
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFree and self-trapped exciton emission in perovskite CsPbBr3 microcrystals
Fang Pana,
Jinrui Li b,
Xiaoman Mac,
Yang Niea,
Beichen Liua and
Honggang Ye*a
b,
Xiaoman Mac,
Yang Niea,
Beichen Liua and
Honggang Ye*a
aDepartment of Applied Physics, MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, Xi'an Jiaotong University, Xi'an, 710049, People's Republic of China. E-mail: hgye@mail.xjtu.edu.cn
bElectronic Materials Research Laboratory, Key Laboratory of the Ministry of Education and International Center for Dielectric Research, School of Electronic Science and Engineering, Xi'an Jiaotong University, Xi'an, 710049, People's Republic of China
cSchool of Physical Science and Technology, Xinjiang University, Urumqi, 830046, People's Republic of China
First published on 5th January 2022
Abstract
The all-inorganic perovskite CsPbBr3 has been capturing extensive attention due to its high quantum yield in luminescence devices and relatively high stability. Its luminescence is dominated by free exciton (FE) recombination but additional emission peaks were also commonly observed. In this work, a CsPbBr3 microcrystal sample in the orthorhombic phase was prepared by the chemical vapor deposition method. In addition to the FE peak, a broad emission peak was found in this sample and it was attributed to self-trapped excitons (STEs) based on its photophysical properties. The STE emission can only be observed below 70 K. The derived Huang–Rhys factor is ∼12 and the corresponding phonon energy is 15.3 meV. Its lifetime is 123 ns at 10 K, much longer than that of FE emission. The STE emission is thought to be an intrinsic property of CsPbBr3.
1. Introduction
All-inorganic perovskite semiconductors have been capturing extensive attention due to their unique optical and electronic properties, such as high light absorption coefficient,1 strong carrier transport capacity,2 highly efficient photoluminescence (PL),3 tunable band gap,4 high defect tolerance,5 and relatively high stability.6 Caesium lead halides CsPbX3 (X = Cl, Br, I) are the most studied all-inorganic perovskite semiconductors, which exhibit significant potential for applications in light emitting devices such as lasers and light emitting diodes.7–11 These applications are closely associated with their luminescence properties. The PL spectrum of CsPbX3 usually manifests as a single Gaussian fluorescence peak from the free exciton (FE) recombination. However, PL spectra with additional peaks and fine structures were also obtained in some conditions, indicating the existence of complex underlying physical processes in the materials.In some reports the CsPbX3 perovskites display dual PL peaks and a series of illustrations have come up for this phenomenon, including free and trapped excitons,12 surface and volume emission,13,14 existence of two phases,15 the Rashba effect16 and the reabsorption effect.17 Similar phenomenon in hybrid halide perovskites such as CH3NH3PbX3 (X = Br, I) was advised to be from free carriers and exciton recombination,18 the transverse inhomogeneity and structure coexistence,19–21 and the inelastic exciton–exciton scattering or biexciton emission.22 Emission spectra with three peaks were assigned to two BEs and an FE emission.23,24 Even without observation of two or more luminescence peaks, the low-temperature PL spectrum of CsPbX3 usually reveals an asymmetric tail in the low-energy side of the FE peak, implying the existence of some weak components.12,24–27 In some reports the weak subcomponent was suggested to be from the trap states in the band gap induced by either defects, e.g. Br vacancy, or surface states.28,29 In a recent report this phenomenon was attributed to a local dipole moment induced by the off-centre localization of Cs+ cations.30 None of these hypotheses to understand the multiple emission peaks have been confirmed and the occurrence conditions for them have not been explored clearly.
The exciton–phonon coupling is a significant interaction in polar semiconductors and has great influence on the luminescence spectrum. It may lead to a low energy tail of the excitonic peak due to the appearance of phonon replicas. Such a result was observed in CsPbBr3 nanocrystals at cryogenic temperature and a longitudinal optical (LO) phonon in energy of ∼18 meV was derived.31–33 The strong exciton–phonon coupling also may induce the appearance of self-trapped excitons (STEs), which represent a type of excitons trapped by transient local lattice deformation in the excited state. It can be extrinsic or intrinsic. The former is dependent on doping and the latter is independent on any impurity or defect. STE was rarely discussed in the conventional semiconductors but it has received many attentions in the perovskites due to the strong ionic bond and soft lattice nature. STE emission is usually characterized by a broad below-band-gap emission band with relatively long lifetime. White-light emitting devices have been developed based on the broad STE emission and a series of halide perovskite materials have been designed and synthesised for highly efficient STE emission.34–39 The double perovskites and low-dimensional perovskites were found to have strong STE emission.40–48 Since the lattice deformation in these structures is easy to occur. In the fully inorganic Cs–Pb–Br systems, the STE emission was mainly discussed in zero-dimensional Cs4PbBr6,49–51 in which the [PbBr6]− octahedrons are isolated. An STE band was also detected from the low energy wing of the PL spectra of CsPbBr3 and CsPb(Br1−xIx)3 quantum dots under weak excitation condition,52 but this conclusion has not been widely accepted. In spite of the difference in intensity and bandwidth, the STE emission was sometimes thought to be universal in the halide perovskites.
In this paper, we prepared CsPbBr3 microcrystals (MCs) by chemical vapour deposition (CVD) method. Steady and time-resolved PL spectra in the temperature range from 10 to 300 K were measured and a broad band besides sharp peaks was observed in the sample. The FE and STE emissions were mainly discussed based on their power dependence and temperature dependence of PL intensity, line width and lifetime. The STE was found to be an intrinsic emission channel in CsPbBr3. This conclusion is contributory to get a fully understanding to the optical properties of CsPbBr3 semiconductor.
2. Materials and methods
The CsPbBr3 MCs were prepared by the CVD method. CsBr and PbBr2 powder were adopted as raw materials and (100) silicon wafer as the substrate. The mixture of raw materials was placed in the centre of a quartz tube furnace and the substrate was placed with 15 cm departure in the downstream side. The quartz tube was vacuumed by a mechanical pump and the high-purity argon in flown of 30 sccm was supplied as the carrier gas in the whole process. The furnace was heated to 600 °C in 30 minutes and then maintained for 15 minutes. The sample was obtained after the furnace cooled naturally to room temperature.The morphology of the samples was characterized by the scanning electronic microscopy (SEM) (JEOL-7800F). The crystal structure of the samples was characterized using X-ray diffraction (XRD) (Bruker Advance D8) with Cu Kα radiation (λ = 0.15406 nm). The PL spectra were obtained by a home-made system composed of a monochromator (ZOLIX, Omni-λ 500i) and a photomultiplier detector (R928). The monochromator has a focal length of 500 mm and was equipped with a 1200 l mm−1 grating blazing at 500 nm. A standard lock-in amplification technique was employed to remove the noise and background signal. The samples were mounted with silver paint on the cold finger of a CRYO closed-cycle cryostat controlled by a Lakeshore temperature controller. A diode pumped solid state lasers in 473 nm wavelength was used as the excitation light. The time-resolved PL spectra were measured by using the time-correlated single photon counting (TCSPC) technology. The detection system was produced by Edinburgh Instruments (LifeSpec II), with the detector mounting on another exit port of the same monochromator for steady fluorescence. A 372 nm pulse laser diode with pulse width of 60 ps was adopted as excitation light for lifetime measurement.
3. Results and discussion
The SEM images of the CsPbBr3 MCs shown in Fig. 1a and b display regular cubes distributed as islands on the silicon substrate. The edge length ranges from a few microns to dozens of microns and the surface is smooth. XRD pattern shown in Fig. 1c provides the information of MC samples' crystal structure. The main diffraction peaks correspond to the (002), (110), (004) and (220) planes of orthorhombic phase (space group Pnma, ICSD # 97851),53 which is the most stable perovskite phase at room temperature or lower. The sharp peaks demonstrate the high-quality crystallinity of the CsPbBr3 MCs, which is advantageous to get a deep insight into its PL processes. | ||
| Fig. 1 Morphology and structure analysis of CsPbBr3 MCs. (a and b) SEM images of CsPbBr3 MCs grown by CVD method at low and high magnification, respectively. (c) X-ray diffraction pattern of CsPbBr3 MCs (red) at room temperature and reference data (black) of orthorhombic phase (ICSD #97851). | ||
The PL spectra of CsPbBr3 MCs are shown in Fig. 2, excited by 473 nm laser and measured within a wide temperature range (from 10 to 300 K). The fluorescence image in Fig. 2b presents luminescent MCs in bright green. The PL spectra manifests multiple emission peaks at 10 K but one Gaussian shaped peak at 300 K. As shown in Fig. 2c, we have performed a careful fit to decompose the spectrum at 10 K into four Gaussian components peaked at 2.254, 2.294, 2.348, and 2.405 eV, which are in turn named as peak-1, 2, 3, and 4 hereinafter, respectively. Peak-1 is the main component of the spectrum at 10 K. It has a full width at half maximum (FWHM) of 123.7 meV, much broader than the other components. However, its intensity declines quickly with increasing temperature and is hardly seen above 70 K. Peak-2 is the strongest one within the whole temperature range and only it survives above 110 K. With increasing temperature, peak-2 blue-shifts from 2.294 eV at 10 K to 2.394 eV at 300 K, which is one of the typical optical characteristics of perovskite materials.54 It is essentially determined by the special band structure of perovskite semiconductors whose valence band is composed of antibonding state,55–58 whereas the valence band of normal semiconductors such as CdSe and GaAs is made up of bonding state.59,60 The intensity of peak-2 first increases (from 10 to 70 K) then quickly decreases (above 70 K).
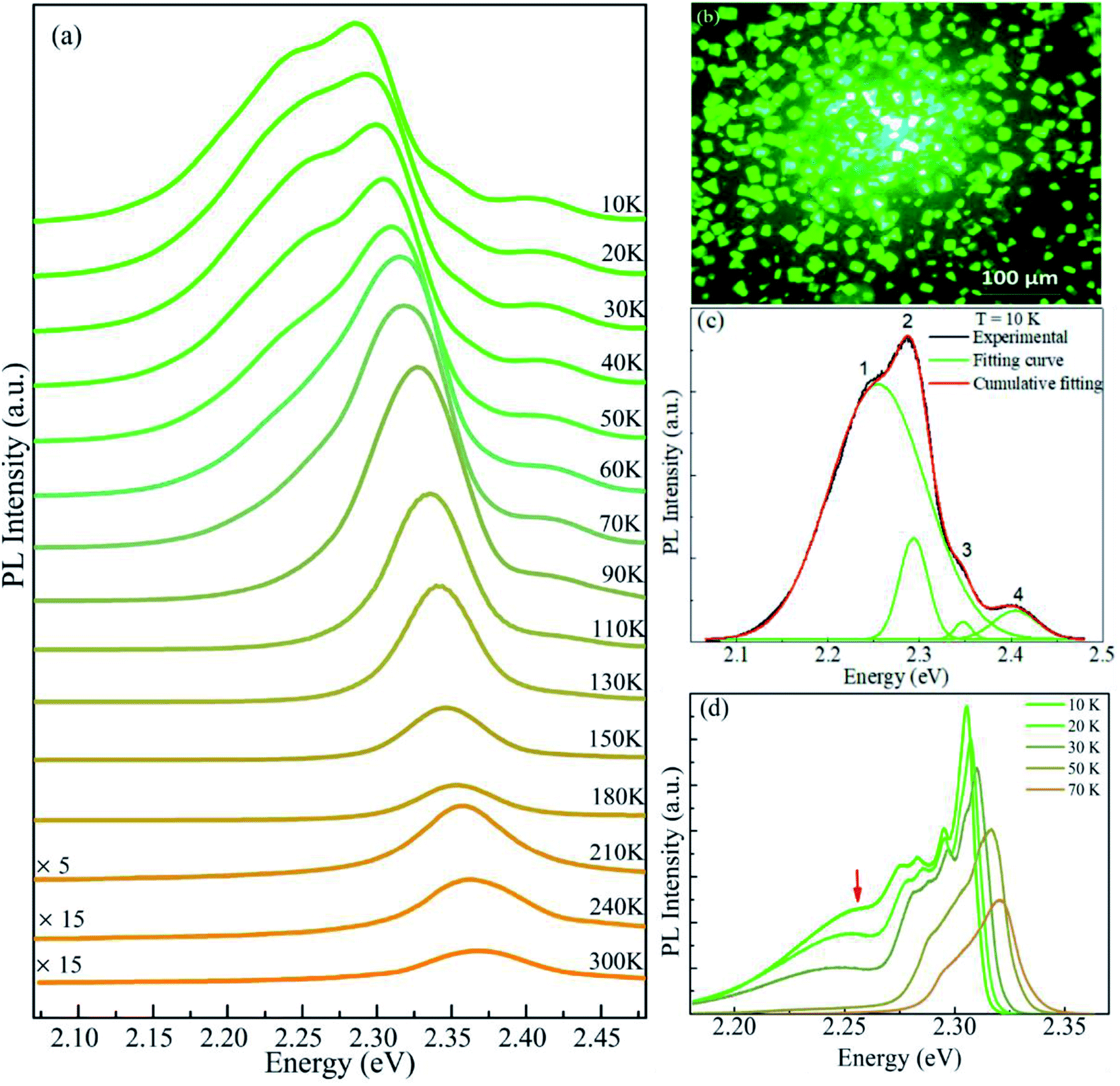 | ||
| Fig. 2 PL spectra of CsPbBr3 MCs under the excitation of 473 nm laser. (a) Temperature evolution of the PL spectra from 10 to 300 K with a constant vertical offset. (b) Fluorescence image of the sample. (c) The decomposition of the spectrum at 10 K, the obtained four Gaussian-shaped peaks locate at 2.254, 2.294, 2.348, and 2.405 eV, respectively. (d) PL spectra of the sample obtained after six months of exposure at ambient condition. | ||
Peak-3 and peak-4 are two weak components existing only at low temperatures (below 110 K). They locate at higher energy and their peak centers are insensitive to temperature. When the sample was exposed at ambient condition for six months, the PL measurement was performed again and the result is shown in Fig. 2d. In this case, the broad band (marked by red arrow) and the highest peak correspond to peak-1 and peak-2, respectively. Although the relative intensity of them varied the two peaks maintain at the same positions as that in Fig. 2c. The significant change is that peak-3 and peak-4 disappear absolutely and some additional peaks emerge between peak-1 and peak-2. This result indicates that peak-1 and peak-2 are intrinsic luminescence in the CsPbBr3 MCs and the other emission peaks may be concerned with imperfections of the crystals, such as Pb–Br clusters adsorbed on surface or defects induced by corrosion at ambient condition. The small clusters on surface have larger band gaps and are more easily destroyed by the water and oxygen molecules in air so that the corresponding peak-3 and peak-4 vanish. Hereafter, we focus mainly on peak-1 and peak-2.
Peak-2 is the strongest emission in the whole temperature range and the only peak at temperatures above 110 K, so it is naturally attributed to the FE recombination. To confirm this proposition, an absorption spectrum was derived from the diffuse reflection measurement at room temperature. As shown in Fig. 3a, it can be seen clearly that the emission peak (2.368 eV) almost overlaps with the excitonic absorption peak (2.386 eV), with a small Stokes shift of about 18 meV. Additionally, the power dependence of peak-1 and peak-2 was obtained at 10 K and shown in Fig. 3b. The integrated PL intensity of peak-1 and peak-2 are linearly dependent on the excitation power in a wide range. These results fit well with the features of FE emission.12,24,26,27 Therefore, peak-2 can be undoubtedly attributed to FE recombination.
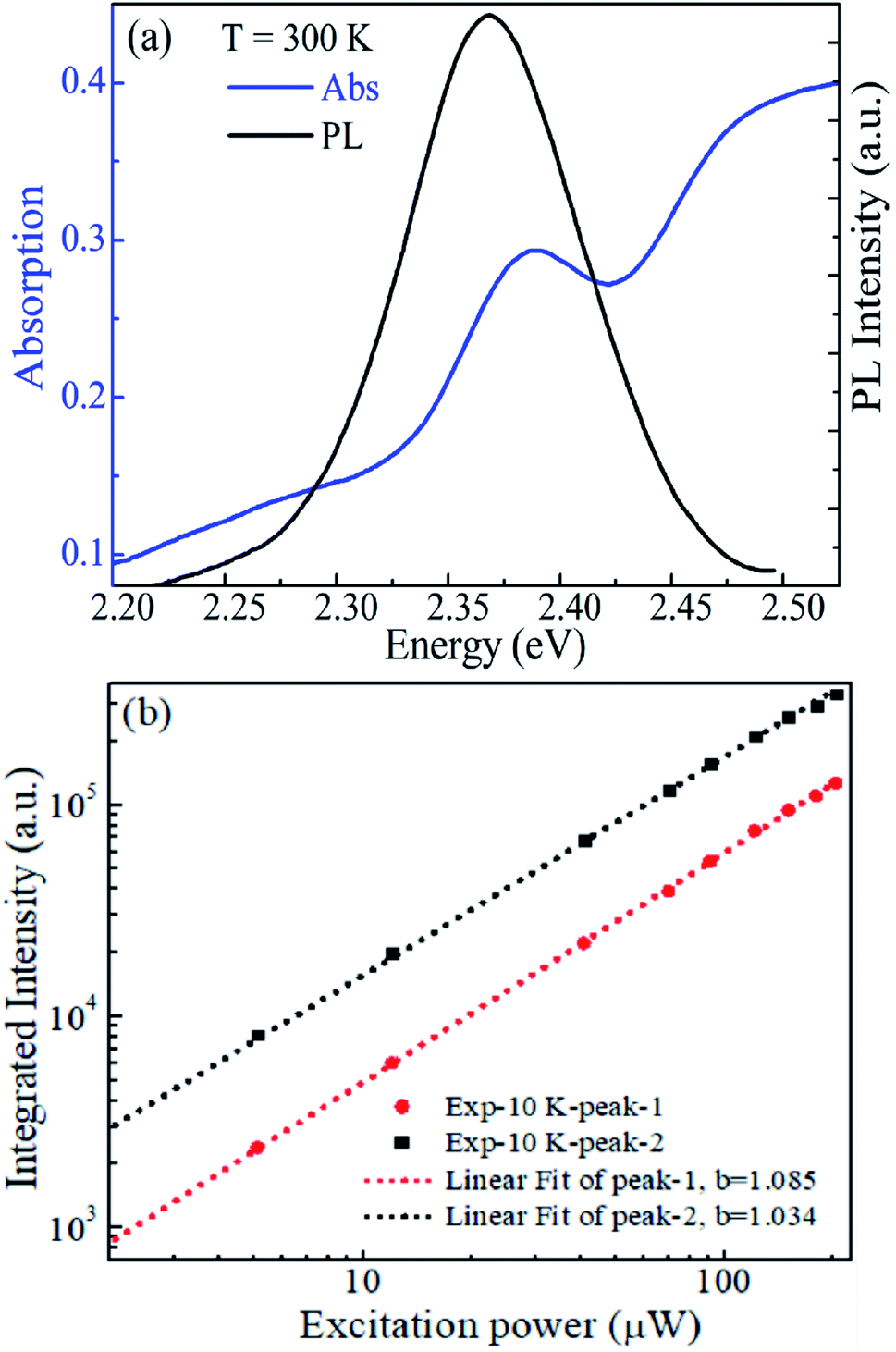 | ||
| Fig. 3 (a) Optical absorption (blue line) and emission PL (black line) spectra of CsPbBr3 MCs at 300 K. (b) Integrated PL intensity of peak-1 and peak-2 with respect to the excitation power. | ||
Peak-1 locates at energy 40 meV lower than peak-2 at 10 K. The large energy separation excludes it to be a phonon replica of the FE peak since a typical LO-phonon in CsPbBr3 was reported to be ∼18 meV.31–33 Emission peak with similar character was also proposed to be from the trapped excitons such as the BEs induced by impurities or defects in the crystal.3,24,26,27,61 Nevertheless, there remains controversy since it is very challenging to explore the trapping center definitely. The halide perovskites are thought to have high defect tolerance,5 so that defect or impurity generally does not introduce deep energy levels in the band gap. Additionally, the linear power dependence shown in Fig. 3b also denies it to be concerned with defects. Therefore, by considering the broad width characterization,62 peak-1 is proposed to be from STEs. STE was less reported in perovskite CsPbBr3 since the lattice is constrained in the three-dimensional structure. But it was still thought to be possible because of its soft lattice and strong ionic bonds, even though it may not be as strong as that in other perovskites with substituted components or low-dimensional structures.
As peak-1 and peak-2 are proposed to be from STEs and FEs, respectively, their intensity variation with rising temperature can be understood as the thermal transformation from STEs to FEs.52 The integrated PL intensity of peak-1 and peak-2 with respect to temperature are summarized in Fig. 4a and b, respectively. It can be seen clearly that the increase in peak-2 is almost equal to the decrease in peak-1 from 10 to 70 K, which indicates that the thermal activation from STEs are FEs has a high efficiency. This transformation is a thermal quenching process to STE emission but a negative thermal quenching process to FE emission, therefore the two processes share the same energy barrier. When the data are fitted using the Arrhenius equation,63,64 the activation energy obtained from the STE decrease process is 31.0 meV and 30.8 meV from the FE increase process. The equivalence of the two data gives a further support to the thermal transformation model. Consistent with previous reports,65 the intensity of FE peak decreases rapidly when the temperature is above 70 K. It may be induced by the dissociation of FEs and the activation of non radiative recombination centers. It may be noted that the temperature evolution of the PL intensity is different in Fig. 2d, where the intensity of both FE and STE peak decreases with increasing temperature. This seems to be in contrast to above discussion but it is understandable. The additional peaks locating between the FE and STE peak can be attributed to defect emission. The emergence of defects may induce thermal quenching of the FE emission and prevent the thermal transformation from STEs to FEs. So the enhancement section with temperature rising of FE peak was not observed in Fig. 2d.
 | ||
| Fig. 4 Photophysical properties of CsPbBr3 MCs. (a and b) The integrated PL intensity of peak-1 and peak-2, (c) the shift of peak centers and (d) FWHM of peak-1 with respect to temperature. | ||
The physical process of STE emission is usually described by the configuration coordinate model with a relatively large Huang–Rhys factor. This model indicates that the STE emission must be a broad band. According to previous theoretical analysis, the FWHM of STE emission is temperature dependent and it can be quantitatively given by66–68
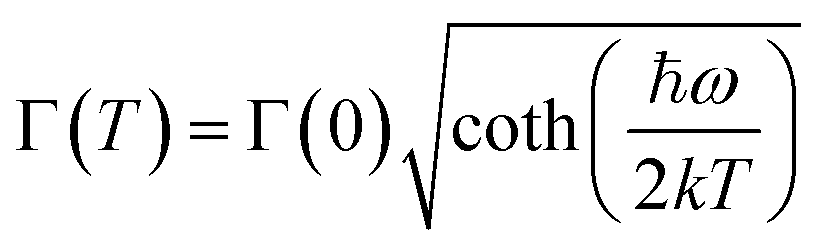 | (1) |
 | (2) |
Another key feature of STE emission is that it usually has a relative long lifetime. It has been reported to be 1.62 μs in Sb3+-doped Cs2NaInCl6 double perovskites70 and 1.44 μs in one-dimentional C4N2H14PbBr4.71 On the contrary, the lifetime of FE emission in perovskite CsPbBr3 is generally reported to be several nanoseconds25,54,72 and the lifetime in single CsPbBr3 quantum dot can be as short as tens of picoseconds.73 The lifetimes of both peak-1 and peak-2 were measured by fixing the detection wavelength at peak center under different temperatures and the results are shown in Fig. 5a and b. It can be noticed that the lifetime of peak-1 is obviously longer than that of peak-2. The lifetime as a function of temperature exhibits a rather complex feature but it can be well understood by considering the thermal transformation from STEs to FEs, which also lead to the non-single-exponential decay. All the decay curves (Fig. 5a and b) are fitted with a biexponential function.
| It = a1e−t/τ1 + a2e−t/τ2 | (3) |
 | ||
| Fig. 5 Temperature evolution of the time-resolved PL spectra of (a) peak-1 and (b) peak-2, and the lifetimes obtained by biexponential fitting for (c) peak-1 and (d) peak-2. The inset in (b) is an enlargement of the fast decay process. | ||
Which includes a fast (τ1) and a slow (τ2) decay process with amplitude represented by a1 and a2, respectively. The fitting results are revealed in Fig. 5c and d for peak-1 and peak-2, respectively. The lifetime τ1 spans from 1.1 to 2.3 ns in the concerned temperature range, consistent with previous reports for FE emission,25,54,72 whereas the value of τ2 can be as large as 123 ns at 10 K, consistent with the prediction for STE emission. The coexistence of fast and slow processes in each decay curve is due to the overlap of peak-1 and peak-2 (see Fig. 2c). In Fig. 5c, the lifetime τ2 decreases from 123 at 10 K to 32 ns at 100 K. That is because the probability of thermal transformation from STEs to FEs increases with rising temperature, which accelerates the depletion of STEs. This transformation also leads to the enhancement of FE emission in a short time (several nanoseconds) so the lifetime τ1 increases with increasing temperature. Except for the decrease section of τ1 from 10 to 50 K, the lifetimes of peak-2 shown in Fig. 5d both increase as the temperature goes up. This result is consistent with previous reports where the lifetime of FE in CsPbBr3 increases with rising temperature and many possible mechanisms such as dark-state occupation, Rashba splitting induced direct-to-indirect band gap, self-absorption induced photon recycle and trap-state activation, have been proposed to understand this abnormal phenomenon.54,72 Here we would like to attribute it to the thermal transformation from STEs to FEs. The decrease section of τ1 from 10 to 50 K is consistent with the conventional semiconductors. It is generally attributed to the defect-induced non radiative recombination, whose effect increases with increasing temperature. We do not discuss the lifetimes measured above 100 K here, since the peaks merge into one so that the PL signal does no longer have exclusive characters.
Above discussion all support that peak-1 is from STEs. It is necessary and significant to figure out the conditions to observe STE emission in CsPbBr3. Firstly, it can only be detected at low temperature. When the temperature is higher than 70 K, the STEs have been fully transformed to FEs. Secondly, the sample must have high crystalline quality, with low defect and impurity densities. This conclusion can be obtained by comparing the spectra in Fig. 2a and d. It can be seen that the relative intensity of STE emission (the ratio of peak-1 to peak-2) decreases greatly after the sample was left in air for six months; at the same time some defect concerned emission peaks appear between the FE and STE peak. It indicates that the STE emission can be eliminated by defect or impurity.
4. Conclusion
The steady and time-resolved PL spectra of CsPbBr3 MCs were measured within the temperature range from 10 to 300 K. The two main emission peaks were attributed to FE and STE recombination, respectively. The STE emission can only be observed below 70 K. The PL intensity variation of the two peaks with respect to temperature can be understood as the thermal transformation from STEs to FEs. The FWHM of the STE peak is ∼124 meV at 10 K and its temperature evolution follows a theoretical rule. The derived Huang–Rhys factor is ∼12 and the corresponding phonon energy is 15.3 meV. The lifetime of STEs is 123 ns at 10 K, much longer than that of FEs. These characterizations conform that of STE emission.Conflicts of interest
There are no conflicts to declare.References
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- D. Fröhlich, K. Heidrich, H. Künzel, G. Trendel and J. Treusch, J. Lumin., 1979, 18/19, 385–388 CrossRef.
- K. Nitsch, V. Hamplová, M. Nikl, K. Polák and M. Rodová, Chem. Phys. Lett., 1996, 258, 518–522 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi and E. Mosconi, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- C. Bao, J. Yang, S. Bai, W. Xu, Z. Yan, Q. Xu, J. Liu, W. Zhang and F. Gao, Adv. Mater., 2018, 30, 1–8 Search PubMed.
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef CAS PubMed.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- X. Zhang, B. Xu, J. Zhang, Y. Gao, Y. Zheng, K. Wang and X. W. Sun, Adv. Funct. Mater., 2016, 26, 4595–4600 CrossRef CAS.
- B. Cai, X. Li, Y. Gu, M. Harb, J. Li, M. Xie, F. Cao, J. Song, S. Zhang, L. Cavallo and H. Zeng, Sci. China Mater., 2017, 60, 811–818 CrossRef CAS.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- X. Lao, Z. Yang, Z. Su, Z. Wang, H. Ye, M. Wang, X. Yao and S. Xu, Nanoscale, 2018, 10, 9949–9956 RSC.
- B. Wu, H. T. Nguyen, Z. Ku, G. Han, D. Giovanni, N. Mathews, H. J. Fan and T. C. Sum, Adv. Energy Mater., 2016, 6, 1–9 Search PubMed.
- B. Murali, S. Dey, A. L. Abdelhady, W. Peng, E. Alarousu, A. R. Kirmani, N. Cho, S. P. Sarmah, M. R. Parida, M. I. Saidaminov, A. A. Zhumekenov, J. Sun, M. S. Alias and E. Yengel, ACS Energy Lett., 2016, 1, 1119–1126 CrossRef CAS.
- M. Fu, P. Tamarat, H. Huang, J. Even, A. L. Rogach and B. Lounis, Nano Lett., 2017, 17, 2895–2901 CrossRef CAS PubMed.
- M. Isarov, L. Z. Tan, M. I. Bodnarchuk, M. V Kovalenko, A. M. Rappe and E. Lifshitz, Nano Lett., 2017, 17, 5020–5026 CrossRef CAS PubMed.
- B. Wu, H. Yuan, Q. Xu, J. A. Steele, D. Giovanni, P. Puech, J. Fu, Y. F. Ng, N. F. Jamaludin, A. Solanki, S. Mhaisalkar, N. Mathews, M. B. J. Roeffaers, M. Grätzel, J. Hofkens and T. C. Sum, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- Y. Qiu, F. Nan, Q. Wang, X. Liu, S. Ding, Z. Hao, L. Zhou and Q. Wang, J. Phys. Chem. C, 2017, 121, 6916–6923 CrossRef CAS.
- W. Kong, Z. Ye, Z. Qi, B. Zhang, M. Wang, A. Rahimi-iman and H. Wu, Phys. Chem. Chem. Phys., 2015, 17, 16405–16411 RSC.
- M. I. Dar, G. Jacopin, S. Meloni, A. Mattoni, N. Arora, A. Boziki, S. M. Zakeeruddin, U. Rothlisberger and M. Grätzel, Sci. Adv., 2016, 2, 1601156 CrossRef PubMed.
- F. Panzer, S. Baderschneider, T. P. Gujar, T. Unger, S. Bagnich, M. Jakoby, H. Bässler, S. Hüttner, J. Köhler, R. Moos, M. Thelakkat, R. Hildner and A. Köhler, Adv. Opt. Mater., 2016, 4, 917–928 CrossRef CAS.
- H. Kunugita, Y. Kiyota, Y. Udagawa, Y. Takeoka, Y. Nakamura, J. Sano, T. Matsushita, T. Kondo and K. Ema, Jpn. J. Appl. Phys., 2016, 55, 060304 CrossRef.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- K. Shibata, J. Yan, Y. Hazama, S. Chen and H. Akiyama, J. Phys. Chem. C, 2020, 124, 18257–18263 CrossRef CAS.
- Q. Han, W. Wu, W. Liu, Q. Yang and Y. Yang, J. Lumin., 2018, 198, 350–356 CrossRef CAS.
- M. Sebastian, J. A. Peters, C. C. Stoumpos, J. Im, S. S. Kostina, Z. Liu, M. G. Kanatzidis, A. J. Freeman and B. W. Wessels, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 1–9 CrossRef.
- H. H. Fang, R. Raissa, M. Abdu-Aguye, S. Adjokatse, G. R. Blake, J. Even and M. A. Loi, Adv. Funct. Mater., 2015, 25, 2378–2385 CrossRef CAS.
- A. Dey, P. Rathod and D. Kabra, Adv. Opt. Mater., 2018, 6, 1–9 CAS.
- F. Gabelloni, F. Biccari, G. Andreotti, D. Balestri, S. Checcucci, A. Milanesi, N. Calisi, S. Caporali and A. Vinattieri, Opt. Mater. Express., 2017, 7, 4367 CrossRef CAS.
- A. Boziki, M. I. Dar, G. Jacopin, M. Grätzel and U. Rothlisberger, J. Phys. Chem. Lett., 2021, 12, 2699–2704 CrossRef CAS PubMed.
- C. M. Iaru, J. J. Geuchies, P. M. Koenraad, D. Vanmaekelbergh and A. Y. Silov, ACS Nano, 2017, 11, 11024–11030 CrossRef CAS PubMed.
- S. V. Myagkota, A. V. Gloskovskii and A. S. Voloshinovskii, Opt. Spektrosk., 2000, 88, 598–601 Search PubMed.
- X. Lao, Z. Yang, Z. Su, Y. Bao, J. Zhang, X. Wang, X. Cui, M. Wang, X. Yao and S. Xu, J. Phys. Chem. C, 2019, 123, 5128–5135 CrossRef CAS.
- K. M. Mccall, C. C. Stoumpos, S. S. Kostina, M. G. Kanatzidis and B. W. Wessels, Chem. Mater., 2017, 9, 4129–4145 CrossRef.
- M. Cong, B. Yang, F. Hong, T. Zheng, Y. Sang, J. Guo, S. Yang and K. Han, Sci. Bull., 2020, 65, 1078–1084 CrossRef CAS.
- G. Zhou, B. Su, J. Huang, Q. Zhang and Z. Xia, Mater. Sci. Eng. R Rep., 2020, 141, 100548 CrossRef.
- S. Li, J. Luo, J. Liu and J. Tang, J. Phys. Chem. Lett., 2019, 10, 1999–2007 CrossRef CAS PubMed.
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS PubMed.
- B. Dhanabalan, G. Biffi, A. Moliterni, V. Olieric, C. Giannini, G. Saleh, L. Ponet, M. Prato, M. Imran, L. Manna, R. Krahne, S. Artyukhin and M. P. Arciniegas, Adv. Mater., 2021, 33, 1–9 CrossRef PubMed.
- J. Li, H. Wang and D. Li, Front. Optoelectron., 2020, 13, 225–234 CrossRef.
- X. Li, X. Lian, J. Pang, B. Luo, Y. Xiao, M. De Li, X. C. Huang and J. Z. Zhang, J. Phys. Chem. Lett., 2020, 11, 8157–8163 CrossRef CAS PubMed.
- B. Luo, D. Liang, S. Sun, Y. Xiao, X. Lian, X. Li, M. De Li, X. C. Huang and J. Z. Zhang, J. Phys. Chem. Lett., 2020, 11, 199–205 CrossRef CAS PubMed.
- Z. Qi, Y. Chen, Y. Guo, X. Yang, F. Q. Zhang, G. Zhou and X. M. Zhang, J. Mater. Chem. C, 2021, 9, 88–94 RSC.
- Y. Zhang, X. Liu, H. Sun, J. Zhang, X. Gao, C. Yang, Q. Li, H. Jiang, J. Wang and D. Xu, Angew. Chem., 2021, 133, 7665–7670 CrossRef.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- L. Zdražil, S. Kalytchuk, M. Langer, R. Ahmad, J. Pospíšil, O. Zmeškal, M. Altomare, A. Osvet, R. Zbořil, P. Schmuki, C. J. Brabec, M. Otyepka and Š. Kment, ACS Appl. Energy Mater., 2021, 4, 6445–6453 CrossRef.
- R. Ahmad, L. Zdražil, S. Kalytchuk, A. Naldoni, A. L. Rogach, P. Schmuki, R. Zboril and Š. Kment, ACS Appl. Mater. Interfaces, 2021, 13, 47845–47859 CrossRef CAS PubMed.
- R. Ahmad, L. Zdražil, S. Kalytchuk, A. Naldoni and E. Mohammadi, Appl. Mater. Today, 2022, 26, 1–13 Search PubMed.
- B. M. Benin, D. N. Dirin, V. Morad, M. Wörle, S. Yakunin, G. Rainò, O. Nazarenko, M. Fischer, I. Infante and M. V. Kovalenko, Angew. Chem., Int. Ed., 2018, 57, 11329–11333 CrossRef CAS PubMed.
- J. Yin, J. L. Brédas, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2020, 32, 5036–5043 CrossRef CAS.
- S. Bhaumik, ACS Omega, 2020, 5, 22299–22304 CrossRef CAS PubMed.
- X. Ma, F. Pan, H. Li, P. Shen, C. Ma, L. Zhang, H. Niu, Y. Zhu, S. Xu and H. Ye, J. Phys. Chem. Lett., 2019, 10, 5989–5996 CrossRef CAS PubMed.
- Y. Wang, F. Yang, X. Li, F. Ru, P. Liu, L. Wang, W. Ji, J. Xia and X. Meng, Adv. Funct. Mater., 2019, 29, 1–9 Search PubMed.
- X. Yuan, P. Jing, J. Li, M. Wei, J. Hua, J. Zhao, L. Tian and J. Li, RSC Adv., 2016, 6, 78311–78316 RSC.
- R. Ahmed, F. -e-Aleem, S. J. Hashemifar and H. Akbarzadeh, Phys. B: Condens. Matter, 2008, 403, 1876–1881 CrossRef CAS.
- G. R. Yettapu, D. Talukdar, S. Sarkar, A. Swarnkar, A. Nag, P. Ghosh and P. Mandal, Nano Lett., 2016, 16, 4838–4848 CrossRef CAS PubMed.
- K. Heidrich, H. Künzel and J. Treusch, Solid State Commun., 1978, 25, 887–889 CrossRef CAS.
- D. N. Dirin, L. Protesescu, D. Trummer, I. V. Kochetygov, S. Yakunin, F. Krumeich, N. P. Stadie and M. V. Kovalenko, Nano Lett., 2016, 16, 5866–5874 CrossRef CAS PubMed.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468–471 CrossRef CAS.
- A. N. Chaika, V. A. Grazhulis, A. M. Ionov, P. K. Kashkarov, S. L. Molodtsov, A. M. Shikin and C. Laubschat, Surf. Sci., 1999, 433, 352–356 CrossRef.
- C. C. Stoumpos and M. G. Kanatzidis, Acc. Chem. Res., 2015, 48, 2792–2802 CrossRef PubMed.
- D. Lee, A. Mysyrowicz, A. V. Nurmikko and B. J. Fitzpatrick, Phys. Rev. Lett., 1987, 58, 1475–1478 CrossRef CAS PubMed.
- Z. Shi, Y. Li, Y. Zhang, Y. Chen, X. Li, D. Wu, T. Xu, C. Shan and G. Du, Nano Lett., 2017, 17, 313–321 CrossRef CAS PubMed.
- R. Saran, A. Heuer-Jungemann, A. G. Kanaras and R. J. Curry, Adv. Opt. Mater., 2017, 5, 1–18 Search PubMed.
- V. B. Mykhaylyk, H. Kraus, V. Kapustianyk, H. J. Kim, P. Mercere, M. Rudko, P. Da Silva, O. Antonyak and M. Dendebera, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed.
- T. G. Mack, L. Jethi and P. Kambhampati, J. Phys. Chem. C, 2017, 121, 28537–28545 CrossRef CAS.
- D. Pal and D. N. Bose, J. Appl. Phys., 1995, 78, 5206–5208 CrossRef CAS.
- S. Bhushan and M. V. Chukichev, J. Mater. Sci. Lett., 1988, 7, 319–321 CrossRef CAS.
- R. K. Dawson, Phys. Status Solidi, 1969, 95, 95–105 CrossRef.
- R. Zeng, L. Zhang, Y. Xue, B. Ke, Z. Zhao, D. Huang, Q. Wei, W. Zhou and B. Zou, J. Phys. Chem. Lett., 2020, 11, 2053–2061 CrossRef CAS PubMed.
- Z. Yuan, C. Zhou, Y. Tian, Y. Shu, J. Messier, J. C. Wang, L. J. Van De Burgt, K. Kountouriotis, Y. Xin, E. Holt, K. Schanze, R. Clark, T. Siegrist and B. Ma, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- X. Y. Zhang, G. T. Pang, G. C. Xing and R. Chen, Mater. Today Phys., 2020, 15, 1–7 Search PubMed.
- B. T. Diroll, H. Zhou and R. D. Schaller, Adv. Funct. Mater., 2018, 28, 1–7 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |