
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical insight into mercury species adsorption on graphene-based Pt single-atom catalysts†
Wenchao Ji a,
Xiuhua Xiaoa,
Feiyue Lia,
Xingjun Fana,
Yuanyuan Meng*b and
Maohong Fan*c
a,
Xiuhua Xiaoa,
Feiyue Lia,
Xingjun Fana,
Yuanyuan Meng*b and
Maohong Fan*c
aCollege of Resource and Environment, Anhui Science and Technology University, Fengyang 233100, China
bCollege of Chemistry & Chemical Engineering, Taiyuan University of Technology, Taiyuan 030024, P. R. China. E-mail: mengyuanyuan@tyut.edu.cn
cDepartments of Chemical and Petroleum Engineering, University of Wyoming, Laramie, WY 82071, USA. E-mail: mfan@uwyo.edu
First published on 16th February 2022
Abstract
Mercury emission from coal-fired flue gases is environmentally crucial. Revealing the interaction between mercury (Hg) and functional materials is significant to controlling emission. We conducted an investigation into the adsorption mechanism of mercury species onto graphene-based Platinum (Pt) single-atom catalysts (SACs). Single-atom Pt is the active center for Hg species chemisorption, with an adsorption energy range of 0.555–3.792 eV. In addition, Hg species adsorbed preferentially at lower temperatures. Pt/3N-GN exhibits a higher adsorption ability than Pt/SV-GN. The strong interaction of Hg0 with Pt SACs contributed to atomic-orbital hybridization between them. Further analysis revealed that s, p orbitals of Hg contribute significantly to orbital hybridization with Pt SACs. Moreover, the charge decomposition analysis confirmed that s, p orbitals of Hg hybridized with d, s orbitals of Pt SACs. The net charge transfer from Hg0 to Pt/SV-GN and Pt/3N-GN are 0.059 and 0.097 e−, respectively. The higher the charge transfers, the more intense the electron and orbital interaction between Hg and the surface. Consequently, Pt/3N-GN is a highly effective catalyst for Hg adsorption.
1. Introduction
Mercury (Hg) is neurotoxic, bioaccumulative, and environmentally persistent, arousing huge concern worldwide.1–3 The United Nations Environment Programme (UNEP) has been conducting a global assessment of Hg and its compounds for many years. UNEP has adopted an international, legally binding instrument, the Minamata Convention on Hg (entered into force on August 16, 2017), aiming at minimizing its anthropogenic emissions to the environment.4,5 The burning of fossil fuels (primarily from coal-fired power plants) is the largest single anthropogenic emission source of Hg.6 Hg in coal-fired flue gas exists as elemental mercury (Hg0), oxidized mercury (Hg2+), and particle mercury (Hgp).7,8 Hg2+ and Hgp can be effectively removed from flue gas using wet flue gas desulfurization and electrostatic precipitators.9,10 Due to its high vapor pressure and low water solubility, Hg0 is the most tedious to remove by these techniques.11,12 However, adsorption has sufficed as the most effective approach for Hg0 removal, along with HgCl and HgCl2.13,14 Therefore, it is essential to develop high-performance adsorption materials for Hg pollution control.Because of their high reactivity, long-term stability, and reusability, noble metals (such as Pt, Au and Ag) effectively adsorb and catalytically oxidize Hg0.15 In the absence of halogen species and oxygen in the flue gas, noble metals adsorb Hg0 even at temperatures up to 400 °C.16 Density functional theory (DFT) calculations also show that Hg binds relatively strongly on Pt surfaces, with binding energies up to ∼1 eV.17 However, noble metal catalysts (especially Pt-based) represented by platinum exhibit deactivation over time in the presence of halogens and oxygen. Besides, the high cost of Pt greatly limits its practical applications. To prevent deactivation and reduce the cost of noble metal catalysts, surface atoms should be maximized. For instance, nanoscience has upped the catalytic activity of catalysts by increasing the specific surface area, reactivity, and selectivity of surface atoms.
Single-atom catalysts (SACs) are derived from an innovative concept based on reaching the limit of catalyst particle size, and it has aroused global research interest.18 The SAC of Pt1/FeOx was first prepared, proving that the single-atom Pt could serve as an active site for CO oxidation.19 Later, more SACs have been synthesized and applied accordingly.20 For instance, single-atom Pt efficiently photocatalyzed hydrogen evolution with high activity and unique selectivity.21 Elsewhere, thermally stable, single-atom Pt doping on mesoporous Al2O3 was fabricated for selective hydrogenation and CO oxidation.22 Yang and co-workers23 prepared 3D-ordered mesoporous iron oxide-supported single-atom platinum with high activity for benzene combustion. Moreover, it has been reported that Pt1–Co3O4 catalyst exhibits high efficiency for the complete oxidation of methanol.24 Because Pt based SACs have a strong oxidation effect in many reactions, they are easily associated with mercury removal from flue gas. Adsorption is an essential prerequisite in catalysis. Therefore, adsorption performance is a good evaluation critical for mercury removal.
Generally, transition metal atoms embedded in graphene-based substrates are widely used to prepare SACs.25,26 Graphene, tethered with N atoms, exhibits more excellent properties than other supports.27,28 This design concept is also applied to novel carbon-based adsorption materials for removing Hg0 from gas streams.29 N-doped graphene material has been successfully used for oxygen reduction reaction.30,31 However, it is still challenging to confirm Hg0 adsorption on Pt SACs.
The research on the adsorption of mercury species in flue (coal-fired) gas on N-doped graphene-based Pt SACs is lacking, and it has tremendous inherent economic value. Here, using DFT calculations, we investigated Hg species adsorption on Pt SACs. Functionalized graphene was selected as the support for single-atom Pt. Valance defects graphene and rich-N graphene anchored the single-atom Pt, namely Pt/SV-GN and Pt/3N-GN. The adsorption energy, electronic structures, wave functional, thermodynamic properties were calculated to establish the adsorption mechanism. This research is of great significance to balancing the cost and efficiency of noble metal catalysts for Hg removal.
2. Theoretical methods
We carried out spin-unrestricted calculations for Hg species and Pt SACs (Pt/SV-GN and Pt/3N-GN) using the DMol3 package.32,33 Gradient-corrected functionals of Perdew–Burke–Ernzerhof treated the electronic exchange and correlation effects.34 DFT semi-core pseudopots method35 treated the core electrons of Pt and Hg, while all electrons were considered for C, N, and O atoms. A double numeric with polarization (DNP 4.4) basis set and a global orbital cutoff of 4.6 Å were adopted in all calculations. Brillouin zone integrations were performed with k-point generated with Monkhorst–Pack grid. A 5 × 5 × 1 grid of Monkhorst–pack points was used for geometry optimizations. The self-consistent field tolerance energy and convergence criteria were the same as in our earlier study.36 A 5 × 5 graphene supercell was constructed as catalyst support, and a vacuum thickness of 18 Å was set to avoid interaction of between layers. The slabs of Pt/SV-GN and Pt/3N-GN were fabricated according to pervious report.37 Hg species and isolated Pt atoms were placed in a cubic cell (length 18 Å) for energy calculation.The binding energy (Eb) of Pt/xN-GN (x = 0, 3) slabs was calculated using eqn (1), which EPt/xN-GN is the total energy of Pt/xN-GN (x = 0, 3), while EPt and ExN-GN are total energy of Pt atom and N doped graphene surface, respectively.
| Eb= EPt/xN-GN − (EPt + ExN-GN) | (1) |
The adsorption energy of Hg species on Pt/xN-GN (x = 0, 3) SAC surfaces was defined as follows:
| Eads = EHg-species + Eslab − EHg-species/slab | (2) |
The frequency was calculated to investigate the temperature effect on the favorability of spontaneous adsorption and to check the stability of Pt SACs, for which the numerical displacements are calculated sequentially.38 Thermodynamic data (including translational, rotational, and vibrational components) were used to calculate equilibrium constants (Keq). 250–1000 K temperature range was considered for adsorption of various Hg species on Pt/xN-GN (x = 0, 3) surfaces. The computational methods are based on the following equations:
| ln(Keq) = −ΔG/RT | (3) |
ΔG ≈ ΔEads + ΔE0 + T(ΔSvib + ΔStrans,rot) − kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(P/P0) ln(P/P0)
| (4) |
Gaussian 1639 and Multiwfn software40 carried out wave function analysis. Based on the optimized structures of Hg0 on Pt SACs by DMol3, the non-period structures were molded in Gaussian 16, and hydrogen atoms saturated the dangling bonds. The single point energy was calculated by B3LYP functional. Each system was on a GENECP basis sets the level of theory with cc-pvdz for C, H, N, and sdd for Pt.41 This ensures the consistency of the surface structure and the active centre. The graphene (5 × 5) area is large enough for Hg0 adsorption and eliminate the boundary effect. Hence, the results of DMol3 and Gaussian 16 can maintain the consistency. After obtaining the wave function of each system, we conducted the Charge Decomposition Analysis (CDA),42 which decomposed the charge transfer between molecular fragments into orbital contributions toward understanding the nature of charge transfer more clearly.
3. Results and discussion
3.1 Catalyst models
The single-atom catalysts originate from a graphene structure. Based on the selected DFT method, the calculated C–C bond of graphene is 1.42 Å, consistent with the experimental results.43 The equilibrium geometry structures of Pt anchored on defected graphene and 3N doped graphene are shown in Fig. 1.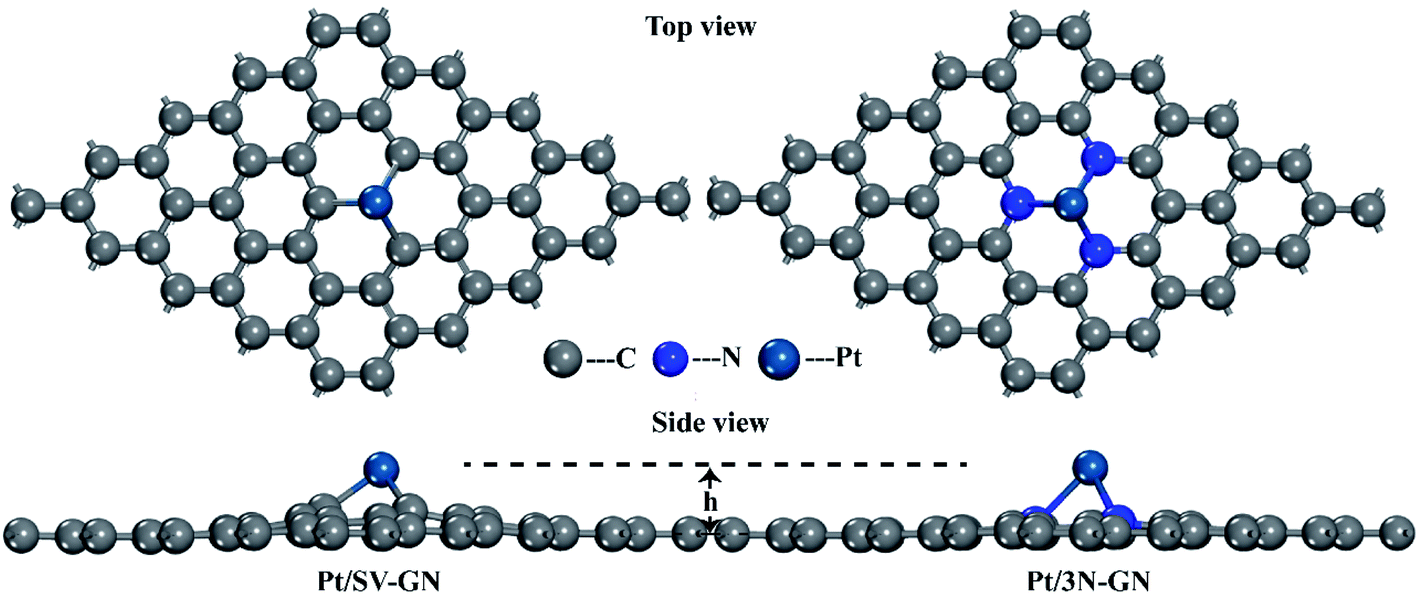 | ||
| Fig. 1 The optimized geometry structures Pt/SV-GN and Pt/3N-GN. | ||
Both Pt/SV-GN and Pt/3N-GN can maintain a two-dimensional plane structure. Graphene doped with 3N has a flatter structure. Nitrogen and carbon are almost on the same plane. The height of anchored Pt, the minimum bond length of Pt–C and Pt–N, the charge transfer of Pt atom, and the binding energies for Pt SACs are summarized in Table 1.
The Pt atom cannot be anchored in the plane of pristine GN because of the different atomic sizes among Pt, N, and C atoms. Furthermore, the geometry and local sp2 hybridization interaction of GN are destroyed by N and Pt atoms. The height of the Pt atom ranged 1.786–1.837 Å for Pt/SV-GN and Pt/3N-GN, having an uptrend with N atom introduction. Moreover, the minimum bond length of Pt–C and Pt–N are 1.946 Å in Pt/SV-GN and 2.153 Å in Pt/3N-GN, similar to those reported by Liu and Huang.44 According to the Hirshfeld charge analysis results, the charge transfer of the Pt atom is 0.392 e− in Pt/SV-GN and 0.242 e− in Pt/3N-GN. Liu et al.45 investigated a monodisperse Pt atom on 3N/GN for CO oxidation, and Hirshfeld charge transfer of Pt on the surface was 0.20, similar to that of Pt/3N-GN in the current study. The Eb of the Pt atom anchored on single C-defected graphene and 3N-doped graphene supporters is 7.536 and 2.945 eV, respectively, similar to those in related literature.44,46 Moreover, frequency calculations47 identified the stability of Pt/SV-GN and Pt/3N-GN slabs. The IR spectrums of two Pt SACs are shown in Fig. S1 (ESI).† No imaginary frequencies were found for Pt SACs, in which the lowest frequency of >60 cm−1, suggesting that the equilibrium structures are dynamically stable. The optimized structure parameters and vibrational analysis confirmed that the selected DFT method is reliable and the Pt SACs are stable for further research.
3.2 Adsorption of Hg species onto Pt SACs
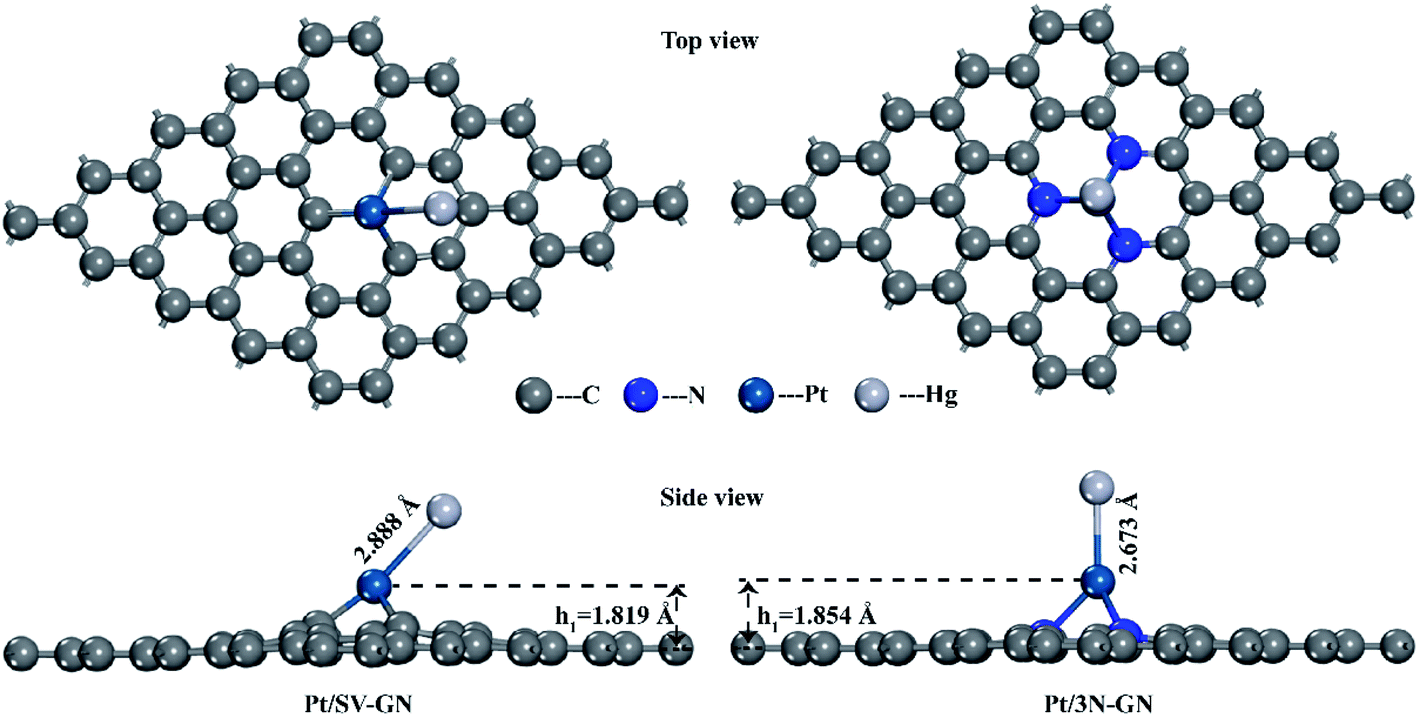 | ||
| Fig. 2 The top and side views of optimized geometry of Hg0 adsorption on Pt SACs. | ||
The surface Pt atom is the most favorable site for Hg0 adsorption, serving as the active center. The Hg–Pt (DHg–Pt) bond length is 2.888 and 2.637 Å for Pt/SV-GN and Pt/3N-GN, respectively. A new bond was formed between Hg and Pt atoms on the surfaces. The adsorption energy of Hg0 is −0.555 eV and −1.058 eV in Pt/SV-GN and Pt/3N-GN, respectively, indicative of chemisorption. Compared to Pt/SV-GN, 3N-doping GN support improves the overall interaction performance with Hg0 two-fold. Take Pt/3N-GN for example, the interaction is stronger than that of typical metal oxides, such as MnO2 (−0.812 eV),48 CeO2 (−0.093 eV)38 and Co3O4 (−0.767 eV).49 We also observed that Pt/3N-GN has a high potential for Hg0 removal.
Based on Hirshfeld charge analysis, electrons transfer from Hg0 to Pt SACs occurred with 0.121 and 0.138 e− for Pt/SV-GN and Pt/3N-GN, respectively. The remarkable electron transfer leads to a strong interaction between Hg0 and Pt SACs. Furthermore, the partial density of states (PDOS), calculated to obtain further understanding of Hg0 adsorption on Pt SACs, is plotted in Fig. 3.
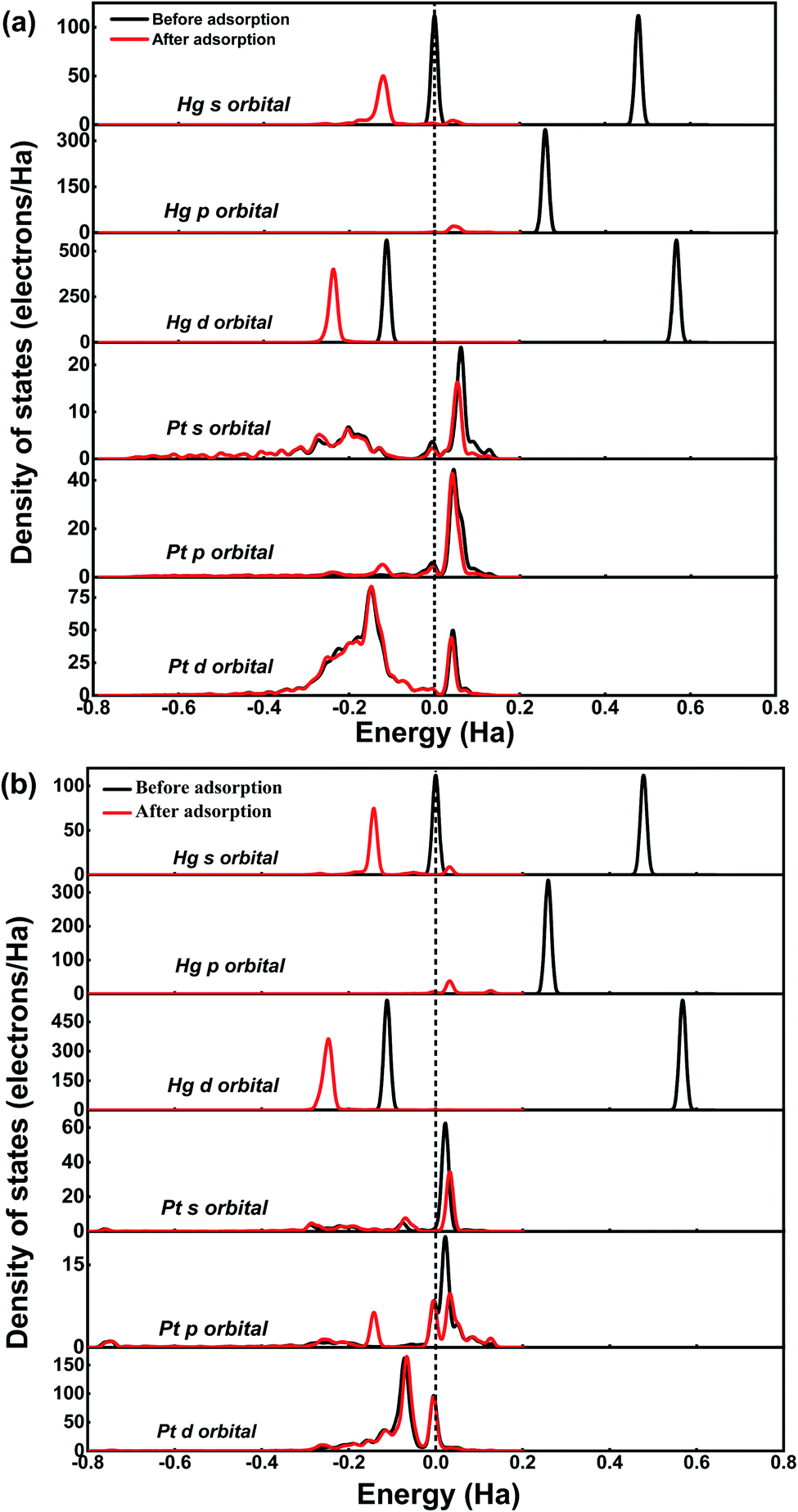 | ||
| Fig. 3 The PDOS of Hg0 and Pt before and after adsorption on Pt SACs (a) Hg0 adsorption on Pt/SV-GN (b) Hg0 adsorption on Pt/3N-GN (The black and red lines are the situations before and after adsorption, respectively. Fermi level is set as a dash line). | ||
Generally, all forms of Hg atom shift downward lower energy areas after adsorption, indicating electrons transfer from Hg0 to the catalysts. For Hg0 adsorption on Pt/SV-GN (regarding Hg atom), the s orbital is mainly distributed near Fermi level and 0.48 Ha energy level, with ≈111.96 electrons/Ha. However, it shifts to −0.12 Ha after adsorption with a PDOS value of 50.09 electrons per Ha. The p orbit is located near 0.26 Ha, with 335.09 electrons/Ha, before moving to 0.04 Ha (22.00 electrons per Ha). Moreover, the d orbital is in −0.11 and 0.57 Ha energy before adsorption and changes to −0.24 Ha after adsorption. The PDOS reduction of s- and p-orbitals is significant, which may contribute to the complex formation. No apparent change in single-atom Pt after adsorption; only a decrease of 7.34 electrons per Ha for s orbital in PDOS value occurs. In this system, Hg's s and p orbitals respectively interact with d- and s- orbitals of the single-atom Pt at −0.15 and −0.04 Ha.
Moreover, Hg0 adsorption on Pt/3N-GN has a similar trend in PDOS change, where all orbitals decrease and shift to lower energy areas with more obvious reduction. This reduction indicates that a stronger electrons interaction occurs between Hg0 and the surface. Based on the superposition of the PDOS, hybridization ensures between Hg atom's s-orbital and Pt's p-orbital atom at −0.17 Ha, while Hg-p orbital hybridization occurs with Pt-s orbital near the Fermi level. More so, the electronic orbital of the surface atom of Pt did not change significantly from that of the Hg0 before adsorption. This observation is attributable to the fact that the former is smaller than the latter. Therefore, the electronic orbital of the Pt atom is challenging to polarize.50 Hence, the Pt SACs exhibit high adsorption capacity and can maintain Hg stability in the adsorbed state.
Based on fragment orbital, charge decomposition analysis proposes a method to decompose charge transfer between molecular fragments into regular contributions to understand charge transfer better. We, therefore, analyzed Hg0 adsorption on Pt/SV-GN and Pt/3N-GN in detail. The orbital interaction diagram of Hg–Pt/SV-GN is shown in Fig. 4. Here, the Hg0 and single-atom Pt/SV-GN are defined as two fragments. The results show that orbital 159 of the complex has the most significant contribution to the electron transfer from Hg0 to Pt/SV-GN surface. Hg0 denotes 0.070 net electrons to Pt/SV-GN by forming the complex orbital 159. The 6 s orbital of Hg0 and 5d orbital of Pt/SV-GN contribute 35% and 28% for complex orbital 159, respectively. The 6px, 6py and 6pz orbitals interact with the s orbital of the 6s orbital of Pt in Pt/SV-GN surface. Frontier orbital theory is important for understanding the interaction between molecules. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), LUMO+1, LUMO+2, LUMO+3 and LUMO+4 orbitals of Hg have strong hybridization interaction with Pt/SV-GN surface.
 | ||
| Fig. 4 Hg0 adsorption on Pt-SV/GN (Frag.1, Frag.2, and complex refer to Hg0, Pt/SV-GN, and Hg–Pt/SV-GN, respectively. Solid and dash lines represent occupied orbitals and unoccupied orbitals, respectively). | ||
Since Hg0 adsorption on Pt/3N-GN corresponds to the open-shell system, alpha and beta orbitals interaction diagrams are plotted in Fig. 5. As shown in Fig. 5(a), 154 alpha orbital of the complex contributed most significantly to the electron transfer from Hg0 to Pt/3N-GN surface. Hg0 denotes 0.059 net electrons to Pt/SV-GN by forming the orbital 154 complex. In addition, the 6 s orbital of the Hg0 and 5d orbitals contribute 51% and 10% for orbital 154, respectively. The 6px, 6py and 6pz orbitals interact with the 6s orbital of Pt in Pt/3N-GN surface. The HOMO, LUMO, LUMO+1, LUMO+2, LUMO+3 and LUMO+4 orbitals of Hg have strong hybridization interaction with Pt/3N-GN surface. The LUMO+1 of Hg has the same orbital phase as the 6s orbital of Pt/3N-GN; they are strongly mixed. Compared with beta orbitals between Hg0 and Pt/3N-GN in Fig. 5(b), the interaction between the related orbitals is weaker than that of alpha orbitals, with predominant anti-bonding orbital interactions. The net electrons obtained by Pt/3N-GN is 0.038, less than electron transfer among the alpha orbitals. By combining the alpha and beta orbital interactions, the net electron transfer corresponds to 0.097.
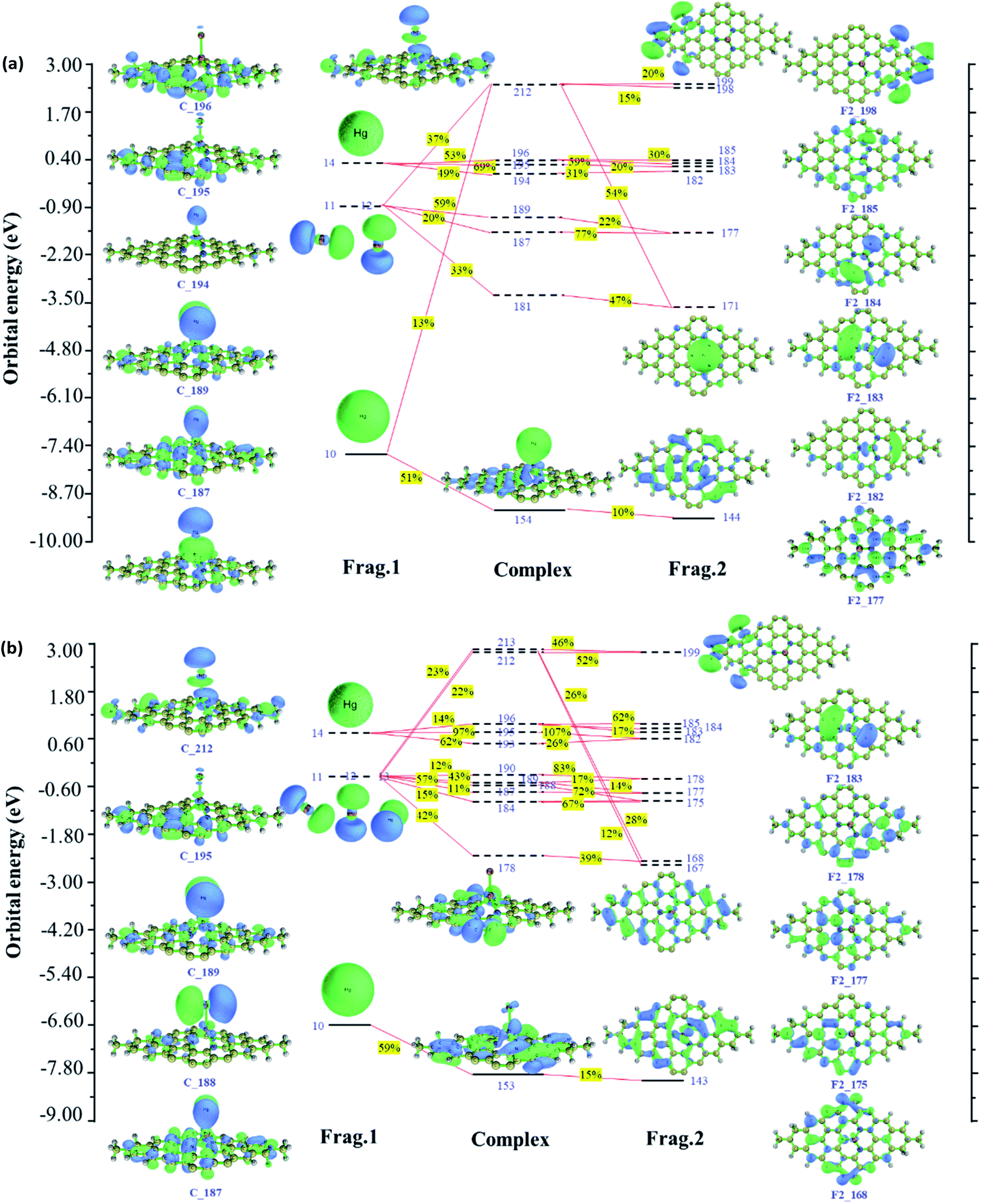 | ||
| Fig. 5 Schematic of orbital interaction mechanism for Hg0 adsorption on Pt–3N/GN (a) alpha orbitals (b) beta orbitals (Frag.1, Frag.2, and complex refer to Hg0, Pt/3N-GN and Hg–Pt/3N-GN, respectively. Solid and dash lines represent occupied and unoccupied orbitals, respectively). | ||
Fig. 5 shows the orbital interactions between Hg0 and Pt SACs. Generally, the CDA results are consistent with PDOS, having the same trend in electron transfer and orbital hybridization. This result can be used as a powerful supplement to PDOS analysis to understand the interaction of orbitals in the adsorption process.
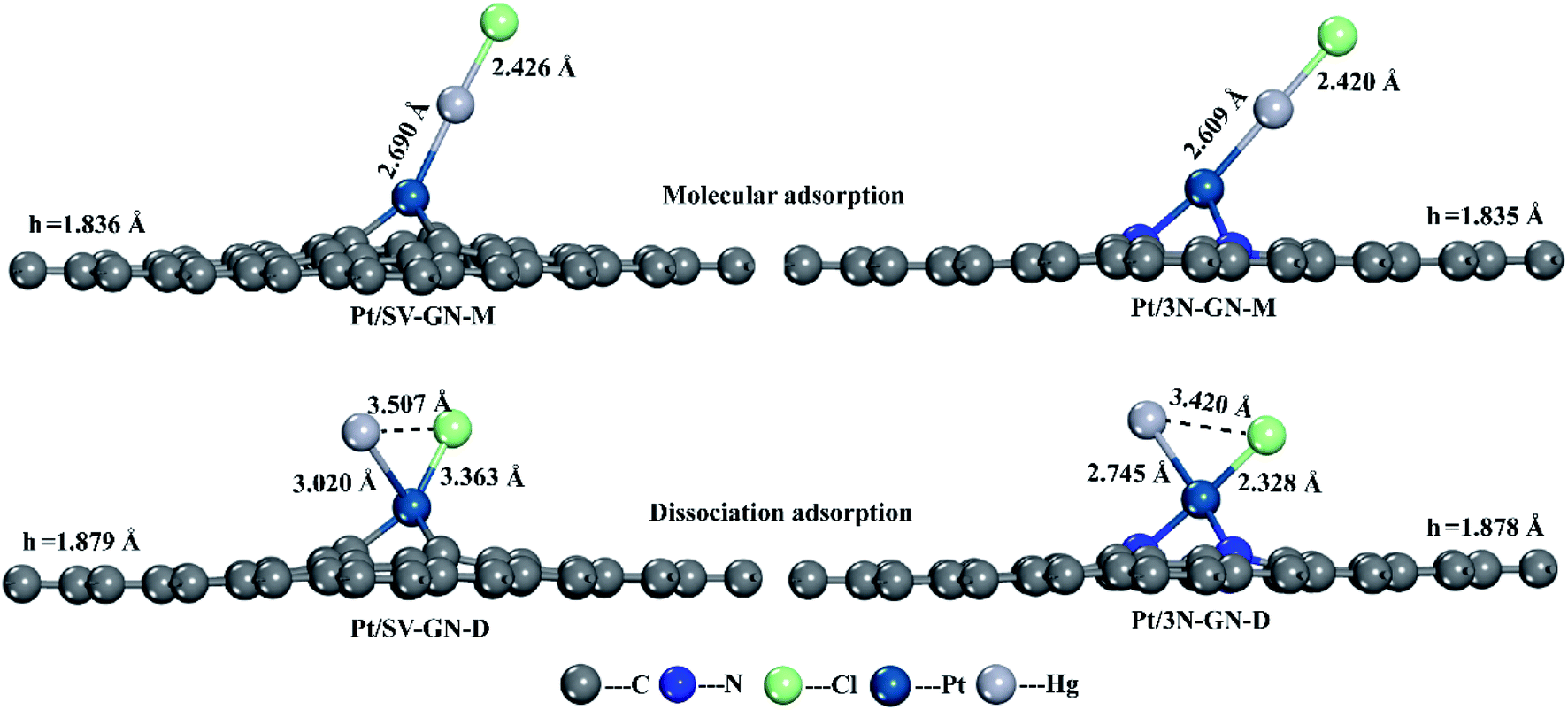 | ||
| Fig. 6 The optimized geometry structures of HgCl adsorption on Pt/SV-GN and Pt/3N-GN. | ||
To further investigating the desorption properties of HgCl on Pt SACs, we calculated the potential energy of HgCl on different catalysts. In Fig. 7, Hg desorbs faster as Hg0 because the energy needed in this process is only 0.116 and 0.470 eV for Pt/SV-GN and Pt/3N-GN, respectively. The greater the adsorption capacity, the greater the energy needed for adsorbed HgCl, consistent with the general adsorption fundamental rules. In molecular adsorption, desorption for HgCl* to Hg + Cl* from the Pt SACs is tedious due to the high energy barrier. Hence, the HgCl molecule adsorbs refractorily on the surface of the Pt SACs. According to the principle of exothermic reactions, dissociation adsorption is more favorable than molecular adsorption of HgCl. This inference is consistent with the reported adsorption of HgCl on Fe2O3,52 MnFe2O4,53 ZnO54 and Fe SACs.55 Generally, HgCl is one of the intermediates in Hg0 oxidation. It cannot be stably adsorbed on Pt SACs, indicating that direct oxidation may occur rather than gradual oxidation.
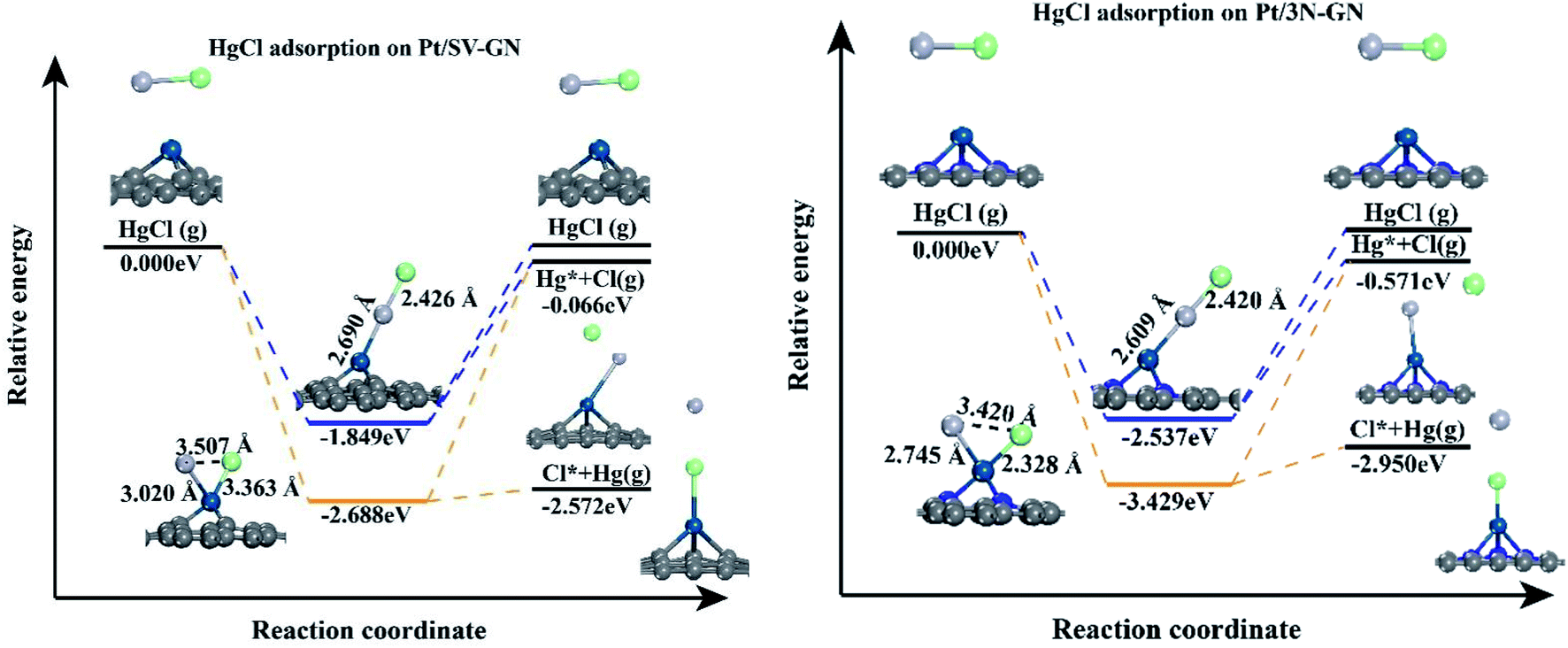 | ||
| Fig. 7 The potential energy diagrams of HgCl on Pt SACs. | ||
 | ||
| Fig. 8 The optimized geometry structures of HgCl2 adsorption on Pt SACs. | ||
In vertical adsorption, all Hg–Cl bond lengths are elongated in varying degrees compared to gas phase HgCl2 (2.32 Å, reported value of 2.42 Å51). The height of the Pt atom increases in Pt SACs after HgCl2 adsorption, in which Pt/3N-GN has a more noticeable change. HgCl2 has a stronger interaction (−1.555 eV) with Pt/3N-GN than Pt/SV-GN. In bending adsorption, HgCl2 dissociates to HgCl and Cl and bonding with the surface Pt atom after adsorption. The adsorption energy is higher than that of the vertical model, especially with HgCl2–Pt/3N-GN (−3.231 eV) system. Also, the height of Pt atom shows a positive correlation with the adsorption strength.
The potential energy diagrams were plotted to understand the desorption behaviors of HgCl2 on Pt SACs (Fig. 9). In vertical adsorption, adsorbed HgCl2 is more inclined to desorb from the surface, forming HgCl. The energies needed for Pt/3N-GN are only 0.968 eV, while HgCl2 desorption to form HgCl is a spontaneous process on Pt/SV-GN. The adsorbed HgCl2 can be thoroughly desorbed on Pt SACs because desorption energies are ≤ 1.389 eV, which can be realized in the actual coal-fired flue gas environment.
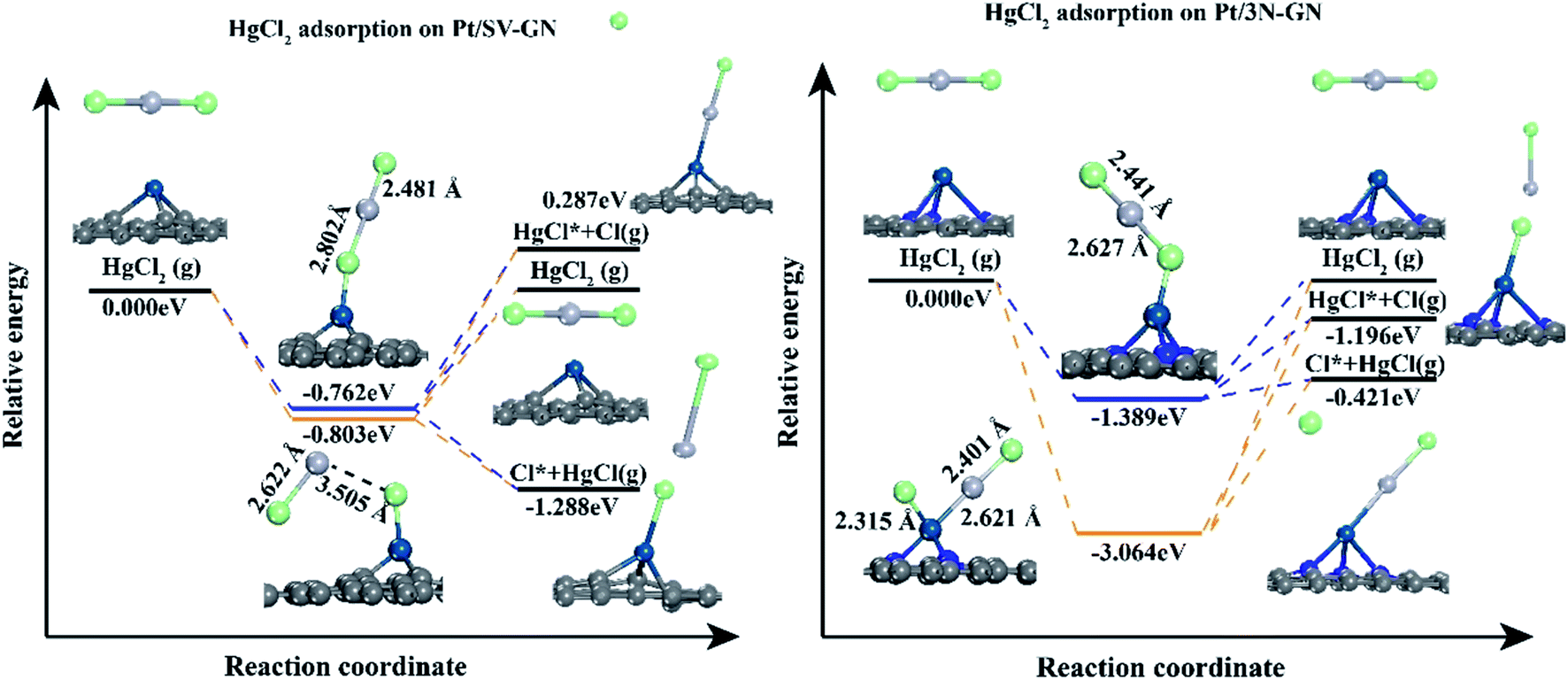 | ||
| Fig. 9 The potential energy diagram of HgCl2 on Pt SACs. | ||
As for bending adsorption, HgCl2 can be stably adsorbed on the Pt SACs. The energies needed for gas-phase HgCl2 formation are 0.803 and 3.064 eV on Pt/SV-GN and Pt/3N-GN, respectively. This process depends on the actual environmental conditions of the coal-fired flue gas. It has been reported that HgCl2 is the main existing form when the temperature is lower than 725 K.56 When the temperature is >975 K, >99% of Hg (as Hg0) exists in coal-fired plants' flue gas.
3.3 Thermodynamic analysis
In controlling Hg from coal-fired flue gas, the Hg species distribution varies with flue gas temperature. Temperature is key to understanding Hg adsorption on the catalyst. Here, we studied the equilibrium constants of Hg species on Pt SACs. Based on DFT thermodynamic analysis, the Gibbs free energy change and the adsorption equilibrium constants are calculated in corresponding systems at 250–1000 K. Changes in the Gibbs free energies are listed in the ESI (Table S1).† Generally, the change of Gibbs free energies is negative, indicating that Hg adsorption on Pt SACs is spontaneous. Further, the relationship between equilibrium constants and temperature is shown in Fig. 10.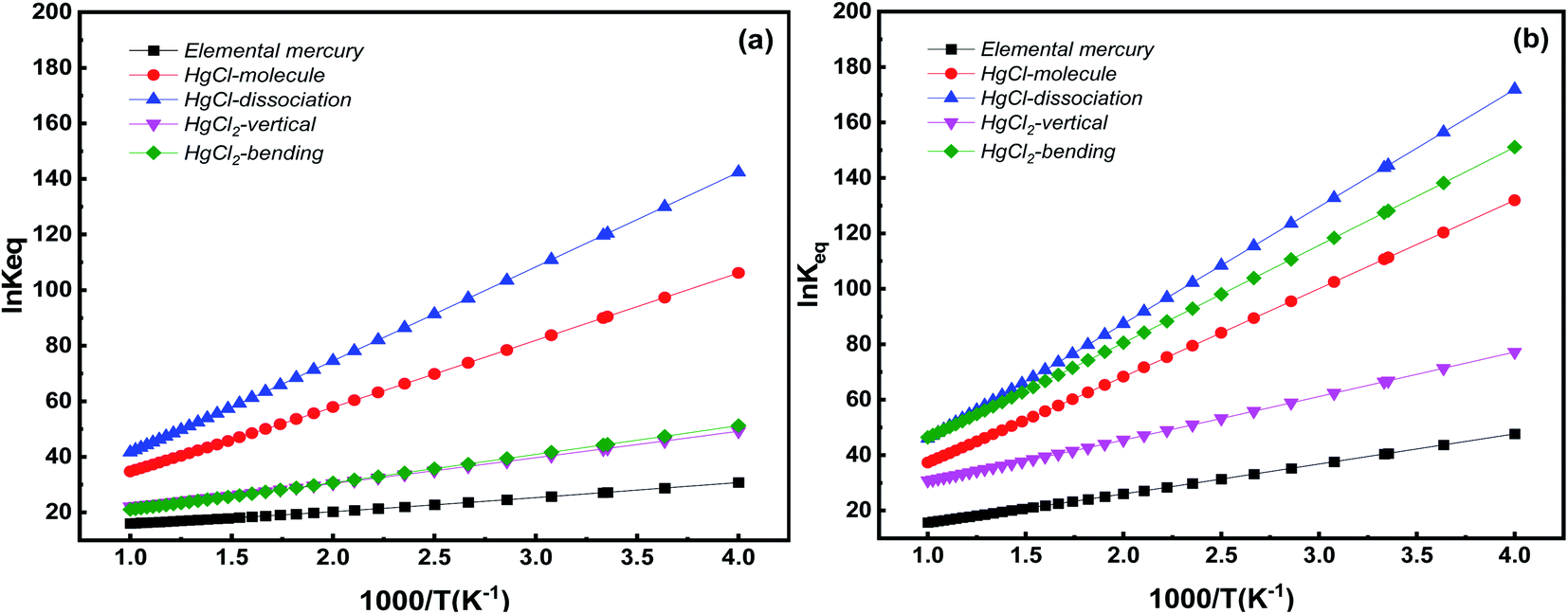 | ||
| Fig. 10 Equilibrium constants for various Hg species adsorption on Pt SACs as a function of temperature (a) Pt/SV-GN (b) Pt/3N-GN. | ||
Hg species adsorption equilibrium constants decrease inversely with temperature in all adsorption systems, i.e., low temperature favors Hg adsorption on Pt SACs. This conclusion is consistent with the previous theoretical calculation and experimental results.49,57 Specifically, Pt/SV-GN is the most favorable catalyst for HgCl dissociation adsorption, followed by HgCl molecular adsorption, then HgCl2 adsorption in both bending and vertical models, and finally, Hg0 adsorption. On the Pt/3N-GN surface, the dissociation adsorption of HgCl has the highest adsorption equilibrium constant in all the selected temperature ranges, followed by HgCl2 adsorption in the bending model, HgCl in the molecular model, HgCl2 in the vertical model. By comparing the two Pt SACs, the adsorption equilibrium constants of Hg0 on Pt/3N-GN is slightly higher than that on Pt/SV-GN. We can improve the adsorption equilibrium constants by doping N atoms on Pt-based graphene. At the high-temperature area, two Pt SACs evinced similar adsorption equilibrium constants for all Hg species. However, Pt/3N-GN exhibits higher equilibrium constants than Pt/SV-GN in the low-temperature area. Therefore, Pt/3N-GN is more suitable for the adsorption Hg species at low temperatures, as previous observed.58 Hence, the equilibrium constant of HgCl dissociation adsorption is more sensitive to temperature change than other adsorption processes.
4 Conclusions
Spin-polarized DFT calculations were conducted to evaluate Hg adsorption behaviors on graphene-based Pt SACs. All Hg species are chemisorption on Pt SACs surface where Pt provides more active adsorption sites. Pt/3N-GN shows higher adsorption activity for Hg than that of Pt/SV-GN. PDOS and wave functional analysis show that s, p orbitals of Hg contribute significantly to forming complex orbitals with Pt SACs. We identify that sd and ps hybridization occur between Hg0 and surface Pt atom of Pt SACs. In addition, CDA results show that the net electron transfer from Hg0 to Pt/SV-GN and Pt/3N-GN are 0.070 and 0.097 e−, respectively. Dissociation adsorption of HgCl and HgCl2 on Pt SACs is dominant. The Pt/3N-GN has higher adsorption equilibrium constants than that of Pt/SV-GN for Hg species, having a better adsorption effect at a lower temperature range. The adsorption capacity of Pt SACs for Hg0 is stronger than that of traditional bulk materials, which is controlled by a chemical interaction with atom orbital hybridization. Finally, Pt SACs exhibit tremendous potential for Hg species adsorption.Author contributions
Wenchao Ji: conceptualization, calculations, formal analysis, writing – original draft preparation. Xiuhua Xiao: formal analysis, writing – review & editing. Feiyue Li: formal analysis, writing – review & editing. Xingjun Fan: formal analysis, writing – review & editing. Yuanyuan Meng: conceptualization, software, writing – review & editing. Maohong Fan: conceptualization, formal analysis, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52100114), the Natural Science Foundation of Anhui Province (No. 2108085QB56 and 2108085MD140), University Science Research Project of Anhui Province (No. KJ2020A0049), Talent Projects of Anhui Science and Technology University (No. ZHYJ201802), and Anhui Zhenhao Environmental Protection Technology Co., Ltd (No. ZHEPST2020003). We also thank to International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.Notes and references
- L. Chen, S. Liang, Y. Zhang, M. Liu, J. Meng, H. Zhang, X. Tang, Y. Li, Y. Tong and W. Zhang, Environ. Sci. Technol., 2018, 52, 13792–13800 CrossRef CAS PubMed.
- D. Raj and S. K. Maiti, Environ. Monit. Assess., 2019, 191, 566 CrossRef CAS PubMed.
- Z. Fu and S. Xi, Toxicol. Mech. Methods, 2020, 30, 167–176 CrossRef CAS PubMed.
- D. M. Meyer, L. M. Kaste, K. M. Lituri, S. Tomar, C. H. Fox and P. E. Petersen, Dent. Clin., 2016, 60, 921–942 Search PubMed.
- M. A. Coulter, Int. Leg. Mater., 2016, 55, 582–616 CrossRef.
- J. M. Pacyna, E. G. Pacyna, K. Sundseth, J. Munthe, S. Wilson and J. Leaner, 2020.
- Q. Tang, W. Ji, W. Guo, Z. Shen and M. Fan, Ind. Eng. Chem. Res., 2019, 58, 11101–11110 CrossRef CAS.
- C. Li, Y. Duan, H. Tang, C. Zhu, Y.-n. Li, Y. Zheng and M. Liu, Fuel, 2018, 211, 621–628 CrossRef CAS.
- S. Zou, Y. Liao, S. Xiong, N. Huang, Y. Geng and S. Yang, Environ. Sci. Technol., 2017, 51, 3426–3434 CrossRef CAS PubMed.
- W. Yang, Y. Shan, S. Ding, X. Han, Y. Liu and J. Pan, Environ. Technol., 2019, 40, 1923–1936 CrossRef CAS PubMed.
- X. Zhang, Q. Shi, B. Shen, Z. Hu and X. Zhang, J. Hazard. Mater., 2020, 381, 121003 CrossRef CAS PubMed.
- R. Hao, Z. Wang, X. Mao, Y. Gong, B. Yuan, Y. Zhao, B. Tian and M. Qi, J. Hazard. Mater., 2019, 374, 120–128 CrossRef CAS PubMed.
- H. Zhao, S. Yin, L. Lu, Z. Rui, C. Zheng, C. Hu, X. Gao and T. Wu, J. Hazard. Mater., 2020, 381, 121037 CrossRef CAS PubMed.
- X. Zhang, L. Cui, Y. Li, Y. Zhao, Y. Dong and S. Cao, Energy Fuels, 2019, 33, 7852–7860 CrossRef CAS.
- E. J. Granite, H. W. Pennline and R. A. Hargis, Ind. Eng. Chem. Res., 2000, 39, 1020–1029 CrossRef CAS.
- B.-A. Dranga, L. Lazar and H. Koeser, Catalysts, 2012, 2, 139–170 CrossRef CAS.
- J. A. Steckel, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 115412 CrossRef.
- S. Liang, C. Hao and Y. Shi, ChemCatChem, 2015, 7, 2559–2567 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65 CrossRef CAS.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang and T. Zhang, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- K. Yang, Y. Liu, J. Deng, X. Zhao, J. Yang, Z. Han, Z. Hou and H. Dai, Appl. Catal., B, 2019, 244, 650–659 CrossRef CAS.
- Z. Jiang, X. Feng, J. Deng, C. He, M. Douthwaite, Y. Yu, J. Liu, Z. Hao and Z. Zhao, Adv. Funct. Mater., 2019, 1902041 CrossRef.
- H. Li, H.-x. Zhang, X.-l. Yan, B.-s. Xu and J.-j. Guo, New Carbon Mater., 2018, 33, 1–11 CrossRef CAS.
- L.-L. Liu, C.-P. Chen, L.-S. Zhao, Y. Wang and X.-C. Wang, Carbon, 2017, 115, 773–780 CrossRef CAS.
- J. Liu, Q. Ma, Z. Huang, G. Liu and H. Zhang, Adv. Mater., 2019, 31, 1800696 CrossRef PubMed.
- M. Narreddula, R. Balaji, K. Ramya, N. Rajalakshmi and A. Ramachandraiah, Int. J. Hydrogen Energy, 2019, 44, 4582–4591 CrossRef CAS.
- D. Liu, C. Li, J. Wu and Y. Liu, Chem. Eng. J., 2019, 123514 Search PubMed.
- J. Oh, S. Park, D. Jang, Y. Shin, D. Lim and S. Park, Carbon, 2019, 145, 481–487 CrossRef CAS.
- X. Wen, Z. Duan, L. Bai and J. Guan, J. Power Sources, 2019, 431, 265–273 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 78, 1396 CrossRef.
- D. R. Hamann, M. Schlüter and C. Chiang, Phys. Rev. Lett., 1979, 43, 1494 CrossRef CAS.
- W. Ji, Q. Tang, Z. Shen, M. Fan and F. Li, Appl. Surf. Sci., 2020, 501, 144233 CrossRef CAS.
- Z. Gao, X. Li, A. Li, C. Ma, X. Liu, J. Yang and W. Yang, Appl. Organomet. Chem., 2019, 33, e5079 CrossRef.
- B. Zhang, J. Liu and F. Shen, J. Phys. Chem. C, 2015, 119, 15047–15055 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- F. C. Tian Lu, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- S. I. Gorelsky and E. I. Solomon, Theor. Chem. Acc., 2008, 119, 57–67 Search PubMed.
- H. Sun, S. Mukherjee, M. Daly, A. Krishnan, M. H. Karigerasi and C. V. Singh, Carbon, 2016, 110, 443–457 CrossRef CAS.
- S. Liu and S. Huang, Carbon, 2017, 115, 11–17 CrossRef CAS.
- X. Liu, Y. Sui, T. Duan, C. Meng and Y. Han, Catal. Sci. Technol., 2015, 5, 1658–1667 RSC.
- M. Rafique, Y. Shuai, M. Xu, G. Zhang and Y. Guo, Physica E, 2017, 93, 26–38 CrossRef CAS.
- J.-x. Zhao, Y. Chen and H.-g. Fu, Theor. Chem. Acc., 2012, 131, 1242 Search PubMed.
- B. Zhang, J. Liu, C. Zheng and M. Chang, Chem. Eng. J., 2014, 256, 93–100 CrossRef CAS.
- W. Ji, Z. Shen, Q. Tang, B. Yang and M. Fan, Chem. Eng. J., 2016, 289, 349–355 CrossRef CAS.
- Y. Yang, J. Liu, B. Zhang and F. Liu, J. Hazard. Mater., 2017, 321, 154–161 CrossRef CAS PubMed.
- Z. Xu, X. Lv, J. Chen, L. Jiang, Y. Lai and J. Li, Chem. Eng. J., 2017, 308, 1225–1232 CrossRef CAS.
- J.-E. Jung, S. Liguori, A. D. Jew, G. E. Brown Jr and J. Wilcox, J. Air Waste Manage., 2018, 68, 39–53 CrossRef CAS PubMed.
- Y. Yang, J. Liu, B. Zhang and F. Liu, Chem. Eng. J., 2017, 308, 897–903 CrossRef CAS.
- L. Ling, S. Zhao, P. Han, B. Wang, R. Zhang and M. Fan, Chem. Eng. J., 2014, 244, 364–371 CrossRef CAS.
- W. Yang, Z. Gao, X. Ding, G. Lv and W. Yan, Appl. Surf. Sci., 2018, 455, 940–951 CrossRef CAS.
- C. L. Senior, A. F. Sarofim, T. Zeng, J. J. Helble and R. Mamani-Paco, Fuel Process. Technol., 2000, 63, 197–213 CrossRef CAS.
- Z. Tan, S. Su, J. Qiu, F. Kong, Z. Wang, F. Hao and J. Xiang, Chem. Eng. J., 2012, 195, 218–225 CrossRef.
- B. Zhang, J. Liu, C. Zheng and M. Chang, Chem. Eng. J., 2014, 256, 93–100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08891b |
| This journal is © The Royal Society of Chemistry 2022 |