
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA scalable, ecofriendly, and cost-effective lithium metal protection layer from a Post-it note†
Yeonsong Kima,
Jun Choib,
Ji Ho Youkc,
Byoung-Sun Lee*d and
Woong-Ryeol Yu *a
*a
aDepartment of Materials Science and Engineering (MSE), Research Institute of Advanced Materials (RIAM), Seoul National University, Seoul 08826, Republic of Korea. E-mail: woongryu@snu.ac.kr
bHuman Convergence Technology R&D Department, Korea Institute of Industrial Technology (KITECH), Ansan, 15588, Republic of Korea
cDepartment of Chemistry and Chemical Engineering, Education and Research Center for Smart Energy and Materials, Inha University, Incheon 22212, Republic of Korea
dSchool of Polymer System/Department of Fiber Convergence Materials Engineering, Dankook University, Yongin 16890, Republic of Korea. E-mail: bslee2020@dankook.ac.kr
First published on 21st December 2021
Abstract
Although there have been many studies addressing the dendrite growth issue of lithium (Li)–metal batteries (LMBs), the Li–metal anode has not yet been implemented in today's rechargeable batteries. There is a need to accelerate the practical use of LMBs by considering their cost-effectiveness, ecofriendliness, and scalability. Herein, a cost-effective and uniform protection layer was developed by simple heat treatment of a Post-it note. The carbonized Post-it protection layer, which consisted of electrochemically active carbon fibers and electrochemically inert CaCO3 particles, significantly contributed to stable plating and stripping behaviors. The resulting protected Li anode exhibited excellent electrochemical performance: extremely low polarization during cycling (<40 mV at a current density of 1 mA cm−2) and long lifespan (5000 cycles at 10 mA cm−2) of the symmetric cell, as well as excellent rate performance at 2C (125 mA h g−1) and long cyclability (cycling retention of 62.6% after 200 cycles) of the LiFePO4‖Li full cell. The paper-derived Li protection layer offer a facile and scalable approach to enhance LMB electrochemical performance.
1. Introduction
In addition to stimulating the growth of the electric vehicle industry, the concept of carbon neutrality strongly influences the development of highly advanced energy storage systems. Currently, the most important target of the most popular energy storage system (i.e., lithium (Li) rechargeable batteries) is high energy density (500 W h kg−1), which can be achieved by optimization of many factors; these include adjusting the electrolyte amount to achieve 2.4 g (A h)−1, minimizing the porosity to 25%, maximizing the cathode capacity at 252 mA h g−1, and maintaining a tight negative electrode capacity-to-positive electrode capacity ratio of 1.1 An Li–metal anode with extra-high capacity (3860 mA h g−1) and low negative electrochemical potential (−3.040 V vs. the standard hydrogen electrode) addresses the high energy density issue.2,3 However, the poor cycling performance of Li–metal batteries (LMBs) prevents application of the Li–metal anode to commercial rechargeable batteries. The inferior cycling curves can be categorized into two types: (i) gradual degradation by electrolyte consumption to form the solid electrolyte interphase (SEI) on the dendrites4 and (ii) sudden failure by short-circuiting.5 In both scenarios, the main cause of cell failure is dendrite growth during Li plating and stripping. Accordingly, management of these processes is fundamental and crucial for addressing cycling performance issues.Many attempts have been made to control plating and stripping behaviors while suppressing dendrite growth. These include Li–metal host design,6–9 protection layer formation,9–12 electrolyte composition change,13–16 and functional separator design.17–19 Various materials have been used as the protection layer. Polymer (poly(ethylene oxide) (PEO) segments and ureido-pyrimidinone (UPy))20 and organic/inorganic composites such as polyvinylidene fluoride/LiF21 and lithiated Nafion/LiCl22 were coated on the Li metal surface to slow down and homogenize the fast Li+ flux to the surface of the Li metal. Carbonaceous materials were also used as the protection layers: directly grown graphene on copper foil was used to homogenize charge distribution,23 three-dimensional graphene on nickel foam was employed to reduce the effective electrode current density,24 and manganese ceramic containing carbon nanofibers were used to accommodate the volume expansion of Li metal and facilitate more Li insert.25 Briefly, previous efforts to improve plating stability and stripping of the Li metal have significantly enhanced electrochemical performance. Nevertheless, LMBs have not been adopted in applications that require high energy density. A simple, scalable, cost-effective, and ecofriendly process that uses low-cost raw materials is needed to accelerate the practical use of LMBs. The use of a protection layer on Li metal could be the simplest and most effective approach to suppress dendrite growth and avoid side reactions.26 Electrochemically active carbon is a good candidate material for the protection layer because of its mechanical robustness and underlying controlled Li deposition27
Paper, made of natural fibers that contain cellulose and lignin,28 is a low-cost and ecofriendly two-dimensional material with uniform thickness. Like other plant-derived materials, such as coffee husks and sawdust, paper can be transformed into an energy storage material via thermal treatment because the cellulose and lignin can be carbonized.29,30 Thus, it is conceivable that carbonized paper could be used as the protection layer of the Li metal anode, provided that dimensional uniformity is maintained during the carbonization process. Herein, a carbon nonwoven mat was prepared from the ubiquitous Post-it note through a simple thermal process and used as the protection layer of the Li–metal anode to improve the electrochemical performance of an LMB. The morphologies and structures of the carbonized Post-it note were systematically examined; the electrochemical performances of the protection layer and protected Li anode were thoroughly evaluated using various electrochemical tests. The protected Li anode exhibited much better rate and cycling performances than did the bare Li anode.
2. Experimental
2.1 Protection layer preparation
Canary yellow Post-it notes, purchased from 3M, were used as the carbonaceous nonwoven mat precursor. Length and width of the Post-it notes used in this work is commonly 76 mm, and the width of the adhesive part is 15 mm. The adhesive-free part was used to get the carbon non-woven mat. The Post-it notes were fixed on an alumina plate to prevent random shrinkage during heat treatment after cutting and removing adhesive part. Before heat treatment, the thermal oxidation behavior of the Post-it note from room temperature to 800 °C under air atmosphere was examined by thermogravimetric analysis (TA Instruments) at a ramping rate of 10 °C min−1. The Post-it note was thermally stabilized at 350 °C for 1 h under air atmosphere, then carbonized at 1000 °C for 1 h under reducing gas atmosphere (Ar/H2 at 400/100 sccm v/v) to minimize the oxygen content after thermal treatment. The ramping rate was 10 °C min−1. The areal specific mass of carbonized Post-it note was 2.7 mg cm−2 and the porosity was calculated as 76%.2.2 Characterization
Morphologies of the carbonized Post-it note were observed by field-emission scanning electron microscopy and energy dispersive X-ray spectrometry (SUPRA 55VP; Carl Zeiss). Carbonized microstructures were examined by wide-angle XRD (D8 Advance; Bruker Miller) with Cu radiation (λ = 0.154 nm) and by Raman spectroscopy (DXR2xi; Thermo Scientific). Chemical bonds and atomic compositions were investigated by X-ray photoelectron spectroscopy (AXIS Nova; Kratos Analytical). Specific surface area was assessed by N2 adsorption at 77 K using Brunauer–Emmett–Teller analysis (TriStar II 3020; Micromeritics Instruments).2.3 Electrochemical testing
Electrochemical performances were evaluated by galvanostatic charge–discharge testing. The bare and protected Li anodes were assembled in 2032-type coin cells (MTI) with various counter electrodes and polypropylene–polyethylene separator (Celgard 2400) in an Ar-filled glove box. Free-standing carbonized Post-it notes without further treatment was physically placed in between the Li metal and separator during the cell assembly process. This work focuses only on to show the newly developed protective layer effects to the electrochemical performances, so that excess amount of electrolyte was used for preventing the electrolyte as a rate-determining factor. It should be mentioned that excess amount of electrolyte beyond certain level does not affect to the electrochemical performances.31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in N-methyl-2-pyrrolidone solvent using a mixer (Thinky) at 2000 rpm for 30 min. The slurries were pasted onto an Al foil current collector and dried at 80 °C under vacuum to remove residual N-methyl-2-pyrrolidone. Rate and cycling tests were conducted using only the constant-current mode over the voltage range of 2.5–4.2 V.
:1:1 in N-methyl-2-pyrrolidone solvent using a mixer (Thinky) at 2000 rpm for 30 min. The slurries were pasted onto an Al foil current collector and dried at 80 °C under vacuum to remove residual N-methyl-2-pyrrolidone. Rate and cycling tests were conducted using only the constant-current mode over the voltage range of 2.5–4.2 V.3. Results and discussion
Flattening the bare Li surface enables stable Li plating with minimal dendrite growth; such flattening and smoothing is typically performed before cell assembly. Nevertheless, dendrite growth is inevitable because Li nuclei formation during plating is an inherent cause of Li–metal roughening. The potential difference (Δφ = φt − φs) between the base (φs) and the nuclei tip (φt) becomes the driving force for dendrite growth by lower-overpotential-driven selective Li deposition on the tip.32 Accordingly, dendrites form on the bare Li–metal surface (Fig. 1a top). To prevent this, the approach of stepwise lithiation (i.e., intercalation and subsequent deposition) by the nonwoven carbon protection layer was implemented (Fig. 1a bottom). In this scheme, Li ions are electrochemically reduced on the carbon fibers; the reduced Li atoms are intercalated into the carbon matrix; and Li atoms precipitate from the saturated carbon, then deposit underneath the carbon fibers.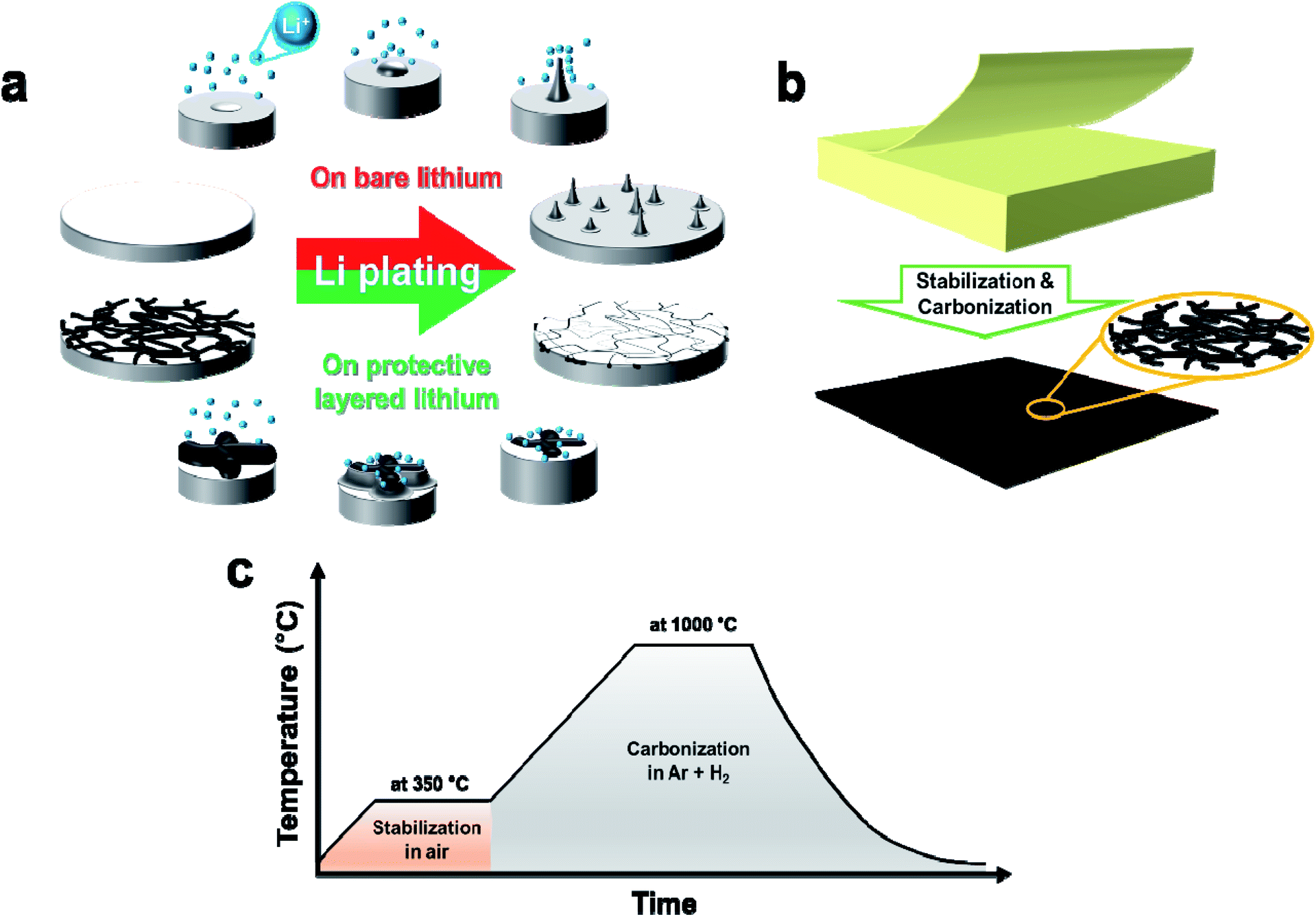 | ||
| Fig. 1 Schematic illustrations of (a) plating on bare lithium surface (red) vs. on the carbonaceous non-woven fiber protected Li surface (green), (b) protection layer from the canary yellow Post-it, and (c) thermal processing profile. | ||
A canary yellow Post-it note ca. 76 μm-thick with carbonizable nonwoven natural fibers and uniform thickness (<100 μm) was chosen as the precursor material for the carbon nonwoven fabric protection layer. The thermal decomposition behavior of the Post-it note was examined by thermogravimetric analysis under air atmosphere (Fig. S1†) to determine the optimal heat-treatment condition. Three main weight-reduction temperatures were observed at ca. 339 °C, 451 °C, and 620 °C; the residual weight after full thermal decomposition was less than 4 wt% of the initial weight. The first two thermal decompositions were attributed to degradation of cellulose (280–400 °C) and lignin (320–450 °C),33 while the weight loss at ca. 620 °C resulted from thermal decomposition of the inorganic salt used to improve the mechanical properties of the paper.34,35 Based on the thermogravimetric analysis results, stabilization was carried out at 350 °C for 1 h under air atmosphere to increase the carbon yield from the cellulose and lignin. This was followed by carbonization at 1000 °C for 1 h under reducing gas atmosphere (i.e., nitrogen and hydrogen gas mixed at 5:1 v/v) to minimize the oxygen functional groups for better reversible electrochemical behavior (Fig. 1c). Notably, the simple precursor shape and thermal treatment make this approach highly scalable (Fig. S2†).
The morphologies of the carbonized Post-it note protection layer are shown in Fig. 2a. The atomic compositions of the carbonized Post-it note determined from energy dispersive X-ray spectrometry were as follows: carbon 53.89 at%, oxygen 34.79 at%, and calcium 10.04 at%. The fibers were randomly distributed (mapped with orange color in Fig. 2b), while the interfiber regions were filled with inorganic salts (Fig. S3b–f†). Small amounts of silicon, magnesium, aluminum, chlorine, and sulfur were also observed; these were likely from pigments and other additives present in the uncarbonized paper. Structural changes caused by the thermal treatment were investigated using wide-angle X-ray diffraction (XRD; Fig. 2c and S4†). The XRD curve of the as-received Post-it note displayed peaks at 2θ = 17.9°, 28.5°, 34.0°, 47.1°, 50.7°, and 54.2°, all of which were attributed to Ca(OH)2.36 The carbonized Post-it note XRD curve displayed a turbostratic carbon peak (black star) at ca. 2θ = 23.1°;37,38 the calculated (002) interlayer spacing was 0.385 nm. Polymorphic CaCO3 crystals were formed by the thermal treatment. Calcite CaCO3 crystal formation was confirmed by XRD peaks (orange triangles) at 2θ = 29.5°, 36.0°, 39.4°, 43.1°, 47.6°, and 48.5°;39 vaterite CaCO3 crystal formation was confirmed by XRD peaks (green squares) at 2θ = 24.9°, 27.2°, and 32.8°.40 The carbonized microstructure was then examined by Raman spectroscopy (Fig. 2d). Peaks corresponding to turbostratic carbon (i.e., D-peak at ca. 1340 cm−1 and G-peak at ca. 1590 cm−1) were observed41 with a peak-intensity ratio (ID/IG) of 0.991. Despite the substantial oxygen content, the intensity of the D-peak originating from defects and sp3 carbon atoms was comparable with the intensity of the G-peak attributed to sp2 carbon atoms because of the reducing atmosphere used during the thermal treatment. A minor peak at 1084 cm−1 corresponded to symmetric stretching of the C–O bonds of CaCO3.42
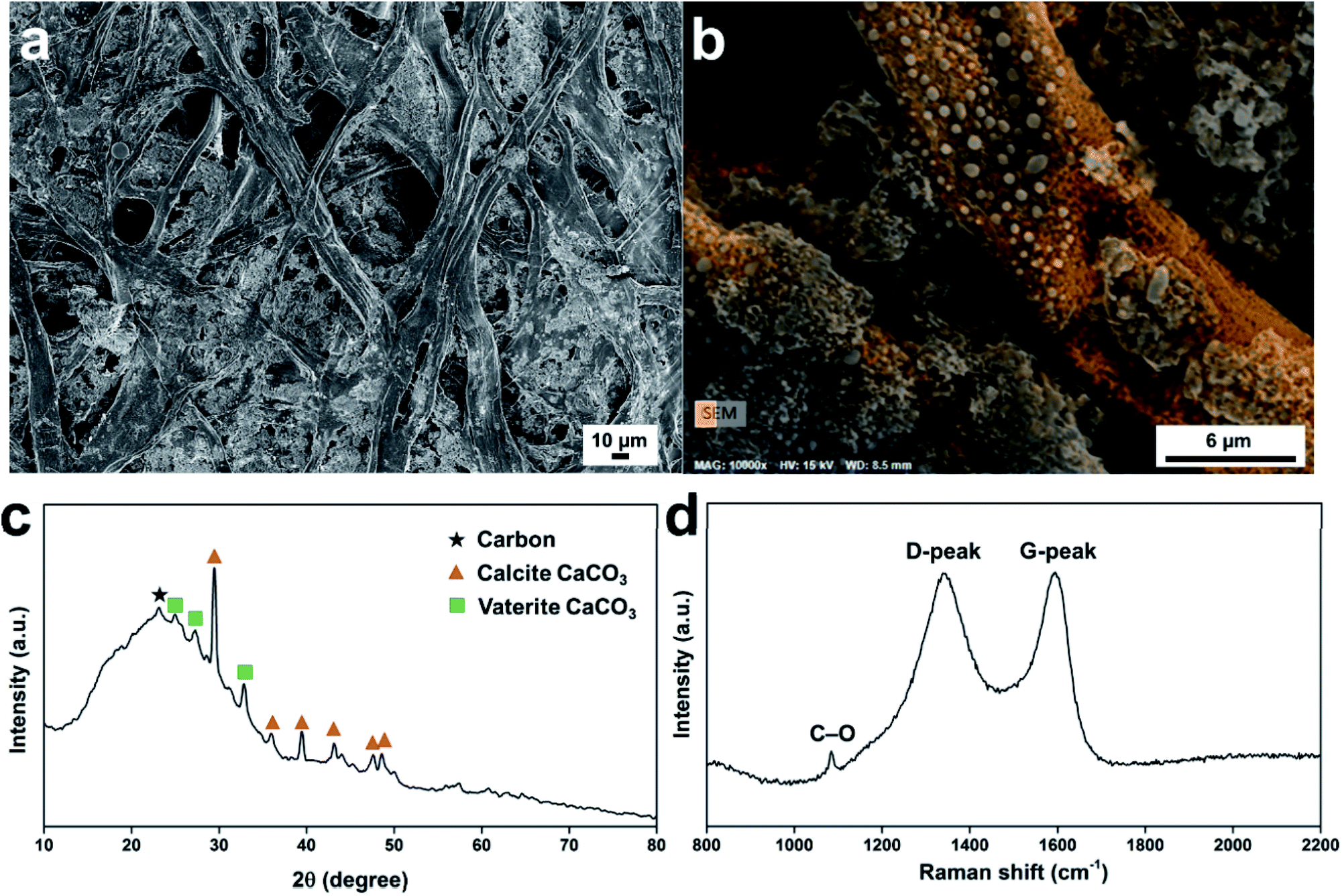 | ||
| Fig. 2 Characterizations of the carbonized Post-it note protection layer. (a) Low magnification scanning electron microscope image, (b) energy dispersive X-ray spectrometry based carbon-atom mapping of high scanning electron microscopy image, (c) wide-angle X-ray diffractogram, and (d) Raman spectrum. | ||
The types of chemical bonds of the carbonized Post-it note were investigated using X-ray photoelectron spectroscopy. The wide-scan X-ray photoelectron spectroscopy spectrum presented in Fig. 3a showed peaks corresponding to carbon (C 1s), oxygen (O 1s), and calcium (Ca 2p). The high-resolution C1s scan (Fig. 3b) confirmed the presence of C–C (284.5 eV) and C–O bonds (286.1 eV), as well as CO3 (288.3 eV) from the CaCO3.43 The O 1s peaks assigned to bonds in CaCO3 appeared at 531.2 eV for O–C bonds and 532.9 eV for O–Ca bonds (Fig. 3c). The Ca 2p peaks of CaCO3 appeared at 350.5 (2p1/2) and 347.1 eV (2p3/2) (Fig. 3d).43 Thus, the microstructure of the carbonized Post-it note consisted of turbostratic carbon and polymorphic CaCO3.
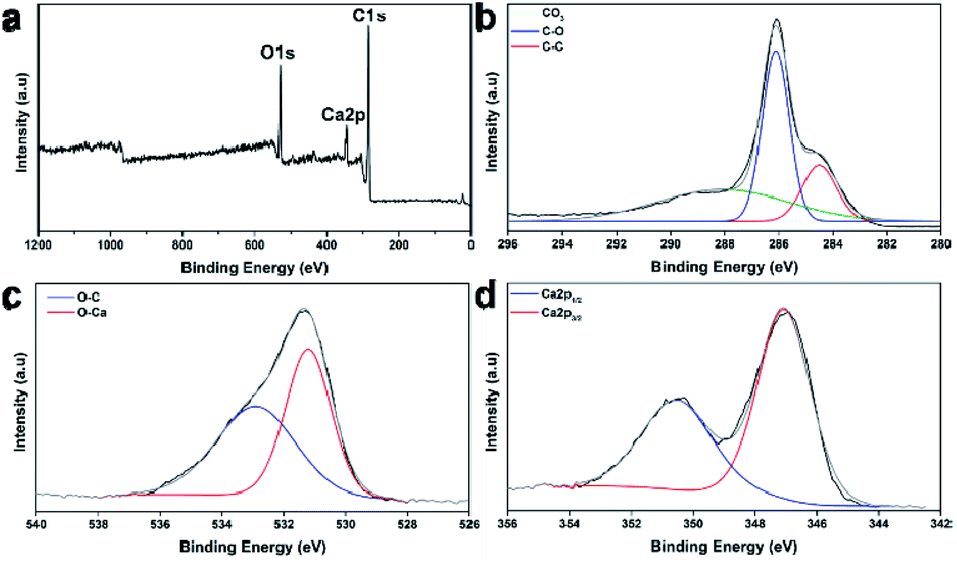 | ||
| Fig. 3 Chemical bond analysis according to X-ray photoelectron spectroscopy. (a) Wide scan and high-resolution spectra of (b) C 1s, (c) O 1s, and (d) Ca 2p regions. | ||
The electrochemical performances of the carbonized Post-it note and the protected Li were thoroughly investigated by galvanostatic charge–discharge testing. First, a Li‖carbonized Post-it note half-cell plating/stripping test was carried out to understand the electrochemical performance of the carbonized Post-it note protection layer (Fig. 4a). The carbonized Post-it the electrochemical performances of the carbonized Post-it note and the protected Li were thoroughly investigated by galvanostatic charge–discharge testing. First, a Li‖carbonized Post-it note half-cell plating/stripping test was carried out to understand the electrochemical performance of the carbonized Post-it note protection layer (Fig. 4a). The carbonized Post-it note was lithiated at a current density of 2 mA cm−2 to the capacity of 5 mA h cm−2, then delithiated at the same current density to 1.5 V. The initial inflection at ca. 0.75 V corresponded to lithiation of pores and SEI formation;44 the subsequent slope was attributed to lithiation of hard carbon with some oxygen.45 The areal discharge capacity above 0 V because of intercalation into the carbonaceous structure was 0.83 mA h cm−2. Note that CaCO3 is considered electrochemically inert toward Li; hence, there was no CaCO3-driven lithiation behavior.46 Next, the plateau below 0 V (ca. −0.1 V) was caused by plating, where the plating capacity was 4.17 mA h cm−2. Delithiation mainly occurred at ca. 40 mV, where the delithiation capacity was 3.71 mA h cm−2. Based on the delithiation capacity from the slope (0.34 mA h cm−2) between 0.04 and 1.5 V, the irreversible areal capacity by carbon redox was 0.49 mA h cm−2. Inherently present oxygen and the Type IV isotherm caused by the high surface area (222.7 m2 g−1) were the main sources of the irreversible capacity (Fig. S4†).45,47 The irreversible capacity from the plating/stripping was 0.46 mA h cm−2; the coulombic efficiency was 74.4%. It could be understood that Li was irreversibly trapped in the narrow pore inducing the slow diffusion of nitrogen during adsorption and desorption and following incomplete equilibrium of the nitrogen isotherm curves.48
 | ||
| Fig. 4 Electrochemical characterization of the carbonized Post-it note and the protected lithium (Li). (a) Voltage profile of Li‖carbonized Post-it note half-cell plating/stripping test. Nyquist plots of (b) Li‖Li symmetric cell and (c) protected Li‖protected Li symmetric cell, symmetric cell cycling test results at the current densities of (d and e) 2, (f and g) 5, (h and i) 10 mA cm−2. | ||
The electrochemical performance of the protected Li was first examined for the symmetric cell. Electrolyte bulk resistance (Rsol), ionic resistance in surface roughness (Rion), and charge-transfer resistance (Rct) were evaluated by electrochemical impedance spectroscopy.49 Fig. 4b shows that the as-assembled bare Li symmetric cell displayed a depressed semicircular electrochemical impedance spectroscopy curve with Rsol of 2.3 Ω and Rct of ca. 534 Ω. In contrast, the cycled bare Li symmetric cell (at a current density of 1 mA cm−2 for 1 h each of plating and stripping) showed increased Rsol of 8.9 Ω and slightly developed Rion, represented as 45° from 8.9 to 13.8 Ω; it also showed decreased Rct of ca. 313 Ω because of dendritic surface formation after cycling. This high Rct of the as-assembled bare symmetric cell was comparable with the findings of previous research using carbonate electrolyte.50 Additionally, the electrochemical impedance spectroscopy curves of the protected Li symmetric cells displayed depressed semicircles with significantly reduced cell resistances; the Rsol and Rct of the as-assembled cell were ca. 3.5 and 2.9 Ω, respectively, while the Rsol and Rct of the cycled cell were ca. 2.5 and 2.5 Ω, respectively.
The cycling performances of the symmetric cells were evaluated at current densities of 2, 5, and 10 mA cm−2 (Fig. 4d–i). The areal capacities were set at 1 mA h cm−2, regardless of current density. Polarizations of the protected Li symmetric cells at all current densities were much smaller than polarizations of the bare Li symmetric cells. The bare Li symmetric-cell polarization at 2 mA cm−1 fluctuated from ca. 0.2 to 1.4 V; cycling suddenly terminated at cycle 771 (771 h) because of increased resistance. Note that the cell did not short-circuit during cycling; cell polarization abruptly increased because of electrolyte depletion resulting from repetitive SEI formation.51 In contrast, the protected Li symmetric-cell polarization remained below 40 mV after stabilization, and the cell was successfully cycled up to 1000 times. Fig. 4e shows that the protected Li symmetric cell demonstrated a stable and flat voltage plateau with a much smaller polarization of <34 mV at 30 h; the bare Li symmetric cell showed the typical voltage profile, implying dendrite formation on the cathode and pit formation on the anode, with a substantial polarization of >250 mV.52
The symmetric cells displayed higher polarizations at the current density of 5 mA cm−2 (Fig. 4f). The polarization of the bare Li symmetric cell gradually increased to ca. 2.5 V, and the cycling ended at cycle 1766 (707 h) because of electrolyte depletion. However, the protected Li symmetric-cell polarization slightly increased as cycling proceeded, although the final polarization remained below 235 mV. Fig. 4g at 30 h shows the same trend as Fig. 4e; the polarizations of the bare and protected Li symmetric cells were ca. 560 and 79 mV, respectively. At the current density of 10 mA cm−2, the bare Li symmetric-cell cycling quickly ended at cycle 207 (42 h), while the protected symmetric cell cycled up to 5000 times with high polarization of 400 mV (Fig. 4h). The voltage profiles at 30 h (Fig. 4i) indicate substantial bare and protected Li symmetric-cell polarizations of ca. 2.1 V and 156 mV, respectively. Photographs of the cycled bare and protected Li electrodes are presented in Fig. S6.† The blackish and grayish color on the cycled bare Li surface indicates an uneven surface with substantial dendrites, pits, and SEI layer. In contrast, the protected Li after 1000 h of cycling at a current density of 1 mA cm−2 maintained the robust carbon protection layer. Densely plated Li-containing shiny chunks formed during cycling at higher current densities. After 5000 cycles at the current density of 10 mA cm−2, surface and cross-sectional scanning electron microscopy images confirmed densely plated Li on the surface and surrounding the carbon fibers in the protection layer (Fig. S7†). Thus, the protection layer guided Li plating without random dendrite growth and SEI formation; this resulted in stable cycling.
Full-cell testing with an LiFePO4 (LFP) cathode was carried out to demonstrate the practical use of the protected Li (Fig. 5). The areal loadings of the LFP cathode were set at ca. 1 mA h cm−2. Rate performances of the LFP‖bare Li and LFP‖protected Li full cells (Fig. 5a) were determined over the range of 2.5–4.2 V. The full cells were cycled at the charging current densities of 0.1C, 0.2C, 0.5C, 1C, and 2C for five cycles each; the discharging current density during the rate test was set at 0.1C for the first five cycles to complete the formation, then maintained at 0.2C. The reversible discharge capacities of both cells at low rates (up to 1C) remained constant at ca. 156 and 153 mA h g−1. The reversible capacities of the LFP‖bare Li cell at 2C suddenly decreased to 1.8 mA h g−1 (almost electrochemically deactivated), while the LFP‖protected Li cell at 2C remained electrochemically active with reversible capacity of 125 mA h g−1. Fig. 5b and c present the voltage profiles of the LFP‖bare Li and LFP‖protected Li cells at cycles 1 (0.1C charging/0.1C discharging), 6 (0.2C/0.2C), 11 (0.5C/0.2C), 16 (1C/0.2C), and 21 (2C/0.2C). The voltage profiles had shapes identical to the shapes of previously reported LFP‖Li cells.53 The polarizations of the cells during charging increased with increasing current density. At the charging current density of 2C, the increased polarization of the LFP‖bare Li cell did not allow delithiation from the LFP cathode; in contrast, the protected Li suppressed the polarization increase, which enabled the high reversible capacity of 125 mA h g−1 at 2C. The discharge profiles did not change substantially because of the low current densities during discharge. After the rate test, the full cells were cycled with charging current density of 0.1C and discharging current density of 0.2C (Fig. S8†). The reversible specific capacity of the LFP‖protected Li cell after cycle 160 was 145.8 mA h g−1 with extremely high retention of 96.4%, while the reversible specific capacity of the LFP‖bare Li cell was 119.4 mA h g−1 (77.2% of the initial reversible capacity). Thus, the protection layer enhanced rate performance and cycling performance at low current density.
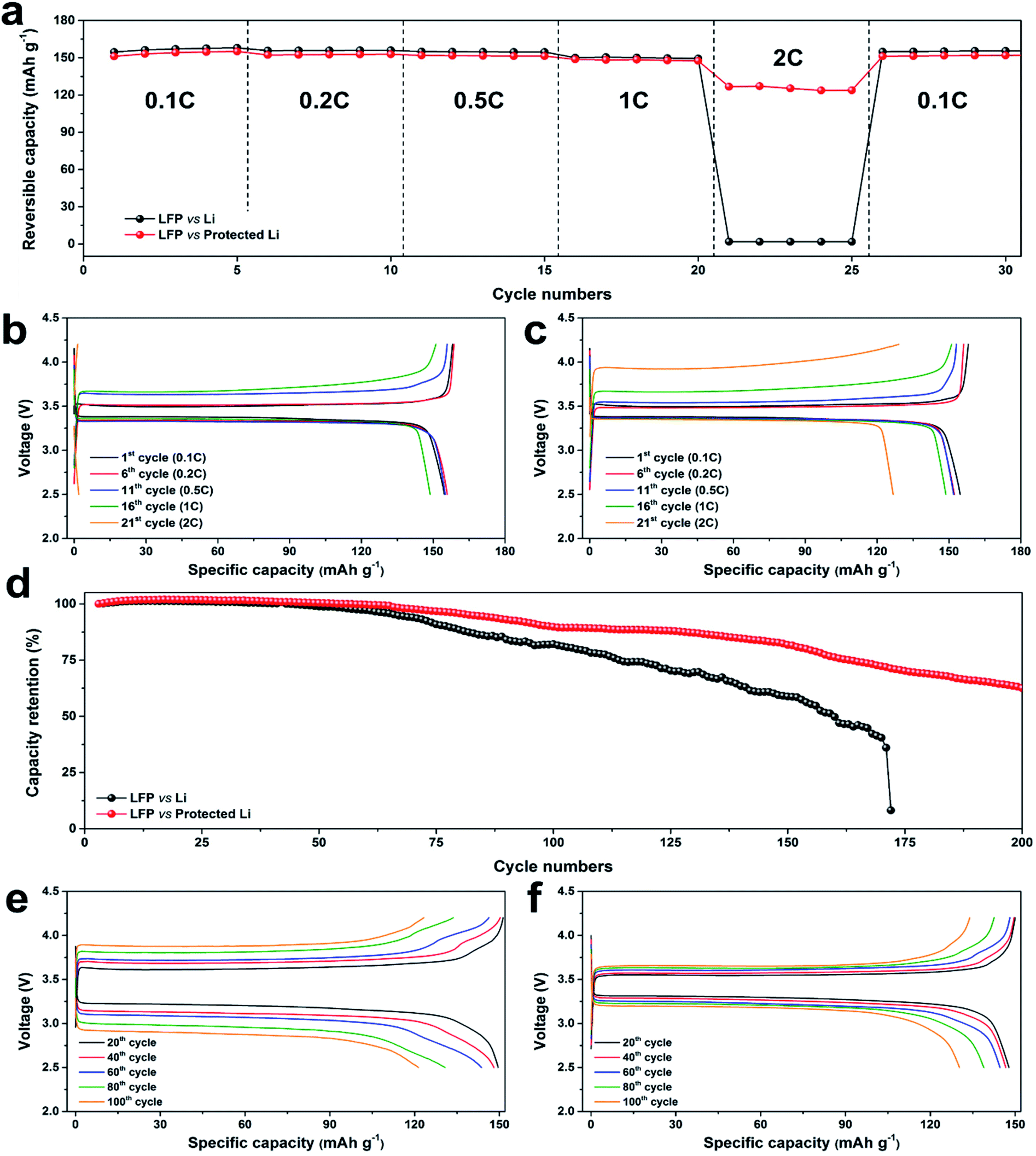 | ||
| Fig. 5 LiFePO4 (LFP)‖Li full cell performance tests. (a) Rate capabilities at charging current densities of 0.1C, 0.2C, 0.5C, 1C, 2C, and 0.1C. Voltage profiles of (b) LFP‖bare Li cell and (c) LFP‖protected Li cell during the rate test. (d) Cycling test at the current density of 0.5C. Voltage profiles of the (e) LFP‖bare Li cell and (f) LFP‖protected Li cell during the cycling test. | ||
Cycling performances of the full cells were investigated at the current density of 0.5C (Fig. 5d and S9†). The specific capacities of both cells were stable to 60 cycles, but they subsequently degraded at different rates. The capacity retentions of the LFP‖bare Li and LFP‖protected Li cells at cycle 100 were 82.1% and 89.8%, respectively. The LFP‖bare Li cell cycling abruptly ended at cycle 172, while the LFP‖protected Li cell was successfully cycled up to 200 times with cycling retention of 62.6%. The voltage profiles of the cells at cycles 20, 40, 60, 80, and 100 are presented in Fig. 5e and f. The redox potential differences of both cells gradually increased as cycling proceeded, but the voltage profile hysteresis of the LFP‖protected Li cell as cycling increased was much smaller than the hysteresis of the LFP‖bare Li cell. Quantitatively, the increasing rate of the LFP‖protected Li cell between cycles 20 and 100 (2.9 mV cycle−1) was significantly lower than the increasing rate of the LFP‖bare Li cell (7.6 mV cycle−1) because of the stable plating and stripping behaviors. The carbonized Post-it note protection layer enabled better rate capability and longer cycle life by guiding stable Li plating and stripping of the Li–metal anode.
4. Conclusions
A cost-effective and uniform protection layer was prepared through simple heat treatment of the canary yellow Post-it note without using a non-ecofriendly shaping process. The carbonized note consisted of electrochemically active carbon fibers and electrochemically inert CaCO3. The carbonized note served as a protection layer, which led to stable Li plating and stripping behaviors. Compared with the bare Li anode, the protected Li anode exhibited outstanding symmetric-cell performance, including extremely low Rct (2.5 Ω after 1 cycle) and polarization during cycling (<40 mV at a current density of 1 mA cm−2), more stable voltage profile, and longer cycle life (5000 cycles at 10 mA cm−2). The effect of the carbonized Post-it note was evident in LFP‖Li full-cell testing where the LFP‖protected Li cell displayed excellent rate performance at 2C (125 mA h g−1) and longer lifespan (cycling retention of 62.6% after 200 cycles), compared with the LFP‖bare Li full cell.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (2021R1F1A1049123). This work was also supported by Korea Evaluation institute of Industrial Technology Through Development of Carbon Industrial Cluster Foundation Program, funded by Ministry of Trade, Industry & Energy (10083615, Development of T700/48K grade carbon fiber manufacturing technology with spinning speed of over 250 m min−1). The Institute of Engineering Research at Seoul National University provided research facilities for this work.References
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- B.-S. Lee, Polymers, 2020, 12, 2035 CrossRef CAS PubMed.
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2019, 4, 551–559 CrossRef CAS.
- Y. Zhu, J. Xie, A. Pei, B. Liu, Y. Wu, D. Lin, J. Li, H. Wang, H. Chen, J. Xu, A. Yang, C.-L. Wu, H. Wang, W. Chen and Y. Cui, Nat. Commun., 2019, 10, 2067 CrossRef PubMed.
- S.-S. Chi, Y. Liu, W.-L. Song, L.-Z. Fan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1700348 CrossRef.
- D. Lin, Y. Liu, Z. Liang, H.-W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 CrossRef CAS PubMed.
- F. Shen, F. Zhang, Y. Zheng, Z. Fan, Z. Li, Z. Sun, Y. Xuan, B. Zhao, Z. Lin, X. Gui, X. Han, Y. Cheng and C. Niu, Energy Storage Mater., 2018, 13, 323–328 CrossRef.
- W. Zhang, H. L. Zhuang, L. Fan, L. Gao and Y. Lu, Sci. Adv., 2018, 4, eaar4410 CrossRef PubMed.
- X. Kong, P. E. Rudnicki, S. Choudhury, Z. Bao and J. Qin, Adv. Funct. Mater., 2020, 30, 1910138 CrossRef CAS.
- S. Kim, J. Park, A. Friesen, H. Lee, Y. M. Lee and M.-H. Ryou, Electrochim. Acta, 2018, 282, 343–350 CrossRef CAS.
- T. Yang, Y. Sun, T. Qian, J. Liu, X. Liu, F. Rosei and C. Yan, Energy Storage Mater., 2020, 26, 385–390 CrossRef.
- H. Liu, H. Zhou, B.-S. Lee, X. Xing, M. Gonzalez and P. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 30635–30642 CrossRef CAS PubMed.
- H. Zheng, Y. Xie, H. Xiang, P. Shi, X. Liang and W. Xu, Electrochim. Acta, 2018, 270, 62–69 CrossRef CAS.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- D. W. Kang, J. Moon, H.-Y. Choi, H.-C. Shin and B. G. Kim, J. Power Sources, 2021, 490, 229504 CrossRef CAS.
- T. Zhang, J. Yang, Z. Xu, H. Li, Y. Guo, C. Liang and J. Wang, ACS Appl. Energy Mater., 2020, 3, 506–513 CrossRef CAS.
- H. Lee, X. Ren, C. Niu, L. Yu, M. H. Engelhard, I. Cho, M.-H. Ryou, H. S. Jin, H.-T. Kim, J. Liu, W. Xu and J.-G. Zhang, Adv. Funct. Mater., 2017, 27, 1704391 CrossRef.
- Y. Liu, S. Xiong, J. Wang, X. Jiao, S. Li, C. Zhang, Z. Song and J. Song, Energy Storage Mater., 2019, 19, 24–30 CrossRef.
- G. Wang, C. Chen, Y. Chen, X. Kang, C. Yang, F. Wang, Y. Liu and X. Xiong, Angew. Chem., 2020, 132, 2071–2076 CrossRef.
- R. Xu, X.-Q. Zhang, X.-B. Cheng, H.-J. Peng, C.-Z. Zhao, C. Yan and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1705838 CrossRef.
- S. Li, L. Fan and Y. Lu, Energy Storage Mater., 2019, 18, 205–212 CrossRef.
- K. Yan, H.-W. Lee, T. Gao, G. Zheng, H. Yao, H. Wang, Z. Lu, Y. Zhou, Z. Liang, Z. Liu, S. Chu and Y. Cui, Nano Lett., 2014, 14, 6016–6022 CrossRef CAS PubMed.
- K. Xie, W. Wei, K. Yuan, W. Lu, M. Guo, Z. Li, Q. Song, X. Liu, J.-G. Wang and C. Shen, ACS Appl. Mater. Interfaces, 2016, 8, 26091–26097 CrossRef CAS PubMed.
- Q. Qin, N. Deng, L. Wang, L. Zhang, Y. Jia, Z. Dai, Y. Liu, W. Kang and B. Cheng, Chem. Eng. J., 2019, 360, 900–911 CrossRef CAS.
- Z. Han, C. Zhang, Q. Lin, Y. Zhang, Y. Deng, J. Han, D. Wu, F. Kang, Q.-H. Yang and W. Lv, Small Methods, 2021, 5, 2001035 CrossRef CAS PubMed.
- G. Zheng, S. W. Lee, Z. Liang, H.-W. Lee, K. Yan, H. Yao, H. Wang, W. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- E. Małachowska, M. Dubowik, P. Boruszewski, J. Łojewska and P. Przybysz, Sci. Rep., 2020, 10, 19998 CrossRef PubMed.
- Y. J. Hwang, S. K. Jeong, K. S. Nahm, J. S. Shin and A. M. Stephan, J. Phys. Chem. Solids, 2007, 68, 182–188 CrossRef CAS.
- J. H. Kwak, J. C. Hyun, S. B. Moon, H.-J. Jin, H.-D. Lim and Y. S. Yun, Macromol. Res., 2020, 28, 1204–1210 CrossRef CAS.
- S. J. An, J. Li, D. Mohanty, C. Daniel, B. J. Polzin, J. R. Croy, S. E. Trask and D. L. Wood III, J. Electrochem. Soc., 2017, 164, A1195 CrossRef CAS.
- Y. Liu, Q. Liu, L. Xin, Y. Liu, F. Yang, E. A. Stach and J. Xie, Nat. Energy, 2017, 2, 17083 CrossRef CAS.
- J. Gug, D. Cacciola and M. J. Sobkowicz, Waste Manage., 2015, 35, 283–292 CrossRef CAS PubMed.
- Q. Zhang, M. U. Khan, X. Lin, W. Yi and H. Lei, J. Cleaner Prod., 2020, 262, 121251 CrossRef CAS.
- R. Babou-Kammoe, S. Hamoudi, F. Larachi and K. Belkacemi, Can. J. Chem. Eng., 2012, 90, 26–33 CrossRef CAS.
- X. Zhu, D. Tang, K. Yang, Z. Zhang, Q. Li, Q. Pan and C. Yang, Constr. Build. Mater., 2018, 175, 467–482 CrossRef CAS.
- B.-S. Lee, S.-B. Son, K.-M. Park, W.-R. Yu, K.-H. Oh and S.-H. Lee, J. Power Sources, 2012, 199, 53–60 CrossRef CAS.
- B.-S. Lee, K.-M. Park, W.-R. Yu and J. H. Youk, Macromol. Res., 2012, 20, 605–613 CrossRef CAS.
- Q. Q. Zhou, X. Y. Chen and B. Wang, Microporous Mesoporous Mater., 2012, 158, 155–161 CrossRef CAS.
- D. B. Trushina, T. V. Bukreeva, M. V. Kovalchuk and M. N. Antipina, Mater. Sci. Eng., C, 2014, 45, 644–658 CrossRef CAS PubMed.
- B.-S. Lee, J. Yoon, C. Jung, D. Y. Kim, S.-Y. Jeon, K.-H. Kim, J.-H. Park, H. Park, K. H. Lee, Y.-S. Kang, J.-H. Park, H. Jung, W.-R. Yu and S.-G. Doo, ACS Nano, 2016, 10, 2617–2627 CrossRef CAS PubMed.
- T. M. DeCarlo, Nat. Commun., 2018, 9, 1–3 CrossRef CAS PubMed.
- D. H. Chu, M. Vinoba, M. Bhagiyalakshmi, H. Baek II, S. C. Nam, Y. Yoon, S. H. Kim and S. K. Jeong, RSC Adv., 2013, 3, 21722–21729 RSC.
- B.-S. Lee, H.-S. Yang, H. Jung, S. K. Mah, S. Kwon, J.-H. Park, K. H. Lee, W.-R. Yu and S.-G. Doo, Eur. Polym. J., 2015, 70, 392–399 CrossRef CAS.
- L. Fan and B. Lu, Small, 2016, 12, 2783–2791 CrossRef CAS PubMed.
- K. Zhang, S. Liang, X. Yin, P. Gao, R. Ding and E. Liu, J. Electroanal. Chem., 2019, 851, 113476 CrossRef CAS.
- Y. Li, F. Wang, J. Liang, X. Hu and K. Yu, New J. Chem., 2016, 40, 325–329 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- N. Ogihara, S. Kawauchi, C. Okuda, Y. Itou, Y. Takeuchi and Y. Ukyo, J. Electrochem. Soc., 2012, 159, A1034–A1039 CrossRef CAS.
- Z. Liang, K. Yan, G. Zhou, A. Pei, J. Zhao, Y. Sun, J. Xie, Y. Li, F. Shi, Y. Liu, D. Lin, K. Liu, H. Wang, H. Wang, Y. Lu and Y. Cui, Sci. Adv., 2019, 5, eaau5655 CrossRef CAS PubMed.
- L. Fan, S. Li, L. Liu, W. Zhang, L. Gao, Y. Fu, F. Chen, J. Li, H. L. Zhuang and Y. Lu, Adv. Energy Mater., 2018, 8, 1802350 CrossRef.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K.-H. Chen, J.-G. Zhang, K. Thornton and N. P. Dasgupta, ACS Cent. Sci., 2016, 2, 790–801 CrossRef CAS PubMed.
- S. Park, B.-S. Lee, T.-H. Kang, I.-S. Choi and W.-R. Yu, Mater. Des., 2019, 166, 107620 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08310d |
| This journal is © The Royal Society of Chemistry 2022 |