
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiviral activities of natural compounds and ionic liquids to inhibit the Mpro of SARS-CoV-2: a computational approach†
Kandhan Palanisamy a,
S. M. Esther Rubavathya,
Muthuramalingam Prakash*a,
Ramasamy Thilagavathib,
Maryam S. Hosseini-Zarec and
Chelliah Selvam*c
a,
S. M. Esther Rubavathya,
Muthuramalingam Prakash*a,
Ramasamy Thilagavathib,
Maryam S. Hosseini-Zarec and
Chelliah Selvam*c
aDepartment of Chemistry, Faculty of Engineering and Technology, SRM Institute of Science and Technology, Kattankulathur-603203, Chennai, Tamil Nadu, India. E-mail: prakashspm@gmail.com; prakashm4@srmist.edu.in
bDepartment of Biotechnology, Faculty of Engineering, Karpagam Academy of Higher Education, Coimbatore, India
cDepartment of Pharmaceutical and Environmental Health Sciences, College of Pharmacy and Health Sciences, Texas Southern University, Houston, TX-77004, USA. E-mail: selvam.chelliah@tsu.edu
First published on 28th January 2022
Abstract
The recalcitrant spread of the COVID-19 pandemic produced by the novel coronavirus SARS-CoV-2 is one of the most destructive occurrences in history. Despite the availability of several effective vaccinations and their widespread use, this line of immunization often faces questions about its long-term efficacy. Since coronaviruses rapidly change, and multiple SARS-CoV-2 variants have emerged around the world. Therefore, finding a new target-based medication became a priority to prevent and control COVID-19 infections. The main protease (Mpro) is a salient enzyme in coronaviruses that plays a vital role in viral replication, making it a fascinating therapeutic target for SARS-CoV-2. We screened 0.2 million natural products against the Mpro of SARS-CoV-2 using the Universal Natural Product Database (UNPD). As well, we studied the role of ionic liquids (ILs) on the structural stabilization of Mpro. Cholinium-based ILs are biocompatible and used for a variety of biomedical applications. Molecular docking was employed for the initial screening of natural products and ILs against Mpro. To predict the drug-likeness features of lead compounds, we calculated the ADMET properties. We performed MD simulations for the selected complexes based on the docking outcomes. Using MM/PBSA approaches, we conclude that compounds NP-Hit2 (−25.6 kcal mol−1) and NP-Hit3 (−25.3 kcal mol−1) show stronger binding affinity with Mpro. The hotspot residues of Thr25, Leu27, His41, Met49, Cys145, Met165, and Gln189 strongly interacted with the natural compounds. Furthermore, naproxenate, ketoprofenate, and geranate, cholinium-based ILs strongly interact with Mpro and these ILs have antimicrobial properties. Our findings will aid in the development of effective Mpro inhibitors.
Introduction
The emergence of the highly transmissible disease SARS-CoV-2 has given rise to the pandemic COVID-19, causing widespread dread and a severe impact on the world economy.1 An unexpected outbreak of COVID-19, triggered by the highly contagious SARS-CoV-2 virus, has affected millions of people worldwide. A large number of deaths are reported every day at the hands of this devastating disease. It is the seventh coronavirus to afflict humans, and because of its transmissible nature, it has a greater fatality rate.2–4 The World Health Organization (WHO) proclaimed the COVID-19 outbreak a Public Health Emergency of International Concern (PHEIC) in March 2020.5 Elderly persons with diabetes, pulmonary issues, and lung injury who require hospitalization or succumb to COVID-19 can suffer severe sickness, dry cough, and respiratory-related diseases.6 According to a recent study, viral infection leads to long-term detrimental consequences such as kidney impairment and myocardial ischemia.Currently, seven COVID-19 vaccines have received approval for the Emergency Use Authorization (EUA) by WHO. The global immunization campaign is underway, but the infection rate continues to rise due to the emergence of COVID-19 mutations.7 The most promising treatment option for patients with COVID-19 is convalescent plasma therapy. Nonetheless, numerous COVID-19 variations have emerged due to mutation, posing a new challenge to humankind.8 Alternative immunization strategies are urgently required. COVID-19 strains are more infectious and have a higher fatality rate. As a result, new effective antiviral medications are essential to control the rising number of COVID-19 patients. Previous studies have shown that people with low immunity and pre-existing health problems like cancer, diabetes, hypertension, and respiratory disorders, among other things, are more vulnerable to severe infection.6
The coronavirus latches onto the human spike protein and interacts with the human angiotensin-converting enzyme 2 (hACE2). SARS-CoV-2 is a beta coronavirus belonging to the Coronaviridae family, single-strand positive RNA virus ((+) ssRNA), capable of infecting animals to humans (i.e., zoonotic).9 It contains two cysteine proteases and four structural proteins. The structural proteins of spike glycoprotein (S protein) are involved in virus generation and replication, as well as membrane (M), envelope (E), and nucleocapsid (N) proteins.10 The acid-base forms of quercetin and its metabolites potential inhibitors against the targets of furin and S protein of SARS-CoV-2.11 Furthermore, the replication processes are mediated by the two proteases 3C-like protease (3CLpro) or main protease (Mpro) and Papain-like protease (PLpro).12 The viral polyproteins are cleaved into 11 nonstructural proteins (NPs) by Mpro, while three nonstructural proteins are cleaved by PLpro. Other proteolytic enzymes, which are more similar to human proteases, are more complicated to target. SARS CoV-2 Mpro is a promising drug target for developing inhibitors for COVID-19 infections.13,14 Domain I (10–99), domain II (100–182), and domain III (198–303) amino acids constitute the Mpro structure. Mpro cysteine protease's active site consists of catalytic dyads, His-41 and Cys-145, located between domains I and II. On the other hand, domain III has five helices and forms dimerization of Mpro through salt–bridge interactions.15 Mutations occurred in 282 out of 306 residues in the Mpro. Remaining 24 residues had no reported mutations, these non-mutational residues are called as cold spot residues.16
According to recent research, vaccination options for COVID-19 mutations are limited. To counteract and reduce COVID-19 infection rates, we need to focus on antiviral medications from a wide range of sources that can effectively inhibit Mpro. Antiviral drugs must meet the following criteria: high binding affinity for the target protein, bioavailability, low manufacturing costs, and minimal risk of side effects.17 Natural products and herbs have been extensively examined for their antiviral properties and shown to have significant activity against coronavirus.18 Michael acceptor inhibitors, α-ketoamides, aldehydes, ketones, and their derivatives, among others, have a strong interaction with Mpro's active sites.19,20 Recently, Marković and his co-workers studied, adamantane-isothiourea derivatives have strong inhibition potency towards Mpro and mutation of SARS-CoV-2 Spike protein D614G.21 Coumarin derivatives and their Pd(II) complexes have stronger binding affinity with Mpro of SARS-CoV-2 compared to the FDA approved drugs.22 These studies explored that natural compounds and their derivatives potent inhibitors for Mpro of SARS-CoV-2. With human cells, these natural compounds have potent antifungal, antibacterial, anti-inflammatory, and antiviral properties.23 We have screened the natural products from the universal natural product database with the Mpro of SARS-CoV-2.
Ionic liquids (ILs) have recently gained prominence in biological applications as solvents, stabilizers, and destabilizers.24,25 Because of their antibacterial properties, ILs are used in biomedical applications.26 The physicochemical attributes of designer solvent ILs, such as viscosity, solubility, toxicity, and hydrophobicity, can all be calibrated. Juhi and her co-workers investigated the antiviral activity of ionic liquids, herbs, and natural compounds against Mpro. As a result, ILs have a higher inhibitory action against SARS-CoV-2.27 Bio-degradation, toxicity, inadequate physicochemical qualities, and expense have been issued with classic ILs like imidazolium,28 pyrrolidinium, and pyridinium.29 Cholinium-based ILs have several specific characteristics, including nontoxicity and biodegradability.30 Recent developments have utilized cholinium-based ILs for many drug delivery applications due to their biocompatibility and benign nature.31 Insulin structure is strongly stabilized by cholinium-based ILs which are used in biomedical applications.32,33 Cholinium-based ILs are chosen against Mpro based on their antibacterial and anti-inflammatory properties.
The structure, stability and interactions of protein-ligand complexes were determined using integrated computational methods of molecular docking, atomistic molecular dynamics (MD) simulations, MM/PBSA binding free energy calculations, and ADMET properties. Virtual screening methods are widely used for drug designs and development.34–37 Large library of natural products (UNPD) and cholinium-based ILs screened using molecular docking. As a result, we chose the most stable complexes of MD simulations to interpret the stability and interaction mechanism between natural products and ILs. Our research will contribute further insights into the inhibitory potential of natural compounds and ILs. Based on these findings, we can look for potential inhibitors of the SARS-CoV-2 Mpro.
Computational methodologies
Molecular docking simulations
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 358 molecules that were conformationally analyzed. The X-ray crystal structure of Mpro was retrieved from the protein data bank (PDB ID: 6xr3) which is shown in Fig. 1. The receptor grid was created with the OEDOCKING's pdb2receptor tool. The FRED (Fast Rigid Exhaustive Docking) module of OpenEye was used to construct the virtual screen.38 FRED performs a rigid docking of ligand into the protein active site. To generate an ensemble of poses, FRED performs a systematic and comprehensive search. Omega2 was used to generate numerous conformers for the specified compounds. To exhibit the various conformations of selected library compounds, OMEGA (OpenEye) was employed. It is a rapid conformer generation tool developed by OpenEye that optimizes geometry using a modified Merck Molecular Force Field (MMFF).39 During the conformation generation, the OMEGA default parameters were employed. The binding poses that resulted from docking were examined. We chose four compounds (UNPD68644, UNPD19759, UNPD75460 and UNPD23673) from the top hits and performed molecular dynamics simulations on them. The natural products of UNPD68644, UNPD19759, UNPD75460 and UNPD75473 are denoted as NP-Hit1, NP-Hit2, NP-Hit3 and NP-Hit4, respectively. The resulting binding modes of selected compounds were analyzed and discussed. The virtual screening of natural compounds are displayed in Fig. 2.
358 molecules that were conformationally analyzed. The X-ray crystal structure of Mpro was retrieved from the protein data bank (PDB ID: 6xr3) which is shown in Fig. 1. The receptor grid was created with the OEDOCKING's pdb2receptor tool. The FRED (Fast Rigid Exhaustive Docking) module of OpenEye was used to construct the virtual screen.38 FRED performs a rigid docking of ligand into the protein active site. To generate an ensemble of poses, FRED performs a systematic and comprehensive search. Omega2 was used to generate numerous conformers for the specified compounds. To exhibit the various conformations of selected library compounds, OMEGA (OpenEye) was employed. It is a rapid conformer generation tool developed by OpenEye that optimizes geometry using a modified Merck Molecular Force Field (MMFF).39 During the conformation generation, the OMEGA default parameters were employed. The binding poses that resulted from docking were examined. We chose four compounds (UNPD68644, UNPD19759, UNPD75460 and UNPD23673) from the top hits and performed molecular dynamics simulations on them. The natural products of UNPD68644, UNPD19759, UNPD75460 and UNPD75473 are denoted as NP-Hit1, NP-Hit2, NP-Hit3 and NP-Hit4, respectively. The resulting binding modes of selected compounds were analyzed and discussed. The virtual screening of natural compounds are displayed in Fig. 2.
 | ||
| Fig. 1 A) Schematic representation of SARS-CoV-2 and (B) crystal structure of Mpro, blue color indicates the active site of catalytic dyads (i.e., His41 and Cys142). | ||
 | ||
| Fig. 2 Virtual screening of natural compounds from Universal Natural Product Database (UNPD). | ||
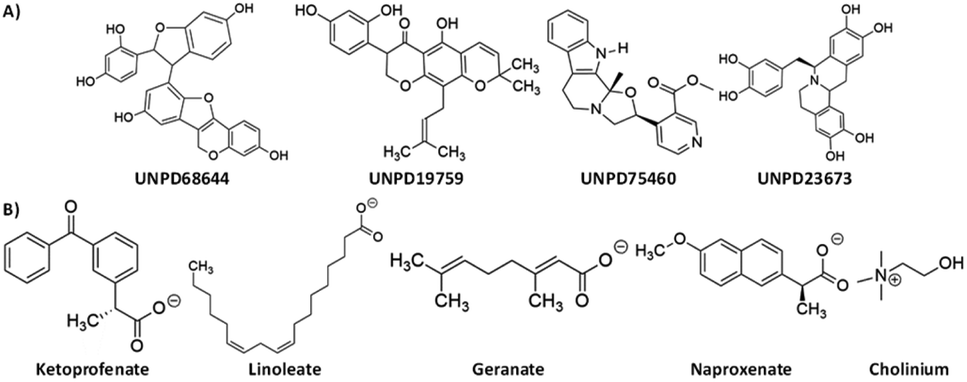 | ||
| Fig. 3 The virtual screening hits of (A) Natural compounds and (B) Cholinium-based ILs. | ||
ADMET analysis
ADMET characteristic analysis used to predict the pharmacokinetic properties of all selected classes of compounds were discussed in this work, such as adsorption, distribution, metabolism, excretion, and toxicity (ADMET). SwissADME was used to predict the pharmacokinetic characteristics of a selected class of ionic liquids and natural compounds.46 Veber's rule, Ghose's rule, and Lipinski's rule of five (ROF) were calculated for our selected compounds. The drug-likeness can be determined using ROF, which states that the molecular weight, hydrophobicity, number of hydrogen bond (nHB) acceptors, and nHB donors should not exceed 500 kDa, 5, 10, and 5, respectively, to be considered a possible drug-like molecule. Veber's rule said that the topological polar surface area (TPSA) and the number of rotatable bonds should not exceed 140 Å2 and 10, respectively, to be considered a prospective drug-like molecule. Another foremost aspect of drug design is determining the toxicity of a specific class of chemicals. The computational approach to toxicity evaluation aids in calculating the toxic dose level in an animal model. It decreases the number of animal model trials and reduces the probability of failure in the experimental technique.Molecular dynamics simulations
We ran MD simulations for Mpro complexes of natural products and cholinium-based ILs using Gromacs 2016.3.47 The complexes of Mpro and natural products were placed in a cubic box with a volume of 9.8 nm3. The system is solvated with the SPC/E water model and four Na+ ions were added to maintain neutrality.48 For Mpro and ligands, force filed parameters of Amber ff99SB-ILDN and general amber force field (GAFF) were used, respectively.49,50 The atomic partial charges for ligands were calculated the B3LYP/6-31+G** level of electronic calculations using the RESP method.51 For all three dimensions, periodic boundary conditions (PBC) were used. The steepest descent algorithm was used to achieve energy minimization of the system. At 300 K and 1 bar pressure, the system was equilibrated with isothermal-isochoric (NVT) and isothermal-isobaric (NPT) ensembles using velocity-rescaling and Berendsen coupling, respectively. For long-range electrostatic interactions, the Particle Mesh Ewald technique has been utilized with a cut-off distance of 1.2 nm.52 The LINCS algorithm was utilized to limit all hydrogen and heavy atom bonds.53 The integration time step of 2 fs was applied using the leap-Frog algorithm.54 The molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) approaches were used for binding free energy calculations.55We solvated Mpro with a 3 M concentration of cholinium-based ILs to better understand its stability in the presence of ILs medium. At 3 M concentration, the effect of stabilization and destabilization of protein was determined.56 The GAFF parameters were used for cholinium-based ILs. Further, the above-mentioned MD protocol was applied to the system. The basic analysis was done with the gromacs suite of programs. VMD and pyMOL tools were used to visualize and analyze the trajectories.57,58 The production run was performed for 200 ns, and the time series with energies were plotted to establish the equilibration.
Results and discussion
The importance of natural compounds as antiviral agents has already been explored. We performed molecular docking investigations involving naturally occurring compounds (UNPD), and ILs in addition to the control drugs for treating SARS-CoV-2 in this work. On protein targets, the binding methods and binding sites of ligand were determined. The most stable conformations were sorted according to their highest binding energy, and the results were then used to interpret the results individually.Docking studies
358 molecules) of natural compounds against Mpro. Table 1 shows the results of the top hits and their interactions. NP-Hit1, NP-Hit2, NP-Hit3 and NP-Hit4 are the natural compounds with a binding score range of −11.87 to −10.91 kcal mol−1. Complex formation is associated with negative binding energy, implying that the complex formation is spontaneous. These four compounds exhibit a higher affinity for binding than lopinavir (−9.9 kcal mol−1), a potent Mpro inhibitor that has been explored in vitro and preclinical studies. Molecular docking, ADMET and MD simulations on Mpro-lopinavir complex is shown in ESI.† In molecular docking, FRED and Autodock 4.2 were used for natural compounds and cholinium-based ILs, respectively. Natural compounds interact with the active sites (His41 and Cys145) of the Mpro, as shown by the docked poses. The docked conformations of Mpro with natural compounds are shown in Fig. 4. NP-Hit1 compound forms five H-bonding interactions with Cys44, Glu143, His164, Asp187, and Gln189. NP-Hit2 also has six H-bonding interactions with Tyr54, Phe140, Ser144, His163, Glu166, and Asp187. The amino acids involved in the H-bonding interactions of NP-Hit3 are Phe140, Leu141, Asn142, His163, and Arg188. The residues in contact with the NP-Hit4 through H-bond are Thr25, Cys44, Phe140, Ser144, His163, Glu166, His172, and Arg188. Aside from this, the compounds NP-Hit1, NP-Hit2, NP-Hit3, NP-Hit4 forms π–π interactions with His41. NP-Hit4 shows anion-π stacking interactions with the amino acid residue Gln189.
| Selected NP compounds | Docking Score (in kcal mol−1) | H-bonding interactions |
|---|---|---|
| a The docking score and H-bonding interactions are shown. | ||
| NP-Hit1 | −11.87 | Cys44, Gly143, His164, Asp187, and Gln189 |
| NP-Hit2 | −11.16 | Tyr54, Phe140, Ser144, His163, Glu166, and Asp187 |
| NP-Hit3 | −11.51 | Phe140, Leu141, Asn142, His163, and Arg188 |
| NP-Hit4 | −10.91 | Thr25, Cys44, Phe140, Ser144, His163, Glu166, His172, and Arg188 |
| Choline naproxenate | −10.76 | Lys5, Lys102, Lys137, Asp153, Asp155, and Glu288 |
| Choline ketoprofene | −10.28 | Arg4, Lys5, Lys102, Asp153, Asp155, and Glu288 |
| Choline linoleate | −9.35 | Phe3, Arg4, Lys5, Lys102, Asp153, Asp155, and Glu288 |
| Choline geranate | −9.05 | Lys12, Lys102, Ile152, Asp153 and Asp155 |
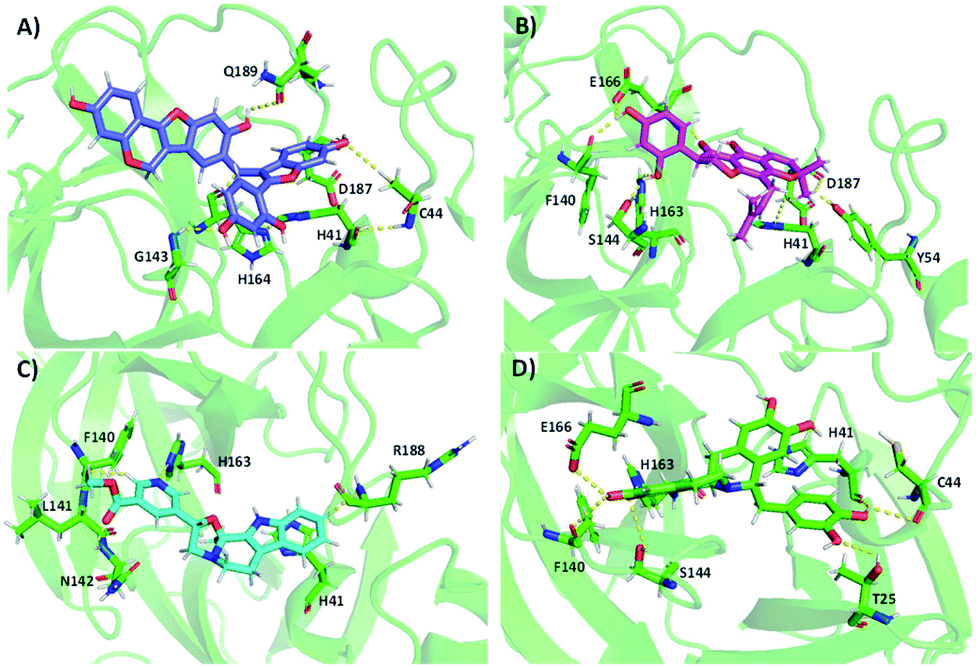 | ||
| Fig. 4 Docking conformations of selected natural compounds with Mpro. (A) NP-Hit1 (B) NP-Hit2 (C) NP-Hit3 and (D) NP-Hit4. | ||
The conformers for four ILs with various anion moieties, choline naproxenate, choline ketoprofenate, choline linoleate, and choline geranate, were docked using the minimum energy approach.59–61 The binding scores of cholinium-based ionic liquids include choline naproxenate (−10.76 kcal mol−1), choline ketoprofenate (−10.28 kcal mol−1), choline linoleate (−9.35 kcal mol−1), and choline geranate (9.05 kcal mol−1). The binding energies and interactions of Mpro-ILs are shown in Table 1.
It was observed that the targeted protein and ILs solely form H-bonding interactions. The docking conformations and interaction sites of Mpro-ILs are displayed in Fig. 5. Choline cation interacts with Lys102, Tyr154, Cys156, Asp 153, and Asp155 amino acids. Especially, Lys102, Asp153, and Asp155 form H-bonding interactions with choline. Positive amino acids of Lys5 and Lys137 have implicated H-bonding interactions at 1.7 and 1.8 Å distances with choline naproxenate.
 | ||
| Fig. 5 Docking conformations of selected choline-based ILs with Mpro. (A) Naproxenate (B) ketoprofenate (C) linoleate (D) geranate and (E) cholinium. | ||
With choline ketoprofenate, the residues Arg4, Lys5, and Glu288 make six H-bonding interactions. The amino acid residues Phe3, Arg4, Lys5, and Glu288 create H-bond interactions with choline linoleate. Arg4 forms stronger interactions (NH⋯O) at 1.7 Å with linoleate. At last, choline geranate forms H-bond interaction with the following amino acid residues of the targeted protein Pro9, Lys12, Glu14, and Ile152. Lys12 makes stronger H-bonding interactions at 1.9 Å distances. These interactions stabilize the Mpro-IL complexes.
Drug-likeness prediction and PAINS filtration
We have calculated the ADMET properties for the stable compounds. The ADMET analysis evaluates drug-likeness properties of natural compounds as well ILs which is displayed in Table 2. The drug-likeness parameter is influenced by the way the drug is administered. Aqueous solubility and intestinal permeability are associated with these pharmacokinetic parameters. Lipinski's rule of five (RO5) aids in predicting the medicinal and combinatorial chemistry of a given class of compounds. The predicted value of various properties such as size, lipophilicity, polarity, etc. The value of molecular weight describes the size of the molecule. The logP value represents the lipophilicity of the molecules, which is connected to the drug molecule's solubility in an aqueous environment.
| Compounds | MW | nHB acceptors | nHB donors | nRotB | nVio | TPSA (Å2) | GI absorption | Heptatoxicity | Carcinogenicity | PAINS |
|---|---|---|---|---|---|---|---|---|---|---|
| a ADMET properties of MW = molecular weight (g mol−1), nHB donors = number of H-bond donors, nHB acceptors = number of H-bond acceptors, nRotB = number of rotatable bonds, nVio = number of Lipinski's rule violations, TPSA = topological polar surface area, and GI absorption = gastrointestinal absorption. | ||||||||||
| NP-Hit1 | 508.4 | 8 | 5 | 3 | 1 | 136.66 | Low | Inactive(0.79) | Inactive(0.58) | 0 |
| NP-Hit2 | 422.4 | 6 | 3 | 3 | 0 | 96.22 | High | Inactive(0.78) | Inactive(0.64) | 0 |
| NP-Hit3 | 363.4 | 5 | 1 | 3 | 0 | 67.45 | High | Inactive(0.81) | Inactive(0.53) | 0 |
| NP-Hit4 | 421.4 | 7 | 6 | 2 | 1 | 124.62 | High | Active(0.69) | Inactive(0.62) | 0 |
| Choline | 104.1 | 1 | 1 | 2 | 0 | 20.23 | Low | Inactive(0.94) | Inactive(0.78) | 0 |
| Naproxenate | 230.2 | 3 | 1 | 3 | 1 | 49.36 | High | Inactive(0.58) | Inactive(0.57) | 0 |
| Ketoprofen | 254.2 | 3 | 1 | 4 | 0 | 54.37 | High | Inactive(0.54) | Inactive(0.75) | 0 |
| Linoleate | 280.4 | 2 | 1 | 14 | 1 | 37.30 | High | Inactive(0.55) | Inactive(0.64) | 0 |
| Geranate | 167.7 | 2 | 0 | 4 | 0 | 40.13 | High | Inactive(0.54) | Inactive(0.79) | 0 |
The RO5 implies that when there are more than 5 H-bond donors, 10 H-bond acceptors, the molecular weight is greater than 500 Da, and the computed logP is greater than 5, poor absorption is more likely. TPSA, number of rotatable bonds, and molar refractivity were calculated and applied Veber and Ghose rules to assess drug likeness. PAINS (Pan-assay interference) analysis was carried out on the lead compounds generated by the virtual screening and has shown distinct structural features, which can result in false-positive results in some cases when used in virtual screenings and can interact non-specifically with random targets rather than specific targets. As a result, it's critical to rule out compounds having PAINS characteristics to avoid false positives. Based on the ADMET results, natural compounds have high GI (Gastro-Intestinal tract) absorption. Furthermore, ionic liquids follow the Lipinski rule of five and have high absorption in the GI system. From Table 2, we conclude that our natural compounds and cholinium-based ILs have good drug-like properties. Hepatotoxicity and carcinogenicity are found inactive in both natural compounds and ionic liquids. Hepatotoxicity refers to liver damage caused by chemicals. The liver is a vital organ for converting and removing toxins, and it is vulnerable to their toxicity.
Molecular dynamics simulations
 | ||
| Fig. 6 A) and (C) shows the RMSD and (B) and (D) shows the radius gyration (RR) of complexes of Mpro with natural compounds and choline-based ILs. | ||
The average RMSD values of Mpro with natural compounds and ILs were displayed in ESI (Table S4 and Table S5†). The natural compounds NP-Hit3 and NP-Hit4 have observed the lowest RMSD values of 1.77 and 1.78 Å, respectively. While the other compound NP-Hit1 has a higher RMSD value of 2.04 Å. We considered IL medium for the stabilization of Mpro structure, cholinium-based ILs used for various drug delivery applications.
The RMSD plots of Mpro structure in the presence of IL medium are shown in Fig. 6C. The average RMSD values of Mpro in the IL medium are displayed in ESI (Table S5†). Cholinium-based ILs were tightly bound with the protein and conserved the structural stability of Mpro. The system density is represented by the radius of gyration (Rg), which has an impact on the folding rate and protein compactness. Rg was used to determine the compactness of the complexes, and it was found that Rg of all systems was consistent with the RMSD system. This shows that the protein remained stable and compacted throughout 200 ns. The radius of gyration (Rg) of protein and natural compound complexes was found to be between 2.20 and 2.21 nm. Also, the radius of gyration for the protein and ionic liquids complexes is found to be between 2.16 and 2.19 nm. Fig. 6B and D reveal the compactness and unfolding of Mpro structure binding with the natural products and ILs. There is no large difference in the average Rg values. Thereby, we conclude that both natural compounds and ILs maintain structural stability. Also, the flexibility of C-alpha atoms of Mpro is plotted in ESI (Fig. S3†). Thereby, the flexibility of the Mpro structure is minimized in the presence of natural compounds and ILs. All these results conclude that both natural products and ILs equally stabilize the Mpro structure. Further, the average number of H-bonding interactions calculated for Mpro-natural compounds and Mpro-ILs are is shown in ESI Table S6.† NP-Hit4 compound form ∼2 H-bonding interactions with Mpro, whereas compound NP-Hit3 has very less probability of forming H-bonding interactions. Compound NP-Hit1 and NP-Hit2 are stabilized by one H-bonding interaction with Mpro. On the other hand, 3 M concentration of ILs forms more number of H-bonded interactions. Choline ketoprofenate and naproxenate form more H-bonding interactions, while linoleate forms less number of H-bonds. It is interesting to note that the stability of Mpro-ligand complex highly depend on the number of H-bonded interactions with natural compounds and ILs.
We computed the spatial distribution functions (SDF) of the individual ion species surrounding the protein to investigate local ion coordination. We calculated the SDF of choline cation with naproxenate, ketoprofenate, linoleate, and geranate anions to better understand the coordination of cholinium-based ILs around Mpro, as shown in Fig. 7. It is found from calculations that the naproxenate anions are strongly accumulated around the protein surface than the choline cations. The close inspection reveals that the anions form H-bonding interactions with the protein structure and choline cation interacted with the negatively charged amino acids. The density of all the other anions is higher around the Mpro except linoleate. It has fewer interactions with the structure of the Mpro.
 | ||
| Fig. 7 The SDF of Mpro with (A) choline naproxenate (B) choline ketoprofen (C) choline linoleate and (D) choline geranate. Blue color denotes the choline cation and red color indicates the various anions. | ||
Binding free energy calculations (MM-PBSA)
g_mmpbsa is used to calculate the binding free energy of protein-ligand complexes using the Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) approach. Free binding energy estimated using the MM-PBSA approach have been successful in improving in silico ligand-protein affinity predictions. In MM-PBSA calculations, nonpolar solvation energy, van der Waals interactions, and electrostatic interactions contribute negative values to the total potential of the system, whereas polar solvation energy contributes positive values. These energies were plotted in Fig. 8 MM-PBSA estimates the ΔE (binding free energy) of four natural compounds for the last 200 ns. The four top-ranked natural compounds demonstrated lower binding energy and higher binding affinities toward their respective targeted protein. The observation shows that the binding energy of the compounds NP-Hit2 (−25.6 kcal mol−1) and NP-Hit3 (−25.3 kcal mol−1) revealed more binding affinity than the other compounds, whereas the binding energy of NP-Hit4 is −24.1 kcal mol−1. The binding free energies of natural products and cholinium-based ILs with Mpro were shown in ESI Table S7 and S8.† | ||
| Fig. 8 Calculated MM-PBSA binding free energies of natural compounds with Mpro. vdW = van der Waals, Ele = Electrostatic, Pol = polar solvation, and SASA = solvent accessible surface area are shown. | ||
The binding energies with time series are plotted in ESI (Fig. S4†). A comparison of MM-PBSA calculations to molecular docking revealed greater binding energies, indicating the compounds will likely be potential hits. The binding free energy of NP-Hit1 is −19.2 kcal mol−1, which is very less compared to other compounds. Also, the GI absorption is very less. It concludes that the other three compounds NP-Hit2, NP-Hit3, and NP-Hit4 have a higher binding affinity, and hence, we can consider these are potential inhibitors against Mpro of SARS-CoV-2.
Using MM-PBSA, we performed a per-residue decomposition analysis to understand the impact of appropriate amino acid interactions on binding free energy. The hot spot residues that contribute to protein-ligand binding interactions were determined using a free energy decomposition approach. For each system, we extracted 2000 snapshots from the MD trajectories and estimated the average ligand–residue interaction energies (ΔElig–res). The hot spot residues have (ΔElig–res) greater than −1.0 kcal mol−1 which is shown in Table 3. These residues interact with the ligands and form stronger H-bond and hydrophobic interactions. The most significant hotspot residue is Thr25, Leu27, His41, Met49, Cys145, Met165, and Gln189. Natural compounds NP-Hit2 and NP-Hit3 have strongly interacted with the hot spot residues. These results indicate that NP-Hit2 and NP-Hit3 are potent inhibitors for the Mpro of SARS-CoV-2.
| Residue name | NP-Hit1 | NP-Hit2 | NP-Hit3 | NP-Hit4 |
|---|---|---|---|---|
| a The most significant hot spot residues are shown. | ||||
| Thr25 | −0.89 | −0.27 | −0.34 | −0.77 |
| Leu27 | −0.89 | −0.37 | −1.59 | −0.76 |
| His41 | −1.30 | −0.38 | −0.43 | −1.40 |
| Met49 | −1.35 | −1.42 | −1.32 | −1.61 |
| Cys145 | −0.73 | −0.28 | −1.17 | −0.79 |
| Met165 | −0.58 | −1.68 | −0.59 | −0.29 |
| Gln189 | −0.27 | −1.45 | −0.88 | −0.53 |
Conclusion
SARS-CoV-2 is a highly contagious disease that has spread over the world, increasing the fatality rate. Seeking new antiviral drugs for SARS-CoV-2 is a top priority. Computer-aided drug design techniques can assist in identifying a viable COVID-19 medication and management at a lower cost and in lesser time. Existing drugs have adverse effects, and the mechanisms of their interactions are yet unknown. We have selected top-ranked natural compounds and a series of biocompatible ILs for the drug-likeness study. The top-ranked natural compounds and ionic liquids were screened using docking studies based on their binding energies. For both natural compounds and ionic liquids, ADMET properties are used to study the drug-likeness attributes. For the four natural compounds whose binding affinity is −11 kcal mol−1 and also for the ionic liquids the binding affinity ranges from −10 to −9 kcal mol−1 are chosen for further screening in which MD simulation was used. As a result, protein-ligand complexes remain stable throughout the simulation time. The binding free energy for the top-ranked natural compounds was calculated using the MM-PBSA method. The most significant hotspot residues Thr-25, Leu-27, His-41, Met-49, Cys-145, Met-165, and Gln-189 have made significant contributions to the protein-ligand binding, which may be useful in drug development for Mpro target interactions. In conclusion, the natural compounds of NP-Hit2 and NP-Hit3 have higher binding affinity and obey the ADMET properties. Additionally, NP-Hit2 and NP-Hit3 interact with the cold spot residues, which is resist the mutations of Mpro. The active site residues His-41, Phe-140, Cys-145, Glu-166, and His-172 have low mutation frequencies, cold spot residue interactions with natural products will be studied in the future. Also, the ionic liquids choline ketoprofenate and choline geranate strongly enhance the stability of Mpro. In conclusion, we screened top-ranked natural compounds among 0.2 million compounds and top-ranked ionic liquids have made significant contributions to the protein-ligand binding, which may be useful in drug development for COVID-19.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KP and SMER thank the SRM Institute of Science and Technology (SRM-IST) Research Fellowship for his research work. MP thanks the Department of Science and Technology-Science and Engineering Research Board (DST-SERB), New Delhi, India for the financial support. The authors also thank SRM-IST for providing the supercomputing facility and financial support.References
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 3405, 1–9 Search PubMed.
- J. Liu, X. Zheng, Q. Tong, W. Li, B. Wang, K. Sutter, M. Trilling, M. Lu, U. Dittmer and D. Yang, J. Med. Virol., 2020, 92, 491–494 CrossRef CAS PubMed.
- H. Wei, H. Yin, M. Huang and Z. Guo, J. Neurol., 2020, 267, 1550–1553 CrossRef CAS PubMed.
- M. S. Hosseini-zare, R. Thilagavathi and C. Selvam, RSC Adv., 2020, 10, 28287–28299 RSC.
- C. Sohrabi, Z. Alsafi, N. O'Neill, M. Khan, A. Kerwan, A. Al-Jabir, C. Iosifidis and R. Agha, Int. J. Surg., 2020, 76, 71–76 CrossRef PubMed.
- C. Wu, X. Chen, Y. Cai, J. Xia, X. Zhou, S. Xu, H. Huang, L. Zhang, X. Zhou, C. Du, Y. Zhang, J. Song, S. Wang, Y. Chao, Z. Yang, J. Xu, X. Zhou, D. Chen, W. Xiong, L. Xu, F. Zhou, J. Jiang, C. Bai, J. Zheng and Y. Song, JAMA Intern. Med., 2020, 180, 934–943 CrossRef CAS PubMed.
- O. S. Amamuddy, G. M. Verkhivker and Ö. T. Bishop, J. Chem. Inf. Model., 2020, 60, 5080–5102 CrossRef PubMed.
- A. S. Lauring and E. B. Hodcroft, JAMA, J. Am. Med. Assoc., 2021, 325, 529–531 CrossRef CAS PubMed.
- B. Hu, H. Guo, P. Zhou and Z. L. Shi, Nat. Rev. Microbiol., 2021, 19, 141–154 CrossRef CAS PubMed.
- S. Satarker and M. Nampoothiri, Arch. Med. Res., 2020, 51, 482–491 CrossRef CAS PubMed.
- Ž. B. Milanović, M. R. Antonijević, A. D. Amić, E. H. Avdović, D. S. Dimić, D. A. Milenković and Z. S. Marković, RSC Adv., 2021, 11, 2838–2847 RSC.
- S. Rajpoot, M. Alagumuthu and M. S. Baig, Curr. Res. Struct. Biol., 2021, 3, 9–18 CrossRef PubMed.
- H. M. Mengist, X. Fan and T. Jin, Signal Transduction Targeted Ther., 2020, 5, 2–3 CrossRef PubMed.
- D. Suárez and N. Díaz, J. Chem. Inf. Model., 2020, 60, 5815–5831 CrossRef PubMed.
- C. A. Ramos-Guzmán, J. J. Ruiz-Pernía and I. Tuñón, ACS Catal., 2021, 11, 4157–4168 CrossRef PubMed.
- I. d. A. Santos, V. R. Grosche, F. R. G. Bergamini, R. Sabino-Silva and A. C. G. Jardim, Front. Microbiol., 2020, 11, 1818 CrossRef PubMed.
- N. Krishnamoorthy and K. Fakhro, IUBMB Life, 2021, 73, 670–675 CrossRef CAS PubMed.
- O. Motwalli and M. Alazmi, J. Mol. Model., 2021, 27, 160 CrossRef CAS PubMed.
- W. Dai, B. Zhang, X. M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L. K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed.
- K. Arafet, N. Serrano-Aparicio, A. Lodola, A. J. Mulholland, F. V. González, K. Świderek and V. Moliner, Chem. Sci., 2021, 12, 1433–1444 RSC.
- J. Đ. Jovanović, M. Antonijević, A. A. El-Emam and Z. Marković, ChemistrySelect, 2021, 6, 8603–8610 CrossRef PubMed.
- D. A. Milenković, D. S. Dimić, E. H. Avdović and Z. S. Marković, RSC Adv., 2020, 10, 35099–35108 RSC.
- L. Angiolella, G. Sacchetti and T. Efferth, Evidence-Based Complementary Altern. Med., 2018, 2018, 1–3 CrossRef PubMed.
- K. S. Egorova, E. G. Gordeev and V. P. Ananikov, Biological Activity of Ionic Liquids and Their Application in Pharmaceutics and Medicine, Chem. Rev., 2017, 117, 7132–7189 CrossRef CAS PubMed.
- M. Reslan and V. Kayser, Biophys. Rev., 2018, 10, 781–793 CrossRef CAS PubMed.
- A. Jordan and N. Gathergood, Chem. Soc. Rev., 2015, 44, 8200–8237 RSC.
- J. Saraswat, P. Singh and R. Patel, J. Mol. Liq., 2021, 326, 115298 CrossRef CAS PubMed.
- A. Thomas, R. Ahamed and M. Prakash, RSC Adv., 2020, 10, 39160–39170 RSC.
- J. Flieger and M. Flieger, Int. J. Mol. Sci., 2020, 21, 1–41 Search PubMed.
- M. Petkovic, J. L. Ferguson, H. Q. N. Gunaratne, R. Ferreira, M. C. Leitão, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Green Chem., 2010, 12, 643–664 RSC.
- J. M. M. Araújo, C. Florindo, A. B. Pereiro, N. S. M. Vieira, A. A. Matias, C. M. M. Duarte, L. P. N. Rebelo and I. M. Marrucho, RSC Adv., 2014, 4, 28126–28132 RSC.
- K. Palanisamy and M. Prakash, Phys. Chem. Chem. Phys., 2021, 23, 25298 RSC.
- V. Sundaram, R. N. Ramanan, M. Selvaraj, R. Vijayaraghavan, D. R. MacFarlane and C. W. Ooi, J. Mol. Liq., 2021, 322, 114501 CrossRef CAS.
- B. K. Shoichet, Nature, 2004, 432, 862–865 CrossRef CAS PubMed.
- G. Schneider and H. J. Böhm, Drug Discovery Today, 2002, 7, 64–70 CrossRef CAS PubMed.
- C. Selvam, N. Oueslati, I. A. Lemasson, I. Brabet, D. Rigault, T. Courtiol, S. Cesarini, N. Triballeau, H. O. Bertrand, C. Goudet, J. P. Pin and F. C. Acher, J. Med. Chem., 2010, 53, 2797–2813 CrossRef CAS PubMed.
- C. Selvam, C. D. Mock, O. P. Mathew, K. Ranganna and R. Thilagavathi, Mol. Inf., 2020, 39, 1–5 Search PubMed.
- M. McGann, J. Chem. Inf. Model., 2011, 51, 578–596 CrossRef CAS PubMed.
- T. A. Halgren, J. Comput. Chem., 2000, 17, 520–552 CrossRef.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2021, 49, D1388–D1395 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Phys. Chem. Chem. Phys., 2005, 7, 2701–2705 RSC.
- M. Prakash, V. Subramanian and S. R. Gadre, J. Phys. Chem. A, 2009, 113, 12260–12275 CrossRef CAS PubMed.
- K. Palanisamy, M. Prakash and V. Rajapandian, New J. Chem., 2020, 44, 17912–17923 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- N. T. Nguyen, T. H. Nguyen, T. N. H. Pham, N. T. Huy, M. Van Bay, M. Q. Pham, P. C. Nam, V. V. Vu and S. T. Ngo, J. Chem. Inf. Model., 2020, 60, 204–211 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS PubMed.
- K. G. Sprenger, V. W. Jaeger and J. Pfaendtner, J. Phys. Chem. B, 2015, 119, 5882–5895 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- W. F. Van Gunsteren and H. J. C. Berendsen, Mol. Simul., 1988, 1, 173–185 CrossRef.
- R. Kumari, R. Kumar and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- Y. Yu, J. Wang, Q. Shao, J. Shi and W. Zhu, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- E. W. Ricker, J. Fish. Res. Board Can., 1954, 11, 559–623 CrossRef.
- W. L. DeLano, Protein Crystallogr., 2002, 40, 82–92 Search PubMed.
- A. M. O. Azevedo, S. P. F. Costa, A. F. V. Dias, A. H. O. Marques, P. C. A. G. Pinto, K. Bica, A. K. Ressmann, M. L. C. Passos, A. R. T. S. Araújo, S. Reis and M. L. M. F. S. Saraiva, J. Mol. Liq., 2017, 232, 20–26 CrossRef CAS.
- K. N. Ibsen, H. Ma, A. Banerjee, E. E. L. Tanner, S. Nangia and S. Mitragotri, ACS Biomater. Sci. Eng., 2018, 4, 2370–2379 CrossRef CAS PubMed.
- M. K. Ali, R. M. Moshikur, R. Wakabayashi, Y. Tahara, M. Moniruzzaman, N. Kamiya and M. Goto, J. Colloid Interface Sci., 2019, 551, 72–80 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Docking energies of ILs with Mpro, the average values of RMSD, RMSF, Rg and hydrogen bonding interactions, RMSF plots and binding free energies of Mpro with cholinium-based ILs and natural compounds. See DOI: 10.1039/d1ra08604a |
| This journal is © The Royal Society of Chemistry 2022 |