
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of cyclic stability and volume expansion on graphite–SiOx–C hierarchical structure for Li-ion battery anodes†
Jae Hyeon Yun‡
ab,
Tae Kyung Whang‡ab,
Won Jun Ahnab,
Young-Seak Leeb and
Ji Sun Im *ac
*ac
aC1 Gas & Carbon Convergent Research, Korea Research Institute of Chemical Technology (KRICT) 141 Gajeong-ro, Yuseong-Gu, Daejeon, 34114, Republic of Korea. E-mail: jsim@krict.re.kr
bDepartment of Chemical Engineering and Applied Chemistry, Chungnam University, 99 Daehak-ro, Yuseong-gu, Daejeon, 34134, Republic of Korea
cAdvanced Materials and Chemical Engineering, University of Science and Technology (UST), 217 Gajeong-ro, Yuseong-gu, Daejeon, 34113, Republic of Korea
First published on 24th February 2022
Abstract
To increase the energy density of today's batteries, studies on adding Si-based materials to graphite have been widely conducted. However, adding a Si-based material in the slurry mixing step suffers from low distribution due to the self-aggregation property of the Si-based material. Herein, a hierarchical structure is proposed to increase the integrity by using APS to provide a bonding effect between graphite and SiOx. Additionally, to endow a protection layer, carbon is coated on the surface using the CVD method. The designed structure demonstrates enhanced integrity based on electrochemical performance. The MSG (methane decomposed SiOx@G) electrode demonstrates a high ICE of 85.6% with 429.8 mA h g−1 initial discharge capacity. In addition, the MSG anode has superior capacity retention (89.3%) after 100 cycles, with enhanced volumetric expansion (12.7%) after 50 cycles. We believe that the excellent electrochemical performance of MSG is attributed to increased integrity by using APS (3-aminopropyltrimethoxysilane) with a CVD carbon coating.
1. Introduction
As the electric vehicle market surges, researchers have been striving to increase battery energy density. Because the energy density of lithium-ion batteries (LIBs) is determined by the anode capacity, increasing the anode material's capacity is crucial.1–4 For this reason, the introduction of alloy-type materials (Si, Sn, Ge etc.) with high energy density to graphite, a commonly used anode material, is being studied.5–7 Among these materials, silicon is a major candidate for improving energy density, with a high theoretical capacity of 3572 mA h g−1 (Li15Si4), a low operating voltage (∼0.4 V vs. Li/Li+), and abundant resources.4,5 However, during lithiation/delithiation, silicon suffers large volume changes (∼300%) and has poor electric conductivity.8,9 Furthermore, the volumetric expansion leads to atomization of particles, exposing new surfaces, which increases lithium consumption during lithiation/delithiation, and results in a continuous SEI layer with low coulombic efficiency.6,10Compared to silicon, SiOx has been studied due to a lower volumetric pulverization (<200%) and relatively high cyclic stability during the lithiation/delithiation process.11,12 The lower volume expansion of SiOx is caused by products (Li2O, Li4SiO4) that result from the oxygen content of SiOx reacting with lithium, which acts as a buffer during the lithiation process.11,13–15 However, the generation of Li2O and Li4SiO4, which is an inactive material, consumes a lot of lithium, which leads to a large initial irreversible capacity of SiOx anodes.15–17 For these reasons, the initial coulombic efficiency (ICE) of SiOx anodes is about 70%.17
To improve the ICE of SiOx anodes, the application of a carbon coating on the surface has been studied. Many methods have been applied to prepare carbon coatings, including mechanical milling,18 spray drying,9,19 polymer pyrolysis,20 and chemical vapour deposition (CVD).21 The coating layer of the surface can help build a stable solid electrolyte interface (SEI) layer because of the formation of a uniform surface, as well as improving the electrical conductivity.22 The stable SEI layer can increase the ICE of the SiOx anodes.9,23
In spite of these efforts, the SiOx anodes should be blended with commercial graphite anodes due to the low cyclic performance against graphite for industrial applications.18,24–26 But, in slurry mixing step, characteristic of the SiOx that tends to be aggregated itself interrupts to form uniform slurry. Thus, “integrity” in graphite and SiOx anodes must be considered importantly when constructing electrodes.25,27 Otherwise, low integrity between graphite and SiOx leads to uneven electrode construction, and the composed anode consequently experiences severe volume effects. As a result, the electrode has not only poor electrical contact but also low cyclic stability. To resolve these effects, increasing integrity between graphite and SiOx is important for high stability in cycling.
In this article, we developed a hierarchical structure (G–SiOx–C) to increase the integrity. This type of structure has a main core of graphite and a SiOx shell in the subsequent layer and the outermost layer is carbon layer, where SiOx is attached to graphite to increase the integrity between graphite and SiOx. A simple wet-stirring method with 3-aminopropyltrimethoxy-silane (APS) was employed to enhance the integrity of graphite and SiOx. This could give amino groups on SiOx, leading to chemical bonding with graphite28–30 (hereafter the SiOx loaded graphite is denoted as SG). As a result, SiOx was homogeneously loaded on graphite. To further increase the integrity of graphite and SiOx, CVD carbon coating using methane was performed (the methane decomposed SG is hereafter denoted as MSG). The carbon coating of the outermost layers allows the formation of a stable SEI layer and also increases the electrical conductivity. Consequently, we discussed the advantages of this designed structure focusing on (1) improving integrity using APS and (2) surface protection using CVD carbon coating.
2. Experimental
2.1 Material preparation
2.2 Material characterization
The morphology of the prepared samples was evaluated by scanning electron microscopy (SEM, Tescan Mira 3 LMU FEG) with energy dispersive X-ray spectroscopy (EDX, Bruker Quantax 200 XFlash4010) at 10 kV and transmission electron microscopy (TEM, JEM-ARM200F). Structural investigation of the samples was carried out with an X-ray diffractometer (XRD, Rigaku Ultima IV) using Cu Kα radiation (λ = 1.5418 Å) and Raman spectra were obtained using a Nanophoton Ramanforce Raman spectrometer with a laser wavelength of 532 nm. The particle size distribution (PSD) of the samples was obtained by a laser particle size distribution analyzer (Microtrac Bluewave). Thermogravimetric analysis (TGA, SDT Q600 V20.9 Build 20) was carried out in air within a temperature range of 25–900 °C. X-ray photoelectron spectrometry (XPS, AXIS SUPRA) was used for measuring the binding energy of the samples with Al Kα radiation. To demonstrate the variation of the electrode after the cycling test, the electrode was washed by dimethyl carbonate in an argon-filled glove box. The electrode before and after cycling was analyzed by scanning electron microscopy – plasma focused ion beam (SEM-PFIB, Helios G4 PFIB CXe DualBeam) with Xe plasma.2.3 Electrochemical measurements
The electrode was prepared by a slurry mixing method on a Cu current collector. The slurry was composed of the active materials (SG, MSG, G/SiOx etc.), conductive (Super P, Imerys Graphite & Carbon), carboxymethyl cellulose (CMC, MTI Corp.), and styrene butadiene rubber (SBR, MTI Corp.) and was uniformly mixed by a Thinky mixer in a mass ratio of 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2.5:2.5 and the loading level of the electrode was 7–8 mg cm−2. For the G, the slurry was mixed in a mass ratio of 95:2.5:2.5 (active:CMC:SBR). The electrode was dried at room temperature for 2 h, and then at 80 °C for 12 h in a vacuum oven and subsequently punched into a circular electrode with a diameter of 13.5 mm. The electrode was calendared for 1.2–1.3 g cc−1 of electrode density by a calendaring process. In the cell test, a CR 2032 coin-type cell was used for assembling the cell. The electrolyte was 1.0 M LiPF6 in a mixture of ethylene carbonate (EC):diethyl carbonate (DEC) = 1:1 volume ratio and 12 μm of polyethylene (PE) was used as separator. All electrode fabrication was carried out in an argon-filled glove box. The electrochemical properties were measured in a voltage range of 0.01 to 1.5 V vs. Li/Li+, and all electrochemical tests were estimated with a Wonatech WBCS3000. The cyclic voltammetry (CV) curves were also obtained on a WBCS3000 electrochemical workstation, and the scan rate was 0.1 to 1.0 mV s−1 at a voltage range of 0.01 to 1.5 V.
:3:2.5:2.5 and the loading level of the electrode was 7–8 mg cm−2. For the G, the slurry was mixed in a mass ratio of 95:2.5:2.5 (active:CMC:SBR). The electrode was dried at room temperature for 2 h, and then at 80 °C for 12 h in a vacuum oven and subsequently punched into a circular electrode with a diameter of 13.5 mm. The electrode was calendared for 1.2–1.3 g cc−1 of electrode density by a calendaring process. In the cell test, a CR 2032 coin-type cell was used for assembling the cell. The electrolyte was 1.0 M LiPF6 in a mixture of ethylene carbonate (EC):diethyl carbonate (DEC) = 1:1 volume ratio and 12 μm of polyethylene (PE) was used as separator. All electrode fabrication was carried out in an argon-filled glove box. The electrochemical properties were measured in a voltage range of 0.01 to 1.5 V vs. Li/Li+, and all electrochemical tests were estimated with a Wonatech WBCS3000. The cyclic voltammetry (CV) curves were also obtained on a WBCS3000 electrochemical workstation, and the scan rate was 0.1 to 1.0 mV s−1 at a voltage range of 0.01 to 1.5 V.
In the full cell test, the cathode electrode was fabricated by the slurry mixing method on a Al current collector with lithium cobalt(III) oxide (LCO, Alfa Aesar), conductive, and polyvinylidene fluoride (PVdF, Kynar HSV900) in a mass ratio of 94:3:3 with a solvent of NMP and the loading level of the electrode was ∼17.5 mg cm−2. The electrode was dried at 110 °C for 6 h in a vacuum oven, and then cut into a disk with a diameter of 13.5 mm. The electrode was pressed until ∼3.5 g cc−1 electrode density. The N/P ratio of the prepared electrode was ∼1.1 and an electrochemical analysis was carried out at a voltage range of 2.7 to 4.3 V. The CR 2032 coin-type cell was used for assembling the full cell. The electrolyte and separator were the same as previously described. All electrochemical tests were performed with a Wonatech WBCS3000.
3. Results and discussion
3.1 Morphological analysis of hierarchical structure
Fig. 1a describes the overall synthesis process of MSG. First, the graphite and SiOx were acid-treated to endow carboxylic groups and hydroxyl groups, respectively. Subsequently, the surface treated SiOx was stirred in solution to give an amine group by using APS. After that, the SiOx was added to graphite and complexed through a condensation reaction between amine and the carboxylic group on the surface of SiOx and graphite, respectively. The surface modification of SiOx using APS suppresses the tendency of aggregating with each other, allowing it to be loaded regularly into graphite. The MSG was then prepared using the CVD method to form a carbon layer on the SG surface. The carbon coating process was implemented at 900 °C, which was determined by the amount of carbon deposited at different temperatures. When the SiOx was reacted at 900 °C, about 5.8 wt% carbon was formed, 13.3 wt% carbon formed at 950 °C, and 24.3 wt% carbon formed at 1000 °C in the TG analysis (Fig. S2†). | ||
| Fig. 1 (a) Scheme of the fabrication process of the MSG particles. SEM images of (b) artificial graphite (c) SG, (d) MSG particles. SEM-EDS mapping images of MSG (e–h). | ||
To analyze the morphology of the as-prepared samples, SEM was used. As shown in Fig. 1c, in the case of SG, it can be seen that the surface modification of SiOx using APS formed a hierarchical structure (G–SiOx) through a condensation reaction between the amine and carboxyl groups. In Fig. 1d, we can also see that the carbon coated MSG maintains a hierarchical structure (G–SiOx–C). However, without surface modification, it was found that the SiOx was not well placed on the graphite (Fig. S3†). The designed structure was confirmed by SEM-EDS analysis, which showed that the SiOx surface was covered by a carbon layer (Fig. 1e–h). The detailed morphology of MSG was elucidated through TEM. In Fig. 2a, as shown in the previous SEM results, we confirmed that the SiOx of MSG was well loaded on the graphite surface. From Fig. 2b and c, the deposited carbon coating layer of MSG has about 10 nm thickness and has a graphitic shape with about 0.3879 nm of d-spacing, representing the (002) plane. In addition, the SiOx showed crystalline Si embedded in an amorphous matrix, and the d-spacing of Si is 0.297 nm, representing the (111) plane. The SiOx was composed with crystalline silicon and amorphous SiOx. To investigate the oxygen content of SiOx, XPS analysis was applied as shown in Fig. S4.† The Si 2p peak of the SiOx was deconvoluted based on oxidated state of Si4+, Si3+, Si2+, and Si1+, which corresponds SiO2.0, SiO1.5, SiO1.0, and SiO0.5. Based on XPS spectra, the oxygen content was calculated by the areal ratio of each oxidated state curve. As a result of estimation, the oxygen content of SiOx can be exhibited as 1.14 (Table S1†).31,32
 | ||
| Fig. 2 (a–c) TEM images of MSG. (d) Particle size distribution of G, MSG. (e) Raman spectra of G, MSG, SG, SiOx. (f) Thermogravimetric analysis of G, SiOx, SG. | ||
To verify the compositional characterization of the designed structure, we further confirmed it by various analysis methods. As shown in Fig. 2d, the particle size of the as-prepared MSG is D50 = 13.06 μm, which confirmed that the SiOx of MSG was well loaded on graphite without agglomeration considering the size of graphite and SiOx. However, in the case of samples that did not undergo surface modification, the curves show two peaks at 1.03 μm and 11.13 μm, indicating that surface modification played a major role in forming the composite using APS (Fig. S5†). In the X-ray photoelectron spectroscopy (XPS) analysis, it can be seen that a N 1s peak appears in the SG, whereas in the case of the simple mixed G/SiOx, the N 1s peak does not appear. This is caused by a condensation reaction using APS (Fig. S6†). To confirm the chemical bonding of obtained samples, FT-IR analysis was conducted. As shown in Fig. S7(a),† it was observed that a lot of functional groups exist in the APS, 3-amino-propyltrimethoxysilane ((CH3O)3–Si–CH2–CH2–NH2). Most of all, the Si–O peak was exhibited at 1192 cm−1 and 1086 cm−1 and these peaks were found in SG and MSG (Fig. S7(b)†). However, these peaks were not found in G sample. Therefore, it is noted that the as-prepared samples was well bound by APS.33 X-ray diffraction (XRD) reveals the crystallinity of G, SiOx, SG, and MSG (Fig. S8†). It can be seen that pristine SiOx has a wide peak of amorphous SiOx around 20°, which is the same as seen in SG and MSG, indicating SiOx is added to the graphite. Raman spectra are presented in Fig. 2e to further examine the features of as-obtained samples. As shown in the graph of SG, the peak of SiOx appears at around 500 cm−1. In the graph of MSG, this peak did not appear because the deposited carbon covered the surface. Moreover, the generated carbon characteristics were identified by comparing two peaks that appear at about 1350 cm−1 (D band) and 1600 cm−1 (G band), which are typically expressed as ID/IG. Compared to graphite, the ID/IG ratio of SG is 0.98, indicating an increase in the D band. This is the result of the increased defects causing by modifying the graphite surface. In the MSG, the ID/IG ratio reduces to 0.8, verifying the deposited carbon is graphitic carbon, which is consistent with the HR-TEM results. To verify the SiOx content in SG, a thermogravimetric analysis (TG) was conducted in air (Fig. 1f). The amount of loaded SiOx was confirmed as around 10 wt%, considering the increased weight causing by oxidation of SiOx. The amount of carbon coating was calculated as the amount of carbon on SiOx (Fig. S2†). In the case of SiOx, the amount of carbon coating was approximately 5 wt% of its weight, considering the amount of carbon coating and the D50 of MSG, and the thickness of carbon coating was around 11.66 nm, which is consistent with HR-TEM.
3.2 Structural benefits of MSG in electrochemical characterization
To verify the effect of increased integrity of the MSG electrode, an electrochemical analysis in half cells was tested. As indicated in Fig. 3a, G exhibited a first cycle discharge capacity of 320.9 mA h g−1 with an ICE of 91.9%. The first cycle discharge capacity of SG, G/SiOx (physically blended in slurry mixing step, G:SiOx = 9:1 in mass ratio) exhibited 412.2 and 369.2 mA h g−1, respectively. The ICE of SG and G/SiOx exhibited high performance (82.2% and 80.3% respectively), compared to the ICE of SiOx (70%, Fig. S9†). In the case of G/SiOx with low integrity, a large amount of Li consumption occurred due to the high reactivity with the electrolyte, leading to a decrease in ICE. In addition, it appears that the capacity was reduced because internal SiOx could not participate in the reaction due to self-aggregation between SiOx. On the other hand, SG increased in both ICE and the capacity, and this is ascribed to the improved integrity of the composite by using APS. Moreover, carbon-coated MSG displayed a higher ICE of 85.6% compared to SG, with an improved discharge capacity of 429.8 mA h g−1. This improvement in capacity and ICE originated from the surface carbon coating, which could form a stable SEI layer in the initial charge/discharge process with increased integrity. To increase the discharge capacity of anode material, electrochemical testing of MSG-20 that is prepared with 20 wt% of SiOx, was conducted. MSG-20 exhibited a first discharge capacity of 526.6 mA h g−1, with ICE of 80.6% (Fig. S10†). The slightly lower ICE compared to MSG is due to the small amount of covered carbon layer. The rate performance of the as-prepared samples was measured by varying the discharge rates from 0.2C to 5C (Fig. 3b). In the case of MSG, retention of 5C/0.2C exhibits high capability with an average of 43.4%, compared to 35.3% retention of SG. However, for the simple blended G/SiOx, retention was very low and similar to that of G (G/SiOx: 27.0%, G: 23.7%). This also increased the electrical contact between G and SiOx using APS, indicating excellent rate capability. In addition, owing to the carbon coating preventing surface exposure between SiOx and the electrolyte, MSG exhibits the highest rate capability among the other electrodes. For G/SiOx, the dispersion was not performed smoothly during the slurry mixing step, resulting in electrochemical activation with increased electrode capacity in the initial five cycles. This was similar in the evaluation of cyclic stability, which was stabilized after about five cycles. The cycle performance was estimated at 0.5C (Fig. 3c). Cycle retention of MSG exhibited high stability, about 89.3% at 100 cycles (84.8% at 150 cycles), which was lower than that of G (94.0%). For the SG and G/SiOx, cyclic stability was relatively low (SG: 83.8%, G/SiOx: 67.8%). Notably, this indicated that an increase in integrity also affected cycle performance. Furthermore, the carbon coating through the CVD method covered the SG surface, forming a stable SEI layer that acted to increase the cyclic stability. In Fig. 3d, we describe CE during the cycles. In the case of MSG, the electrode was quickly stabilized and exhibited a high CE of 99.5% after 5 cycles. However, for SG, the electrode reached a CE of 99.5% after 10 cycles, and G/SiOx reached a CE of 99.5% after 30 cycles with difficulty. It is expected that this is due to a uniform carbon coating on the surface. In the case of MSG-20, the capacity retention exhibited 83.6% after 50 cycles (Fig. S10†). The lower cyclic stability compared to MSG is due to the increase of SiOx, implying that the surface carbon layer was insufficient to control the volume expansion of SiOx.
 | ||
| Fig. 3 Electrochemical performance in Li-ion half batteries. (a) Galvanostatic charge/discharge voltage profiles of G, G/SiOx, SG, MSG measured at 0.1C. (b) Rate capabilities of G, G/SiOx, SG, MSG under increasing C-rates from 0.2C to 5C. (c) Discharge capacity of 100 cycles at current density of 1C. (d) Magnified CEs of G, G/SiOx SG, MSG for 50 cycles. (e) CV curves of MSG at a scan rate of 0.1 mV s−1. (f) log(i)/log(v) plots of MSG and SG at various scan rates from 0.1 to 1.0 mV s−1. | ||
In Fig. 3e, cyclic voltammetry graphs at a scan rate of 0.1 mV s−1 for five cycles are shown. At the first cycle, MSG exhibits a broad cathodic peak at 0.7 V, indicating SEI formation. On the other hand, this peak did not appear in SG, due to the formation of an unstable SEI layer. After the first cycle, the cathodic peak of MSG exhibited two peaks, which corresponded to Li lithiation of graphite (at 0.01 V) and SiOx (at 0.15 V, alloying of LixSi).24 In the anodic peaks at 0.25 V corresponding to the delithiation peak of the graphite,34 the delithiation peak of the SiOx did not appear. It is expected that the direct reaction of SiOx is obscured by the carbon layer on the surface.24 For SG, lithiation peaks appear after the first cycle (at 0.01 V of graphite, at 0.15 V of SiOx), with delilthiation peaks of SiOx (at 0.68 V) (Fig. S11†).35,36 In both cases of MSG and SG, the peak intensity increased as the cycle proceeded and stabilized after four cycles. Moreover, we investigated the lithium storage kinetics of SG and MSG. A CV test was implemented while keeping the scan rates of lithiation at 0.1 mV s−1, with variation of the scan rates of delithiation from 0.1 to 1.0 mV s−1 (Fig. S12†). Typically, the peak current and scan rate can be indicated as i = avb; by taking logs and plotting log(i) versus log(v), we can determine that the electrode is dominated by diffusion or surface reaction (b = 0.5 indicating diffusion-controlled reaction and b = 1.0 indicating surface controlled reaction).37 As a result of plotting log(i) versus log(v), MSG and SG were 0.61 and 0.67, respectively. This means that the electrode reaction is controlled by diffusion, and a lower value of MSG than SG is a result of graphitic carbon generated on the surface, which is dominantly controlled by diffusion. In other words, this is because graphite is lithiated through Li intercalation while SiOx is lithiated through Li alloying.
To demonstrate the effect on the electrode resistance of the carbon coating, we conducted an EIS analysis. The EIS analysis was implemented in the following circuit, in a completely discharged state after the 1st and 50th cycles (Fig. 4a and b). The Rb, which indicates the electrode bulk resistance, is an index of the State of Health (SOH), representing the sum of the resistance of the electrode, separator, and electrolyte.38 The Rb of G, SG, and MSG increased 2.28, 4.04, and 1.93 times over cycles, respectively. Interestingly, the highest rate of increase was exhibited by SG, which appears to be related to two effects. First, SiOx exposed to the electrolyte continues to be atomized with cycling, causing a new SEI; that is, resistance is increased due to electrolyte depletion as the electrolyte is continuously consumed.39 Second, the atomized SiOx gave rise to a reduced contact area in the internal electrode, which raises the contact resistance, leading to an increase in the electrode resistance. The RSEI, which indicates the SEI resistance, of the G, SG, and MSG increased 3.69, 5.83, and 4.48 times over cycles, respectively. Likewise in RSEI, the increased resistance of SG came from excessive SEI formation caused by electrolyte exposure of SiOx. On the other hand, for MSG, relatively low SEI formation results from low exposure to electrolytes and inhibition of atomization caused by carbon coating. The Rct, which indicates the electrode charge transfer resistance, showed the highest increase rate in SG. This difference suggests that carbon coating cannot only alleviate volume expansion but also maintains a single structure during repeated cycles, contributing to stable charge transfer. This can be obtained through the Warburg resistance of the straight line, and it can be seen that the diffusion in MSG is higher than in SG (MSG: 3.63 × 10−9, SG: 1.04 × 10−9 at 1st cycle after) (Table S2†), which means that as the surface is covered with the carbon layer, Li diffusion coefficient increases, and this is consistent with the improvement of the rate performance of the MSG electrodes.
 | ||
| Fig. 4 Nyquist plot of (a) G, SG, MSG after 1st cycle and (b) 50th cycles. Charting the Nyquist plot of (c) G, SG, MSG after the 1st cycle and (d) 50th cycles. | ||
3.3 Swelling behavior
To identify the volume deformation of the electrode during lithiation/delithiation, we measured the thickness change of the G, SG, and MSG electrodes using scanning electron microscopy – plasma focused ion beam (SEM-PFIB). In the cycle tests, the electrode was charged at 0.2C rates and discharged at 0.5C rates for 50 cycles. In Fig. 5a and d, G exhibits a volume expansion of 11.4% (71.61 μm after a cycle from 64.27 μm before a cycle), which is similar to the general expansion of graphite in volume of 10%.40 However, the electrode of SG exhibited 25.2% swelling (from 57.84 μm to 61.91 μm), which implied the continuous formation of SEI with the surface caused by exposure to the electrolyte. The electrode of MSG, where the surface was covered by a carbon layer, exhibited 12.7% swelling (from 54.92 μm to 61.91 μm), and it increased by only 1.3% compared to that of G. Many trials have been studied to alleviate volume expansion of the electrode such as using porous structure. However, in this experiment, electrode integrity was increased using APS, which leads to alleviate volume expansion.41–44 This indicates that volume expansion was successfully alleviated due to the well-designed structure. As shown in the electrode top view before and after the cycles, it can be seen that the degree of damage to the electrode after the cycles is greater than in G and MSG (Fig. S13†). Fig. S14 and S15† present the XPS data of the SG and MSG electrodes after 50 cycles. As shown in the previous electrode expansion results, SG forms an unstable and a thick SEI layer because of electrolyte contact on a new surface. It is confirmed that the Si peak does not appear in the XPS data of the electrode after the cycles (Fig. S14†). On the other hand, in the MSG electrode, the surface carbon layer formed a thin SEI, which was confirmed by a Si peak in the XPS data (Fig. S15†). | ||
| Fig. 5 SEM-PFIB images of (a and d) G, (b and e) SG, (c and f) MSG (a–c) before cycling and (d–f) after the 50th cycle, respectively. | ||
3.4 Practical application evaluation
With the superior electrochemical performance of MSG, we further investigated practical application of the MSG as an anode in a lithium-ion full fabrication using LiCoO2 (LCO) as a cathode. Fig. 6a displays the differential capacity of LCO, indicating general cathodic/anodic peaks of LCO. The capacity of the LCO/SG and LCO/MSG was adjusted to 4.0 mA h with 1.1 of the N/P ratio. The initial discharge capacity of the full cell was 139.9 mA h g−1 with the ICE of 84.9% (Fig. 6b). In the case of SG full cell, the initial discharge capacity exhibited 137.4 mA h g−1 with an ICE of 82.3%, which is similar to the half cell results. As a result of the cycle test at 0.2C, after 100 cycles, LCO/MSG presented a higher capacity retention of 80.6% than LCO/SG of 74.7%, with stable coulombic efficiency, which was due to stable SEI formation caused by the carbon coating (Fig. 6c and d). It is expected that the increased integrity by using APS can lead to excellent cycling stability. Moreover, carbon coating on the surface can lead to the formation of a stable SEI layer that reduces side reactions caused by the electrolyte with the newly exposed surface. This indicates that the fabricated LCO/MSG electrode can be used in practical applications considering its high performance in cycling.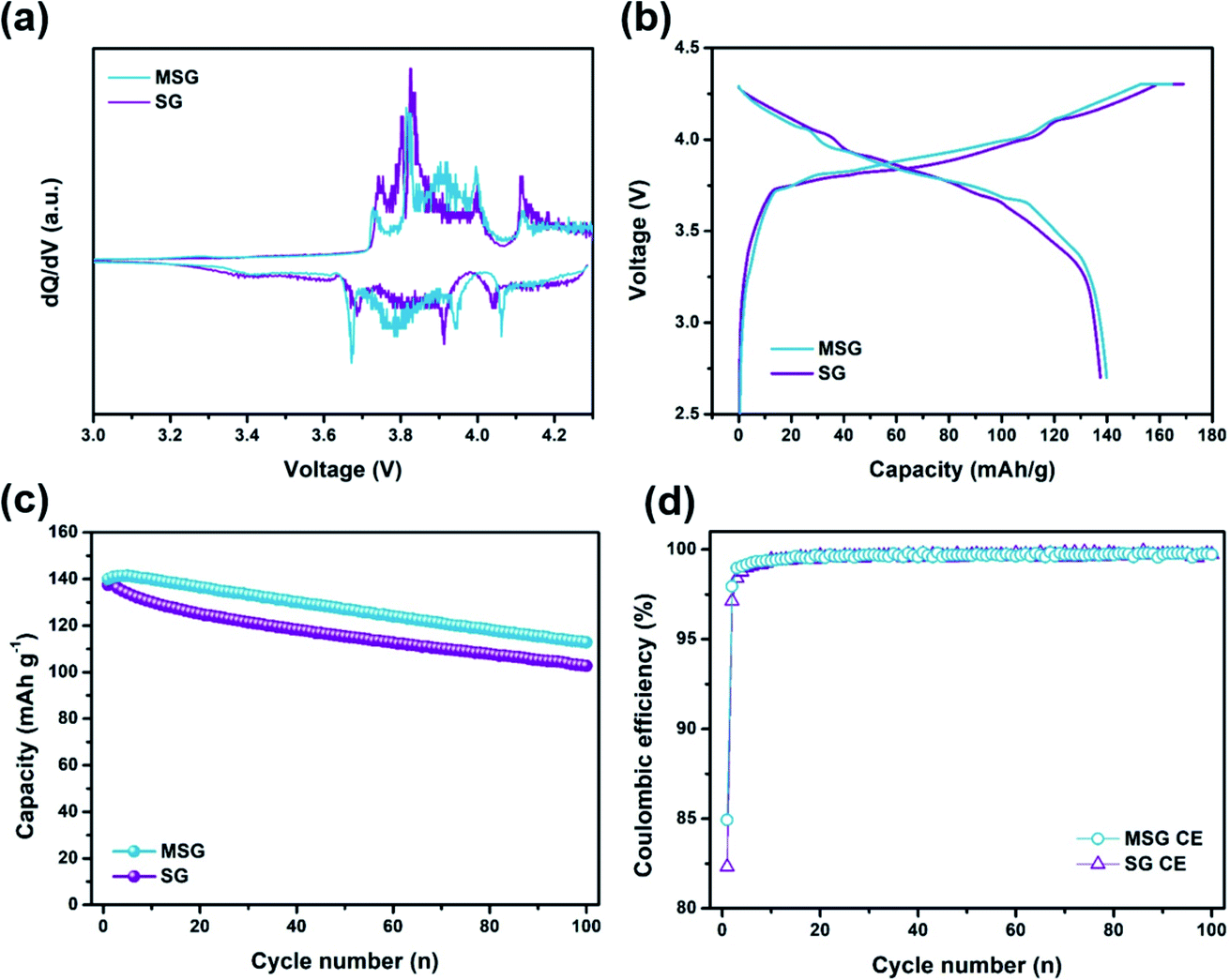 | ||
| Fig. 6 Electrochemical performance in Li-ion full batteries. (a) dQ/dV plots of SG, MSG at 1st cycle. (b) Galvanostatic charge/discharge voltage profiles of SG, MSG measured at 0.2C. (c) Discharge capacity of 100 cycles and (d) CEs of G, SG, MSG. | ||
4. Conclusions
In conclusion, we conducted complexation of graphite and SiOx through a condensation reaction by surface modification using APS. These new strategies increased the integrity of graphite and SiOx, which was further increased by carbon coating using the CVD method. This led to a high discharge capacity of MSG (429.8 mA h g−1) compared to that of graphite (320.9 mA h g−1) and a high ICE of MSG (85.6%) compared to that of SiOx (70%). In addition, the MSG electrode exhibits excellent cycling retention of 89% for 100 cycles with the lowest electrode resistance among the measured electrodes, which shows it was well-fabricated in terms of forming a SEI layer. Furthermore, electrode swelling of MSG was 12.7% after 100 cycles, and only increased by 1.3% compared to that of G, indicating controlled volume expansion. Moreover, the fabricated LCO/MSG full cell presents excellent capacity retention of 80.6% for 100 cycles. This is the result of achieving two purposes: (1) preventing electrolyte exposure and (2) maintaining electrical contact by combining graphite and SiOx through APS and CVD to form a composite. This attempt fully maintained the high capacity performance of SiOx and led to improvement of the cycle performance, which has been vulnerable. Thus, it is considered a strong candidate for introducing a new LIB anode material that exceeds the limitations of existing graphite anodes. We believe that these results are due to the increased integrity of the MSG electrodes. For the commercial use, the ICE of full-cell should be more than 90%. Prelithiaion strategy, to compensate for irreversible Li loss before full-cell fabrication, could be achieve more than 90% of ICE.45–48 This is expected to be helpful in practical applications in the future.Author contributions
J. Yun: Conceptualization, Investigation, Data curation, Writing – original draft. T. Whang: Investigatioon, Formal analysis, Writing – original draft. W. Ahn: Formal analysis, Investigation. Y. S. Lee: Supervision, Validation. J. Im: Conceptualization, Supervision, Writing – review & editing. All authors contributed to the scientific discussion and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Evaluation Institute of Industrial Technology (KEIT, No. 10083621 and No. 20007171).Notes and references
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. Speirs, M. Contestabile, Y. Houari and R. Gross, Renewable Sustainable Energy Rev., 2014, 35, 183–193 CrossRef.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- D. Liu, Z. Liu, X. Li, W. Xie, Q. Wang, Q. Liu, Y. Fu and D. He, Small, 2017, 13, 1702000 CrossRef PubMed.
- W. J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- M. Ko, S. Chae, J. Ma, N. Kim, H. W. Lee, Y. Cui and J. Cho, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- G. Li, J. Y. Li, F. S. Yue, Q. Xu, T. T. Zuo, Y. X. Yin and Y. G. Guo, Nano Energy, 2019, 60, 485–492 CrossRef CAS.
- J. Entwistle, A. Renni and S. Patwardhan, J. Mater. Chem. A, 2018, 6, 18344–18356 RSC.
- J. I. Lee and S. Park, Nano Energy, 2013, 2, 146–152 CrossRef CAS.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia and J. Y. Xie, Solid State Ionics, 2002, 152, 125–129 CrossRef.
- R. Fu, K. Zhang, R. P. Zaccaria, H. Huang, Y. Xia and Z. Liu, Nano Energy, 2017, 39, 546–553 CrossRef CAS.
- W. S. Chang, C. M. Park, J. H. Kim, Y. U. Kim, G. Jeong and H. J. Sohn, Energy Environ. Sci., 2012, 5, 6895–6899 RSC.
- Q. Xu, J. K. Sun, Z. L. Yu, Y. X. Yin, S. Xin, S. H. Yu and Y. G. Guo, Adv. Mater., 2018, 30, 1707430 CrossRef PubMed.
- H. Zhao, W. Yuan and G. Liu, Nano Today, 2015, 10, 193–212 CrossRef CAS.
- J. Zhao, H. W. Lee, J. Sun, K. Yan, Y. Liu, W. Liu, Z. Lu, D. Lin, G. Zhou and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7408–7413 CrossRef CAS PubMed.
- J. Nzabahimana, Z. Liu, S. Guo, L. Wang and X. Hu, ChemSusChem, 2020, 13, 1923–1946 CrossRef CAS PubMed.
- D. Lee, A. Kondo, S. Lee, S. Myeong, S. Sun, I. Hwang, T. Song, M. Naito and U. Paik, J. Power Sources, 2020, 457, 228021 CrossRef CAS.
- Q. Ma, H. Xie, J. Qu, Z. Zhao, B. Zhang, Q. Song, P. Xing and H. Yin, ACS Appl. Energy Mater., 2019, 3, 268–278 CrossRef.
- K. Fu, L. Xue, O. Yildiz, S. Li, H. Lee, Y. Li, G. Xu, L. Zhou, P. D. Bradford and X. Zhang, Nano Energy, 2013, 2, 976–986 CrossRef CAS.
- L. Luo, P. Zhao, H. Yang, B. Liu, J. G. Zhang, Y. Cui, G. Yu, S. Zhang and C. M. Wang, Nano Lett., 2015, 15, 7016–7022 CrossRef CAS PubMed.
- Q. Xu, J. K. Sun, Y. X. Yin and Y. G. Guo, Adv. Funct. Mater., 2018, 28, 1705235 CrossRef.
- M. Xu, J. Ma, G. Niu, H. Yang, M. Sun, X. Zhao, T. Yang, L. Chen and C. Wang, ACS Omega, 2020, 5, 16440–16447 CrossRef CAS PubMed.
- Y. Xiong, H. Xing, Y. Fan, Y. Wei, J. Shang, Y. Chen and J. Yan, RSC Adv., 2021, 11, 7801–7807 RSC.
- A. Guerfi, P. Charest, M. Dontigny, J. Trottier, M. Lagacé, P. Hovington, A. Vijh and K. Zaghib, J. Power Sources, 2011, 196, 5667–5673 CrossRef CAS.
- H. Xue, Y. Cheng, Q. Gu, Z. Wang, Y. Shen, D. Yin, L. Wang and G. Huang, Nanoscale, 2021, 13, 3808–3816 RSC.
- J. Jung, J. Jang, O. B. Chae, T. Yoon, J. H. Ryu and S. M. Oh, J. Power Sources, 2015, 287, 359–362 CrossRef CAS.
- Y. Lin, Y. Chen, Y. Zhang, J. Jiang, Y. He, Y. Lei, N. Du and D. Yang, Chem. Commun., 2018, 54, 9466–9469 RSC.
- Y. Wen, Y. Zhu, A. Langrock, A. Manivannan, S. H. Ehrman and C. Wang, Small, 2013, 9, 2810–2816 CrossRef CAS PubMed.
- Y. Cao, J. C. Bennett, R. A. Dunlap and M. N. Obrovac, Chem. Mater., 2018, 30, 7418–7422 CrossRef CAS.
- J. Lee, S. A. Han, S. M. Lee, M. S. Park and J. H. Kim, Composites, Part B, 2019, 174, 107024 CrossRef CAS.
- M. Gueye, T. Gries, C. Noël, S. Migot-Choux, S. Bulou, E. Lecoq, P. Choquet, K. Kutasi and T. Belmonte, Plasma Chem. Plasma Process., 2016, 36, 1031–1050 CrossRef CAS.
- X. Zhu, H. Chen, Y. Wang, L. Xia, Q. Tan, H. Li, Z. Zhong, F. Su and X. S. Zhao, J. Mater. Chem. A, 2013, 1, 4483–4489 RSC.
- J. S. Kim, C. C. Nguyen, H. J. Kim and S. W. Song, RSC Adv., 2014, 4, 12878–12881 RSC.
- J. J. Wu and W. R. Bennett, 2012 IEEE Energytech, 2012, pp. 1–5 Search PubMed.
- P. Li, J. Y. Hwang and Y. K. Sun, ACS Nano, 2019, 13, 2624–2633 CAS.
- W. Ren, H. Chen, R. Qiao, Y. Lin and F. Pan, J. Mater. Chem. A, 2017, 5, 22598–22606 RSC.
- W. Choi, H. C. Shin, J. M. Kim, J. Y. Choi and W. S. Yoon, J. Electrochem. Sci. Technol., 2020, 11, 1–13 CrossRef CAS.
- L. Mickelson, H. Castro, E. Switzer and C. Friesen, J. Electrochem. Soc., 2014, 161, A2121 CrossRef CAS.
- E. Park, J. Kim, D. J. Chung, M. S. Park, H. Kim and J. H. Kim, ChemSusChem, 2016, 9, 2754–2758 CrossRef CAS PubMed.
- J. Lee, J. Moon, S. A. Han, J. Kim, V. Malgras, Y. U. Heo, H. Kim, S. M. Lee, H. K. Liu, S. X. Dou, Y. Yamauchi, M. S. Park and J. H. Kim, ACS Nano, 2019, 13, 9607–9619 CrossRef CAS PubMed.
- H. Yoo, E. Park, J. Bae, J. Lee, D. J. Chung, Y. N. Jo, M. S. Park, J. H. Kim, S. X. Dou, Y. J. Kim and H. Kim, Sci. Rep., 2018, 8, 6904 CrossRef PubMed.
- C. Huang, A. Kim, D. J. Chung, E. Park, N. P. Young, K. Jurkschat, H. Kim and P. S. Grant, ACS Appl. Mater. Interfaces, 2018, 10, 15624–15633 CrossRef CAS PubMed.
- J. Choi, H. Jeong, J. Jang, A. R. Jeon, I. Kang, M. Kwon, J. Hong and M. Lee, J. Am. Chem. Soc., 2021, 143, 9169–9176 CrossRef CAS PubMed.
- K. S. Kim, J. U. Hwang, J. S. Im, J. D. Lee, J. H. Kim and M. I. Kim, Carbon letters, 2020, 30, 545–553 CrossRef.
- H. S. Jeong, J. Kim, K. I. Jo, J. Kee, J. H. Choi and J. Koo, Carbon letters, 2021, 31, 277–285 CrossRef.
- S. E. Lee, J. H. Kim, Y. S. Lee, B. C. Bai and J. S. Im, Carbon letters, 2021, 31, 911–920 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08901c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |