
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid pollutant degradation by peroxymonosulfate via an unusual mediated-electron transfer pathway under spatial-confinement†
Siting Shaoa,
Jiahao Cuia,
Lina Lia,
Mingqi Wanga,
Peng Zhang a,
Jianguo Cuib,
Chun Hua and
Yubao Zhao*a
a,
Jianguo Cuib,
Chun Hua and
Yubao Zhao*a
aKey Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, Institute of Environmental Research at Greater Bay Area, Guangzhou University, 510006 Guangzhou, P. R. China. E-mail: ybzhao@gzhu.edu.cn
bBaotou Research Institute of Rare Earths, 014030 Baotou, P. R. China
First published on 11th February 2022
Abstract
Nano-confinement systems offer various extraordinary chemical/physical properties, due to the spatial restriction and the electronic interaction between the confined species and the surrounding medium. They are, therefore, providing rich opportunities for the design of efficient catalytic reaction systems for pollutant removal. Herein, a highly efficient mediated-electron transfer pathway is identified on a spatially-confined zero valent cobalt for abatement of the organic pollutants by PMS. The catalyst showed efficient catalytic performance in both batch and a flow reactor for degradation of various pollutants, e.g., a degradation reaction constant of 0.052 s−1 for sulfamethoxazole and 0.041 s−1 for BPA. Regulated by the spatial-confinement, a distinctive inverse relationship between PMS decomposition rate and the electron density of the pollutant molecule was experimentally substantiated, e.g., in the presence of the electron-rich sulfamethoxazole, PMS decomposed slower than that with BPA, while in the presence of electron deficient diphenhydramine, PMS decomposed faster than that with BPA. The unique reaction mechanism endows the spatially-confined cobalt with the capability of eliminating the priority pollutants in the complex water matrix with pervasive halide ions and natural organic matter (NOM) via PMS activation.
1. Introduction
Peroxymonosulfate (PMS) based advanced oxidation processes (AOPs) have drawn great attention as potential technology for abatement of the refractory organic pollutants in water and soil.1–6 Among various methods for the activation of PMS, cobalt catalysis was found to be an efficient approach, in which the reactive sulfate radicals (SO4˙−) and hydroxyl radicals (˙OH) were reckoned as the predominant reactive species.7–9 With the aim of efficient water remediation, various cobalt based catalysts were designed; and, notably, the nano-structure/coordination configuration of cobalt species were found to be sensitive to the catalytic performance for PMS activation. The understanding on the insights of PMS activation mechanism by cobalt species consequently underwent constant evolution. On the cobalt nanoparticles, PMS was reductively activated, generating SO4˙− and ˙OH species which are predominant reactive species for pollutants degradation.10 While in the cases of single-atom cobalt catalysts with Co–Nx configurations, singlet oxygen (1O2) was identified to be the predominant reactive species from PMS activation.11,12 Nevertheless, SO4˙− and ˙OH were identified to contribute predominantly to the pollutant degradation in the reaction system wherein PMS was activated by pyridine-coordinated Co single atoms embedded in a polyaromatic macrostructure.13 In Co(II) catalyzed PMS activation process, high-valent cobalt-oxo species, Co(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, was proposed to be active for methyl phenyl sulfoxide oxidation.14 Concurrently, Co(II)-PMS complex (Co2+–OOSO3−) was proposed to be the predominant reactive species according to the analysis on its capability of conducting both one-electron-transfer and oxygen-atom-transfer reactions.15
O, was proposed to be active for methyl phenyl sulfoxide oxidation.14 Concurrently, Co(II)-PMS complex (Co2+–OOSO3−) was proposed to be the predominant reactive species according to the analysis on its capability of conducting both one-electron-transfer and oxygen-atom-transfer reactions.15
PMS activation mechanism varies, and the performance of the reactive species is sensitively impacted by the complexity in the water matrix as well.16–24 For instance, the radical pathways involve highly oxidative SO4˙− and ˙OH (E0 (SO4˙−/SO42−) = 2.6–3.1 V vs. NHE, E0 (˙OH/OH−) = 1.9–2.7 V vs. NHE), and the reaction systems are consequently vulnerable to the natural organic matters (NOMs), and the halide ions;17,25–27 the singlet oxygen has relatively low oxidation power (E0 (1O2/O2˙−) = 0.81 V vs. NHE), therefore presenting substrate-specific reactivity, such as the minimal interference from the NOMs, and selectivity towards the biomolecules.28–31 Therefore, it is highly desirable to control the PMS activation pathways, so as to meet the specific requirements in a real water treatment scenario. It is, however, challenging, due to the fact that the current understanding on the PMS activation mechanism is still insufficient and controversial.16,25 Further understanding on the structural dependence of the reaction mechanism is highly desirable.
Due to the spatial restriction and the electronic interaction between the confined species with the surrounding medium. Spatial-confinement system offers various extraordinary chemical/physical properties.32,33 Pan et al. experimentally substantiated that the Fenton reaction, an archetypal radical reaction, proceeded via a novel non-radical pathway with singlet oxygen as the major reactive species under nanoconfinement.34 The strategy of nanoconfinement is offering rich opportunities to the design of efficient catalytic reaction system for pollutants removal.35–37 By various nanostructure engineering strategies, the radical and singlet oxygen involved reaction pathways are hitherto realized in the cobalt catalyzed PMS activation reaction systems.10–12 The mediated-electron transfer reaction pathway, which usually governs the carbon and noble metals catalyzed reaction systems, is sporadically mentioned in the cobalt based catalysis system. Herein, an efficient pollutants degradation reaction system was developed based on the strategy of spatial-confinement, and an unusual mediated-electron transfer mechanism on the spatially confined Co nanocrystal is demonstrated.
