
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic hydrogen generation using tripod containing pyrazolylborate-based copper(II), nickel(II), and iron(III) complexes loaded on a glassy carbon electrode†
Mohamed M. Ibrahima,
G. A. M. Mersala,
Ahmed M. Fallataha,
Khaled Althubeiti a,
Hamdy S. El-Sheshtawyb,
Manal F. Abou Talebcd,
Manash R. Dasef,
Rabah Boukherroubg,
Mohamed S. Attia*h and
Mohammed A. Amin*a
a,
Hamdy S. El-Sheshtawyb,
Manal F. Abou Talebcd,
Manash R. Dasef,
Rabah Boukherroubg,
Mohamed S. Attia*h and
Mohammed A. Amin*a
aDepartment of Chemistry, College of Science, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia. E-mail: mohamed@tu.edu.sa
bChemistry Department, Faculty of Science, Kafrelsheikh University, Kafr El Sheikh 33516, Egypt
cDepartment of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Al-Kharj, Saudi Arabia
dPolymer Chemistry Department, National Center for Radiation Research and Technology (NCRRT), Egyptian Atomic Energy Authority, Cairo, Egypt
eAdvanced Materials Group, Materials Sciences and Technology Division, CSIR-North East Institute of Science and Technology, Jorhat 785006, Assam, India
fAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
gUniv. Lille, CNRS, Centrale Lille, Univ. Polytechnique Hauts-de-France, UMR 8520 – IEMN, F59000 Lille, France
hChemistry Department, Faculty of Science, Ain Shams University, Abbassia 11566, Cairo, Egypt. E-mail: Mohd_mostafa@sci.asu.edu.eg
First published on 11th March 2022
Abstract
Three transition metal complexes (MC) namely, [TpMeMeCuCl(H2O)] (CuC), [TpMeMeNiCl] (NiC), and [TpMeMeFeCl2(H2O)] (FeC) {TpMeMe = tris(3,5-dimethylpyrazolyl)borate} were synthesized and structurally characterized. The three complexes CuC, NiC, and FeC-modified glassy carbon (GC) were examined as molecular electrocatalysts for the hydrogen evolution reaction (HER) in alkaline solution (0.1 M KOH). Various GC-MC electrodes were prepared by loading different amounts (ca. 0.2–0.8 mg cm−2) of each metal complex on GC electrodes. These electrodes were used as cathodes in aqueous alkaline solutions (0.1 M KOH) to efficiently generate H2 employing various electrochemical techniques. The three metal complexes' HER catalytic activity was assessed using cathodic polarization studies. The charge-transfer kinetics of the HER at the (GC-MC)/OH− interface at a given overpotential were also studied using the electrochemical impedance spectroscopy (EIS) technique. The electrocatalyst's stability and long-term durability tests were performed employing cyclic voltammetry (repetitive cycling up to 5000 cycles) and 48 h of chronoamperometry measurements. The catalytic evolution of hydrogen on the three studied MC surfaces was further assessed using density functional theory (DFT) simulations. The GC-CuC catalysts revealed the highest HER electrocatalytic activity, which increased with the catalyst loading density. With a low HER onset potential (EHER) of −25 mV vs. RHE and a high exchange current density of 0.7 mA cm−2, the best performing electrocatalyst, GC-CuC (0.8 mg cm−2), showed significant HER catalytic performance. Furthermore, the best performing electrocatalyst required an overpotential value of 120 mV to generate a current density of 10 mA cm−2 and featured a Tafel slope value of −112 mV dec−1. These HER electrochemical kinetic parameters were comparable to those measured here for the commercial Pt/C under the same operating conditions (−10 mV vs. RHE, 0.88 mA cm−2, 108 mV dec−1, and 110 mV to yield a current density of 10 mA cm−2), as well as the most active molecular electrocatalysts for H2 generation from aqueous alkaline electrolytes. Density functional theory (DFT) simulations were used to investigate the nature of metal complex activities in relation to hydrogen adsorption. The molecular electrostatic surface potential (MESP) of the metal complexes was determined to assess the putative binding sites of the H atoms to the metal complex.
1. Introduction
In the worldwide energy demand plan, the generation of hydrogen (H2) gas, a non-polluting multi-purpose energy carrier with a variety of properties and prospective industrial applications, has become a top priority.1–4 For efficient generation of H2 from fossil fuels, several conventional technologies were used.5 Natural gas was utilized to manufacture H2 through an industrial process called natural gas steam reforming, because it was plentiful, inexpensive, and contained the greatest H2 content of all fossil fuels.5 Nonetheless, there are two key downsides to this industrial process: high energy use and CO2 emissions.6–8 As a result, scientists around the world have been working to create safe, cost-effective (low-energy) ways for producing CO2-free H2 from non-fossil fuel sources.8In terms of sustainability and environmental impact, water electrolysis is the most promising technique for creating high purity H2 from renewable energy sources, as it solely produces oxygen as a by-product and emits no greenhouse gases.1–3 The rate of the cathodic half-reaction of water splitting, viz. the hydrogen evolution reaction (HER), determines the efficiency of the water electrolysis.9 A highly efficient and robust heterogeneous electrocatalyst is required to accelerate the HER kinetics.10 The efficient electrocatalyst should be able to reduce the HER overpotential below 100 mV. The greater the activity of a catalyst, the lower its overpotential.11
The efficient electrocatalyst should also have the highest possible electrochemically active surface area (ECSA), which is the active region on the catalyst's surface that is accessible for the charge transfer process, as well as a high faradaic efficiency, which is calculated as the ratio of the experimentally determined quantity of H2 to the calculated (theoretical) quantity of H2 assuming 100% faradaic efficiency.11 In addition to the electrochemical parameters mentioned above that can be used to evaluate the electrocatalyst's performance, the coveted electrocatalyst should have a high turnover frequency (TOF), which is defined as the amount of reactants that a catalyst can convert per catalytic site per unit time into the required product, as well as good stability.11
All of the above-mentioned conditions for a desirable electrocatalyst were met using platinum (Pt) and Pt-based materials. As a result, among active electrocatalysts reported in the literature, such materials remain the most efficacious electrocatalysts for the HER.11,12 However, because of their high cost and rarity, their usage in industry has been severely limited.11,12 To meet the industry's need for H2 generation, substantial scientific research has been performed to develop and manufacture non-Pt-based cost-effective and efficient electrocatalysts.12,13 For example, according to Jaramillo et al.,14 MoS2 demonstrated exceptional HER catalytic performance. There has recently been a lot of interest in studies on carbon-based catalysts with low overpotentials for the HER, such as N, B, or S-doped carbons.15,16
H2 may also be produced efficiently using molecular electrocatalysts, such as transition-metal complexes, which are the most stable materials and are used to store energy in chemical bonds as well as generate electricity through hydrogen and oxidation production.17 For instance, recently, two complexes with bipyridine ligand, namely [Co(bpy)2(SCN)2] 1 and [Ni(bpy)2(SCN)2] 2 were synthesized and characterized by C.-L. Wang et al.18 Both showed promising catalytic activity for the hydrogen evolution reaction (HER). In an aqueous solution of neutral buffer (pH 7.0), at an overpotential of 837.6 mV, complexes 1 and 2 electro-catalyzed the HER with turnover frequency (TOF) values of 699.6 and 1469 mol H2 per mol catalyst per h, respectively.
In another study, Z. Niu et al.19 prepared three molecular catalysts with square planar geometries based on mononuclear nickel(II) complexes, and investigated their catalytic activity for H2 generation in aqueous and non-aqueous solutions using cyclic voltammetry. The hydrogen production in both solutions was confirmed by controlled-potential electrolysis using gas chromatography. R. Li et al.20 synthesized two Co(II)/Ni(II) complexes based on nitrogenous heterocyclic ligand and explored them as stable electrocatalysts for the HER.
Incorporating transition metal (TM) complexes into cluster-based chalcogenide frameworks is a useful synthetic method for inducing structural diversity and controlling optoelectronic characteristics, which may boost electrocatalytic performance even further.21 For instance, J. Li et al.22 solvothermally synthesized and structurally characterized three novel isostructural neutral inorganic–organic open frameworks of gallium thioantimonate that are covalently bound with certain TM complexes. Thermal and chemical stability were good in the synthesized materials. The title compounds have tunable photocatalytic H2 evolution activity thanks to an adjustable TM center.
The hydrogenase enzymes23,24 are depicted as naturally occurring microorganisms that efficiently catalyze the reversible interconversion of protons to hydrogen using earth-abundant metals (Ni and Fe) with high turnover rates 6000–9000 ([Fe–Fe]) and 700 ([Ni–Fe]) moles of H2 per mole of enzyme per second for the HER.25 However, enzymes are difficult to produce in sufficient quantities for commercial uses, and their stability outside of their native system is generally limited.24,26 For these reasons, the structural and functional mimicking of hydrogenase's active site has recently acquired attention.27–29 As a result, the development of highly efficient, stable, and robust HER molecular electrocatalysts for the production of hydrogen, which are inexpensive and made of earth-abundant transition metal elements and different ligand combinations, has attracted the community's interest.27–29
Herein, three mononuclear metal complexes (MC) namely, [TpMeMeCuCl(H2O)] (CuC), [TpMeMeNiCl] (NiC), and [TpMeMeFeCl2(H2O)] (FeC) {TpMeMe = tris(3,5-dimethylpyrazolyl)borate} were reported as molecular catalysts supported on glassy carbon (GC) for the HER in an alkaline electrolyte (0.1 M KOH). The stability and catalytic mechanism of these molecular catalysts were investigated by chemical and electrochemical methods. Based on the achieved experimental data, we established a relationship between the functional pyrazolyl nitrogen of the pyrazolylborate ligand and the electrocatalytic water splitting stability of the corresponding CuC, NiC, and FeC complexes. The catalytic evolution of hydrogen on the three supported complexes was further studied using density functional theory (DFT) simulations.
2. Experimental
2.1 Materials and general methods
All chemicals were of commercial grade and were used without extra purification. The ligand potassium hydrotris(3-methyl-5-phenylpyrazolyl)borate (KTpMePh) and its metal complexes MC (Scheme 1) were synthesized following a previously published method,30 which is fully described in Section S1 (ESI†).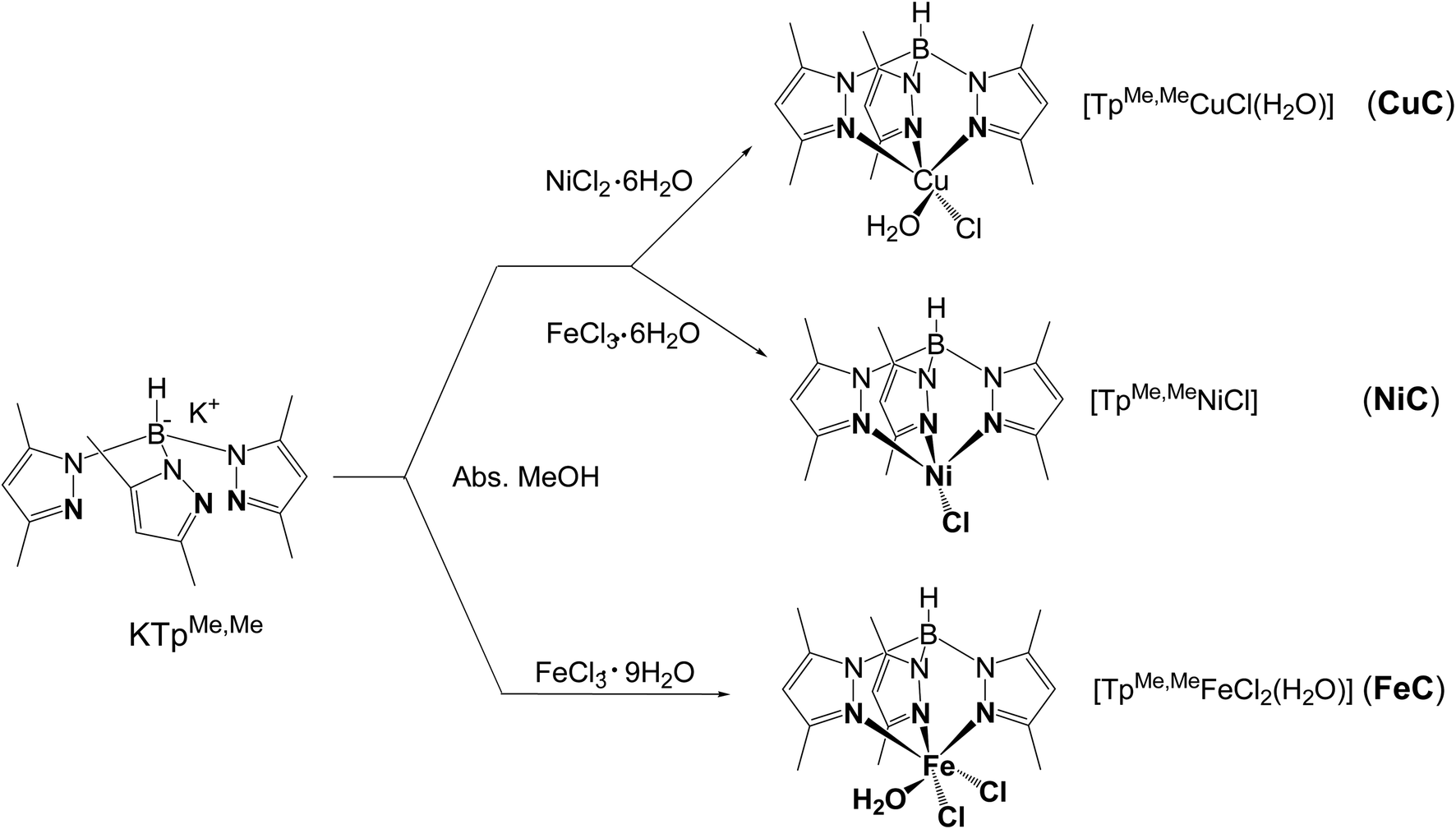 | ||
| Scheme 1 The structures of ligand KTpMeMe and its metal complexes MC. | ||
2.2 Electrochemical measurements
Electrochemical measurements were conducted in 0.1 M KOH electrolyte using a standard double-jacketed three electrodes electrochemical cell. Various electrochemical techniques were applied to evaluate HER electrocatalytic activity and stability of the tested materials, as fully reported in Section S2 (ESI†).3. Results and discussion
3.1 Characterization of the ligand and its metal complexes
The syntheses of TpMeMeCuCl(H2O)] (CuC), [TpMeMeNiCl] (NiC), and [TpMeMeFeCl2(H2O)] (FeC) (TpMeMe− = tris(3,5-dimethylpyrazolyl)borate) were carried in absolute methanol by the reaction of the tripod ligand KTpMeMe with an equal amount of the corresponding MCln salt (M = Cu, Ni, and Fe). The sterically hindered methyl substituents at the pyrazolyl rings were found to have a major requirement for avoiding more chelate formation and stabilization of the formed complexes.31 The obtained metal complexes MC were stable in the solid state as well as in solution under atmospheric conditions. Analytical spectral features confirmed that all complexes MC have a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (metal:ligand) stoichiometry. The structure of FeC was octahedral whereas CuC was square pyramidal and NiC complex has a quaternary surface arrangement (Scheme 1).
:1 (metal:ligand) stoichiometry. The structure of FeC was octahedral whereas CuC was square pyramidal and NiC complex has a quaternary surface arrangement (Scheme 1).
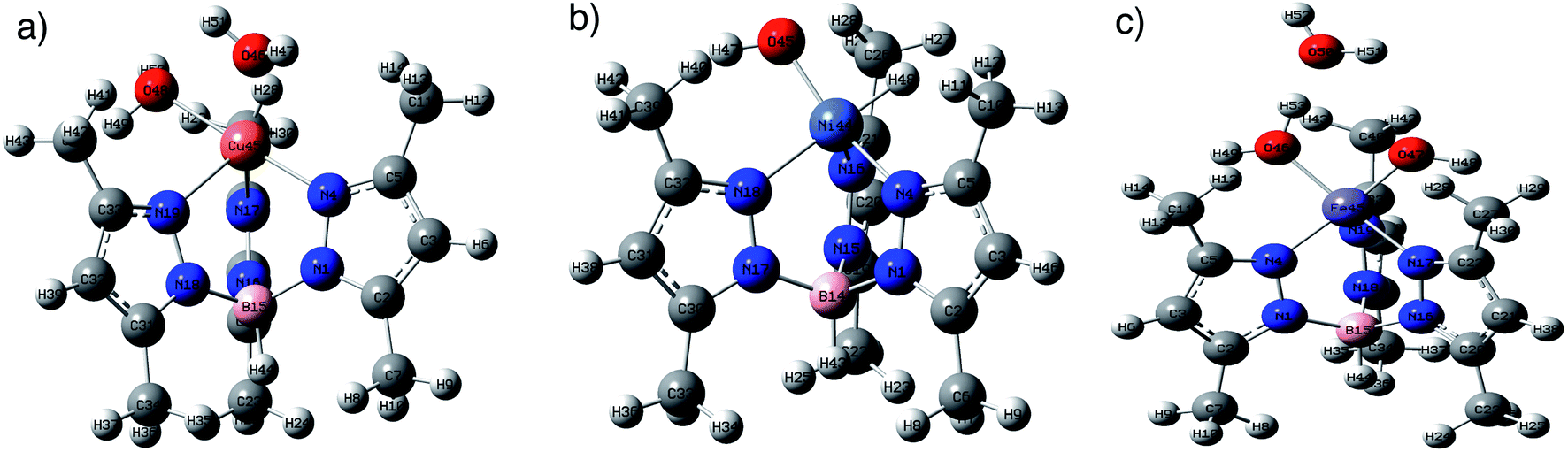
DFT calculations using B3LYP/lanl2dz level of theory were used to explore the structures of metal complexes MC. Fig. 1 shows the optimized structures of the metal complexes in alkaline medium, where the chloride anions (Cl−) were substituted by hydroxyl groups (OH−). The structures of the metal complexes showed that each metal ion is coordinated to three nitrogen atoms from the anionic ligand TpMeMe−, forming symmetrical structures. The stable copper(II) complex CuC binds with one more water molecule (Cu–O48H2 = 2.16 Å), besides the OH− groups (Cu–O46 = 1.94 Å). While the iron(III) complex FeC binds with one water molecule (Fe–O50H2 = 2.05 Å) and two hydroxyl groups (Fe–O47 = 1.78 Å and Fe–O46 = 1.78 Å). These results were established by the TGA data and confirmed by the stable DFT structures. On the other hand, the neutral nickel(II) complex NiC binds with only one OH− group (Ni–O46 = 1.80 Å).
