
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying surface degradation, mechanical failure, and thermal instability phenomena of high energy density Ni-rich NCM cathode materials for lithium-ion batteries: a review
Fikadu Takele Geldasa
 a,
Mesfin Abayneh Kebede
*bd,
Megersa Wodajo Shura
a and
Fekadu Gashaw Hone
*c
a,
Mesfin Abayneh Kebede
*bd,
Megersa Wodajo Shura
a and
Fekadu Gashaw Hone
*c
aAdama Science and Technology University, Department of Applied Physics, P. O. Box 1888, Adama, Ethiopia
bEnergy Centre, Smart Places, Council for Scientific and Industrial Research (CSIR), Pretoria 0001, South Africa. E-mail: mkebede@csir.co.za
cAddis Ababa University, Department of Physics, P. O. Box: 1176, Addis Ababa, Ethiopia. E-mail: fekeye@gmail.com
dMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg 2050, South Africa
First published on 16th February 2022
Abstract
Among the existing commercial cathodes, Ni-rich NCM are the most promising candidates for next-generation LIBs because of their high energy density, relatively good rate capability, and reasonable cycling performance. However, the surface degradation, mechanical failure and thermal instability of these materials are the major causes of cell performance decay and rapid capacity fading. This is a huge challenge to commercializing these materials widely for use in LIBs. In particular, the thermal instability of Ni-rich NCM cathode active materials is the main issue of LIBs safety hazards. Hence, this review will recapitulate the current progress in this research direction by including widely recognized research outputs and recent findings. Moreover, with an extensive collection of detailed mechanisms on atomic, molecular and micrometer scales, this review work can complement the previous failure, degradation and thermal instability studies of Ni-rich NMC. Finally, this review will summarize recent research focus and recommend future research directions for nickel-rich NCM cathodes.
 Fikadu Takele Geldasa | Fikadu Takele was born in Welega, Ethiopia, in 1993. He received his BSc degree in Applied Physics from Hawassa University in 2015 and his MSc degree in Solid State Physics from Dilla University, in 2017. Currently he is a PhD student at Adama Science and Technology University under the supervision of Dr Fekadu Gashaw, Dr Mesfin Abayneh and Dr Megersa Wodajo. His research interest is on energy storage electrode materials specifically on lithium ion batteries. |
 Mesfin Abayneh Kebede | Mesfin Kebede obtained his PhD in Metallurgical Engineering/Applied Physics from Inha University, South Korea. He is currently a Principal Research Scientist at the Energy Centre of Council for Scientific and Industrial Research (CSIR), South Africa. In addition, he holds a Honorary Associate Professor position at the University of the Witwatersrand, South Africa. His current research mainly focuses on the development and characterization of electrode materials for energy storage and fabrication of coin cells and pouch cells for full cells and improving the electrochemical performance of electrode materials for energy storage systems such as lithium-ion batteries, sodium-ion batteries, and supercapacitors. |
 Megersa Wodajo Shura | Megersa Wodajo Shura earned his BSc degree in Physics at Asmara University, Eritrea in 1990. After serving at different institutions for a while, he joined Addis Ababa University for his MSc Study in 1997. Then, he joined Adama Teachers College until he moved to Adama Science and Technology University in September 2006. Then, he joined Nelson Mandela University, South Africa for his PhD study in March 2009. Upon completion of his PhD, he returned to ASTU and served as an Assistance and Associate Professor of Physics. His research interest is dealing with the various optoelectrical properties of inorganic semiconductors. |
 Fekadu Gashaw Hone | Fekadu Gashaw is an Associate Professor of Materials Physics and is currently working at Addis Ababa University Department of Physics as a researcher and lecturer. He obtained his first degree in Physics from Debub University in 2001 and his MSc degree from AAU in Laser Spectroscopy (2006). He got his PhD degree from KNUST (Ghana) (2015). Fekadu was also a postdoctoral researcher at the University of the Free State, (South Africa) from 2016 to 2020. His research mainly focuses on the development and characterization of novel nanomaterials for various applications such as energy storage and conversion, gas sensors and antimicrobial activities. |
1 Introduction
Today's electricity generation and transportation depend heavily on fossil fuels, thus becoming the two major sources of CO2 emissions that lead to global warming.1 Ecofriendly renewable energy sources such as wind and solar must be increasingly used to reduce or eliminate fossil fuel utilisation.2,3 The energy produced by these renewable energies must be stored and due to their power and energy density greatly exceeding those of other battery systems lithium-ion batteries (LIBs) are the most preferred energy storage devices.4–6 Compared to conventional secondary batteries, LIBs are advantageous in terms of low environmental pollution, high energy density, good rate performance, and long cycle life.7 LIBs have been used successfully in a variety of applications, from portable electronics and electric vehicles (EVs) to energy storage systems.8 Fig. 1a9 shows the relationship between specific energy density and volumetric energy density for the different rechargeable battery technologies. Specifically, the general goal of battery development technology is to increase energy and power densities, while minimizing volumetric and mass constraints (i.e., move to the upper right of Fig. 1a).9 | ||
| Fig. 1 (a) Diagram comparing the rechargeable battery technologies as a function of volumetric and specific energy densities. The arrows indicate the direction of development to reduce battery size and weight.9 (b) Schematic diagram of the charge/discharge principle of a LIB cell.10 | ||
Historically, the lithium-based rechargeable battery was first demonstrated in the 1970s, which was proposed by M. S. Whittingham while working at Exxon in 1976.11 Apparently, LIBs have two different current collectors, two electrodes (anode and cathode), and the separator/electrolyte main components, and Whittingham's battery was made with layered TiS2 as the cathode and Li metal as the anode. The positive cathode worked well, whereas the lithium metal anode was shown to have uneven dendrite growth during charging and discharging.11 Apparently dendrite is the main enemy of LIBs, it could penetrate the separation layer and reach the opposite electrode, resulting in a short circuit and a potential fire hazard.12 The dendrite growth of Whittingham's battery was proved difficult to solve, and the commercialization of such type of batteries became a failure. Because of this safety problem, researchers have increasingly proposed for both electrodes that can accommodate ions. In 1980 John B. Goodenough and his co-workers at Oxford University, UK, discovered the layered LixCoO2 cathode materials.8,9,13 Due to the absence of suitable anode materials and electrolytes that match the discovered positive electrode, it took nearly ten years to commercialize LIBs.7 That is, the lack of safe anode materials limited the application of layered oxide cathode LiCoO2 in LIBs.14 In 1980, Rachid Yazami examined lithium intercalation compounds in graphite and discovered the graphite anode.15
In the early 1980s, Akira Yoshino conceived the LIB and he completed a practical prototype in 1986.16 He replaced the negative electrode with carbon that provided greater capacity without causing decomposition of the electrolyte. The secondary battery that he successfully fabricated consist carbon coke as anode and LiCoO2 as cathode and enabled stable charging and discharging over many cycles for a long period. Yoshino also carried out the first safety test on LIBs to validate their enhanced safety features, by dropping iron lumps on the battery cells, in contrast to that of metallic lithium batteries which caused fire and explosion. Based on these effective efforts, the first commercial LIB was released by Sony company in 1991.17 LIB commercialized by Sony is still governing of the electronic market today.7 Moreover, the commercialization of LIB was successful by the effort of the three scientists Whittingham, Goodenough, and Yoshino. To acknowledge their pioneering contribution, they were awarded the 2019 Nobel Prize in Chemistry.18
(Fig. 1b)10 shows the working principle of the LIBs cell consisting of graphite as an anode and layered lithium oxide as cathode materials. The full cell battery undergoes both discharge and charge processes. On charging, Li-ions are moving from the cathode to anode through electrolyte and electrons flow through an external circuit to anode. On discharging, electrons are moving from the anode through the copper current collector and, simultaneously, Li-ion extracted from the anode into the electrolyte and migrates toward the cathode. Then, the removed electron enters the cathode from an external circuit through the aluminum current collector. Discharging process is a spontaneous process, whereas charging needs external power sources and is non-spontaneous.
Currently, LIBs have been successfully applied in portable electronics, but researchers need them to electrify the world. However, conventional LIBs could not meet the required capacity to electrify the world.19 Especially, compared to the anode material, the cathode material capacity is lower, which greatly hinders the further development of LIBs.20 Therefore, to improve energy storage materials, researchers around the world are trying to improve the capacity of existing cathode materials and discovering new cathode materials. More recently, LIBs have been considered to be the most promising energy storage devices for electric vehicles (EVs) and have been used by Nissan (Leaf) and Tesla to manufacture electric vehicles.19,20 For the sake of environmental protection and energy conservation, electric vehicles are considered as the future means of transportation, which do not emit pollutants and can efficiently utilize energy.21,22 Despite their environmental advantages, electric vehicles have limited popularity due to inferior battery performance and higher cost compared to vehicles with internal combustion engine.20 For more development of EVs and their market-dominant, LIBs having high energy density, long life, good safety, fast charging, and low price are needed. In this sense, EV batteries require cathode materials with high energy density to achieve these properties because the most commonly used anode, graphite, can deliver a much higher specific capacity (372 mA h g−1) than available cathodes.19 Because of their high energy density, Ni-rich cathode materials are promising next-generation batteries both for the improvement of portable electronics and for the development of EVs. Ni-rich cathodes can be either nickel–cobalt–manganese (NCM) or nickel–cobalt–aluminum (NCA) which has similar crystal structures.23 However, these cathode materials are commercialized currently still there are remaining challenges in cycle life, surface degradation and thermal stability that hindered them from widely used for EVs. Thus, LIBs and LIB-based systems can fail catastrophically causing fire and/or explosion, while it is important to gain an understanding of the energetics and dynamics of such failure causes to mitigate their future occurrence.24 So, before trying to solve these challenges, it is more important to understand the mechanisms of these challenges. Many review papers have been published on the degradation mechanisms of Ni-rich cathode materials. However, to the best of our knowledge, there is no reported review article emphasized on the well organized and identified surface degradation, mechanical failure and thermal instability of Ni-rich NCM cathode materials, specifically including upper cutoff voltage. The challenges discussed in this review are also dependent on each other but not extremely identified by scientific investigation. In this paper, we present a comprehensive review to summarized related research out puts on NCMs challenges as well as how either of the three degradation mechanisms focused in this review affect one another. Hence, we believe that this review could provide an overall understanding for researchers interested in the currently hot issue of the degradation mechanisms of Ni-rich cathode materials and finding well-developed mitigation strategies for these challenges.
