
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the effect of distal histidine and water hydrogen bonding on NO ligation to ferrous and ferric heme: a DFT study†
Fatemeh Fateminasaba,
Aurelien de la Lande b and
Reza Omidyan*a
b and
Reza Omidyan*a
aDepartment of Chemistry, University of Isfahan, 81746-73441 Isfahan, Iran. E-mail: r.omidyan@sci.ui.ac.ir; rezaomidyan51@gmail.com; Fax: +98 31 3668 9732
bUniversité Paris-Saclay, CNRS, Institut de Chimie Physique, UMR8000, 91405, Orsay, France
First published on 8th February 2022
Abstract
The effect of distal histidine on ligation of NO to ferrous and ferric-heme, has been investigated with the high-level density functional theoretical (DFT) method. It has been predicted that the distal histidine significantly stabilizes the interaction of NO ferrous-heme (by −2.70 kcal mol−1). Also, water hydrogen bonding is quite effective in strengthening the Fe–NO bond in ferrous heme. In contrast in ferric heme, due to the large distance between the H2O and O(NO) and lack of hydrogen bonding, the distal histidine exhibits only a slight effect on the binding of NO to the ferric analogue. Concerning the bond nature of FeII–NO and FeIII–NO in heme, a QTAIM analysis predicts a partially covalent and ionic bond nature in both systems.
1. Introduction
Hemoproteins are a vital component of the human body where they play a crucial role.1 Hemes, the iron complexes of porphyrins have an extensive variety of structures and functions, including electron and proton transfer,2 oxidation of substrates,3 transport and storage of metal ions,4 and aerobic breathing.5 Moreover, ligation of dual and more atomic ligands such as O2, N2, NO, CO and H2O affects the function of hemoproteins such as detection, tracking, transport and storage.1,6 Myoglobin (MB) and hemoglobin (HB) transport molecular oxygen in vertebrate blood and muscle cells respectively. The active site of these hemeproteins is the heme B, prosthetic group. Naturally in biological systems, especially MB and HB, iron is found in the common oxidation states including ferrous (FeII) and ferric (FeIII). HB and MB can coordinate CO, NO, O2 in the reduced state, FeII, and also in an oxidized state, FeIII, binds H2O and small anionic ligands (e.g. CN−, SCN−, F−).6 In MB, four N atoms at the equatorial position, one or two ligand groups at the axial sites, could be coordinated the iron cation.7 In the proximal site of MB, the fifth coordination site is accomplished by the imidazole ring of a histidine residue (HIS93) and thus the heme group binds to this protein. Moreover, a distal histidine residue (HIS64) is located near the opposite side in the right position. The distal HIS group is not directly bound to iron but can stabilize the sixth ligand by hydrogen bonding. This hydrogen bond plays a key role in the selectivity of the heme group to adsorb different ligands.6,8The diatomic NO˙ radical, being toxic and corrosive, is an important molecule that is produced, sensed, and detoxified by heme proteins. It plays a vital role in mammals both as a means of immune defense versus pathogens and as a signaling molecule in the cardiovascular organ system and the brain.9 The effect of native distal group (HIS64) and other residues as a distal group into NO, has been studied experimentally and computationally by different groups.10–15 It has been shown that the FeII–N–O angle is bent (∼140°), while FeIII–N–O is linear (∼180°).16 Olson et al.10 reported that ligation of NO to heme is stabilized several times using hydrogen bonding to HIS64. Moreover, by replacing of distal group from HIS64 to apolar residues and the association and dissociation rate constants of NO to iron exhibit slight alterations.10
From a computational perspective, Blomberg and co-workers13 investigated the geometric structures for the binding of O2, NO, and CO to ferrous-heme in three models by Density Functional Theory (DFT) calculations with the B3LYP exchange-correlation (XC) functional. They calculated the Gibbs free energy of binding of O2, NO, and CO for three models (i.e. free porphyrin, myoglobin and cytochrome oxidase). Spiro proposed that the distal HIS64 is allowed to rotate out of its plane, thus it moves closer to the N atom of ligated NO and both the O, and N atoms of NO could be involved in hydrogen bonds with H of HIS64.16 Praneeth et al.17 investigated the interaction between 5-coordinated ferric-hemes with bound NO and axial imidazole ligands both theoretically and experimentally. By DFT calculations, the low spin (LS) FeIII–NO(radical) (S = 0), LS FeII–NO+ (S = 1/2) and high spin (HS) FeIII–NO(radical) (S = 2) states were compared. These results indicated that the properties of these low spin states have differed from each other and also the HS FeIII–NO(radical) state has a weak Fe–NO bond.18 These results were confirmed by Hunt and Lehnert.18 The binding energies of FeP(Im)-AB (AB = NO, O2 and CO, imidazole) evaluated with different density functional models by Liao et al.19 The results showed different values for bonding energy in these systems.19 In the other work, Liao and coworkers15 investigated the binding of NO, and O2 to heme in heme-nitric oxide proteins with DFT and dispersion-corrected DFT methods. Also, the local protein environment has been determined by considering the six nearest surrounding residues in the investigated systems. Particularly, the effects of the distal HIS64 (MB) have been also investigated on the proximal Fe–HIS binding. The heme-AB (AB = O2, NO) binding energies in iron porphyrin FeP(His)(AB) and myoglobin Mb(AB) were determined. However, and to the best of our knowledge, the effects of neighboring amino acids (e.g. histidine) and solvent on the energy and nature of NO interaction to heme are not fully understood yet. In this study, a comprehensive density functional theory (DFT) and QTAIM analysis, have been employed to investigate the mentioned issues as well. As stated in previous reports on the interaction of NO to Fe-porphyrin and Mb (in absence and presence of distal His), the protein environment in Mb has slight effect on the heme-NO binding strength.15,20,21
In this study, we have considered a simplified porphyrin-iron model to decrease the computation costs. This model has been examined in our previous work (comparing to experiment) and it has been established the model is capable enough to describe the binding of small ligands to heme.22 Also, it retains the central feature of heme and has been used widely in previous computational and theoretical studies,24,37,38 and it has been emphasized that this model is accurate enough for investigation of ligand to heme interactions. The notations of [FeIIP], [FeIIIP]+, [FeIIP-NO], [FeIIIP-NO]+, [MI-FeIIP-NO], [MI-FeIIIP-NO]+ respectively for four coordinated heme, NO ligated and six coordinated heme-NO systems will be used hereafter (P states to porphyrin and MI refers to 5-methyl imidazole).
2. Computational methods
2.1. The heme-model systems
To investigate environment effect on NO binding to ferrous and ferric-heme, we have considered three models:(i) The iron–porphyrin–NO in five- and six coordinated states.
(ii) Histidine interacted model in five- and six coordinated states from distal position.
(iii) Mono-hydrated systems in five- and six coordinated states (see Fig. 1).
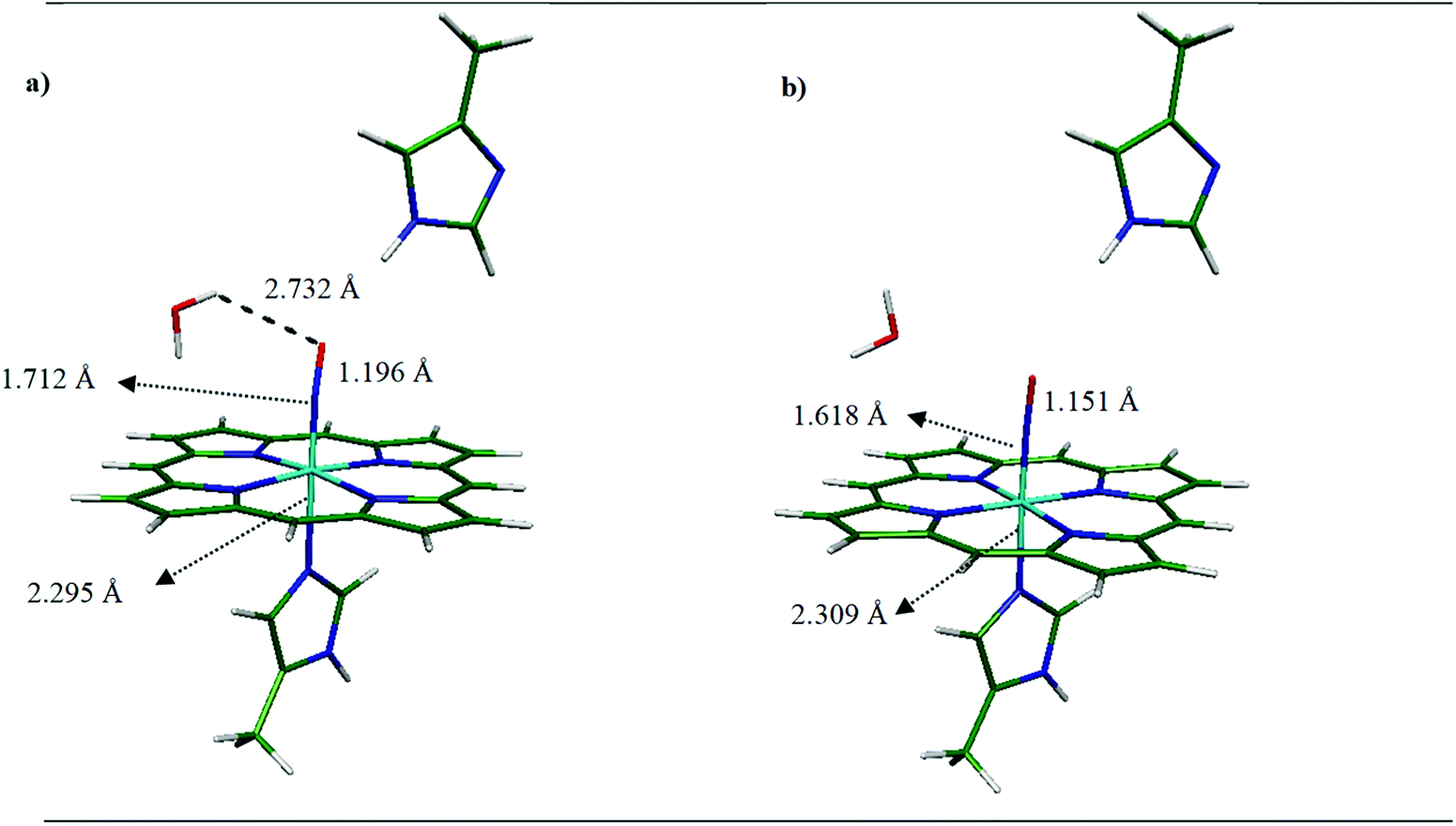 | ||
| Fig. 1 Optimized structure and model system for (a) [MI-FeIIP-NO⋯MI] and (b) [MI-FeIIIP-NO⋯MI]+ determined at the OPBE/DZVP-GGA/GEN-A2* level of theory. | ||
In this regard, the 3D coordinates of the crystal structure of oxyMb at 1.0 Å resolution has been selected as the heme model (PDB ID 1A6M)23 then O2 was replaced by NO in the next step, for further studies and to investigate the effect of histidine (HIS64), the side chains of the heme systems were replaced all by hydrogen atoms. We previously showed that this system is a suitable minimal model to investigate binding interactions.22 Furthermore, following Kepp et al.,24 the distal MI was modeled as a HIS64 (wild type) for the FeII and FeIII systems. Based on Kepp and Dasmeh,24 in distal imidazole, the α carbon was replaced by a hydrogen atom and then kept fixed in all geometry optimizations to prevent system deformation and change in system conformation. Also, in line with our previous studies,22,25 the 5-methyl imidazole (MI) group was considered as the histidine residue in axial position. As shown in Fig. 1, the NO is positioned bent to the FeII and linear to the FeIII and the methyl-imidazole is placed on the 6th coordinated axial ligand to FeII and FeIII.
Moreover, the effect of MI as the model of HIS64 on NO, with FeII and FeIII-porphyrins in 5 and 6 coordinated states in the presence and absence of water molecule with water hydrogen binding role have been also investigated. It is worth mentioning that the most stable spin states have been considered for heme-FeII and heme-FeIII complexes, namely the doublet state for 5 and 6 coordinated heme-FeII-NO18,26 and the singlet state for 5 and 6 coordinated heme-FeIII-NO.17,18 We have used the deMon2k software27 in the auxiliary density functional theory (ADFT) framework for geometry optimizations of considered systems. ADFT is a fast and robust method and also computationally cost-effective DFT methodology that resorts to variationally fitted densities to evaluate classical coulombic and exchange-correlation (XC) contributions.28
2.2. Methodology: ADFT and geometry optimization
We used OPBE as a XC functional developed by Swart and co-workers,29 that combines the OPTX exchange functional to the Perdew–Burke–Ernzerhof (PBE) model of electronic correlation. A double zeta valence polarization basis set calibrated for the first row transition metals have been employed for application with generalized gradient approximation (GGA) functionals have been chosen (DZVP-GGA). We have worked with the spin-unrestricted open shell Kohn–Sham (SR UKS) formalism for all calculations. ADFT relied on fitted electronic densities that are expanded on automatically generated auxiliary basis sets. We have considered the GEN-A2, which contains s, p and d auxiliary functions for C, H, and the GEN-A2* auxiliary function sets, that includes, p, d, f, and g auxiliary functions, for Fe, N, and O in NO as a ligand. Moreover, an empirical dispersion energy correction has been used due to the importance of van der Waals interaction between CO and considered systems.30,31To integrate the XC energy and potential, an adaptive grid of fine accuracy has been used (10−6 Ha). SCF iterations and geometry optimizations were also performed with convergence criteria of 10−7 Ha and 10−5 Ha bohr−1 respectively (Ha stands for Hartree).
Vibrational frequencies were calculated on the optimized structures at PBE/DZVP/GEN-A2* level of theory. These analyses confirmed that all structures were at their lowest energy state and no imaginary frequencies appeared.
2.3. Binding energy and QTAIM studies
We have determined binding energies of NO to heme (FeII, FeIII) in the presence and absence of environmental species. As explained above ground state geometry optimizations have been carried out at the OPBE/DZVP-GGA level, and subsequently, binding energies have been determined in gas phase or in solvent conductor-like polarizable continuum (CPCM) at the B3LYP/def2-TZVP level for all atoms using Gaussian 16 program.32 CPCM is used to model the solvent effect in quantum chemical calculations, where the solvent is exhibited as a dielectric polarizable continuum and the solute is located inside the molecular shape cavity. The solvent reaction field is described using polarization charges on the cavity surface. The cavity is created by the GEPOL algorithm by a solvent-excluding or solvent-accessible surface.33 The binding energy for all structures was corrected by basis set superposition error (BSSE) and also the empirical dispersion (DJ). The BSSE was evaluated with the counterpoise methodology of Boys and Beranrdi.34 Empirical dispersion was added using B3LYP/def2-TZVP/GDBJ (D3 version of Grimme with Becke-Johnson damping factors).35 Additionally, the binding energy of NO complexation to penta (5) and hexa (6) coordinated of FeIIP and FeIIIP were calculated at B3LYP/def2-TZVP level of theory and are compared with the values of binding energy from previous experimental and computational methods13,36–38 (see Table S1 in ESI File).†Bader's Quantum Theory of Atoms in Molecules, (QTAIM), analyzes a various interaction of inter- and intramolecular, specially atom–atom interaction e.g. covalent bonds, or hydrogen bonds as an intermolecular bonding interactions.39 In this study, QTAIM has been carried out using AIMAll software.40
3. Results and discussions
3.1. Ferrous-heme
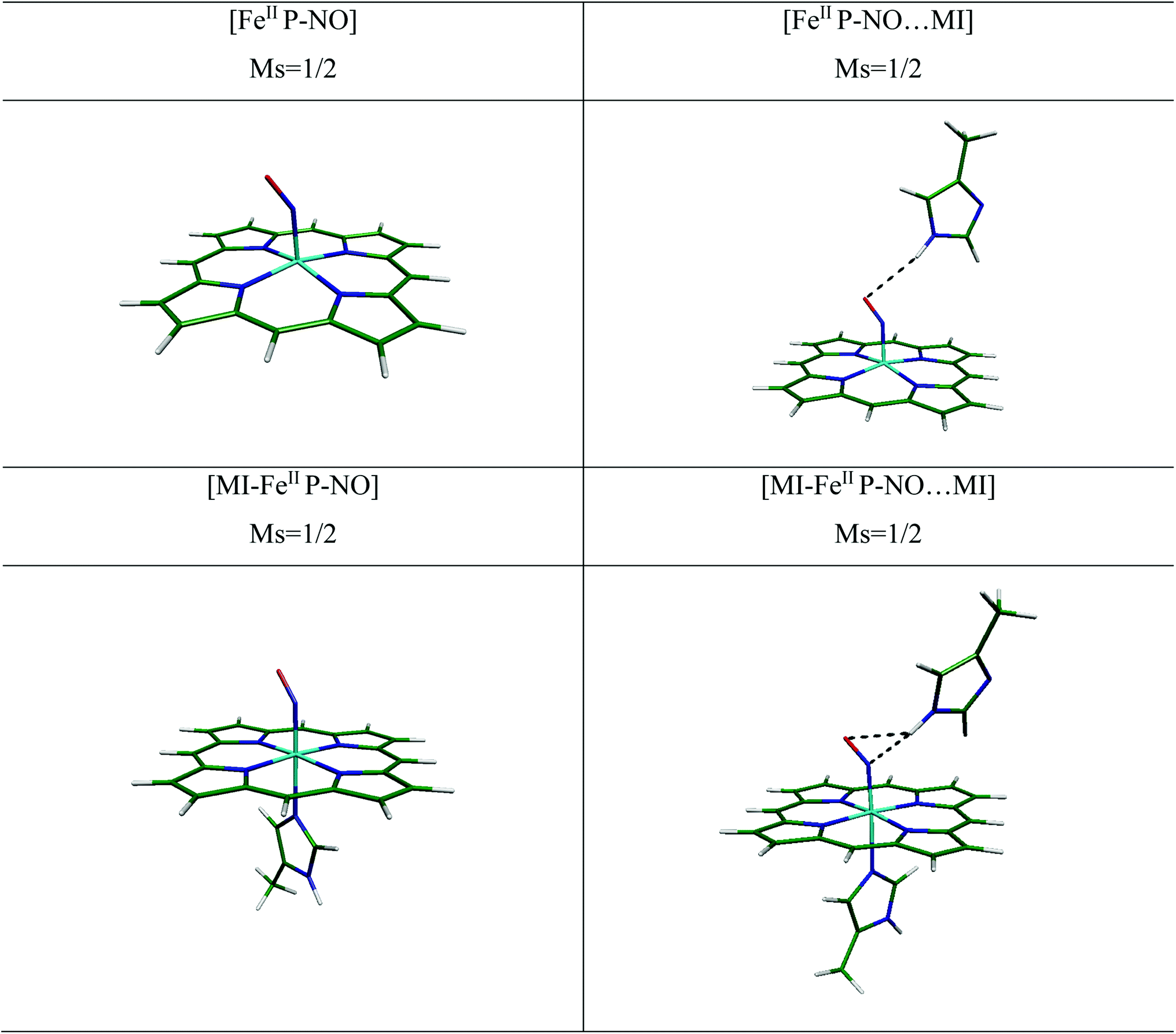 | ||
| Fig. 2 The optimized structures of complexes of ferrous with NO ligand investigated with/without HIS64 (MI) distal group at the OPTX-PBE/DZVP-GGA/GEN-A2* level of theory. Ms indicates to spin quantum number. | ||
| Complex | Ms | Fe–NNO (Å) | N–O (Å) | Fe-Nax (Å) | Doming | ∠FeNO | O (N)⋯Hdistal (Å) | ν(Fe–N) cm−1 | ν(N–O) cm−1 |
|---|---|---|---|---|---|---|---|---|---|
| [FeIIP-NO] | 2 | 1.690 | 1.183 | — | 0.262 | 145.294 | — | 634.1 | 1779.90 |
| Experiment41 | 1.717 | 1.122 | 144.4 | ||||||
| Calculated37 | 1.69 | 1.19 | 146 | ||||||
| [FeIIP-NO⋯MI] | 2 | 1.690 | 1.183 | — | 0.268 | 144.611 | 3.415 (O⋯H) | 632.9 | 1760.30 |
| 2.974 (N⋯H) | |||||||||
| [MI-FeIIP] | 5 | — | — | 2.125 | −0.252 | — | — | — | — |
| Exp.23 | 2.141 | −0.365 | |||||||
| [MI-FeIIP-NO] | 2 | 1.724 | 1.185 | 2.212 | 0.082 | 140.1 | — | 624.0 | 1765.8 |
| Experiment44 | 1.743 | 1.144 | 2.180 | 142.2 | |||||
| Calculated37 | 1.720 | 1.200 | 2.220 | 138.0 | |||||
| [MI-FeIIP-NO⋯MI] | 2 | 1.716 | 1.191 | 2.252 | 0.083 | 140.19 | 2.693 (N⋯H) | 634.0 | 1732.7 |
| Calculated15 | 1.728 | 1.195 | 2.182 | −0.071 | 140.90 | 3.013 (O⋯H) | |||
| 3.06 (O⋯H) | |||||||||
| 2.357 (N⋯H) | |||||||||
| [FeIIP-NO]⋯H2O | 2 | 1.684 | 1.186 | — | 0.268 | 145.110 | 2.737 (O⋯Hw) | 644.4 | 1765.40 |
| [FeIIP-NO⋯MI]⋯H2O | 2 | 1.681 | 1.192 | — | 0.264 | 143.894 | 2.590 (O⋯HMI) | 647.2 | 1741.10 |
| 2.511(O⋯Hw) | |||||||||
| [MI-FeIIP]⋯H2O | 5 | — | — | 2.115 | −0.232 | — | — | — | — |
| [MI-FeIIP-NO]⋯H2O | 2 | 1.710 | 1.192 | 2.304 | 0.074 | 140.126 | 2.903 (N⋯Hw) | 652.9 | 1727.2 |
| 2.687 (O⋯Hw) | |||||||||
| [MI-FeIIP-NO⋯MI]⋯H2O | 2 | 1.712 | 1.196 | 2.295 | 0.108 | 139.232 | 3.090 (O⋯H) | 636.7 | 1727.2 |
| 3.045 (N⋯Hw) | |||||||||
| 2.732 (O⋯Hw) |
NO complexation patterns to [MI-FeIIP] in free and in presence of MI are collected in Table 1. In six-coordination complexes, the planarity of porphyrin ring is well-preserved. In the 6-coordinated complexes (6C), binding of NO to [MI-FeIIP] complex leads to stability of the Fe–NO interaction and also the trans-repulsive effect of the NO ligand on axially coordinated N-donor MI ligand, which induces elongated FeII–Nax bond length.16 As a result, the MI in the proximal position is almost displaced and gets away from Fe. Moreover, the Fe–Nax bond length (2.212 and 2.252 Å) are longer than corresponding bonds in the decarboxylase (2.125 Å).
In the [MI-FeIIP-NO] complex, the Fe–NO and N–O bond lengths and also the Fe–N–O bond angle has been evaluated to 1.724, 1.185 Å, and 140.1° respectively. Moreover, the Fe–NO and N–O distances in [MI-FeIIP-NO] complex are longer than the corresponding bond lengths in [FeIIP-NO] complex and the bending of Fe–N–O angle in six coordinated complex (∼5°) is more than the corresponding bond angle in the 5 coordinated complex (5C). Also, the Fe–N–O bond angle (140.1°) is in agreement with experimental X-ray data for [FeII(TPP)(NO)(1-MeIm)] complex,44 however the Fe–NO, N–O, and Fe–Nax bond lengths (1.743, 1.144, and 12.180 Å) have been only slightly differed by 0.019, 0.041 and 0.032 Å with [MI-FeIIP-NO] complex in this work. Furthermore, our calculated minimum structural parameters of [MI-FeIIP-NO] are almost in agreement with literature.14,15 Therefore, the consistency of geometry parameters with experimental results confirms the validity of our theoretical results for the investigation of considered heme models.
In the [MI-FeIIP-NO⋯MI] complex (Fig. 2), in presence of distal MI the Fe–NO bond has been predicted to be slightly shortened (from 1.724 Å to 1.716 Å), and other bond lengths (N–O, Fe–Neq, and Fe–Nax) exhibit no significant change. Furthermore, the calculated Fe–NO and N–O bond lengths of 1.716 and 1.191 Å, are comparable with the corresponding experimental X-ray values of 1.889 and 1.154 Å (ref. 11) respectively which are also in line with Liao's report.15
Moreover, an inspection of the vibrational frequency of normal modes in [MI-FeIIP-NO], compared to [MI-FeIIP-NO⋯MI] exhibits that the vibrational frequency of Fe–NO stretching-mode is 624 cm−1 (in the absence of distal MI) and 634 cm−1 (in presence of MI). In addition, in the [MI-FeIIP-NO⋯MI] complex, the N(NO)⋯HN(MI) and O(NO)⋯HN(MI) distances have been determined to be 2.693 and 3.013 Å (see Table 1) which indicate to increase in π-back-donation from the occupied dπ (dxz and dyz) orbitals of iron to unoccupied π*–NO orbitals. Since slight alteration in geometry parameters and vibrational frequency has been predicted, it could be concluded that distal MI (in absence of hydration) only slightly affects the FeII–NO interaction in heme system. In comparison NO ligation of heme in Mb, the NO interaction with cytochrome c' (AXCP) has been investigated by Marti et al.45 and the optimized parameters have been reported there. In the presence of distal group (LEU16) of AXCP, the Fe–NO, N–O and Fe–Nax bonds have been reported to 1.74, 1.21, and 2.33 Å (in protein media) by QM/MM and also 1.75, 1.21, and 2.24 Å (in gas phase) by QM method. In the ligation of NO to AXCP, due to the negative NO trans effect, the Fe–Nax has been reported to be weakened. In other work by Marti (2008),46 the NO interaction with HB has been investigated by molecular dynamics (MD) simulations. The results demonstrated that due to trans effect of N(NO)–Fe–Nax, the Fe–Nax is weakened. These results are in agreement with the optimized geometry parameters of [MI-FeIIP-NO⋯MI] in our work.
| ΔEbinding = E[FeIIP-NO⋯MI] (LS, S = 1/2) − (E[FeIIP⋯MI] (HS, S = 1) + E[NO] (S = 1/2)) | (1) |
| ΔEbinding = E[MI-FeIIP-NO⋯MI] (LS, S = 1/2) − (E[MI-FeIIP⋯MI] (HS, S = 2) + E[NO] (S = 1/2)) | (2) |
| Complex | ΔE (kcal mol−1) (gas) | ΔE (kcal mol−1) (solvent) |
|---|---|---|
| [FeIIP-NO] | −16.6 | −15.8 |
| Experiment:47 −26.6 ± 0.7 | ||
| Calculated:38 −27.5 | ||
| [FeIIP-NO⋯MI] | −16.5 | −14.8 |
| [MI-FeIIP-NO] | −17.9 | −16.9 |
| Calculated:49 −16.3 | ||
| [MI-FeIIP-NO⋯MI] | −19.1 | −16.7 |
| Experiment38 (Mb-NO): −22.8 | ||
| [FeIIP-NO]⋯H2O | −17.2 | −14.1 |
| [FeIIP-NO⋯MI]⋯H2O | −19.9 | −17.0 |
| [MI-FeIIP-NO]⋯H2O | −19.8 | −16.8 |
| [MI-FeIIP-NO⋯MI]⋯H2O | −19.6 | −16.3 |
As presented in Table 2, The binding energy of [FeIIP-NO] has been reported experimentally to be −26.6 and −28.9 kcal mol−1 ([FeII-Tpyr-PH2-NO]2+) by Chen et al.47
Furthermore, NO-binding energy to ferrous heme has been determined theoretically by several groups15,37,38 being reported within the broad range of –(5.5–32.0) kcal mol−1 based on the different theoretical levels. In the present study, the binding energy for heme-NO has been determined to −16.6 kcal mol−1 at B3LYP/def2-TZVP level of theory. It can be concluded that the binding energy values are quite sensitive to the choice of DFT method, nevertheless, the selected theoretical model of this work has been previously examined for heme systems and gave reliable results.22,25
In the presence of the MI in distal position (i.e., in [FeIIP-NO⋯MI] complex), we have determined the binding energy of NO to 4C heme-NO to −16.5 and −14.8 kcal mol−1 in the gas phase and implicit solvent medium respectively. In the 6-coordinated [MI-FeIIP-NO] complex, the binding energy has been determined to −17.9 kcal mol−1. This is while the experimental binding energy of NO in [MI-FeIIP-NO] complex has been reported to −22.8 kcal mol−1 by Olson10 and Springer.48 Nevertheless, the corresponding theoretical value has been reported to lie within the range of –(12.45–36.0) kcal mol−1 by different theoretical groups.13,37,38,49,50 Thus, our theoretical result (−16.5 kcal mol−1) is in good agreement with experimental- and previous theoretical reports as well.
Moreover, in Table 2, water H-bond effect, in addition to distal group, has been presented. In the [FeIIP-NO]⋯H2O complex, the formation of the HOH⋯ON hydrogen bond, results in a slight shortening in the Fe–NO bond length and increases the ν(Fe–NO) vibrational frequency. The binding energies are determined to −17.2 kcal mol−1. Moreover, in the [FeIIP-NO⋯MI]⋯H2O complex, the interaction of NO to [MI-FeIIP⋯MI]⋯H2O is strengthened by the formation a hydrogen bond.
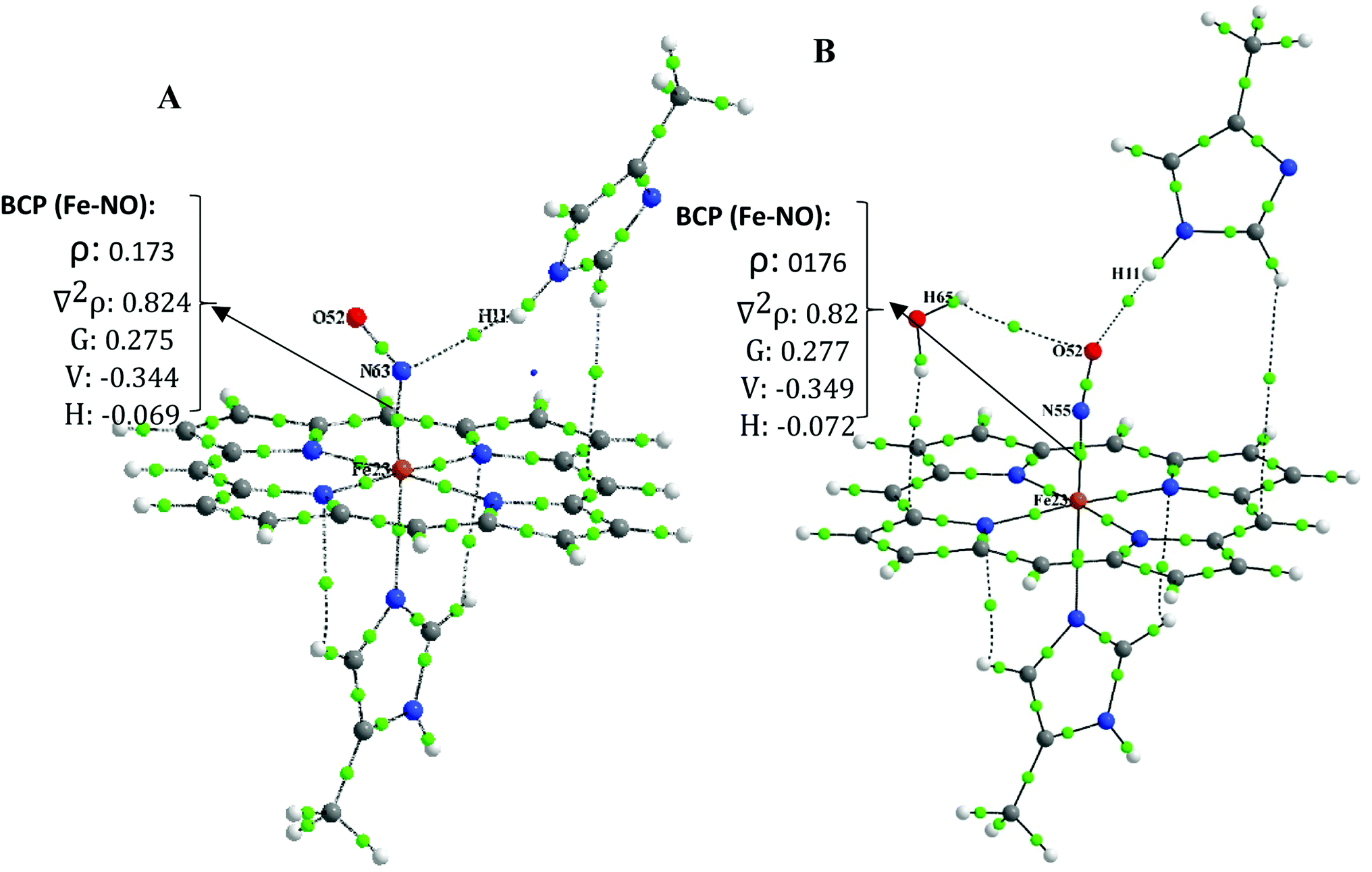 | ||
| Fig. 3 The QTAIM molecule graph of (A) [MI-FeIIP-NO⋯MI] complex, (B) [MI-FeIIP-NO⋯MI]⋯H2O. | ||
To simplify, we follow our comparative studies to investigate the variation in topological properties with a change in the interatomic distances, d(Fe–NO). Concerning the NO–Fe bond, the ρBCP is significantly large (0.169 a.u. < ρ(r) < 0.194 a.u.) and ∇2ρBCP is positive (0.784 a.u. < ∇2ρ(r) < 0.830 a.u.) indicating a “partially covalent” character of the coordination bonds.51 Furthermore, the values of local electron potential energy density, V(r) (−0.339 a.u. < V(r) < −0.387 a.u.), is larger than the local electron kinetic energy density G(r) (0.268 a.u. < G(r) < 0.289 a.u.). The |V(r)|/G(r) ratio is another useful parameter, the 1 <| V(r)|/G(r) < 1.325 and |V(r)|/G(r) < 1 is characteristic of an ionic interaction and |V(r)|/G(r) > 2 indicates to a pure covalent bond. Therefore, our QTAIM analysis indicates that the Fe–NO bond is not only a pure covalent bond but also it has both the ionic/covalent characters.
Moreover, from Table S3,† it is seen that with shortening Fe–NO bond length in [FeIIP-NO⋯MI]⋯H2O compared to its individual analogue (([MI-FeIIP-NO]) to [MI-FeIIP NO⋯MI]⋯H2O) the ρBCP increases and also VBCP becomes more negative when d(Fe–NO) decreases.
Due to this fact that the BCP with 0.002 < ρ(r) < 0.035, ∇2ρ(r) > 0, |V(r)|/G(r) < 1 and H(r) > 0 is indicative of the presence of closed-shell (non-covalent bond such as hydrogen bond, van der walls, ionic interaction).39,51 In the distal interacted [FeIIP-NO⋯MI] complex, the ρBCP and ∇2ρBCP at BCP of N⋯HMI bond are calculated to be 0.006 and 0.021, respectively. These values are both in the range of hydrogen bond interaction. Also, in monohydrated [FeIIP-NO⋯MI]⋯H2O complexes, the ρBCP and ∇2ρBCP values at BCP of O⋯Hw and O⋯HMI bonds are within 0.004–0.006 and 0.014 < ∇2ρBCP < 0.023 which are both relatively significant, being in the range of strong hydrogen bonds. In the six coordinated complex of [MI-FeIIP-NO]⋯H2O, the ρBCP and ∇2ρBCP values at BCP of O⋯Hw bond are evaluated to 0.005 a.u. and 0.017 a.u. respectively, confirming the existence of a hydrogen bond between H (H2O)⋯O (NO).
Due to the large ρBCP (0.169 a.u. < ρ(r) < 0.194 a.u.) and positive ∇2ρBCP (0.784 a.u. < ∇2ρ(r) < 0.830 a.u.), it can be concluded that the FeII–NO bond has an ionic-covalent property.
3.2. Ferric-heme
| Complex | Ms | Fe–NNO (Å) | N–O (Å) | Fe-Nax (Å) | Dominga | ∠FeNO | O (N)⋯Hdistal (Å) | ν(Fe–N) cm−1 | ν(N–O) cm−1 |
|---|---|---|---|---|---|---|---|---|---|
| a Displacement of the Fe from the porphyrin and heme plane that is defined as doming.b [FeIII(NO)(TPP)]ClO4. | |||||||||
| [FeIIIP-NO]+ | 1 | 1.599 | 1.159 | — | 0.335 | 179.955 | — | 435.6 | 2002.9 |
| Calculated55 | 1.614 | 1.145 | — | 180.0 | |||||
| [FeIIIP-NO⋯MI]+ | 1 | 1.605 | 1.164 | — | 0.314 | 172.384 | 2.900 | 425.8 | 1961.20 |
| [MI-FeIIIP]+ | 6 | — | — | 2.075 | −0.394 | — | — | — | — |
| Calculated22 | 2.100 | −0.430 | |||||||
| [MI-FeIIIP-NO]+ | 1 | 1.620 | 1.150 | 2.017 | 0.066 | 179.300 | — | 627.1 | 2027.00 |
| Experimentb,53 | 1.628 | 1.148 | 1.973 | — | 176.300 | ||||
| [MI-FeIIIP-NO⋯MI]+ | 1 | 1.618 | 1.151 | 2.039 | 0.035 | 179.394 | 3.575 | 629.3 | 2022.10 |
| Calculated56 | 1.650 | 1.130 | 2.020 | 175.80 | — | ||||
| [FeIIIP-NO]+⋯H2O | 1 | 1.600 | 1.156 | — | 0.323 | 179.440 | 3.201 (O⋯O) | 433.5 | 2012.80 |
| [FeIIIP-NO⋯MI]+⋯H2O | 1 | 1.606 | 1.161 | — | 0.325 | 166.543 | 3.298 (O⋯O) | 417.0 | 1966.50 |
| 2.607 (O⋯H) | |||||||||
| [MI-FeIIIP]+⋯H2O | 6 | — | — | 2.066 | 0.376 | — | — | — | — |
| [MI-FeIIIP-NO]+⋯H2O | 1 | 1.618 | 1.146 | 2.043 | 0.034 | 179.416 | 3.068 (O⋯O) | 630.8 | 2060.30 |
| [MI-FeIIIP-NO⋯MI]+⋯H2O | 1 | 1.620 | 1.147 | 2.042 | 0.036 | 176.979 | 3.463(O⋯H) | 621.8 | 2041.50 |
| 3.070 (O⋯O) | |||||||||
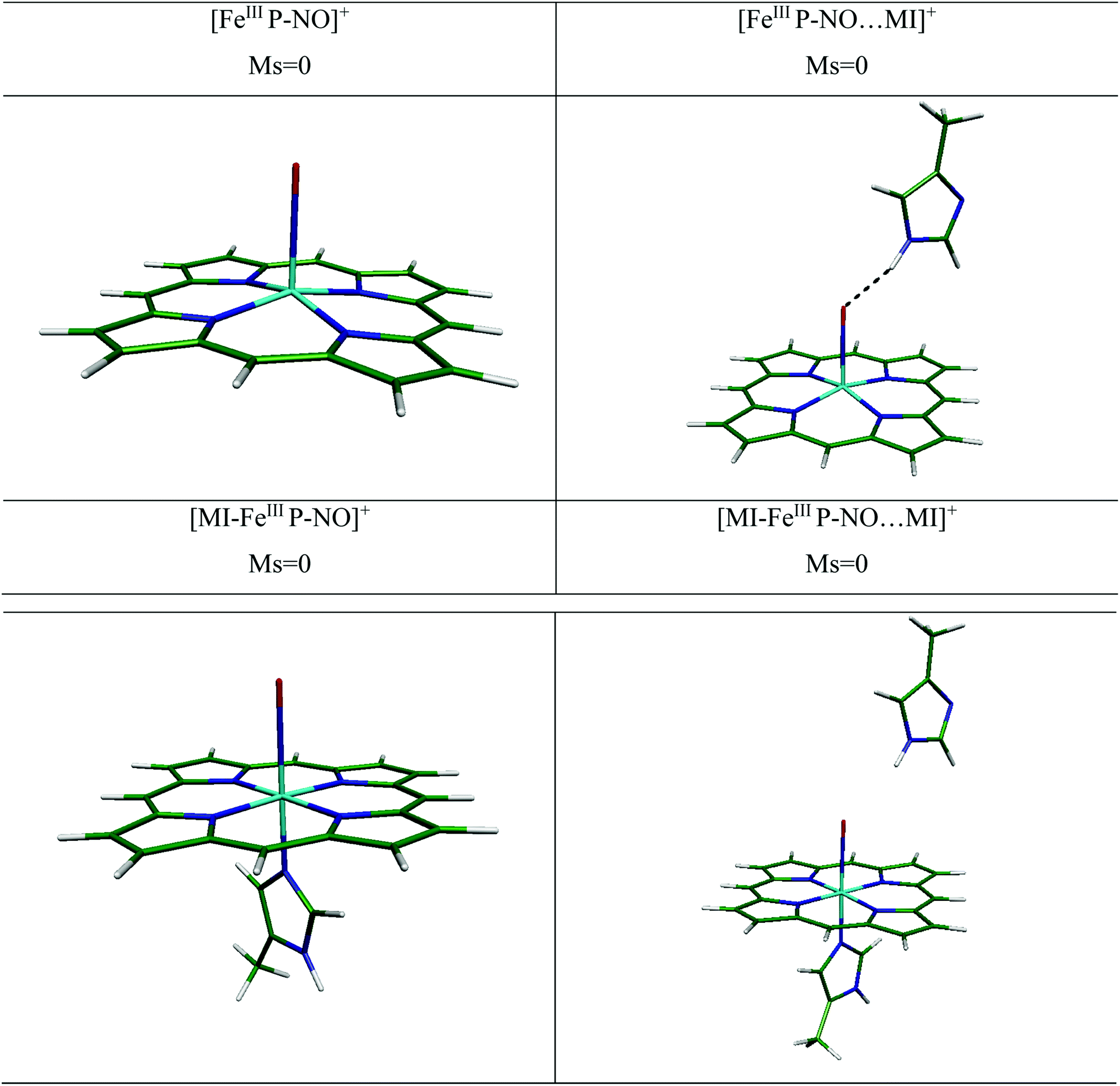 | ||
| Fig. 4 The optimized structures of complexes of ferric with NO ligand investigated with/without HIS64 (MI) distal group at the OPTX-PBE/DZVP-GGA/GEN-A2* level of theory. Ms indicates to spin quantum number. | ||
(i) In the bare and distal interacted [FeIIIP-NO]+ and [MI-FeIIIP-NO]+. We have investigated the NO ligation to [FeIIIP]+ in free and presence of distal MI. We have presented the optimized structures for several [FeIIIP-NO]+ complexes in Fig. 4. In the presence of distal MI, the planarity of porphyrin ring in the [FeIIIP-NO]+ and [FeIIIP-NO⋯MI]+ complexes have been significantly preserved and the out-of-plane distortion of Fe from the porphyrin plane (doming) is in the range of 0.314–0.335 Å.
The experimental and theoretical studies of geometry and electronic structure of ferric heme-NO have been investigated by several groups.22,52,53 The experimental values of Fe–NO and N–O bond lengths have been reported to be in the range of 1.640–1.644 Å and 1.110–1.153 Å, respectively.52,53 In the [FeIIIP-NO⋯MI]+ complex, in the presence of distal group, the Fe–NO bond length slightly elongates (0.006 Å), the FeIII out-of-plane doming (0.021 Å) and Fe–N–O angle (7.6°) decrease, compared to its individual analogue. Furthermore, the vibrational frequencies of ν(Fe–NO) has been poorly changed from 435.6 cm−1 to 425.8 cm−1 in the presence of MI (9.8 cm−1). Consequently, it could be concluded that the distal MI residue has a negligible effect on NO bonding to 5C ferric-heme.
We have also determined the optimized structure of the 6C [MI-FeIIIP-NO]+ systems, in the presence and absence of distal MI (see Table 3). In [MI-FeIIIP-NO]+ and [MI-FeIIIP-NO⋯MI]+, the planarity of porphyrin ring is preserved. The strong π-back bonding and weak σ-bond explain the short Fe–NO bond in [MI-FeIIIP-NO]+ and insignificant trans-effect of NO in ligation of NO with ferric-heme.17,18,54 In the [MI-FeIIIP-NO]+ complex, the Fe–NO and N–O are determined to be 1.620 and 1.150 Å, respectively. Also, the Fe–Nax bond length and Fe–N–O angle are 2.017 Å and 179.30°. In the [Fe(TPP)(NO)(MI)]PO2F2 complexes,53 the Fe–NO bond lengths have been evaluated in the range 1.628 and N–O 1.148. Å, in agreement with the obtained structural parameters (Fe–NO: 1.620 Å, N–O 1.150 Å). Other calculated structural parameters of [MI-FeIIIP-NO]+ contain the Fe–Nax (2.00 Å) and Fe–N–O angle (179.300°) and the vibrational frequency of ν(Fe–NO) is calculated 627.10 cm−1. In the [MI-FeIIIP-NO⋯MI]+ complex, the calculated values of Fe–NO and N–O bond lengths are 1.618 and 1.151 Å being comparable with the corresponding computational results of 1.650 and 1.130 Å. In the presence of distal MI residue, the Fe–NO has been predicted to be slightly shortened (from 1.620 Å to 1.614 Å). However, since the alteration in geometry parameters and vibrational frequency is poorly significant, it could be concluded that distal MI has not a significant alteration effect on the FeIII–NO bonding in heme system. We will investigate this issue in the next sections by considering binding energy values and QTAIM results.
(ii) Water H-bond effect. Due to the significant solvent effect on NO binding, in this section we have investigated the effect of explicit water on NO ligation of ferric-heme system. The optimized geometry parameters of hydrated systems have been tabulated in Table 3. Also, the optimized structures have been presented in Fig. S2 in ESI file.† As shown, water H-bond affects the geometry parameters of [FeIIIP-NO⋯MI]+ complex, the FeIII out-of-plane doming (0.011 Å) increases, the Fe–N–O angle decreases from 172.82° in [FeIIIP-NO⋯MI]+ complex to 166.54° in [FeIIIP-NO⋯MI]+⋯H2O system. In this system, an H-bond between distal MI and O(NO) have been predicted to form. Thus, the vibrational frequencies of ν(Fe–NO) has been red-shifted from 425.8 cm−1 in the absence of water molecule to 417.0 cm−1 in the presence of that (∼8.8 cm−1). Consequently, the distal MI and also water H-bond have no significant effect on binding of NO to 5C ferric heme.
By comparing individual [MI-FeIIIP-NO⋯MI]+ complex with water H-bond [MI-FeIIIP-NO⋯MI]+⋯H2O analogue, poorly change on Fe–NO (1.620 Å) and N–O (1.147 Å) bond distances have been predicted. Also, the vibrational frequencies of Fe–NO and N–O have been slightly changed (from 629.3 cm−1 in [MI-FeIIIP-NO⋯MI]+ complex to 621.8 cm−1 in [MI-FeIIIP-NO⋯MI]+⋯H2O complex (by 7.5 cm−1)) and there is no hydrogen bond between H (distal MI) and H2O with NO. We will more investigate the distal histidine microhydration effects on binding energy of NO to ferric heme in the next sections.
Additionally, more details of optimized geometry parameters and XYZ coordinates can be found in ESI file (see Table 3, Fig. 3, S2 and Table S6 in ESI file).†
| ΔEbinding = E[FeIIIP-NO⋯MI]+ (LS, S = 0) − (E[FeIIIP⋯MI]+ (HS, S = 3/2) + E[NO] (S = 1/2)) | (3) |
| ΔEbinding = E[MI-FeIIIP-NO⋯MI]+ (LS, S = 0) − (E[MI-FeIIIP⋯MI]+ (HS, S = 5/2) + E[NO] (S = 1/2)) | (4) |
For [FeIIIP-NO]+ complex, there is π-back bonding between a single two-electron low-spin FeIII dπ (dxz, dyz) orbital and unoccupied NO π* orbital. Furthermore, the other dπ orbital is formed between unoccupied FeIII dπ (dxz, dyz) orbital and half-filled NO π* orbital, and thus a more covalent bond with the lone π*-electron in the HOMO of NO can be organized.55 As detailed in Table 4, the [FeIII-heme-NO]+ binding energy has been reported experimentally to be −24.88 ± 0.71 kcal mol−1 in gas phase22 and −34.4 kcal mol−1 based on the CPMD (Car–Parrinello Molecular Dynamics) theoretical model.57 We have determined the binding energy for [FeIIIP-NO]+ to −11.2 kcal mol−1 at B3LYP/def2-TZVP level of theory.
| Complex | ΔE (kcal mol−1) (gas) | ΔE (kcal mol−1) (solvent) |
|---|---|---|
| Ferric-heme | ||
| [FeIIIP-NO]+ | −11.2 | −4.2 |
| Experiment:47 −24.88 ± 0.71 | ||
| [FeIIIP-NO⋯MI]+ | −8.0 | 0.5 |
| [MI-FeIIIP-NO]+ | −12.3 | −10.1 |
| Calculated:17 −12.8 | ||
| Calculated:17 −12.3 | ||
| [MI-FeIIIP-NO⋯MI]+ | −13.3 | −7.2 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Ferric-heme (water H-bond effect) | ||
| [FeIIIP-NO]+⋯H2O | −8.1 | −0.7 |
| [FeIIIP-NO⋯MI]+⋯H2O | −4.9 | 2.6 |
| [MI-FeIIIP-NO]+⋯H2O | −13.1 | −8.9 |
| [MI-FeIIIP-NO⋯MI]+⋯H2O | −11.2 | −6.3 |
Moreover, the increasing of Fe–NO bond length in [FeIIIP-NO⋯MI]+, from 1.599 Å into 1.605 Å in the presence of MI distal position is in line with decreasing of the calculated binding energy from −11.2 kcal mol−1 to −8.0 kcal mol−1. In [FeIIIP-NO]+⋯H2O complex, in the presence of water molecule, the calculated binding energy has diminished from −11.2 kcal mol−1 to −8.1 kcal mol−1 while there is a slight alteration compared to [FeIIIP-NO⋯MI]+ (1.605 Å), but the binding energy has been decreased from −8.0 kcal mol−1 to −4.9 kcal mol−1.
In the 6 coordinated [MI-FeIIIP-NO]+ complex, we have determined the binding energy of NO to −12.3 kcal mol−1 in gas phase and −10.1 kcal mol−1 in solvent medium. The result of binding energy is in good agreement with computational binding energy (−12.3 kcal mol−1 at BP86/TZVP level of theory) for [FeIII(P)(MI)(NO)]+ in LS state.17 In presence of distal MI, the Fe–NO bond changes from 1.620 Å ([MI-FeIIIP-NO]+) to 1.618 Å ([MI-FeIIIP-NO⋯MI]+ complex), and the NO binding energy has been determined to −13.3 kcal mol−1. The results indicate that the distal MI group slightly stabilizes the ligation of NO to 6C ferric-heme (∼1.0 kcal mol−1).
Moreover, insertion of a water molecule close to NO in ferric-heme results to no significant alteration in the FeIII–NO bond length. In [MI-FeIIIP-NO]+⋯H2O complex, the Fe–NO bond is determined to be 1.618 Å, being similar to that of [MI-FeIIIP-NO]+ complex. Furthermore, in [MI-FeIIIP-NO⋯MI]+⋯H2O complex, a small increase in Fe–NO bond distance (from 1.618 to 1.620 Å) results in the slight decrease of the binding energy from −13.3 kcal mol−1 (in absence of water molecule) to −11.2 kcal mol−1 (in presence of water molecule). As a result, the MI as a distal group slightly stabilizes the interaction of NO ligand to 6C ferric-heme while no significant change has been predicted to take place in the 5C system.
3.3. QTAIM results: Fe–NO bond analysis
(i) In the 6C ferric-heme complexes, due to the relatively large ρBCP (0.197 < ρ(r) < 0.202 a.u.) and positive ∇2ρBCP (1.406 a.u. < ∇2ρ(r) < 1.445 a.u.), it can be concluded that the FeIII–NO bond has a polar-covalent property.
(ii) In the 5 and 6C ferric-heme complexes, when both of the MI and H2O have been considered, the ρBCP and ∇2ρBCP values at BCP of FeIII–NO bond in ligation of NO to ferric heme only slightly change (0.003–0.005 a.u. for ρBCP) and (0.039–0.118 au for ∇2ρBCP).
It is worth noting that the FeIII–NO bond in the 5 and 6C ferric-heme complexes is not a pure covalent bond, instead it has both the ionic and covalent characters.51 As a consequence, in both 5C and 6C complexes, these results are in well agreement with calculated binding energy values and it seems that the distal MI and water do have a slight effect on strengthening of the NO to ferric heme.
4. Conclusion
Density functional theory has been employed to investigate the effect of distal histidine, water hydrogen bond, and bulk solvent model on the interaction of NO with ferrous and ferric-heme. It has been predicted that water hydrogen bond and distal histidine stabilize NO binding by ∼2.7 kcal mol−1 in the 6-coordinated ferrous heme-NO system [MI-FeIIP-NO]. The results exhibit a synergic effect of water and distal MI interactions on the stabilization of NO–FeII bond in ferrous heme system, since of the formation of strong hydrogen bonds. This is while the distal MI and water exhibit a less pronounced effect on stabilization of FeIII–NO interaction in ferric heme.In addition, we have investigated the Fe–NO bond nature based on the Quantum Theory of Atom in Molecule (QTAIM). It has been shown that the Fe–NO bond has partially ionic nature in addition to its covalent feature in both of ferric and ferrous heme.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support of research council of University of Isfahan is appreciated. We also acknowledge extended discussions and valuable comments received from Professor Niloufar Shafizadeh and Professor Benoit Soep (University of Paris Saclay). We thank GENCI for a generous time allocation on the CINES supercomputers (project number hcp6830).References
- C. J. Reedy and B. R. Gibney, Chem. Rev., 2004, 104, 617–650 CrossRef CAS PubMed.
- H. B. Gray and J. R. J. Winkler, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS PubMed.
- H. B. Dunford, Peroxidases and catalases: biochemistry, biophysics, biotechnology and physiology, John Wiley & Sons, 2010 Search PubMed.
- P. Harrison and E. J. Huehns, Nature, 1979, 279, 476–477 CrossRef CAS PubMed.
- S. Yoshikawa, in Handbook of Copper Pharmacology and Toxicology, Springer, 2002, pp. 131–152 Search PubMed.
- B. A. Springer, S. G. Sligar, J. S. Olson and G. N. J. J. Phillips, Chem. Rev., 1994, 94, 699–714 CrossRef CAS.
- J. B. Wittenberg and B. A. J. Wittenberg, J. Exp. Biol., 2003, 206, 2011–2020 CrossRef CAS PubMed.
- S.-i. Ozaki, M. P. Roach, T. Matsui and Y. J. Watanabe, Acc. Chem. Res., 2001, 34, 818–825 CrossRef CAS PubMed.
- N. Lehnert, M. G. I. Galinato, F. Paulat, G. B. Richter-Addo, W. Sturhahn, N. Xu and J. Zhao, Inorg. Chem., 2010, 49, 4133–4148 CrossRef CAS PubMed.
- J. S. Olson and G. N. J. Phillips Jr, J. Biol. Inorg Chem., 1997, 2, 544–552 CrossRef CAS.
- E. A. Brucker, J. S. Olson, M. Ikeda-Saito and G. N. Phillips Jr, Proteins: Struct., Funct., Bioinf., 1998, 30, 352–356 CrossRef CAS.
- C. R. Andrew, S. J. George, D. M. Lawson and R. R. Eady, Biochem, 2002, 41, 2353–2360 CrossRef CAS PubMed.
- L. M. Blomberg, M. R. Blomberg and P. E. J. Siegbahn, J. Inorg. Biochem., 2005, 99, 949–958 CrossRef CAS PubMed.
- L. E. Goodrich, F. Paulat, V. Praneeth and N. Lehnert, Inorg. Chem., 2010, 49, 6293–6316 CrossRef CAS PubMed.
- M.-S. Liao, M.-J. Huang and J. D. Watts, J. Phys. Chem. B, 2013, 117, 10103–10114 CrossRef CAS PubMed.
- T. G. Spiro, A. V. Soldatova and G. Balakrishnan, Coord. Chem. Rev., 2013, 257, 511–527 CrossRef CAS PubMed.
- V. Praneeth, F. Paulat, T. C. Berto, S. D. George, C. Näther, C. D. Sulok and N. Lehnert, J. Am. Chem. Soc., 2008, 130, 15288–15303 CrossRef CAS PubMed.
- A. P. Hunt and N. Lehnert, Acc. Chem. Res., 2015, 48, 2117–2125 CrossRef CAS PubMed.
- M.-S. Liao, M.-J. Huang and J. D. J. Watts, Mol. Phys., 2011, 109, 2035–2048 CrossRef CAS PubMed.
- V. E. Berryman, R. J. Boyd and E. R. Johnson, J. Chem. Theory Comput., 2015, 11, 3022–3028 CrossRef CAS PubMed.
- V. E. Walker, N. Castillo, C. F. Matta and R. J. Boyd, J. Phys. Chem. A, 2010, 114, 10315–10319 CrossRef CAS PubMed.
- M. Aarabi, R. Omidyan, S. Soorkia, G. Grégoire, M. Broquier, M.-E. Crestoni, A. de La Lande, B. Soep and N. Shafizadeh, Phys. Chem. Chem. Phys., 2019, 21, 1750–1760 RSC.
- J. Vojtěchovský, K. Chu, J. Berendzen, R. M. Sweet and I. J. Schlichting, Biophys. J., 1999, 77, 2153–2174 CrossRef.
- K. P. Kepp and P. J. Dasmeh, J. Phys. Chem. B, 2013, 117, 3755–3770 CrossRef CAS PubMed.
- M. Aarabi, S. Soorkia, G. Grégoire, M. Broquier, A. de la Lande, B. Soep, R. Omidyan and N. J. Shafizadeh, Phys. Chem. Chem. Phys., 2019, 21, 21329–21340 RSC.
- V. Praneeth, F. Neese and N. Lehnert, Inorg. Chem., 2005, 44, 2570–2572 CrossRef CAS PubMed.
- A. M. Koster, G. Geudtner, P. Calaminici, M. E. Casida, V. D. Dominguez, R. Flores-Moreno, G. U. Gamboa, A. Goursot, T. Heine, A. Ipatov, F. Janetzko, J. M. del Campo, J. U. Reveles, A. Vela, B. Zuniga-Gutierrez, and D. R. Salahub, deMon2k, Version 6.1.7, the deMon developers, Cinvestav, México, D.F Search PubMed.
- M. Krack and A. M. J. Köster, J. Chem. Phys., 1998, 108, 3226–3234 CrossRef CAS.
- M. Swart, A. W. Ehlers and K. J. Lammertsma, Mol. Phys., 2004, 102, 2467–2474 CrossRef CAS.
- A. Goursot, T. Mineva, R. Kevorkyants and D. J. Talbi, J. Chem. Theory Comput., 2007, 3, 755–763 CrossRef CAS PubMed.
- E. R. Johnson and A. D. J. Becke, J. Chem. Phys., 2017, 146, 211105 CrossRef PubMed.
- M. Frisch, M. Head-Gordon and J. Pople., J. Chem. Phys., 1990, 141, 189–196 Search PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- S. F. Boys and F. J. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. J. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- K. Falahati, H. Tamura, I. Burghardt and M. J. Huix-Rotllant, Nat. Commun., 2018, 9, 1–8 CrossRef CAS PubMed.
- C. Rovira, K. Kunc, J. Hutter, P. Ballone and M. J. Parrinello, J. Phys. Chem. A, 1997, 101, 8914–8925 CrossRef CAS.
- M. Radon and K. J. Pierloot, J. Phys. Chem. A, 2008, 112, 11824–11832 CrossRef CAS PubMed.
- R. Bader and T. Nguyen-Dang, in Adv. Quantum Chem., Elsevier, 1981, vol. 14, pp. 63–124 Search PubMed.
- T. A. Keith, AIMAll (Version10.05.04), 2010, aim.tkgristmill.com Search PubMed.
- W. R. Scheidt and M. E. Frisse, J. Am. Chem. Soc., 1975, 97, 17–21 CrossRef CAS PubMed.
- W. R. Scheidt and P. L. Piciulo, J. Am. Chem. Soc., 1976, 98, 1913–1919 CrossRef CAS PubMed.
- W. R. Scheidt, H. F. Duval, T. J. Neal and M. K. Ellison, J. Am. Chem. Soc., 2000, 122, 4651–4659 CrossRef CAS.
- G. R. Wyllie, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2003, 42, 5722–5734 CrossRef CAS PubMed.
- M. A. Martí, L. Capece, A. Crespo, F. Doctorovich and D. A. Estrin, J. Am. Chem. Soc., 2005, 127, 7721–7728 CrossRef PubMed.
- M. A. Marti, L. Capece, A. Bidon-Chanal, A. Crespo, V. Guallar, F. J. Luque and D. A. Estrin, Methods Enzymol., 2008, 437, 477–498 CAS.
- O. Chen, S. Groh, A. Liechty and D. P. Ridge, J. Am. Chem. Soc., 1999, 121, 11910–11911 CrossRef CAS.
- B. A. Springer, K. Egeberg, S. Sligar, R. Rohlfs, A. Mathews and J. J. Olson, J. Biol. Chem., 1989, 264, 3057–3060 CrossRef CAS.
- P. E. Siegbahn, M. R. Blomberg and S.-L. Chen, J. Chem. Theory Comput., 2010, 6, 2040–2044 CrossRef CAS PubMed.
- V. Praneeth, C. Näther, G. Peters and N. Lehnert, Inorg. Chem., 2006, 45, 2795–2811 CrossRef CAS PubMed.
- S. J. Grabowski, Chem. Rev., 2011, 111, 2597–2625 CrossRef CAS PubMed.
- W. R. Scheidt, Y. J. Lee and K. Hatano, J. Am. Chem. Soc., 1984, 106, 3191–3198 CrossRef CAS.
- A. B. McQuarters, J. W. Kampf, E. E. Alp, M. Hu, J. Zhao and N. Lehnert, Inorg. Chem., 2017, 56, 10513–10528 CrossRef CAS PubMed.
- F. Paulat and N. Lehnert, Inorg. Chem., 2007, 46, 1547–1549 CrossRef CAS PubMed.
- D. P. Linder, K. R. Rodgers, J. Banister, G. R. Wyllie, M. K. Ellison and W. R. Scheidt, J. Am. Chem. Soc., 2004, 126, 14136–14148 CrossRef CAS PubMed.
- A. V. Soldatova, M. Ibrahim, J. S. Olson, R. S. Czernuszewicz and T. G. Spiro, J. Am. Chem. Soc., 2010, 132, 4614–4625 CrossRef CAS PubMed.
- B. Chiavarino, M. E. Crestoni, S. Fornarini and C. Rovira, Inorg. Chem., 2008, 47, 7792–7801 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08398h |
| This journal is © The Royal Society of Chemistry 2022 |