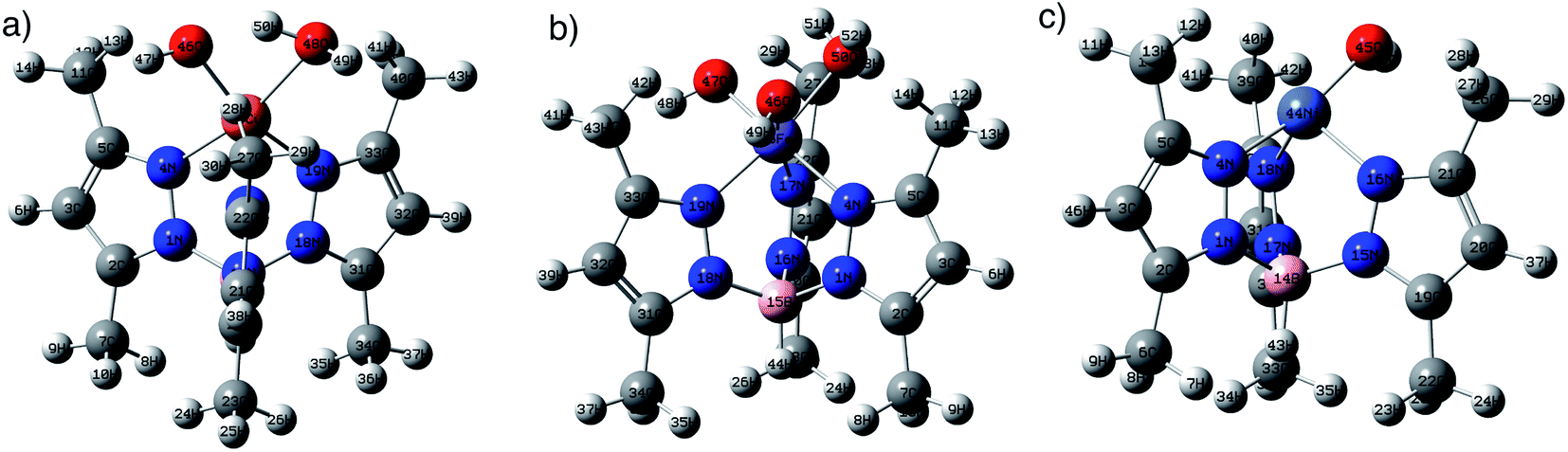 | ||
| Fig. 1 The optimized structures of (a) CuC, (b) NiC, and (c) FeC at B3LYP/lanl2dz level of theory in the gas phase. | ||
The formation of metal complexes (MC) was further evidenced from the high energy shift of ν(B–H) stretching vibration near 2500 cm−1 in the IR spectra of the metal complexes compared to that of the free ligand KTpMeMe, which appeared at 2436 cm−1 (Fig. 2). Similar shifts were reported for other Tp complexes.32 The IR spectra also showed other characteristic stretching and bending vibration bands at ∼2920, 1680, and 1150 cm−1, assigned to ν(C–H), ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), and δ(C–H), respectively. Raman spectra of the ligand and its metal complexes (Fig. 3) revealed almost identical vibration bands with those found in their IR spectra. The non-electrolytic chloride ion in all complexes exhibits vibration bands, characteristics of the terminally-coordinated chloride ions.33 Additionally, the three metal complexes all featured weak bands at around 442, 449, and 452 cm−1, suggesting the existence of M–N bonds.34
N), and δ(C–H), respectively. Raman spectra of the ligand and its metal complexes (Fig. 3) revealed almost identical vibration bands with those found in their IR spectra. The non-electrolytic chloride ion in all complexes exhibits vibration bands, characteristics of the terminally-coordinated chloride ions.33 Additionally, the three metal complexes all featured weak bands at around 442, 449, and 452 cm−1, suggesting the existence of M–N bonds.34
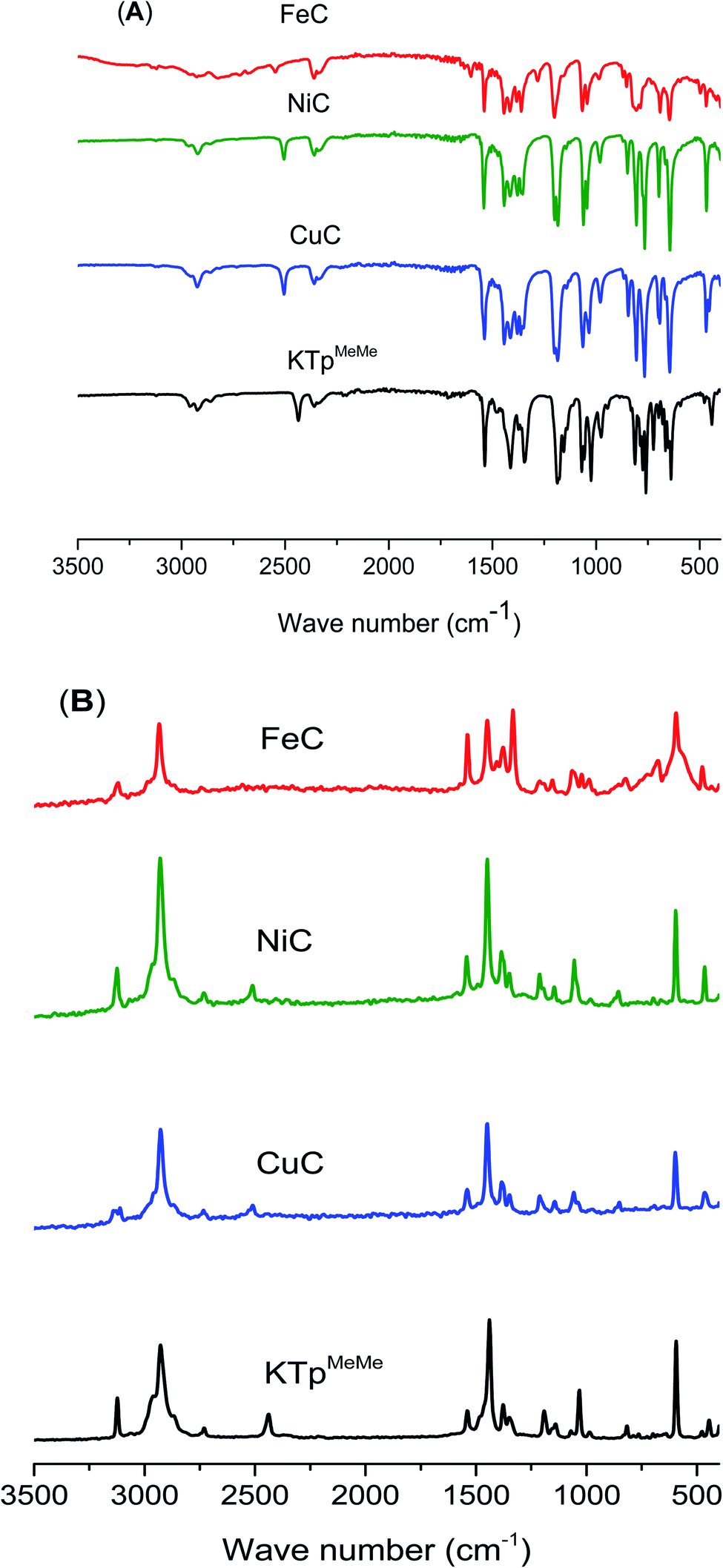 | ||
| Fig. 2 (A) FT-IR and (B) Raman spectra of the ligand KTpMeMe and its metal complexes MC. | ||
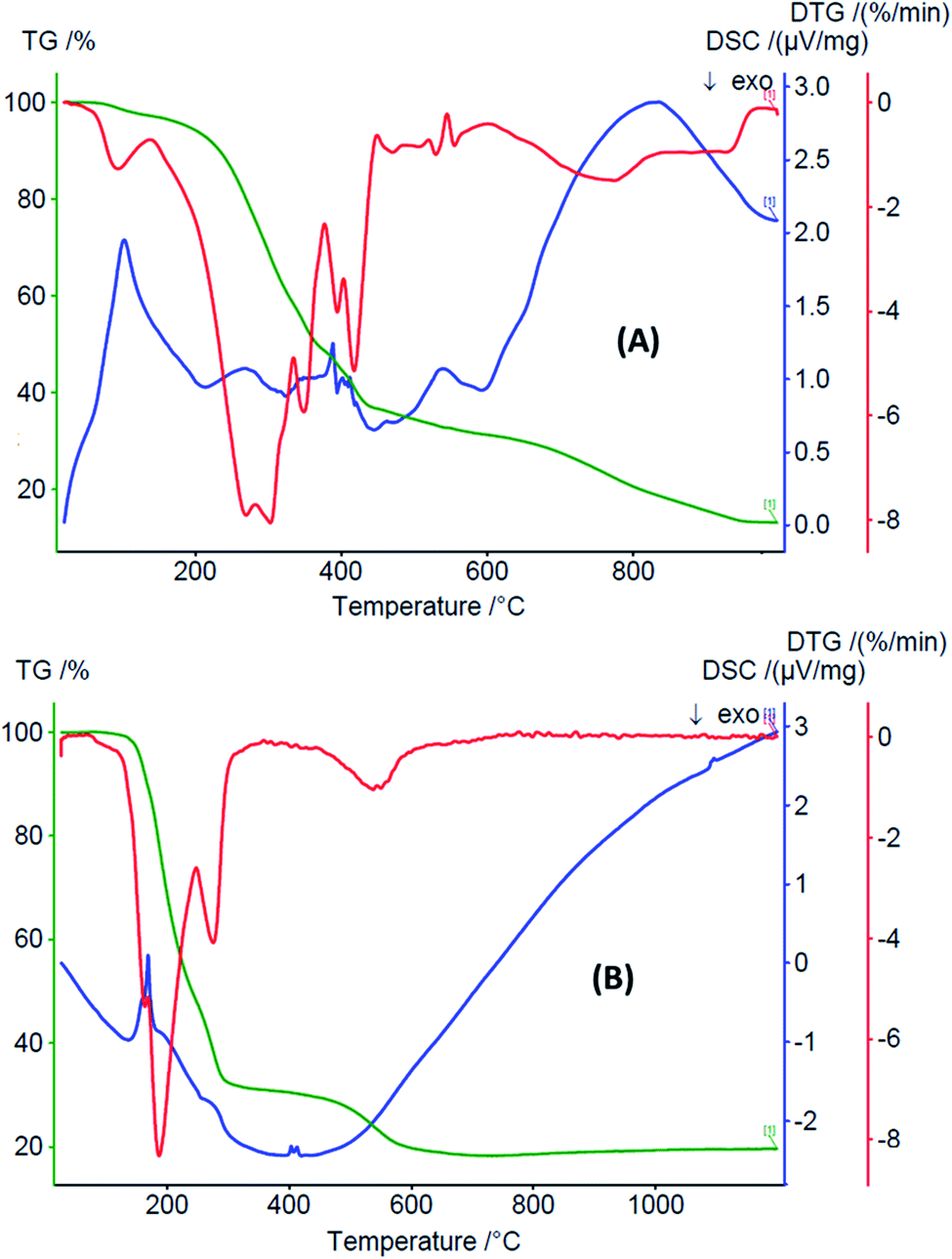 | ||
| Fig. 3 TGA-DTG-DSC curves of (A) [TpMeMeFeCl2(H2O)] (FeC) and (B) [TpMeMeCuCl(H2O)] (CuC). | ||
The thermogram TGA-DTG-DSC of FeC (Fig. 3A) shows that the complex dehydrates one water molecule with an exothermic peak at 98 °C. It decomposes in the range of 120–900 °C (endothermic peak, DSC: 410 °C) to eliminate the organic part with a mass loss of 63.86% (calc. 63.27%), forming a mixture of ferrous(II) oxide and potassium oxide. The thermal analysis of the copper(II) complex CuC indicates that its decomposition takes place in several stages (Fig. 3B). The thermogram shows that the complex decomposes to eliminate the organic part in the range of 105–820 °C (endothermic peak, DSC: 461 K) with a mass loss of 81.98% (calc. 81.52%), generating a mixture of copper(II) and potassium oxide (found 19.64%; calc. 18.73%).
The electronic absorption spectrum of CuC in methanol (Fig. 4)35 exhibited weak bands in the lowest energy visible region attributable to d–d transitions of the Cu(II) ion in a square-pyramidal environment.36 The absorption spectrum of NiC displays three absorption bands at 330, 560, and 635 nm. An examination of these bands indicates that the complex has a tetrahedral geometry with ground state 3T1.37,38 In the regular tetrahedral nickel(II) complexes, only one d–d transition is observed in the visible region. The observation of the three transitions in the spectrum of NiC complex may be attributed to the distortion in the tetrahedral symmetry around the nickel(II) center. This spectral behavior is comparable with analogous tetrahedral nickel(II) complexes.39,40 Hence the nickel(II) ion has an sp3 hybridization with tetrahedral geometry. The intensity of the UV band at around 330 nm is consistent with a ligand-centered π → π* transition or a charge-transfer transition. The electronic spectrum of FeC exhibits four d–d transitions at 325, 374, 490, and 588 nm, assignable to 6A1g → 4T1g (G), 6A1g → 4T2g (G), 6A1g → 4Eg (G) and 6A1g → 4Eg (D), respectively in the octahedral stereochemistry.41 The magnetic moment value of CuC was determined to be 1.98 BM, confirming the presence of magnetically dilute Cu(II) complex,42 whereas the magnetic moment for FeC was close to the spin-only value of 5.63 and 5.44 μB, which is characteristic of high spin of d5 configuration.43,44
 | ||
| Fig. 4 Absorption spectra of MC complexes in methanol. | ||
The quantitative elemental analysis of the three complexes CuC, NiC, and FeC were also determined using energy dispersive X-ray fluorescence (EDXRF) spectroscopy at atmospheric pressure and the obtained spectra are illustrated in Fig. 5. The Ar-Kα was found in all the spectra at 2.957 keV and it has no influence on the characteristic lines of Cu, Ni and Fe. Additional impurities of Ca and Zn were also found, which were originated form the used Mylar foil. In the case of complex NiC, the characteristic radiation Ni-Kα and Ni-Kβ was measured at 7.473 keV and 8.265 keV respectively. Addition escape peak of Ni-Kα was found at 5.733. The spectrum belongs to complex CuC, the Cu-Kα and Kβ were detected successfully at 8.042 keV and 8.906 keV respectively. Whereas in the case of complex FeC, the characteristic radiation of Fe–K lines could be easily detected free from interference at 5.895 keV and 6.491 keV for Kα and Kβ of Fe respectively. Escape peak of Fe-Kα could be recognized also at 4.155 keV. The % of the coordinated TpMe,Me in all complexes were calculated and omitted from the fundamental parameter program. This is due to the limitation of EDXRF setup whereas there is not possibility to detected elements of atomic number less than 11 (Z < 11). The weight% of complexes CuC, NiC, and FeC were found to be 15.27 ± 0.18, 15.22 ± 0.18, and 12.55 ± 0.18, respectively.
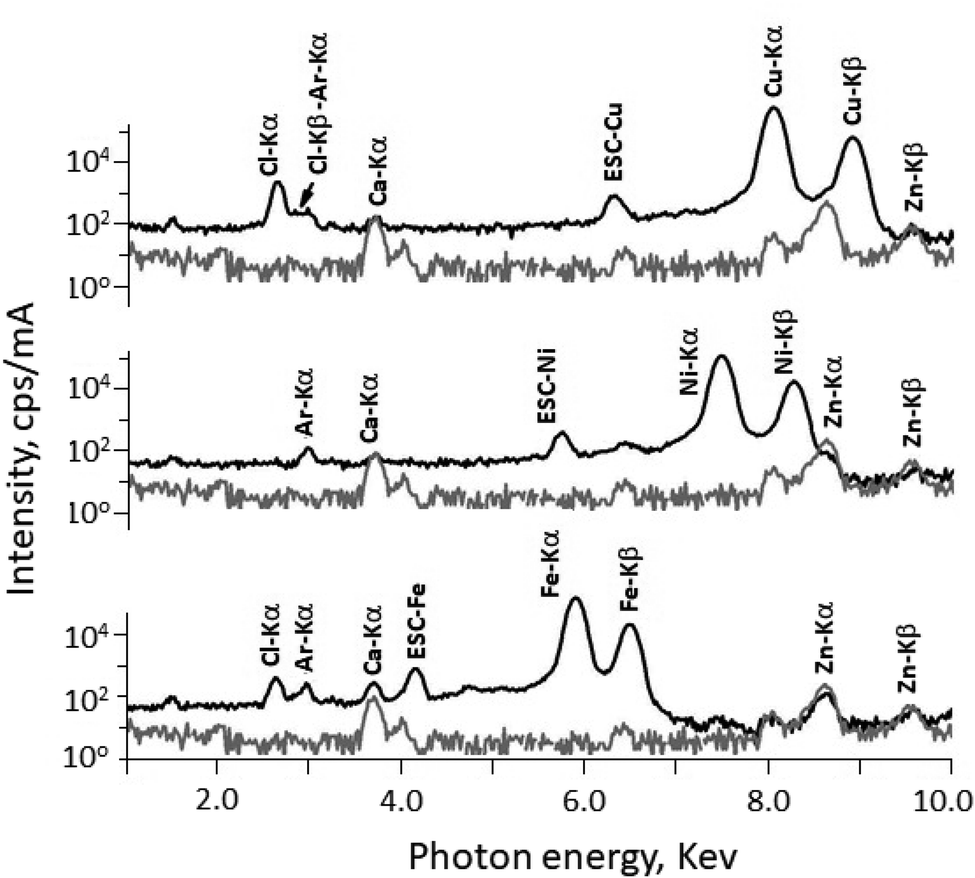 | ||
| Fig. 5 EDXRF spectra of complexes CuC, NiC, and FeC with the background (sample holder with Mylar window) using direct excitation of X-ray tube. | ||
3.2 HER electrocatalytic studies
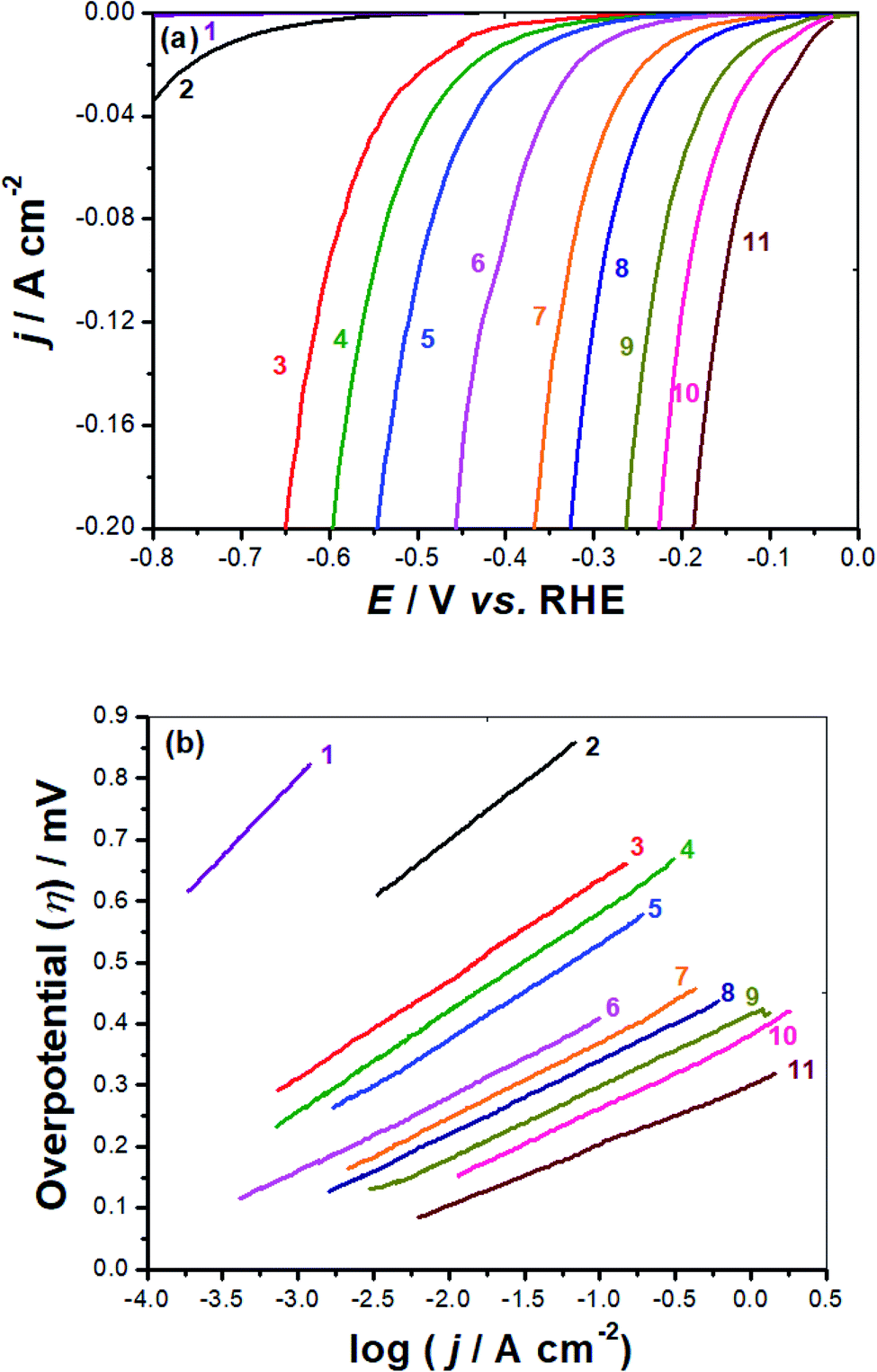 | ||
| Fig. 6 IR-Corrected cathodic polarization measurements for the HER (a) and the corresponding Tafel plots (b) recorded for our synthesized catalysts. Measurements were conducted in deaerated KOH solution (0.1 M) at a scan rate of 5 mV s−1 at room temperature. (1) Bare GC electrode; (2) GC-FeC (0.2 mg cm−2); (3) GC-FeC (0.4 mg cm−2); (4) GC-FeC (0.8 mg cm−2); (5) GC-NiC (0.2 mg cm−2); (6) GC-NiC (0.4 mg cm−2); (7) GC-NiC (0.8 mg cm−2); (8) GC-CuC (0.2 mg cm−2); (9) GC-CuC (0.4 mg cm−2); (10) GC-CuC (0.8 mg cm−2); (11) Pt/C. | ||
Current densities in this work are obtained by dividing currents by the electrochemical active surface area (EASA), calculated from cyclic voltammetry measurements, see later. The various HER electrochemical kinetic parameters (Table 1) associated with such polarization measurements were derived from Tafel plots, Fig. 6(b).
| Tested cathode | EHER/mV (RHE) | −βc/mV dec−1 | j0/mA cm−2 | η10/mV |
|---|---|---|---|---|
| Bare GC electrode | — | 254(6) | 6.3(0.15) × 10−4 | — |
| GC-FeC (0.2 mg cm−2) | 305(5) | 162(3.3) | 1.1(0.05) × 10−2 | 467(8) |
| GC-FeC (0.4 mg cm−2) | 255(3.4) | 160(3.2) | 2.5(0.07) × 10−2 | 423(7.6) |
| GC-FeC (0.8 mg cm−2) | 230(3.2) | 158(3) | 3.7(0.1) × 10−2 | 372(5) |
| GC-NiC (0.2 mg cm−2) | 180(2.6) | 157(2.8) | 4.3(0.3) × 10−2 | 278(4.2) |
| GC-NiC (0.4 mg cm−2) | 107(2.2) | 160(2.6) | 10(0.12) × 10−2 | 250(4.6) |
| GC-NiC (0.8 mg cm−2) | 80(1.7) | 155(2.2) | 14(0.35) × 10−2 | 213(4) |
| GC-CuC (0.2 mg cm−2) | 45(1.5) | 118(1.8) | 29(0.6) × 10−2 | 176(4.4) |
| GC-CuC (0.4 mg cm−2) | 30(1.1) | 114(2.1) | 60(0.9) × 10−2 | 140(2.6) |
| GC-CuC (0.8 mg cm−2) | 25(0.5) | 112(1.6) | 70(1.1) × 10−2 | 120(2.2) |
| Pt/C | 10(0.2) | 108(1.2) | 88(1.2) × 10−2 | 110(1.4) |
Such parameters, which include exchange current density (j0), cathodic Tafel slope (βc), and the overpotential required to deliver a current density of 10 mA cm−2 (η10), are essential to evaluate and compare the HER catalytic activity of the studied metal complexes 1–3.
Included also in Table 1 is another important HER electrochemical kinetic parameter, namely the HER onset potential (EHER), the potential at which the reduction of water molecules commences to liberate H2, followed by a rapid growth in the cathodic current. This parameter is directly estimated from the linear polarization plot, Fig. 6(a). It is one of the key electrochemical parameters employed to evaluate and compare electrocatalysts' performance towards the HER, since any alteration in the EHER's location on the linear polarization results in considerable changes in the exchange current density value.45 The literature has demonstrated that the anodic shift in EHER prompts hydrogen production with higher exchange currents at lower overpotentials, referring to enhanced electrocatalytic activity.45 Table 1 reveals, at any loading density, a considerable anodic shift in EHER, corresponding to improved catalytic performance, ongoing from GC-FeC to GC-CuC with the GC-NiC's EHER value locates in between. This anodic drift in EHER, which increases upon increasing the loading density, enhances following the sequence: GC-FeC (−255 mV vs. RHE @ 0.4 mg cm−2) < GC-NiC (−107 mV vs. RHE @ 0.4 mg cm−2) < GC-CuC (−30 mV vs. RHE @ 0.4 mg cm−2) < GC-CuC (−25 mV vs. RHE @ 0.8 mg cm−2).
The overpotential the electrocatalyst must acquire to attain a current density of 10 mA cm−2 (η10), another important HER electrochemical kinetic parameter employed for comparing and evaluating the catalytic performance of electrocatalysts,46 exhibits the same trend as for EHER values anodic drift. Here again, at any tested loading density, the lowest η10 value is estimated for the GC-CuC catalyst. These findings demonstrate the outstanding HER catalytic performance of the GC-CuC catalyst as it recorded the lowest EHER (25 mV vs. RHE) and η10 (120 mV) values among the other studied catalysts.
The GC-CuC catalyst's low EHER value, together with its steep polarization curve beyond its EHER, led to a j0 value of 0.29 mA cm−2 at 0.2 mg cm−2 loading density calculated by the Tafel extrapolation method, Table 1. This GC-CuC catalyst's j0 value increased with increase in loading density attaining a j0 value of 0.7 mA cm−2 at a loading density of 0.8 mg cm−2, approaching that of the commercial Pt/C catalyst performed here under the same operating conditions (0.88 mA cm−2).
These findings highlight the catalyst's loading density role in catalyzing the HER. The population of the HER active catalytic sites is expected to increase with increase in the loading density of the catalyst, as evidenced from the catalyst surface's active sites (n, mol) calculations (see later). This, in turn, expedites efficacious transfer of charge during the water molecules reduction process thus, boosts the HER kinetics.
 | ||
| Fig. 7 Complex-plane impedance plots recorded for the HER on the surfaces of (1) GC-FeC (0.2 mg cm−2); (2) GC-FeC (0.4 mg cm−2); (3) GC-FeC (0.8 mg cm−2); (4) GC-NiC (0.2 mg cm−2); (5) GC-NiC (0.4 mg cm−2); (6) GC-NiC (0.8 mg cm−2); (7) GC-CuC (0.2 mg cm−2); (8) GC-CuC (0.4 mg cm−2); (9) GC-CuC (0.8 mg cm−2). Measurements were conducted in deaerated 0.1 M KOH solution at an overpotential of 0.5 V vs. RHE at room temperature. | ||
Two distinct depressed semicircles can be seen in the Nyquist plots of all materials investigated. The electrocatalytic HER on Ni,47 Co,48 and some of their alloys13,49–51 in alkaline solutions exhibited such impedance behavior. Similar impedance results were reported earlier in our lab for HER investigations on Co50Ni23Ga26Al1.0 magnetic shape memory alloy,52 reduced graphene oxide nanosheets, some metallic nanoparticles-decorated titanium substrates, and cathodically activated carbon felt samples.53–56
At high frequency values, a first small semicircle (with diameter R1) was visible, whereas a large second semicircle (with diameter R2) appeared at low frequencies. Each of them can be assigned to the resistance–capacitance (RC) network. Each RC network at the catalyst/KOH interface corresponds to the charge-transfer resistance (R1 for the first semicircle and R2 for the second, Rtotal or Rct = R1 + R2) and associated double-layer capacitance (C1 for the first semicircle and C2 for the second, Ctotal = C1 + C2).56 The two semicircles appear as conspicuous humps in the related Bode graphs, Fig. S10 (ESI†). It is obvious from Fig. 7 that the size of the two semicircles varies according to the type the tested catalyst and its loading density. Also, referring to Fig. S10,† at a fixed loading density, both semicircles are clearly potential-dependent. These findings indicate that the two semicircles' size is connected to the HER kinetics.57
For comparison, EIS responses of bare GC and Pt/C electrodes with a single capacitive loop (Fig. S11, ESI†) were also recorded. The RC networks' elements were estimated using the equivalent circuit illustrated in Fig. S12 (ESI†), allowing us to gain a better grasp of the HER kinetics. This electrical equivalent circuit was described in more detail elsewhere.54
Based on the impedance data fitting, when the loading density of the catalyst was raised, the values of both R1 and R2, and hence the Rct value (Rct = R1 + R2), reduced considerably, indicating higher HER catalytic activity. For example, the Rct values recorded for the NiC catalyst decreased from 315 Ω cm2 at a loading density value of 0.2 mg cm−2 to 248 and 145 Ω cm2 at loading densities 0.4 and 0.8 mg cm−2, respectively. The CuC electrocatalyst showed the lowest Rct value among the other studied samples at any studied loading density thus, confirming the excellent HER catalytic activity of the CuC electrocatalyst. For instance, at a loading density value of 0.8 mg cm−2, the CuC electrocatalyst recorded an Rct value of 51 Ω cm2. This value is very close to that of the Pt/C (47 Ω cm2), and lower than those measured for GC-NiC (145 Ω cm2) and GC-FeC (450 Ω cm2) at the same loading density.
Another critical electrochemical parameter, namely the Tafel slope (βc), is also utilized to further assess the studied electrocatalysts' HER catalytic leverage towards the HER. The βc value recorded here for the commercial Pt/C catalyst is found to be 108 mV dec−1, close to that reported in the literature under the same operating conditions, 111 mV dec−1,58 meaning that the electrochemical measurements employed here are reliable.
The alkaline HER step mechanism can be represented by eqn (1)–(3):59
| H2O + e− → Hads + HO− (Volmer step) | (1) |

The Volmer step is followed by either a combination of adsorbed species to form molecular H2:
| Hads + Hads → Hads (Tafel step) | (2) |
or the desorption Heyrovsky step:
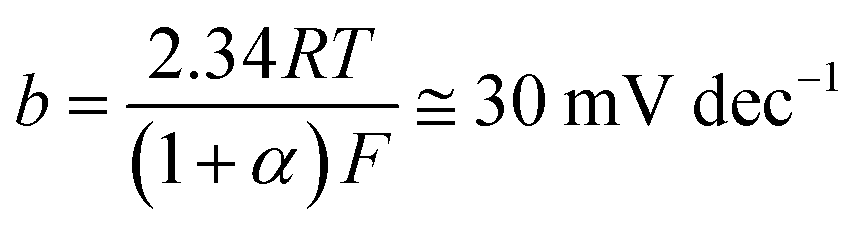
| H2O + e− + Hads → H2 + HO− (Heyrovsky) | (3) |

The Pt/C catalyst's βc value obtained here (108 mV dec−1), which is comparable with that reported in the literature (111 mV dec−1) under the same operating conditions,58 is close to the standard one (120 mV dec−1) reported in eqn (1), meaning that the alkaline HER mechanism comprises the Volmer step as the rate-limiting.60
The Tafel lines of FeC and NiC catalysts are parallel to each other, irrespective of the value of the catalyst's loading density, with βc values close to 160 mV dec−1. Also, the three studied CuC catalysts, namely CuC (0.2 mg cm−2), CuC (0.4 mg cm−2), and CuC (0.8 mg cm−2) exhibited parallel Tafel lines, but with reduced βc values (118, 114, and 112 mV dec−1, respectively). These findings indicate that the kinetics of the HER on the CuC catalyst's surface is faster than on the surfaces of FeC and NiC catalysts. This is evidenced from the marked drop in the Tafel slope values of the CuC catalyst since lower Tafel slopes often allude to a large number of catalytic active sites being available.60,61
The CuC (0.8 mg cm−2) catalyst's βc value (112 mV dec−1) is close to that of the Pt/C (108 mV dec−1) thus advocating a mechanism for the HER on the surface of that catalyst controlled by the Volmer step.60 The results show that the CuC catalyst can efficiently catalyze HER in 0.1 M KOH solution, which is similar to the most active HER electrocatalysts reported in the literature, Table S1 (ESI†).
The electrochemical active surface area (EASA) is another essential parameter used to compare different electrocatalysts' activity, as the kinetics of the HER over electrocatalysts is directly linked to their ECSA. Being directly related to ECSA, double layer capacitance (Cdl), determined from cyclic voltammetry measurements in non-faradic region, Fig. S1 (ESI†), is used to calculate ECSA.62–64 From the slopes of the current density versus scan rate (v) plots, Fig. S1 (ESI†), for any studied meal complex, Cdl values were found to increase with increase in the loading density. At any investigated loading density, this increase in Cdl enhances following the sequence: FeC < NiC < CuC. These findings confirm the high catalytic performance of the CuC electrocatalyst since electrocatalysts with higher Cdl values can provide higher adsorption density of reactive sites, and therefore promote efficient charge transfer, resulting in enhanced catalytic activity.
Eqn (4) is used to calculate the ECSA from Cdl.65
| EASA = Cdl/Cs | (4) |
The catalyst surface's active sites (n, mol) are estimated using eqn (5):66
| n = Qnet/2F | (5) |
485 C mol−1), and number 2 refers to the number of electrons transferred during the HER's progress. Qnet is the net voltammetry charge of the catalyst derived from subtracting charges produced from the bare GC electrode from those obtained from the studied catalyst (Qnet = QGC-catalyst − Qbare GC electrode). Such Q values were obtained from CV measurements recorded for the investigated electrocatalysts, Fig. S1–S9 (ESI†).
For any studied catalyst, the value of n is found to enhance with the catalyst's loading density. At any tested loading density, the value of n increases obeying the order: FeC < NiC < CuC. These findings confirm the CuC's highest HER catalytic performance.
To further evaluate the catalytic performance of the investigated catalysts at a given overpotential, the turnover frequency (TOF) was calculated, which is the number of hydrogen molecules created per second per active site.66 The TOF values for the tested catalysts were computed using the EASA values in Table 2 at an overpotential of 500 mV, as discussed previously.52 Table 2 depicts the obtained TOF values, which feature a dramatic increase in the value of TOF as the loading density of the examined catalyst increases. The TOF value is always higher for the CuC at any studied loading density. To our knowledge, the TOF value recorded for the best catalyst, namely CuC, is comparable to the most efficient molecular catalysts and greater than many others described in the literature for electrochemical H2 production. Table S1 (ESI†) summarizes these comparisons.
| Tested cathode | Cdl/mF cm−2 | ECSA/cm2 | Q × 103/C | n × 108/mol | TOF @ 0.5 V |
|---|---|---|---|---|---|
| GC-FeC (0.2 mg cm−2) | 11.26 | 375.3 | 4.2 | 2.2 | 872 |
| GC-FeC (0.4 mg cm−2) | 20.27 | 675.7 | 6.8 | 3.5 | 988 |
| GC-FeC (0.8 mg cm−2) | 30.55 | 1018.3 | 11.2 | 5.8 | 1124 |
| GC-NiC (0.2 mg cm−2) | 15.55 | 518.3 | 7.4 | 3.8 | 927 |
| GC-NiC (0.4 mg cm−2) | 28.32 | 944.0 | 12.4 | 6.4 | 1192 |
| GC-NiC (0.8 mg cm−2) | 44.43 | 1481.0 | 19.8 | 10.3 | 1338 |
| GC-CuC (0.2 mg cm−2) | 21.2 | 706.7 | 11.6 | 6.0 | 1154 |
| GC-CuC (0.4 mg cm−2) | 36.71 | 1223.7 | 17.8 | 9.2 | 1510 |
| GC-CuC (0.8 mg cm−2) | 53.02 | 1767.3 | 26.9 | 13.9 | 1820 |
Origin of catalytic activity (DFT calculations). The DFT calculations were performed to explore the nature of the metal complex activities towards the hydrogen adsorption process. In order to assess the possible binding sites of the H atoms to the metal complexes, the molecular electrostatic surface potential (MESP) values of the metal complexes were calculated (Fig. 8). The calculated MESP shows the presence of electrostatic negative potential on the metal atom, which was found to be higher in Ni (−16.31 kcal mol−1), while the N atoms on pyrazole ring carry negative electrostatic potential (−13.8 kcal mol−1). On the other hand, the complex FeC owns a positive electrostatic potential. Accordingly, the metal complexes show two possible sites for the adsorption of the hydrogen atom to initiate the process. The DFT binding energy of the H atom (Fig. 9) to the available sites on the metal complexes reveals that the binding to the metal complexes is more stable than to the N atom by 5.1, 7.67, and 8.52 kcal mol−1 for complexes CuC, NiC, and FeC, respectively.
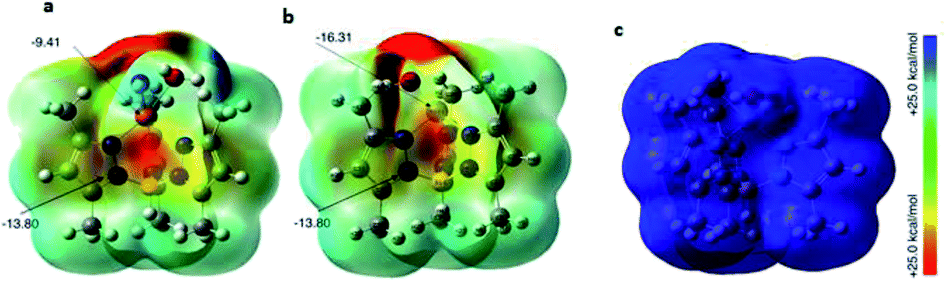 | ||
| Fig. 8 Calculated MESP of the (a) CuC, (b) NiC, and (c) FeC metal complexes at B3LYP/lanl2dz in the gas phase. | ||
 | ||
| Fig. 9 Optimized structures of the H atom adsorbed on the (a) CuC, (b) NiC, and (c) FeC metal complexes at B3LYP/lanl2dz in the gas phase. | ||
To retain the molecular nature of the metal complex MC catalysts under the alkaline conditions, it is necessary for the ligand to be tightly bound to the metal center. These catalysts were generated by treating metal(II) chloride (MCln) with one equivalent of ligand in absolute methanol, in which one tris(pyrazolyl)borate anion (TpMeMe−) is bound to the metal center with the fourth coordination site, occupied by a chloride in the case of NiC, whereas in CuC and FeC, the ligand is bound to the metal center with one and two more water molecules, respectively. Upon the addition of one equivalent of strong base, the chloride ligand in all complexes is substituted by hydroxide (OH−) anion, and the resulting Cu(II)-bound hydroxide complex, for example, adopts a square pyramidal structure with the OH-donor. Upon the addition of one equivalent of hydrochloric acid, the OH ligand can be protonated to regenerate the aqua complex. This reversible protonation of the ligand is an attractive feature for a water-oxidation catalyst, as it may allow for proton-coupled electron transfer (PCET), a common reaction step in water oxidation.
The actual catalyst for the current catalytic systems is generated in an alkaline medium, where the hydroxide anion must bind to the central metal ion of the catalyst. Thus, the initiation step of the present catalytic reaction involves binding of OH− to the central metal ion of the catalyst. In this context, the octahedral Fe(III) complex is coordinately saturated, and thus has no vacant coordination sites, which can be utilized to bind any reacting species during the catalytic process. The ability of such coordinately saturated complexes to dissociate (ML6 ⇌ ML5 + L) and consequently the energy of dissociation is one of the key factors in imparting catalytic properties to these metal complexes. On the other hand, the five coordinated square pyramidal Cu(II) complex and the four coordinated tetrahedral Ni(II) complex already have a vacant coordination site for the substrate binding and thus need no energy for ligand dissociation as in the case of the octahedral Fe(III) complex.
Although Cu(II) and Ni(II) complexes are both coordinatively unsaturated, Cu(II) complex exhibits higher catalytic activity. The superiority of Cu(II) can be explained by the possible mechanistic implication of the six coordinate intermediate [H2–ML–OH]. Based on Jahn–Teller theorem, this hexacoordinated species is highly unstable in the case of Cu(II) complex as compared to Ni(II) complex. According to the Jahn–Teller theorem, the hexa-coordinated Cu(II) complexes are usually associated with a tetragonal distortion reducing the symmetry from Oh to Dh.67,68 Thus, we propose that rapid structural reorganization of this active intermediate leads to rapid product formation and release. These structural changes will reduce the coordination number from six to five and return the catalyst to its original, stable form. As for the Ni(II) complex does not suffer from the Jahn–Teller effect, its catalytic reaction proceeds more slowly than it does in the case of the Cu(II) complex.
Copper and nickel are more labile than iron. This sequence of the lability degree could also be due to the electronic configuration of the metal center in these metal complexes and can be illustrated from the following discussion. The 3d-electronic configuration (electron density) of the metal ion is a key factor for determining the degree of lability or inertness of transition metal complexes.69 According to valence bond theory (VBT), the electron density of MC complexes is increasing in the following order CuC (4 lone pairs) > NiC (three lone pairs) > FeC (no lone pair). This is in accordance with the HER catalytic performance, in which the three tested metal complexes' activity follows the sequence: CuC > NiC > FeC.
Stability test. The best performing electrocatalyst, namely GC-CuC (0.8 mg cm−2), was further evaluated for stability and long-term durability using CV measurements. Fig. 10 shows the first cycle of the catalyst in comparison to the 6000th cycle.
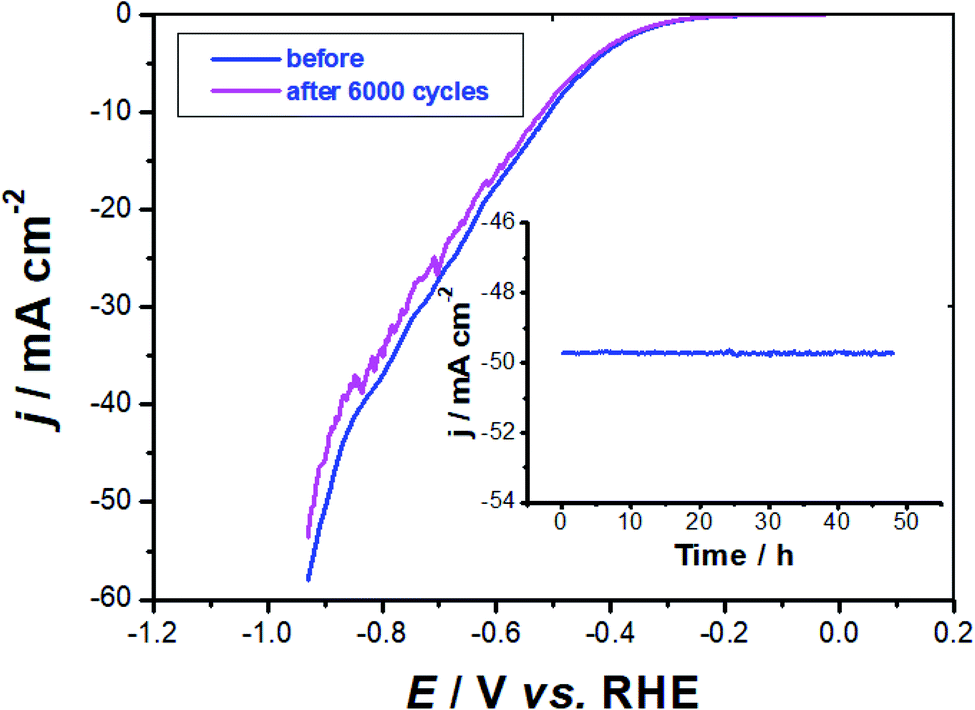 | ||
| Fig. 10 IR-Corrected long-term stability test recorded for the best performing electrocatalyst, namely GC-CuC (0.8 mg cm−2) in 0.1 M KOH solution at room temperature for the HER. LSV measurements were conducted at a scan rate of 50 mV s−1. Inset – chronoamperometry measurements performed on the catalyst at a constant applied potential of −0.9 V vs. RHE. | ||
In these solutions, the catalyst is particularly stable, as indicated by the humble loss in current after 6000 continuous potential cycles. The Tafel slope obtained after the last cycle (115 mV dec−1) is close to that recorded after the first cycle (112 mV dec−1), suggesting that catalyst potential cycling has a negligible influence on the mechanism of the HER. Because the current remains nearly constant throughout the operation, the chronoamperometry measurements (inset of Fig. 10) give an additional corroboration for the catalyst stability.
4. Conclusion
Three tripod-containing ligand metal complexes namely, [TpMeMeCuCl(H2O)] (CuC), [TpMeMeNiCl] (NiC), and [TpMeMeFeCl2(H2O)] (FeC)-modified glassy carbon (GC) electrodes were fabricated and examined as electrocatalysts for the efficient generation of H2 at different loading densities (0.2, 0.4, and 0.8 mg cm−2) in 0.1 M KOH. HER electrocatalytic activity of these materials was investigated using LSV and EIS techniques. The HER electrocatalytic activity of the investigated catalysts was found to increase with increase in their loading density. Among the tested catalysts, at any studied loading density, the GC-CuC exhibited the highest catalytic activity for the HER. At a loading density of 0.8 mg cm−2, the GC-CuC recorded a low HER onset potential (EHER) of −25 mV vs. RHE and a high exchange current density of 0.7 mA cm−2. Further, to achieve a current density of 10 mA cm−2, the most performing electrocatalyst required an overpotential of 120 mV and recorded a Tafel slope of −112 mV dec−1. Under the same operating conditions, the best catalyst's HER electrochemical kinetic parameters were close to those of the commercial Pt/C (−10 mV vs. RHE, 0.88 mA cm−2, 108 mV dec−1, and 110 mV to yield a current density of 10 mA cm−2) thus, highlighting its outstanding HER electrocatalytic activity. The nature of metal complex activities in relation to hydrogen adsorption was investigated using DFT simulations. To examine the likely binding sites of the H atoms to the metal complex, the molecular electrostatic surface potential (MESP) of the metal complexes was calculated. The estimated MESP reveals the presence of electrostatic negative potential on the metal atom, which was higher in Ni (−16.31 kcal mol−1). The negative electrostatic potential on the N atoms on the pyrazole ring was calculated to be (−13.8 kcal mol−1). As a result, the metal complexes exhibit two viable sites for hydrogen atom adsorption to initiate the process. The cationic iron(III) complex, on the other hand, has a positive electrostatic potential which may be the reason behind its low HER catalytic activity compared with Cu and Ni complexes.Author contributions
All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the University of Taif, Saudi Arabia, Deanship of Scientific Research, project no. 1-439-6070.References
- S. P. Badwal, S. S. Giddey, C. Munnings, A. I. Bhatt and A. F. Hollenkamp, Emerging electrochemical energy conversion and storage technologies, Front. Chem., 2014, 2(1–28), 79, DOI:10.3389/fchem.2014.00079.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution, Chem. Soc. Rev., 2014, 43(18), 6555–6569, 10.1039/C3CS60468C.
- X. Chen, D. Wang, Z. Wang, P. Zhou, Z. Wu and F. Jiang, Molybdenum phosphide: a new highly efficient catalyst for the electrochemical hydrogen evolution reaction, Chem. Commun., 2014, 50(79), 11683–11685, 10.1039/C4CC05936K.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution, J. Am. Chem. Soc., 2005, 127(15), 5308–5309, DOI:10.1021/ja0504690.
- M. Steinberg and H. C. Cheng, Modern and prospective technologies for hydrogen production from fossil fuels, Int. J. Hydrogen Energy, 1989, 14(11), 797–820, DOI:10.1016/0360-3199(89)90018-9.
- J. A. Turner, Sustainable Hydrogen Production, Science, 2004, 305(5686), 972–974, DOI:10.1126/science.1103197.
- S. Bhavsar, M. Najera, R. Solunke and G. Veser, Chemical looping: to combustion and beyond, Catal. Today, 2014, 228, 96–105, DOI:10.1016/j.cattod.2013.12.025.
- W. Yan and S. K. Hoekman, Production of CO2-free hydrogen from methane dissociation: a review, Environ. Prog. Sustainable Energy, 2014, 33(1), 213–219, DOI:10.1002/ep.11746.
- I. Roger, M. A. Shipman and M. D. Symes, Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting, Nat. Rev. Chem., 2017, 1(1), 0003, DOI:10.1038/s41570-016-0003.
- R. B. Gordon, M. Bertram and T. E. Graedel, Metal stocks and sustainability, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(5), 1209–1214, DOI:10.1073/pnas.0509498103.
- S. Sarkar and S. C. Peter, An overview on Pd-based electrocatalysts for the hydrogen evolution reaction, Inorg. Chem. Front., 2018, 5(9), 2060–2080, 10.1039/C8QI00042E.
- A. Eftekhari, Electrocatalysts for hydrogen evolution reaction, Int. J. Hydrogen Energy, 2017, 42(16), 11053–11077, DOI:10.1016/j.ijhydene.2017.02.125.
- F. Safizadeh, E. Ghali and G. Houlachi, Electrocatalysis developments for hydrogen evolution reaction in alkaline solutions – a review, Int. J. Hydrogen Energy, 2015, 40(1), 256–274, DOI:10.1016/j.ijhydene.2014.10.109.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts, Science, 2007, 317(5834), 100–102, DOI:10.1126/science.1141483.
- W. Zhou, J. Jia, J. Lu, L. Yang, D. Hou, G. Li and S. Chen, Recent developments of carbon-based electrocatalysts for hydrogen evolution reaction, Nano Energy, 2016, 28, 29–43, DOI:10.1016/j.nanoen.2016.08.027.
- L. Zhang, J. Xiao, H. Wang and M. Shao, Carbon-Based Electrocatalysts for Hydrogen and Oxygen Evolution Reactions, ACS Catal., 2017, 7(11), 7855–7865, DOI:10.1021/acscatal.7b02718.
- A. Mazzeo, S. Santalla, C. Gaviglio, F. Doctorovich and J. Pellegrino, Recent progress in homogeneous light-driven hydrogen evolution using first-row transition metal catalysts, Inorg. Chim. Acta, 2021, 517, 119950, DOI:10.1016/j.ica.2020.119950.
- C.-L. Wang, H. Yang, J. Du and S.-Z. Zhan, Effect of metal centers of complexes bearing bipyridine ligand for electrochemical- and photochemical-driven hydrogen evolution, Appl. Organomet. Chem., 2022, 36, e6453, DOI:10.1002/aoc.6453.
- L. Zhang, J. Xiao, H. Wang and M. Shao, Carbon-Based Electrocatalysts for Hydrogen and Oxygen Evolution Reactions, ACS Catal., 2017, 7(11), 7855–7865, DOI:10.1007/s12678-022-00708-8.
- R. Li, H. Zhang, M. Hong, J. Shi, X. Liu and X. Feng, Two Co(II)/Ni(II) complexes based on nitrogenous heterocyclic ligand as high-performance electrocatalyst for hydrogen evolution reaction, Dalton Trans., 2022, 51, 3970, 10.1039/D1DT03814A.
- G. Giuffredi, T. Asset, Y. Liu, P. Atanassov and F. D. Fonzo, Transition Metal Chalcogenides as a Versatile and Tunable Platform for Catalytic CO2 and N2 Electroreduction, ACS Mater. Au, 2021, 1(1), 6–36, DOI:10.1021/acsmaterialsau.1c00006.
- J. Li, C. Liu, X. Wang, Y. Ding, Z. Wu, P. Sun, J. Tang, J. Zhang, D.-S. Li, N. Chen and T. Wu, Stable 3D neutral gallium thioantimonate frameworks decorated with transition metal complexes for a tunable photocatalytic hydrogen evolution, Dalton Trans., 2022, 51, 978–985, 10.1039/D1DT03255K.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Investigating and exploiting the electrocatalytic properties of hydrogenases, Chem. Rev., 2007, 107, 4366–4413, DOI:10.1021/cr050191u.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases, Chem. Rev., 2007, 107, 4273–4303, DOI:10.1021/cr050195z.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, Synthetic Models for the Active Site of the [FeFe]-Hydrogenase: Catalytic Proton Reduction and the Structure of the Doubly Protonated Intermediate, J. Am. Chem. Soc., 2012, 134, 18843–18852, DOI:10.1021/ja309216v.
- T. Goris, A. F. Wait, M. Saggu, J. Fritsch, N. Heidary, M. Stein, I. Zebger, F. Lendzian, F. A. Armstrong, B. Friedrich and O. Lenz, A unique iron-sulfur cluster is crucial for oxygen tolerance of a [NiFe]-hydrogenase, Nat. Chem. Biol., 2011, 7, 310–318, DOI:10.1038/nchembio.555.
- M. Gómez-Gallego and M. A. Sierra, Deuteration mechanistic studies of hydrogenase mimics, Inorg. Chem. Front., 2021, 8, 3934–3950, 10.1039/D1QI00505G.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Mimicking hydrogenases: from biomimetics to artificial enzymes, Coord. Chem. Rev., 2014, 270–271, 127–150, DOI:10.1016/j.ccr.2013.12.018.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides, Chem. Rev., 2016, 116(15), 8693–8749, DOI:10.1021/acs.chemrev.6b00180.
- G. Parkin, Synthetic analogues relevant to the structure and function of zinc enzymes, Chem. Rev., 2004, 104, 699–767, DOI:10.1021/cr0206263.
- J. Perkinson, S. Brodie, K. Yoon, K. Mosny, P. J. Carroll, T. V. Morgan and S. J. N. Burgmayer, Preparations and properties of transition-metal pterin complexes. Models for the metal site in phenylalanine hydroxylase, Inorg. Chem., 1991, 30, 719–727, DOI:10.1021/ic00004a023.
- K. Nakamoto, in Infrared Spectra of Inorganic and Coordination Compounds, ed. K. Nakamoto, J. Wiley and Sons, New York, 1986, p. 256 Search PubMed.
- S. Y. Shaban, A. M. Ramadan, M. M. Ibrahim and M. A. R. Mohamedvan Eldik, Spectroscopic, thermodynamic, kinetic studies and oxidase/antioxidant biomimetic catalytic activities of tris(3,5-dimethylpyrazolyl)borate Cu(ii) complexes, Dalton Trans., 2015, 44, 14110–14121, 10.1039/C5DT01817J.
- O. O. E. Onawumi, O. O. P. Faboya, O. A. Odunola, T. K. Prasad and M. V. Pajasekharan, Synthesis, structure and spectral studies on mixed ligand copper(II) complexes of diimines and acetylacetonate, Polyhedron, 2008, 27, 113–117, DOI:10.1016/j.poly.2007.08.041.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 2nd edn, 1984 Search PubMed.
- M. Amirnasr, A. H. Mahmoudkhani, A. Gorji, S. Dehghanpour and H. R. Bijanzadeh, Polyhedron, 2002, 21, 2733–2742, DOI:10.1016/S0277-5387(02)01277-9.
- H. A. Bayoumi, A. M. A. Alaghaz and M. S. Aljahdali, Cu(II), Ni(II), Co(II) and Cr(III) Complexes with N2O2-Chelating Schiff's Base Ligand Incorporating Azo and Sulfonamide Moieties: Spectroscopic, Electrochemical Behavior and Thermal Decomposition Studies, Int. J. Electrochem. Sci., 2013, 8, 9399–9413 CAS.
- B. N. Figgis and J. Lewis, The Magnetic Properties of Transition Metal Complexes, Prog. Inorg. Chem., 1964, 6, 37, DOI:10.1002/9780470166079.ch2.
- R. L. Dutta and A. Syamal, Elements of Magnetochemistry, Affiliated East-West Press, Delhi, 2nd edn, 2007, pp. 893–930 Search PubMed.
- R. S. Drago, Physical Methods in Chemistry, Saunders, Philadelphia, 1977, vol. 481, pp. 494–502 Search PubMed.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, Recent Development in Hydrogen Evolution Reaction Catalysts and Their Practical Implementation, Phys. Chem. Lett., 2015, 6, 951–957, DOI:10.1021/acs.jpclett.5b00306.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, Amorphous Molybdenum Sulfide Catalysts for Electrochemical Hydrogen Production: Insights Into the Origin of Their Catalytic Activity, ACS Catal., 2012, 2, 1916–1923, DOI:10.1021/cs300451q.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Electrocatalysts for Hydrogen Evolution in Alkaline Electrolytes: Mechanisms, Challenges, and Prospective Solutions, Adv. Sci., 2018, 5, 1700464, DOI:10.1002/advs.201700464.
- J. Durst, A. Siebel, C. Simon, F. Hasche, J. Herranz and H. A. Gasteiger, New Insights into the Electrochemical Hydrogen Oxidation and Evolution Reaction Mechanism, Energy Environ. Sci., 2014, 7, 2255–2260, 10.1039/C4EE00440J.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Insight on Tafel Slopes from A Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion, Sci. Rep., 2015, 5, 13801, DOI:10.1038/srep13801.
- E. A. Franceschini, G. I. Lacconi and H. R. Corti, Kinetics of the hydrogen evolution on nickel in alkaline solution: new insight from rotating disk electrode and impedance spectroscopy analysis, Electrochim. Acta, 2015, 159, 210–218 CrossRef CAS.
- D. Delgado, G. Hefter and M. Minakshi, Hydrogen Generation, in Alternative Energies, ed. G. Ferreira, Springer, Berlin, 2013, pp. 141–161 Search PubMed.
- C. Lupi, A. Dell'Era and M. Pasquali, Nickel–Cobalt Electrodeposited Alloys for Hydrogen Evolution in Alkaline Media, Int. J. Hydrogen Energy, 2009, 34, 2101–2106 CrossRef CAS.
- F. Rosalbino, S. Delsante, G. Borzone and E. Angelini, Electrocatalytic Behaviour of Co–Ni–R (R = Rare Earth Metal) Crystalline Alloys as Electrode Materials for Hydrogen Evolution Reaction in Alkaline Medium, Int. J. Hydrogen Energy, 2008, 33, 6696–6703 CrossRef CAS.
- P. Elumalai, H. N. Vasan, N. S. Munichandraiah and A. Shivashankar, Kinetics of Hydrogen Evolution on Submicron size Co, Ni, Pd and Co–Ni Alloy Powder Electrodes by D. C. Polarization and A. C. Impedance Studies, J. Appl. Electrochem., 2002, 32, 1005–1010 CrossRef CAS.
- M. A. Amin, N. El-Bagoury, M. H. H. Mahmoud, M. M. Hessien, S. S. Abd ElRehim, J. Wysocka and J. Ryl, Catalytic Impact of Alloyed Al on the Corrosion Behavior of Co50Ni23Ga26Al1.0 magnetic Shape Memory Alloy and Catalysis Applications for Efficient Electrochemical H2 Generation, RSC Adv., 2017, 7, 3635–3649 RSC.
- G. Darabdhara, M. A. Amin, G. A. M. Mersal, E. M. Ahmed, M. R. Das, M. B. Zakaria, V. Malgras, S. M. Alshehri, Y. Yamauchi, S. Szunerits and R. Boukherroub, Reduced graphene oxide nanosheets decorated with Au, Pd and Au–Pd bimetallic nanoparticles as highly efficient catalysts for electrochemical hydrogen generation, J. Mater. Chem. A, 2015, 3, 20254–20266 RSC.
- M. A. Amin, S. A. Fadlallah and G. S. Alosaimi, In Situ Aqueous Synthesis of Silver Nanoparticles Supported on Titanium as Active Electrocatalyst for the Hydrogen Evolution Reaction, Int. J. Hydrogen Energy, 2014, 39, 19519–19540 CrossRef CAS.
- M. A. Amin, S. A. Fadlallah, G. S. Alosaimi, F. Kandemirli, M. Saracoglu, S. Szunerits and R. Boukherroub, Cathodic activation of titanium-supported gold nanoparticles: an efficient and stable electrocatalyst for the hydrogen evolution reaction, Int. J. Hydrogen Energy, 2016, 41, 6326–6341 CrossRef CAS.
- M. Ifires, A. Addad, A. Barras, T. Hadjersi, R. Chegroune, S. Szunerits, R. Boukherroub and M. A. Amin, Cathodic pre-polarization studies on the carbon felt/KOH interface: an efficient metal-free electrocatalyst for hydrogen generation, Electrochim. Acta, 2021, 375, 137981 CrossRef CAS.
- A. P. Murthy, J. Theerthagiri, J. Madhavan and K. Murugan, Highly active MoS2/carbon electrocatalysts for the hydrogen evolution reaction – insight into the effect of the internal resistance and roughness factor on the Tafel slope, Phys. Chem. Chem. Phys., 2017, 19, 1988–1998 RSC.
- T. Tang, W. J. Jiang, S. Niu, N. Liu, H. Luo, Y. Y. Chen, S. F. Jin, F. Gao, L. J. Wan and J. S. Hu, Electronic and Morphological Dual Modulation of Cobalt Carbonate Hydroxides by Mn Doping toward Highly Efficient and Stable Bifunctional Electrocatalysts for Overall Water Splitting, J. Am. Chem. Soc., 2017, 139(24), 8320–8328, DOI:10.1021/jacs.7b03507.
- J. Kibsgaard and T. F. Jaramillo, Molybdenum Phosphosulfide: An Active, Acid-Stable, Earth-Abundant Catalyst for the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2014, 53, 14433–14437, DOI:10.1002/anie.201408222.
- Y.-R. Liu, X. Shang, W.-K. Gao, B. Dong, J.-Q. Chi, X. Li, K.-L. Yan, Y.-M. Chai, Y.-Q. Liu and C.-G. Liu, Ternary CoS2/MoS2/RGO Electrocatalyst with CoMoS Phase for Efficient Hydrogen Evolution, Appl. Surf. Sci., 2017, 412, 138–145, DOI:10.1016/j.apsusc.2017.03.245.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Engineering the surface structure of MoS2 to preferentially expose active edge sites for electrocatalysis, Nat. Mater., 2012, 11, 963–969, DOI:10.1038/nmat3439.
- N. Liu, Y. Guo, X. Yang, H. Lin, L. Yang, Z. Shi, Z. Zhong, S. Wang, Y. Tang and Q. Gao, Microwave-Assisted Reactant-Protecting Strategy toward Efficient MoS2 Electrocatalysts in Hydrogen Evolution Reaction, ACS Appl. Mater. Interfaces, 2015, 7(42), 23741–23749 CrossRef CAS PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, Controllable Disorder Engineering in Oxygen-Incorporated MoS2 Ultrathin Nanosheets for Efficient Hydrogen Evolution, J. Am. Chem. Soc., 2013, 135(47), 17881–17888 CrossRef CAS PubMed.
- T. Tang, W. J. Jiang, S. Niu, N. Liu, H. Luo, Y. Y. Chen, S. F. Jin, F. Gao, L. J. Wan and J. S. Hu, Electronic and Morphological Dual Modulation of Cobalt Carbonate Hydroxides by Mn Doping toward Highly Efficient and Stable Bifunctional Electrocatalysts for Overall Water Splitting, J. Am. Chem. Soc., 2017, 139(24), 8320–8328 CrossRef CAS PubMed.
- J. Kibsgaard and T. F. Jaramillo, Molybdenum Phosphosulfide: An Active, Acid-Stable, Earth-Abundant Catalyst for the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2014, 53(52), 14433–14437 CrossRef CAS PubMed.
- Y.-R. Liu, X. Shang, W.-K. Gao, B. Dong, J.-Q. Chi, X. Li, K.-L. Yan, Y.-M. Chai, Y.-Q. Liu and C.-G. Liu, Ternary CoS2/MoS2/RGO electrocatalyst with CoMoS phase for efficient hydrogen evolution, Appl. Surf. Sci., 2017, 412, 138–145 CrossRef CAS.
- M. M. Ibrahim, S. Y. Shaban, A. A. Amer, S. F. Mohamed, A. M. Fathy, S. A. Al-Harbi and A. M. Ramadan, Tuning the structural and catalytic properties of copper(II)-based complexes containing pyridine-2,6-diimines, J. Biomol. Struct. Dyn., 2021, 39(15), 1–19, DOI:10.1080/07391102.2021.1947379.
- M. M. Ibrahim, M. A. El-Kemary, S. A. Al-Harbi, H. M. Al-Saidi, S. A. Sallam and A. M. Ramadan, Synthesis and Structural Characterization of Pyridine-based Mn(III), Fe(III), and Co(III) Complexes as SOD Mimics and BSA Binding Studies, J. Mol. Struct., 2021, 1228, 129706, DOI:10.1016/j.molstruc.2020.129706.
- J. Wang, S. Dou and X. Wang, Structural tuning of heterogeneous molecular catalysts for electrochemical energy conversion, Sci. Adv., 2021, 7, 1–13, DOI:10.1126/sciadv.abf3989.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08530a |
| This journal is © The Royal Society of Chemistry 2022 |