B. Hanna and A. Punnoose, J. Appl. Phys., 2013, 113, 17C308 CrossRef.
- E. Pragna, M. Ramanadha, A. Sudharani and K. S. Kumar, J. Supercond. Novel Magn., 2021, 34, 1507–1516 CrossRef CAS.
- R. Priya, P. Sahay, N. Saxena, P. Rajput, V. Chawla, R. Sharma, O. P. Sinha and R. Krishna, J. Mater. Sci.: Mater. Electron., 2021, 32, 2011–2025 CrossRef CAS.
- A. GuruSampath Kumar, X. Li, Y. Du, Y. Geng and X. Hong, Appl. Surf. Sci., 2020, 509, 144770 CrossRef CAS.
- V. Fauzia, A. Yudiana, Y. Yulizar, M. A. Dwiputra, L. Roza and I. Soegihartono, J. Phys. Chem. Solids, 2021, 154, 110038 CrossRef CAS.
- R. Beaulac, P. I. Archer, S. T. Ochsenbein and D. R. Gamelin, Adv. Funct. Mater., 2008, 18, 3873–3891 CrossRef CAS.
- R. W. Meulenberg, T. van Buuren, K. M. Hanif, T. M. Willey, G. F. Strouse and L. J. Terminello, Nano Lett., 2004, 4, 2277–2285 CrossRef CAS.
- J. A. Hachtel, A. R. Lupini and J. C. Idrobo, Sci. Rep., 2018, 8, 5637 CrossRef PubMed.
- A. Kumar, J. N. Baker, P. C. Bowes, M. J. Cabral, S. Zhang, E. C. Dickey, D. L. Irving and J. M. LeBeau, Nat. Mater., 2021, 20, 62–67 CrossRef CAS PubMed.
- H. Li, C. Yoo, T.-J. Ko, J. H. Kim and Y. Jung, Mater. Adv., 2022, 3, 1401–1414 RSC.
- Y. Kotaka, Appl. Phys. Lett., 2012, 101, 133107 CrossRef.
- J. Y. Zhang, J. Hwang, B. J. Isaac and S. Stemmer, Sci. Rep., 2015, 5, 12419 CrossRef PubMed.
- C. Bazioti, A. Azarov, K. M. Johansen, B. G. Svensson, L. Vines, A. Y. Kuznetsov and Ø. Prytz, J. Phys. Chem. Lett., 2019, 10, 4725–4730 CrossRef CAS PubMed.
- C. Ophus, Microsc. Microanal., 2019, 25, 563–582 CrossRef CAS PubMed.
- D. Reifsnyder Hickey and K. A. Mkhoyan, APL Mater., 2020, 8, 070902 CrossRef CAS.
- L. Tang, R. Xu, J. Tan, Y. Luo, J. Zou, Z. Zhang, R. Zhang, Y. Zhao, J. Lin, X. Zou, B. Liu and H. Cheng, Adv. Funct. Mater., 2021, 31, 2006941 CrossRef CAS.
- H. Jiang, J. Qi, D. Wu, W. Lu, J. Qian, H. Qu, Y. Zhang, P. Liu, X. Liu and L. Chen, Nano Res., 2021, 14, 4802–4807 CrossRef CAS.
- Y. Wen, C. Ophus, C. S. Allen, S. Fang, J. Chen, E. Kaxiras, A. I. Kirkland and J. H. Warner, Nano Lett., 2019, 19, 6482–6491 CrossRef CAS PubMed.
- Y.-H. Kim, S. Kim, K. Kim, C. Kim, J. H. Jang, Y.-M. Kim and H. Lee, J. Mater. Chem. A, 2020, 8, 25345–25354 RSC.
- D. Loche, L. M. Morgan, A. Casu, G. Mountjoy, C. O'Regan, A. Corrias and A. Falqui, RSC Adv., 2019, 9, 6745–6751 RSC.
- Q. Yang, Q. Wu, Y. Liu, S. Luo, X. Wu, X. Zhao, H. Zou, B. Long, W. Chen, Y. Liao, L. Li, P. K. Shen, L. Duan and Z. Quan, Adv. Mater., 2020, 32, 2002822 CrossRef CAS PubMed.
- Y. Guo, S. V. Kalinin, H. Cai, K. Xiao, S. Krylyuk, A. V. Davydov, Q. Guo and A. R. Lupini, npj Comput. Mater., 2021, 7, 180 CrossRef.
- S. Kondo, A. Ishihara, E. Tochigi, N. Shibata and Y. Ikuhara, Nat. Commun., 2019, 10, 2112 CrossRef PubMed.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- T. E. Williams, D. Ushizima, C. Zhu, A. Anders, D. J. Milliron and B. A. Helms, Chem. Commun., 2017, 53, 4853–4856 RSC.
- B. A. Helms, T. E. Williams, R. Buonsanti and D. J. Milliron, Adv. Mater., 2015, 27, 5820–5829 CrossRef CAS PubMed.
- M. O'Keeffe, Chem. Soc. Rev., 2009, 38, 1215 RSC.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- Z. Lu, M. Ye, N. Li, W. Zhong and Y. Yin, Angew. Chem., Int. Ed. Engl., 2010, 49, 1862–1866 CrossRef CAS PubMed.
- J. Yin, Y. Huang, S. Hameed, R. Zhou, L. Xie and Y. Ying, Nanoscale, 2020, 12, 17571–17589 RSC.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS PubMed.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- R. Buonsanti, T. E. Pick, N. Krins, T. J. Richardson, B. A. Helms and D. J. Milliron, Nano Lett., 2012, 12, 3872–3877 CrossRef CAS PubMed.
- I. E. Rauda, V. Augustyn, B. Dunn and S. H. Tolbert, Acc. Chem. Res., 2013, 46, 1113–1124 CrossRef CAS PubMed.
- A. Corma, P. Atienzar, H. García and J.-Y. Chane-Ching, Nat. Mater., 2004, 3, 394–397 CrossRef CAS PubMed.
- A. Deshpande, N. Pinna, B. Smarsly, M. Antonietti and M. Niederberger, Small, 2005, 1, 313–316 CrossRef CAS PubMed.
- D. E. Gómez, I. Pastoriza-Santos and P. Mulvaney, Small, 2005, 1, 238–241 CrossRef PubMed.
- Y. Lin, A. Böker, H. Skaff, D. Cookson, A. D. Dinsmore, T. Emrick and T. P. Russell, Langmuir, 2005, 21, 191–194 CrossRef CAS PubMed.
- A. Dong, X. Ye, J. Chen, Y. Kang, T. Gordon, J. M. Kikkawa and C. B. Murray, J. Am. Chem. Soc., 2011, 133, 998–1006 CrossRef CAS PubMed.
- Y. Zhao, K. Thorkelsson, A. J. Mastroianni, T. Schilling, J. M. Luther, B. J. Rancatore, K. Matsunaga, H. Jinnai, Y. Wu, D. Poulsen, J. M. J. Fréchet, A. Paul Alivisatos and T. Xu, Nat. Mater., 2009, 8, 979–985 CrossRef CAS PubMed.
- P. K. Stoimenov, R. L. Klinger, G. L. Marchin and K. J. Klabunde, Langmuir, 2002, 18, 6679–6686 CrossRef CAS.
- I. Kim, K. Viswanathan, G. Kasi, S. Thanakkasaranee, K. Sadeghi and J. Seo, Food Rev. Int., 2020, 1–29 CrossRef.
- T. Gordon, B. Perlstein, O. Houbara, I. Felner, E. Banin and S. Margel, Colloids Surf., A, 2011, 374, 1–8 CrossRef CAS.
- N. Thakur, P. Manna and J. Das, J. Nanobiotechnol., 2019, 17, 84 CrossRef PubMed.
- N. M. Zholobak, V. K. Ivanov and A. B. Shcherbakov, in Nanobiomaterials in Antimicrobial Therapy, Elsevier, 2016, pp. 419–450 Search PubMed.
- S. K. Kannan and M. Sundrarajan, Int. J. Nanosci., 2014, 13, 1450018 CrossRef CAS.
- B. Abebe, E. A. Zereffa, A. Tadesse and H. C. A. Murthy, Nanoscale Res. Lett., 2020, 15, 190 CrossRef CAS PubMed.
- B. Das, S. K. Dash, D. Mandal, T. Ghosh, S. Chattopadhyay, S. Tripathy, S. Das, S. K. Dey, D. Das and S. Roy, Arabian J. Chem., 2017, 10, 862–876 CrossRef CAS.
- S. Jiang, K. Lin and M. Cai, Front. Chem., 2020, 8, 1–5 CrossRef PubMed.
- J. Cui, Y. Liang, D. Yang and Y. Liu, Sci. Rep., 2016, 6, 21423 CrossRef CAS PubMed.
- S. Adhikari, A. Banerjee, N. K. Eswar, D. Sarkar and G. Madras, RSC Adv., 2015, 5, 51067–51077 RSC.
- Y. Zhang, X. Gao, L. Zhi, X. Liu, W. Jiang, Y. Sun and J. Yang, J. Inorg. Biochem., 2014, 130, 74–83 CrossRef CAS PubMed.
- A. Nel, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- S. Jagadeeshan and R. Parsanathan, Advanced Nanostructured Materials for Environmental Remediation, 2019, pp. 59–90 Search PubMed.
- A. Raghunath and E. Perumal, Int. J. Antimicrob. Agents, 2017, 49, 137–152 CrossRef CAS PubMed.
- W. Fang, C. X. Chaofa Xu, J. Zheng, G. Chen and K. Jiang, RSC Adv., 2015, 5, 39612–39619 RSC.
- L. Wang, Z. Z. Wang, Z. Z. Wang, C. Zhang, Y. Wu and H. Zheng, RSC Adv., 2021, 11, 33883–33889 RSC.
- M. M. Zare, K. Namratha, S. Alghamdi, Y. H. E. Mohammad, A. Hezam, M. M. Zare, Q. A. Drmosh, K. Byrappa, B. N. Chandrashekar, S. Ramakrishna and X. Zhang, Sci. Rep., 2019, 9, 8303 CrossRef PubMed.
- I. Matai, A. Sachdev, P. Dubey, S. Uday Kumar, B. Bhushan and P. Gopinath, Colloids Surf., B, 2014, 115, 359–367 CrossRef CAS PubMed.
- A. Khalid, P. Ahmad, A. I. Alharthi, S. Muhammad, M. U. Khandaker, M. R. I. Faruque, I. U. Din, M. A. Alotaibi and A. Khan, PLoS One, 2021, 16, e0251082 CrossRef CAS PubMed.
- Q. Ma, X. Yang, X. Lv, H. Jia and Y. Wang, J. Mater. Sci.: Mater. Electron., 2019, 30, 2309–2315 CrossRef CAS.
- K. S. Ahmad and S. B. Jaffri, Open Chem., 2018, 16, 556–570 CAS.
- X. Yang, L. Qiu and X. Luo, RSC Adv., 2018, 8, 4890–4894 RSC.
- R. Kumar, D. Rana, A. Umar, P. Sharma, S. Chauhan and M. S. Chauhan, Talanta, 2015, 137, 204–213 CrossRef CAS PubMed.
- R. S. Sabry, W. J. Aziz and M. I. Rahmah, J. Inorg. Organomet. Polym. Mater., 2020, 30, 4533–4543 CrossRef CAS.
- K. V. Karthik, A. V. Raghu, K. R. Reddy, R. Ravishankar, M. Sangeeta, N. P. Shetti and C. V. Reddy, Chemosphere, 2022, 287, 132081 CrossRef CAS PubMed.
- S. Kuriakose, B. Satpati and S. Mohapatra, Phys. Chem. Chem. Phys., 2015, 17, 25172–25181 RSC.
- I. Okeke, K. Agwu, A. Ubachukwu, M. Maaza and F. Ezema, J. Nanopart. Res., 2020, 22, 272 CrossRef CAS.
- V. Shanmugam and K. S. Jeyaperumal, Appl. Surf. Sci., 2018, 449, 617–630 CrossRef CAS.
- J. M. Yang, R. J. Ying, C. X. Han, Q. T. Hu, H. M. Xu, J. H. Li, Q. Wang and W. Zhang, Dalton Trans., 2018, 47, 3913–3920 RSC.
- X. Zhao, F. Gong, Y. Zhao, B. Huang, D. Qian, H.-E. Wang, W. Zhang and Z. Yang, Chem. Eng. J., 2020, 392, 123675 CrossRef CAS.
- W. Zhang, Z.-Y. Wu, H.-L. Jiang and S.-H. Yu, J. Am. Chem. Soc., 2014, 136, 14385–14388 CrossRef CAS PubMed.
- E. L. Runnerstrom, A. Llordés, S. D. Lounis and D. J. Milliron, Chem. Commun., 2014, 50, 10555–10572 RSC.
- H. Shiozawa, B. C. Bayer, H. Peterlik, J. C. Meyer, W. Lang and T. Pichler, Sci. Rep., 2017, 7, 2439 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |