
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoluminescent polymer micelles with thermo-/pH-/metal responsibility and their features in selective optical sensing of Pd(II) cations†
Haruka Takebuchi and
Ren-Hua Jin *
*
Department of Material and Life Chemistry, Kanagawa University, 3-2-7 Rokkakubashi, Yokohama 221-8686, Japan. E-mail: rhjin@kanagwa-u.ac.jp
First published on 15th February 2022
Abstract
Photoluminescent polymers can be divided into two types of structures: one is the well-known conventional π-conjugated rigid chain polymers bearing π-conjugated chromophores in their side chains, and the other is the common flexible polymers without π-conjugated chromophores in their main or side chains but with a feature of clustering electron-rich and/or dipole groups in their main and/or side chains. In this work, we found a new photoluminescent polymer comprising theophylline (T) and imidazole (I) residues in a suitable ratio in the side chains on the common polystyrenic block (PVB-T/I). We synthesized a block copolymer (denoted as P2) consisting of hydrophobic PVB-T/I and hydrophilic poly(N-isopropylacrylamide), and we investigated its self-assembly into micelles and their micellar features, such as thermo-responsibility, fluorescence emission, pH, and metal ion-dependent photoluminescence, in detail. Especially, the micelles self-assembled from P2 showed intrinsic blue emission which was emitted from the charge transfer association between T and I residues in the intra-chains. Weakening the association by adjustment of the pH or addition of metal ions could evidently reduce the photoluminescence in the micellar state. Very interestingly, among many metal cations, only Pd2+, which can chelate strongly with theophylline, strongly quenched the photoluminescence from the micelles. Therefore, the polymer micelles functioned as an optical sensor for Pd(II) ion not only by spectroscopy but also with the naked eye.
Introduction
Amphiphilic macromolecules have great potential for accessing functional nanomaterials because their hydrophilic and hydrophobic components prefer self-organization in aqueous media, affording nanostructured entities such as spherical micelles, rods, worms, vesicles and other higher-level hierarchical structures.1a–e Amphiphilic macromolecules used in functional nanomaterials are also called smart polymers, which can respond to heat,2–5 pH,6–8 light,9–13 and solvents.14–16 These smart polymers can be applied in the fields of nanosensors, bioimaging, catalysts, etc. Among them, temperature-responsive copolymers with a lower critical solution temperature (LCST)2–5 and upper critical solution temperature (UCST)17,18 have been studied most widely. As is well known, poly(N-isopropylacrylamide) (PNIPAM) is often used as an LCST-type polymer, and it shows a sharp coil-globule phase transition at temperatures near 32 °C.19–23 The design of copolymers possessing PNIPAM blocks or segments in different structural dimensions is a very attractive and desirable process to approach thermo-responsive materials with additional responsivities.19 Light-responsive polymers are interesting molecules that can be combined with thermoresponsive polymers.There are many reports about luminescent polymers with π-conjugated moieties24–31 such as pyrene,24,25 porphyrin,26 and rhodamine27,28 in their side chains and/or end groups. Although these luminescent polymers have high potential for sensors, fluorescence probes and imaging materials, they possess poor hydrophilicity and/or readily cause aggregation quenching. Therefore, they are not good candidates for use in the aggregated state in aqueous systems. As a useful tool for adapting photofunctional polymers to aqueous media, the polymers are often constructed as amphiphiles possessing hydrophobic luminescent segments and hydrophilic moieties, by which it is feasible to respond not only to light but also to environmental changes such as heat and pH.32–39 These designed polymers can enhance the effect of the luminescence emission and thus make it possible to control the ON/OFF switching of their sensing power, which exerts its function in a certain environment. For example, introducing some moieties with the property of aggregation-induced emission (AIE) in the hydrophobic block could enhance the fluorescence emission of the amphiphilic block copolymer in aqueous media due to the appearance of self-assembled entities driven by hydrophobic interactions between the AIE moieties.40 Different from such AIE systems, in the case of aggregation-caused quenching (ACQ), π-conjugated polymers such as poly[fluorenyl-alt-p-phenyleneethynylene] (PFPE) backbones with pendant carboxylic acid groups can function very well as fluorescent sensors for distinguishing aliphatic and aromatic amines in aqueous media; aliphatic amines associated with pendant groups of carboxylic acid cause extension of the π-conjugated polymer chains to overcome ACQ, while aromatic amines interact with the π-conjugated polymer chains with π–π interactions, forcing ACQ.31
In the issue of π-conjugated polymers, the imidazole-conjugated structure is also very effective for developing photoluminescent materials.41 Imidazole is a heterocyclic compound with an imine group as a proton acceptor and an amine group as a proton donor in the same aromatic ring, and imidazole itself is also sensitive to pH and/or metal ions. The architecture of π-conjugated polymers containing imidazole rings or imidazole-condensed rings has attracted much attention because of their photonic properties as charge transfer chromophores and the unique photophysical process of excited-state intramolecular proton transfer (ESIPT).42–44 Imidazole-conjugated polymers bearing proton-donating phenol side groups can cause effective ESIPT by photoexcitation when they form intramolecular hydrogen bonds between the imidazole and phenol groups.45 In this system, however, the covalent conjugation between the electron donor and electron acceptor (often an imidazole moiety) is indispensable. Although there are remarkable achievements of polymers possessing imidazole-condensed rings as side chains on their photoactive main chains, such as polythiophene and polycabazole, it is rare that luminescent polymers contain imidazole rings but possess no π-conjugation backbone structures.
Recently, nonconventional fluorescent polymers without π-conjugation structures have also drawn great research interest.46–48 Among them, water-soluble hyperbranched polyethyleneimine (h-PEI) is especially interesting. Luo and coworkers reported that h-PEI reacted with aldehydes could function as light emission polymers due to the formation of Schiff base bonds.49–51 In this hyperbranched structure, the Schiff base bonds could crowd into clusters which are very similar to AIE to increase the radiative decay when excited.50,51 They also demonstrated that the emission of the Schiff base bond-based h-PEI could respond to pH or acidic compounds and metal ions in aqueous media.49 In practice, many polymers, even those without conventional chromophores (only have groups of cyano, imine, carbonyl, tertiary amine, etc.), are also potentially photoluminescent materials, as the polymers afford definite aggregates through which space conjugation occurs.52 Polyacrylonitrile is a typical nonconventional chromophore which can emit blue fluorescence in the solid state when excited by UV light.53
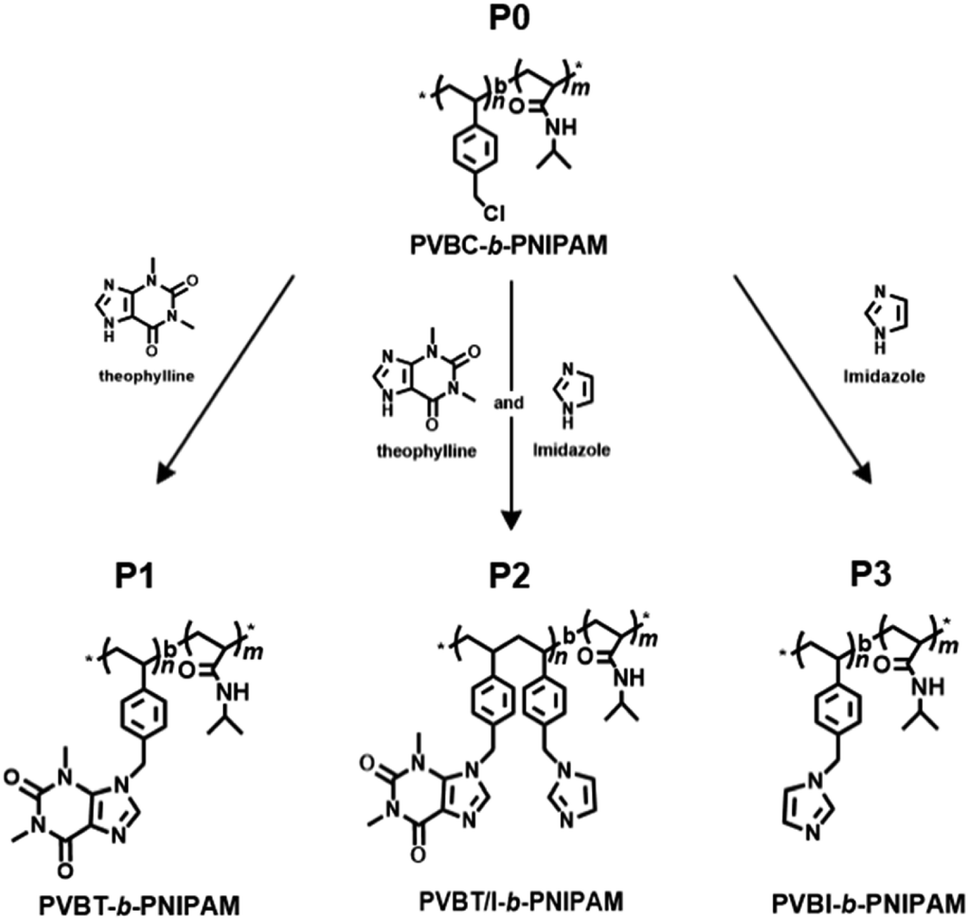
Very recently, we revealed that theophylline, an imidazole-condensed xanthine derivative, is a strong receptor of Pd(II) ions due to the formation of an extraordinary stable complex with Pd(II), and we found that polymers bearing residues of the theophylline coordinated-Pd(II) complex could function as good catalysts in C–C coupling reactions.54,55 Herein, we focused our attention on multiple stimuli-responsive issues in the design of an amphiphilic block copolymer comprising a pH/photo-responsive non-conjugated hydrophobic polystyrenic block with pendant rings of a xanthine derivative (theophylline) and imidazole and a thermo-responsive hydrophilic block of PNIPAM. We firstly synthesized a block copolymer that consisted of a poly(vinylbenzyl chloride) (PVBC) block and PNIPAM block via RAFT polymerization, and then we introduced theophylline (xanthine derivative) and/or imidazole pendants into the PVBC block. By changing the ratio of theophylline (T) and imidazole (I) in the pendants, we prepared targeting polymers of PVB(T/I)-b-PNIPAM, where T/I was altered to 100/0, 77/23 and 0/100. It was found that PVBT-b-PNIPAM (P1, T/I = 100/0) and PVBT/I-b-PNIPAM (P2, T/I = 77/23) appeared to be amphiphiles that could easily form micelles in aqueous media, while PVBI-b-PNIPAM (P3, T/I = 0/100) became a double-hydrophilic block copolymer which dissolved in water. All the polymers showed thermo-responding ability due to possessing a PNIPAM block. Very interestingly, polymer P2 with T/I in a definite ratio in its side groups showed remarkably blue emission, although P1 and P3 themselves are weakly or not emissive under the same conditions. We revealed that intra-polymer chain associations between theophylline and imidazole in the side chains of P2 functioned as an intricate emitter to afford blue emission. This emitter acted as an excellent optical sensor for Pd(II) ion because Pd ion strongly chelates with the theophylline moiety, resulting in dissociation of the emitter and thus in quenching of the photoluminescence.
Results and discussion
Preparation and characterization of the PVB(T/I)-b-PNIPAM block copolymers
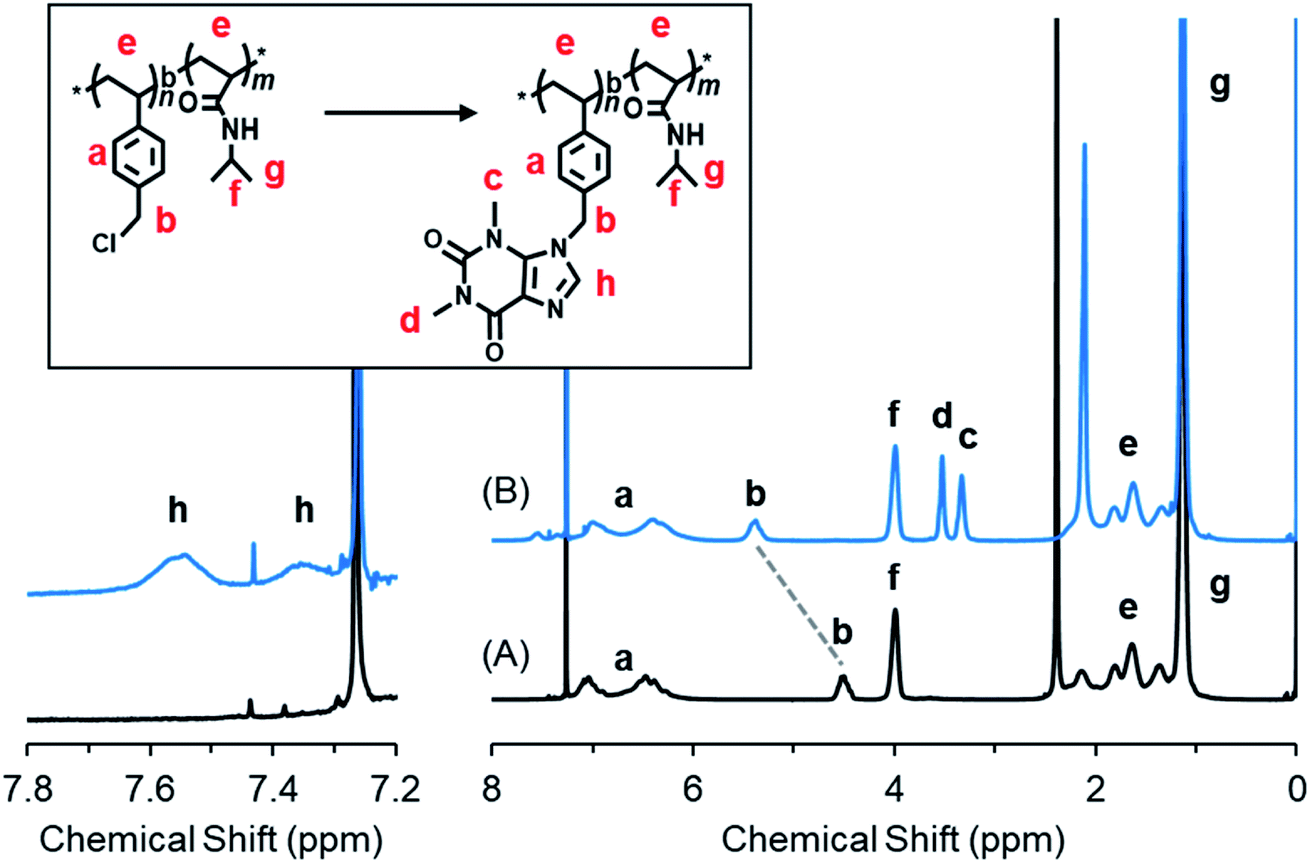
Scheme 1 shows the strategy to synthesize our target PVB(T/I)-b-PNIPAM polymers possessing theophylline and/or imidazole pendants in the polystyrenic block. The block copolymer of PVBC-b-PNIPAM, in which the side group –CH2Cl on PVBC is very active to react easily with nucleophiles, was synthesized by our previously reported method via a standard RAFT polymerization procedure.56,57 Firstly, VBC was polymerized using cyanomethyl benzodithioate (CMB) as a chain transfer agent and AIBN as the initiator. Then, the PVBC obtained was used as a macro-chain transfer agent in copolymerization with NIPAM for preparation of block copolymer PVBCn-b-PNIPAMm-SC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)Ph. (see Scheme S1†). Finally, the end groups of dithioate on PVBCn-b-PNIPAMm-SC(S)Ph were removed to obtain the end-deactivated block copolymer PVBCn-b-PNIPAMm (P0). The polymerization degrees of n and m in P0 are 74 and 383, respectively, which were estimated by 1H NMR end analysis of the integral value method (for details, see Fig. S1†). The related GPC profiles before and after copolymerization are displayed in Fig. S2,† and the polydispersity Mw/Mn values are summarized in Table S1.† We performed the introduction of theophylline and imidazole compounds onto P0 by dissolving P0, theophylline and/or imidazole in DMF and heating the solution at 80 °C for 24 h. The three isolated polymers of P1, P2 and P3 are very soluble in chloroform, so we characterized the polymers by 1H NMR spectroscopy using CDCl3 as the solvent. It was found that the chemical shift due to –CH2Cl at 4.48 ppm for P0 completely disappeared after reaction with theophylline (T) or imidazole (I) or a mixture of T/I, indicating the high reactivity of P0 with imidazole ring compounds (Fig. 1 and S3†). As shown in Fig. 1, P1 showed four new peaks due to theophylline residues compared to P0. The signals at δ 3.33 ppm and 3.53 ppm are ascribed to the two methyl groups on –NCH3 of theophylline, and the weak peaks at δ 7.35 ppm and 7.55 ppm are due to the condensed imidazole ring (–NCH). The peaks due to PNIPAM at δ 4.00 ppm of the methine group (N–CHMe2) and δ 1.34 ppm of the two methyl groups on the isopropyl group did not change after the reaction. In contrast, the methylene peak (–CH2Cl) at 4.51 ppm on the PVBC block shifted to 5.39 ppm (–CH2N) after introducing theophylline. The introduction ratio was ca. 99%, which was calculated from the ratio of the integral values of peaks c (–NCH3, theophylline residue) and b (–CH2N–, methylene in the PVBC block) (see Fig. S3†). P2 and P3 were characterized similarly, and their 1H NMR spectra are shown in Fig. S4.† The reaction of P0 with T/I (feeding molar ratio of 70
S)Ph. (see Scheme S1†). Finally, the end groups of dithioate on PVBCn-b-PNIPAMm-SC(S)Ph were removed to obtain the end-deactivated block copolymer PVBCn-b-PNIPAMm (P0). The polymerization degrees of n and m in P0 are 74 and 383, respectively, which were estimated by 1H NMR end analysis of the integral value method (for details, see Fig. S1†). The related GPC profiles before and after copolymerization are displayed in Fig. S2,† and the polydispersity Mw/Mn values are summarized in Table S1.† We performed the introduction of theophylline and imidazole compounds onto P0 by dissolving P0, theophylline and/or imidazole in DMF and heating the solution at 80 °C for 24 h. The three isolated polymers of P1, P2 and P3 are very soluble in chloroform, so we characterized the polymers by 1H NMR spectroscopy using CDCl3 as the solvent. It was found that the chemical shift due to –CH2Cl at 4.48 ppm for P0 completely disappeared after reaction with theophylline (T) or imidazole (I) or a mixture of T/I, indicating the high reactivity of P0 with imidazole ring compounds (Fig. 1 and S3†). As shown in Fig. 1, P1 showed four new peaks due to theophylline residues compared to P0. The signals at δ 3.33 ppm and 3.53 ppm are ascribed to the two methyl groups on –NCH3 of theophylline, and the weak peaks at δ 7.35 ppm and 7.55 ppm are due to the condensed imidazole ring (–NCH). The peaks due to PNIPAM at δ 4.00 ppm of the methine group (N–CHMe2) and δ 1.34 ppm of the two methyl groups on the isopropyl group did not change after the reaction. In contrast, the methylene peak (–CH2Cl) at 4.51 ppm on the PVBC block shifted to 5.39 ppm (–CH2N) after introducing theophylline. The introduction ratio was ca. 99%, which was calculated from the ratio of the integral values of peaks c (–NCH3, theophylline residue) and b (–CH2N–, methylene in the PVBC block) (see Fig. S3†). P2 and P3 were characterized similarly, and their 1H NMR spectra are shown in Fig. S4.† The reaction of P0 with T/I (feeding molar ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) produced the corresponding polymer P2 with a ratio of T/I of 77/23. The introduction ratios estimated by the 1H NMR spectra are summarized in Table S2.†
:30) produced the corresponding polymer P2 with a ratio of T/I of 77/23. The introduction ratios estimated by the 1H NMR spectra are summarized in Table S2.†
 | ||
| Scheme 1 Representation of the synthesis of the PVB(T/I)-b-PNIPAM block copolymers. | ||
 | ||
| Fig. 1 1H NMR spectra of (A) PVBC74-b-PNIPAM383 (P0) and (B) P1 in CDCl3 at 25 °C. | ||
Imidazole and theophylline dissolved in chloroform showed absorption peaks at 241 nm and at 273 nm, respectively (Fig. S5†). With this as a reference, we measured the UV-Vis absorption of polymers P0–P3. As shown in Fig. 2A, there are no absorption peaks at 273 nm for P0 or P3, which contain no theophylline moiety. In contrast, P1 and P2 showed absorption at 273 nm due to the presence of theophylline residues in the pendant. In the expansion of the spectral line from 300 to 400 nm (Fig. 2A′), we can see that P3 with only imidazole in its pendants gives a tailing line, while P2, which contains both imidazole and theophylline in its side chains, exhibits an obvious shoulder at around 340 nm, although P1 has no absorption in this area.
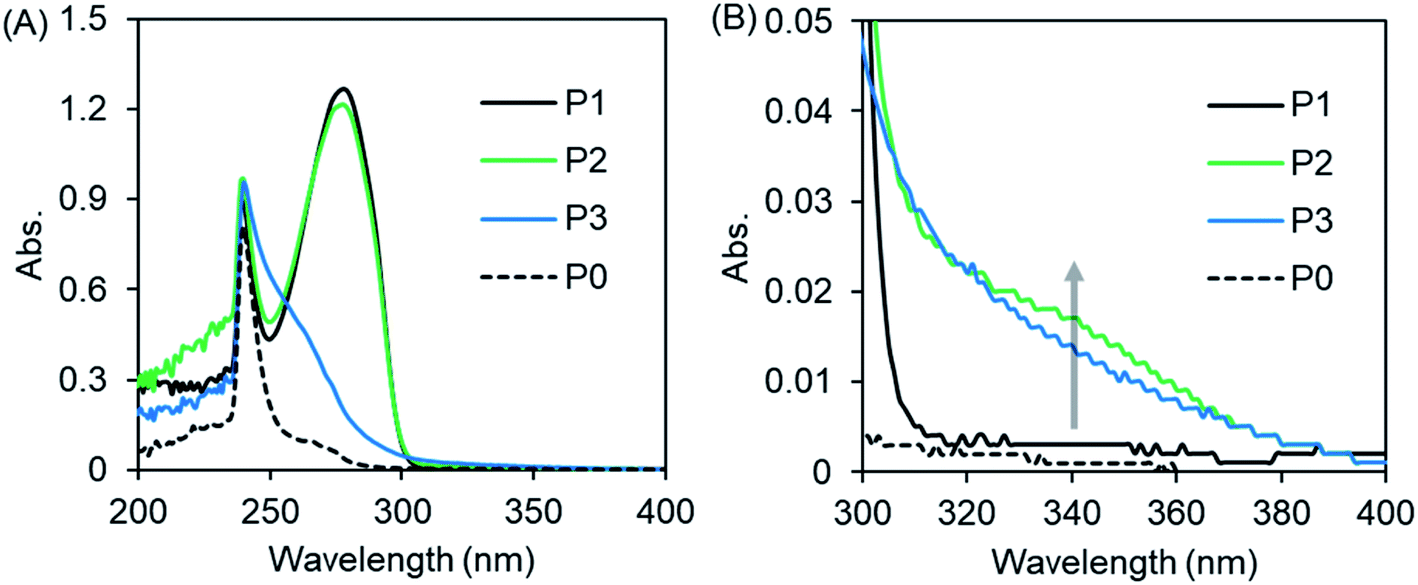 | ||
| Fig. 2 (A) UV-vis spectra of PVB(T/I)-b-PNIPAM in CHCl3 measured at 25 °C. (B) is the enlarged area around 300–400 nm of (A). The polymer concentration is 2 g L−1. | ||
To understand the thermophysical properties of the polymers, we performed differential scanning calorimetry (DSC) measurements for the block copolymers of P0–P3 (Fig. 3A) and the homopolymers of PVBC74, PVBT74 and PNIPAM334. As can be seen from Fig. 3B, the glass transition temperatures (Tg) for the homopolymers of PVBC74 and PNIPAM334 appeared at 89.2 °C and 124.8 °C, respectively. After the introduction of theophylline into PVBC74, the corresponding PVBT74 showed a Tg of 180.3 °C, indicating that the pendant theophylline makes the polymer chains rigid and enhances the interactions between the polymer chains. In comparison, the block copolymers P0–P3, which consisted of PNIPAM383 and polystyrenic blocks possessing different side chains, showed similar Tg values of around 141–145 °C, which are higher than the Tg values of the homopolymers PVBC74 and PNIPAM334 but lower than the Tg of the homopolymer PVBT74. It seems that there is no formation of individual phases from the two blocks of polystyrenic derivatives (with smaller DP 74) and PNIPAM (with larger DP 383). Probably, the good compatibility between the two blocks enhances the miscibility of the polymer chains, leading to a homogeneous phase with a new glass transition temperature (Tg).
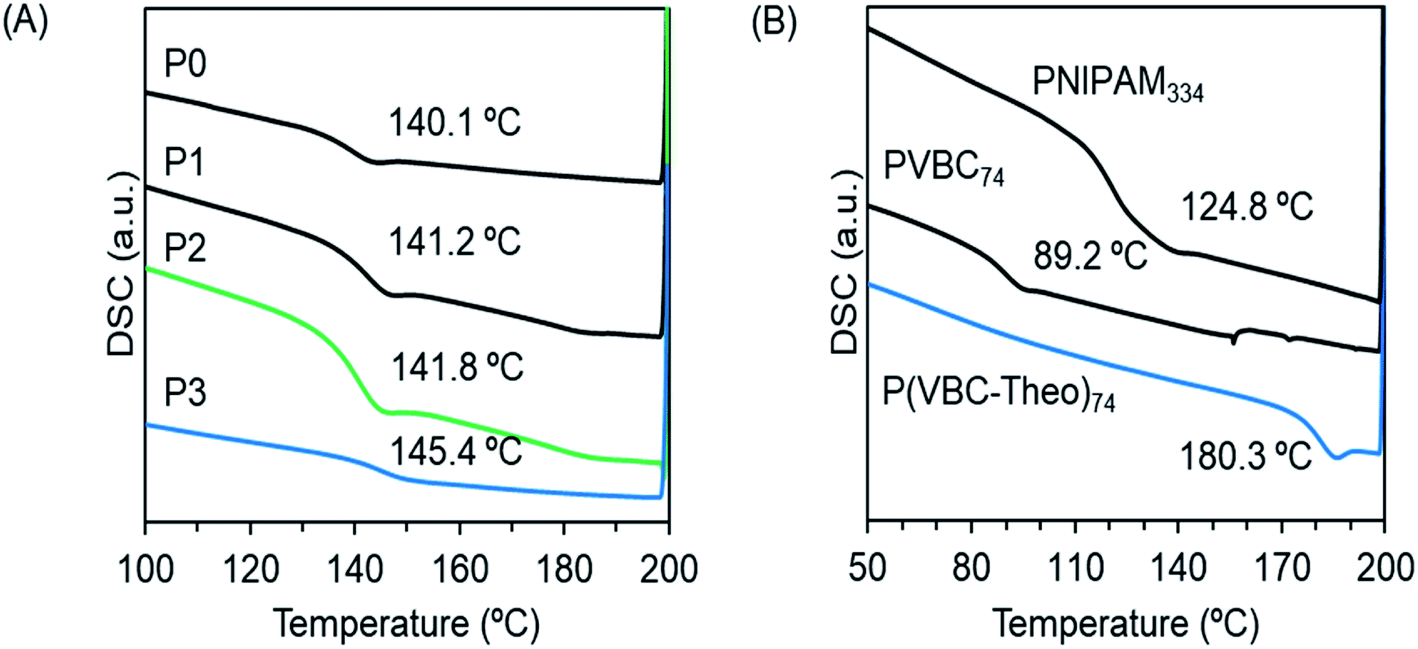 | ||
| Fig. 3 DSC curves of the second heating of the (A) block copolymers and (B) homopolymers. | ||
Micelle formation and temperature-responsiveness
Theophylline itself is easily soluble in water and methanol. However, the homopolymer PVBT74 with theophylline pendants was not soluble in water, even after heating to 100 °C. Moreover, unexpectedly, this homopolymer with a high Tg was not easily soluble in DMF or methanol, but was very soluble in chloroform at room temperature and also soluble in DMSO upon heating. Therefore, we think that in the polystyrenic chain with theophylline pendants, the inter-chain and/or intra-chain π–π interactions between the theophylline moieties and between theophylline and phenyl residues in the side chains may feature in its physical performance. This idea would be helpful in understanding the behavior of block copolymers in aqueous systems. We confirmed that the block copolymers of P1 and P2 with the hydrophilic PNIPAM block are easily soluble in chloroform and DMSO but do not show good solubility in water, methanol or DMF, while P3 is soluble in water and chloroform. Therefore, P1 and P2 could be described as amphiphilic block copolymers, while P3 should be considered to be a double-hydrophilic block copolymer. Here, we performed micellization of P0, P1 and P2 by the solvent selection method. After adding a small amount of a DMSO solution of P0, P1 and P2, respectively, to an excess amount of water, it was found that P1 and P2 resulted in micellar dispersion at 2 mg mL−1 concentration, but P0 at the same concentration led to precipitation. In contrast, direct dissolution of P3 in water resulted in a transparent aqueous solution (2 mg mL−1).Since the block copolymers possess longer PNIPAM blocks, they should exhibit thermo-responsibility in aqueous media. Therefore, we conducted micellar dispersions of P1 and P2 and the solution of P3 in a thermo-heater-equipped DLS and UV-Vis spectrometer to investigate the thermo-responsivity of the polymers in aqueous media. At room temperature, the P1 and P2 dispersions showed hydrodynamic diameters of 89 and 71 nm, respectively, indicating the formation of polymeric micelles, while the P3 solution did not show a light scattering signal because of the molecular solution (see Fig. 4A). As shown in Fig. 4B, when the dispersions were heated to 45 °C, the hydrodynamic diameter increased to 380 nm for P1 but decreased to 24 nm for P2. In comparison, at 45 °C, heating-triggered micellization occurred from the molecular solution of P3 due to dehydration of the PNIPAM block, showing a hydrodynamic diameter of 200 nm. On the other hand, at a wavelength of 500 nm, we compared the temperature-dependent transparences of the samples. As can be seen from Fig. 4C, below 30 °C, the transparence of the dispersion of P1 was over 30%, but it decreased to near zero as the temperature increased to 35 °C. The transparence of the P3 dispersion decreased from 92% to 50% as it was heated to 50 °C, and the micelles of P2 only showed a slight change from 87% to 77% even when heated to 50 °C. This change is in agreement with the heat-induced change of the micellar size. The smaller the micellar size, the higher the transparency, and the larger the micellar size, the lower the transparency. Apparently, each polymer that possessed the PNIPAM block indicated an LCST but at different temperatures. The LCSTs for P1, P2 and P3 appeared at 30 °C, 30 °C and 34 °C, respectively. Here, the relationship between the transmittance and heating/cooling cycles for the micellar dispersion of P1 were conducted in the temperature range between 25 °C and 45 °C with 10 min intervals. As shown in Fig. 4D, the thermo-response is highly reversible in each recycle. P2 and P3 also responded well with reversibility on heating and cooling recycles (data not shown). The thermo-response of P1 was also confirmed by 1H NMR spectroscopy in D2O (see Fig. S6†). At 25 °C, only the peaks due to the isopropyl group of PNIPAM appeared. This is because of the formation of core–shell micelles in which the polystyrenic block with theophylline pendants formed a core (non-solvated solid state) while the PNIPAM block formed a shell layer (solvated coil state). When heated to 45 °C, the peaks due to the chemical shifts of PNIPAM disappeared, indicating the phase-transition of the PNIPAM shell into dehydrated globules from hydrated coils. Photographs of the samples in vials at room temperature and under heated conditions are displayed in Fig. S7.† The solutions of P1 and P3 became remarkably opaque, while that of P2 did not show distinguishable cloudiness. These findings are also in agreement with the argument based on the results shown in Fig. 4.
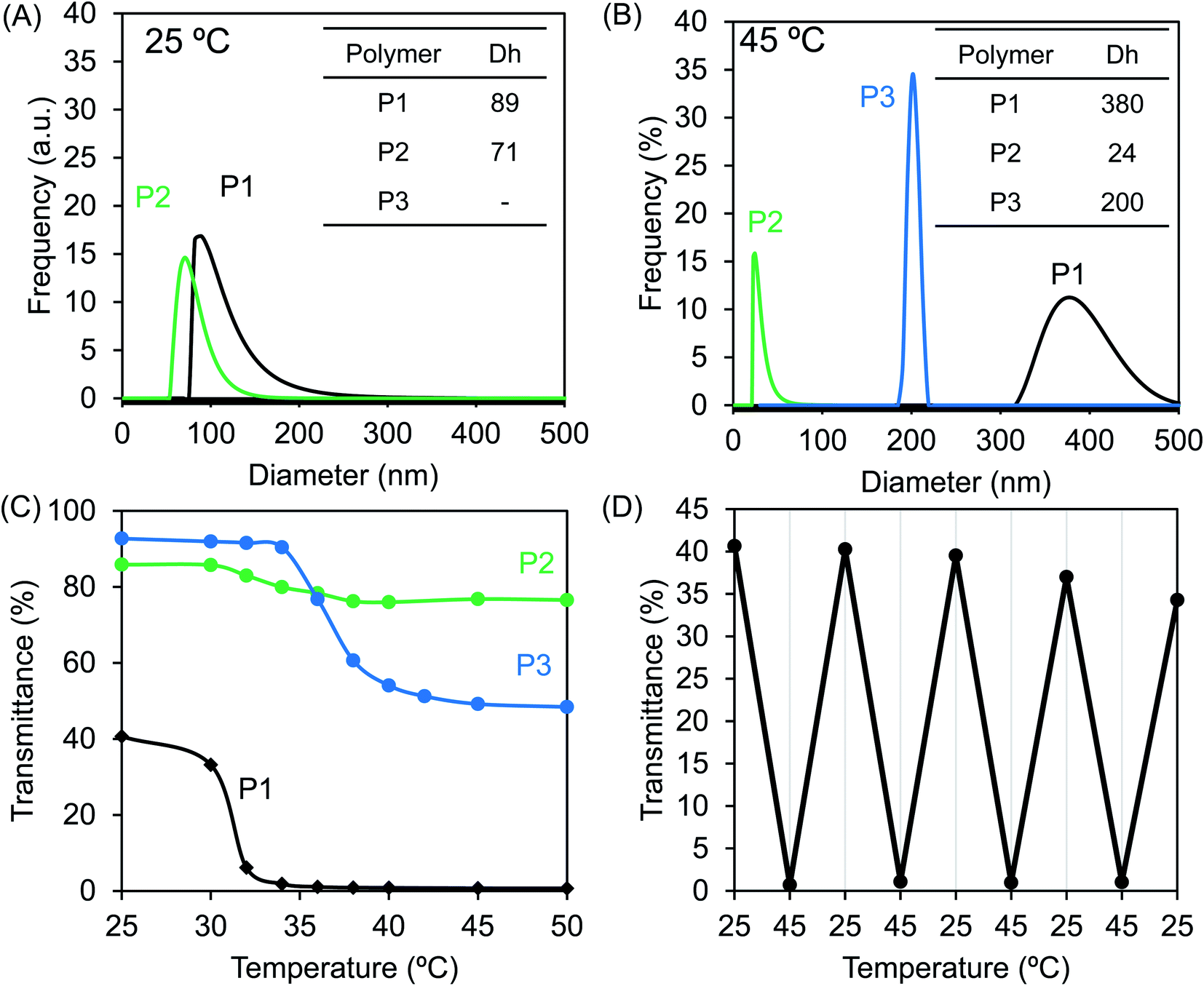 | ||
| Fig. 4 DLS traces of aqueous dispersions of P1–P3 (concentration: 2 g L−1) at (A) 25 °C and (B) 45 °C. (C) Temperature-dependent transmittance of the aqueous dispersions of P1–P3 (2 g L−1) at λ = 500 nm. (D) Transmittance variations for the dispersion of P1 (2 g L−1) depending on the heating–cooling cycle between 25 °C and 45 °C at λ = 500 nm. | ||
Based on the above results, the thermo-responsivity of the micellar dispersions can be explained as illustrated in Fig. 5. Below the LCST, P1 exists as core–shell micelles with a PVBT core and PNIPAM shell. Above the LCST, the shell of the micelles from P1 loses water due to dehydration of PNIPAM, and some inter-micellar interactions take place to form larger micellar aggregates. This is reversible with heating and cooling. Similarly, P2 forms core–shell micelles below the LCST. However, the micelles of P2 do not associate with each other above the LCST but form smaller micelles. This should arise from the contributions of intra-micellar interactions: that is, the shrinking of the PNIPAM shell leads the PNIPAM chains to collapse onto the micellar core and enhances the intra-micellar polymer-to-polymer interactions between PNIAPM and the theophylline/imidazole moieties existing in the core surface. Compared to P1, the thermo-response of P2 micelles is restricted only to intra-micelle contact. It seems that the pathway of thermo-response towards inter-micellar or intra-micellar is determined by the core components; the core gathered with theophylline alone causes inter-micellar association of the shells, while the core consisting of random arrangements of theophylline/imidazole leads only to intra-micellar collapse. In contrast to both the amphiphilic polymers of P1 and P2, the double hydrophilic block copolymer P3 is a unimer coil below the LCST but becomes core–shell micelles with the formation of the PNIPAM core and PVBI shell above the LCST.
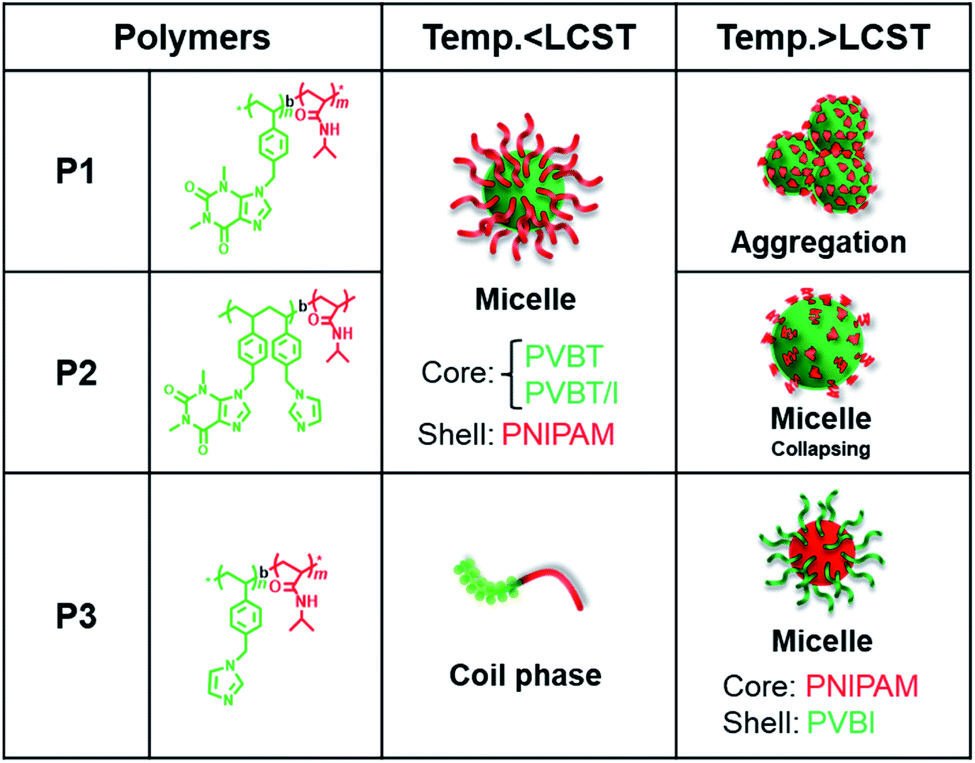 | ||
| Fig. 5 Schematic of the self-assembly of the temperature-responsive PVB(T/I)-b-PNIPAM series in water. | ||
Emission and pH-response of amphiphilic block copolymers
As we mentioned above (Fig. 2B), P2 with imidazole and theophylline pendants exhibits an obvious shoulder above 300 nm in its UV-Vis spectrum in chloroform. This attracted our attention to examine the possibility of using the polymer as a chromophore because the shoulder might be related to charge transfer associations between imidazole and theophylline. Therefore, we firstly examined the micellar and unimer solutions of P1–P3 by fluorescence spectroscopy. As shown in Fig. 6A, P2 emitted noticeable fluorescence at 425 nm when excited at 365 nm. In comparison, the emission intensity of P1 is about 1/3 that of P2, while the emission is very faint for P3. As the micellar dispersion of P2 was heated to 45 °C, the emission intensity decreased slightly from 905 to 726 (Fig. 6B). This also suggests the above description, in which the collapse of the PNIPAM shell onto the micellar core due to heating causes interactions between PNIPAM and the theophylline/imidazole moieties in the core, which somewhat weakens the interactions between theophylline and imidazole. It was also confirmed that P2 emits fluorescence emission at the same concentration in chloroform solution, but the emission intensity is lower than that in the micellar state (Fig. S8A†). Here, it should be noted that compared to P2, block copolymers with components of T/I of about 30/70 or 13/87 show inferior fluorescence emission (Fig. S8E†). It seems that the components of T/I (in molar) near 70/30 in the polymer pendants are better for emitting photoluminescence and favor retention of the isolated micellar state without inter-micellar aggregation in its thermo-response. This is a desirable characteristic for use as smart micelles.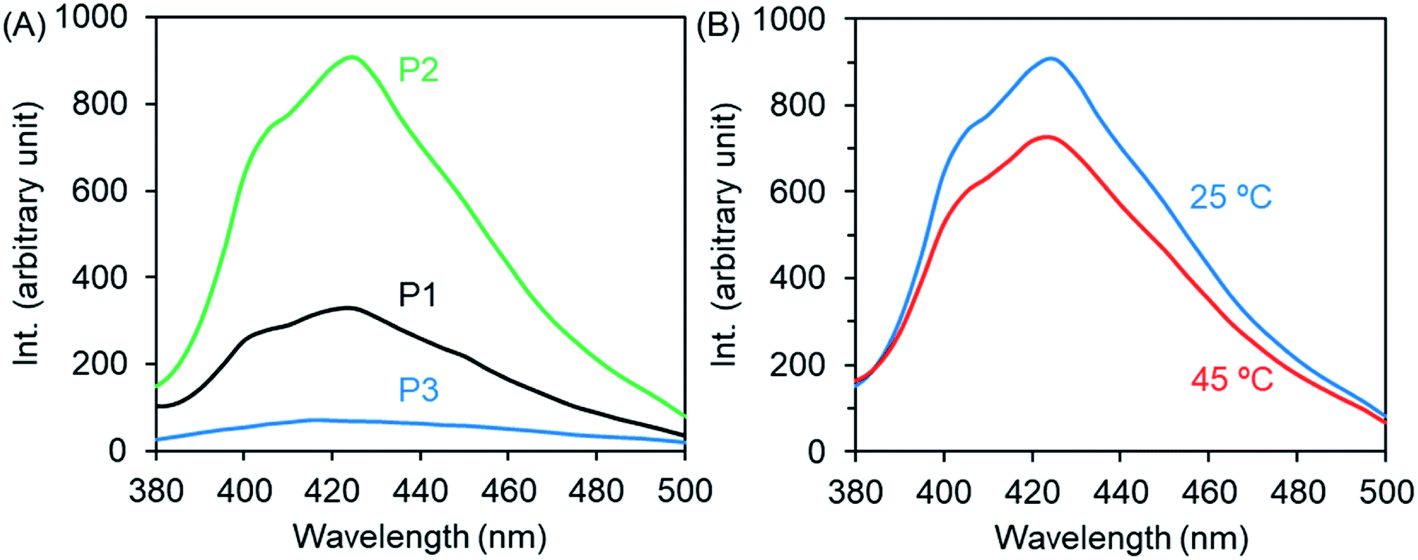 | ||
| Fig. 6 Fluorescence emission spectra (λex = 365 nm) of the micellar and unimer solutions of P1–P3 (2 g L−1) at room temperature (A) and at 45 °C (B). | ||
In the above emission spectrum measurements, the amounts of the two residues of T and I in P2 solution (2 mL of 2 g L−1) are equal to 3.4 mmol and 1.0 mmol, respectively. Therefore, we prepared a solution containing P1 (only theophylline pendants) and P3 (only imidazole pendants) with a ratio of T/I of 77/23 (in molar) and subjected the solution to fluorescence measurements to confirm whether the mixture could function as an emitter similar to P2. As shown in Fig. S8D,† it is obvious that there is no enhancement of the emission from the mixture. We also performed comparative fluorescence measurements by using two aqueous solutions; one contained monomeric theophylline and imidazole in a molar ratio of 77/23 (3.4 mmol (0.60 mg)/1.0 mmol (0.07 mg) in 2 mL water), [T] + [I] = 0.22 mmol mL−1, and the other was a mixture of imidazole with P1 micelles with a ratio of T/I of 77/23. In both cases, however, no fluorescence increment appeared (Fig. S8B and C†). These results indicate that the emission could not be strengthened from the mixtures containing theophylline and imidazole residues either as low molecular state or as polymeric pendants in the given conditions. Only P2, which has random copolymeric structures containing theophylline and imidazole in a suitable ratio in its pendants, emits fluorescence strongly at low concentration. Accordingly, the appearance of remarkable emission from P2 should be assigned to the synergy effect of the intramolecular (intra-chain) interactions between the moieties of theophylline and imidazole in the pendants, which are crowded in one polymer chain. As a derivative of xanthine, the ring in the theophylline plane condensed with imidazole would be more electron-poor than the imidazole ring itself. Therefore, the