
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid hydrogels derived from renewable resources as a smart stimuli responsive soft material for drug delivery applications†
Vandana Singha,
Yadavali Siva Prasadad,
Arun Kumar Rachamallac,
Vara Prasad Rebakac,
Tohira Banooc,
C. Uma Maheswari a,
Vellaisamy Sridharanb,
Krishnamoorthy Lalitha*a and
Subbiah Nagarajan*ac
a,
Vellaisamy Sridharanb,
Krishnamoorthy Lalitha*a and
Subbiah Nagarajan*ac
aSchool of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur-613401, Tamil Nadu, India. E-mail: lalitha@scbt.sastra.edu
bDepartment of Chemistry and Chemical Sciences, Central University of Jammu, Rahya-Suchani (Bagla), District-Samba, Jammu-181143, J&K, India
cDepartment of Chemistry, National Institute of Technology Warangal, Warangal-506004, Telangana, India. E-mail: snagarajan@nitw.ac.in; Tel: +91-9940430715
dDepartment of Biomedical Engineering, Saveetha School of Engineering, Saveetha Nagar, Thandalam, Tamil Nadu, India
First published on 12th January 2022
Abstract
The design and synthesis of amphiphilic molecules play a crucial role in fabricating smart functional materials via self-assembly. Especially, biologically significant natural molecules and their structural analogues have inspired chemists and made a major contribution to the development of advanced smart materials. In this report, a series of amphiphilic N-acyl amides were synthesized from natural precursors using a simple synthetic protocol. Interestingly, the self-assembly of amphiphiles 6a and 7a furnished a hydrogel and oleogel in vegetable oils. Morphological analysis of gels revealed the existence of a 3-dimensional fibrous network. Thermoresponsive and thixotropic behavior of these gels were evaluated using rheological analysis. A composite gel prepared by the encapsulation of curcumin in the hydrogel formed from 7a displayed a gel–sol transition in response to pH and could act as a dual channel responsive drug carrier.
Introduction
Self-assembly is a natural process accomplishing a key role in the origin, maintenance, and growth of life.1–5 Inspired by natural phenomena, the interest in the assembly of bio-macromolecules and low molecular weight gelators (LMWGs) to fabricate biomimetics, and functional materials for practical applications has increased significantly.1–5 Gels are a kind of smart material possessing a 3D supramolecular network displaying visco-elastic behaviour via the self-organization of amphiphiles in a suitable solvent involving reversible non-covalent interactions such as van der Waals forces, hydrogen bonding, metal–ligand coordination, anionic-π, cationic-π, and CH–π interactions, and π–π stacking.3 In assembled materials, a finite tuning in molecular level interaction is observed upon the exposure to external stimuli, which enable them to behave as a prospective soft material suitable for a wide range of applications such as wound healing, bone-repair, drug delivery, sensors, imaging, to name a few.1–5 Owing to the potential applications of these soft materials, we are interested in the design and synthesis of the amphiphiles with an amide bond from the natural molecules, that could display molecular self-assembly. It is worth mentioning that, an effective balance of hydrophilicity and lipophilicity (HLB) is essential for construction of 3-D supramolecular architecture.6,7 Generally, amphiphiles derived from amino alcohols, amino acids, hydrazides, carbohydrates, peptides, to name a few, display various degree of HLB to furnish different supramolecular architecture. Among these building blocks, amino alcohols are of much industrial importance on account of their potential usefulness in the manufacture of pharmaceuticals, surfactants, agrochemicals and lubricating oil additives.8 In the past 2 decades, gelators derived by molecular self-assembly of amino alcohol such as bis(amino acid)oxalamides,9 bis(amino alcohol)oxalamides,9,10 bis(aminoalcohol)diglycolamides,10 bis(phenylglycinol)malonamide,11 lipopeptides were extensively studied and their potential application in biology and medicine were explored.12,13 It is worth mentioning that dopamine an important amino alcohol largely explored in the field of medicine, coatings, water purification, sensors energy storage, and disease treatment.14–25 However, there is not much research on the use of dopamine for the generation of amphiphilic molecules. By looking at the broad applications of soft materials derived from biomolecules and renewable resources, in this paper, we have selected amino alcohols like propanol amine, tris and dopamine as a moiety rendering hydrophilicity, and cardanol, an alkyl phenolic oil extracted from the cashew nutshell, as one of the renewable value-added raw materials for the synthesis of amphiphilic N-acyl amide derivatives.Cardanol is a bio-based phenolic lipid mixture comprising 50% of 3-(8Z-pentadecenyl)phenol, 16% of 3-(8Z,11Z-pentadecadienyl)phenol, 29% of 3-(8Z,11Z,14-pentadecatrienyl)phenol and 5% of 3-(n-pentadecyl)phenol.26–30 The presence of a phenolic moiety and the long alkyl chain with varying degrees of unsaturation at meta-position provides the unique characteristics to cardanol and enables the self-assembly via π–π stacking and hydrophobic interactions.26–30 In this line, John and co-workers reported the synthesis of phenoxyacyl-ethanolamides from this CNSL and studied their amidohydrolase activity of fatty acid amide hydrolase (FAAH).31 In this paper, we report a facile synthesis of amphiphilic N-acyl amides by the judicious combination of various amino alcohols such as dopamine, propanol amine, and tris, and the phenolic esters derived from cardanol and explored their molecular self-assembly in a wide variety of solvents and vegetable oils. We have also demonstrated the potential application of one of the amphiphilic N-acyl amides as a dual channel stimuli-responsive delivery system for the natural hydrophobic drug, curcumin. Further, supramolecular architecture and stimuli responsive behaviour were investigated with respect to the molecular structure.
Results and discussion
Inspired by the natural self-assembly of proteins, amino acids, nucleic acids, and lipids in biological systems, the interest in the design and synthesis of amphiphiles to fabricate functional soft materials by exploiting their molecular self-assembly has raised significantly. Recently, we have reported the synthesis and self-assembly of cardanol-based functional molecules and explored their potential applications in the field of biomaterials and electronics.32–37 The excellent antioxidant property and self-assembling characteristics of largely explored fatty acid amides elicited us to design and synthesize the amphiphilic N-acyl amides from cardanol and amino alcohols such as propanol amine 4, tris 5, and dopamine 8. Amphiphilic N-acyl amides (6, 7) bearing aliphatic OH group as a hydrophilic moiety were synthesized via the nucleophilic substitution of 3-alkyl phenols 1a,b with methyl bromoacetate 2, followed by amidation with amino alcohols 4, and 5 (Scheme 1).31,37 Amphiphilic N-(3-hydroxypropyl)-2-(3-alkyl phenoxy)acetamides 6a,b bearing an aliphatic OH group was prepared by following the procedure reported in the literature.31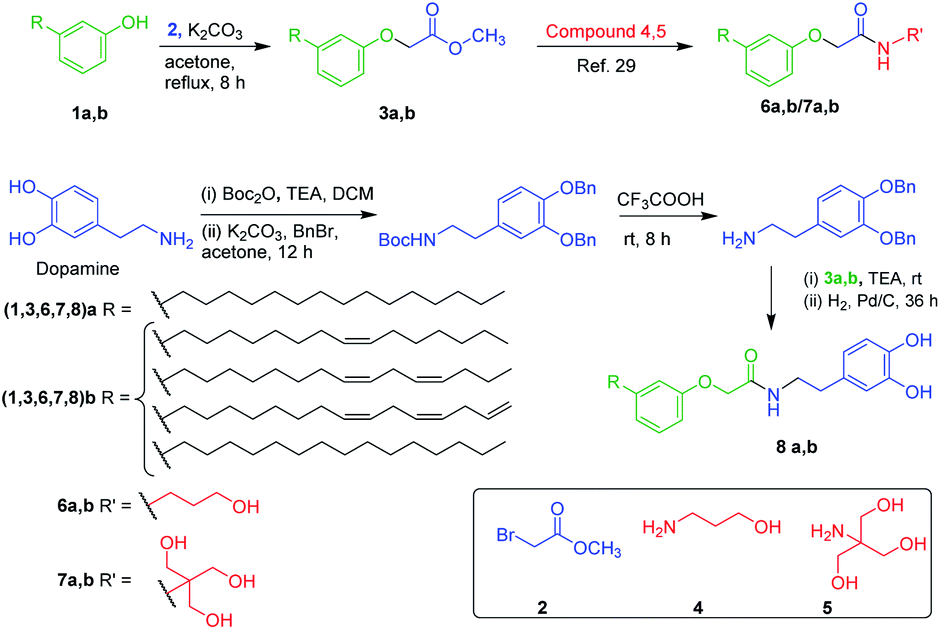 | ||
| Scheme 1 Synthesis of N-acyl amides-based amphiphiles (6–8)a,b. | ||
The formation of the desired product was confirmed using NMR and mass spectral analysis. In 1H NMR spectrum of compound 6a, the peak at δ 6.94 and 4.51 ppm represent –NH and –OCH2– protons, and in 13C NMR spectra, the peak at δ 169.6 and 67.1 ppm represent the carbonyl and –OCH2– group respectively. A separate reaction condition suitable for the synthesis of amphiphilic N-(1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl)-2-(3-alkyl phenoxy)acetamides, 7a,b bearing three aliphatic OH groups was optimized using solvents such as MeOH, EtOH, DCM, DMSO, and THF by varying the temperature and reaction time (Table S1†). Amphiphilic N-acyl amides (8a,b) bearing aromatic OH moiety were synthesized by following Scheme 1. Direct amidation of dopamine with 3a,b resulted in the polymerization of dopamine.25 In order to avoid the polymerization during the synthesis of compounds 8a,b we have adopted the protected/deprotection strategy as given in Scheme 1. The formation of compounds 8a,b was identified using NMR and mass spectral analysis (Fig. S17–S25, ESI†).
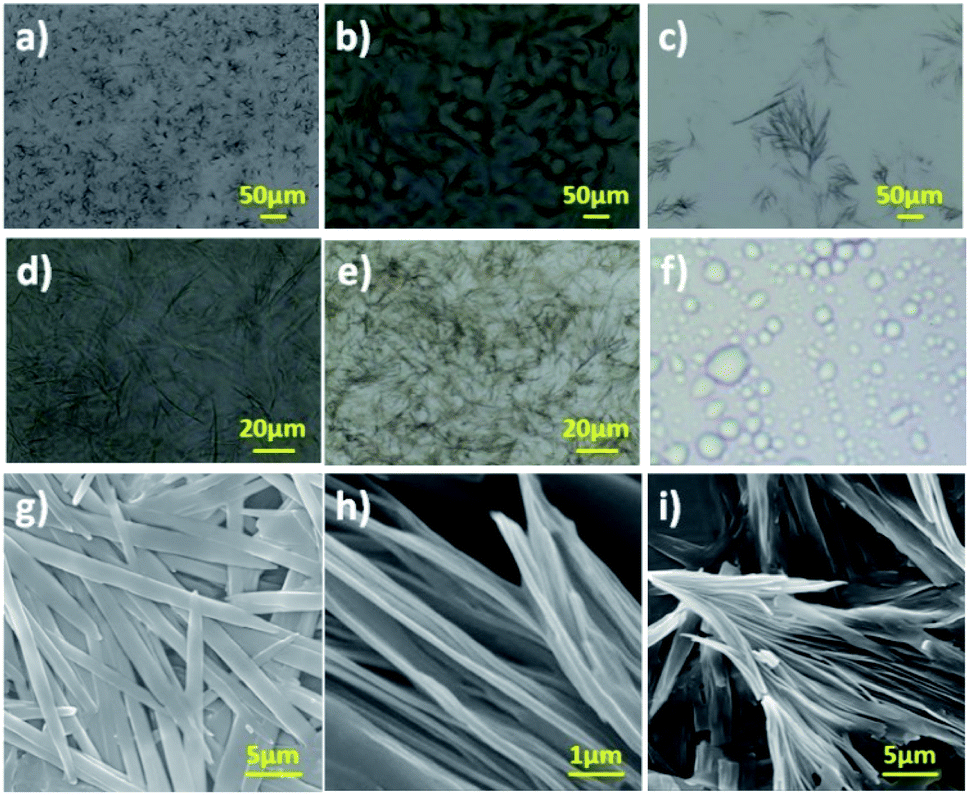
By having amphiphilic N-acyl amides bearing various degree of hydrophilic groups, we further moved on investigating the gelation efficiency in various solvents and vegetable oils using the “stable to inversion” method.32–40 Most interestingly, compounds 6a and 7a displayed oleogelation in light paraffin oil with the critical gelation concentration (CGC) of 1.0 and 0.9% (w/v) respectively. In addition, compounds 6a and 7a also formed organogel in PEG with a CGC of 1.0 and 0.85% (w/v), and hybrid hydrogel in water–DMSO (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with a CGC of 1.3% (w/v) and 1.0% (w/v) respectively (Table 1). The existence of a kink in the hydrophobic part of the amphiphilic N-acyl amides 6b and 7b belittled their gelation behaviour with respect to the corresponding saturated moieties. N-Acyl amides, 8a and 8b bearing aromatic OH as hydrophilic part did not show any gelation in the tested solvent and vegetable oil, instead formed micelles in an ethanol–water mixture (1:9) (Fig. 2a(iv)). It is worth mentioning that, compounds 8a and 8b bearing aromatic OH (pKa: ∼9) did not form gel even at basic pH levels, for the reason that the phenolic OH can dissociate into ionic form, there by tune the hydrophilicity. Overall, to our surprise, N-acyl amides bearing aliphatic OH group as hydrophilic part displayed gelation in both highly hydrophobic solvent (paraffin oil) and hydrophilic solvent (polyethylene glycol and water), whereas compound bearing aromatic OH group as hydrophilic part did not show molecular self-assembly to form a gel. In order to identify the thermal stability of gels, we have determined gel-to-sol transition temperature (Tgel) of the gels formed by 6a and 7a.39,41 Tgel of 6a in light paraffin oil, PEG and DMSO:water mixture was observed as 84, 87 and 75 °C respectively. Surprisingly, compound 7a, which is having more hydroxyl group displayed increased Tgel in light paraffin oil, PEG and DMSO:water system such as 82, 89 and 85 °C respectively. The presence of more hydroxyl groups in compound 7a might enhance the intensity of H-bonding in DMSO:water system, thereby increasing the gel-to-sol transition temperature.
:1) with a CGC of 1.3% (w/v) and 1.0% (w/v) respectively (Table 1). The existence of a kink in the hydrophobic part of the amphiphilic N-acyl amides 6b and 7b belittled their gelation behaviour with respect to the corresponding saturated moieties. N-Acyl amides, 8a and 8b bearing aromatic OH as hydrophilic part did not show any gelation in the tested solvent and vegetable oil, instead formed micelles in an ethanol–water mixture (1:9) (Fig. 2a(iv)). It is worth mentioning that, compounds 8a and 8b bearing aromatic OH (pKa: ∼9) did not form gel even at basic pH levels, for the reason that the phenolic OH can dissociate into ionic form, there by tune the hydrophilicity. Overall, to our surprise, N-acyl amides bearing aliphatic OH group as hydrophilic part displayed gelation in both highly hydrophobic solvent (paraffin oil) and hydrophilic solvent (polyethylene glycol and water), whereas compound bearing aromatic OH group as hydrophilic part did not show molecular self-assembly to form a gel. In order to identify the thermal stability of gels, we have determined gel-to-sol transition temperature (Tgel) of the gels formed by 6a and 7a.39,41 Tgel of 6a in light paraffin oil, PEG and DMSO:water mixture was observed as 84, 87 and 75 °C respectively. Surprisingly, compound 7a, which is having more hydroxyl group displayed increased Tgel in light paraffin oil, PEG and DMSO:water system such as 82, 89 and 85 °C respectively. The presence of more hydroxyl groups in compound 7a might enhance the intensity of H-bonding in DMSO:water system, thereby increasing the gel-to-sol transition temperature.
| S. No. | Solvent/oil | Observationa | |||||
|---|---|---|---|---|---|---|---|
| 6a | 6b | 7a | 7b | 8a | 8b | ||
| a Critical gelation concentration is given in % w/v within parenthesis, S – soluble; I – insoluble; G – gelation. | |||||||
| 1 | Olive oil | S | S | S | S | S | S |
| 2 | Heavy paraffin oil | S | S | S | S | S | S |
| 3 | Light paraffin oil | G (1.0%) | S | G (0.9%) | S | S | S |
| 4 | Sesame oil | S | S | S | S | S | S |
| 5 | Linseed oil | S | S | S | S | S | S |
| 6 | Water | I | I | I | I | I | S |
| 7 | Water + DMSO (3:1) |
G (1.3%) | S | G (1.0%) | S | S | S |
| 8 | DMF | S | S | S | S | S | S |
| 9 | Ethyl acetate | I | S | I | S | S | S |
| 10 | Cyclohexane | S | S | S | S | S | S |
| 11 | Jojoba oil | S | S | S | S | S | S |
| 12 | Castor oil | S | S | S | S | S | S |
| 13 | Xylene | S | S | S | S | S | S |
| 14 | Glycerol | S | S | S | S | S | S |
| 15 | Ethylene glycol | S | S | S | S | S | S |
| 16 | Soya bean oil | S | S | S | S | S | S |
| 17 | Eucalyptus oil | S | S | S | S | S | S |
| 18 | Buffer | I | I | I | I | S | S |
| 19 | Hazel nut oil | S | S | S | S | S | S |
| 20 | Poly ethylene glycol | G (1.0%) | S | G (0.85%) | S | S | S |
| 21 | Methanol | S | S | S | S | S | S |
| 22 | 1,2-Dichlorobenzene | S | S | S | S | S | S |
| 23 | Lauryl alcohol | S | S | S | S | S | S |
Morphological analysis
Generally, aggregation of a gelator to generate supramolecular architecture in micro or nanoscale is based on the molecular structure of the gelator, solvent used, temperature and pressure.32–42 However, properties of soft materials were governed by the molecular assembly, which requires a clear understanding across length scales controlled by intermolecular interactions. In order to probe gel morphology and gelation mechanism, microscopic analysis of xerogel formed by amphiphilic N-acyl amides 6a and 7a was performed using optical microscopy, and scanning electron microscopy (SEM).42 Optical microscopy images of gel formed by 6a and 7a were given in Fig. 1a–e. Gel formed by 6a in DMSO–water (1:3), polyethylene glycol and light paraffin oil display a micro or nanoscale fibrillar structure generating a 3D entangled network (Fig. 1a–d). Assembly pattern of compound 7a in DMSO–water (1:3) furnishes bundled fibers with entanglement, whereas 8a in DMSO–water (1:3) did not form gel instead display a micelle like morphology (Fig. 1e and f). The average size of the micelles were analysed using a particle size analyser, which displayed an average size of 400 nm (Fig. S27†).
 | ||
| Fig. 1 (a–f) Optical micrograph of gel formed by (a and b) 6a in DMSO + H2O (1:3), (c) 6a in poly(ethylene glycol), (d) 6a in light paraffin, (e) 7a in DMSO + H2O (1:3). (f) Micellar structure formed by 8a in DMSO–water (1:3). (g–i) SEM images of the gel formed by (g) 6a and (h and i) 7a in DMSO + H2O (1:3) respectively. Gel formed in DMSO + H2O (1:3) is referred as hydrogel. | ||
 | ||
| Fig. 2 a) Gel images of (i) 6a in light paraffin oil, (ii) hydrogel derived from 6a, (iii) hydrogel derived from 7a (iv) 8a in DMSO–water (1:3); (b) ATR-IR spectra of the compound (i) 6a and (ii) 7a in amorphous and xerogel respectively; (c) SAXRD of xerogel formed by 6a and 7a; (d) pictorial representation of molecular self-assembly of compound 6a (top) and 7a (bottom) respectively. Intermolecular H-bonding is represented in red dotted line. | ||
Optical microscopic analysis clearly revealed the existence of distinct morphology, which is based on the molecular structure and solvent used. Further, we have performed SEM analysis to get a clear cut morphology of hydrogel formed by 6a and 7a (Fig. 1g–i and S26†). Hydrogel obtained from compound 6a displayed lamellar morphology of width ranging from 1.0–1.5 μm, whereas compound 7a rendered a bundled fibrillar architecture of width ranging from 500–800 nm (Fig. 1g–i). It is worth discussing that the small tuning in the hydrophilicity of N-acyl amides drastically change the self-assembly behaviour and morphology. The replacement of aliphatic OH in compound 6 and 7 with aromatic OH (8a,b) resulted in the formation of micelles (Fig. S27†). In addition to the molecular tuning, change in solvent also influence the aggregation pattern of amphiphilic N-acyl amides (Fig. 1).43,44
Interesting results obtained on the gelation and morphology further inspired us to look into the detailed packing pattern of gelator using intermolecular interactions. ATR-IR spectroscopic analysis of compounds 6a and 7a in amorphous and self-assembled form (xerogel) is given in Fig. 2b. Compound 6a in amorphous state displayed absorption band at ν = 3443 (–OH, H-bonded), 3305 (–NH), 2955 (–CH, alkane), 2917 (–CH, alkane), 2847 (–CH, alkane), 1648 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, amide), 1589 (–CC bending, aromatic), and 1550 (–NH bending) cm−1, whereas in xerogel state showed absorption band at ν = 3451 (–OH, H-bonded), 3302 (–NH), 2969 (–CH, alkane), 2916 (–CH, alkane), 2847 (–CH, alkane), 1648 (–CO, amide), 1592 (–CC bending, aromatic), and 1551 (–NH bending) cm−1. Analysis of IR data revealed that the existence of random molecular aggregation in amorphous state facilitated by intermolecular H-bonding, whereas in xerogel state, in addition to intermolecular H-bonding, π–π stacking and hydrophobic interactions in long alkyl chain are clearly visualized (Fig. 2b).45,46 Compound 7a displayed IR absorption band at ν = 3365, 3340, 3239, 2960, 2920, 2846, 1662, 1591 and 1540 cm−1 in amorphous form, and 3450, 2917, 2846, 1654, 1628, 1535 cm−1 in the xerogel state. The appreciable shift in bands corresponding to –OH, –NH, –CH (alkane), –CC (Ar), –CH (Ar), –CO (amide) clearly reveal the establishment of strong intermolecular interaction during the self-assembly process.46 IR data analysis of 6a and 7a in amorphous and xerogel state reveal that compound 7a upon self-assembly establish a strong intermolecular interactions than 6a, which could be clearly visualized from the significant shift in absorption frequency (cm−1).
O, amide), 1589 (–CC bending, aromatic), and 1550 (–NH bending) cm−1, whereas in xerogel state showed absorption band at ν = 3451 (–OH, H-bonded), 3302 (–NH), 2969 (–CH, alkane), 2916 (–CH, alkane), 2847 (–CH, alkane), 1648 (–CO, amide), 1592 (–CC bending, aromatic), and 1551 (–NH bending) cm−1. Analysis of IR data revealed that the existence of random molecular aggregation in amorphous state facilitated by intermolecular H-bonding, whereas in xerogel state, in addition to intermolecular H-bonding, π–π stacking and hydrophobic interactions in long alkyl chain are clearly visualized (Fig. 2b).45,46 Compound 7a displayed IR absorption band at ν = 3365, 3340, 3239, 2960, 2920, 2846, 1662, 1591 and 1540 cm−1 in amorphous form, and 3450, 2917, 2846, 1654, 1628, 1535 cm−1 in the xerogel state. The appreciable shift in bands corresponding to –OH, –NH, –CH (alkane), –CC (Ar), –CH (Ar), –CO (amide) clearly reveal the establishment of strong intermolecular interaction during the self-assembly process.46 IR data analysis of 6a and 7a in amorphous and xerogel state reveal that compound 7a upon self-assembly establish a strong intermolecular interactions than 6a, which could be clearly visualized from the significant shift in absorption frequency (cm−1).
Small angle X-ray diffraction (SAXRD) studies
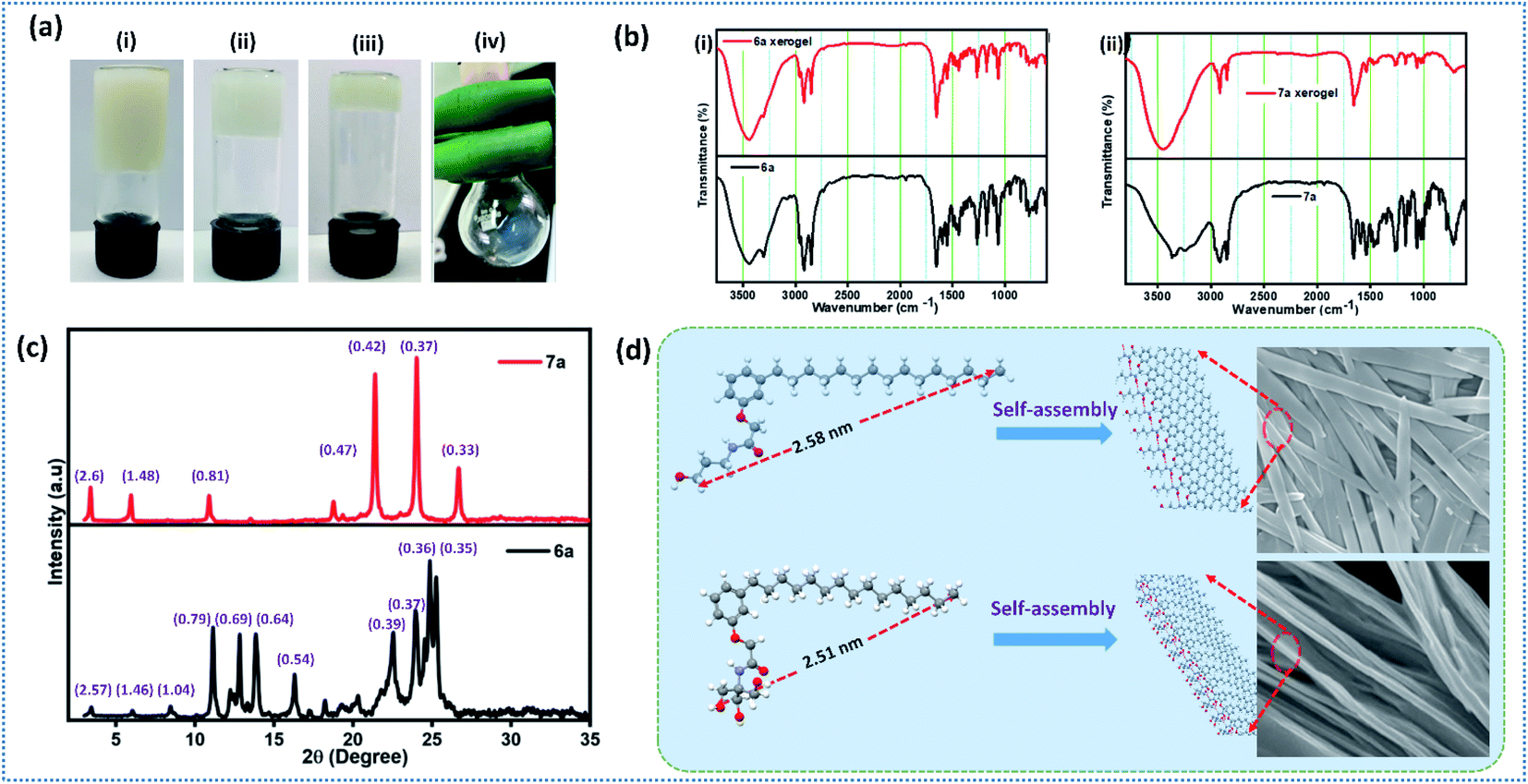
To explore the dynamic gelation due to molecular packing, low angle powder XRD was used. SAXRD pattern of the xerogel obtained from compound 6a and 7a is given in Fig. 2c. Xerogel of the 6a displayed Bragg's reflection at 2θ = 3.44°, 6.05°, 8.46°, 11.14°, 12.82°, 13.85°, 16.31°, 22.54°, 23.97°, 24.86°, 25.26° corresponding to the d-spacing of 2.57, 1.46, 1.04, 0.79, 0.69, 0.64, 0.54, 0.39, 0.37, 0.36, 0.35 nm (Fig. 2c), whereas 7a displayed a well distinct peaks at 2θ = 3.40°, 5.96°, 10.90°, 18.77°, 21.40°, 24.03°, 26.69°, with d-spacing of 2.6, 1.48, 0.81, 0.47, 0.42, 0.37, 0.33 nm respectively. It is worth mentioning that peaks of 6a and 7a are not at the same position indicating the different packing pattern. However, peaks of 7a is well resolved and distinct in nature when compared to 6a. In order to establish the packing arrangement involving various inter and intermolecular interactions, energy minimized structure were obtained and their molecular length were calculated and compared with experimental XRD diffraction data. XRD of compound 6a and 7a furnished d-spacing value of 2.57 and 2.6 nm, which is almost equal to their corresponding molecular length of 2.58 and 2.51 nm respectively. The observed d-spacing value clearly reveals the existence of monolayer arrangement forming the fibrillar structure.47 Further, small tuning in the hydrophilic part of an amphiphile display different packing arrangement in molecular level generating drastic change in morphological structure in gels.Spectral and morphological studies clearly revealed that the tuning of intermolecular interaction, especially H-bonding in the hydrophilic part can change the self-assembly. pKa value of primary alcohol is around 16.0 and generally considered as weak Brønsted acid and base.48 However, the dissociation of alcohol can be achieved by tuning the pH of the medium.48 Since the system selected for our investigation is having amide linkage and varying degree of hydroxyl group, we were curious to check the pH responsive behaviour of self-assembled monolayer fibrils formed from 6a and 7a under varying pH. Acidic buffer of pH 4.0 and basic buffer of pH 9.0 was added slowly over the hydrogel and observed their influence. Interestingly, a rapid gel to sol was observed in basic buffer, whereas a slow and steady trend of gel to sol transition was observed in an acidic buffer (Fig. 3a and b), which clearly reveals the disassembly of fibrils formed by monolayer arrangement.49 Percolation of acidic and basic buffer into the gel further tune the intermolecular H-bonding interaction, facilitating gel-to-sol transition, which could be visualized by the naked eye (Fig. 3a and b). It is worth mentioning that the gel formed by 6a and 7a were highly stable in neutral pH for a long period. Molecular packing and pH responsive behaviour of hydrogel prompted us to investigate the practical utility of these gels in stimuli responsive delivery of the natural hydrophobic drug, curcumin. While forming hydrogel, we have encapsulated a hydrophobic drug, curcumin in the ratio of 2:1 and subsequently studied the drug release profile under acidic and basic pH. On to the curcumin encapsulated composite hydrogel formed from compound 7a in the ratio of 2:1 (ratio of gelator to curcumin load), we have added 15 mL of buffer solution and monitored their release profile using UV-vis spectrometer by withdrawing 1 mL supernatant solution at 10 minutes intervals. Fig. 3c–e reveals the absorbance of curcumin and drug release profile of gel under the influence of acidic and basic buffer respectively. Aliquot collected at 5 minutes time intervals displayed the absorbance of curcumin and an increase in absorption intensity with respect to time was also observed. It should be noted that acidic buffer as an external stimulus released yellow solution from the encapsulated gel, whereas the use of basic buffer generated light brown solution. However, there is no drastic change in λmax of released curcumin. Stimuli-responsive release of the encapsulated drug by acidic and basic buffer could be attributed to H-bonding triggered assembly and disassembly of monolayer fibres, which address the problems associated with the solubilisation of hydrophobic drugs and formulation of dual channel stimuli responsive delivery systems.
 | ||
| Fig. 3 (a) Images of (i) curcumin encapsulated gel formed by compound 7a, (ii–v) addition of buffer of pH 4.0 and inverted vial displaying the natural drug, curcumin release with respect to time respectively, (b) images of (i) addition of buffer of pH = 9.0 (ii–iv) pH responsive curcumin release after 10 and 30 minutes respectively, (c) UV-vis spectrum of curcumin dissolved in MeOH, (d and e) curcumin release profile of gel formed by 7a under the influence of acidic and basic buffer. | ||
Rheological analysis
A defined extent of material deformation from its original state in response to a force applied is generally narrated by rheological measurements. In particular, a deep insight into a flow and viscoelastic behaviour of a soft material, which belongs to a unique category neither behave like ideal solid and nor ideal liquid were obtained by measuring the storage modulus G′ (elastic) and the loss modulus G′′ (viscous) in response to the strain.32–37,50,51 Such an interesting deformation of a soft material in response to stress or strain is due to the internal morphology drew up with the supra-molecular arrangement of small or macromolecules and colloidal organic/inorganic particles. However, till to date there is no direct correlation were derived between the internal morphology and viscoelastic behaviour of soft materials, in particular, many of the questions related to the micro/nanostructure and viscoelastic behaviour remain unanswered. In order to realize the practical utility of these gels in medicine or drug formulation, a clear cut understanding of soft material rheology is needed. Rheological measurement of hydrogel formed by compound 6a and 7a is given in Fig. 4. Initially, the response of G′ and G′′ with respect to strain sweep and frequency sweep was investigated in detail. Strain amplitude sweep measurements revealed that within the range of linear viscoelastic region (LVE), G′ > G′′ suggesting the existence of gel like structure with good mechanical strength (Fig. 4a and c). Interestingly, in the entire range of frequency sweep test, the storage modulus G′ is greater than loss modulus G′′ clearly reveal the stability and good tolerance towards the external forces of the hydrogels formed by 6a and 7a (Fig. 4b and d). It is worth mentioning that the surface of the soft material derived from 6a and 7a form a slight depression or fold in its smoothness beyond the critical strain (γc) value of 0.83 and 0.35% and complete gel to sol transformation was observe at γ = 7.92%, and 4.8% respectively (Fig. 4a and c). Curcumin encapsulated composite hydrogel prepared from compound 7a displayed good stability, and no physical change were observed even after 120 days. When compared to hydrogel of 7a, composite hydrogel displayed an increase in critical strain (γc) value of from 0.35 to 1.5% and gel to sol transition strain from 4.8% to 12.1%, which clearly reveal the uniform intercalation of hydrophobic drug curcumin in the self-assembled fibrillar monolayer structure, thereby enhancing the mechanical behaviour (Fig. 4e). Nevertheless the storage modulus G′ of the hydrogel and composite gel of compound 7a is greater than 6a indicating the enhanced stiffness.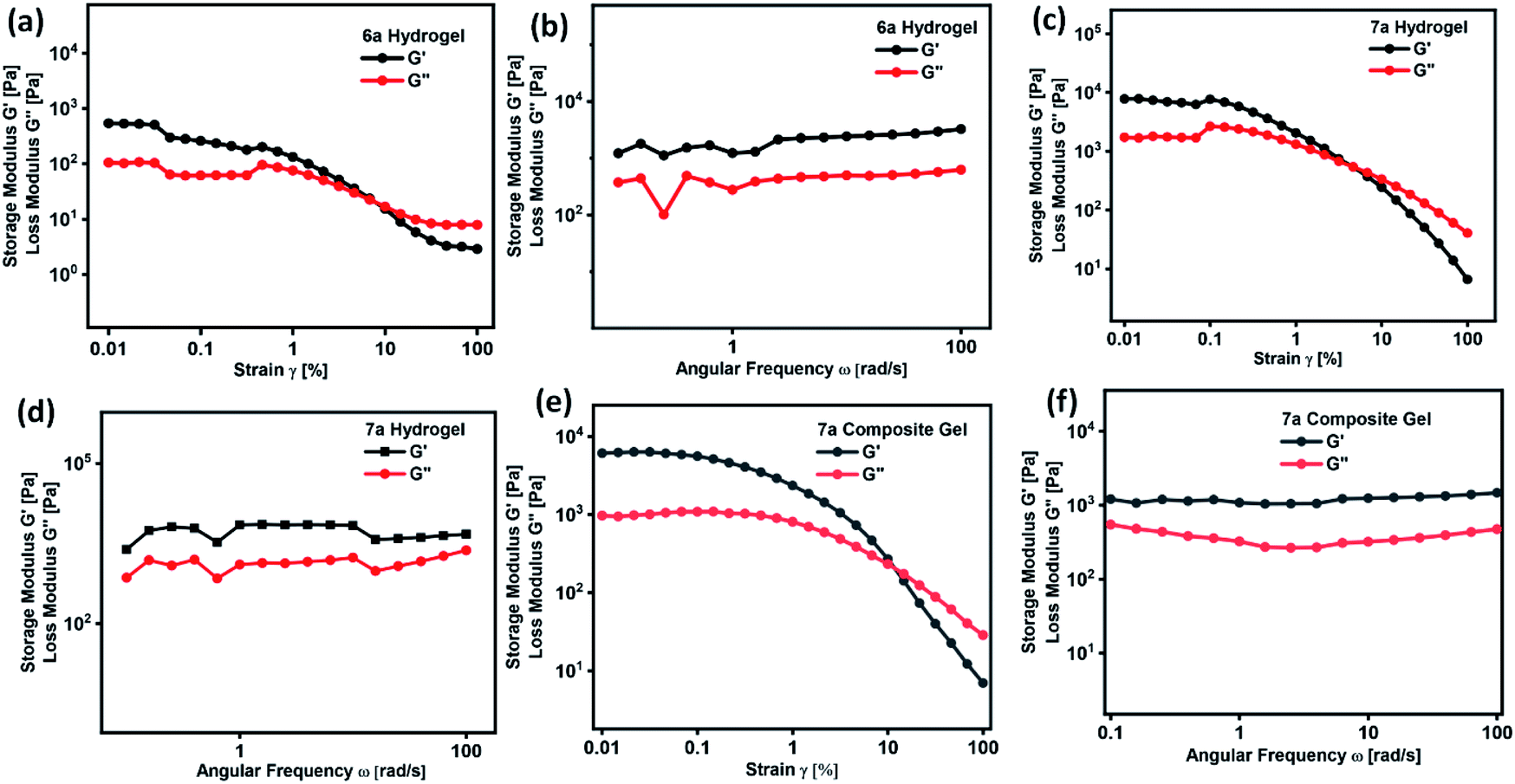 | ||
| Fig. 4 Strain amplitude and angular frequency dependence of storage (G′) and loss modulus (G′′) of hydrogel formed by (a and b) 6a, (c and d) 7a and (e and f) composite hydrogel of 7a respectively. | ||
In the development of materials science and technology, plenty of assembled systems derived from polymers, small molecules and inorganic nanoparticles were studied for their delivery of biotherapeutics. However, thermo-reversible hydrogels derived from renewable raw materials have received great attention because of their resemblance with living tissues and biocompatibility.52 In order to validate the thermal processability of hydrogel in the field of drug formulation, bioengineering and drug delivery, we have performed continuous temperature ramp-up and ramp-down experiments. Hydrogel and composite gel derived from 7a displayed good thermal stability and processability even after several temperature ramp up and down cycles (Fig. 5a and c). Thixotropic behaviour of hydrogels were studied by a continuous step-strain experiment (Fig. 5b and d). Upon the application of high strain (100%), storage modulus G′ is found to be lower than the loss modulus G′′ indicating the development of folds followed by the destruction of 3D-network in hydrogel. Once the strain is reduced to 1%, a higher G′ value is observed, which clearly reveal the rapid recovery of gel to its original structure by the reconstruction of fibrillar network (Fig. 5b and d). Thixotropic behaviour of hydrogels and composite gel were consistent even after 4 cycles of step-strain experiment.
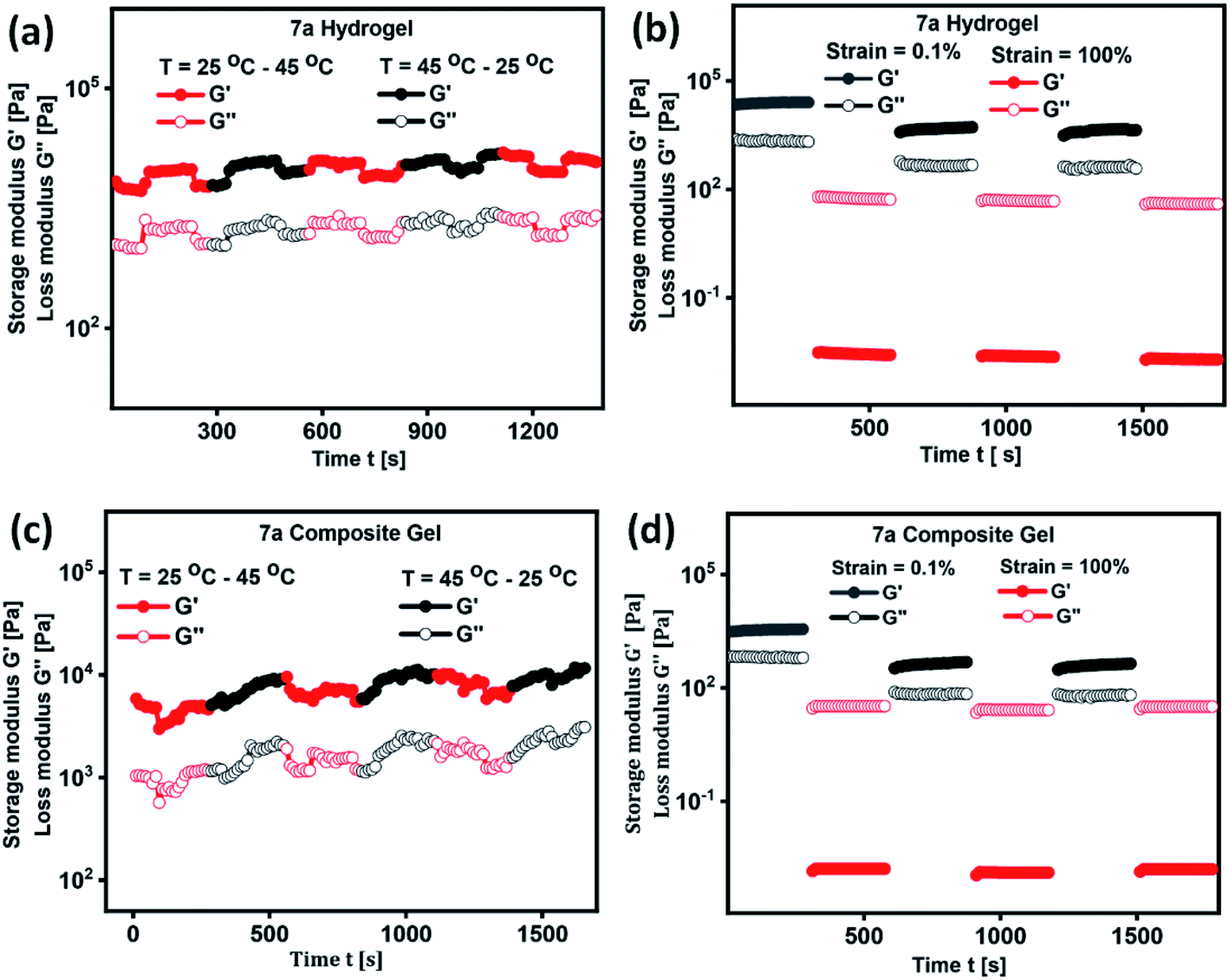 | ||
| Fig. 5 Temperature dependence and time course change of G′ and G′′ in the continuous step-strain measurements of (a and b) hydrogel of 7a and (c and d) composite hydrogel of 7a respectively. | ||
Conclusion
In summary, a set of three structurally similar amphiphilic-N-acyl amides were synthesized from renewable raw materials and biologically significant natural substrates and well characterized by NMR and mass spectral techniques. A simple protocol was adopted to synthesize amphiphilic N-acyl amides and the self-assembly of two interesting amphiphiles 6a and 7a furnished hydrogel and oleogel in vegetable oils. Morphological analysis of gels revealed the existence of lamellar and bundled fibrillar architecture. Interestingly, a fine tuning of hydrophilicity in N-acyl amides drastically change the self-assembly behaviour and morphology, which clearly reveal the existence of different degree of intermolecular interactions. Self-assembly mechanism has been proposed based on gelation studies, XRD and FT-IR techniques. Rheological studies clearly revealed the details related to the deformation of gel with respect to stress or strain, which is solely depend on the internal morphology drew up with the supra-molecular arrangement of gelators. The existence of thermoresponsive and thixotropic behaviour of these gels were established by rheological studies. Interesting hydrogel formed from 7a exhibited gel to sol transition in response to pH. In order to realize the practical application of hydrogel formed from 7a, a composite gel was prepared by the encapsulation of curcumin and gel–sol transition in response to pH were studied in detail. We envision that this dual channel responsive drug carrier could play a significant role in medical and pharmaceutical sciences, in particular for the development of advanced stimuli responsive drug delivery systems.Experimental section
Materials and general methods
All the essential reagents and solvents needed for the synthesis of N-acyl amides-based amphiphiles were purchased from Alfa Aesar, Sigma Aldrich, Merck, Avra chemicals, Loba, SRL and used as such without any further purification. LR grade solvents and distilled solvents were used when necessary. Thin-layer chromatography using pre-coated silica gel plates purchased from Merck was employed to monitor the reaction progress and visualized by UV detection or p-anisaldehyde stain or molecular iodine. Column chromatography was performed with silica gel (60–120 mesh) purchased from Avra chemicals. 1H- and 13C-NMR spectra for amphiphilic N-acyl amides and their precursors were recorded on a Bruker Avance 300 MHz instrument in CDCl3 at room temperature. Chemical shifts (δ) are reported in parts per million (ppm) using TMS as internal standard and coupling constants (J) are given in Hz. Proton multiplicity is assigned using the following abbreviations: singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). Electrospray ionization mass spectra (ESI-MS) were carried out in positive mode with a Thermo Fisher LCQ Advantage Max. instrument by dissolving the solid sample in methanol.Synthesis
Compound 6a. Isolated as white solid; melting point = 48–50 °C; yield = 82%; 1H NMR (300 MHz, CDCl3) δ = 7.22 (t, J = 7.8 Hz, 1H, Ar–H), 6.94 (s (br), 1H, –NH), 6.86 (d, J = 7.8 Hz, 1H, Ar–H), 6.75–6.70 (m, 2H, Ar–H), 4.51 (s, 2H, –O–CH2–), 3.65 (t, J = 5.6 Hz, 2H, –CH2–), 3.53 (q, J = 6.3 Hz, 2H, –CH2), 2.58 (t, J = 7.7 Hz, 2H, –CH2), 1.77–1.70 (m, 2H, –CH2), 1.62–1.54 (m, 2H, –CH2), 1.29–1.24 (m, 24H, –CH2), 0.88 (t, J = 6.9 Hz, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ = 169.6, 157.1, 145.1, 129.4, 122.3, 114.8, 111.6, 67.1, 59.3, 35.9, 35.8, 32.1, 31.9, 31.3, 29.7, 29.6, 29.6, 29.5, 29.3, 29.3, 22.6, 14.1; HRMS(ESI) m/z calculated for C26H45NO3 = 420.3477 [M + H]+; observed = 420.3482.
Compound 6b. Isolated as yellow semi-solid; yield = 78%; 1H NMR (300 MHz, CDCl3) δ = 7.22 (t, J = 7.7 Hz, 1H, Ar–H), 6.95 (s (br), 1H, –NH), 6.86 (d, J = 7.5 Hz, 1H, Ar–H), 6.74–6.70 (m, 2H, Ar–H), 5.39–5.33 (m, 2H, –CH
CH), 4.52 (s, 2H, –O–CH2), 3.65 (t, J = 5.7 Hz, 2H, –CH2), 3.53 (q, J = 6.3 Hz, 2H, –CH2), 2.58 (t, J = 7.8 Hz, 2H, –CH2–Ar), 2.09–1.96 (m, 2H, –CH2), 1.78–1.69 (m, 4H, –CH2), 1.65–1.57 (m, 4H, –CH2), 1.30–1.22 (m, 14H, –CH2), 0.88 (t, J = 6.6 Hz, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ = 169.5, 157.1, 145.0, 129.9, 129.7, 129.4, 122.3, 114.8, 111.6, 67.1, 59.3, 36.0, 35.9, 32.5, 32.0, 31.8, 31.7, 31.6, 31.3, 29.6, 29.5, 29.3, 29.1, 28.9, 27.2, 27.1, 22.6, 14.1.
Compound 7a. Isolated as white solid; melting point = 85–87 °C; yield = 72%; 1H NMR (300 MHz, CDCl3) δ = 7.69 (s, 1H, –NH–), 7.21 (t, J = 7.8 Hz, 1H, Ar–H), 6.86 (d, J = 7.8 Hz, 1H, Ar–H), 6.76–6.72 (m, 2H, Ar–H), 4.51 (s, 2H, –O–CH2CO), 3.99 (s, 3H, –OH), 3.69 (s, 6H, –CH2OH), 2.57 (t, J = 7.8 Hz, 2H, –CH2–Ar), 1.61–1.54 (m, 2H, –CH2), 1.30–1.06 (m, 24H, –CH2), 0.0.88 (t, J = 6.5 Hz, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ = 168.7, 155.6, 143.9, 128.2, 121.2, 113.8, 110.4, 66.0, 64.0, 61.0, 60.4, 51.0, 34.7, 30.6, 30.1, 30.0, 28.4, 28.4, 28.3, 28.2, 28.1, 28.0, 21.4, 12.8. HRMS (ESI) m/z calculated for C27H47NO5 = 466.3532 [M + H]+; observed = 466.3557.
Compound 7b. Isolated as yellow viscous liquid; yield = 70%; 1H NMR (300 MHz, CDCl3) δ = 7.69 (s, 1H, –NH), 7.22 (t, J = 7.5 Hz, 1H, Ar–H), 6.86 (d, J = 8.1 Hz, 1H, Ar–H), 6.78–6.72 (m, 2H, Ar–H), 5.40–5.30 (m, 2H, –CH
CH), 4.51 (s, 2H, –OCH2), 3.69 (s, 6H, –CH2–OH), 2.58 (t, J = 7.8 Hz, 2H, –CH2–Ar), 2.02–1.90 (m, 2H, –CH2), 1.81–1.57 (m, 6H, –CH2), 1.40–1.27 (m, 14H, –CH2), 0.88 (t, J = 6.5 Hz, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ = 167.8, 154.7, 142.8, 127.7, 127.5, 127.1, 120.2, 112.8, 109.5, 64.9, 59.8, 58.8, 33.7, 30.3, 29.6, 29.5, 29.5, 29.1, 27.4, 27.4,27.2, 27.1, 27.1, 26.7, 24.9, 20.4, 11.8.
Compound 8a. Isolated as a pale brown solid; mp: 65–67 °C; yield: 78%;1H NMR (300 MHz, CDCl3) δ = 7.20 (t, J = 7.8 Hz, 1H, Ar–H), 6.85 (d, J = 7.8 Hz, 1H, Ar–H), 6.78 (d, J = 8.1 HZ, 1H, Ar–H), 6.72–6.64 (m, 3H, 1H, Ar–H), 6.54 (d, J = 8.1 HZ, 1H, Ar–H), 4.46 (s, 2H, –OCH2), 3.54 (q, J = 6.6 Hz, 2H, –CH2–NH), 2.71 (t, J = 6.9 Hz, 2H, –NHCH2CH2–Ar), 2.57 (t, J = 7.8 Hz, 2H, –CH2), 1.59–1.50 (m, 2H, –CH2), 1.25–1.11 (m, 24H, –CH2), 0.88 (t, J = 6.6 Hz, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ = 169.2, 157.0, 145.1, 144.1, 143.0, 130.4, 129.5, 122.3, 120.7, 115.5, 115.3, 114.8, 111.6, 67.0, 40.5, 35.9, 34.9, 31.9, 31.3, 29.7, 29.6, 29.6, 29.5, 29.3, 22.7, 14.1. C31H47NO4 [M + H]+ = 498.3583; found 498.3622.
Compound 8b. Isolated as brown viscous liquid; yield: 75%; 1H NMR (300 MHz, CDCl3) δ = 7.16–7.10 (m, 1H, Ar–H), 6.80–6.58 (m, 5H, Ar–H), 6.48 (d, J = 8.4 Hz, 1H, Ar–H), 4.39 (s, 2H, –OCH2), 3.51–3.44 (m, 2H, –CH2–NH), 2.67–2.61 (m, 2H, –NHCH2CH2–Ar), 2.50 (t, J = 7.2 Hz, 2H, –CH2), 1.55–1.48 (m, 4H, –CH2), 1.22–1.13 (m, 18H, –CH2), 0.88–0.75 (m, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ = 169.4, 157.0, 145.1, 144.3, 143.1, 139.9, 130.4, 129.5, 122.4, 120.6, 115.6, 114.9, 111.6, 111.5, 67.0, 40.6, 35.9, 34.8, 31.9, 31.7, 31.4, 29.7, 29.6, 29.6, 29.5, 29.4, 29.3, 22.7, 22.6, 22.6, 21.4, 14.1; C31H45NO4 [M + H]+ = 496.3427; found 496.6.
:3 ratio) by the action of heating, which on subsequent cooling to room temperature furnish the gel. The formation of the gel is confirmed by “stable to inversion” method. The heating and cooling were repeated to check the thermo-reversibility and stability of these gels.35–42Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Science and Engineering Research Board, Department of Science and Technology, India (sanction order no: CRG/2018/001386) and SPARC, Ministry of Human Resource Development, India (SPARC/2018–2019/P263/SL) is gratefully acknowledged. The authors thank SASTRA Deemed University and National Institute of Technology Warangal for the infrastructure facilities. SN thank INYAS, INSA, Govt of India for membership.References
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- J. Zhou, J. Li, X. Du and B. Xu, Biomaterials, 2017, 129, 1–27 CrossRef CAS PubMed.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Mater. Today Adv., 2019, 3, 100021 CrossRef.
- E. R. Triboni, T. B. F. Moraes and M. J. Politi, in Nano Design for Smart Gels, Elsevier, 2019, pp. 35–69 Search PubMed.
- S. Panja, A. Panja and K. Ghosh, Mater. Chem. Front., 2021, 5, 584–602 RSC.
- M. C. Muñoz, G. Blay, I. Fernández, J. R. Pedro, R. Carrasco, M. Castellano, R. Ruiz-Garcia and J. Cano, CrystEngComm, 2010, 12, 2473–2484 RSC.
- D. Ghosh, M. T. Mulvee and K. K. Damodaran, Molecules, 2019, 24, 3472 CrossRef CAS PubMed.
- M. Frauenkron, J.-P. Melder, G. Ruider, R. Rossbacher and H. Höke, Ethanolamines and Propanolamines, in Ullmann’s Encyclopedia of Industrial Chemistry, 2001, vol. 31, pp. 405–431, DOI:10.1002/14356007.a10_001.
- L. Frkanec and M. Žinić, Chem. Commun., 2010, 46, 522–537 RSC.
- M. Çolak, D. Bariş, N. Pirinççioğlu and H. Hoşgören, Turk. J. Chem., 2017, 41, 658–671 CrossRef.
- M. Jokić, V. Čaplar, T. Portada, J. Makarević, N. Š. Vujičić and M. Žinić, Tetrahedron Lett., 2009, 50, 509–513 CrossRef.
- M. Löfman, J. Koivukorpi, V. Noponen, H. Salo and E. Sievänen, J. Colloid Interface Sci., 2011, 360, 633–644 CrossRef PubMed.
- A. Valkonen, M. Lahtinen, E. Virtanen, S. Kaikkonen and E. Kolechmainen, Biosens. Bioelectron., 2004, 20, 1233–1241 CrossRef CAS PubMed.
- M. Zaboli, H. Raissi, N. R. Moghaddam and F. Farzad, J. Mol. Liq., 2020, 301, 112458 CrossRef CAS.
- S. Zhang, J. Hou, Q. Yuan, P. Xin, H. Cheng, Z. Gu and J. Wu, Chem. Eng. J., 2020, 392, 123775 CrossRef CAS.
- S. Loganathan, Y. Guo, W. Jiang, T. Radovits, M. Ruppert, A. A. Sayour, M. Brune, P. Brlecic, P. Gude, A.-I. Georgevici, B. Yard, M. Karck, S. Korkmaz-Icöz and G. Szabó, Pharmacol. Res., 2019, 150, 104503 CrossRef CAS PubMed.
- N. Alipour and H. Namazi, Mater. Sci. Eng. C, 2020, 108, 110459 CrossRef CAS PubMed.
- S. Lee, S. Kim, J. Park and J. Y. Lee, Int. J. Biol. Macromol., 2020, 151, 1314–1321 CrossRef CAS PubMed.
- G. Zeng, K. Wei, D. Yang, J. Yan, K. Zhou, T. Patra, A. Sengupta and Y.-H. Chiao, Colloids Surf., A, 2020, 586, 124142 CrossRef CAS.
- J. Ding, H. Wu and P. Wu, J. Membr. Sci., 2020, 598, 117658 CrossRef CAS.
- Y. Lv, H.-C. Yang, H.-Q. Liang, L.-S. Wan and Z.-K. Xu, J. Membr. Sci., 2015, 476, 50–58 CrossRef CAS.
- H. Li, H. Lin, X. Wang, W. Lv and F. Li, ACS Appl. Mater. Interfaces, 2019, 11, 36469–36475 CrossRef CAS PubMed.
- S. Zhao, Z. Bai, B. Wang, T. Tian and Z. Hu, Sep. Purif. Technol., 2020, 241, 116633 CrossRef CAS.
- Z. Yan, Y. Zhang, H. Yang, G. Fan, A. Ding, H. Liang, G. Li, N. Ren and B. Van der Bruggen, Chem. Eng. Res. Des., 2020, 157, 195–214 CrossRef CAS.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- V. S. Balachandran, S. R. Jadhav, P. K. Vemula and G. John, Chem. Soc. Rev., 2013, 42, 427–438 RSC.
- J. R. Silverman, M. Samateh and G. John, Eur. J. Lipid Sci. Technol., 2016, 118, 47–55 CrossRef CAS PubMed.
- P. Anilkumar, Cashew nut shell liquid: A goldfield for functional materials, 2017 Search PubMed.
- T. G. Barclay, K. Constantopoulos and J. Matisons, Chem. Rev., 2014, 114, 10217–10291 CrossRef CAS PubMed.
- M. S. Behalo, E. Bloise, L. Carbone, R. Del Sole, D. Lomonaco, S. E. Mazzetto, G. Mele and L. Mergola, J. Exp. Nanosci., 2016, 11, 1274–1284 CrossRef CAS.
- L. Faure, S. Nagarajan, H. Hwang, C. L. Montgomery, B. R. Khan, G. John, P. Koulen, E. B. Blancaflor and K. D. Chapman, J. Biol. Chem., 2014, 289, 9340–9351 CrossRef CAS PubMed.
- Y. S. Prasad, S. Miryala, K. Lalitha, K. Ranjitha, S. Barbhaiwala, V. Sridharan, C. U. Maheswari, C. S. Srinandan and S. Nagarajan, ACS Appl. Mater. Interfaces, 2017, 9, 40047–40058 CrossRef CAS PubMed.
- K. Lalitha, V. Sridharan, C. U. Maheswari, P. K. Vemula and S. Nagarajan, Chem. Commun., 2017, 53, 1538–1541 RSC.
- G. P. Sarvepalli, D. K. Subbiah, K. Lalitha, S. Nagarajan and J. B. B. Rayappan, J. Mater. Sci.: Mater. Electron., 2020, 31, 1594–1603 CrossRef CAS.
- K. Lalitha, Y. S. Prasad, C. U. Maheswari, V. Sridharan, G. John and S. Nagarajan, J. Mater. Chem. B, 2015, 3, 5560–5568 RSC.
- K. Lalitha and S. Nagarajan, J. Mater. Chem. B, 2015, 3, 5690–5701 RSC.
- K. Lalitha, Y. S. Prasad, V. Sridharan, C. U. Maheswari, G. John and S. Nagarajan, RSC Adv., 2015, 5, 77589–77594 RSC.
- K. Lalitha, K. Gayathri, Y. Prasad, R. Saritha, A. Thamizhanban, C. Maheswari, V. Sridharan and S. Nagarajan, Gels, 2017, 4, 1 CrossRef PubMed.
- Y. S. Prasad, B. Saritha, A. Tamizhanban, K. Lalitha, S. Kabilan, C. U. Maheswari, V. Sridharan and S. Nagarajan, RSC Adv., 2018, 8, 37136–37145 RSC.
- A. Thamizhanban, S. Balaji, K. Lalitha, Y. Prasad, R. Vara Prasad, R. Arun Kumar, C. U. Maheswari, V. Sridharan and S. Nagarajan, J. Agric. Food Chem., 2020, 68, 14896–14906 CrossRef CAS PubMed.
- O. A. Zhikol, S. V. Shishkina, V. V. Lipson, A. N. Semenenko, A. V. Mazepa, A. V. Borisov and P. V. Mateychenko, New J. Chem., 2019, 43, 13112–13121 RSC.
- E. R. Draper and D. J. Adams, Langmuir, 2019, 35, 6506–6521 CrossRef CAS PubMed.
- J. Wang and X. Yan, Nano/Micro-Structured Materials for Energy and Bio-medical Applications 2018, pp. 205–226 Search PubMed.
- B. Hu, W. Sun, B. Yang, H. Li, L. Zhouu and S. Li, AAPS PharmSciTech, 2018, 19, 2288–2300 CrossRef CAS.
- V. R. Aldilla, R. Chen, A. D. Martin, C. E. Marjo, A. M. Rich, D. S. Black, P. Thordarson and N. Kumar, Sci. Rep., 2020, 10, 770 CrossRef CAS PubMed.
- A. Pasc, F. O. Akong, S. Cosgun and C. Gérardin, Beilstein J. Org. Chem., 2010, 6, 973–977 CrossRef CAS PubMed.
- B. Adhikari, G. Palui and A. Banerjee, Soft Matter, 2009, 5, 3452–3460 RSC.
- C. A. Baladasare and P. G. Seybold, J. Phys. Chem. A, 2021, 125, 3600–3605 CrossRef PubMed.
- N. Goyal, H. P. R. Mangunuru, B. Parikh, S. Shrestha and G. Wang, Beilstein J. Org. Chem., 2014, 10, 3111–3121 CrossRef PubMed.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- V. M. P. Vieira, L. L. Hay and D. K. Smith, Chem. Sci., 2017, 8, 6981–6990 RSC.
- H. Huang, X. Qi, Y. Chen and Z. Wu, Saudhi Pharm. J., 2019, 27, 990–999 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08447j |
| This journal is © The Royal Society of Chemistry 2022 |