2. Experimental section
2.1 Catalysts synthesis
20 grams of 2-cyanoguanidine was dissolved in 100 mL H2O in a reflux condensation system. 9.88 g Co(NO3)2·6H2O (8.487 mmol) was added in the solution and refluxed until dissolution, followed by adding 17.6 mL formaldehyde solution (36–38 wt%). The mixture was refluxed for 20 h to obtain dicyandiamide–formaldehyde polymer with Co2+ coordinated in the framework. The mixture was then heated on a hot-plate for removing water. Dried powder was calcined at 900 °C in argon flow, and the temperature ramping rate was 2.5 °C min−1. The as obtained sample was then pulverized and refluxed in 1 M sulfuric acid at 80 °C for 12 h to remove the cobalt particles outside the carbon nanotube. Due to the changed redox properties of the metals encapsulated inside the curved surface, the spatially-confined cobalt crystals were preserved in the tube after acid treatment.38–40 The sample was then washed until the filtrate is at circumneutral pH. The dried sample was Co-NC for catalytic reactions. Control sample without Co (denoted by NC) was synthesized in the absence of Co(NO3)2·6H2O via otherwise the same method to that for Co-NC. Co metal powder (denoted by Co-P) was purchased from Mecklin with purity of 99.9% and particle size of 300 mesh. Co3O4 was purchased from Mecklin as a bench mark catalyst.2.2 Batch and flow reaction procedures
Unless otherwise specified, all the batch reactions were conducted in a 100 mL reactor; the catalyst loading was 0.15 g L−1, PMS concentration was 1 mM, and pollutants concentration was 20 ppm. In a typical batch reaction experiment, 7.5 mg Co-NC was dispersed in 50 mL BPA solution (20 ppm), and the reaction was started by adding 1 mL PMS (50 mM) solution. The suspension was magnetically stirred and sampled at specified time intervals; the samples were filtered and mixed with equal volume of methanol for analysis on HPLC.2.3 Flow reaction conditions
7.5 mg Co-NC was mixed with silica sand and packed into a silica tube with inner diameter of 1 cm. The solution with 10 ppm BPA and 0.75 mM PMS was feed into the reactor at the flow rate of 96 mL h−1 by peristaltic pump. The effluent was sampled at specified interval and mixed with equal amount of methanol for analysis.The pollutants degradation processes were analyzed by Shimadzu SIL-20A HPLC with Shim-pack GIST C18 column (4.6 × 250 mm, 5 μm). Mobile phase and detection wavelength setting for the pollutants: bis phenol A (BPA), methanol/water (70/30) and λ = 225 nm; sulfamethoxazole, methanol/water (55/45) and λ = 266 nm; 2-chloro-phenol and diphenhydramine, phosphate acid solution (0.08%)/acetonitrile (50/50) and λ = 221 nm; phenytoin, phosphate acid solution (0.08%)/acetonitrile (60/40) and λ = 220 nm; carbamazepine, methanol/water (70/30) and λ = 285 nm; methyl phenyl sulfoxide (MPSO), phosphate acid solution (0.08%)/acetonitrile (70/30) and λ = 215 nm.
3. Results and discussion
3.1 Materials synthesis and characterizations
Co-NC is synthesized by coordinative dispersion of the Co2+ in the polymer of melamine and formaldehyde, followed by pyrolysis of the compound (Fig. 1a). The cobalt catalysis under high temperature leads to the growth of the nitrogen-doped carbon nanotube, wherein the cobalt nanocrystals is capsulated inside the dead-end of the multilayers carbon nanotube. As shown in the TEM image (Fig. 1b), the nanotube is around 50 nm in diameter, and the encapsulated cobalt crystal with size of 20–30 nm is obviously observed by TEM. HRTEM image clearly shows the lattice of cobalt and the interface between few layers carbon and the cobalt crystal. In the X-ray diffraction (XRD) pattern (Fig. 1c), the diffraction peaks at 2θ of 44.2°, 51.2° and 75.8° state that the cobalt nanocrystals are in face-centered cubic (fcc) structure.41 As determined by nitrogen physical adsorption method, BET surface area of Co-NC is 303.2 m2 g−1, and pore sized is distributed mainly at around 2–6 nm (Fig. S1†). As shown in the X-ray photo-electron spectra (Fig. S2†), the N 1s signal is deconvoluted into three peaks at 398.3 eV, 400.6 eV, and 403.5 eV, which are attributed to pyridinic N, pyridonic N, and pyridine-N-oxide N, respectively.42,43 The C 1s signal could be deconvoluted into three peaks at 284.4 eV, 285.1 eV, and 288.5 eV, which are attributed to the carbon atoms in sp2 configuration, C–O group, and the adventitious carbon, respectively.44 O 1s peaks are deconvoluted into two peaks at 530.1 and 532.1 eV, which are assigned to surface Co–O species and the oxygen atom in organic groups.45,46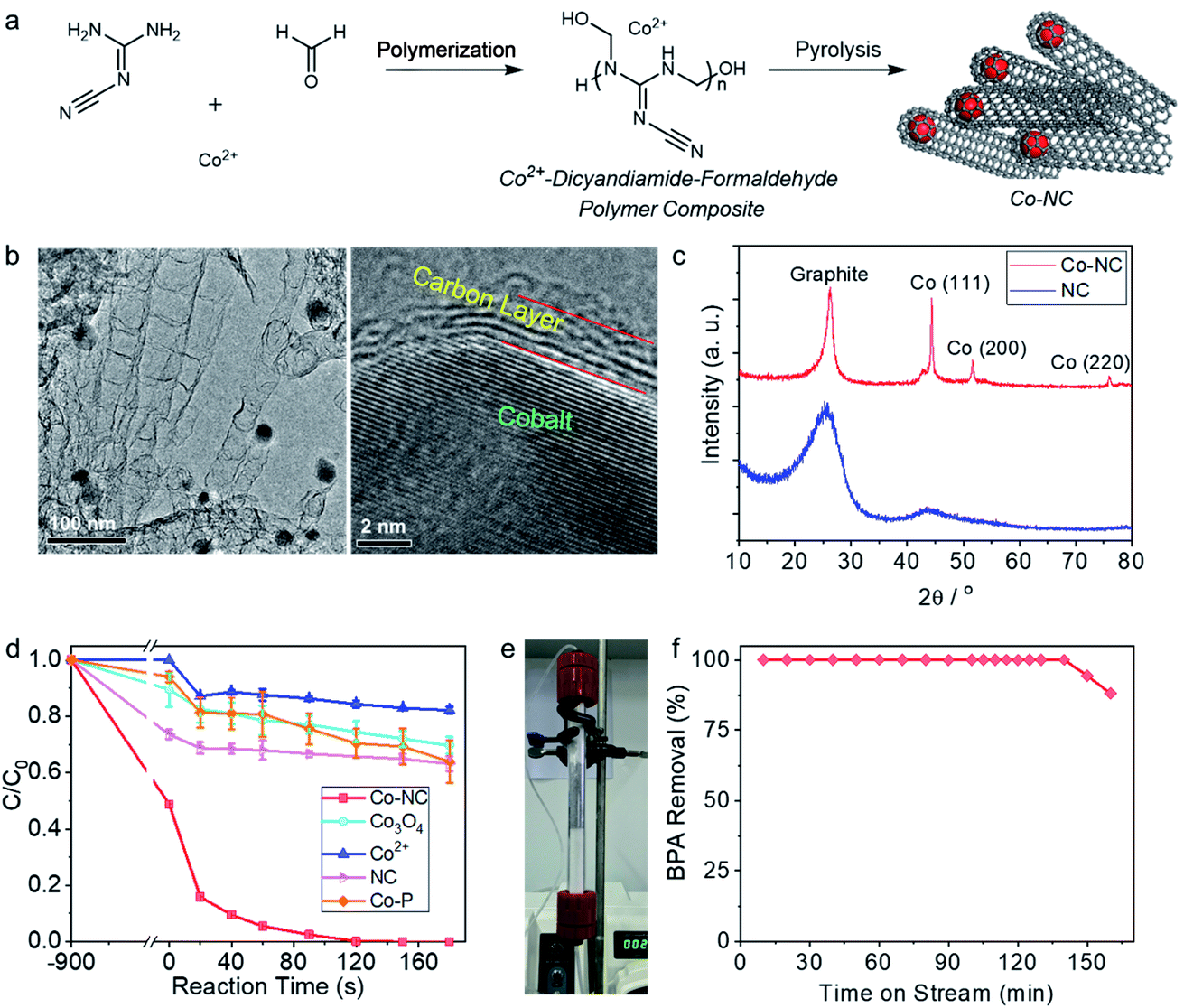 | ||
| Fig. 1 (a) Synthesis of Co-NC. (b) Transmission electron microscopy (TEM) images of Co-NC. (c) X-ray diffraction profiles of NC and Co-NC. (d) Degradation of bisphenol A (BPA) in the reaction systems with various catalyst. Reaction conditions: BPA, 20 ppm; PMS, 1 mM; initial pH, 6.5; temperature, 25 °C; catalyst loadings: Co2+, 0.4 mg L−1; Co3O4, NC, and Co-NC, 0.15 g L−1. (e) Photograph of the fixed bed flow reactor. (f) BPA removal performance in a fixed bed flow reactor with Co-NC as the catalyst. Reaction conditions: catalyst loading, 7.5 mg; BPA, 10 ppm; PMS, 0.75 mM; flow rate, 96 mL h−1. | ||
3.2 Pollutants degradation reaction performance
BPA, which is a commonly used plastic additive, but a pervasive endocrine disruptor in the aquatic systems, was employed as one of the target pollutants.47 Under optimized reaction conditions (Fig. S3†), 20 ppm BPA could be rapidly degraded within 120 s on Co-NC (0.15 g L−1) in the presence of 1 mM PMS, presenting a remarkable pseudo-first order reaction kinetic constant of 0.040 s−1 (Fig. 1d). The concentration of the leached Co2+ at 120 s was 0.26 ppm; and the catalytic performance of the leached Co2+ played a minor role in the overall catalytic reaction, due to the fact that 18% of the BPA was removed in the presence of 0.4 mg L−1 Co2+ in 180 s. Under the same reaction conditions, the reaction systems with Co3O4, a benchmark catalyst for PMS activation, showed only 25% BPA removal within 120 s. NC was synthesized without the addition of the cobalt ion via otherwise the same method to that for Co-NC, while the cobalt free NC showed poor catalytic efficiency, removing only 37% BPA in 180 s.Humic acid is a one of the typical natural organic matters (NOMs) in the natural water matrix, and the impact of the humic acid on the catalytic performance of Co-NC was thus investigated. As shown in Fig. S4a,† the presence of humic acid slightly impeded the BPA degradation performance; in the presence of 100 mg L−1 humic acid, >90% of BPA could be removed within 180 s reaction. It is worth noting that the presence of humic acid in the solution decreased the BPA absorption on Co-NC, e.g., 68.2 μg per mg Co-NC in the absence of humic acid and 58.0 μg per mg Co-NC in the presence of 100 ppm humic acid (Fig. S4b†); the surface adsorption of BPA on Co-NC is thus tentatively believed to be related to the degradation process. The mineral anions, which are commonly presenting in the natural water system, were added in the reaction system for checking their potential impacts on the pollutants degradation performance. BPA was efficiently eliminated in the presence of 5 mM of Cl−, CO32−, H2PO4−, and NO3− (Fig. S5†). Carbonate anion, in particular, showed relatively obvious negative impact on the BPA degradation reaction. Inspired by the case of humic acid influence, an investigation on the effect of CO32− in the BPA adsorption was conducted. Similarly, the presence of CO32− in the solution reduced the surface adsorption of BPA, e.g., the amount of BPA adsorbed on Co-NC in the presence of 5 mM CO32− and in the absence of CO32− was, respectively, 36.1 and 68.2 μg per mg Co-NC. We thus surmise that it is the surface catalytic reaction mechanism wherein the BPA surface adsorption property matters, rather than the free radical mechanism, that is governing the pollutant degradation process in this case. The mechanism will be analyzed in the following radical quenching experiments.
For further exploring the durability of Co-NC in the BPA degradation reaction, the catalytic performance of Co-NC was evaluated with a fixed-bed flow reactor (Fig. 1e). As shown in Fig. 1f, at flow rate of 12.8 L h−1 (for 1 g Co-NC), the reactor run efficiently for 140 min with 100% removal of BPA, and the adsorption accounts for a small part in the BPA removal (Fig. S6†). For the benchmark catalyst of Co3O4, there is no stable BPA degradation performance observed; and the Co-free NC deactivated in 50 min in the flow reactor (Fig. S7†).
The catalyst was then employed for degrading a couple of selected refractory organic pollutants concerned by the public, such as sulfamethoxazole, carbamazepine, phenytoin, ibuprophen, and diphenhydramine, which are pharmaceuticals, and 2-chloro-phenol, a representative organohalide pollutant (Fig. 2a).47 As shown in Fig. 2b and S8,† sulfamethoxazole and 2-chloro-phenol can be completely removed within 120 s. Diphenhydramine, phenytoin, and carbamazepine were also vulnerable to be rapidly degraded, and more than 80% removal could be achieved in 180 s. However, ibuprofen, with electron deficient molecular structure, was unable to be degraded in this reaction system. The selectivity towards the pollutants was indicative of a non-radical reaction mechanism.
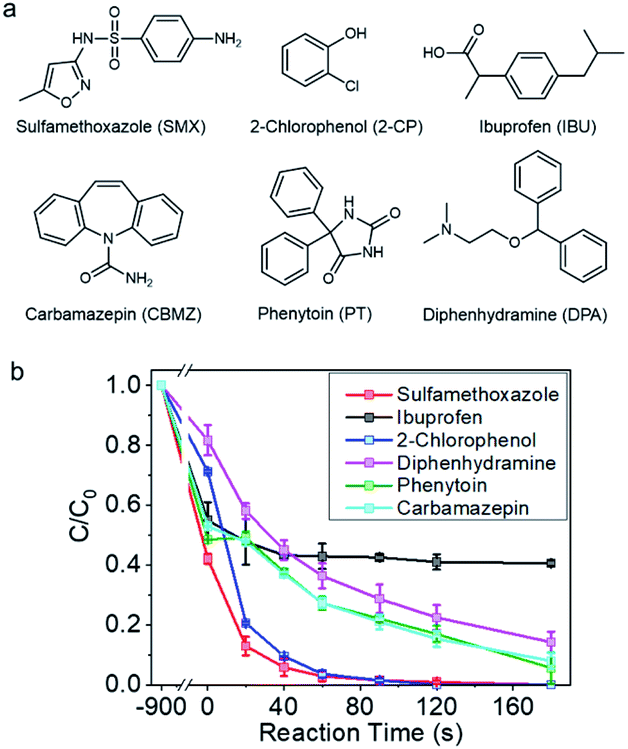 | ||
| Fig. 2 (a) The structures of selected refractory organic pollutants in water. (b) Catalytic degradation of various refractory organic pollutants by Co-NC. Reaction conditions: pollutants concentration, 20 ppm; PMS concentration, 1 mM; catalyst loading, 0.15 g L−1; initial pH, 6.5. | ||
3.3. Mechanistic insights
Methanol and tert-butyl alcohol (TBA) are capable of trapping the free radicals due to the rapid reaction rate, e.g., k (˙OH/CH3OH) = (1.2–2.8) ×109 M−1 s−1, k (SO4˙−/CH3OH) = (1.6–7.8) ×107 M−1 s−1, and k (˙OH/TBA) = (3.8–7.6) × 108 M−1 s−1.48,49 Methanol and TBA were thus employed to examine the contribution of the free radicals to the pollutant degradation process in Co-NC catalyzed reaction system. As shown in Fig. 3a, in the presence of 1 M and 2 M methanol (>1 × 105 times of BPA molar concentration), BPA degradation reaction rate is the same to that without methanol. In the presence of 0.2 M TBA, there was no obvious attenuation of the BPA degradation reaction rate, 20 ppm BPA was eliminated efficiently within 180 s; the BPA degradation reaction rate decreased slightly in the presence of 0.5 M TBA (>5 × 104 times of the BPA concentration). The radical quenching experiments demonstrated that the radical pathway played negligible role during BPA degradation.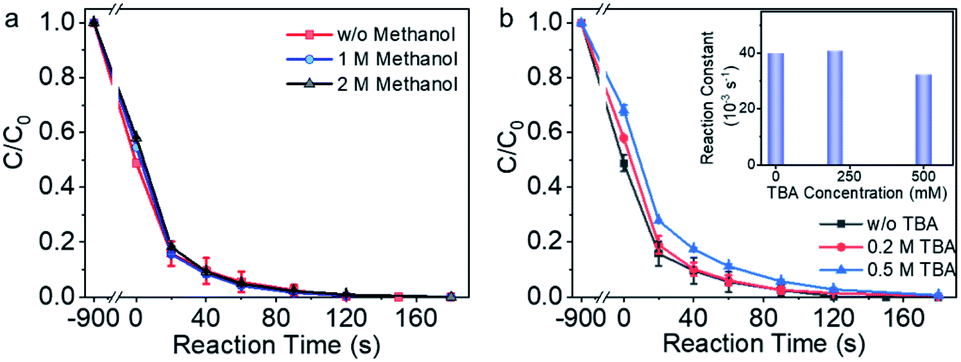 | ||
| Fig. 3 Analyzing the possibility of the radical reaction mechanism. (a) The impact of methanol on the BPA degradation performance. (b) BPA degradation performance in the presence and absence of tert-butyl alcohol (TBA). | ||
Electron spin resonance (ESR) was employed to detect the free radicals, such as SO4˙− and ˙OH, with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the spin-trapping agent.50 However, the concentration of the free radicals might be below the detection limit (Fig. S9†). The ESR trapping experiments, therefore, partially supported the aforementioned hypothetical non-radical reaction mechanism involving a critical step of BPA surface adsorption.
To further explore the possible contribution of singlet oxygen (1O2) to BPA degradation, L-histidine and 2,2,6,6-tetramethylpiperidine (TMP), which are trapping agent for 1O2, were added in the reaction system during BPA degradation.16,34,51 As shown in Fig. 4a, the addition of 5 mM (>50 times of BPA concentration) L-histidine and TMP showed no impact on the BPA degradation rate, and BPA was completely removed in 120 s, tentatively indicating that 1O2 may not contribute to BPA degradation process. For further confirm the role of 1O2 in the reaction system, solvent impact was investigated. The life-time of 1O2 is sensitive to the property of surrounding chemical environment, e.g., kd (H2O) (the decomposition reaction rate constants in H2O) is 2.5 × 105 s−1, kd (D2O) is 1.5 × 105 s−1, and kd (CH3OH) is 1.1 × 105 s−1.52 The BPA degradation performance, on condition that 1O2 was the predominant reactive species, should be higher in D2O and CH3OH than in H2O. However, the BPA degradation rate in D2O was close to that in H2O (Fig. 4b). With CH3OH as the solvent, BPA was degraded rapidly, although the reaction rate was slower than that in H2O and D2O. The experimental investigations on account of solvent-dependent 1O2 life time confirmed that 1O2 did not contribute to the BPA degradation reaction. Instead, the solvent-dependent BPA adsorption behavior on Co-NC was found to be positively related with BPA degradation; and the amount of surface adsorbed BPA per mg Co-NC in H2O, D2O, and CH3OH are 0.135, 0.153, and 0.107 mg, respectively. The above analysis is, repeatedly, indicative of a reaction mechanism with important BPA surface oxidation process.
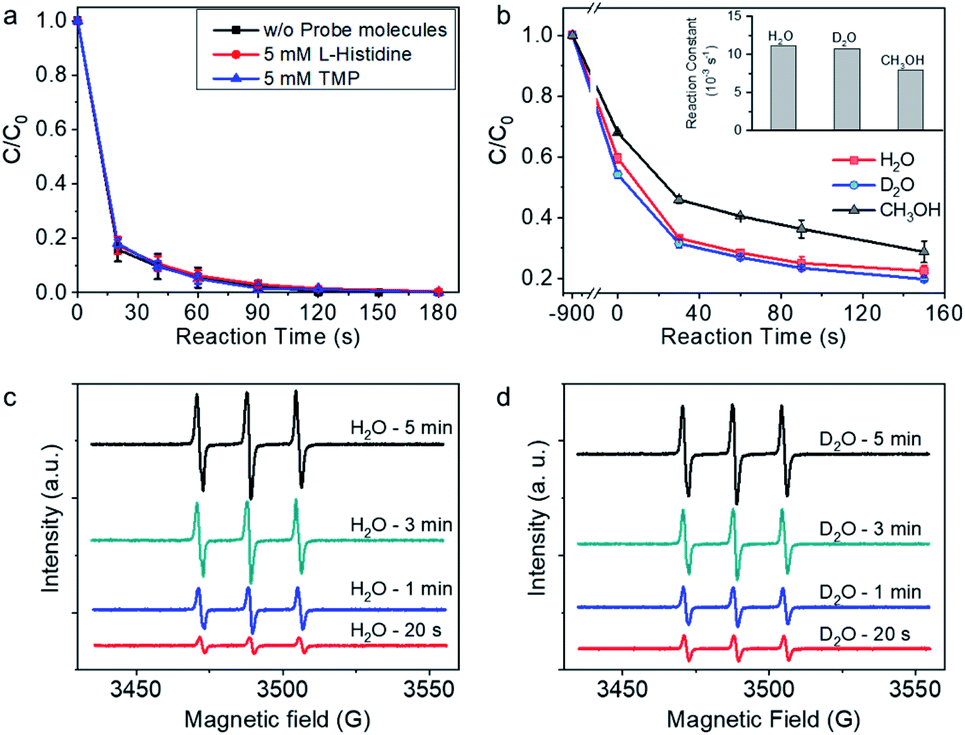 | ||
| Fig. 4 Analyzing the possible role of singlet oxygen (1O2) in the reaction system. (a) Impact of the L-histidine and 2,2,6,6-tetramethylpiperidine (TMP) on the BPA degradation performance. Reaction conditions: Co-NC, 0.15 g L−1; PMS, 1 mM; BPA, 20 ppm; probe molecules, 5 mM; solvent, H2O; initial pH, 6.5. (b) Solvent impact on the BPA degradation performance. Reaction conditions: Co-NC, 0.15 g L−1; PMS, 1 mM; BPA, 50 ppm; H2O, D2O, and CH3OH was respectively employed as solvent in each reaction; initial pH of the aqueous solution, 6.5. (c and d) Electron spin resonance spectra in H2O (c) and D2O (d) with TMP as the spin-trapping agent for 1O2. The scale of the intensity in (c) and (d) are the same. | ||
For further checking the possibility of 1O2 generation during PMS activation, ESR with 2,2,6,6-tetramethylpiperidine (TMP) as the spin-trapping agent for 1O2 was performed. As shown in Fig. 4c, the characteristic triplet resonance peaks (hyperfine split constant, 16.9 G; g factor, 2.0054), which are corresponding to 2,2,6,6-tetramethylpiperidine-N-oxide (TMPO), increased with reaction time. The appearance of TMPO signal was tentatively indicating the possible trapping of 1O2 by TMP.53,54 In lieu of H2O, D2O was used as the solvent for measurement, and intensified signals were expected as long as 1O2 was generated during PMS activation and trapped by TMP. However, the intensity of the spectra was close to that in H2O at the given reaction time, demonstrating that TMPO was produced via an alternative reaction mechanism, rather than 1O2 trapping by TMP.55 There is alternative pathway for TMP to TMPO conversion, which is initiated by one electron oxidation of TMP to TMP˙+. The active TMP˙+ radical cation then goes through the following deprotonation reaction as well as oxidation by dissolved oxygen, and finally converts to TMPO.55,56 In light of the experimental fact that no free radicals generated from PMS activation, it is plausible to proposed that the one-electron transfer process takes place on the surface adsorption site for TMP on Co-NC. The one-electron transfer step initiated TMPO generation on the surface is also indicative of the mediated-electron transfer mechanism.55
In lieu of PMS, peroxydisulfate (PDS) was employed for catalytic activation to degrade BPA. PDS has a more negative reduction potential (Eo (S2O82−/SO42−) = 2.01 V vs. NHE) than that of PMS, and the transition metals, such as Co, are selective for PMS activation.10,57 PDS activation are usually observed via a mediated electron transfer mechanism catalyzed by carbon materials and noble metals.58–61 With a spatial-confinement configuration, Co-NC exhibited a remarkable catalytic capability for BPA degradation via PDS activation, and 20 ppm BPA was eliminated in 2 min. As expected, ionic Co2+ showed negligible catalytic activity for PDS activation to degrade BPA, which is consistent with various reports (Fig. S10†).10,57 The capability of Co-NC in activation of both PMS and PDS, repeatedly, indicated the mediated-electron transfer mechanism.10,16,57
For further understanding the insights of electron transfer on the interfaces in PMS–Co-NC–BPA system, PMS decomposition behavior on Co-NC was investigated. As shown in Fig. 5a, PMS decomposed rapidly on Co-NC in the absence of BPA, and >70% of PMS decomposed within 10 min. While in the presence of 50 ppm BPA in the reaction system, the PMS decomposition reaction was significantly attenuated, and 41% PMS was depleted within 10 min. The impacts of the pollutants on the PMS decomposition behavior on Co-NC were further investigated in the presence of sulfamethoxazole and diphenhydramine, which, respectively, showed faster and slower degradation kinetics than BPA. It is worth noting that in the presence of the electron-rich sulfamethoxazole, PMS decomposed slower than that with BPA; while in the presence of diphenhydramine, PMS decomposed faster than that with BPA (Fig. S11†). The case here is contrary to the known mediated electron transfer process in carbon and noble metal catalyzed reactions wherein the addition of the pollutant accelerates PMS decomposition.10,58,60–62 The sharp contrast herein indicates that the spatial-confinement of Co nanocrystals may alters the electron transfer process from pollutants to PMS.
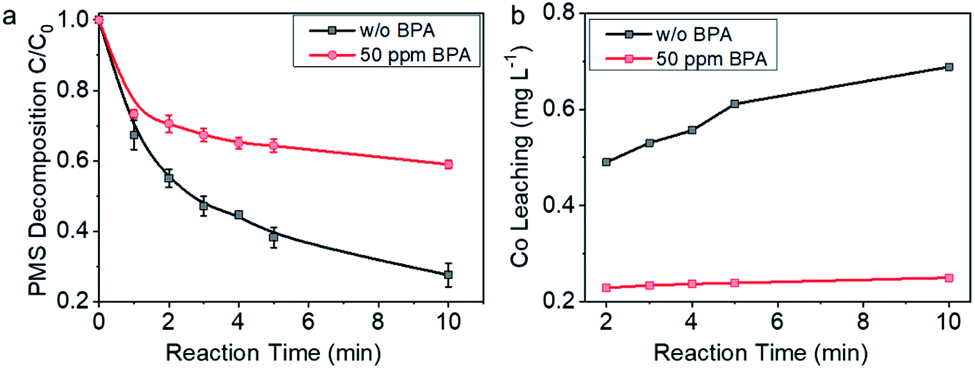 | ||
| Fig. 5 Confirmation on the mechanism of mediated electron transfer from BPA to PMS. (a) PMS decomposition behavior on Co-NC in the presence and absence of BPA. (b) Co2+ leaching with reaction time in the presence and absence of BPA. Reaction conditions: PMS concentration, 1 mM; 50 mL BPA solution with concentration of 50 ppm; Co-NC concentration, 0.15 g L−1. | ||
Co2+ leaching behavior was monitored for further understanding the electron transfer process. As shown in Fig. 5b, in the absence of BPA, Co2+ concentration increased with reaction time, and the concentration reached 0.69 ppm with 10 min reaction. In contrast, Co2+ concentration in the reaction system with 50 ppm BPA was almost stable, e.g., 0.23 ppm at 2 min and 0.25 ppm at 10 min. These data are indicating that the spike of BPA changed the electron transfer pathway: in the absence of BPA, there was direct interaction between Co0 and PMS, and electron transfer process started upon contacting, leading to the diffusion of Co2+ into the solvent. While in the case with 50 ppm BPA, Co nanocrystal was no longer the electron donor, but played a distinct role of conductor, transferring the electrons coming from BPA to PMS. We thus tentatively propose that BPA was oxidized by PMS with Co-NC as an electron transfer platform, which favorably improved the efficiency of PMS. For such an efficient electron transfer process, detailed insights into the relationship between microstructure and the unique electron transfer behavior is highly desired and expected to inspire the design of high-performance catalysts. It drives us to further explore the details about the catalytic process on Co-NC.
In the Co2+ catalyzed reaction systems, the interaction between cobalt and PMS generated cobalt active species; via oxygen transfer reaction with the cobalt active species, methyl phenyl sulfoxide (MPSO) was converted to methyl phenyl sulfone (MPSO2) (Fig. 6a).14,15 The observation of MPSO to MPSO2 conversion demonstrated the accessibility of reactive species by MPSO.19,63 As shown in Fig. 6b, Co-P showed high catalytic activity for MPSO to MPSO2 conversion, and 5 min reaction afforded both high conversion of 71.9% and remarkable selectivity of 86.9%. During MPSO oxidation, cobalt leached gradually into the solution, and the concentration of Co2+ reached 2.1 ppm at the end of the reaction (Fig. S12†). To figure out the role of Co0 for MPSO oxidation, a reaction catalyzed by 3.8 ppm Co2+ was carried out for a comparison; notwithstanding with a high Co2+ concentration (∼2 times of the final Co2+ concentration in Co-P catalyzed reaction system), 5 min reaction afforded a relatively lower conversion of 67.4%. Zero valent cobalt was thus proven to be capable of catalyzing MPSO oxidation by PMS to generate MPSO2.
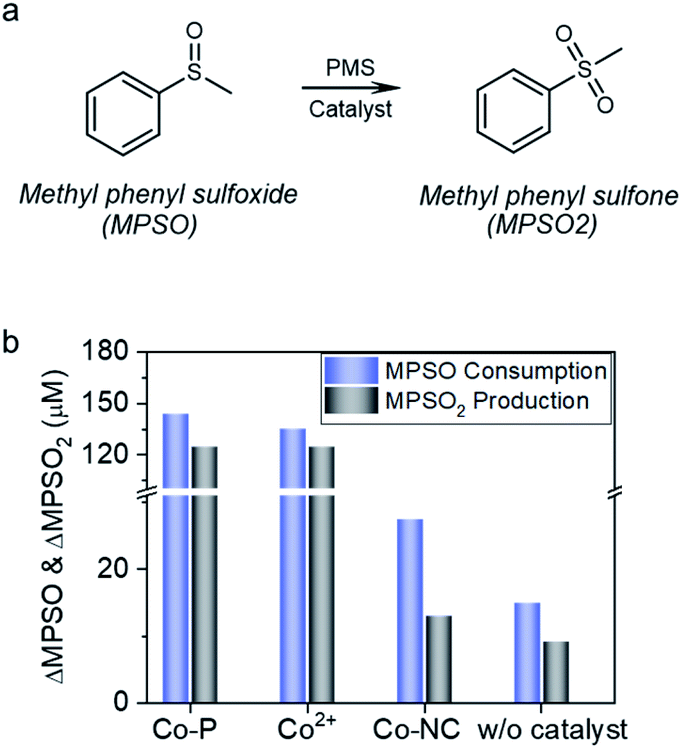 | ||
| Fig. 6 (a) Reaction equation of MPSO to MPSO2 probe reaction. (b) MPSO to MPSO2 conversion under different reaction conditions. Reaction conditions, 50 mL solution with 1 mM PMS and 0.2 mM MPSO; Co-P, 8.0 mg L−1; Co2+, 3.8 mg L−1; Co-NC, 150 mg L−1 (Co, 15.6 mg L−1); circumneutral pH; 5 min reaction at 25 °C. | ||
In stark contrast to the catalytic performance of Co-P, Co-NC is almost inactive for MPSO to MPSO2 conversion. Specifically, Co-NC catalysis for 5 min offered a low conversion of 13.7%, and poor MPSO2 selectivity of 47.3%. In light of the facts that a blank reaction without catalyst presented 7.5% conversion and 61.4% MPSO2 selectivity, the contribution of Co-NC catalysis in MPSO to MPSO2 conversion is negligible. In other words, PMS direct oxidation reaction contributes predominantly to the MPSO2 production in Co-NC case, indicating that MPSO oxidation on Co-NC proceeded through an alternative electron transfer pathway wherein MPSO has no access to the spatially-confined cobalt specie, thus very low selectivity towards MPSO2.
In light of the aforementioned experimental evidences of (1) BPA adsorption being critical for degradation, (2) TMPO formation initiated by one-electron oxidation, (3) capability in both PMS and PDS activation, and (4) negligible MPSO2 production by Co-NC catalysis, it is plausible to propose that mediated-electron transfer mechanism is governing the MPSO oxidation reaction on Co-NC. However, the details about the interaction between carbon nanotube and spatially-confined cobalt crystal, and its role in tuning the electronic properties of the cobalt crystal are still unknown and deserve further investigations.
4. Conclusions
Cobalt catalyzed PMS activation is reckoned as a highly efficient process for refractory organic pollutants depletion. Mechanistic understanding on the insights of the nano-structure dependent PMS activation process is instructive to the design of efficient catalysts for dealing with the specific requirements from the complex water matrix. Under spatial-confinement, the reaction process in cobalt catalysis system was adjusted to the mediated electron transfer mechanism, rather than the known radicals or singlet oxygen involved reaction. This endows the reaction system with competitively favorable capability in selectively abatement of the priority pollutants in complex water matrix with NOMs and halide ions.Author contributions
S. Shao: methodology, formal analysis, investigation. J. Cui: methodology, formal analysis. L. Li: methodology, writing – original draft. M. Wang: methodology, formal analysis. P. Zhang: methodology, formal analysis, writing – review & editing. J. G. Cui: formal analysis. C. Hu: resources, writing – review & editing, funding acquisition. Y. Zhao: conceptualization, formal analysis, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (21976041, 51538013, and 51838005).Notes and references
- C. Liu, Y. Zhang, F. Dong, A. H. Reshak, L. Ye, N. Pinna, C. Zeng, T. Zhang and H. Huang, Chlorine intercalation in graphitic carbon nitride for efficient photocatalysis, Appl. Catal., B, 2017, 203, 465–474 CrossRef CAS.
- X. Duan, H. Sun and S. Wang, Metal-free carbocatalysis in advanced oxidation reactions, Acc. Chem. Res., 2018, 51, 678–687 CrossRef CAS PubMed.
- H. Liu, T. A. Bruton, F. M. Doyle and D. L. Sedlak, In situ chemical oxidation of contaminated groundwater by persulfate: decomposition by Fe(III)- and Mn(IV)-containing oxides and aquifer materials, Environ. Sci. Technol., 2014, 48, 10330–10336 CrossRef CAS PubMed.
- C. Wang, J. Kang, P. Liang, H. Zhang, H. Sun, M. O. Tadé and S. Wang, Ferric carbide nanocrystals encapsulated in nitrogen-doped carbon nanotubes as an outstanding environmental catalyst, Environ. Sci.: Nano, 2017, 4, 170–179 RSC.
- S. Liu, B. Jing, C. Nie, Z. Ao, X. Duan, B. Lai, Y. Shao, S. Wang and T. An, Piezoelectric activation of peroxymonosulfate by MoS2 nanoflowers for the enhanced degradation of aqueous organic pollutants, Environ. Sci.: Nano, 2021, 8, 784–794 RSC.
- R. R. Solís, Ö. Dinc, G. Fang, M. N. Nadagouda and D. D. Dionysiou, Activation of inorganic peroxides with magnetic graphene for the removal of antibiotics from wastewater, Environ. Sci.: Nano, 2021, 8, 960–977 RSC.
- G. P. Anipsitakis and D. D. Dionysiou, Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- Q. Yang, H. Choi and D. D. Dionysiou, Nanocrystalline cobalt oxide immobilized on titanium dioxide nanoparticles for the heterogeneous activation of peroxymonosulfate, Appl. Catal., B, 2007, 74, 170–178 CrossRef CAS.
- Q. Yang, H. Choi, Y. Chen and D. D. Dionysiou, Heterogeneous activation of peroxymonosulfate by supported cobalt catalysts for the degradation of 2,4-dichlorophenol in water: The effect of support, cobalt precursor, and UV radiation, Appl. Catal., B, 2008, 77, 300–307 CrossRef CAS.
- Y.-Y. Ahn, H. Bae, H.-I. Kim, S.-H. Kim, J.-H. Kim, S.-G. Lee and J. Lee, Surface-loaded metal nanoparticles for peroxymonosulfate activation: Efficiency and mechanism reconnaissance, Appl. Catal., B, 2019, 241, 561–569 CrossRef CAS.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang, J. Gao, J. Wang, Y. Huang, T. Zhang and B. Liu, Single cobalt atoms anchored on porous N-doped graphene with dual reaction sites for efficient fenton-like catalysis, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS PubMed.
- X. Mi, P. Wang, S. Xu, L. Su, H. Zhong, H. Wang, Y. Li and S. Zhan, Almost 100% peroxymonosulfate conversion to singlet oxygen on single-atom CoN2+2 sites, Angew. Chem., Int. Ed., 2020, 60, 4588–4593 CrossRef PubMed.
- C. Chu, J. Yang, X. Zhou, D. Huang, H. Qi, S. Weon, J. Li, M. Elimelech, A. Wang and J.-H. Kim, Cobalt single atoms on tetrapyridomacrocyclic support for efficient peroxymonosulfate activation, Environ. Sci. Technol., 2021, 55, 1242–1250 CrossRef CAS PubMed.
- Y. Zong, X. Guan, J. Xu, Y. Feng, Y. Mao, L. Xu, H. Chu and D. Wu, Unraveling the overlooked involvement of high-valent cobalt-oxo species generated from the cobalt(II)-activated peroxymonosulfate process, Environ. Sci. Technol., 2020, 54, 16231–16239 CrossRef CAS PubMed.
- H. Li, Z. Zhao, J. Qian and B. Pan, Are free radicals the primary reactive species in Co(II)-mediated activation of peroxymonosulfate? new evidence for the role of the Co(II)–peroxymonosulfate complex, Environ. Sci. Technol., 2021, 55, 6397–6406 CrossRef CAS PubMed.
- J. Lee, U. von Gunten and J.-H. Kim, Persulfate-based advanced oxidation: critical assessment of opportunities and roadblocks, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- C. Chen, Z. Wu, S. Zheng, L. Wang, X. Niu and J. Fang, Comparative study for interactions of sulfate radical and hydroxyl radical with phenol in the presence of nitrite, Environ. Sci. Technol., 2020, 54, 8455–8463 CrossRef CAS PubMed.
- S. Hou, L. Ling, D. D. Dionysiou, Y. Wang, J. Huang, K. Guo, X. Li and J. Fang, Chlorate formation mechanism in the presence of sulfate radical, chloride, bromide and natural organic matter, Environ. Sci. Technol., 2018, 52, 6317–6325 CrossRef CAS PubMed.
- N. Jiang, H. Xu, L. Wang, J. Jiang and T. Zhang, Nonradical oxidation of pollutants with single-atom-Fe(III)-activated persulfate: Fe(V) being the possible intermediate oxidant, Environ. Sci. Technol., 2020, 54, 14057–14065 CrossRef CAS PubMed.
- P. Hu, H. Su, Z. Chen, C. Yu, Q. Li, B. Zhou, P. J. J. Alvarez and M. Long, Selective degradation of organic pollutants using an efficient metal-free catalyst derived from carbonized polypyrrole via peroxymonosulfate activation, Environ. Sci. Technol., 2017, 51, 11288–11296 CrossRef CAS PubMed.
- K. H. Chan and W. Chu, Degradation of atrazine by cobalt-mediated activation of peroxymonosulfate: different cobalt counteranions in homogenous process and cobalt oxide catalysts in photolytic heterogeneous process, Water Res., 2009, 43, 2513–2521 CrossRef CAS PubMed.
- S. Wang, Y. Liu and J. Wang, Peroxymonosulfate activation by Fe–Co–O-codoped graphite carbon nitride for degradation of sulfamethoxazole, Environ. Sci. Technol., 2020, 54, 10361–10369 CrossRef CAS PubMed.
- Y. Zhou, X. Wang, C. Zhu, D. D. Dionysiou, G. Zhao, G. Fang and D. Zhou, New insight into the mechanism of peroxymonosulfate activation by sulfur-containing minerals: role of sulfur conversion in sulfate radical generation, Water Res., 2018, 142, 208–216 CrossRef CAS PubMed.
- G. Wang, X. Nie, X. Ji, X. Quan, S. Chen, H. Wang, H. Yu and X. Guo, Enhanced heterogeneous activation of peroxymonosulfate by Co and N codoped porous carbon for degradation of organic pollutants: the synergism between Co and N, Environ. Sci.: Nano, 2019, 6, 399–410 RSC.
- H. V. Lutze, S. Bircher, I. Rapp, N. Kerlin, R. Bakkour, M. Geisler, C. von Sonntag and T. C. Schmidt, Degradation of chlorotriazine pesticides by sulfate radicals and the influence of organic matter, Environ. Sci. Technol., 2015, 49, 1673–1680 CrossRef CAS PubMed.
- W.-D. Oh, Z. Dong and T.-T. Lim, Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: current development, challenges and prospects, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- G. Liu, S. You, Y. Tan and N. Ren, In situ photochemical activation of sulfate for enhanced degradation of organic pollutants in water, Environ. Sci. Technol., 2017, 51, 2339–2346 CrossRef CAS PubMed.
- F. Manjón, L. Villén, D. García-Fresnadillo and G. Orellana, On the factors influencing the performance of solar reactors for water disinfection with photosensitized singlet oxygen, Environ. Sci. Technol., 2008, 42, 301–307 CrossRef PubMed.
- J. Lee, Y. Mackeyev, M. Cho, D. Li, J.-H. Kim, L. J. Wilson and P. J. J. Alvarez, Photochemical and antimicrobial properties of novel C60 derivatives in aqueous systems, Environ. Sci. Technol., 2009, 43, 6604–6610 CrossRef CAS PubMed.
- W. H. Koppenol, D. M. Stanbury and P. L. Bounds, Electrode potentials of partially reduced oxygen species, from dioxygen to water, Free Radical Biol. Med., 2010, 49, 317–322 CrossRef CAS PubMed.
- A. D. Bokare and W. Choi, Singlet-oxygen generation in alkaline periodate solution, Environ. Sci. Technol., 2015, 49, 14392–14400 CrossRef CAS PubMed.
- D. Muñoz-Santiburcio and D. Marx, Chemistry in nanoconfined water, Chem. Sci., 2017, 8, 3444–3452 RSC.
- J. Ko, R. Berger, H. Lee, H. Yoon, J. Cho and K. Char, Electronic effects of nano-confinement in functional organic and inorganic materials for optoelectronics, Chem. Soc. Rev., 2021, 50, 3585–3628 RSC.
- Z. Yang, J. Qian, A. Yu and B. Pan, Singlet oxygen mediated iron-based Fenton-like catalysis under nanoconfinement, Proc. Natl. Acad. Sci. U. S. A., 2019, 110, 14861–14866 Search PubMed.
- S. Zhang, T. Hedtke, Q. Zhu, M. Sun, S. Weon, Y. Zhao, E. Stavitski, M. Elimelech and J.-H. Kim, Membrane-confined iron oxychloride nanocatalysts for highly efficient heterogeneous fenton water treatment, Environ. Sci. Technol., 2021, 55, 9266–9275 CrossRef CAS PubMed.
- S. Zhang, T. Hedtke, X. Zhou, M. Elimelech and J.-H. Kim, Environmental applications of engineered materials with nanoconfinement, ACS EST Engg, 2021, 1, 706–724 CrossRef CAS.
- J. Qian, X. Gao and B. Pan, Nanoconfinement-mediated water treatment: from fundamental to application, Environ. Sci. Technol., 2020, 54, 8509–8526 CrossRef CAS PubMed.
- E. Castillejos, P.-J. Debouttière, L. Roiban, A. Solhy, V. Martinez, Y. Kihn, O. Ersen, K. Philippot, B. Chaudret and P. Serp, An efficient strategy to drive nanoparticles into carbon nanotubes and the remarkable effect of confinement on their catalytic performance, Angew. Chem., Int. Ed., 2009, 48, 2529–2533 CrossRef CAS PubMed.
- Y. V. Fedoseeva, A. S. Orekhov, G. N. Chekhova, V. O. Koroteev, M. A. Kanygin, B. V. Senkovskiy, A. Chuvilin, D. Pontiroli, M. Riccò, L. G. Bulusheva and A. V. Okotrub, Single-walled carbon nanotube reactor for redox transformation of mercury dichloride, ACS Nano, 2017, 11, 8643–8649 CrossRef CAS PubMed.
- W. Chen, X. Pan and X. Bao, Tuning of redox properties of iron and iron oxides via encapsulation within carbon nanotubes, J. Am. Chem. Soc., 2007, 129, 7421–7426 CrossRef CAS PubMed.
- L. D'Souza, L. Jiao, J. R. Regalbuto, J. T. Miller and A. J. Kropf, Preparation of silica- and carbon-supported cobalt by electrostatic adsorption of Co(III) hexaammines, J. Catal., 2007, 248, 165–174 CrossRef.
- T. M. Byrne, X. Gu, P. Hou, F. S. Cannon, N. R. Brown and C. Nieto-Delgado, Quaternary nitrogen activated carbons for removal of perchlorate with electrochemical regeneration, Carbon, 2014, 73, 1–12 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- H. Gao, H. Yang, J. Xu, S. Zhang and J. Li, Strongly coupled g-C3N4 nanosheets-Co3O4 quantum dots as 2D/0D heterostructure composite for peroxymonosulfate activation, Small, 2018, 14, 1801353 CrossRef PubMed.
- B. Wu, L. Zhang, B. Jiang, Q. Li, C. Tian, Y. Xie, W. Li and H. Fu, Ultrathin porous carbon nitride bundles with an adjustable energy band structure toward simultaneous solar photocatalytic water splitting and selective phenylcarbinol oxidation, Angew. Chem., Int. Ed., 2021, 60, 4815–4822 CrossRef CAS PubMed.
- S. D. Richardson and S. Y. Kimura, Water analysis: emerging contaminants and current issues, Anal. Chem., 2016, 88, 546–582 CrossRef CAS PubMed.
- J. Ma and N. J. D. Graham, Degradation of atrazine by manganese-catalysed ozonation—influence of radical scavengers, Water Res., 2000, 34, 3822–3828 CrossRef CAS.
- L. Wang, H. Xu, N. Jiang, Z. Wang, J. Jiang and T. Zhang, Trace cupric species triggered decomposition of peroxymonosulfate and degradation of organic pollutants: Cu(III) being the primary and selective intermediate oxidant, Environ. Sci. Technol., 2020, 54, 4686–4694 CrossRef CAS PubMed.
- F. A. Villamena, in Reactive Species Detection in Biology, ed. F. A. Villamena, Elsevier, Boston, 2017, pp. 163–202, DOI:10.1016/B978-0-12-420017-3.00004-9.
- A. L. Boreen, B. L. Edhlund, J. B. Cotner and K. McNeill, Indirect photodegradation of dissolved free amino acids: the contribution of singlet oxygen and the differential reactivity of DOM from various sources, Environ. Sci. Technol., 2008, 42, 5492–5498 CrossRef CAS PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution. An expanded and revised compilation, J. Phys. Chem. Ref. Data, 1995, 24, 663–677 CrossRef CAS.
- X. Cheng, H. Guo, Y. Zhang, X. Wu and Y. Liu, Non-photochemical production of singlet oxygen via activation of persulfate by carbon nanotubes, Water Res., 2017, 113, 80–88 CrossRef CAS PubMed.
- P. Liang, C. Zhang, X. Duan, H. Sun, S. Liu, M. O. Tade and S. Wang, An insight into metal organic framework derived N-doped graphene for the oxidative degradation of persistent contaminants: formation mechanism and generation of singlet oxygen from peroxymonosulfate, Environ. Sci.: Nano, 2017, 4, 315–324 RSC.
- E.-T. Yun, J. H. Lee, J. Kim, H.-D. Park and J. Lee, Identifying the nonradical mechanism in the peroxymonosulfate activation process: singlet oxygenation versus mediated electron transfer, Environ. Sci. Technol., 2018, 52, 7032–7042 CrossRef CAS PubMed.
- G. Nardi, I. Manet, S. Monti, M. A. Miranda and V. Lhiaubet-Vallet, Scope and limitations of the TEMPO/EPR method for singlet oxygen detection: the misleading role of electron transfer, Free Radical Biol. Med., 2014, 77, 64–70 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Radical generation by the interaction of transition metals with common oxidants, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- Y.-Y. Ahn, E.-T. Yun, J.-W. Seo, C. Lee, S. H. Kim, J.-H. Kim and J. Lee, Activation of peroxymonosulfate by surface-loaded noble metal nanoparticles for oxidative degradation of organic compounds, Environ. Sci. Technol., 2016, 50, 10187–10197 CrossRef CAS PubMed.
- H. Lee, H.-J. Lee, J. Jeong, J. Lee, N.-B. Park and C. Lee, Activation of persulfates by carbon nanotubes: oxidation of organic compounds by nonradical mechanism, Chem. Eng. J., 2015, 266, 28–33 CrossRef CAS.
- T. Zhang, Y. Chen, Y. Wang, J. Le Roux, Y. Yang and J.-P. Croué, Efficient peroxydisulfate activation process not relying on sulfate radical generation for water pollutant degradation, Environ. Sci. Technol., 2014, 48, 5868–5875 CrossRef CAS PubMed.
- E.-T. Yun, H.-Y. Yoo, H. Bae, H.-I. Kim and J. Lee, Exploring the role of persulfate in the activation process: radical precursor versus electron acceptor, Environ. Sci. Technol., 2017, 51, 10090–10099 CrossRef CAS PubMed.
- J. Miao, W. Geng, P. J. J. Alvarez and M. Long, 2D N-doped porous carbon derived from polydopamine-coated graphitic carbon nitride for efficient nonradical activation of peroxymonosulfate, Environ. Sci. Technol., 2020, 54, 8473–8481 CrossRef CAS PubMed.
- Z. Wang, J. Jiang, S. Pang, Y. Zhou, C. Guan, Y. Gao, J. Li, Y. Yang, W. Qiu and C. Jiang, Is sulfate radical really generated from peroxydisulfate activated by iron(II) for environmental decontamination?, Environ. Sci. Technol., 2018, 52, 11276–11284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08954d |
| This journal is © The Royal Society of Chemistry 2022 |