imidazole plane could act as an electronic donor to interact with the theophylline plane via π–π interactions to form charge-transfer-like associates. The appearance of a shoulder around 340 nm in the UV-Vis spectrum of P2 (Fig. 2B) could be related to the associates caused by π–π interactions. From the excitation spectrum of the micellar dispersion of P2 (Fig. S8A†), we observed that P2 showed a remarkable absorption peak at 365 nm, which is a maximum excitation wavelength, either in the aqueous micellar state or in chloroform solution. This excitation peak should be attributed to emitters that may be charge transfer (or excimer- and exiplex-like) associates in the clusters of theophylline and imidazole residues introduced densely in the polymer side chains. In these associates, the bond of imine –CN– in theophylline or imidazole is basic and is thus able to function as a proton acceptor and/or ligand to metal cations. Therefore, we expect that the fluorescence emission from the associates could respond to pH and/or metal cations because the imine –CN– is capable of accepting protons or of chelating with metal cations.49
Here, we prepared P2 micelles at different pH values by adding a DMSO solution of P2 to excess water pH-adjusted in advance with HCl or ammonia solution, and then we examined their fluorescence emissions. As shown in Fig. 7A, the emission intensity is the same in the cases of pH 7.0 and 10.2 but became very weak at pH 2.2. This unambiguously indicates that the forces supporting the emitters formed via π–π interactions from theophylline and imidazole residues are weakened due to competitive interactions arising from protonation of the imine bond –CN–. We also compared the diameters of the micelles prepared in the different pH conditions. In Fig. 7B, it can be seen that the micellar diameters become larger by nearly ten times (278 nm) in the acidic conditions (pH 2.2) compared to the neutral (33 nm) and basic (25 nm) conditions. It needs to be noted here that the emission and diameter of the micelles did not change as the pH was lowered to 4 from 7. However, as the pH values changed in the order of 3.2, 2.9 and 2.7, the micellar sizes and the fluorescence emissions increased and decreased, respectively, in a stepwise fashion (see Fig. S9†). We think that during the formation of core–shell micelles, the protonated –NC– bonds in the hydrophobic block weaken the hydrophobic and π–π interactions between the hydrophobic blocks and thus cause the core sediment to lack close packing, resulting in core-loose micelles with larger diameters. The thermo- and excitation-responsibility were also examined for the pH-adjusted P2 micelles. As shown in Fig. S10A,† the light transparence at a wavelength of 500 nm is very low (about 15%) for the micelles formed at pH 2.2 but high (about 90%) for the micelles formed at both pH 7.0 and 10.2 at 25 °C. As they were heated, the micelles formed at pH 2.2 turned opaque at 31 °C, while the transparence decreased slightly to 74% at 38 °C for the micelles formed at pH 7.0 and to 84% at 36 °C for the micelles formed at pH 10.2. This means that the smaller micelles formed at the higher pH values showed higher LCSTs than the lager micelles formed at lower pH, although the PNIPAM homopolymer itself has no pH-related thermo-response. In addition, in the excitation spectra of the micelles, the absorption at the maximum excitation wavelength is lower for the micelles formed at pH 2.2 than for the micelles formed at pH 7.0 and 10.2 (see Fig. S10B†), which is in agreement with the fluorescence emission response to pH changes (see Fig. 7A). Therefore, it is conclusive that amphiphilic P2 randomly bearing theophylline/imidazole in the side chains on the hydrophobic block is a unique photoluminescent polymer with thermo- and pH-responses, although it contains no π-conjugated polymeric chains. The photoluminescence should arise from the charge-transfer-like π–π association between imidazole and theophylline in adjacent spaces in which clusters crowded by the basic imine bond –CN– would play a central role in the emission.49,52 Once the imine –CN– is protonated, the π–π associations of the theophylline and imidazole residues will be disturbed, and thus the photoluminescence will be reduced. As far as we know, this is a new insight into the architecture of photoluminescent polymers comprising imidazole analogous residues but without rigid imidazole-conjugated polymeric chains. These photoluminescent polymers would have very interesting potential in optical-mediated response to metal ions because the basic –CN– prefers chelation interactions with metal cations and not only with protons.
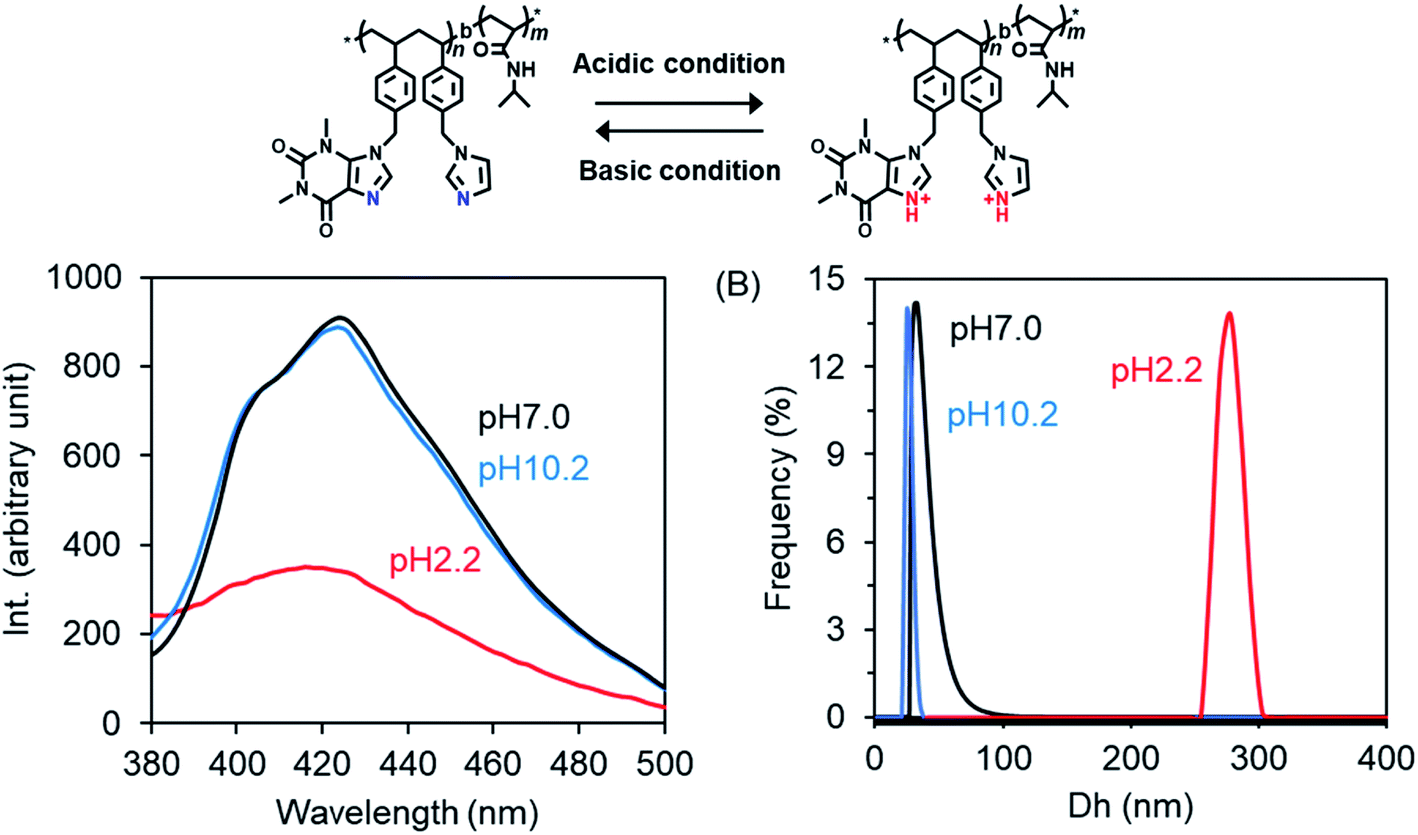 | ||
| Fig. 7 (A) Fluorescence emission spectra (λex = 365 nm) of P2 dispersions with different pH values at 25 °C. (B) DLS traces of P2 dispersions with different pH values at 25 °C (concentration of P2: 2 g L−1). | ||
Palladium sensing by photoluminescence
Recently, we developed a strategy to fabricate nano/micro polymeric materials using theophylline-Pd complexation as a driving force.54,56 In contrast to imidazole, theophylline (T) can chelate with Pd(II) even in strong acidic conditions to form a stable complex consisting of T2Pd. Thus, the moiety of theophylline features selective adsorption of palladium in mixed metal ions. In order to screen the relationship of the photoluminescence of P2 with metal cations, we prepared a series of P2/metal micellar dispersions by the addition of a small amount of P2 solution in DMSO to excess aqueous solutions containing metal acetates (2.5 mM) of Ag+, Ba2+, Zn2+, Cu2+ and Pd2+, and we measured their fluorescence. Fig. 8 shows the emission intensities of the P2/metal mixtures. Interestingly, the emission decreased somewhat in the presence of Ag+, Ba2+, Zn2+ and Cu2+, but was completely quenched in the case of Pd2+. In the micelles containing Ag+, Ba2+, Zn2+ and Cu2+, the micellar turbidity remained without changes, and no sediments or precipitates occurred even if they were placed for a long time (Fig. 8, top). Remarkably different to these metal ions, the dispersion of P2/Pd2+ resulted in yellow sediment within a few minutes. Heating the mixture of P2/Pd2+ to 45 °C caused a quick gel-like dispersion, losing the feature of a smooth liquid (Fig. S11A†). This suggests that even after metal complexing, PNIPAM in the shell layer on the sediment can still express sensitive thermal responsiveness. In other words, P2 can function well as a Pd2+-responding and thermo-responding material. We visualized the sediments that appeared at room temperature by scanning electron microscope (SEM). It can be seen in Fig. S11B† that the sediments are aggregates from rods with thorn structures, where the diameters of the rods are about 200 nm. This suggests that Pd2+ cation could play the role of a cross-linker for the micelles due to chelation with theophylline residues. After mixing P2 with Pd2+ cations, in addition to the self-assembly process of P2, an event of T–Pd–T cross-linking of chelation via intra- and/or inter-polymer chains and via inter- and intra-micelles is probable. Any pathway could weaken and prevent the π–π interactions between theophylline and imidazole and thus result in extinguishment of the photoluminescence. In this sense, it is expectable that P2 could be used as an optical sensor for detection of Pd2+ cations. For the purpose of sensing Pd2+ cations, we prepared a micelle dispersion of P2/Pd2+ with a low concentration of Pd(II) (0.025 mM, 5.6 ppm). This dispersion did not afford sediments/precipitates and remained transparent. As visualized by SEM, the casting sample of a micellar dispersion of P2 (2 mg mL−1)/Pd2+ (0.025 mM) looked like a film, where numerous micellar spheres with a diameter of 100 nm fused with each other (Fig. S11C†). Compared to the case of P2 only, interestingly, the photoluminescence intensity of the micellar dispersion P2 (2 mg mL−1)/Pd2+ (0.025 mM) decreased greatly to 20%. That is, the emission turns “ON” without Pd(II) but turns “OFF” with Pd(II). This “on/off” behavior strongly suggests that the amphiphilic block copolymer of P2 is a good candidate as an optical sensor for Pd2+ cations in aqueous media (Fig. S11D†).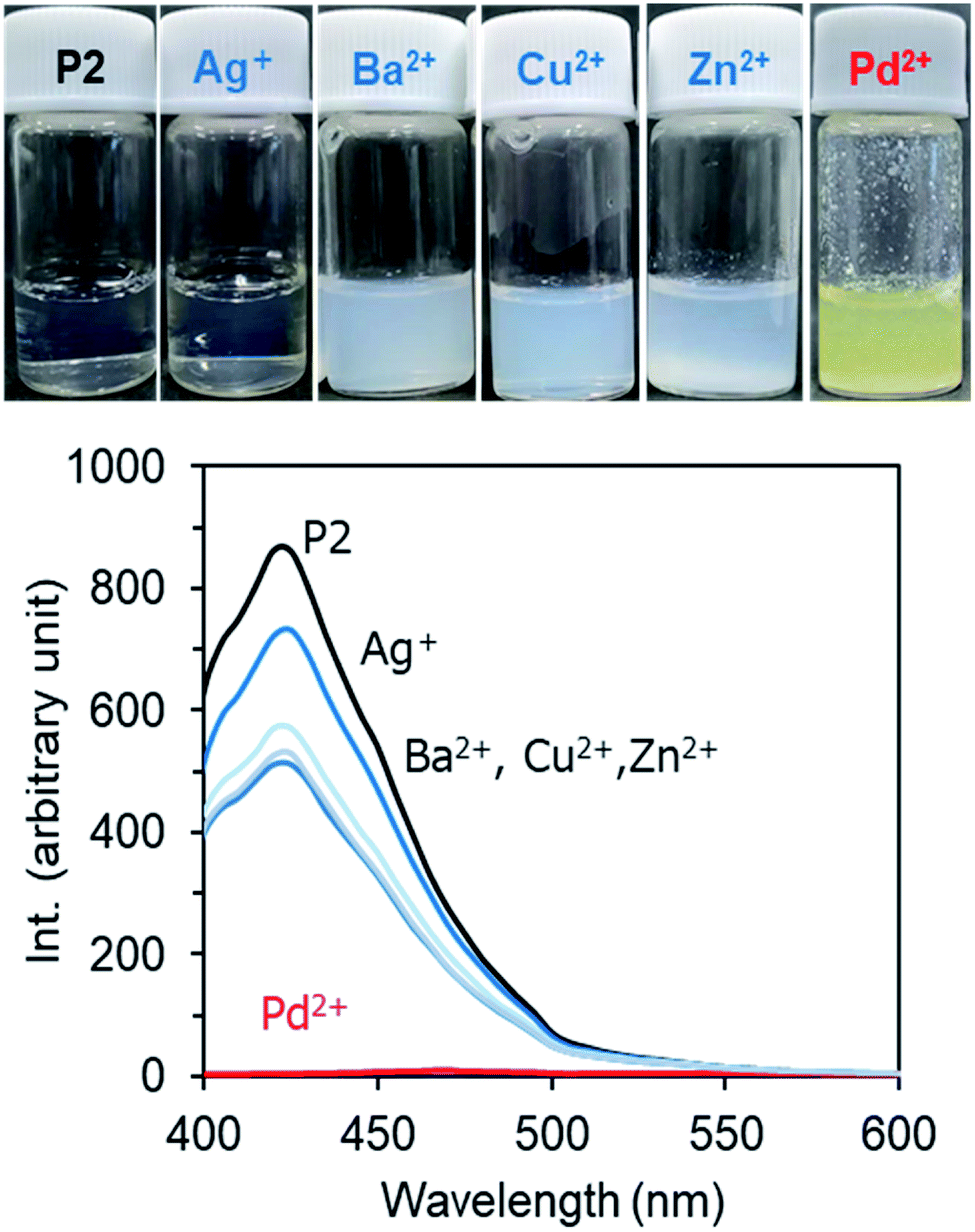 | ||
| Fig. 8 Bottom: Fluorescence emission spectra (λex = 365 nm) of micellar dispersions of P2 (2 g L−1) in the presence of 2.5 mM metal acetate at room temperature. Top: photographs of the dispersions. | ||
In order to further confirm the sensing behavior of P2 micelles, we performed post-addition of Pd2+ cations to a micellar dispersion. In Fig. S12,† the photographs and SEM images are displayed. Compared to the case of P2/Pd2+ micelles formed by simultaneous mixing of P2 and Pd2+ cations, upon the post-addition of Pd2+ cations (2.5 mM), the P2 micelles rapidly became turbid, with the formation of orange sediment (Fig. S12A†). This color suggests the formation of a Pd(II)/theophylline complex which shows absorption at wavelengths around 400–500 nm.55 The sediments visualized by SEM are fibrous network entities which are likely formed by linking of the fibrously fused micelles. Perhaps, the post-added Pd2+ cations chelate with the theophylline and imidazole residues which exist on the core surface and thus promote one-dimensional growth of inter-micellar fusion but do not destroy the micelles. In comparison, when the concentration of the post-added Pd2+ cation was as low as 0.025 mM (5.6 ppm), the P2 micelles remained stable without sediments (Fig. S12B†), accompanying a noticeable reduction of the photoluminescence intensity to 9%. This indicates that Pd2+ cations at a lower concentration chelate with theophylline/imidazole residues via intra-micellar interactions and thus effectively quench the emission (Fig. S12C†). As a comparison, using the opaque mixture of P1 + P3 (T/I = 7/3, 2 g L−1), we performed a similar experiment, as shown in Fig. S13 and 14,† in the presence of Pd cations at 0.25 and 0.025 mM. The emission from the mixture decreased only slightly (about 20%) when Pd2+ cations were added at 0.025 mM, while it decreased almost completely when the concentration of Pd2+ cations added was increased to 0.25 mM, although there was no precipitation. The ability of P2 was well beyond that of the mixture of P1 and P2 to sense a low concentration of Pd2+ cations.
To confirm the selective palladium sensing, we further performed the examination of the photoluminescence of P2 in the case of co-existence of several metal cations. As shown in Fig. 9A, the emission intensity of P2 was reduced to 44% in the co-presence of three ions of Ag+, Zn2+ and Cu2+. Comparatively, in the case of the co-presence of four ions of Pd2+, Ag+, Zn2+ and Cu2+, the emission intensity was reduced greatly to 8.7%, indicating high selectivity of P2 for Pd2+ cations over other metal cations. This quenching effect is equal to the case of Pd2+ cations only. Without a doubt, even when Pd2+ cations are mixed with other metal cations such as Ag+, Zn2+, and Cu2+, the emission quenching is selectively induced by the palladium cations by excluding the other metal cations from moving close to P2. From the photographs in Fig. 9B, it can be seen clearly that the two micellar dispersions coexisting with several metal cations remained in the micellar state without sediments. Under UV light irradiation, the micellar dispersion containing Ag+, Zn2+, and Cu2+ still emits noticeable blue light, while the micellar dispersion in which Pd2+ was added to Ag+, Zn2+, and Cu2+ barely showed the original blue. This suggests that the sensing of Pd2+ cation by P2 can be judged even with the naked eye.
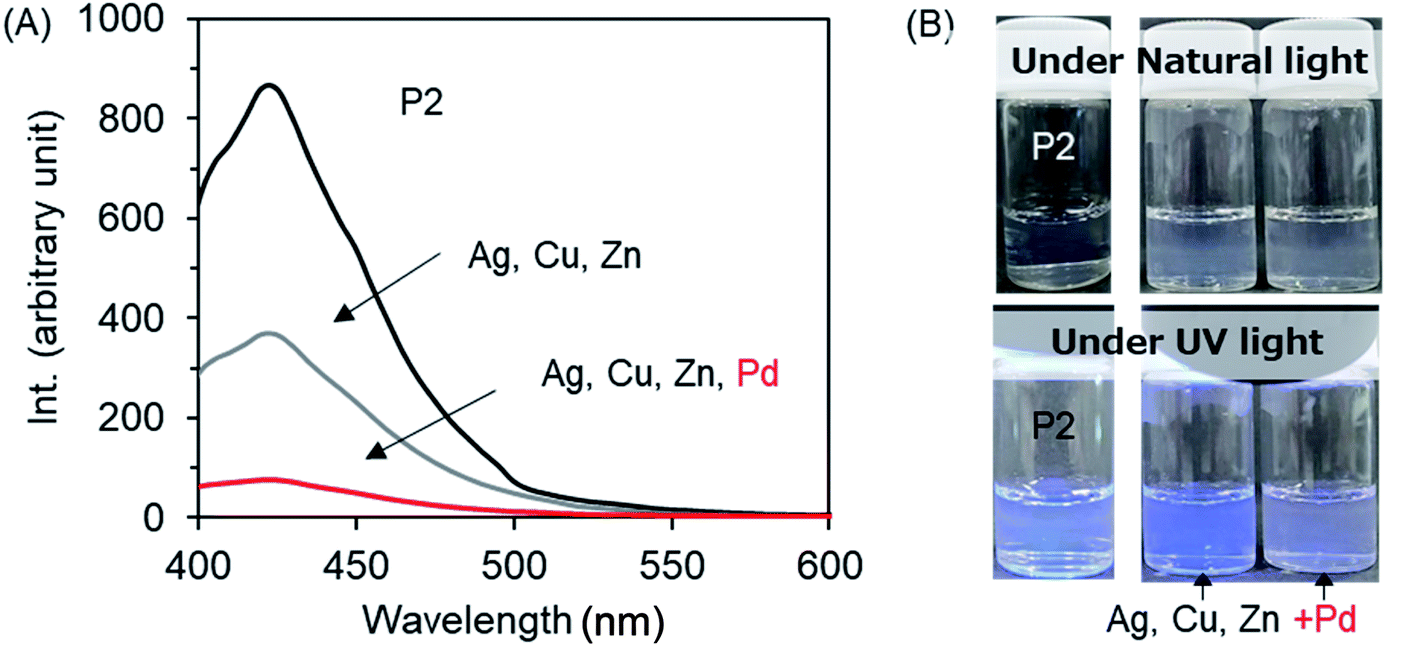 | ||
| Fig. 9 (A) Fluorescence emission spectra (λex = 365 nm) of micellar dispersions of P2 (2 g L−1) containing mixed metal acetates (Ag+, Zn2+, Cu2+ and Pd2+; the concentration of each ion is 0.025 mM) at room temperature. (B) Photographs of the dispersions of P2 with and without metal acetates under ambient light (top) and under UV light irradiation (bottom). | ||
There are many reports of the sensing of Pd2+ cations depending on an optical process using organic fluorescent compounds that can turn their emission “ON” or “OFF” when they chelate with Pd2+ cations.58–65 However, the synthesis of the special fluorescent compounds is expensive. Our polymer reported here with common theophylline and imidazole moieties is not only inexpensive but also simply and effectively useable as an optical sensor for palladium cations.
Conclusions
In summary, we found a new photoluminescent amphiphilic block copolymer featuring thermo-/pH-responsibility and excellent palladium ion response in aqueous media. In this block copolymer, the emitting of blue luminescence is caused by two randomly introduced moieties of theophylline and imidazole in the side chains of the hydrophobic block. That is, the intra-chain π–π interactions between theophylline and imidazole moieties (i.e., in the same polymer chain) is an intrinsic event that promotes the formation of a charge-transfer-like complex which effectively emits blue luminescence under UV-irradiation. Because our amphiphilic block copolymer consisted of a hydrophobic photoluminescent block and hydrophilic thermoresponsive block of PNIPAM, the copolymer can self-assemble into core–shell polymer micelles in aqueous media. The core features photoluminescence with pH and metal cation sensitivity, while the shell displays thermo-sensitivity. Therefore, the micelles can smartly respond to light, heat, pH and metal cations. As an important function, under UV irradiation with a wavelength longer than 350 nm, the micelles exhibit selective optical sensing of Pd2+ cations in a mixture of several metal cations. The photoluminescence from the micelles turns “ON” without Pd2+ cations but turns “OFF” with Pd2+ cations. This is because the theophylline moiety has extraordinary coordination power for Pd2+ cations, by which the emitter (i.e., charge transfer associates of theophylline/imidazole) dissociates. Our finding in this work is the first example of the use of non-π-conjugated imidazole derivatives in polymer side chains to generate photoluminescence. We believe that the polymer found here could be simply applied in sensing, catalysis and vehicles together with thermo-responsivity.Experimental section
Materials
N-Isopropylacrylamide (NIPAM, 98%, Tokyo Kasei Kogyo Co.) was purified by recrystallization from n-hexane. 4-(Chloromethyl)styrene (VBC, 90%, Tokyo Chemical Industry Co.) was passed through a column of activated basic alumina (SIGMA-ALDRICH, neutral, Brockmann) to remove inhibitors. 2,2′-Azobis(isobutyronitrile) (AIBN, 98%, Tokyo Kasei Kogyo Co.) was purified by recrystallization from methanol. Cyanomethyl benzodithioate (CMB, 99%, SIGMA-ALDRICH), benzoyl peroxide (BPO, 75%+ wetted with ca. 25% water, Tokyo Chemical Industry Co.), Theophylline (99.0+%, FUJIFILM Wako Pure Chemical Co.), Imidazole (98.0+%, FUJIFILM Wako Pure Chemical Co.), N,N-dimethylformamide (GODO Co.), 1,4-dioxane (99.5+%, FUJIFILM Wako Pure Chemical Co.), dimethyl sulfoxide (99.5+%, FUJIFILM Wako Pure Chemical Co.), methanol (99.5+%, FUJIFILM Wako Pure Chemical Co.), and metal acetates (palladium, silver, barium, zinc, copper) (FUJIFILM Wako Pure Chemical Co.) were used as received.Synthesis of PVBC-CDB by RAFT polymerization
The PVBC-CDB macro-RAFT agent was prepared by RAFT polymerization as follows. VBC (10.0 g, 65.5 mmol), CMB (0.12 g, 0.66 mmol), AIBN (27 mg, 0.16 mmol), and 1,4-dioxane (4.35 g, 30 wt%) were added to a Schlenk flask. After the mixture was degassed thrice by freeze–pump–thaw cycles by purging with N2 gas, the reaction solution was stirred at 80 °C for 21 hours. The crude product was diluted with THF, precipitated in a large amount of methanol three times and then dried under vacuum at 40 °C. Yield: 7.3 g. GPC: Mn = 10471, Mw = 11998, PDI = 1.15, degree of polymerization (DP) = 74.
Synthesis of PVBC-b-PNIPAM diblock copolymer
A typical procedure of the preparation of the PVBC-b-PNIPAM diblock copolymer and removal of the thiocarbonylthio end-groups is as follows.PVBC74-CDB (1.00 g, equal to 87.1 μmol of CDB end groups), AIBN (4.3 mg, 26 μmol), N-isopropylacrylamide (NIPAM) (9.84 g, 87.1 mmol) and 1,4-dioxane (16.3 g, 60 wt%) were mixed in a Schlenk tube. After the solution was degassed by three freeze–pump–thaw cycles, the sample tube was heated at 65 °C for 6 hours. Then, the crude product diluted with THF was precipitated three times in n-hexane and dried at 40 °C in vacuo. Yield: 4.3 g. GPC: Mn = 60022, Mw = 75649, PDI: 1.260, DP = 383.
The procedure of removal of thiocarbonylthio end-groups from the above diblock copolymer was as follows. A 100 mL round-bottomed flask was charged with PVBC74-b-PNIPAM383-CDB (2.0 g, 37 μmol of CDB end groups, Mn (NMR) = 55 kg mol−1), BPO (0.177 g, 0.73 mmol), and 1,4-dioxane (60 mL). The solution was refluxed for 1 hour at 80 °C under nitrogen atmosphere to remove the end-groups of dithiobenzoate. The crude product was diluted with THF, precipitated in a large amount of n-hexane 4 times and then dried under vacuum. Yield: 1.8 g. GPC: Mn = 61177, Mw = 98649, PDI = 1.61.
Synthesis of PVBT-b-PNIPAM (P1)
PVBC74-b-PNIPAM383 (0.09 g, Mw: 54600, number of molCl: 0.13 mmol), theophylline (24 mg, 0.14 mmol), K2CO3 (37 mg, 0.27 mmol), KI (45 mg, 0.27 mmol) and DMF (10 mL) were mixed in a 30 mL round-bottomed flask and reacted at 80 °C for 24 hours. After cooling to room temperature, the solid fraction was removed by suction filtering and the filtrate was dropped into water/methanol (v/v = 1/1) to precipitate the product. The white precipitates obtained were dried under ambient conditions. Yield: 91 mg.
Synthesis of PVBT/I-b-PNIPAM (P2)
PVBC74-b-PNIPAM383 (0.08 g, Mw: 54600, number of molCl: 0.12 mmol), theophylline (17 mg, 0.094 mmol), imidazole (3 mg, 0.041 mmol), K2CO3 (23 mg, 0.14 mmol), KI (19 mg, 0.14 mmol) and DMF (10 mL) were mixed in a 30 mL round-bottomed flask and reacted at 80 °C for 24 hours. After cooling to room temperature, the reaction mixture was poured into a small amount of water and then dialyzed against water three times. After freeze-drying, a white powder was obtained. Yield: 85 mg.
Synthesis of PVBI-b-PNIPAM (P3)
PVBC74-b-PNIPAM383 (0.09 g, Mw: 54600, number of molCl: 0.13 mmol), imidazole (9 mg, 0.14 mmol), K2CO3 (23 mg, 0.14 mmol), KI (19 mg, 0.14 mmol) and DMF (10 mL) were mixed in a 30 mL round-bottomed flask and reacted at 80 °C for 24 hours. After cooling to room temperature, the reaction mixture was poured into a small amount of water and dialyzed against water three times. After freeze-drying, a white powder was obtained. Yield: 89 mg.
Preparation of the polymer micelles
In 0.2 mL of DMSO, 4 mg of polymer (P1 or P2 or P3) was dissolved with heating. Then, by dropping the solution into 1.8 mL of water [or HCl (aq, pH 2.2) and ammonia (aq, pH 10.2) solutions or metal acetates aqueous solution (2.5 mM and 0.025 mM)] under stirring, a series of micellar dispersions with a concentration of 2 mg mL−1 polymer were prepared. All the micellar dispersions were subjected to measurements with a thermo-equipped DLS spectrophotometer, thermo-equipped UV-Vis spectrometer and spectrofluorometer.Characterizations
1H NMR spectra were recorded on a JEOL JNM-ECA600 NMR and are reported in ppm downfield from tetramethylsilane. GPC profiles were recorded on a HLC-8320 instrument (Tosoh, Japan; column: Shodex Asahipak GF-510 HQ + GF-1G 7B; eluent: DMF containing 10 mM LiBr; flow rate: 0.6 mL min−1). Differential scanning calorimetry (DSC) experiments were performed on a HITACHI X-DSC-7000. UV-vis spectra were recorded on a SHIMADZU UV-2600 spectrometer supplied with a temperature-controlled cell positioner (CPS-100). The diameters of the polymer micelles in water were evaluated with an Otsuka Electronics FPAR-1000 DLS spectrophotometer. Fluorescence studies were carried out with a JASCO FP-8300 spectrofluorometer. The morphology of the nano-micelles was observed using a scanning electron microscope (SEM, Hitachi SU-8010). The SEM samples were prepared by dropping a polymer solution onto a silicon wafer.Conflicts of interest
The authors declare no conflict of interest.References
- (a) Y. Mai and A. Eisenberg, Acc. Chem. Res., 2012, 45, 1657–1666 CrossRef CAS PubMed; (b) I. Dewald and A. Fery, Adv. Mater. Interfaces, 2017, 4, 1600317 CrossRef; (c) B. T. Mai, S. Fernandes, P. B. Balakrishnan and T. Pellegrino, Acc. Chem. Res., 2018, 51, 999–1013 CrossRef CAS PubMed; (d) J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed; (e) Y. Lu, J. Lin, L. Wang, L. Zhang and C. Cai, Chem. Rev., 2020, 120, 4111–4140 CrossRef CAS PubMed.
- W. Chen, L. Su, P. Zhang, C. Li, D. Zhang, W. Wu and X. Jiang, Polym. Chem., 2017, 8, 6886–6894 RSC.
- S. Osawa, K. Osada, S. Hiki, A. Dirisala, T. Ishii and K. Kataoka, Biomacromolecules, 2016, 17, 354–361 CrossRef CAS PubMed.
- M. Karesoja, E. Karjalainen, S. Hietala and H. Tenhu, J. Phys. Chem. B, 2014, 118, 10776–10784 CrossRef CAS PubMed.
- B. Hazer1, E. Ayyıldız1 and F. Bahadır, J. Am. Oil Chem. Soc., 2017, 94, 1141–1151 CrossRef.
- J. Herzberger, D. Kurzbach, M. Werre, K. Fischer, D. Hinderberger and H. Frey, Macromolecules, 2014, 47, 7679–7690 CrossRef CAS.
- M. Rabyk, A. Destephen, A. Lapp, S. King, L. Noirez, L. Billon, M. Hruby, O. Borisov, P. Stepanek and E. Deniau, Macromolecules, 2018, 51, 5219–5233 CrossRef CAS.
- C. Yang, J. Xiao, W. Xiao, W. Lin, J. Chen, Q. Chen, L. Zhang, C. Zhang and J. Guo, RSC Adv., 2017, 7, 27564–27573 RSC.
- C. Sun, X. Jiang, B. Li, S. Li and X. Z. Kong, ACS Sustainable Chem. Eng., 2021, 9, 5166–5178 CrossRef CAS.
- S. Jana, A. Bose, A. Saha and T. K. Mandal, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1714–1729 CrossRef CAS.
- Z. Sun, G. Liu, J. Hu and S. Liu, Biomacromolecules, 2018, 19, 2071–2081 CrossRef CAS PubMed.
- J. Zhang, Z.-H. Zhou, L. Li, Y.-L. Luo, F. Xu and Y. Chen, Mol. Pharmaceutics, 2020, 17, 1100–1113 CrossRef CAS PubMed.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Polym. Chem., 2014, 5, 5403–5411 RSC.
- J. Jiang, Q. Shu, X. Chen, Y. Yang, C. Yi, X. Song, X. Liu and M. Chen, Langmuir, 2010, 26, 14247–14254 CrossRef CAS PubMed.
- M.-C. Tu, J. A. Cheema, U. H. Yildiz, A. Palaniappan and B. Liedberg, J. Mater. Chem. C, 2017, 5, 1803–1809 RSC.
- V. Y. Grinberg, T. V. Burova, N. V. Grinberg, A. P. Moskalets, A. S. Dubovik, I. G. Plashchina and A. R. Khokhlov, Macromolecules, 2020, 53, 10765–10772 CrossRef CAS.
- M. Qi, K. Li, Y. Zheng, T. Rasheed and Y. Zhou, Langmuir, 2018, 34, 3058–3067 CrossRef CAS PubMed.
- M. Tamaki, D. Fukushima and C. Kojima, RSC Adv., 2018, 8, 28147–28151 RSC.
- J. S. Scarpa, D. D. Mueller and I. M. Klotz, J. Am. Chem. Soc., 1967, 89, 6024–6030 CrossRef CAS.
- S. Zhou, S. Fan, S. C. F. Au-Yeung and C. Wu, Polymer, 1995, 36, 1341–1346 CrossRef CAS.
- Y. Qu, X. Chang, S. Chen and W. Zhang, Polym. Chem., 2017, 8, 3485–3496 RSC.
- S. Won, D. J. Phillips, M. Walker and M. I. Gibson, J. Mater. Chem. B, 2016, 4, 5673–5682 RSC.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- G. Wu, S.-C. Chen, C.-L. Liu and Y.-Z. Wang, ACS Nano, 2015, 9, 4649–4659 CrossRef CAS PubMed.
- Y. Niko and G. Konishi, Macromolecules, 2012, 45, 2327–2337 CrossRef CAS.
- J. Tian, L. Xu, Y. Xue, X. Jiang and W. Zhang, Biomacromolecules, 2017, 18, 3992–4001 CrossRef CAS PubMed.
- L. Yan, X. Gu, Z. Wang and Z. Qi, ChemistrySelect, 2018, 3, 3406–3410 CrossRef CAS.
- J. Lee, H. Yang, C. H. Park, H.-H. Cho, H. Yun and B. J. Kim, Chem. Mater., 2016, 28, 3446–3453 CrossRef CAS.
- J. Li, K. Liu, H. Chen, R. Li, M. Drechsler, F. Bai, J. Huang, B. Z. Tang and Y. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 21706–21714 CrossRef CAS PubMed.
- J. D. Harris, C. Mallet, C. Mueller, C. Fischer and K. R. Carter, Macromolecules, 2014, 47, 2915–2920 CrossRef CAS.
- Y.-J. Zhao, K. Miao, Z. Zhu and L.-J. Fan, ACS Sens., 2017, 2, 842–847 CrossRef CAS PubMed.
- P. Tzeng, C.-C. Kuo, S.-T. Lin, Y.-C. Chiu and W.-C. Chen, Macromol. Chem. Phys., 2010, 211, 1408–1416 CrossRef CAS.
- J. Wu, B. Xu, Z. Liu, Y. Yao, Q. Zhuang and S. Lin, Polym. Chem., 2019, 10, 4025–4030 RSC.
- M. Wang, L. Xu, M. Lin, Z. Li and J. Sun, Polym. Chem., 2021, 12, 2825–2831 RSC.
- P. Kumari, M. K. Bera, S. Malik and B. K. Kuila, ACS Appl. Mater. Interfaces, 2015, 7, 12348–12354 CrossRef CAS PubMed.
- Y. Zhao, C. Shi, X. Yang, B. Shen, Y. Sun, Y. Chen, X. Xu, H. Sun, K. Yu, B. Yang and Q. Lin, ACS Nano, 2016, 10, 5856–5863 CrossRef CAS PubMed.
- Y. Zhao, Y. Wu, S. Chen, H. Deng and X. Zhu, Macromolecules, 2018, 51, 5234–5244 CrossRef CAS.
- C. C. Yang, Y. Tian, A. K. Y. Jen and W. C. Chen, J. Polym. Sci., Part A: Polym. Chem., 2006, 10, 5495–5504 CrossRef.
- J. Chen, D. Wang, A. Turshatov, R. Munoz-Espi, U. Ziener, K. Koynov and K. Landfester, Polym. Chem., 2013, 4, 773–781 RSC.
- H. Chen, Y. Fan, N. Zhang, S. Trepout, B. Ptissam, A. Brulet, B. Z. Tang and M.-H. Li, Chem. Sci., 2021, 12, 5495–5504 RSC.
- P. Yu and Y. Xiao, Materials, 2021, 14, 2349 CrossRef CAS PubMed.
- J. Kulhánek and F. B. Beilstein, J. Org. Chem., 2012, 8, 25–49 Search PubMed.
- S. Saxer, C. Marestin, R. Merciera and J. Dupuy, Polym. Chem., 2018, 9, 1927–1933 RSC.
- S. Somasundaram, E. Kamaraj, S. J. Hwang and S. Park, Spectrochim. Acta, Part A, 2018, 191, 325 CrossRef CAS PubMed.
- K. Takagi, K. Sugihara and T. Isomura, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4822–4829 CrossRef CAS.
- M. Sun, C.-Y. Hong and C.-Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581–20584 CrossRef CAS PubMed.
- W. Z. Yuan and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 560–574 CrossRef.
- Q. Zeng, T. Feng, S. Tao, S. Zhu and B. Yang, Light: Sci. Appl., 2021, 10, 142 CrossRef CAS PubMed.
- S. G. Liu, N. Li, Y. Ling, B. H. Kang, S. Geng, N. B. Li and H. Q. Luo, Langmuir, 2016, 32, 1881–1889 CrossRef CAS PubMed.
- S. G. Liu, D. Luo, N. Li, W. Zhang, J. L. Lei, N. B. Li and H. Q. Luo, ACS Appl. Mater. Interfaces, 2016, 8, 21700–21709 CrossRef CAS PubMed.
- S. G. Liu, T. Liu, N. Li, S. Geng, J. L. Lei, N. B. Li and H. Q. Luo, J. Phys. Chem. C, 2017, 121, 6874–6883 CrossRef CAS.
- Z. He, C. Ke and B. Z. Tang, ACS Omega, 2018, 3, 3267–3277 CrossRef CAS PubMed.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586–6592 CrossRef CAS PubMed.
- K. Kaikake, M. Takada, D. Soma and R.-H. Jin, RSC Adv., 2018, 8, 34505–34513 RSC.
- K. Kaikake, N. Jou, S. Go and R.-H. Jin, RSC Adv., 2021, 11, 35311–35320 RSC.
- H. Takebuchi, H. Kubosawa and R.-H. Jin, Chem. Lett., 2019, 48, 647–650 CrossRef CAS.
- H. Takebuchi and R.-H. Jin, Macromol. Chem. Phys., 2021, 222, 2100174 CrossRef CAS.
- D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS PubMed.
- M. Wang, X. Liu, H. Lu, H. Wang and Z. Qin, ACS Appl. Mater. Interfaces, 2015, 7, 1284–1289 CrossRef CAS PubMed.
- J. Xiang, C. Liu, L. Zhou, X. Yang, Y. Li, Y. Jiang, T. Mahmood, P. Zhang, P. Gong and L. Cai, Anal. Chem., 2020, 92, 4721–4725 CrossRef CAS PubMed.
- M. H. Chua, H. Zhou, Q. Zhu, B. Z. Tang and J. W. Xu, Mater. Chem. Front., 2021, 5, 659–708 RSC.
- P. Kumar, V. Kumar and R. Gupta, RSC Adv., 2017, 7, 7734–7741 RSC.
- F. Liu, J. Du, M. Xu and G. Sun, Chem.–Asian J., 2016, 11, 43–48 CrossRef CAS PubMed.
- A. Ren, Y. Zhang, W. Yu, K. Zhao, Z. Hu, Z. Zhang, G. Feng and Z. Song, J. Ind. Eng. Chem., 2021, 99, 292–298 CrossRef CAS.
- J. R. Matthews, F. Goldoni, H. Kooijman, A. L. Spek, A. P. H. J. Schenning and E. W. Meijer, Macromol. Rapid Commun., 2007, 28, 1809–1815 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08756h |
| This journal is © The Royal Society of Chemistry 2022 |