2 Cathode materials
Based on the discovery of John B. Goodenough and successfully charged/discharged over many cycles, LiCoO2 became the primary successfully used positive electrode in LIBs. It has excellent electrochemical performance; however, it poses some problems, such as safety, environmental hazard, and high cost, because of the limited availability of cobalt. During charging, when half of the Li-ions are removed from the host structure, the structural stability of LiCoO2 rapidly deteriorates and researchers improved its cycle performance by limiting the cut-off voltage, which delivers only about 140 mA h g−1 capacity, which is half of the theoretical capacity.7 Other researchers have approximated this value for commercial LiCoO2 to 165 mA h g−1 out of a high theoretical specific capacity of 274 mA h g−1, due to its significant structural instability and severe capacity fading at voltages greater than 4.35 V vs. Li/Li+.25 Also, it could not meet the required energy and power density, especially, for electric vehicles. To solve these issues, Co must be partially or fully replaced by cheap and non-toxic elements. Additionally, the electrochemical performance, price, and safety of LIBs are mainly dependent on cathode materials, it is important to look for materials with high energy density, cheap, and safe.21,22After the discovery of the layered structure LiCoO2 in 1980, other positive electrodes such as the LiMn2O4 spinel structure in 1986 and the LiMPO4 (M = Fe, Mn, etc.) olivine family in 1997 have been discovered.26 The spinel-phase LiMn2O4 is the lowest cost, environmental friendliness, and naturally abundant materials, but it exhibits the problem of Mn2+ ion dissolution, which causes the capacity fading and limits its development.27 Olivine LiFePO4 is another positive electrode material with low cost, excellent electrochemical properties, great thermal stability, and non-toxicity.7 This positive electrode has low Li-ion diffusion, poor conductivity, and poor charge/discharge rate performance. Many researchers have endeavored to resolve these problems, but the discharge platform of LiFePO4 is approximately around 3.4 V and its theoretical specific capacity is 170 mA h g−1.28 Including the aforementioned positive electrodes different layered materials such as LiNiO2, LiMnO2, etc. have been discovered.29 LiNiO2 has the same structure as LiCoO2 and high energy density and low cost; however, at high temperature or high voltage, it exhibits poor structural stability and low cycle reversible, which limits its development.27 The structure and potential profiles of the olivine, spinel, and different layered cathode materials during discharge are shown in (Fig. 2a).30 Because of the drawback of all of the aforementioned cathode materials, the development of LIBs continued and many improvements and alternatives have been reported. Researchers around the world have endeavored to develop a further increasing energy density and reducing the cost of electrode materials, particularly cathode materials, which are still a limiting factor on the total energy density of LIBs and occupy a great weight of LIB cell components (Fig. 2b).31,32 They are mainly aimed at discovering a rechargeable battery with high specific capacity, lower cost, environmental friendliness, and fast charging for transportation and energy storage in addition to for portable electronics.33 Recently, the layered oxide materials, which consist of ternary transition materials, are becoming promising cathode materials. Although single component layered cathode materials, including LiNiO2 and LiMnO2, also suffer from intrinsic problems such as poor cyclability and rate capability, as well as complexity in preparation, layered oxide nickel–cobalt–manganese (NCM) or nickel–cobalt–aluminum (NCA) exhibited promising electrochemical properties, which vary depending on the composition of transition metals (TMs) in the structure.34 Layered oxide nickel–cobalt–manganese (NCM) is a very sensitive area of today's LIBs studies. They can be represented as LiNixCoyMnzO2 with different compositions of TMs. TMs in NCM cathode materials play different roles in terms of crystal structure and electrochemical properties. Generally, Ni provides the majority of the reversible capacity by converting Ni2+ to Ni3+/Ni4+, while Co offers good electronic conductivity and reinforces layered ordering with improved rate capability and additional capacity derived from the Co3+/4+ redox reaction (by converting Co3+ to Co4+). Furthermore, Mn stabilizes the local structure to achieve steady cycling performance, although Mn remains electrochemically inert in the tetravalent state during the charge–discharge process.27 Consequently, the intrinsic structure and performance of LiNixCoyMnzO2 strongly depend on the compositional ratio of Ni, Co, and Mn.19
 | ||
| Fig. 2 (a) Li-ion battery cathodes: important formulae, structures, and potential profiles during discharge30 (b) chart showing the weight fraction occupied by the components of a commercial lithium-ion battery cell.31,32 (c) A map of relationship between discharge capacity, and thermal stability and capacity retention of Li/Li[NixCoyMnz]O2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85).35 (d) Comparison of the capacity retention after 100 cycles achieved by tailoring Umax.36 | ||
After Ohzuku and Makimura reported LiNi1/3Co1/3Mn1/3O2 (NCM111), NCM-based LIBs have become the mainstream with gradual improvements in NCM technology through the steady increase in nickel content in each generation of cathode materials.37 Compared to LiCoO2, LiMn2O4, and LiNiO2, NCM-based materials have outstanding electrochemical properties, such as high energy density, high reversibility, good environmental compatibility, and excellent charge/discharge rate.38 For Ni-rich cathode materials, the nickel content in NCM-based materials exceeds Co and Mn content, which is above 50%.27,28 The theoretical capacity of conventional cathode materials is 170 mA h g−1 for olivine LiFePO4, 140 mA h g−1 for spinel LiMn2O4, 140–160 mA h g−1 for layered LiCoO2 and these materials are limited by their insufficient capacity, which cannot meet the increasing requirements for practical applications.39 The main advantage of Ni-rich materials is their high discharge capacity (200–220 mA h g−1), which represents a large increase in energy density (∼800 W h kg−1) as compared to conventional LiCoO2 (∼570 W h kg−1) and LiMn2O4 spinel (∼440 W h kg−1) materials.28,33 In addition, the enhancement of the capacity of LIBs can be achieved by increasing the Ni content. In the composition of NCM cathode materials, as the content of Ni increases, the capacity of the cathode materials also increases. For example, the Ni-rich LiNi0.6Co0.2Mn0.2O2 has a high specific capacity ∼170 mA h g−1.40 When the Ni content increases to LiNi0.8Co0.1Mn0.1O2 a higher specific capacity of 200 mA h g−1 is achieved.41
Nevertheless, as Ni content increases in layered Ni-rich oxide, some intrinsic problems, such as poor capacity retention and low thermal stability, are revealed. Hyung-Joo Noh et al.35 have investigated the electrochemical and thermal properties of the composition of LiNixCoyMnzO2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85) as a function of the Ni content. They identified that there is a nearly linear decrease in thermal stability and capacity retention as the discharge capacity increases by increasing the relative fraction of Ni in the LiNixCoyMnzO2 electrodes. As shown in (Fig. 2c),35 among the tested compositions; the 1/3 cathode exhibited the best capacity retention and thermal stability; however, its discharge capacity was limited due to the low Ni content in the composition. However, the 85% Ni content cathode exhibits severe capacity fading and the worst thermal stability in spite of the large discharge capacity, while the 1/3 electrode showed the highest safety. However, Lea de Biasi et al.36 have found that Ni-rich NCMs show better capacity retention than those with a low Ni content compared with Hyung-Joo Noh et al. in (Fig. 2d),36 which was achieved by tailoring cut-off voltage (Umax), although they have a structural instability. In addition, poor cycle life due to structural instability upon long-term cycling, particularly under high charge voltage (>4.3 V),42 high defect concentration, and insufficient C-rate performance become more prominent.27 Not only these, the sensitivity to ambient moisture, the formation of micro-cracks15,16 and micro-strain16,18 during cycling, and the thermal instability upon delithiation, which cause capacity fading of Ni-rich oxides have been revealed. Parasitic reactions,43 cation mixing by leading to restructured surface regions,44 active material dissolution,45 and oxygen release46 are the primary factors responsible for the cathode surface degradation mechanism. The high charge voltage would lead to a severe reaction of the Ni-rich surface with the electrolyte, forming a thick cathode-electrolyte interface (CEI) layer to increase the interfacial resistances of electrodes. In addition, highly oxidized Ni3+/4+ ions at deep charge are easily destabilized and reduced as Ni2+ ions, resulting in cation migrations to form surface reconstruction layers consisting of spinel-like and/or NiO-like rock salt phases. This surface structure transition considerably increases the kinetic barrier for Li-ion diffusion, leading to capacity degradation.42 The improved outstanding energy density of Ni-rich cathode material is good while improving cathode structural and thermal stabilities thus safety issues and application range have become the focus of the subsequent research. Many efforts based on different strategies such as dopants, gradient layers, surface coatings, carbon matrixes, and advanced synthesis methods have been devoted so far to improve the electrochemical performance of layered Ni-rich oxide cathode material.39,47 However, it is very important to understand the degradation mechanisms in order to find effective strategies for the aforementioned challenges and to making better batteries.
3 Challenges in NCM materials
To achieve a sufficiently high specific capacity at a given maximum cut-off voltage, the Ni content in the composition of NCM-based cathode materials must be continuously increased. Thus, in LIBs, energy densities can be significantly increased by the inclusion of more Ni content in the NCM cathode materials.48 However, due to surface-related chemical degradations and mechanical failures, Ni-rich NCM cathode materials lead to poor cycling stability.49 The unprotected surface of Ni-rich NCM cathode materials can easily react with electrolyte during battery cycling, causing irreversible structure transformation, composition modification, and surface passivation layer formation, which contribute to capacity decay, and also act as a blocking layer, causing voltage fading. In addition to surface degradation, mechanical failures, such as crack-induced material disintegration, are the major causes of cell performance decay. Mechanical cracks can lead to fragmentations of active materials, which cause poor electronic conduction and exposure of fresh surfaces to electrolytes. Moreover, battery degradation can be occurred during charging, discharging and at rest.50 At high cut-off voltage, these challenges can be caused, and therefore we start our discussions from the upper cut-off voltage and we will continue to the identification of these challenges and their mechanisms.3.1 Upper cutoff voltage
High energy density, high power density, low cost, and longer lifetime are required for LIBs to be widely applied in portable electronics, grid storage systems, and electric vehicles. High energy density can be achieved by increasing the upper cutoff voltage in NCM-based LIBs.20,46 Although the theoretical capacity of NCM is as high as ∼275 mA h g−1, not all of the Li+ can be removed due to the structural instabilities that form when deep Li is removed.44 For deep Li+ removal, the upper cutoff voltage must be increased. However, increasing the upper cut-off voltage can increase undesirable side reactions at the electrode/electrolyte interface, surface film formation, and metal dissolution, which ultimately decrease the battery lifetime.51 For these reasons the operating potential of NCM based cathode materials is limited to approximately 4.3 V; this results in their capacity being much below their theoretical capacity.Having an improved energy density is the very interesting and required one in LIBs. Lea de Biasi et al.36 have proposed two major strategies to improve the energy density of LIBs using NCM cathode active materials. The first is to increase the content of Ni, which allows larger amounts of Li to be extracted at a given cut-off voltage (Umax). The other is to increase Umax, for medium Ni content NCM active materials. They have performed electrochemical tests on NCM111, NCM523, NCM622, NCM721, NCM811, and NCM851005 using coin-type cells with a lithium metal counter electrode. The highest Ni content is the more Li extracted, hence the highest the specific capacity. They have also observed that gravimetric energy density (GED) increases with both increasing Ni content and Umax.
The origin of faster capacity fading at high cutoff potentials is a complex,52 however, the H2–H3 phase change is reported as the origin of capacity fading at a high cutoff potential. The advantages, challenges, and origin of the challenges in increasing the upper cutoff in Ni-rich NCM materials are shown in (Fig. 3).
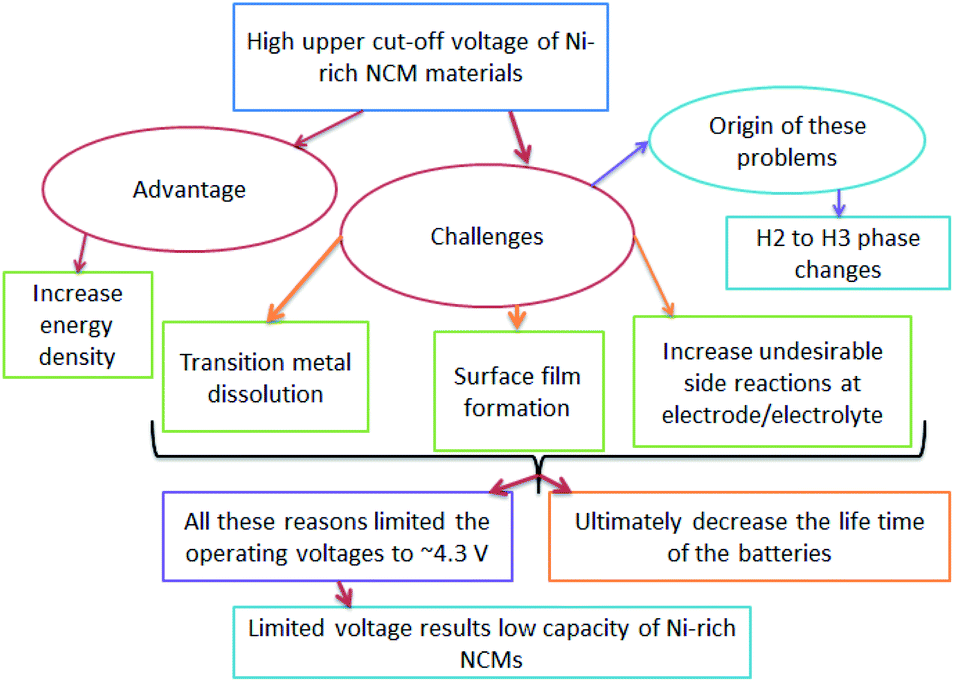 | ||
| Fig. 3 The flow of the advantages, challenges and origin of increasing upper cut-off voltage in Ni-rich NCM cathodes. | ||
Researchers have investigated the effects of increasing and decreasing of upper cut-off voltages for different Ni-rich NCM cathode materials. Yanli Ruan et al.47 have investigated the electrochemical performance of NCM622 at 4.2 V, 4.5 V, and 4.8 V. They have observed that as the upper cutoff voltage increases the charge/discharge capacity also increases, whereas the capacity retention decreases. The cycling stability of this NCM622 active material strongly depends on the cutoff voltage. The discharge voltage plateaus are almost the same in the first discharge whereas decreases after 100 cycles with increasing upper cut-off voltages. After 100 cycles, the discharge capacity decays as the upper cutoff voltage increases. For 4.2 V, all the capacity degradation after 100 cycles is attributed to the increase in cell impedance, implying that the structure of the NMC622 material can be well maintained in low-voltage cycling.
The effects of increasing the upper cut-off voltages on electrochemical properties of Ni-rich NCM cathodes are dependent on the aggregation modes of the primary particles.53 At a high cutoff voltage, a significant contraction and/or expansion of the lattice parameter vector may exist, which results in the changes in lattice volume. The nonuniform accommodation of such a volume change will generate severe local strain, which might result in mechanical failure and inter-granular cracks.
The charge/discharge cycle of Ni-rich NCM-based materials at high upper cutoff voltage can also cause phase transformations. Sung-Kyun Jung et al.34 have proposed the degradation mechanisms of NCM523 at high voltages by comparing 4.5 V and 4.8 V cutoff voltages (see (Fig. 4)).34 Cycling at 4.5 V, the electrode surface suffers from a phase transformation mainly to the spinel phase, with a trace of rocksalt phase formation. When the cutoff voltage increases to 4.8 V, then the highly oxidative environment exhibits the formation of the rocksalt phase. As can be seen in (Fig. 4), the rocksalt phase encircles both the rhombohedral and spinel phases, increasing the impedance of the electrode. Roland Jung et al.44 have compared the electrochemical performance of the NCM111, NCM622, and NCM811 electrodes at different cutoff voltages and they concluded from those results as cutoff voltages increase, the capacity increases, and when Ni content increases capacity also increase with increasing cut-off voltages. The average discharge voltages for both NCM111 and NCM622 are very similar. As the Ni content increases to NCM811, due to the less stable cycling behavior of NCM811, the upper cutoff voltage is limited to low voltages and displays a continuously decreasing mean voltage cathode discharge value, even at the lowest cutoff voltage. Therefore, from this report result, as Ni content increases the onset potential decreases.
 | ||
| Fig. 4 Degradation mechanisms of LiNi0.5Co0.2Mn0.3O2 and phase transformation after cycle tests under high-voltage conditions.44 | ||
A differential capacity plot of the delithiation and lithiation of the three NCM materials in NCM-graphite cells of the 3rd cycle is represented in (Fig. 5).44 This plot investigates the origin of the difference and the reason for the instability for NCM111 and NCM622 at 4.6 V and for NCM811 at 4.1–4.2 V. The voltage region up to 3.8 V is stable for all NCMs, with two anodic peaks between 3.4 V and 3.8 V, which originates from the lithium insertion into the anode and the phase transition from a hexagonal to a monoclinic (H1 → M) lattice of the NCM54 respectively for the two anodic peaks. In the region above 3.8 V, the differential capacity curve for the NCM811 cell deviates from that of the NCM111 and NCM622 cells. NCM811 has a small anodic feature at ∼3.95 V, which belongs to the M → H2 phase transition, and a large anodic peak at ∼4.15 V, which belongs to the H2 → H3 phase transition,48 both of which are not present for the other NCMs. On the contrary, such deviations are not observed for NCM111 and NCM622; however, for NCM622 a broad peak is observed around 4.1 V, which could indicate an M → H2 phase transition. For both NCM111 and NCM622, a clear redox peak is observed at 4.6 V, which could belong to an H2 → H3 phase transition or could also indicate an oxygen redox feature, in analogy to NCM811. The capacity retention of these NCMs cathode active materials is very stable up to the onset of the H2 → H3 phase transition of NCM811 at above 4.0 V and up to the onset of the redox feature at above 4.4 V of NCM111 and NMC622. Therefore, stable cycling is only possible if the cutoff voltage is below the onset of the last peak in the plot. NCM811 cannot be cycled stably at above 4.0 V cutoff voltages due to the early onset of the H2 → H3 phase transition at above 4.0, whereas NCM111 and NCM622 cells show outstanding performance at potentials as high as 4.4 V. Multiphase transitions of hexagonal to monoclinic (H1 → M), monoclinic to hexagonal (M → H2), and hexagonal to hexagonal (H2 → H3) during charging was also reported by Hyung-Joo Noh et al.35
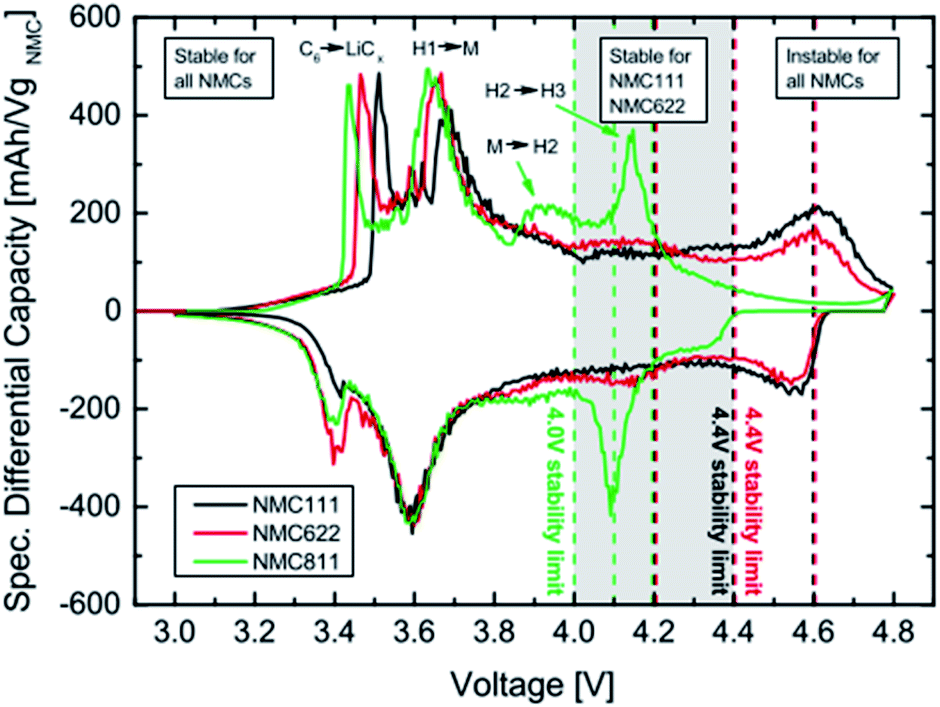 | ||
| Fig. 5 Differential capacity vs. cell voltage of NCM-graphite cells recorded at a 0.1 C-rate (3rd cycle). The vertical dotted lines mark the upper cutoff voltages. The peaks are assigned to their corresponding phase transitions with H1, H2 and H3 representing the three hexagonal phases and M the monoclinic one. C6 → LiCx indicates the lithiation of graphite.44 | ||
3.2 Li+/Ni2+ cation mixing
Cation mixing is the main reason for the capacity decline of the Ni-rich NCM cathode active materials.29,55 As the Ni content in the NCM layered material increases the capacity will increase, which is the result of two-electron of Ni2+/Ni4+ redox reaction but, increasing the Ni content results in a significant degree of mixing of Ni2+ and Li+ cations due to the closeness of their ionic radius (Li+ ∼0.076 nm and Ni2+ ∼0.069 nm).48,49,55 In charged Ni-rich layered materials Ni3+/Ni4+ are unstable and can easily be reduced to Ni2+.50,54 Because of their ionic radius similarity, the low energy barrier Ni2+ migrates from the sites in the transition metal layer to the available Li layer sites when the Li-ion is removed from Li layer sites during charging and promotes the Li+/Ni2+ cation mixing. Thus, during discharge, Li+ occupies the site of the available transition-metal layer. During migration, the Li+ in the transition metal cannot be easily be extracted and the existence of Ni2+ in the Li layer blocks the diffusion of Li+, resulting in both capacity loss and low rate performance.48,51,52 In particular, a large proportion of Li+/Ni2+ cation mixing can lead to increased strain and hinder the diffusion of lithium ions.56 In addition to decreasing the diffusion rate of Li-ion, Li+/Ni2+ cation mixing also reduces the amount of Li-ion participating in the charge/discharge reaction.The structural instability can result from the disordered Li+/Ni2+ cation mixing in NCM cathode materials. The Ni2+ in the Li position can cause the local structure collapse and accelerate the phase transition, hence, reducing the electrochemical performances of Ni-rich NCM materials.57 Thus, the Li+/Ni2+ cation mixing is related to the phase transformation, which may be performed by the kinetic mechanism and the thermodynamic driving force.58 In this process, there are diffusion pathways, which can be divided into two separate processes: the atom-dominant and cell dominant process. Atom-dominant processes are related to Li and Ni atom exchange, in which Li-ions first migrate to a neighboring tetrahedral site, and the Ni-ion located below the tetrahedral Li site moves to the opposite tetrahedral site on the Li layer, forming a so-called Li–Ni dumbbell structure. In the cell-dominant process, Li and Ni ions migrate to a local minimum, forming a distorted five-fold square pyramid and finally, the square pyramids and the transition metal oxide octahedra slightly shuttle relative to each other in order to match the spinel lattice.59 The Li+/Ni2+ cation mixing can be present either during the materials synthesis process or during electrochemical cycling. The similarity of the ionic radius size of Ni2+ and Li+ is the main reason for the formation of Li+/Ni2+ cation mixing during the synthesis process. Li+/Ni2+ exchange during synthesis is related to the layered to spinel transition, in which a Li–Ni dumbbell structure formation at high voltage. Even though such layered-to-spinel phase change is blocked by the cell-dominant pathway, Li–Ni cation mixing can be present as a side effect. Not only during synthesis process the Li+/Ni2+ cation mixing can also occur during electrochemical cycle. In this case, Ni2+ ions first migrate to the tetrahedral positions of the Li layer (dumbbell) during charge and then move to the octahedral positions of the Li layer (Li+/Ni2+ exchange) during discharge. The detailed mechanism of Li+/Ni2+ exchange during electrochemical cycling is as follows: during charge, the enlarged interlayer distance facilitates the diffusion of Ni2+ to the Li layer, forming a dumbbell structure, especially at the lowest energy barrier for dumbbell formation. That is, the anti-site Ni2+ reduces the Li layer space, which decreases the Li+ diffusion.38 Ni2+ ions will then remain in the Li layer during the rest of the charging process owing to their high diffusion barrier in the Li layer. During discharge, Li-ions are inserted back into the Li layer and as well as into the now available Ni positions in the TM layer. The low energy required for Li+/Ni2+ cation mixing at high Li concentration facilitates this process. After one charge/discharge cycle, only a certain amount of Li+/Ni2+ cation mixing is present in the structure. However, as the c lattice parameter increases with Ni concentration, it can be inferred that NCM cathode materials with high Ni content suffer from more severe Li+/Ni2+ cation mixing, which supports the large increase in Li+/Ni2+ cation mixing after repeated charge/discharge cycles.59 Li-ion in the transition metal layer has also a large activation energy barrier of Li-ion diffusion for the extraction of Li-ion from a transition metal, resulting in capacity fading of materials.60,61 Gradually, the inverse Ni2+ in the Li layer can move to the surface of particles, leading to the depletion of Ni2+ in bulk. In other word, the anti-site Ni2+ can migrate to the material surface and destroys the stability of the structure, resulting in rate performance degradation and capacity fading.38 Thus, the cationic mixing region on the particle surface is composed of NiO-like inactive phases, which worsen Li+ diffusion and introduce thermal instability.62
The calcination temperature has also effects on the existence of Li+/Ni2+ cation mixing. Hubert Ronduda et al.63 have reported the effects of calcination temperature on LiNi0.6Mn0.2Co0.2O2 cathode materials and from PXRD results at a higher temperature, this material obtained its well-ordered structure. However, materials calcined at higher temperatures shows a higher Li+/Ni2+ cation mixing, thus deteriorating their electrochemical properties.
According to studies by many researchers, the degree of Li+/Ni2+ cation mixing can be identified from the XRD peak analysis.61 In particular, the XRD peak of the Ni-rich NCM ratio of the intensities 003 and 004 peaks can determine the existence of Li+/Ni2+ cation mixing. When the ratio of I(003)/I(004) is less than 1.2 it shows the degree of cation mixing.64 The process of this ratio reduction is as follows: when a cation disorder is generated, TM-ions occupy the lithium position, which leads to a partial destructive interference of (003) plane's constructive interference at the Bragg angle of this peak by decreasing the intensity of (003) peak whereas, the intensity of (104) peak increases because TM-ions in Li-site also exist on the (104) plane.43 In the XRD peak of Ni-rich NCM materials, the splitting degree of (006)/(012) and (018)/(110) peaks are important, which indicates whether the crystal materials are highly crystalline or not. The clear splitting of (006)/(102) and (018)/(110) peaks shows a high degree of layered structure crystallization.65 As the Ni fraction in NCM increases, the peak splitting of (006)/(012) and (018)/(110) becomes unclear, indicating that the structure's disorder degree increased. If the ratio of I(003)/I(104) showing degree of cation mixing and the peak of (018)/(110) have been combining, the situation of serious cation mixing is occurred leading to some changes of the cell lattice parameters.64 Controversially, researchers also reported that the Li+/Ni2+ cation mixing has a positive effect on Ni-rich layered cathode materials, therefore the ordered mixing of Li+ and Ni2+ can promote the structural stability of the cathode material.66 Un-Hyuck Kim et al.67 reported that the ordered intermixing of the Li and Ni ions structure in Ni-rich layered cathode materials. This ordered structure has the advantages of protecting the particle surface from structural degradation, reducing the voltage and capacity decay of cathode materials, and stabilizing the structure during cycling. Similarly, other studies have suggested that ordered Li+/Ni2+ exchange may be beneficial to the electrochemical performance of batteries. These studies also show: (1) Li+/Ni2+ exchange can mitigate the slab-distance contraction at high states of charge, thereby stabilizing the structure. (2) Although Li+/Ni2+ exchange restricts the diffusion of Li-ions, it benefits the thermal stability of high-Ni NCM materials. Based on the above understandings, a balance between the negative and positive influences should be carefully considered so that an optimal degree of Li/Ni exchange for high-Ni NCM materials can be achieved in order to obtain the best electrochemical performance.68
3.3 Phase transformation
The crystal structures of the layered NCM cathode materials have a general formula LxMO2, which has a close-packed oxygen framework with the alkali L and the transition metal M cations filling alternating layers of interstitial sites.21,28,39 It have an α-NaFeO2 structure with an R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group,7,40,69 which is a repeating O3 structure of oxygen–lithium–oxygen–transition metal–oxygen–lithium–oxygen–transition metal–oxygen along the rhombohedral as shown in (Fig. 6a),43 where the L-ions occupy the site of 3a, M-ions occupy 3b, and O ions occupy 6c. The layers filled by M cations form an MO2 slab consisting of edge-sharing MO6 octahedral. The L cations, which can be shuttled between the MO2 slabs, can occupy octahedral. During the charge, the type of O3 may change to the O1 and the structure of the cathode will transform from the Rm space group to disorder Fdm space group then to the Fmm space group in the end via the successional change of oxygen during cycling70 (Fig. 6b). In addition, for lithium diffusion, the distance of slabs should become small to obtain a higher activation energy barrier.
m space group,7,40,69 which is a repeating O3 structure of oxygen–lithium–oxygen–transition metal–oxygen–lithium–oxygen–transition metal–oxygen along the rhombohedral as shown in (Fig. 6a),43 where the L-ions occupy the site of 3a, M-ions occupy 3b, and O ions occupy 6c. The layers filled by M cations form an MO2 slab consisting of edge-sharing MO6 octahedral. The L cations, which can be shuttled between the MO2 slabs, can occupy octahedral. During the charge, the type of O3 may change to the O1 and the structure of the cathode will transform from the Rm space group to disorder Fdm space group then to the Fmm space group in the end via the successional change of oxygen during cycling70 (Fig. 6b). In addition, for lithium diffusion, the distance of slabs should become small to obtain a higher activation energy barrier.
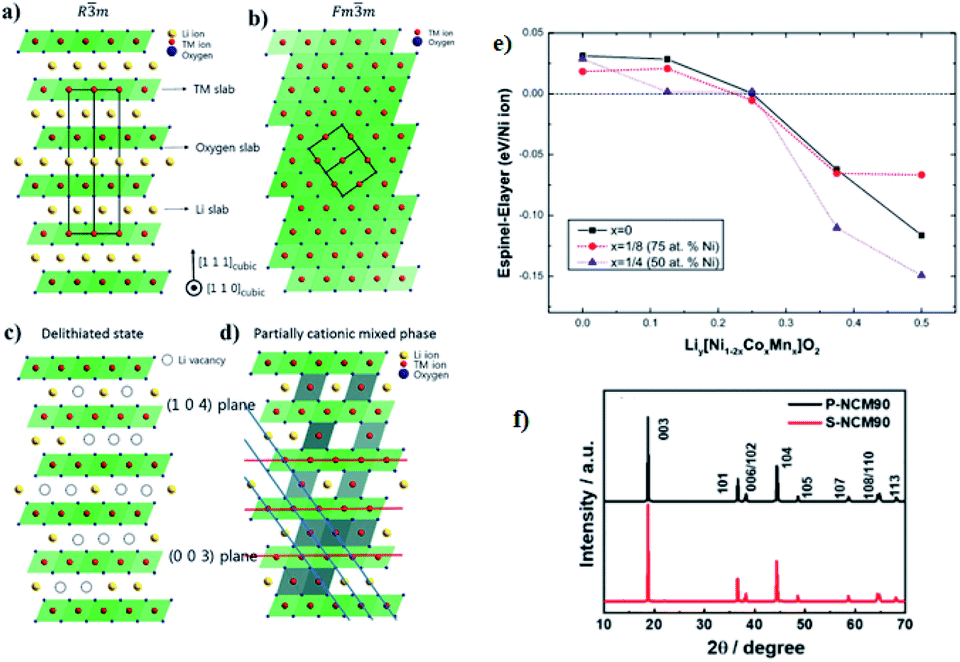 | ||
| Fig. 6 [Illustration of the ordered and disordered phase in layered lithium metal oxides and their structural transformation. (a) Well-ordered Rm structure; (b) the cation disorder or cation mixing phase with Fmm structure; (c) Rm structure with Li vacancies in highly charged state; (d) partially cation mixed phase with TM-ions in Li slab. Li atoms yellow, transition metals red, coordinated oxygen atoms dark blue].43 (e) The relative stability between spinel and layered structure of LiyNi12xCoxMnxO2 (x = 0, 1/8, and 1/4) as a function of Li concentration.59 (f) XRD patterns of S-NCM90 and P-NCM90.71 | ||
During the process of removal of Li-ions from the host structure, NCM materials undergo the phase transition, which involves the cation disordering between TM site (octahedral 3a site) and Li site (octahedral 3b site) by forming Fmm structure as shown in (Fig. 6b).42,70,72,73 In a highly delithiated state, Ni-rich cathode material has an extremely unstable structure due to lithium vacancy (Fig. 6c), and this instability leads to TM-ion migration from the TM layer site to the Li layer site by forming spinel like phase (Fig. 6d).43 In other words, during the removal of Li-ion, Ni-rich NCM materials undergo a series of phase transitions: the original layered structure (H1) transforms to the monoclinic phase (M), the second hexagonal phase (H2), and the third hexagonal phase (H3).74 The difficulty to maintain all Ni-ions at the trivalent state and the ionic radius similarity of Ni2+ and Li+ cations results in cation disorder phenomenon leading to an irreversible phase transition from a layered structure to a rock salt phase, indicating a greater Li-ion diffusion barrier and severe capacity fading.75 This phase transition is also accompanied by the release of oxygen gas, which is an irreversible process and seriously deteriorates the performance of electrodes.41 Specifically, the phase transformation of the layered to rock salt-like phase during repeated lithiation/delithiation causes structural degradation.62 The chemical formula for the phase transition with the release of oxygen is as follows:73
|
3MO2 (Rm) → M3O4 (Fdm) + 2[O]
| (1) |
|
M3O4 (Fdm) → 3MO (Fmm) + 2[O]
| (2) |
In Ni-rich NCM layered materials, phase transformation is usually observed at high voltages but not in bulk NCM.76,77 The layered to spinel phase transition in NCM is reversed after discharge.78 We can easily investigate this reversible phase transition from the thermodynamics of layered-to-spinel phase transition in NCM cathode materials by comparing the total energy of the layered and spinel phases. Chaoping Liang et al.59 have investigated this by comparing the total energy of the two-phase in LiyNi1−2xCoxMnxO2 as observed in (Fig. 6e)59 by using the first-principle calculations within the density functional theory with Hubbard parameter (DFT + U) framework. According to their study, when half of the Li ion (y = 0.5) is extracted from the host structure, the spinel phase is thermodynamically more favorable. However, the layered phase is thermodynamically more stable when y < 0.25 which indicates that the layered-to-spinel phase transition is reversible and the reversible layered-to-spinel transition point would be at y = 0.25. The experimental observation has also supported the fact that the layered-to-spinel phase transition can be reversible during electrochemical cycles.79 Therefore, the layered-to-spinel phase transformations in Ni-rich NCM are often not severe to the electrochemical performance. The spinel phase has much higher voltage than that of its layered phase, which is used to prevent the dissolution of TM ions in the highly delithiated state, and it can be used as a coating material for NCM.80
The phase transition from layered-to-rock salt phase is very serious for the electrochemical performance of Ni-rich cathode materials. Accordingly, after discharge, the phase change on the surface of Ni-rich NCM particles always needs to be reversed.59 However, the rocksalt phase changes occurring on the surface particles are always not reversible and it seriously affects the electrochemical performance of the materials.81 This phase will be accumulated at the electrode/electrolyte interface, forming a NiO surface film. Thus, the resulting NiO surface-film impedes the transport of Li+, and Li+ is irreversibly consumed in its formation.48 The reduced Ni2+ greatly accelerates the formation of an irreversible phase transition from the layered to NiO rocksalt phase.82 At high charge voltage, the deep lithium removal results in enough lithium vacancies and it leads to accelerates this phase transition process, exhibits structural degradation, and causes decreasing of voltage continuously.83 Phase transition causes stacking defects, which increase the diffusion kinetic energy barrier of Li-ion and cause the difference of the diffusion rates of Li-ion in various directions as they pass via the stacking defects.84 The existence of a disordered rock-salt-like structure not only hinders the intercalation of Li+ during discharging for the occupation of TM ions at the Li+ site, leading to severe irreversible capacity loss, but also increases the energy barrier for Li+ diffusion due to its smaller distance between slabs, resulting in a low Li+ diffusion rate.85 The formation of the phase transition in Ni-rich NCM materials may also depend on the cycling rate in addition to involves the Li+/Ni2+ cation mixing. Lianfeng Zou et al.86 have demonstrated that varying the cycling rate changes phase transition products in Ni-rich LiNi0.76Co0.10Mn0.14O2 by using scanning transmission electron microscopy (STEM) and atomic simulation. Fig. 7 shows their pictorial representation of the structure evolution of Ni-rich LiNi0.76Co0.10Mn0.14O2 materials at low and high rates. Varying with the cycling rate, the as-synthesized product layered structure undergoes a variety of depths of discharge upon cycling. The LiNi0.76Co0.10Mn0.14O2 cathode that cycled at a high rate i.e. 2C is generating vacancies, yet accompanied by a large fraction of Li+ remaining in the layered structure which results limited number of Ni2+ to migrate into the lithium position causing spinel phase (Fig. 7d). In contrast, LiNi0.76Co0.10Mn0.14O2 materials with a slow discharge rate, i.e., below 1C, offer the opportunity for adequate draining of Li-ions. The low rate results the disordered rocksalt phase changes (Fig. 7e).
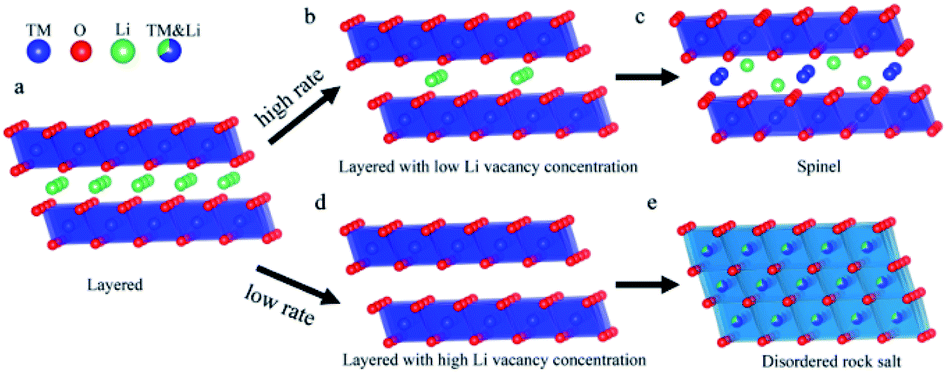 | ||
| Fig. 7 Pictorial illustration of the phase transition occurring at distinct cycling rates. (a) The starting layered structure. (b) The delithiated state under the high cycling rate, creating limited amount of Li vacancies. (c) The structure evolution toward the spinel structure. (d) The delithiated state under the low cycling rate, generating significant amount of Li vacancies. (e) Formation of disordered rock salt structure.86 | ||
The surface degradation of the layered structure of the cubic NiO rocksalt is also accompanied by oxygen evolution and heating. Bak et al.87 have investigated structural changes and gas evolution during the thermal decomposition of charged Ni-rich cathode materials by combining in situ time-resolved X-ray diffraction (TR-XRD) and mass spectroscopy (MS). For structural changes, they reported that the phase transition from the rhombohedral (space group Rm) to disordered spinel starts at 194 °C. This disordered spinel phase gradually transformed to the rocksalt phase as the temperature increased from 275 °C to 500 °C. Due to the irreversible structural degradation of large surfaces, materials are at risk of thermal runaway and loss of active materials.88 From the XRD patterns results, many researchers have proved that the Ni-rich cathode materials can be identified as either it is layered or not. Recently, Hoon-Hee Ryu et al.71 have studied that the powder XRD patterns of both single-crystalline Ni-rich NCM with Ni content 90% (S-NCM90) and polycrystalline NCM90 (P-NCM90) and they observed that as shown in Fig. 6f, which both have a hexagonal α-NaFeO2-type structure belonging to the Rm space group and without impurities detection.
According to some research studies, the orderly accumulated rocksalt on the particle surface can improve the cyclic and thermal stability of materials. Yuefeng Su et al.89 have reported an improvement of the cycling stability of Ni-rich LiNi0.8Co0.1Mn0.1O2 cathode materials by fabricating the surface rock salt phase. They have shown that the fabrication of Ni-rich materials with a surface rock salt phase results in better structural stability and electrochemical performances, in which the rearrangement of the irreversible structure leads to the passivation of the rocksalt phase on the surface and leads to loss of electronic conductivity and structural instability of the materials.72
3.4 Microcrack generation
The structural degradation of the NCM particles and the electrode due to volumetric changes of the rhombohedral structure upon repeated delithiation/lithiation of the NCM crystallites is one of the proposed failure mechanisms of Ni-rich NCM materials.90 Thus, the volumetric changes of the rhombohedral structure causes the microcracks in Ni-rich cathode material that categorized as mechanical failure and largely cause the rapid capacity fading of Ni-rich layered cathodes in the deeply charged state. Microcracks increase the exposed internal surface by creating crack faces and serve as channels through which the electrolyte can penetrate the particle interior.91 Subsequent degradation of the exposed internal surfaces through parasitic electrolyte attack accelerates the accumulation of NiO-like insulating layers at the cathode/electrolyte interfaces that inhibit lithium diffusion. The NiO-like insulating layers accumulated at the cathode/electrolyte interface forms the cathode electrolyte interface (CEI), which is analogous with the solid electrolyte interface (SEI) formed at the anode/electrolyte interface.91,92 As a result, the first formed CEI will be disrupted and then be rebuilt new one by consuming active lithium from the electrolyte.29,44Furthermore, penetration of electrolytes through cracks not only accelerates the formation of the CEI layers, but also causes voltage decay.93 The stable CEI helps for the performance of cathode, but the continuous formation on the newly formed surface of active materials due to crack formation will cause the capacity decline.94 From most of the previous studies for investigation of particle cracking, the electrodes should have to be harvested from the cycled cells and prepared and analyzed by Kr-BET, FIB-SEM, or TEM, which are difficult and furthermore require a large number of repeat analyses to be statistically significant. Stefan Oswald et al.90 have introduced a novel method based on electrochemical impedance spectroscopy (EIS) in blocking conditions to quantify the increase in the active material's surface area upon cycling, utilizing the correlation between the surface area of the electrode and the electrochemical double-layer capacitance that is validated experimentally by comparing the capacitance and BET surface area increase of NCM electrodes upon mechanical compression. To quantify the cracking of the particles after 200 charge/discharge cycles, they perform in situ EIS measurements utilizing a micro-reference electrode and monitor the cathode's impedance response and found surface area increment of up to ∼261%.
Microcracks in cathode particles originate from the local strain build-up caused by their highly anisotropic contraction and expansion during Li+ intercalation/de-intercalation.95 Thus, Li+ intercalation/de-intercalation behavior during charging and discharging of Ni-rich cathode materials can cause significant volume changes, resulting in micro-strain and crack formation of particles, which further leading to the internal and interstitial splitting of crystals.34,76 Lead de Biasi et al.36 have reported the increasing of the volume change of the NCM unit cell with the nickel content by using operand X-ray diffraction combined with detailed Rietveld analysis. They correlated energy density with X-ray diffraction results and they found that irrespective of the Ni contents, all NCM materials are subjected to structural changes during operation. The change of the unit cell is due to the contraction and expansion of both a-axis and the c-axis lattice parameters, which results in changes in the c/a ratio upon repeated charge/discharge that are accompanied by severe stress and strain. This leads them to the assumption that increasing the Ni content results in major challenges in terms of mechanical strain and structural degradation.
Simon Schweidler et al.96 investigated mechanical degradation and fatigue of Ni-rich NCM cathode material (NCM851005) in graphite-based full cells by using galvanostatic charge/discharge tests, electrochemical impedance, XRD and electron microscopy. They found that microcracks formation can be increased by increasing the number of cycles. Fig. 8a–f shows that the SEM examines morphological changes of the primary and secondary particle levels. Particles morphology after 100 and 500 cycles are shown in (Fig. 8c–f) and more fractured secondary particles are observed, some of them having a flattened structure at the top surface.86,96 These particles suffered from cracking due to electrochemical cycling. Finally they suggested that the capacity fading is associated to some with the mechanical degradation of the cathode material. Moreover, anisotropic volume changes during delithiation/lithiation impart stress into the material, leading to particle fracture.
 | ||
| Fig. 8 Top view and cross-sectional SEM images of the NCM851005 cathode in discharged state (a and b) before cycling and (c and d) after 100 and (e and f) 500 cycles at 1C and 45 °C.96 | ||
Hoon-Hee Ryu et al.,97 have also reported, the capacity fading of Ni-rich NCM cathodes, especially above x = 0.8, where x is the Ni content in the NCM composition, largely due to the anisotropic volume change, which is caused by the phase transition. Repeated, nonuniform contraction and expansion during cycling generated internal microcracks that propagated to the particle surface, opening channels for electrolyte infiltration into the particle interior. General speaking, when the content of Ni in the NCM composition increases, this phenomenon becomes more evident.98
The general consequences of particle cracking include: loss of electrical contact NCM active particles, conductive additives and current collectors; increased the growth rate of CEI layer; and electrode pulverizations. All the above phenomena suffer a decrease in electrochemical performance and hinder the widely commercialization of Ni-rich materials.99 Thus, currently the cracking issue has been identified as a major degradation mechanism.
3.5 Electrolyte decomposition
One of the most important issues in Ni-rich cathode materials is the surface damage caused by the parasitic reaction between the cathode material and the organic electrolytes.91 The cathode surface damage causes the capacity decay for full battery cells, which accelerates the electrolyte decomposition. The electrolyte decomposition is more severe the degradation of the cathode materials surface structure, increasing cathode surface impedance. That is, the degradation of the cathode material surface caused by electrolyte erosion is the main reason for the increase in charge resistance during cycling.79 High temperature and high state of charge are the root cause for the decomposition of electrolyte. The battery capacity will gradually decline and the material's structure will change as these side reactions occur, and it will become more severe at high operating voltage.19 At the high state of charge (>4.5 V), Ni2+ will be oxidized to highly reactive Ni4+ form, which reacts with the organic electrolyte, releasing O2 and forming a NiO-like phase.100 Thus, the poor cycling performance is partial because the increased amount of unstable Ni4+ formed during the charging process strongly reacts with the electrolyte and leaves more side products.101 This causes depletes the electrolytes and propagates the undesirable thick cathode electrolyte interface (CEI) layer at the interface, thus increasing the impedance of NCM materials.73,100 This is because the labile Ni4+ species are prostrate to transform to more stable chemical states (Ni3+or Ni2+) by reduction, the electrolyte can be promptly decomposed at the electrolyte/electrode interface, thereby affording electrons to Ni4+.102 The unstable Ni4+ during highly delithiated can cause surface instability. For example, if LiPF6 is used as an electrolyte, it can be decomposed into LiF and PF5 under the high catalytic activity of Ni4+. From the results of the decomposition, LiF play great role in the formation CEI layer and increases the cathode impedance, while PF5 reacts with H2O to form acid HF.88,89 Furthermore, the electrolyte LiPF6 salt is unstable in the presence of H2O molecules and, at high temperature or high operation voltage, is decomposed by the following eqn (3) and the produced PF5 react with water by forming the very dangerous acid that can attack the active materials surfaces eqn (4):69,96| LiPF6 → LiF + PF5 | (3) |
| PF5 + H2O → POF3 + 2HF | (4) |
Furthermore, due to the existence of Ni4+, the surface structure of the material is unstable, and it is easy to be eroded by hydrofluoric acid in the electrolyte during high voltage charging, which makes the capacity of LIBs decay faster.103 Hence, continuous reactions between highly delithiated cathode materials and electrolytes result in structural instability, increasing interfacial resistance, and rapid capacity fading.39
As discussed in Section 3.4 CEI can be formed due to the crack formation of the active materials, in which once crack is formed, new CEI will be formed on each newly formed surface of active materials. Additionally, as discussed in this section, CEI can be formed on the Ni-rich cathode materials by the oxidation of electrolytes.50 Ikuma Takahashi et al.94 have used hard X-ray photoelectron spectroscopy (HAXPES), soft X-ray absorption spectroscopy (S-XAS), and density functional theory (DFT) analyses, to investigate the formation of CEI on Ni-rich cathode surfaces and electrolyte oxidation mechanisms. For the formation of CEI, they have proposed two electrolyte oxidations: (i) oxidation electrolyte solvent which forms Li2CO3 and alkyl carbonates on Ni-rich NCM and when electrolytes proceed continuously during charge/discharge cycles the amount of Li2CO3 and alkyl carbonate increases; (ii) oxidation of electrolyte salt, which produces LixPOyFz and its amount increases during the continuous charge/discharge cycles. Then the oxidizing of the electrolyte solvent and salt continues by Ni-rich cathode, and forms continuously the CEI that consists of Li2CO3, alkyl carbonate, and LixPOyFz during the charge/discharge cycles. From their S-XAS and DFT studies, for the Ni-rich cathode at the delithiated state, they have indicated that the energy of the antibonding hybrid orbital of the TM 3d–O 2p corresponds to the LUMO energy level of the hole for the Ni-rich cathode at the charged state. Thus, the LUMO energy level decreases from the discharged to the charged state and lies close to the HOMO energy level of the electrolyte, and becomes very close to Fermi level as shown in (Fig. 9). They have also observed a high increase in hole concentration. The closeness of the LUMO energy level of a Ni-rich cathode to the HOMO energy level of the electrolyte and the high hole concentration of the Ni-rich cathode produce oxidation of the electrolyte, forming a thick CEI for the Ni-rich cathode.
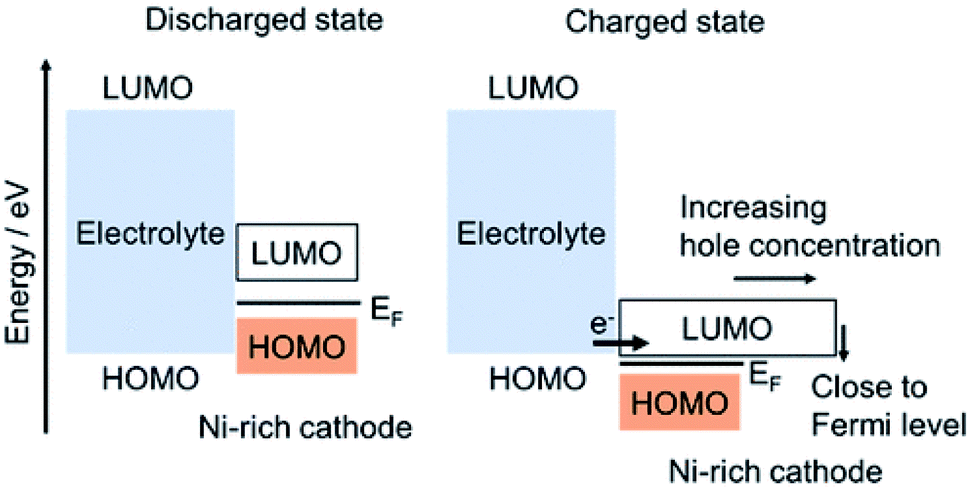 | ||
| Fig. 9 The HOMO and LUMO of electrolyte and Ni-rich cathode forming oxidation of electrolyte and CEI.94 | ||
3.6 Transition metal dissolution
The transition metals (TMs) dissolution also causes the capacity degradation of Ni-rich NCM cathode materials. The root cause of transition metal dissolutions are the high state of charge and high temperature.104 Transition metal dissolution is ascribed to a combined effect of the cation mixing and oxygen evolution reaction, in which the cation mixing accompanies the entire life of the battery while the oxygen evolution occurs mainly in the H3 phase. On the one hand, the cation mixing and the oxygen evolution reaction result in the formation of low-valence transition metal oxide (MO) by the loss of oxygen.73 The gas evolution of CO2 and CO is accompanied by the formation of H2O. As displayed in eqn (5), the formed H2O hydrolyzes the LiPF6 resulting HF (eqn (4)) and then HF reacts with low valence transition metal oxide (MO) which is occurred by the loss of oxygen, resulting in the collapse of the surface structure and the rapid decay of the reversible capacity:27| HF + MO → H2O + MF2 | (5) |
In other word, these acid species react with NCM cathode active materials, resulting in transition metal dissolution ions. These dissolute TMs may include Ni2+/Ni3+/Ni4+, Mn3+/Mn4+ and/or Co3+/Co4+ and they react with electrolyte to form MF2, resulting deposition of MF2 onto the NCM surfaces to form thick layer.50
Electrolyte decomposition also causes the formation of fluoride (F−) in the cell, which seriously accelerates the dissolution of transition metal components from the Ni-rich NCM cathode materials via chemical reactions and these reactions further accelerated at elevated temperature.105 The volume change of Ni-rich cathode materials during the charge/discharge cycles results in lattice parameter distance alter and produces microcracks generation inside or between the primary particles.106 The produced microcracks increase the contact area between the particle interior and the electrolyte, which ultimately accelerates the degradation of the cathode material and results in TMs dissolution.107 The dissolution of transition metals is deposited on the surface electrodes especially on the surface of the anode which hinders the diffusion of Li-ions, resulting in a decrease in the electrochemical performance of the battery.108 When the migration and deposition of dissolved metal ions on anode increases, Li-ion intercalation/de-intercalation between electrolyte and electrode interface disrupts, hence increasing the impedance and it can form a growth of dendrites. The deposition of TMs on the anode surface can be confirmed by X-ray absorption, X-ray fluorescence, magnetic resonance spectroscopy and electron probe microanalysis (EPMA) analyses.107 Dong-Su Ko et al.107 determined the dissolution mechanism and micro-structure origin of TMs from Ni-rich LiNi0.87Co0.9Mn0.4O2 cathode material and its migration into the electrolyte and deposition at the anode. Their results verified that TM dissolution preferentially occurred for particles with high surface area/volume ratios. Furthermore, they recommended that high-resolution chemical composition analysis, providing a powerful technique for investigating the degradation phenomena in lithium-ion batteries.
3.7 Residual lithium compounds
The residual lithium compounds are present as in a manner that is impossible to avoid in a Ni-rich layered oxide and their amount increases with Ni content, which will aggravate the deterioration of the material's electrochemical performance.103 This lithium residual is present initially in the form of Li2O and gradually changes to Li2CO3 and LiOH by reacting with H2O and CO2 in the air.73 Thus, the Ni-rich materials have shown fast moisture and CO2 uptakes in air, originating from the active oxygen species upon the reduction of Ni3+ to Ni2+ and synthetic Li impurities of Li2O/LiOH on the surface.36,104 Such absorption would also result in high pH of Ni-rich NCM particles that cause the gelation of cathode slurry.75 In short, the lithium residual originates from two sources. The first one is due to an excess amount of lithium precursors introduced in the synthesis, which are required to achieve the ordered layered structure that could provide better capacity.109 In this case, lithium will be volatile in the process of high-temperature sintering, the addition of excessive Li precursors can effectively inhibit the Li/TMs cation mixing and this cation mixing could slightly increase to compensate for the loss during the sintering process.110 As a result, extra Li-ions will live on the surface of cathode materials in form of Li2O and reaction of lithium residues with H2O and CO2 in air, meaning that the Li2O on the outer surface of the active materials converted to Li2CO3, LiHCO3, and LiOH layers after exposed to air.57,64,97The lithium residual reaction with CO2 and H2O in formula (6)–(8):111
| Li2O + CO2 → Li2CO3 | (6) |
| Li2O + H2O → 2LiOH | (7) |
| 2LiOH + CO2 → Li2CO3 + H2O | (8) |
The second is from the slow spontaneous reduction of Ni3+ to Ni2+ and the corresponding lattice oxygen O2− will be oxidized to O− and then the active O2− will be generated on the surface of cathode materials. The combination of active O2− and Li-ions in the materials will form Li2O which reacts with H2O and CO2 in the air to form residual lithium compounds on the surface of Ni-rich NCM cathode materials. The Li2CO3/LiOH impurities on the surface of the cathode materials would further react with HF generated from the decomposition of LiPF6 in the electrolyte (eqn (9)), resulting in gas evolution and consequent safety problems.112
Chemical and/or physical adsorption of residual lithium compounds such as Li2O/LiOH on the surface of the materials cause the rapid moisture absorption of Ni-rich cathode materials. Hence, through the LiOH and Li2CO3 titrations, it is confirmed that the Li2O on the active materials induces more formation of LiOH and, in particular, Li2CO3 on the surface of active materials (Fig. 10a).111 The Li2CO3 promotes the gas evolution and increases the moisture of the Ni-rich cathodes and the LiOH increases the powder pH value causing of slurry gelation during electrode fabrication.113 Furthermore, these undesirable LiOH and Li2CO3 species are the main component of residual lithium compound and impede the diffusion of Li+ due to their insulating properties, and deteriorate the electrochemical cycle performance of the Ni-rich cathode materials.114 Sung Wook Doo et al.115 have performed pH titration to measure changes in the amount of residual lithium compounds before and after the storage. As shown in (Fig. 10b), the total amount of residual lithium compounds substantially increased after storage in humid CO2, while negligible increases in residual lithium compounds were measured after storage in dry CO2 and in humid Ar.
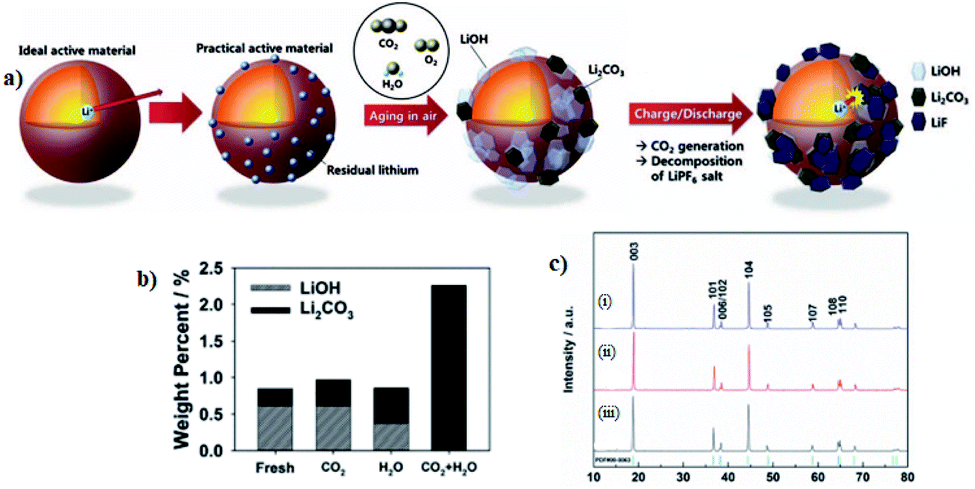 | ||
| Fig. 10 (a) Surface change of LiNi0.7Mn0.3O2 materials after exposure in air and effect of the residual lithium on the surface of LiNi0.7Mn0.3O2.111 (b) The amounts of residual lithium compounds on the LiNi0.8Co0.1Mn0.1O2 surface measured from titration.115 (c) XRD patterns of LiNi0.8Co0.1Mn0.1O2 with Li excesses (i) 20%, (ii) 10%, and (iii) 0%, where some of the minor peaks are residual lithium compound impurities.65 | ||
From eqn (4) the formed HF can react with the residual lithium compound to form CO2 and the formed CO2 react with Li-ions to form CO as illustrated in eqn (9) and (10) respectively:116
| Li2CO3 + 2HF → 2LiF + H2O + CO2 | (9) |
| 2CO2 + 2Li+ + 2e− → Li2CO3 + CO | (10) |
Side reactions at the interface of electrode/electrolyte can also be caused by the residual lithium, accelerating the electrolyte decomposition and forming thick cathode electrolyte interface (CEI), which can affect Li-ions diffusion pathways:57,96
| LiPF6 + 2LiOH → 3LiF + POF3 + H2O | (11) |
| LiPF6 + H2O → LiF + 2HF + POF3 | (12) |
| POF3 + nLi+ + ne− → LiF + LixPOyFz or | (13) |
| 2POF3 + 3Li2O → 6LiF↓ + P2O5↓ (or LixPOyFz) | (14) |
HF produced by the reaction of lithium residue with LPF6 in electrolyte results in high pH of Ni-rich particles, which results in the gelation of cathode slurry.117 The reduction of Ni3+ to Ni2+ on the surface of Ni-rich layered materials occurs spontaneously in the air. The residual lithium on the surface of the Ni-rich NCM absorbs carbon dioxide and water from the ambient air as mentioned before.
The presence of residual lithium compounds can be also detected by powder X-ray diffraction (XRD) measurement, Fourier transformed infrared spectrum (FT-IR) and X-ray photoelectron spectroscopy (XPS) spectra analysis, titration method, and Transmission Electron Microscope (TEM) detecting technique.110 For instant, Feng Wu et al.65 have reported the Li excess LiNi0.8Co0.1Mn0.1O2 that was produced by sintering the Ni0.8Co0.1Mn0.1(OH)2 precursor with different amounts of a lithium source, and from XRD result they have identified that some minor peaks (Fig. 10c), which are assigned to LiOH and Li2CO3 emerged because some amount of LiOH remains and reacts with CO2 in the air to form Li2CO3 during the synthetic process.
Lithium residues can also react with slurry components during mixing and may become gels due to the polymerization of N-methyl-2-pyrrolidone (NMP), which makes it difficult to coat the electrodes on the aluminum foil surface and complicates the preparation of the electrodes.118,133 The washing process is usually used to remove lithium impurities in NCM to avoid lithium residue.118 Although this easy method is attractive in practical applications, it leads to loss of oxygen in the lattice during the immersion of Ni-rich materials, resulting in a NiO defect, which leads to a decrease in the specific capacity of the material after washing.119 In general, two inactive phases can be formed on the surface-active cathode materials in the presence of lithium residue. These are rock-salt NiO phase and Li2CO3/LiOH layer, which are electrochemically inactive for Li-ion intercalation/deintercalation processes and lead to high cathode resistance and severe capacity degradation.124
3.8 Oxygen release and evolution of CO2 and CO
The structural instability of layered oxide cathode materials can lead to the release of oxygen in the form of O2 or forms reactive peroxide species.50 The mechanism of layered oxide cathode material's oxygen release may be related to external local temperature rises, which initiates the undesired reactions that lead to the thermal runaway.120 At the risen temperature, the SEI on the anode can be breakdown and further rises up the temperature.121 The breakdown SEI triggers the side reactions between anode and alkyl carbonate electrolyte by forming combustible gas ethane and methane. Then, the polymer separator between the anode and cathode will melt down causing a short circuit between the two electrodes.122 Ultimately, the layered oxide materials decompose and release a large amount of oxygen.120On the other hand, at a highly delithiated state, Ni-rich cathode materials become unstable and tend to be more stable when oxygen is released,87 which may decompose through phase transitions (MO2 (layered) → M3O4 (spinel) → MO (rock-salt)). The resulting decomposition of the charged cathode materials can result in the release of oxygen-containing species, which are highly reactive.40 Thus, in layered oxide Ni-rich NCM materials the highly oxidized state of Ni4+ in the highly delithiated state, during electrochemical reactions tend to form permanent surface reconstruction layers such as spinel and rock-salt phases, which subsequently cause oxygen release. The released O2 can react with a flammable organic electrolyte, leading to massive heat and severe safety hazards, such as flames and explosions.6,20,65 Oxygen release in Ni-rich cathode materials can be related to phase transition and this phase transition mainly depends on the Ni content related to temperature, that is as Ni content in NCM start to increases, the phase transitions occurred at low temperatures.123 If there is no phase transition, there is no oxygen release, which results in good thermal stability. For low Ni contents, there is no much oxygen release. The existence of nanopores also strongly affects the degradation of Ni-rich NCM cathode materials as the pore surfaces apparently increase oxygen loss and formation of rock-salt regions.124
The cathodes oxygen release can be investigated by thermal analysis, time-resolved X-ray diffraction (TR-XRD), Raman spectroscopy, in situ differential electrochemical mass spectroscopy (DEMS), X-ray absorption spectroscopy (XAS), in situ transmission electron microscopy (TEM), and computational modeling.135 Various study revealed that the O2 evolution is a complex mechanism that is influenced by many parameters such as state of charge (SOC), morphology and size of the particles, chemical composition, and atomic arrangements of the cathodes.120 For instance, Seong-Min Bak et al.97 have been used the combination of in situ time-resolved X-ray diffraction (TR-XRD) and mass spectroscopy (MS) to study the structural change and gas evolution of Ni-rich cathode material. From this combination, they simultaneously observed the structural changes and gas species that are evolved during the thermal decomposition of charged cathode materials. In addition, Chengyu Mao et al.125 have been used gas chromatography-mass spectrometry (GC-MS) and Fourier transform infrared spectroscopy (FTIR) to identify gas species evolved from the full cell of NCM811 cathode and a graphite anode.
The appearance of oxygen vacancies on the surface of cathode materials is a direct consequence of oxygen evolution.126 Similarly, Kyoungmin Min et al.127 have been used first-principles calculations to identify the effects of oxygen vacancy. Thus, when oxygen vacancies are present, Ni can migrate to the Li site during delithiation. For the highly delithiated Ni-rich cathodes, the increase in temperature will induce a series of phase changes and side reactions. As a result, reactive oxygen-containing species are produced, which, together with Ni4+, oxidizes the electrolyte to release gaseous side-products, such as CO2 and CO.44 The evolution of both CO and CO2 in Ni-rich cathode materials can be occurred by two mechanisms:128 (i) electrochemical oxidation of electrolyte, which is either proportional to the sum of the total surface areas of the cathode active material (CAM) and the conductive carbon if it is a noncatalytic process or to only the CAM surface area if catalyzed by transition metals at the layered oxide surface, and (ii) chemical oxidation of electrolyte caused by the release of reactive oxygen from the Ni-rich NCM surface, in which case the oxidation rate would again only depend on the CAM surface area.123,129 When these released reactive [O] also reacts with ethylene carbonate (EC) it forms CO2 and CO and H2O as following reaction:92,127
| EC + [O] → CO2(g) + CO(g) + H2O | (15) |
In Ni-rich NCM materials the oxygen releases that consequence the evolution of both CO2 and CO by reaction with alkyl carbonate electrolyte also depends on temperature. Roland Jung et al.130 have reported that the temperature dependence of oxygen release and evolution of CO2 and CO of NCM622 by using OEMS, hence the amount of CO2 and CO evolution occurred simultaneously with O2 release increase with temperature. Toru Hatsukade et al.131 studied the contributions of different mechanisms to the generation of CO2 for layered NCM cathode materials by using the combinations of isotope labeling, electrolyte substitution, and in situ gas analysis. They found that Li2CO3 decomposition contributes to CO2 evolution and significant during the first cycle but evolution rapidly decreased with subsequent cycles at high potentials. They also reported the major contribution of electrolyte decomposition for CO2 evolution.
Oxygen release in Ni-rich cathode materials can also be related to state-of-charge (SOC). Daniel Pritzl et al.132 have investigated the effect of washing/drying on the gas evolution of a Ni-rich NCM materials in the first charge to high SOC by performing on-line electrochemical mass spectrometry (OEMS) on electrodes with the pristine and the washed NCM-851005. The OEMS in terms of gassing revealed that the release of O2 and CO2 above 80% SOC decreases substantially as the drying temperature of the washed NCM-851005 increased from 25 °C to 300 °C, along with the cathode impedance. In NCMs, the oxygen evolution stops after some cycles, whereas the formation of CO and CO2 from cycle to cycle decreases at a much slower rate. Generally, the evolution of gases raises a serious safety concern on Ni-rich NCM cathode materials.101
3.9 Thermal instability
In several LIBs powered systems ranging from laptops and cell phones to e-cigarettes, headphones, electric vehicles, and even airplanes, thermal runaway events have been observed and have caused serious injuries to the consumers.75,103,108 A series of self-progressive exothermic events triggered by external factors such as ambient temperature rise or mechanical abuse, or caused by internal factors such as a short circuit, overcharge or applied high current rates results in the thermal runaway.109,110,112,113 Thermal runaway is the main cause of safety accidents such as fire and explosion of LIBs.110,112 The occurrence and sequence of these chains of events can vary based on the materials design and the specific conditions that trigger the thermal runaway reaction.103,110 Studies on the thermal stability of Ni-based cathode materials by thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), differential thermal analysis (DTA), thermogravimetric analysis coupled with mass spectrometry (TGA-MS), in situ high-temperature X-ray diffraction (XRD), and accelerating rate calorimetry (ARC) show that the thermal decomposition reaction in charged cathodes starts at lower onset temperatures with increasing Ni content in the cathode materials, which indicates that a high Ni content worsens the thermal stability of the cathode materials.113,114In their charged states, the layered oxide cathode materials are metastable under ambient air, but at elevated temperatures, they begin to decompose and release O2 due to the high effective oxygen partial pressure.133 Specifically, at highly delithiated states in the Ni-rich cathode materials, the reduction of Ni4+ to Ni2+ during heating releases O2 and the released O2 reacts with the flammable electrolytes causing severe thermal runaway and lead to catastrophic failure of the LIBs. In Ni-rich cathode materials, O2-release-related safety concern is the main drawback, despite its obvious advantage in high capacity. Thus, the safety hazards in Ni-rich cathode materials are ascribed to thermodynamic instability of H3 phase, which increases with state of charge and temperature.73 In H3 phase, the delithiated Ni-rich cathodes materials are very thermodynamically unstable, which results in either chemically oxidize electrolyte solvents or spontaneously release O2. These cause safety hazards during charging and storage due to hot conditions. The oxidized electrolyte solvent generates heat and gaseous products. The processes of safety hazard include the O2 evolution reaction intermediate and the dissolved O2 passes through the separator and reach the anode. And then chemically react with the lithiated anode. This is a reaction far more energetic than the reaction with electrolyte solvents, which results in the thermal runaway of LIBs initiating without needing of internal electric shorting.73
Furthermore, in the presence of temperature effects, as Ni content in NCM starts to increases, the phase transitions occurred at low temperatures.123 Fig. 11 shows the phase transitions and cation migration paths of transition metal in the charged NCM cathode materials during thermal decomposition. In the initial layered structure (Fig. 11a), the TM cations occupy octahedral positions (TMoct layer) and the Li+ occupy the alternate layers of octahedral sites (Lioct layer). For the first phase transition from the layered to the disordered spinel, some of TM cations need to migrate from their original sites (labeled “A” in Fig. 11b) to the octahedral positions in the Li layer (labeled “B” in Fig. 11b). This migration will take place through the nearest tetrahedral position via the face-shared neighboring octahedra. Because of its lower energy barrier, it is well-known that this tetrahedral pathway of Oh (the octahedral site in TM layer)–Td (tetrahedral site)–Oh (octahedral site in Li layer) is energetically favorable. This must be accompanied by the movement of Li+ from their original positions to the adjacent tetrahedral positions to complete this first phase transition to the LiMn2O4-type spinel structure, as shown in (Fig. 11c). Such a structural change is accompanied by the reduction of TM cations and O2 release if the cathode is deeply overcharged. With further increases in temperature, the LiMn2O4-type spinel structure changes to M3O4-type spinel structure with an increased partial TM occupancy at the 8a tetrahedral sites as shown in (Fig. 11d).123
 | ||
| Fig. 11 Schematic illustration of phase transition and the possible TM cation migration path in the charged NMC cathode materials: (a) charged LiMO2 (M = Ni, Co, Mn), (b) M cation migration (mostly Ni), (c) 8a tetrahedral site: mostly Li, (d) 8a tetrahedral site: mostly Co or Mn.123 | ||
Thermal stability of the charged NCM materials decreases with increasing Ni content but increases with increasing cobalt and manganese content: the more nickel in the composition, the lower the onset temperature of the phase transition, and the sharper the peak of the oxygen release. That means thermal stability may also depend on the compositions of the transition metals. Seong-Min Bak et al.123 have compared the NCM materials NCM433, NCM532, NCM622, and NCM811 by using combined in situ time-resolved X-ray diffraction and mass spectroscopy (TR-XRD/MS) techniques, and NCM532 composition could be in good thermal stability due to the well-balanced ratio of nickel to manganese and cobalt. Eunkang Lee et al.134 have also investigated the thermal decomposition process of charged Li0.33Ni0.5+xCo0.2Mn0.3−xO2 (x = 0, 0.1, 0.2) cathode materials by using a combination of synchrotron-based XRD, XAS, and a thermoanalytical DSC technique. Their XRD resulted that the charged Ni-rich NCMs cathodes during the heating process exhibit a larger thermal expansion. Moreover, their XAS results showed that before the onset temperature is reached for the layered to a spinel phase transition, the Ni-ions are reduced, which is related to the formation of oxygen vacancies around Ni-ions. In a charged Ni-rich cathode materials, thermal expansion and oxygen vacancy formation facilitate TM migration and lower the onset temperature of the thermal decomposition reaction by activating an energetically favorable pathway for cation migration. Unfortunately, thermal expansion and oxygen vacancy formation are critical factors that affect the thermal stability of charged Ni-rich NCMs cathode materials.
Huaibin Wang et al.135 have also studied the thermal runaway of NCM111, NCM523, and NCM622 cathode batteries by using Extended Volume Accelerating Rate Calorimeter (EV-ACR). According to this report, the main factors that can trigger the thermal runaway of batteries are O2 release, lithium plating at the anode, internal short circuit, and the reaction of the cathode with electrolyte. The process from thermal runaway to a fire is mainly contributed by heat generation, accumulation, and propagation.136 The thermal runaway of LIB goes through nine steps in the EV-ARC test, from low temperature to high temperature,137 including: (1) high-temperature capacity degradation, (2) SEI film decomposition, (3) anode and electrolyte reaction, (4) separator melting, (5) anode decomposition reaction, (6) electrolyte solution decomposition reaction, (7) binder decomposition reaction, (8) vent and smoke, (9) electrolyte combustion. In the same condition, the thermal runaway also occurs in the LIB modules, which are widely used in electric vehicles and energy storage stations for facing the energy shortage and air pollution, and its thermal runaway is shown in (Fig. 12). Fig. 12a shows a pristine sample cell; (Fig. 12b) shows the components of LIB; in stage II SEI decomposes, Mn is dissolved, and gas is released as shown in (Fig. 12c). In stage III, the separator melt and gas releasing is increased as shown in (Fig. 12d); and in stage IV, the separator is completely broken up and an internal short circuit is formed causing smoke and explosion of LIB as shown in Fig. 12e and f.
 | ||
| Fig. 12 The thermal runaway mechanism of the LIB modules in EV-ARC tests: (a) sample cell before test, (b) stage I, (c) SEI film decomposition, (d) separate melting, (e) separate break up, (f) sample test after test.137 | ||
4 Conclusions and future perspectives
In this review, we summarized the surface degradation, mechanical failure and thermal instability of Ni-rich NCM cathode materials for lithium-ion batteries based on the recent findings. The performance degradation and safety hazards are the main problems in Ni-rich NCM cathode material. Performance degradation behaved as declines in capacity and working voltage, and it results from aggressive chemical, structural, and mechanical deterioration, in which there is an increase in volume and impedance of the battery. Because of the thermal instability of Ni-rich NCM materials near fully charged state or at high temperature the safety hazard can occur. This safety hazard is caused by the abuse conditions such as overcharging, overheating and electrical shorting. Li+/Ni2+ cation mixing, particle cracking, phase transformation, electrolyte decomposition, transition metal dissolution, residual lithium compounds, oxygen release, and thermal instabilities are the main problems in Ni-rich cathode active materials. Increasing the upper cut-off voltage can also increase undesirable side reactions at the electrode/electrolyte interface, surface film formation, and metal dissolution, which ultimately decrease the battery lifetime. Commercialization of these cathodes has been proven to be difficult. Hence, further innovations and different state-of-the-art is required to eliminate these issues. Various mitigation strategies such as dopants, gradient layers, surface coatings, carbon matrixes, and advanced synthesis methods proposed by researchers to improve the electrochemical performance of these materials. However, it is very important to understand the degradation mechanisms in order to find effective strategies and to making better batteries. The currently studied degradation mechanisms and the root cause of each challenge in Ni-rich cathode active materials are not widely studied and understood very well. Moreover, the mentioned challenges in this review are also dependent on each other but not extremely identified by scientific investigation. Thus, one degradation mechanism will cause another type of degradation mechanism and the relationship between these degradation mechanisms not studied effectively. Therefore, we recommend that it is good to identify the relationship of each degradation mechanism scientifically and to find a reasonable solution for each challenge to realize the commercialization and high capacity retention of Ni-rich NCM cathode active materials for the next-generation LIBs.Author contributions
Fikadu Takele Geldasa: writing – original draft. Mesfin Abayneh Kebede: supervision, conceptualization, visualization and editing. Megersa Wodajo Shura: provisioning resources. Fekadu Gashaw Hone: conceptualization, supervision, writing – review and editing.Conflicts of interest
There are no interest conflicts.Acknowledgements
We acknowledge Adama Science and Technology University and Oda Bultum University for their financial support.References
- S. S. Zhang, Front. Energy Res., 2013, 1, 1–6 Search PubMed.
- R. Younesi, Front. Energy Res., 2014, 2, 2013–2014 CrossRef.
- T. Kåberger, Global Energy Interconnection, 2018, 1, 48–52 Search PubMed.
- B. Scrosat and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- S. Hwang, D. H. Kim, K. Y. Chung and W. Chang, Appl. Phys. Lett., 2014, 105, 4 CrossRef.
- Y. Ding, D. Mu, B. Wu, R. Wang, Z. Zhao and F. Wu, Appl. Energy, 2017, 195, 586–599 CrossRef CAS.
- W. Cho, S. M. Kim, J. H. Song, T. Yim, S. G. Woo, K. W. Lee, J. S. Kim and Y. J. Kim, J. Power Sources, 2015, 282, 45–50 CrossRef CAS.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 RSC.
- S. Liang, W. Yan, X. Wu, Y. Zhang, Y. Zhu, H. Wang and Y. Wu, Solid State Ionics, 2018, 318, 2–18 CrossRef CAS.
- M. S. Whittingham, Science, 1976, 192, 1126–1127 CrossRef CAS PubMed.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2003, 150, A1377–A1384 CrossRef CAS.
- P. C. J. P. J. W. K. Mizushima and J. B. Goodenough, Jpn. J. Appl. Phys., Part 1, 2003, 42, 6131–6134 CrossRef.
- A. Manthiram, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- R. Yazami and P. Touzain, J. Power Sources, 1983, 9, 365–371 CrossRef CAS.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 2–5 CrossRef PubMed.
- M. V. Reddy, A. Mauger, C. M. Julien, A. Paolella and K. Zaghib, Materials, 2020, 13, 1884 CrossRef CAS PubMed.
- J. L. Brédas, J. M. Buriak, F. Caruso, K.-Sh. Choi, B. A. Korgel, M. R. Palacín, K. Persson, E. Reichmanis, F. Schüth, R. Seshadri and M. D. Ward, Chem. Mater., 2019, 31, 8577–8581 CrossRef.
- Y. Lee, H. Kim, T. Yim, K. Y. Lee and W. Choi, J. Power Sources, 2018, 400, 87–95 CrossRef CAS.
- W. Shan, S. Huang, H. Zhang and X. Hou, J. Alloys Compd., 2021, 862, 158022 CrossRef CAS.
- Y. Lv, X. Cheng, W. Qiang and B. Huang, J. Power Sources, 2020, 450, 227718 CrossRef CAS.
- J. A. Sanguesa, V. Torres-Sanz, P. Garrido, F. J. Martinez and J. M. Marquez-Barja, Smart Cities, 2021, 4, 372–404 CrossRef.
- Y. Xia, J. Zheng, C. Wang and M. Gu, Nano Energy, 2018, 49, 434–452 CrossRef CAS.
- A. O. Said, C. Lee, X. Liu, Z. Wu and S. I. Stoliarov, Proc. Combust. Inst., 2019, 37, 4173–4180 CrossRef CAS.
- Q. Liu, X. Su, D. Lei, Y. Qin, J. Wen, F. Guo, Y. A. Wu, Y. Rong, R. Kou, X. Xiao, F. Aguesse, J. Bareño, Y. Ren, W. Lu and Y. Li, Nat. Energy, 2018, 3, 936–943 CrossRef CAS.
- C. M. Julien, A. Mauger, K. Zaghib and H. Groult, Inorganics, 2014, 2, 132–154 CrossRef CAS.
- B. Zhu, Z. Yu, L. Meng, Z. Xu, C. Lv, Y. Wang, G. Wei and J. Qu, Ionics, 2021, 27, 2749–2784 CrossRef CAS.
- X. Chen, Y. Li and J. Wang, Nanomaterials, 2021, 11, 1–9 Search PubMed.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- C. Liu, Z. G. Neale and G. Cao, Mater. Today, 2016, 19, 109–123 CrossRef CAS.
- A. W. Golubkov, D. Fuchs, J. Wagner, H. Wiltsche, C. Stangl, G. Fauler and G. Voitic, RSC Adv., 2014, 4, 3633 RSC.
- J. P. Pender, G. Jha, D. H. Youn, J. M. Ziegler, I. Andoni, E. J. Choi, A. Heller, B. S. Dunn, P. S. Weiss, R. M. Penner and C. B. Mullins, ACS Nano, 2020, 14, 1243–1295 CrossRef CAS PubMed.
- W. Hua, J. Zhang, Z. Zheng, W. Liu, X. Peng, X. D. Guo, B. Zhong, Y. J. Wang and X. Wang, Dalton Trans., 2014, 43, 14824–14832 RSC.
- S. K. Jung, H. Gwon, J. Hong, K. Y. Park, D. H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1–7 Search PubMed.
- H. J. Noh, S. Youn, C. S. Yoon and Y. K. Sun, J. Power Sources, 2013, 233, 121–130 CrossRef CAS.
- L. De Biasi, A. O. Kondrakov, H. Geßwein, T. Brezesinski, P. Hartmann and J. Janek, J. Phys. Chem. C, 2017, 121, 26163–26171 CrossRef.
- T. Ohzuku and Y. Makimura, Chem. Lett., 2001, 642–643 CrossRef CAS.
- Y. Zhu, Y. Huang, R. Du, M. Tang, B. Wang and J. Zhang, Crystals, 2021, 11, 465 CrossRef CAS.
- P. Zou, Z. Lin, M. Fan, F. Wang, Y. Liu and X. Xiong, Appl. Surf. Sci., 2020, 504, 144506 CrossRef CAS.
- S. W. Lee, M. S. Kim, J. H. Jeong, D. H. Kim, K. Y. Chung, K. C. Roh and K. B. Kim, J. Power Sources, 2017, 360, 206–214 CrossRef CAS.
- L. Wu, X. Tang, X. Chen, Z. Rong, W. Dang, Y. Wang, X. Li, L. Huang and Y. Zhang, J. Power Sources, 2020, 445, 227337 CrossRef CAS.
- J. Yang, M. Hou, S. Haller, Y. Wang, C. Wang and Y. Xia, Electrochim. Acta, 2016, 189, 101–110 CrossRef CAS.
- W. Liu, P. Oh, X. Liu, M. J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef CAS PubMed.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- I. Buchberger, S. Seidlmayer, A. Pokharel, M. Piana, J. Hattendorff, P. Kudejova, R. Gilles and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, A2737–A2746 CrossRef CAS.
- B. Strehle, K. Kleiner, R. Jung, F. Chesneau, M. Mendez, H. A. Gasteiger and M. Piana, J. Electrochem. Soc., 2017, 164, A400–A406 CrossRef CAS.
- Y. Ruan, X. Song, Y. Fu, C. Song and V. Battaglia, J. Power Sources, 2018, 400, 539–548 CrossRef CAS.
- H. H. Sun, H. H. Ryu, U. H. Kim, J. A. Weeks, A. Heller, Y. K. Sun and C. B. Mullins, ACS Energy Lett., 2020, 5, 1136–1146 CrossRef CAS.
- X. Cheng, J. Zheng, J. Lu, Y. Li, P. Yan and Y. Zhang, Nano Energy, 2019, 62, 30–37 CrossRef CAS.
- J. S. Edge, S. O'Kane, R. Prosser, N. D. Kirkaldy, A. N. Patel, A. Hales, A. Ghosh, W. Ai, J. Chen, J. Yang, S. Li, M.-C. Pang, L. Bravo Diaz, A. Tomaszewska, M. Waseem Marzook, K. N. Radhakrishnan, H. Wang, Y. Patel, B. Wu and G. J. Offer, Phys. Chem. Chem. Phys., 2021, 23, 8200 RSC.
- R. S. Arumugam, L. Ma, J. Li, X. Xia, J. M. Paulsen and J. R. Dahn, J. Electrochem. Soc., 2016, 163, A2531–A2538 CrossRef CAS.
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS.
- P. Pang, X. Tan, Z. Wang, Z. Cai, J. Nan, Z. Xing and H. Li, Electrochim. Acta, 2021, 365, 137380 CrossRef CAS.
- H. H. Sun and A. Manthiram, Chem. Mater., 2017, 29, 8486–8493 CrossRef CAS.
- S. S. Jan, S. Nurgul, X. Shi, H. Xia and H. Pang, Electrochim. Acta, 2014, 149, 86–93 CrossRef CAS.
- H. Yu, Y. Qian, M. Otani, D. Tang, S. Guo, Y. Zhu and H. Zhou, Energy Environ. Sci., 2014, 7, 1068–1078 RSC.
- T. He, L. Chen, Y. Su, Y. Lu, L. Bao, G. Chen, Q. Zhang, S. Chen and F. Wu, J. Power Sources, 2019, 441, 227195 CrossRef CAS.
- J. Zheng and F. Wang, Acc. Chem. Res., 2019, 52, 2201–2209 CrossRef CAS PubMed.
- C. Liang, R. C. Longo, F. Kong, C. Zhang, Y. Nie, Y. Zheng, J. S. Kim, S. Jeon, S. A. Choi and K. Cho, J. Power Sources, 2017, 340, 217–228 CrossRef CAS.
- X. Yao, Z. Xu, Z. Yao, W. Cheng, H. Gao, Q. Zhao, J. Li and A. Zhou, Mater. Today Commun., 2019, 19, 262–270 CrossRef CAS.
- J. Zheng, P. Yan, L. Estevez, C. Wang and J. G. Zhang, Nano Energy, 2018, 49, 538–548 CrossRef CAS.
- Y. C. Li, W. Xiang, Y. Xiao, Z. G. Wu, C. L. Xu, W. Xu, Y. D. Xu, C. Wu, Z. G. Yang and X. D. Guo, J. Power Sources, 2019, 423, 144–151 CrossRef CAS.
- H. Ronduda, M. Zybert, A. Szczęsna-Chrzan, T. Trzeciak, A. Ostrowski, D. Szymański, W. Wieczorek, W. Raróg-Pilecka and M. Marcinek, Nanomaterials, 2020, 10, 1–21 CrossRef PubMed.
- P. Li, S. Zhao, Y. Zhuang, J. Adkins, Q. Zhou and J. Zheng, Appl. Surf. Sci., 2018, 453, 93–100 CrossRef CAS.
- F. Wu, J. Tian, Y. Su, J. Wang, C. Zhang, L. Bao, T. He, J. Li and S. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 7702–7708 CrossRef CAS PubMed.
- B. Huang, M. Wang, X. Zhang, Z. Zhao, L. Chen and Y. Gu, J. Alloys Compd., 2020, 830, 154619 CrossRef CAS.
- U. H. Kim, J. H. Kim, J. Y. Hwang, H. H. Ryu, C. S. Yoon and Y. K. Sun, Mater. Today, 2019, 23, 26–36 CrossRef CAS.
- R. Wang, G. Qian, T. Liu, M. Li, J. Liu, B. Zhang, W. Zhu, S. Li, W. Zhao, W. Yang, X. Ma, Z. Fu, Y. Liu, J. Yang, L. Jin, Y. Xiao and F. Pan, Nano Energy, 2019, 62, 709–717 CrossRef CAS.
- J. Zheng, P. Yan, J. Zhang, M. H. Engelhard, Z. Zhu, B. J. Polzin, S. Trask, J. Xiao, C. Wang and J. Zhang, Nano Res., 2017, 10, 4221–4231 CrossRef CAS.
- W. Liu, P. Oh, X. Liu, M. J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef CAS PubMed.
- H.-H. Ryu, B. Namkoong, J.-H. Kim, I. Belharouak, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2021, 6, 2726–2734 CrossRef CAS.
- X. Zhao, L. An, J. Sun and G. Liang, J. Electroanal. Chem., 2018, 810, 1–10 CrossRef CAS.
- S. S. Zhang, Energy Storage Mater., 2020, 24, 247–254 CrossRef.
- J. Chen, H. Yang, T. Li, C. Liu, H. Tong, J. Chen, Z. Liu, L. Xia, Z. Chen, J. Duan and L. Li, Front. Chem., 2019, 7, 1–10 CrossRef PubMed.
- Y. Liu, L. b. Tang, H. x. Wei, X. h. Zhang, Z. j. He, Y. j. Li and J. c. Zheng, Nano Energy, 2019, 65, 104043 CrossRef CAS.
- C. Fu, G. Li, D. Luo, Q. Li, J. Fan and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 15822–15831 CrossRef CAS PubMed.
- P. He, H. Yu, D. Li and H. Zhou, J. Mater. Chem., 2012, 22, 3680–3695 RSC.
- G. Qian, Y. Zhang, L. Li, R. Zhang, J. Xu, Z. Cheng, S. Xie, H. Wang, Q. Rao, Y. He, Y. Shen, L. Chen, M. Tang and Z. F. Ma, Energy Storage Mater., 2020, 27, 140–149 CrossRef.
- S. Hwang, E. Jo, K. Y. Chung, K. S. Hwang, S. M. Kim and W. Chang, J. Phys. Chem. Lett., 2017, 8, 5758–5763 CrossRef CAS PubMed.
- X. Fan, Y. Liu, X. Ou, J. Zhang, B. Zhang, D. Wang and G. Hu, Chem. Eng. J., 2020, 393, 124709 CrossRef CAS.
- H. Jia, W. Zhu, Z. Xu, X. Nie, T. Liu, L. Gao and J. Zhao, Electrochim. Acta, 2018, 266, 7–16 CrossRef CAS.
- F. Lin, I. M. Markus, D. Nordlund, T. C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- F. Wu, N. Liu, L. Chen, Y. Su, G. Tan, L. Bao, Q. Zhang, Y. Lu, J. Wang, S. Chen and J. Tan, Nano Energy, 2019, 59, 50–57 CrossRef CAS.
- H. Yu, Y. G. So, A. Kuwabara, E. Tochigi, N. Shibata, T. Kudo, H. Zhou and Y. Ikuhara, Nano Lett., 2016, 16, 2907–2915 CrossRef CAS PubMed.
- C. Xu, W. Xiang, Z. Wu, Y. Xu, Y. Li, Y. Wang, Y. Xiao, X. Guo and B. Zhong, ACS Appl. Mater. Interfaces, 2019, 11, 16629–16638 CrossRef CAS PubMed.
- L. Zou, W. Zhao, Z. Liu, H. Jia, J. Zheng, G. Wang, Y. Yang, J. G. Zhang and C. Wang, ACS Energy Lett., 2018, 3, 2433–2440 CrossRef CAS.
- S. M. Bak, K. W. Nam, W. Chang, X. Yu, E. Hu, S. Hwang, E. A. Stach, K. B. Kim, K. Y. Chung and X. Q. Yang, Chem. Mater., 2013, 25, 337–351 CrossRef CAS.
- X. Yang, Y. Tang, J. Zheng, G. Shang, J. Wu, Y. Lai, J. Li and Z. Zhang, Electrochim. Acta, 2019, 320, 134587 CrossRef CAS.
- Y. Su, Y. Yang, L. Chen, Y. Lu, L. Bao, G. Chen, Z. Yang, Q. Zhang, J. Wang, R. Chen, S. Chen and F. Wu, Electrochim. Acta, 2018, 292, 217–226 CrossRef CAS.
- S. Oswald, D. Pritzl, M. Wetjen and H. A. Gasteiger, J. Electrochem. Soc., 2020, 167, 100511 CrossRef CAS.
- R. Ning, K. Yuan, K. Zhang, C. Shen and K. Xie, Appl. Surf. Sci., 2021, 542, 148663 CrossRef CAS.
- K. Edström, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397–403 CrossRef.
- J. M. Lim, T. Hwang, D. Kim, M. S. Park, K. Cho and M. Cho, Sci. Rep., 2017, 7, 2–11 CrossRef PubMed.
- I. Takahashi, H. Kiuchi, A. Ohma, T. Fukunaga and E. Matsubara, J. Phys. Chem. C, 2020, 124, 9243–9248 CrossRef CAS.
- H. H. Ryu, N. Y. Park, T. C. Noh, G. C. Kang, F. Maglia, S. J. Kim, C. S. Yoon and Y. K. Sun, ACS Energy Lett., 2021, 6, 216–223 CrossRef CAS.
- S. Schweidler, L. De Biasi, G. Garcia, A. Mazilkin, P. Hartmann, T. Brezesinski and J. Janek, ACS Appl. Energy Mater., 2019, 2, 7375–7384 CrossRef CAS.
- S.-M. Bak, K. W. Nam, W. Chang, X. Yu, E. Hu, S. Hwang, E. A. Stach, K. B. Kim, K. Y. Chung and X. Q. Yang, Chem. Mater., 2013, 25, 337–351 CrossRef CAS.
- W. Li, X. Liu, H. Celio, P. Smith, A. Dolocan, M. Chi and A. Manthiram, Adv. Energy Mater., 2018, 8, 1703154 CrossRef.
- S. J. Sim, S. H. Lee, B. S. Jin and H. S. Kim, J. Power Sources, 2021, 481, 229037 CrossRef CAS.
- S. Gao, X. Zhan and Y. T. Cheng, J. Power Sources, 2019, 410, 45–52 CrossRef.
- W. Tang, Z. Chen, F. Xiong, F. Chen, C. Huang, Q. Gao, T. Wang, Z. Yang and W. Zhang, J. Power Sources, 2019, 412, 246–254 CrossRef CAS.
- H. J. Song, S. H. Jang, J. Ahn, S. H. Oh and T. Yim, J. Power Sources, 2019, 416, 1–8 CrossRef CAS.
- Z. Gan, G. Hu, Z. Peng, Y. Cao, H. Tong and K. Du, Appl. Surf. Sci., 2019, 481, 1228–1238 CrossRef CAS.
- Z. Ruff, C. Xu and C. P. Grey, J. Electrochem. Soc., 2021, 168, 060518 CrossRef CAS.
- H. Ji, S. Ho, Y. Lee, J. Kim and T. Yim, J. Power Sources, 2021, 483, 229218 CrossRef.
- K. Ishidzu, Y. Oka and T. Nakamura, Solid State Ionics, 2016, 288, 176–179 CrossRef CAS.
- D. S. Ko, J. H. Park, S. Park, Y. N. Ham, S. J. Ahn, J. H. Park, H. N. Han, E. Lee, W. S. Jeon and C. Jung, Nano Energy, 2019, 56, 434–442 CrossRef CAS.
- J. Wandt, A. Freiberg, R. Thomas, Y. Gorlin, A. Siebel, R. Jung, H. A. Gasteiger and M. Tromp, J. Mater. Chem. A, 2016, 4, 18300–18305 RSC.
- Y. Ding, D. Mu, B. Wu, Z. Zhao and R. Wang, Ceram. Int., 2020, 46, 9436–9445 CrossRef CAS.
- Y. Su, L. Li, G. Chen, L. Chen, N. Li, Y. Lu, L. Bao, S. Chen and F. Wu, Chin. J. Chem., 2021, 39, 189–198 CrossRef CAS.
- D.-H. Cho, C.-H. Jo, W. Cho, Y.-J. Kim, H. Yashiro, Y.-K. Sun and S.-T. Myung, J. Electrochem. Soc., 2014, 161, A920–A926 CrossRef CAS.
- P. Sun, F. Du, Q. Zhou, D. Hu, T. Xu, C. Mei, Q. Hao, Z. Fan and J. Zheng, J. Power Sources, 2021, 495, 229761 CrossRef CAS.
- J. Kim, H. Lee, H. Cha, M. Yoon, M. Park and J. Cho, Adv. Energy Mater., 2018, 8, 1810023 Search PubMed.
- Y. Zou, X. Yang, C. Lv, T. Liu, Y. Xia, L. Shang, G. I. N. Waterhouse, D. Yang and T. Zhang, Adv. Sci., 2017, 4, 1–8 CrossRef.
- S. W. Doo, S. Lee, H. Kim, J. H. Choi and K. T. Lee, ACS Appl. Energy Mater., 2019, 2, 6246–6253 CrossRef CAS.
- J. H. Park, B. Choi, Y. S. Kang, S. Y. Park, D. J. Yun, I. Park, J. H. Shim, J. H. Park, H. N. Han and K. Park, Energy Technol., 2018, 6, 1361–1369 CrossRef CAS.
- T. Chen, X. Li, H. Wang, X. Yan, L. Wang, B. Deng, W. Ge and M. Qu, J. Power Sources, 2018, 374, 1–11 CrossRef CAS.
- Y. You, H. Celio, J. Li, A. Dolocan and A. Manthiram, Angew. Chem., Int. Ed., 2018, 57, 6480–6485 CrossRef CAS PubMed.
- V. C. Ho, S. Jeong, T. Yim and J. Mun, J. Power Sources, 2020, 450, 227625 CrossRef CAS.
- S. Sharifi-Asl, J. Lu, K. Amine and R. Shahbazian-Yassar, Adv. Energy Mater., 2019, 9, 1–19 Search PubMed.
- T. Cai, A. G. Stefanopoulou and J. B. Siegel, J. Electrochem. Soc., 2019, 166, A2431–A2443 CrossRef.
- M. Kaliaperumal, M. S. Dharanendrakumar, S. Prasanna, K. V. Abhishek, R. K. Chidambaram, S. Adams, K. Zaghib and M. V. Reddy, Materials, 2021, 14, 5676 CrossRef CAS PubMed.
- S.-m. Bak, E. Hu, Y. Zhou, X. Yu, S. D. Senanayake, S.-j. Cho, K.-b. Kim, K. Y. Chung, X.-q. Yang and K.-w. Nam, ACS Appl. Mater. Interfaces, 2014, 6, 22594–22601 CrossRef CAS PubMed.
- S. Ahmed, A. Pokle, S. Schweidler, A. Beyer, M. Bianchini, F. Walther, A. Mazilkin, P. Hartmann, T. Brezesinski, J. Janek and K. Volz, ACS Nano, 2019, 13, 10694–10704 CrossRef CAS PubMed.
- C. Mao, R. E. Ruther, L. Geng, Z. Li, D. N. Leonard, H. M. Meyer, R. L. Sacci and D. L. Wood, ACS Appl. Mater. Interfaces, 2019, 11, 43235–43243 CrossRef CAS PubMed.
- H. Zhang, B. M. May, F. Omenya, M. S. Whittingham, J. Cabana and G. Zhou, Chem. Mater., 2019, 31, 7790–7798 CrossRef CAS.
- K. Min, S. W. Seo, Y. Y. Song, H. S. Lee and E. Cho, Phys. Chem. Chem. Phys., 2017, 19, 1762–1769 RSC.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Phys. Chem. Lett., 2017, 8, 4820–4825 CrossRef CAS PubMed.
- E. J. Lee, Z. Chen, H. J. Noh, S. C. Nam, S. Kang, D. H. Kim, K. Amine and Y. K. Sun, Nano Lett., 2014, 14, 4873–4880 CrossRef CAS PubMed.
- R. Jung, P. Strobl, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, 2869–2879 CrossRef.
- T. Hatsukade, A. Schiele, P. Hartmann, T. Brezesinski and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 38892–38899 CrossRef CAS PubMed.
- D. Pritzl, T. Teufl, A. T. S. Freiberg, B. Strehle, J. Sicklinger, H. Sommer, P. Hartmann and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A4056–A4066 CrossRef CAS.
- Y. Huang, Y. C. Lin, D. M. Jenkins, N. A. Chernova, Y. Chung, B. Radhakrishnan, I. H. Chu, J. Fang, Q. Wang, F. Omenya, S. P. Ong and M. S. Whittingham, ACS Appl. Mater. Interfaces, 2016, 8, 7013–7021 CrossRef CAS PubMed.
- E. Lee, S. Muhammad, T. Kim, H. Kim, W. Lee and W. S. Yoon, Adv. Sci., 2020, 7, 1–12 Search PubMed.
- H. Wang, Z. Du, X. Rui, S. Wang, C. Jin, L. He, F. Zhang, Q. Wang and X. Feng, J. Hazard. Mater., 2020, 393, 122361 CrossRef CAS PubMed.
- M. Chen, O. Dongxu, J. Liu and J. Wang, Appl. Therm. Eng., 2019, 157, 113750 CrossRef CAS.
- H. Li, Q. Duan, C. Zhao, Z. Huang and Q. Wang, J. Hazard. Mater., 2019, 375, 241–254 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |