
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in the site-selective C-5, C-3 and C-2 functionalization of chromones via sp2 C–H activation
Anjitha Theres Benny
and
Ethiraj Kannatt Radhakrishnan
 *
*
Department of Chemistry, Vellore Institute of Technology, Vellore, India. E-mail: ethukr@gmail.com
First published on 26th January 2022
Abstract
In this work, site-selective C–H activation at C-5, C-3 and C-2 positions of chromones for the introduction of structural diversity to the chromone scaffold was studied. The keto group of the chromone moiety acts as the directing group for the selective functionalization of chromones at the C-5 position. Furthermore, the C–H functionalization at the electron-rich C-3 position of the chromone can be achieved using electrophilic coupling partners. The C–H functionalization at the C-2 position can be possible using nucleophilic coupling partners. The direct functionalization methods provide a better pathway for the generation of C-5, C-3 and C-2-substituted chromones with good atom economy than that of classical pre-functionalized reaction protocols.
 Anjitha Theres Benny | Anjitha Theres Benny is currently a doctoral research scholar at Department of Chemistry, School of Advanced Sciences, VIT, Vellore. She has received her Master's degree in Chemistry from St. Berchmans College, Changanassery, Mahatma Gandhi University, Kerala. Her research interests include synthesis of heterocyclic compounds of pharmaceutical interests and medicinal Chemistry. |
 Ethiraj Kannatt Radhakrishnan | Dr Ethiraj K. R., Associate Professor, Dept. of Chemistry, School of Advanced Sciences, VIT, Vellore. He obtained his PhD degree in 2013 in medicinal chemistry. His research interests are in the field of organic synthesis, the synthesis of heterocyclic compounds of pharmaceutical interest and medicinal chemistry, bacterial communication and host/microbe interactions, protein–nanoparticles interactions. He has more than 20 publications in various peer reviewed journals with a h-index 07, i10-index 7. He is a member of International Chemical Biology Society, European Society of Clinical Microbiology and Infectious Diseases. |
1. Introduction
Chromone 1 scaffolds are ubiquitously present in numerous biologically active compounds and, hence, received considerable attention from synthetic chemists in discovering new drugs.1 The importance of chromones in drug discovery and synthetic transformations is evident from a number of publications in the field. Chromones are well documented in the literature owing to their interesting biological applications such as anticancer,2–5 antimicrobial,6–10 anti-inflammatory,11–14 antioxidant,15–18 anti-Alzheimer,19–22 and antidiabetic23–26 activities. Apart from their biological applications, they show promising fluorescent activity, and hence can be used as sensors in detecting various metal ions and in bioimaging.27–34 The promising biological and photophysical activities of chromones make them a valid scaffold of interest among chemists.The area of interest in this review covers the site-selective functionalization of chromones in C-5, C-3 and C-2 positions. Few of the C-2, C-3 and C-5-functionalized chromones with biological applications including anti-asthmatic 2,35 antimalarial 3,36 anticancer 4,37 antiviral 5,38 antimicrobial 6–9,39,40 anti-inflammatory 10,41 antiplatelet 11,42 antioxidant 12,43 and anti-Alzheimer 1344 activities are given in Fig. 1.
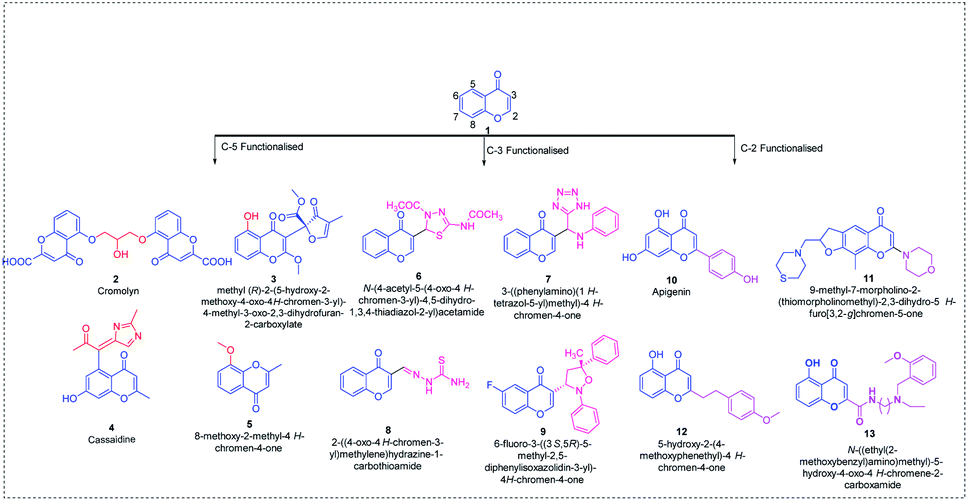 | ||
| Fig. 1 Naturally occurring C-2, C-3 and C-5-functionalized chromones. | ||
Direct C–H bond activation/functionalization has practical and fundamental challenges but it is a highly attractive strategy in the formation of a new functionality or a new C–C bond. The substrate scope by C–H activation is unlimited, including hydrocarbons, complex organic compounds of small molecular weight, and synthetic and biological polymers.45–53
C–H functionalization of chromones is a useful method for the construction of carbon–carbon and carbon–heteroatom bonds. The cross dehydrogenative coupling (CDC) for the formation of Csp2–Csp, Csp–Csp2 and Csp2–Csp2 bonds enables the synthesis of significant building blocks from a simple chromone moiety.54 The C-5 functionalization of chromones by transition metal-catalyzed reactions is reported through chelation-assisted metal-catalyzed reaction transformation. The method proceeds by the effect of the weakly coordinating ability of the keto group on the chromone to the metal catalyst forming a metallacycle.55,56 Similarly, the C-3 position of the chromone is functionalized by a transition metal-catalyzed reaction through a nucleophilic attack of chromones to metals forming a metal–chromone complex. The C-3 functionalization of the chromones occurs predominantly if its C-2 position is already substituted. The C-2 functionalization of chromones occurs when the coupling partners are arylpalladium species or alkyl radicals. The selective functionalization at C-5, C-3 or C-2 positions of chromones by a direct functionalization method acts as a substitution for the classical cross-coupling reactions including Ullmann, Suzuki, Heck, Negishi, Goldberg, Buckwald–Hartwig, Sonogashira, Hiyama, Cadiot–Chodkiewicz, Stille and Kumada coupling, and the carbonylation coupling, which includes pre-functionalization or generation of halogen wastes.57–61 The coupling reaction without pre-functionalization catalyzed by a transition metal or other metal-free methodologies with a directing group containing a substrate are more efficient and atom-economic pathways.62,63 Dahye et al. earlier in 2018 discussed the site-selective C–H functionalization of chromones and coumarins.64 This review covers the selective functionalization of substituted chromones at C-2, C-3 and C-5 positions with recent advancements in the field.
2. C-5 activation of chromones
2.1. Transition metal-catalyzed C-5 activation of chromone
R. Samanta et al. in 2012 introduced Rh(III)-catalyzed C–C bond formation without a pre-functionalized reaction protocol for the synthesis of hybrid molecules of synthetic and medicinal importance. The direct oxidative cross-coupling reaction at the C-5 site of chromones with various olefins (Scheme 1) was carried out in an atom-economic, highly efficient, and concerted reaction pathway.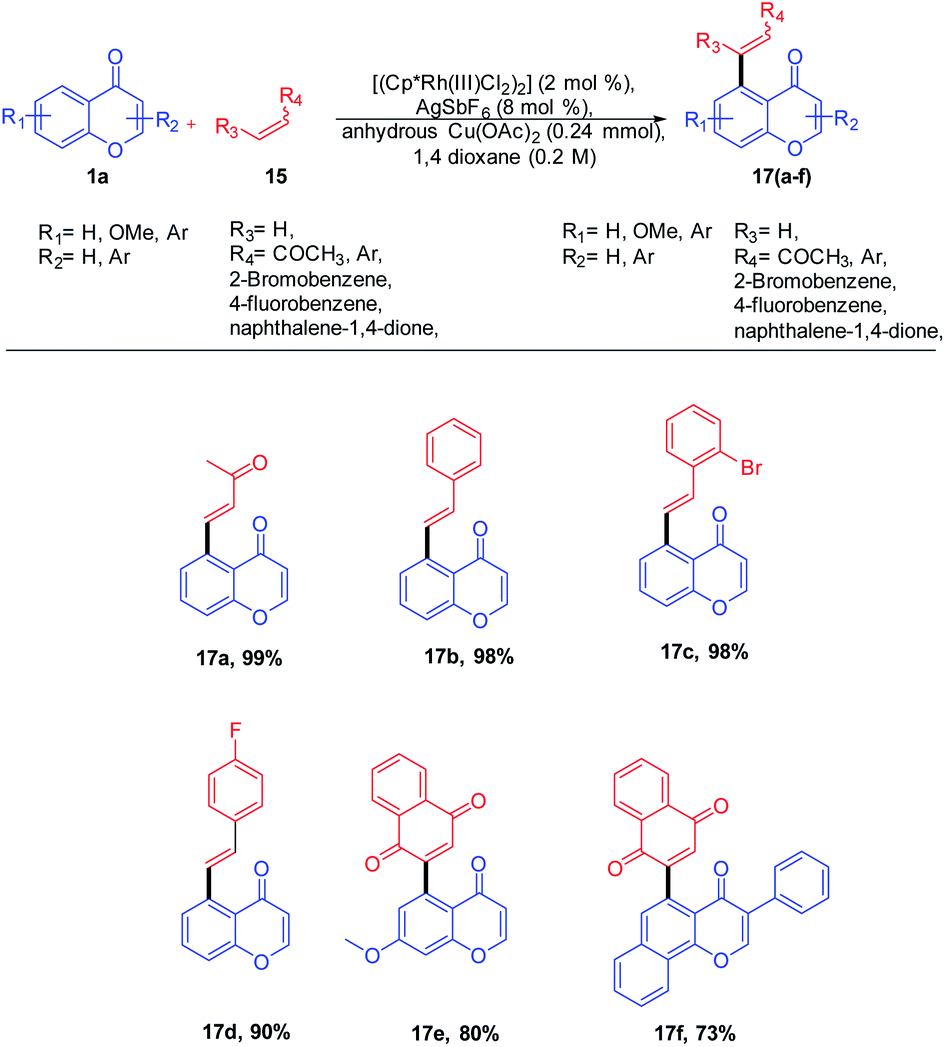 | ||
| Scheme 1 Rhodium(III)-catalyzed C-5 olefination of chromones. | ||
Mechanistically, the transition metal-catalyzed directing group (keto group)-assisted dehydrogenative coupling starts with the C–H bond activation (Scheme 2). Initially, the chloride ligand of the complex [(Rh(III)Cp*Cl2)2] will be removed as AgCl by the action of AgSbF6 to generate an active rhodium complex. The active rhodium species thus generated may contain ligands such as acetate or solvent in addition to pentamethylcyclopentadienyl (Cp*) ligands. Then, the active Rh species coordinates with the keto group of chromone and a concerted metalation/deprotonation (CMD) process activates the ortho C–H. The CMD process yields a five-membered rhodacycle 14 intermediate. The intermediate 14 transforms to a seven-membered rhodacycle 16 via an alkene insertion to the Rh–C bond of 14. The β-H elimination from the seven-membered intermediate afforded desired products 17(a–f) and the reduced inactive [RhCp*] complex. The catalytic cycle was completed by the oxidation of inactive [RhCp*] to [Rh(III)Cp*] by copper acetate.55
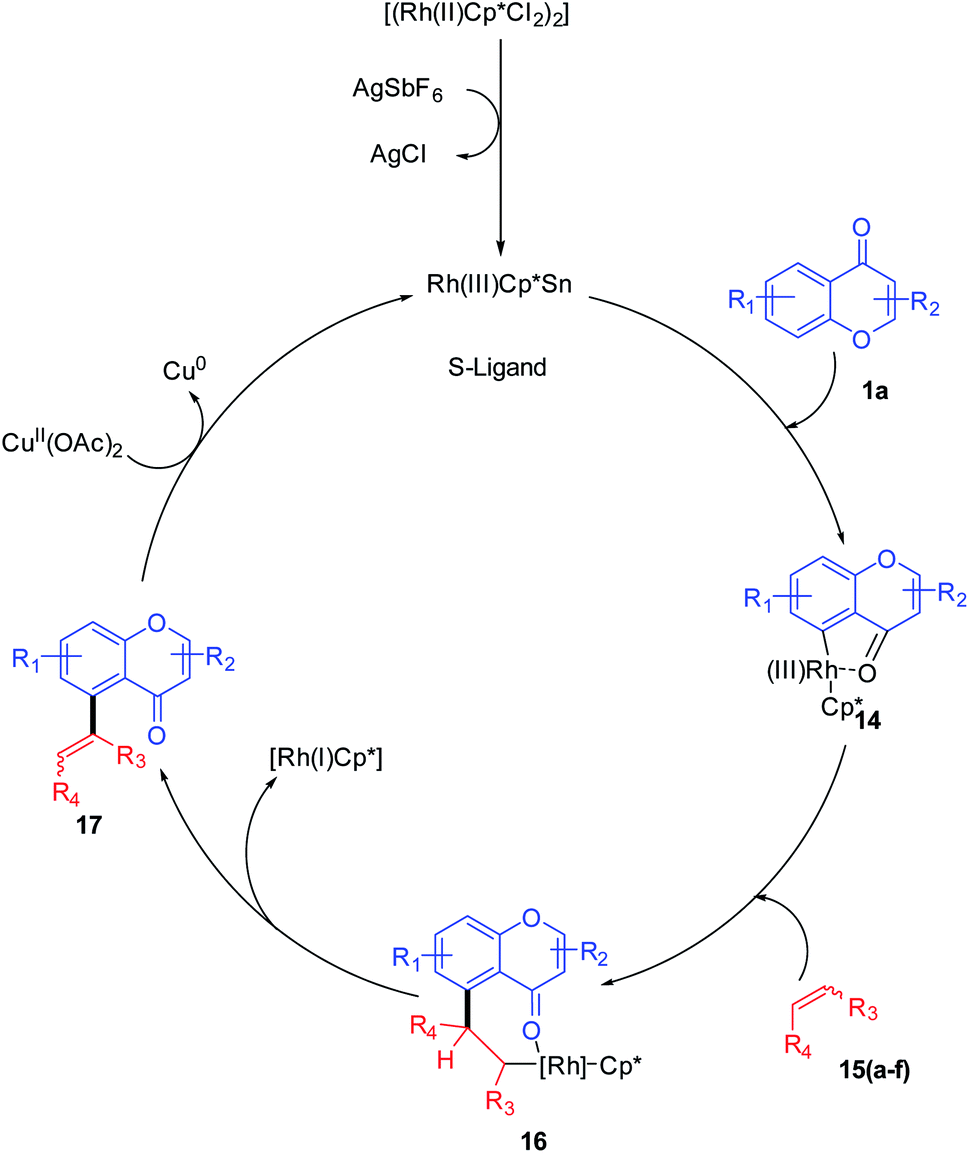 | ||
| Scheme 2 Proposed mechanism for rhodium(III)-catalyzed C-5 olefination of chromones. | ||
S. Kathiravan et al. in 2014 developed a mild Rh(III)-catalyzed reaction protocol for the activation of ortho C–H of aromatic and aliphatic ketones (Scheme 3). The reaction protocol was applicable for the selective perfluoroalkenylation of chromones at its C-5 position. The reaction proceeds under a mild condition with low catalyst loading and very good functional group tolerance and good to excellent yield. The synthetic scope of the method was enhanced by the excellent reactivity of the coupling partners and the regioselectivity of the reaction.
 | ||
| Scheme 3 Rhodium-catalyzed oxidative perfluoroalkenylation at the C-5 position of chromones. | ||
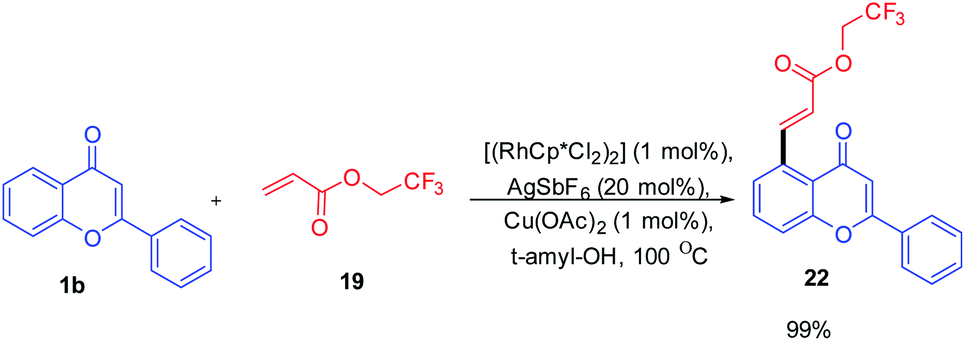
The reaction proceeds by initial electrophilic deprotonation of chromone 1 forming an Ar–Rh(III) intermediate 18. The coordination of the intermediate with CF3-substituted acrylate 19 then activates the olefin for further oxidative addition and β-hydrogen elimination to afford the expected product 2265 (Scheme 4).
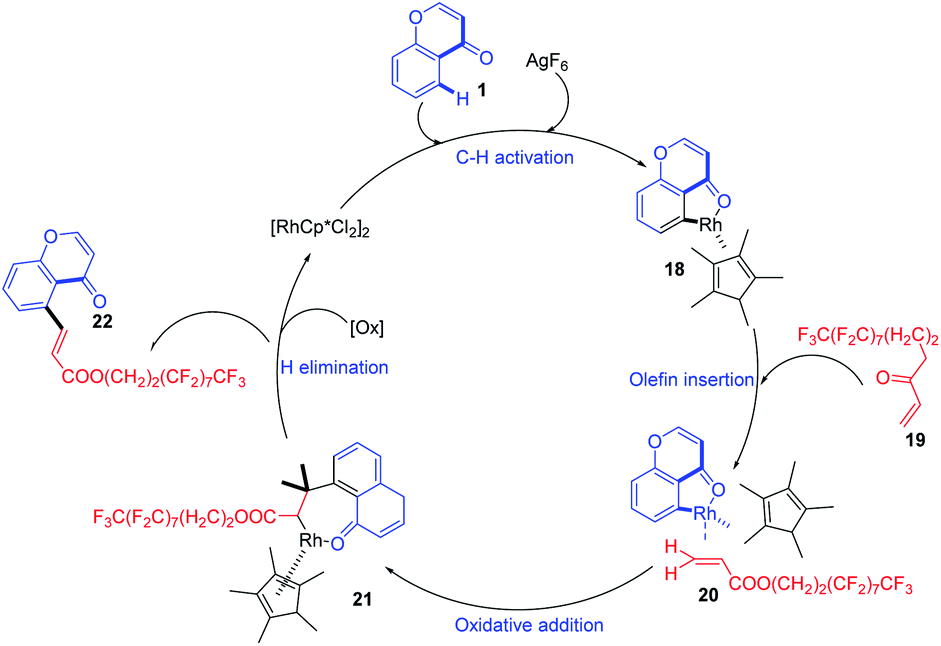 | ||
| Scheme 4 Proposed mechanism for rhodium-catalyzed oxidative perfluoroalkenylation at C-5 position of chromones. | ||
Minsik Min et al. 2014 put forward stereodivergent hydroarylation of chromones with alkynes (Scheme 5). The reaction proceeds in the presence of catalytic amounts of [Ru(p-cymene)Cl2]2 and AgSbF6 was a diastereodivergence in the reaction achieved by the quantitative loading of AgSbF6. The controlled quantitative addition of AgSbF6 to the reaction led to the switching of E/Z isomers in the product instead of the sole E-isomer.
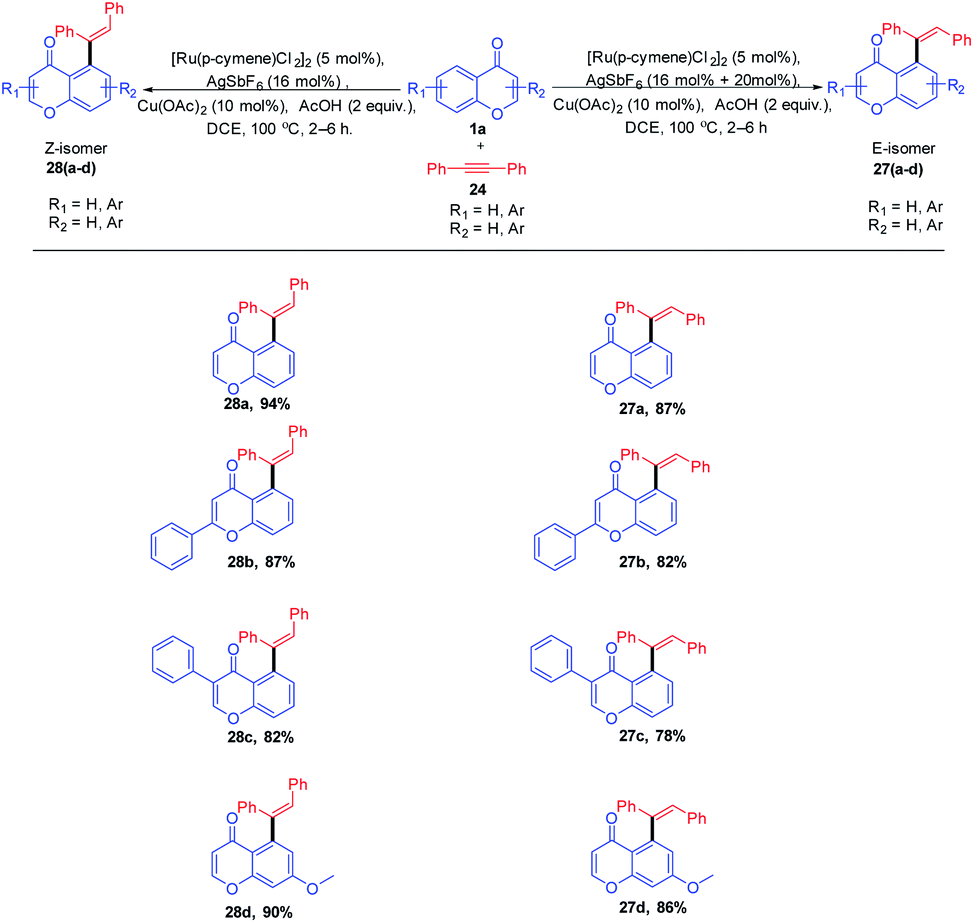 | ||
| Scheme 5 Ruthenium-catalyzed stereodivergent hydroarylation of chromones with alkynes. | ||
Ru(II) for the reaction was generated from [Ru(p-cymene)Cl2]2 upon treatment with AgSbF6 (Scheme 6). A coordinative insertion of alkyne to intermediate 23 yields seven-membered ruthenacycle intermediate 25. Subsequent protonolysis of Ru–C bonds of 25 affords E-alkene 27 regenerating the Ru(II) catalyst, and the rotation of the bond generates thermodynamically stable Z-isomer 28.66
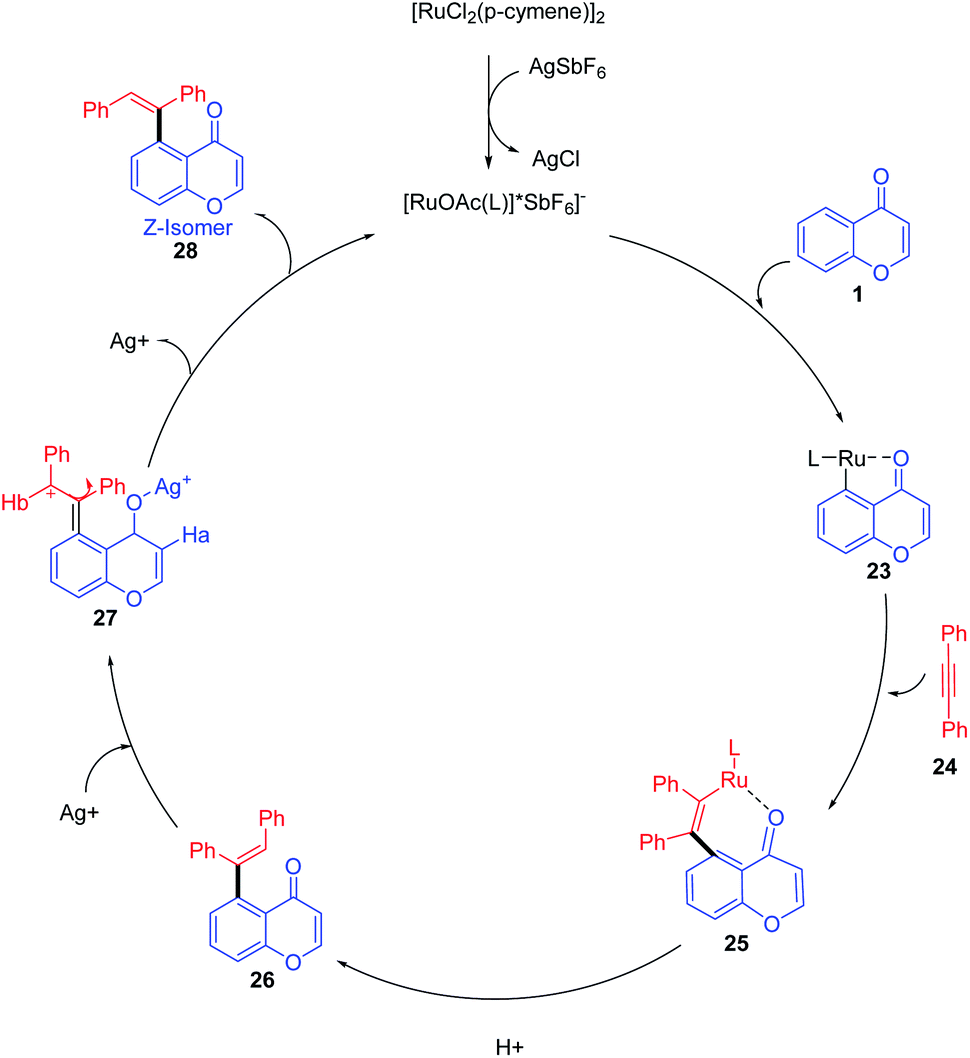 | ||
| Scheme 6 A plausible mechanism for ruthenium-catalyzed stereodivergent hydroarylation of chromones with alkynes. | ||
Kiho Kim et al. 2015 studied the direct C–H oxygenation of chromones using a Ru(II)-catalyzed synthetic protocol for the installation of a hydroxyl group on the C-5 position of chromones (Scheme 7). The oxygenation of chromones was achieved in good yield in the presence of monomeric ruthenium complex [Ru(pcymene)(CF3CO2)2(H2O)].
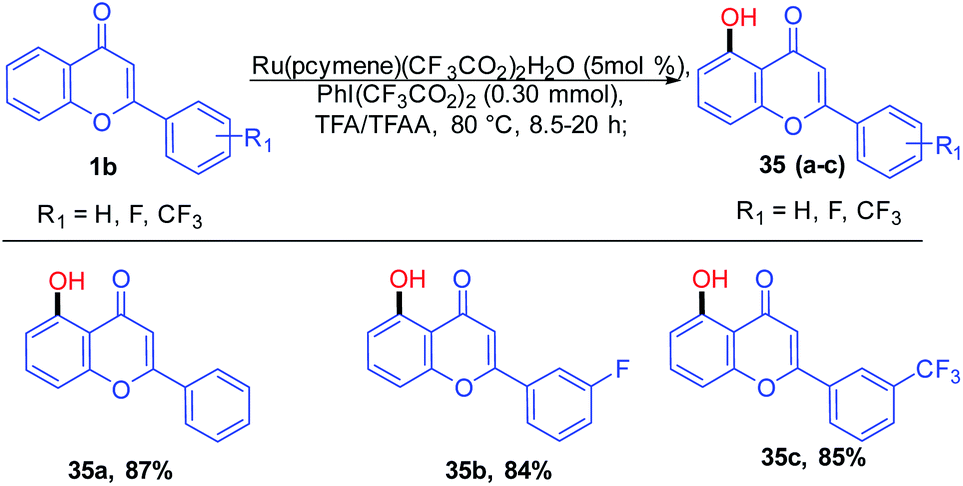 | ||
| Scheme 7 Ruthenium-catalyzed hydroxylation of chromones for the synthesis of 5-hydroxychromones. | ||
Chelate-directed C–H bond activation of chromone generates five-membered ruthenacycle 32, which on oxidation with iodine affords a Ru(IV) 33 species. A subsequent reductive elimination of 33 yields 5-(trifluoroacetyloxy)-flavone 34 which on aqueous workup yields the corresponding C-5-functionalized chromones 3567 (Scheme 8).
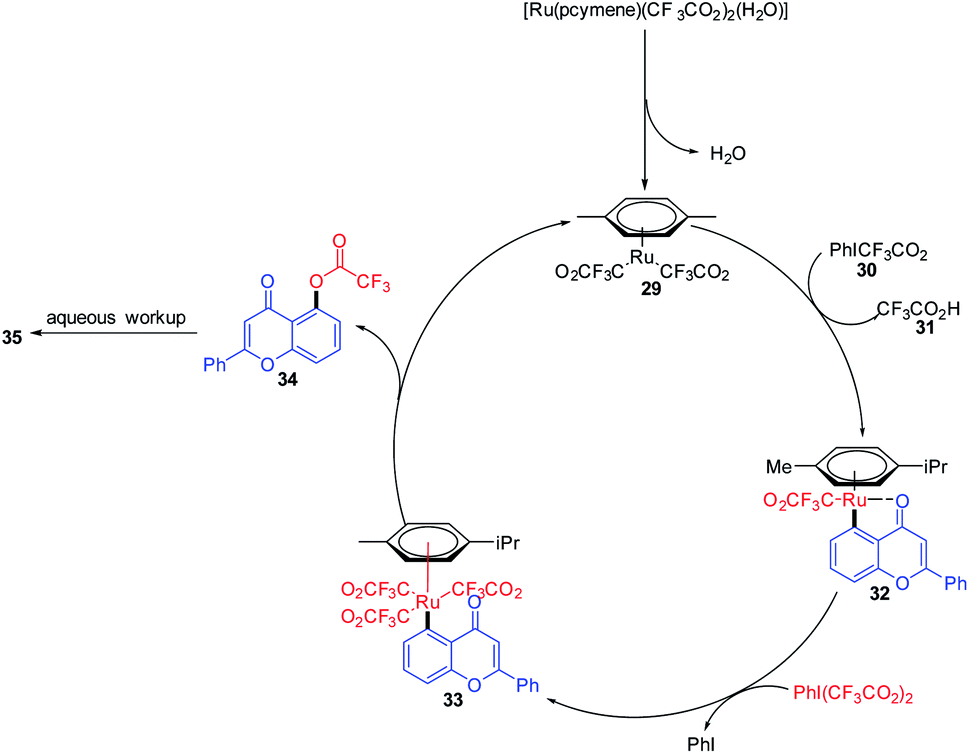 | ||
| Scheme 8 A plausible mechanism for the ruthenium-catalyzed hydroxylation of chromones for the synthesis of 5-hydroxychromones. | ||
Rhodium(II)-catalyzed C–H functionalization of bi- or tricyclic scaffolds such as chromones and xanthones was carried out by Sang Hoon Han et al. in 2016 (Scheme 9). This biologically active compound upon site-selective functionalization with maleimide yields a succinimide substitute at their respective C-5 position with good functional group compatibility and excellent site selectivity. The succinimide generated in the C-5 position of the chromones was obtained by olefin insertion into the metallacycle reaction intermediate followed by a protonation step.68
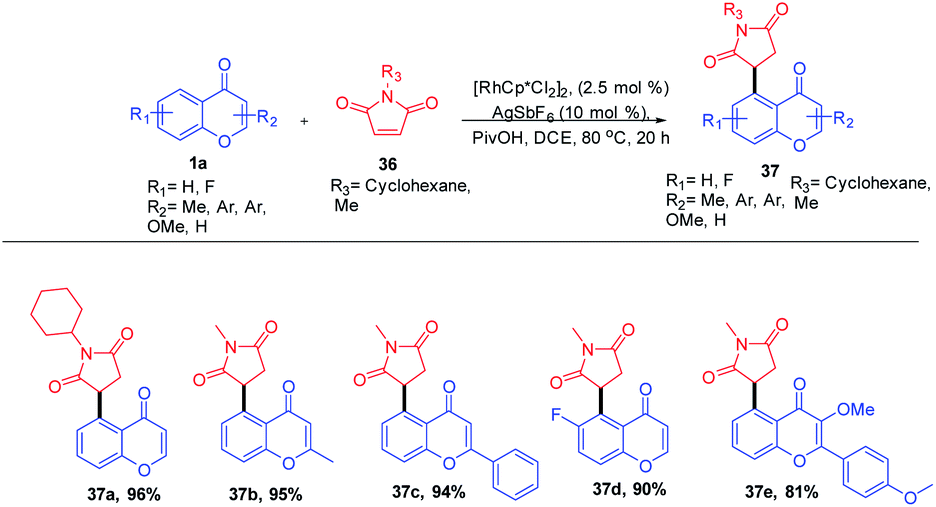 | ||
| Scheme 9 Rhodium-catalyzed synthesis of succinimide-substituted chromones and xanthones. | ||
The C–C bond formation with metal-catalyzed C–H activation of aryl ketones was studied by R. Shang et al. 2016 (Scheme 10). The main attraction of the reaction is the usage of inexpensive methylaluminum as the methylating agent and the economic and environmental merits of iron catalysts. The reaction proceeds by the combined activity of 4-(bis(2-(diphenylphosphanyl)phenyl)phosphanyl)-N,N-dimethylaniline (Me2N-TP) and methylaluminium for the transformation of ortho C–H to C–CH3 under an iron-catalyzed reaction condition. The reaction proceeds well for various functional groups.69
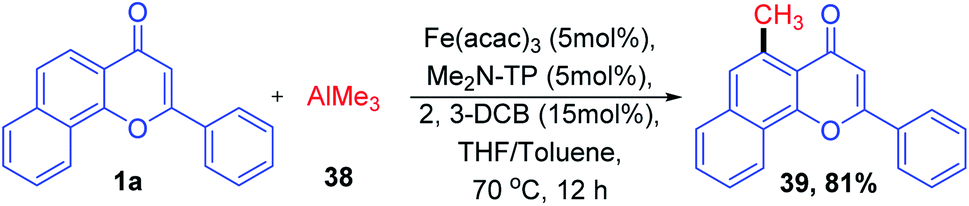 | ||
| Scheme 10 Iron-catalyzed ortho C–H methylation of chromones. | ||
1,2-Diacylbenzene as a valuable precursor for the synthesis of medicinally important compounds such as isobenzofurans, phthalazines, and isoindoles can be generated by decarboxylative acylation of aromatic ketones via a palladium-catalyzed reaction (Scheme 11). The decarboxylative acylation reaction yields 1,2-diacylbenzene in excellent yields with ortho selectivity. This reaction protocol put forward by Wing-Yiu Yu et al. 2017 demonstrates a successful attempt for the direct C–H acylation reaction of aromatic ketones with α-oxocarboxylic acids, which proceeds without pre-derivatization to imines.
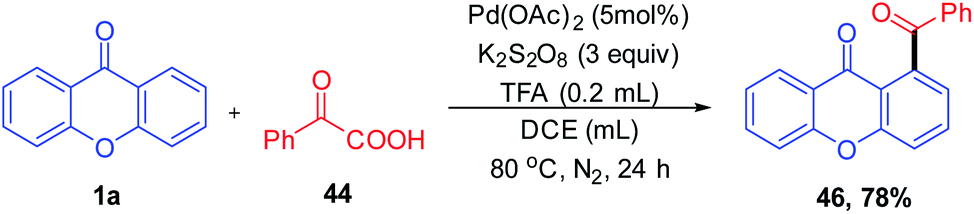 | ||
| Scheme 11 Palladium(II) catalyzed C-5 acylation of aromatic ketones. | ||
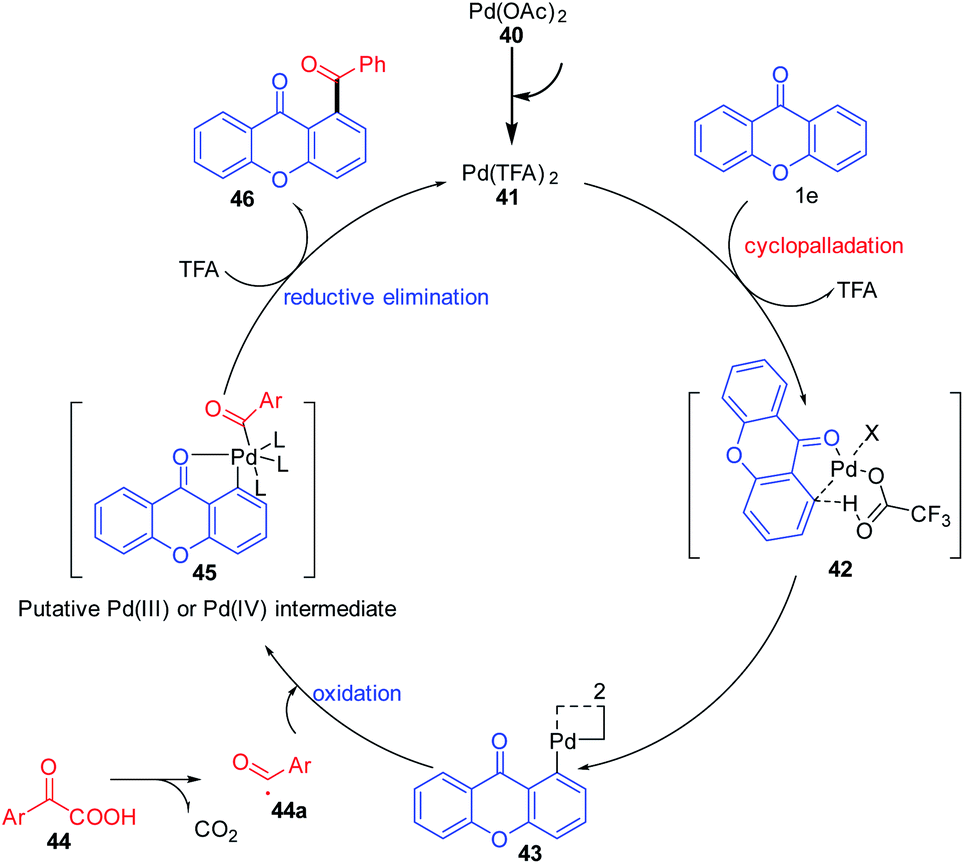
The plausible reaction mechanism for the acylation of chromones under a palladium-catalyzed reaction condition is depicted in (Scheme 12). First, Pd(OAc)2 gets transformed to Pd(TFA)2 in situ by the action of trifluoroacetic acid (TFA). The acylation gets initiated by the directing group (keto) via ortho-selective electrophilic palladation on the olefin by Pd(TFA)2. Palladacycle 43 obtained will then undergo oxidative coupling with acyl radical 44a generated in situ by decarboxylation of α-oxocarboxylic acid. Reductive elimination from Pd(III) or Pd(IV) intermediate would then furnish the final product 1,2-diacylbenzene 46 and an active Pd(II) catalyst.70
 | ||
| Scheme 12 Proposed mechanism for palladium-catalyzed C-5 acylation of aromatic ketones. | ||
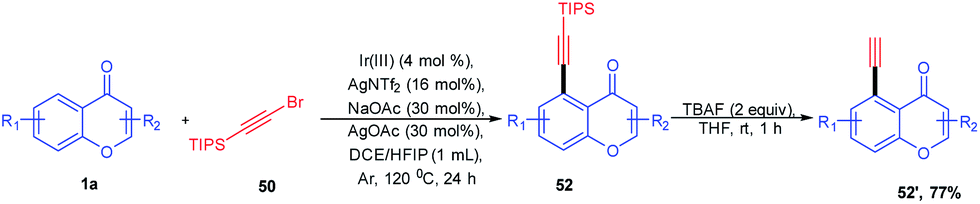
Ir(III)-catalyzed selective C–H bond alkynylation is a useful synthetic transformation of ketones and esters for the construction of molecular complexity and developing drug molecules. The alkynyl functionality introduced with the facilitation of the carbonyl group on ketones and esters was catalyzed by Ir(III) and Ag(I) (Scheme 13). The feasibility of the reaction with both electron-donating and electron-withdrawing substituents was investigated. The yield of the reaction implies the feasibility of electron-rich species over electron-deficient substituents. The substrate scope was also extended with halogen-substituted aromatic rings, which may increase the scope of reaction transformation further. The terminal alkynes were afforded using the desilylation reaction under mild reaction conditions.
 | ||
| Scheme 13 Regioselective C-5 bond alkynylation of chromones via iridium catalysis. | ||
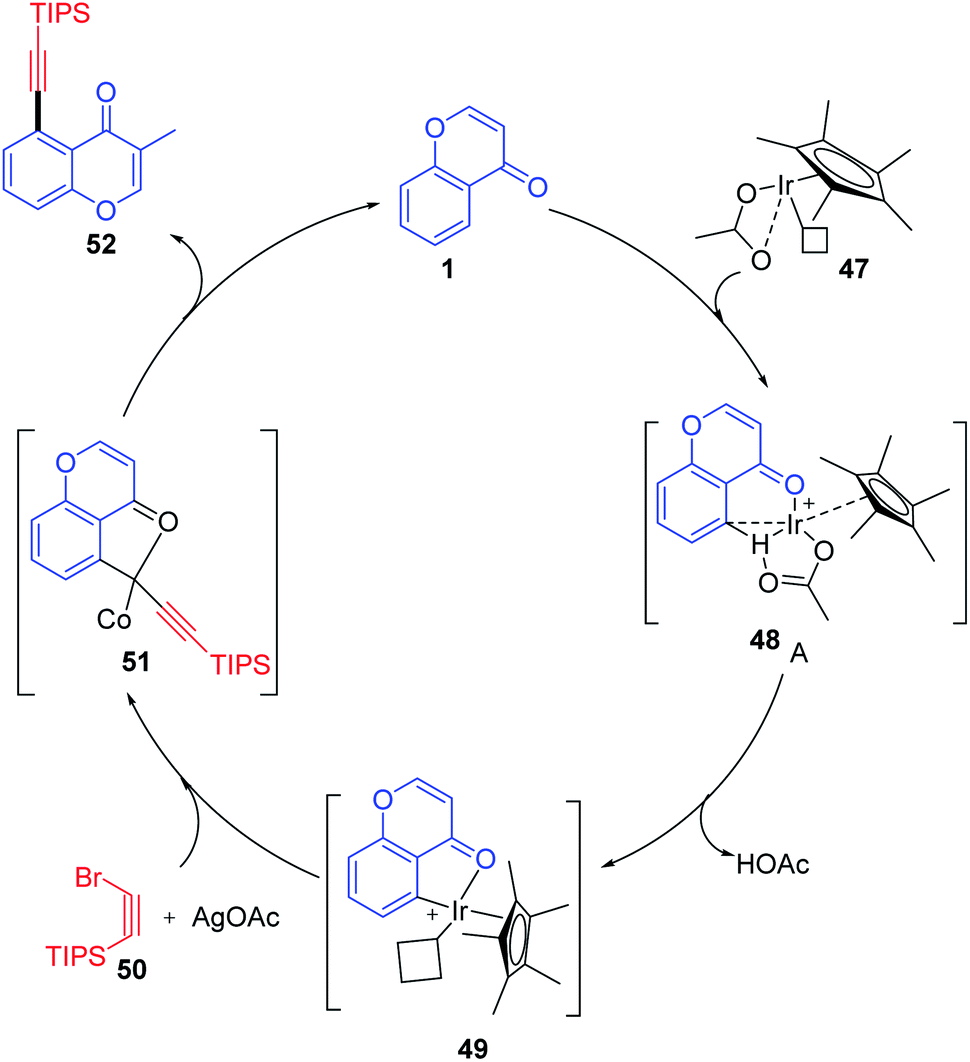
Li, X et al. 2017 explained the mechanism for the alkynylation reaction of chromones, as depicted in Scheme 14. The reactive Ir(III) is initially generated from [Cp*IrCl2]2 and AgNTf2 via ligand exchange. Later, the carbonyl-anchored Ir(III) facilitates the bond cleavage of C-5–H with the aid of acetate ligands. The reaction yields cyclometalated Ir(III) complex 49 the key intermediate of the reaction. The Ir(III) complex obtained undergoes oxidation with Ag(I) and alkynyl bromide in a concerted step or a multistep pathway yielding the product along with the regenerated Ir(III) catalyst.71
 | ||
| Scheme 14 Proposed mechanism for regioselective C-5 bond alkynylation of chromones via iridium catalysis. | ||
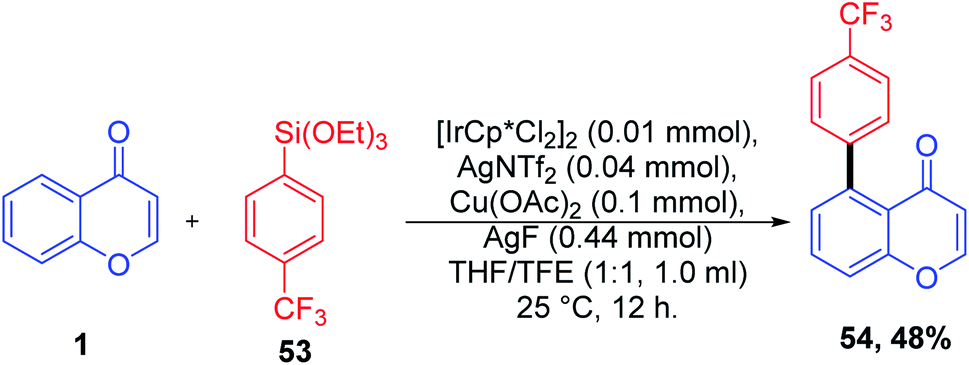
Direct C-5 arylation of chromones catalyzed by [IrCP*Cl2]2 for the synthesis of C-5-functionalized chromones was introduced by K. Shin et al. 2018 (Scheme 15). The reaction afforded C-5 aryl chromones in good yields with functional group tolerance at ambient temperature. The reaction proceeds via C–H cleavage at the C-5 position and subsequent transmetallation. The transmetallated complex forms a high-valent aryl species by one-electron oxidation. The formation of this high-valent species reduces the barrier of reductive elimination by approximately 19 kcal mol−1. Finally, the oxidatively induced reductive elimination yields the C-5 arylated chromones.72
 | ||
| Scheme 15 Iridium-catalyzed arylation of C-5 position of chromones. | ||
Tan, G et al. 2018 discovered a [IrCp*Cl2]-catalyzed C–H arylation of diverse arenes with heteroarenes in a modest yield (Scheme 16). The C–H heteroarylation of aromatic ketones and enamides was carried out by the Cp*Ir catalyst via oxidatively induced reductive elimination. The reaction protocol works well for the synthesis of aryl–heteroaryl and vinyl–heteroaryl scaffolds.73
 | ||
| Scheme 16 Iridium-catalyzed oxidative heteroarylation at C-5 position of chromones. | ||
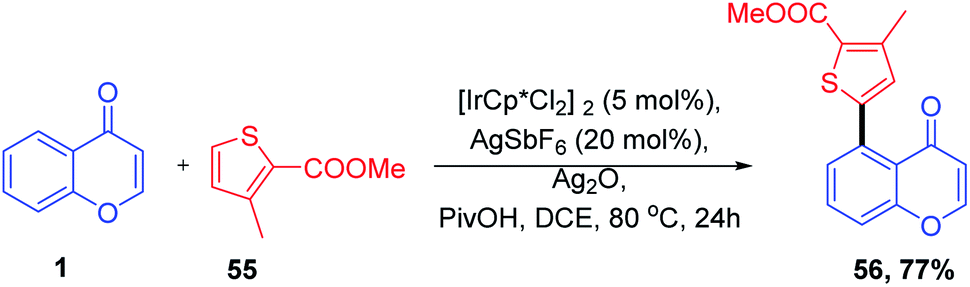
S. Debbarma further studied the wide applicability of chromones. S. Debbarma et al. in 2019 synthesised a set of C-5 substituted chromones by metal-catalyzed C-5 activation of chromones (Scheme 17). The ketone-directed C–H functionalization of C-5 carbon leads to the diversification of chromones by transition metal-catalyzed allylation, amidation, alkenylation, and 1,4-addition reactions.74
 | ||
| Scheme 17 C-5 functionalization of chromones by cobalt and rhodium catalysts. | ||
A C-5 functionalization method for the synthesis of β-aryl- and β-heteroarylethenesulfonyl fluoride was carried out by G. Ncube et al. 2019. The reaction utilizes a catalytic system of tris(acetonitrile)-(pentamethyl cyclopentadienyl)rhodium(III)hexafluoroantimonate, [Cp*Rh(MeCN)3], and (SbF6)2 by oxidative C–H alkenylation of arenes with ethenesulfonyl fluoride (Scheme 18). The directing group strategy allows the synthesis of β-aryl- and β-heteroarylethenesulfonyl fluoride without the pre-installation of halogens, boronic acids, or diazonium functional groups on arenes with modest to good yields for a broad variety of directing groups. The reaction strategy provides an excellent and harmonizing approach for the attainment of novel building blocks in sulfur(IV) exchange click chemistry.75
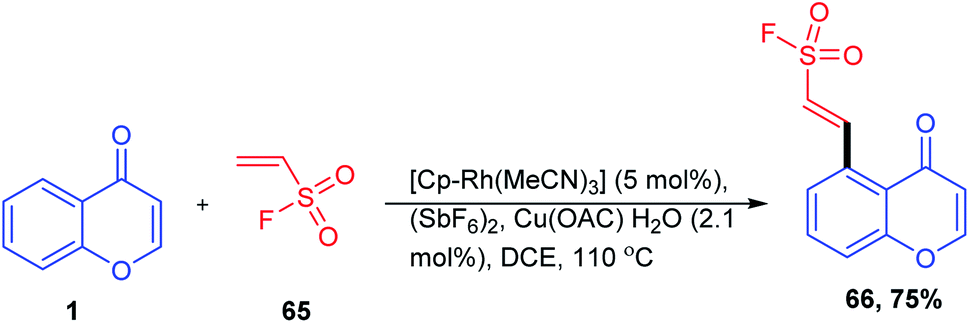 | ||
| Scheme 18 Rhodium-catalyzed C-5 fluorosulfonylvinylation of chromones. | ||
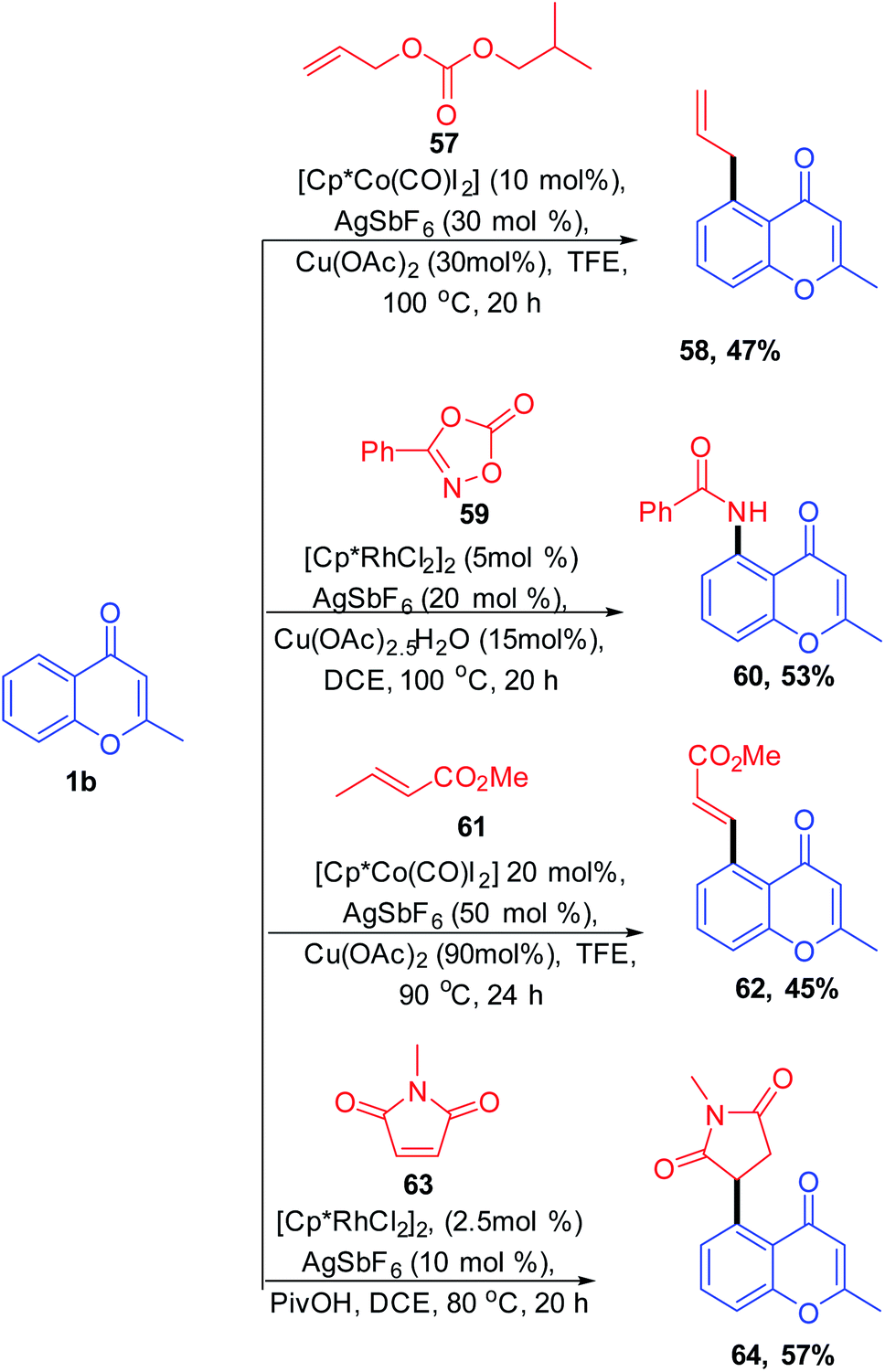
Vinylarenes are structurally important scaffolds due to their applicability as a starting material for various fine chemicals, polymers, and drugs. The synthesis of vinylarenes with ketone-directed C–H vinylation using vinyl acetate was reported by M. R. Sk et al. 2020 (Scheme 19). The vinylation on a set of compounds including diaryls, acetophenones, chalcones, and chromones was carried out by first-row transition metals via ketone-directed C–H bond activation. This is the recently reported reaction for the cobalt-catalyzed carbonyl group-directed C–H vinylation with vinyl acetate.
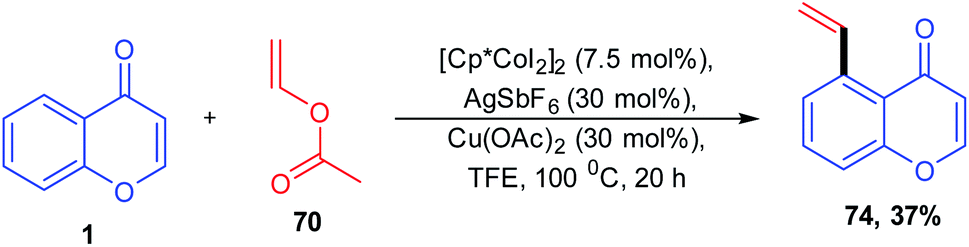 | ||
| Scheme 19 Cobalt(III)-catalyzed C–H vinylation of chromones. | ||
The active catalyst for the vinylation reaction was obtained from [Cp*CoI2]2 complexes in the presence of silver hexafluoroantimonate and acetate by removal of AgI (Scheme 20). The active catalyst and aromatic ketone moiety form a cyclometallated species 69. Subsequently, a 1, 2 migratory insertion of Co–C bond into the vinyl acetate provides the intermediate 72. The β-hydride elimination/reinsertion of the intermediate 72 and the β-acetate elimination of the formed final intermediate 73 gives the desired vinylated product 74.76
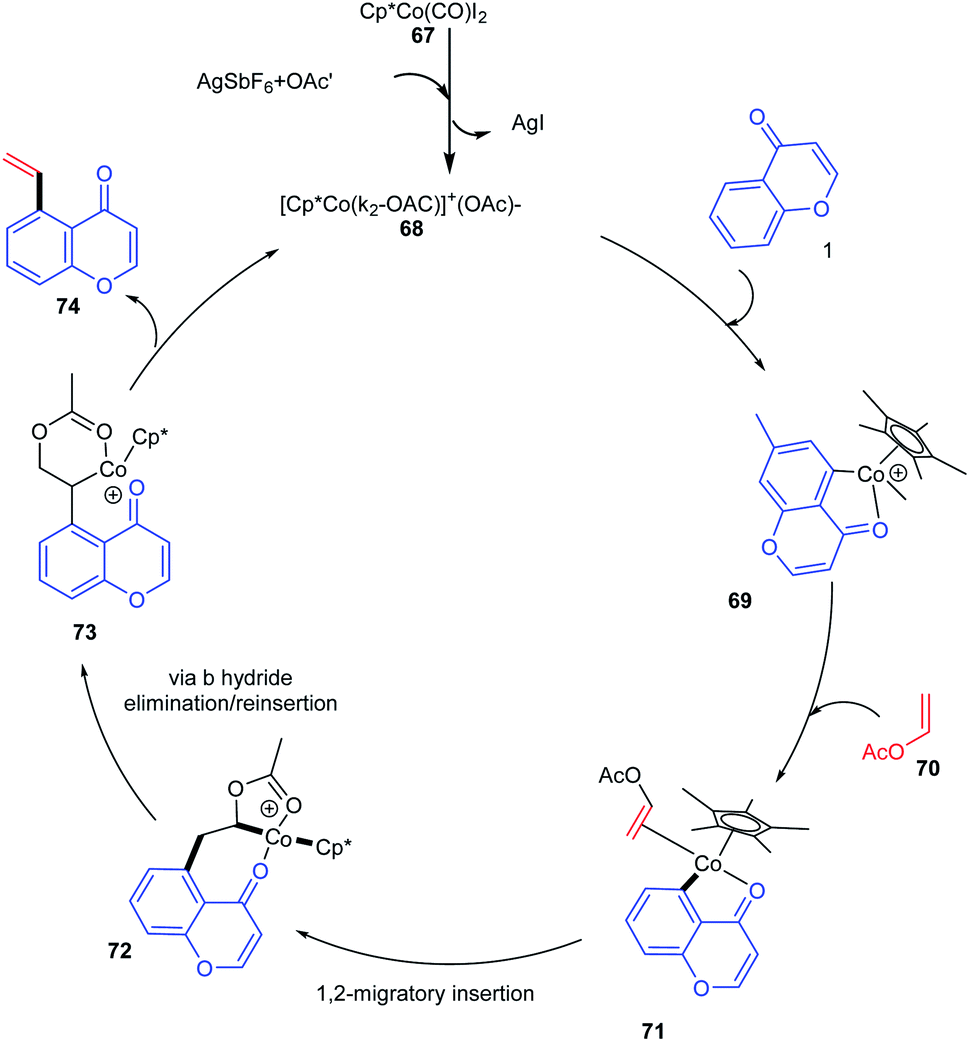 | ||
| Scheme 20 Proposed mechanism for cobalt(III)-catalyzed C–H vinylation of chromones. | ||
Y. Zhou et al. 2020 reported the Ru(II) catalyst for the coupling of chromones with maleimides yielding a C-5-activated chromone by utilizing keto as a weak directing group (Scheme 21). Switching the additives with benzoic acid and silver acetate yields 1,4 addition products and oxidative Heck-type products respectively. The products were obtained in good to excellent yields with good functional group tolerance under a solvent-free reaction condition.
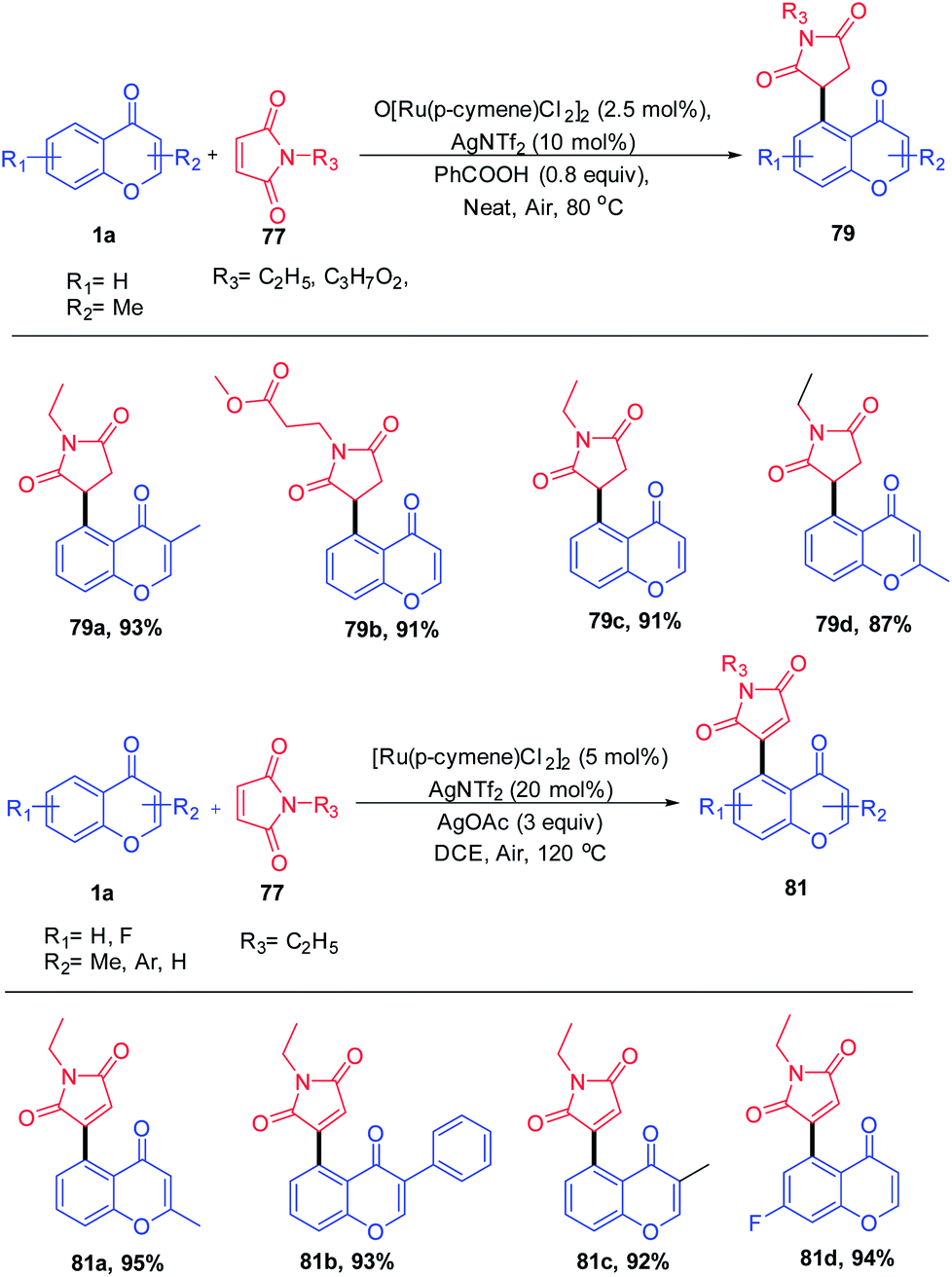 | ||
| Scheme 21 Ruthenium(II)-catalyzed C–H activation of chromones with maleimides to synthesize succinimide/maleimide-containing chromones. | ||
The mechanism of the reaction was studied by deuteration techniques. Initially, the ruthenium complex [Ru(p-cymene)Cl2]2 reacts with AgNTf2 and benzoic acid or AgOAc accordingly for the generation of an active monomeric form or A′ respectively (Scheme 22). The oxygen of the keto group coordinates with the active ruthenium species, which provides a five-membered metallacycle 76. Subsequent insertion of maleimide to the carbon–ruthenium bond of 76 gives a bicyclic intermediate 78. The addition of benzoic acid or silver acetate to the intermediate 78 yields the 1,4-addition product or Heck-type product respectively. The catalyst is regenerated as a Ru(II) species by the deprotonation of β-hydrogen of 78 and subsequent oxidation by silver acetate.77
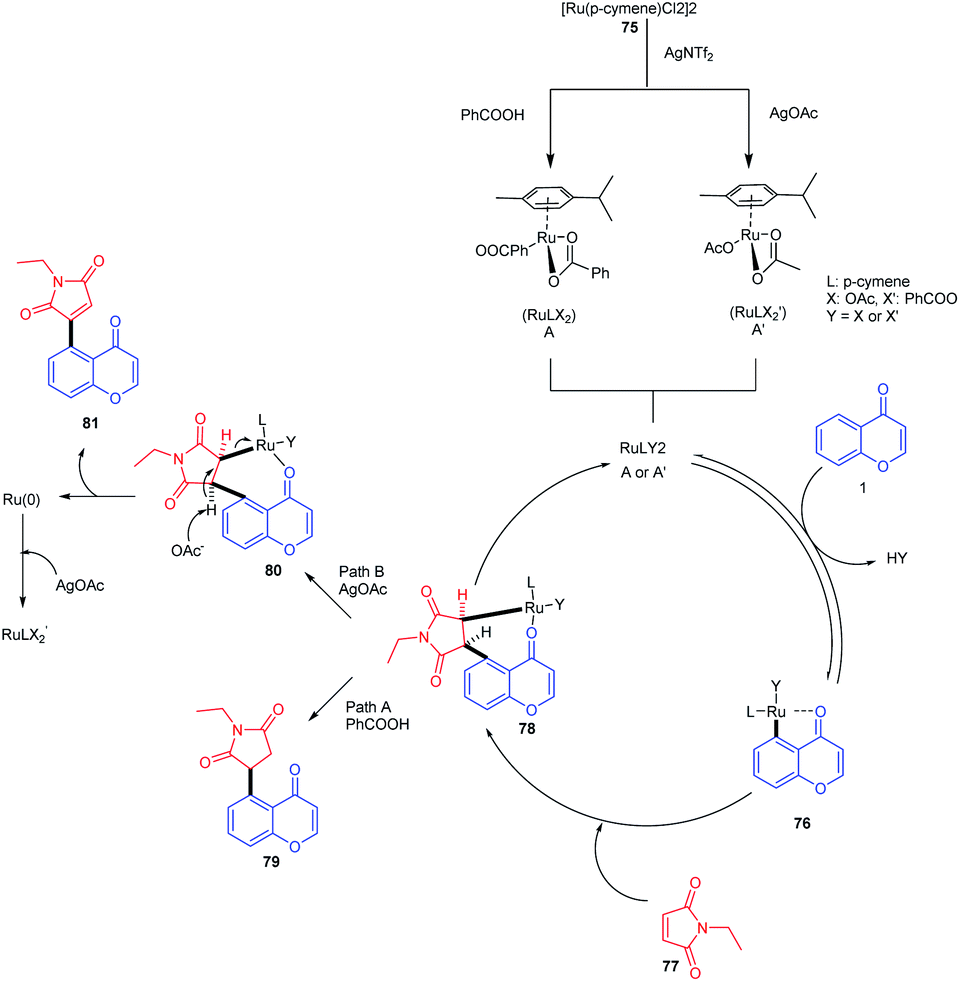 | ||
| Scheme 22 Proposed mechanism for ruthenium(II)-catalyzed C–H activation of chromones with maleimides to synthesize succinimide/maleimide-containing chromones. | ||
2.2. Metal-free C-5 activation of chromones
The C-5 functionalization with the nitro group was carried out by Recanatini, M et al. in 2000 with the traditional nitration method using HNO3 in H2SO4 at 0 °C to room temperature to yield 8-methyl-5-nitro-4H-benzopyran-4-one (Scheme 23). The nitration compound was utilized for further reaction transformation.78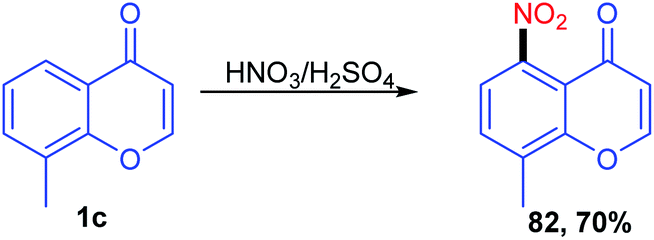 | ||
| Scheme 23 Nitration of C-5 position of chromones. | ||
Tsukasa Ishihara et al. 2004 utilized base-catalyzed C-5 activation of xanthenone by allylbromide (Scheme 24). The C-5 alkylated compounds were synthesized using DMF with allyl bromide at room temperature in excellent yields. The allylated compounds were further utilized for other transformations and biological studies.79
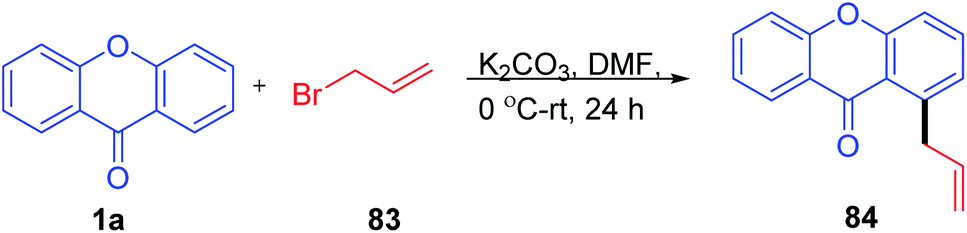 | ||
| Scheme 24 Allylation of C-5 position of xanthenone. | ||
3. C-3 activation of chromones
3.1. Transition metal-catalyzed C-3 activation of chromones
C-3-functionalized chromones were synthesised by Jeganmohan, M et al. 2010 using a tmp4Zr·4MgCl2·6LiCl base in THF (Scheme 25). The base tmp4Zr·4MgCl2·6LiCl initiates the zirconation of heterocycles of pharmaceutical importance like chromones. Chromones upon reaction with the zirconium base at −35 °C yield zirconated chromones in excellent yields. The zirconated chromones upon copper-catalyzed allylation yields a SN2 substituted C-3 allylated product.80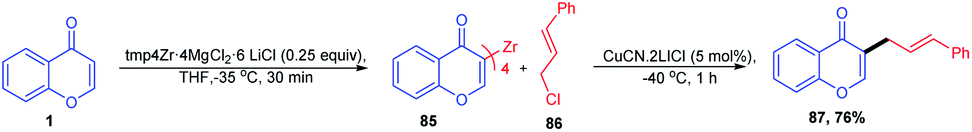 | ||
| Scheme 25 tmp4Zr base for the metalation of functionalized arenes and heteroarenes. | ||
A direct C-3 alkenylation of chromones through a palladium(II) catalyzed methodology was achieved by D. Kim et al. in 2011 (Scheme 26). In this reaction, the reactivity of the coupling reaction between chromones and alkenes was attained by the combined activity of pivalic acid and Cu(OAc)2/Ag2CO3 systems. The cross-coupling method leads to the formation of 3-vinylchromones, which are good structural scaffolds for the generation of biologically and synthetically active compounds. The method represents a C-3 derivatization of chromones with considerable advances over the current two-step methods with a good functional group tolerance and remarkable yield.
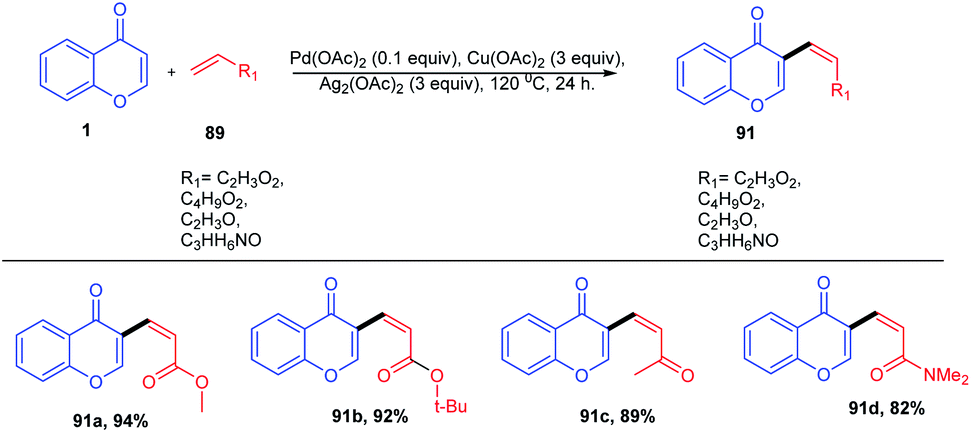 | ||
| Scheme 26 Palladium(II)-catalyzed intermolecular C-3 alkenylation of chromones. | ||
The possible mechanism for the transformation of the chromones to 3-vinylchromones initiates with a nucleophilic attack of chromones on palladium and the subsequent generation of palladium(II) intermediate 88 by the deprotonation by a pivalate ligand (Scheme 27). Next, the C-3 palladated species was inserted into the alkene substrate followed by reductive elimination, which generates the desired C-3 functionalized chromones. The catalyst Pd(II) was then regenerated from Pd(0) using Cu(OAc)2/Ag2CO3 to continue the catalytic cycle further.81
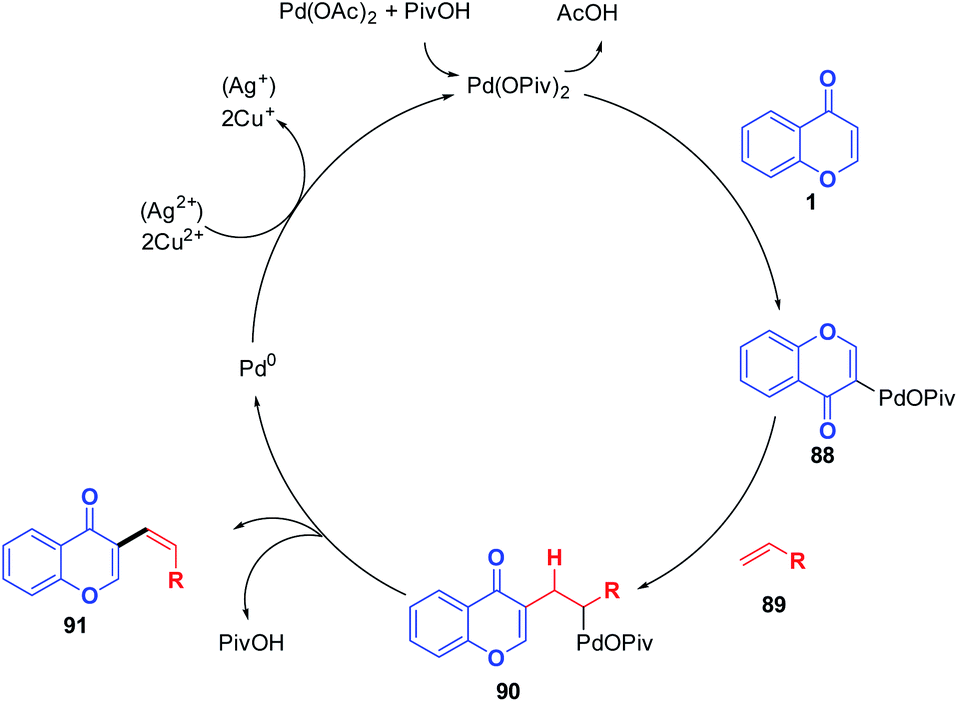 | ||
| Scheme 27 Proposed mechanism for palladium(II)-catalyzed intermolecular C-3 alkenylation of chromones. | ||
Oxidative arylation of chromones catalyzed by a Pd(II) catalyst was conducted by K. H. Kim et al. in 2012 (Scheme 28). The chromone selected for the arylation reaction was C-2, and C-3 was unsubstituted, and hence, the reaction yields a trace amount of C-3 functionalized product.82
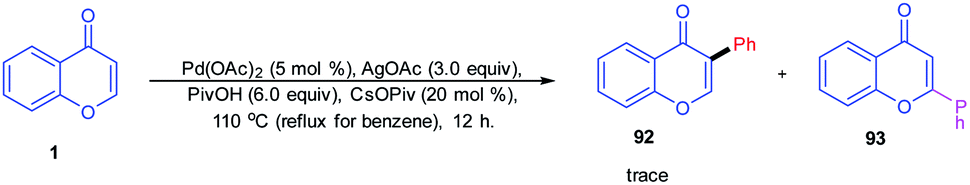 | ||
| Scheme 28 Palladium-catalyzed C-3 arylation of chromones. | ||
F. Chen et al. 2012 investigated the regioselective intermolecular arylation of chromones with polyfluoroarenes using a palladium-catalyzed reaction protocol (Scheme 29). The reaction afforded fluoroarylated enones by a two-fold C–H bond functionalization method. The scope of the reaction extends to C-3 fluoroarylation of quinolones, chromones and pyrimidones in reasonable yields. The proposed reaction mechanism for the cross-coupling reaction proceeds by the formation of a palladated intermediate of both enone and fluoroarene. The intermediates thus formed couple with fluoroarene and enone and the succeeding reductive elimination yields desired products along with the Pd(II) catalyst.83
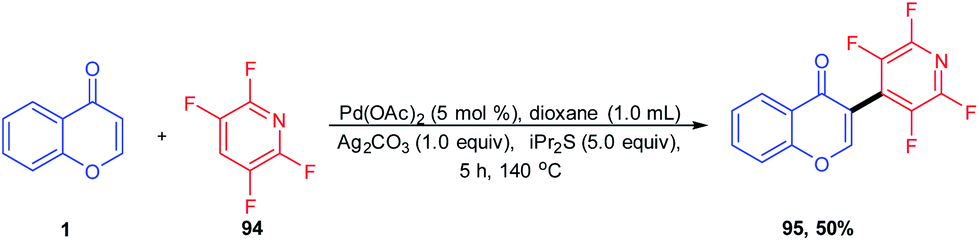 | ||
| Scheme 29 Palladium-catalyzed dehydrogenative coupling of chromones with polyfluoroarenes. | ||
The C-3 functionalization of flavones with tert-butylperoxybenzoate (TBPB)/potassium persulphate (K2S2O8) oxidant combinations catalyzed by an iron(III) catalyst was carried out by B. A. Mir et al. in 2016 (Scheme 30). A Csp2–Csp3 coupling product was afforded for the reaction of flavones in cycloalkanes with exclusive C-3-functionalized cycloalkylated products. The reaction afforded a C-3 amidation product when the solvent was altered from cycloalkane to N,N-dimethylformamide. In both reactions, the solvent itself acts as a coupling partner for the progress of the reaction. The radical forming capability of cycloalkane and N,N-dimethylformamide ensures the formation of a product with the active participation of solvents in reactions. Again, when the solvent was changed to chlorobenzene, the C-3 methylated product was obtained by utilizing tert-butylperoxybenzoate (TBPB) as the source of the methyl group.
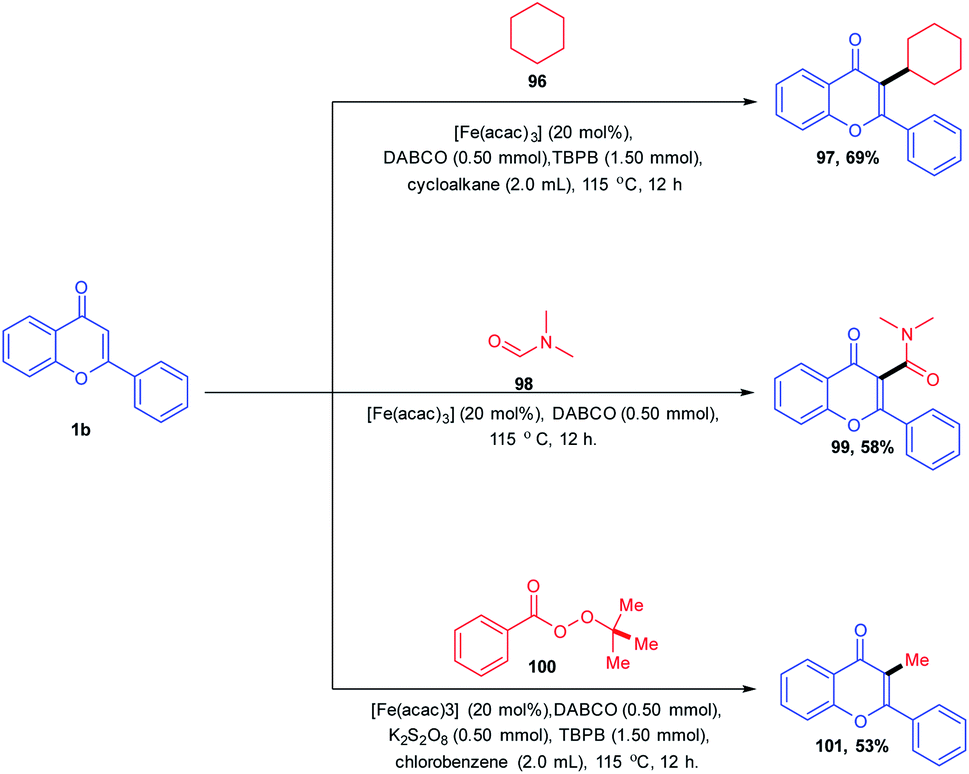 | ||
| Scheme 30 Iron(III)-catalyzed functionalization of the C-3 position of chromones. | ||
The C-3 functionalization reaction is set off by thermal homolytic bond cleavage of peroxide oxidant tert-butyl peroxybenzoate (TBPB) forming peroxybenzoate and tert-butoxy radicals. Both of these radicals generated are capable of scavenging hydrogen from cyclohexane 96 and N,N-dimethylformamide 98. A stable benzyl radical will generate upon the addition of any of these radicals on flavone 1. The radical species 110 generates a cationic intermediate 111 through the reduction of catalyst Fe3+ to Fe2+. Finally, the C-3 proton of the flavone gets abstracted by the base affording the C-3-functionalized product. In the absence of radical-generating solvents, TBPB acts as the source of the CH3 group. The method allows the synthesis of a C-3-functionalized product through cycloalkylation, amidation and methylation by varying the solvents84 (Scheme 31).
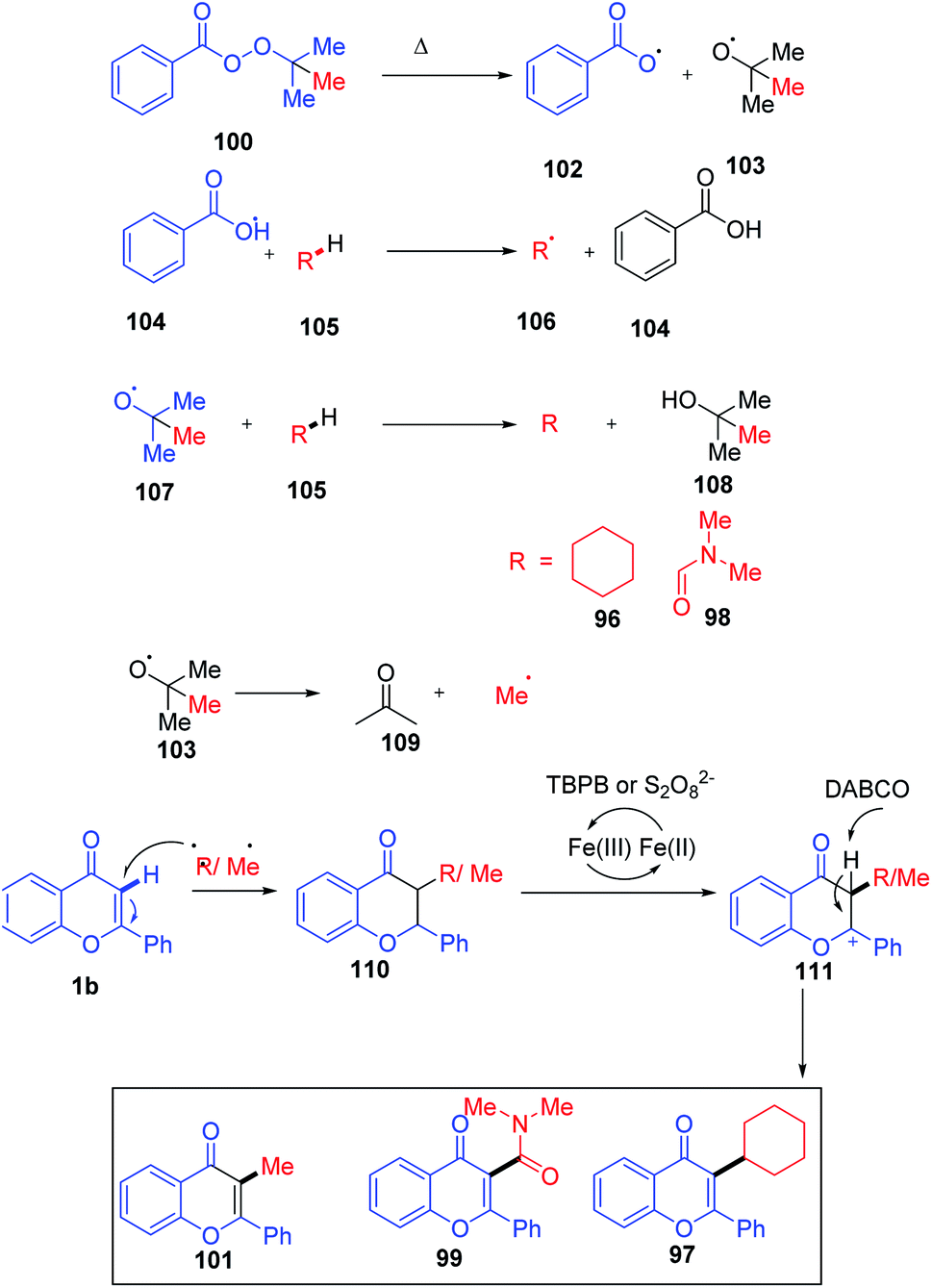 | ||
| Scheme 31 Proposed mechanism for iron(III)-catalyzed functionalization of the C-3 position of chromones. | ||
Youngtaek Moon et al. 2012 put forward an efficient and straightforward methodology for the direct cross-coupling of the C-3 position of chromones with quinones using palladium acetate as the catalyst. The reaction afforded good to excellent yields of quinones with tolerance for a wide range of functional groups on both the chromone and quinone moieties. Later, H. Choi et al. 2016 developed Pd(II)-catalyzed direct arylation of chromones (Scheme 32) by site-selective C-3 functionalization. The C–H activation progresses through a single-step metalation deprotonation method, which selectively activates the C-3 position of chromones.85
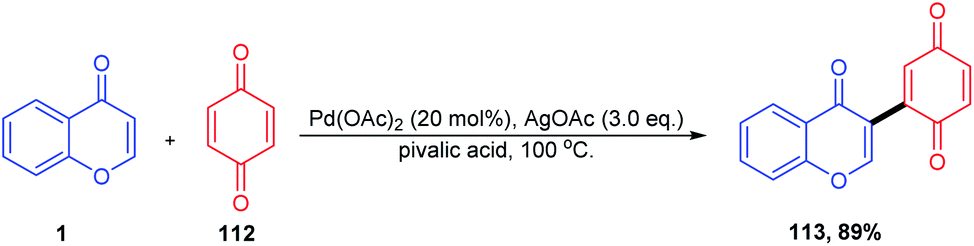 | ||
| Scheme 32 Palladium-catalyzed site-selective C-3 functionalization of chromones. | ||
The synthesis of a C-3 acylated chromone via acylation of 2,3-unsubstituted chromones through decarboxylative C–C cross-coupling with α-keto acid was described by Satenik Mkrtchyan et al. 2018 (Scheme 33). The title transformation was effectively catalyzed by using a set of silver and palladium salts.
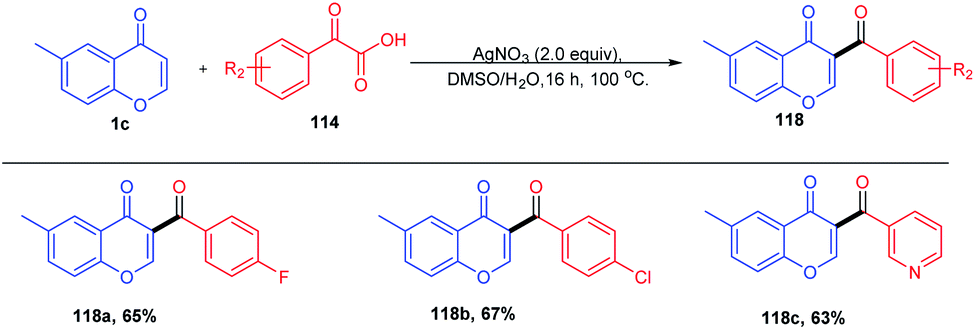 | ||
| Scheme 33 Silver nitrate-catalyzed C-3 acylation of chromones. | ||
The mechanism for the C-3 acylation of chromones involves an initial O-acylated radical-chromone adduct 115 formation (Scheme 34). Subsequently, the adduct formed get oxidised into an O-acyl pyrylium moiety, which further undergoes Fries-type rearrangement followed by deprotonation yielding the preferred product 73 in moderate quantity.86
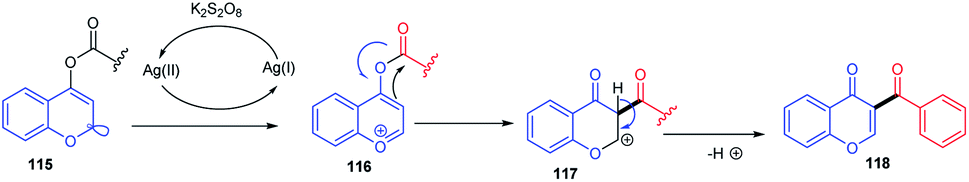 | ||
| Scheme 34 Proposed mechanism for AgNO3-catalyzed C-3 acylation of chromones. | ||
3.2. Metal-free C-3 activation of chromones
Lv, Z et al. 2010 studied the metal-free C-3–H activation of chromones (Scheme 35). The chromones were functionalized at their C-3 position by base-catalyzed iodination under reflux conditions. The iodochromones were further utilized for the generation of a terminal alkyl chain on the C-3 position.87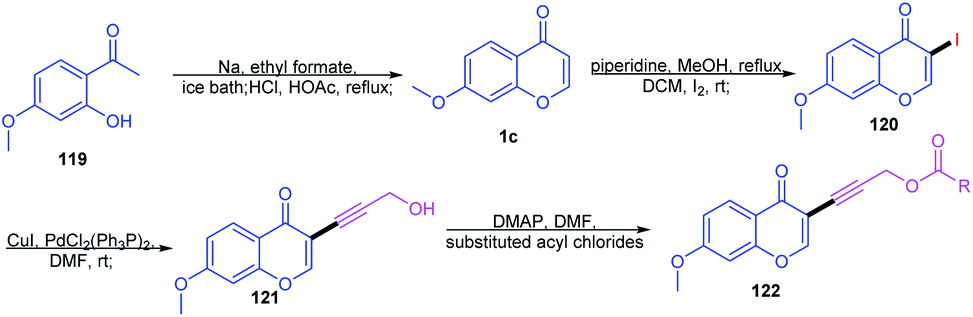 | ||
| Scheme 35 Synthesis of C-3-functionalized chromones. | ||
Lydia Klier et al. in 2012 and 2017 explained the Lewis acid accelerated zincation of chromones and thereby the transformation of the compound to other useful products (Scheme 36). TMPZnCl·LiCl (TMP = 2,2,6,6-tetramethylpiperidyl) leads to the zincation of chromones and quinolones by coordination of TMPZnCl·LiCl to the basic carbonyl oxygen. The zincation yields Zn-coordinated chromones through a complex induced proximity effect (CIPE). The zincation can be done at either the C-3 or C-2 position of chromones depending on the 2,2,6,6-tetramethylpiperidine (TMP) base. A similar zincation product of chromone can be obtained on the C-2 position of the chromone if the TMP base selected is TMP2Zn·2MgCl2·2LiCl. The resulting zinc organometallic intermediate undergoes various reactions including Pd/Cu-catalyzed cross couplings or acylation/allylation reactions.88,89
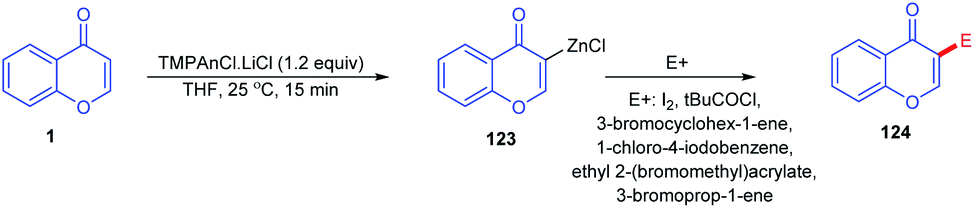 | ||
| Scheme 36 Lewis acid-promoted selective C-3 zincation of chromones. | ||
Y. Ding et al. 2016 put forward ammonium iodide-induced sulfenylation of flavones, arylimidazo[1,2-a]pyridines and indoles for the generation of thioethers (Scheme 37). The reaction proceeds with sodium benzenesulfinates as the sulphur source and odourless ammonium iodide as the reaction inducer. Sodium benzenesulfinate was selected as the sulfur source due to its stability and efficiency in synthesising regioselective products in good yields. The regioselectiveness of the reaction on flavones was studied by substitution with methyl groups at the C-3 and C-2 positions. Once the C-3 position was blocked by the methyl group, no thioether was formed, verifying the regioselectivity of the sulfenylation reaction.90
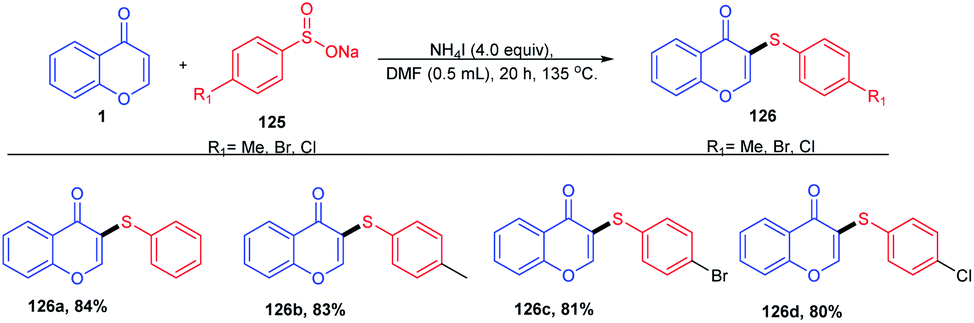 | ||
| Scheme 37 Synthesis of thioethers using benzenesulfinates as a sulfur source. | ||
W. Zhao et al. 2015 introduced arylthiols as a new sulfur source and the same colourless odourless ammonium iodide as the reaction inducer (Scheme 38). The sulfenylation method introduced here was an efficient and regioselective methodology for obtaining organosulfur compounds of flavones in good to excellent yields in a metal-free synthetic protocol.
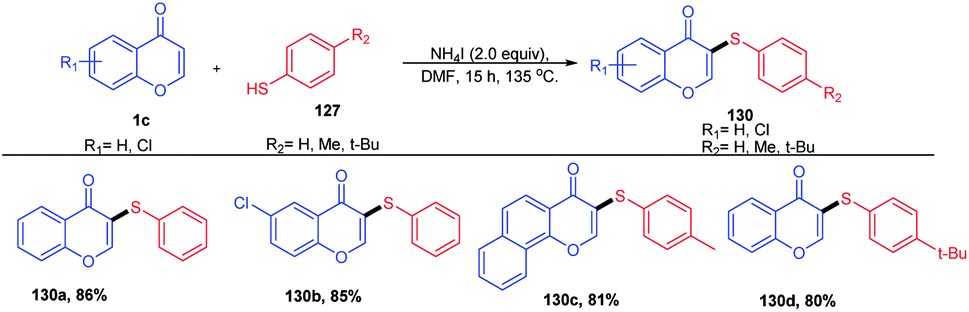 | ||
| Scheme 38 Synthesis of thioethers using aryl thiols as the sulfur source. | ||
At 135 °C, the reaction inducer ammonium iodide was split into NH3 and HI (Scheme 39). The obtained HI gets oxidized into iodine in the air atmosphere. The generated iodine will then react with thiophenol to form an electrophilic species ArS–I. ArS–I upon reaction with flavone 1c at the electron-rich C-3 position gives the corresponding intermediate 128. The intermediate 128 on deprotonation yields the desired thioether 130.91
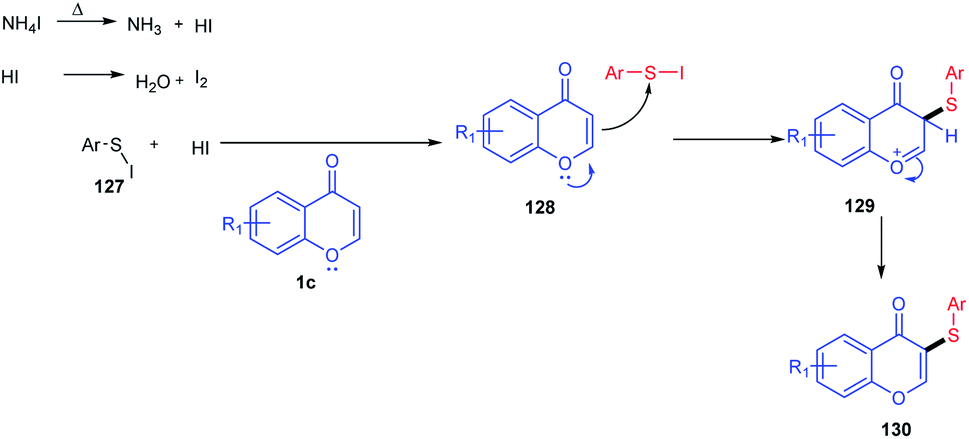 | ||
| Scheme 39 Proposed mechanism for the synthesis of thioethers using aryl thiols as a sulfur source. | ||
Later in 2015, W. Zhao et al. introduced another sulfenylation method with dimethylsulfoxide or benzenesulfonyl hydrazide as the sulfenylating reagents and ammonium iodide as the reaction inducer (Scheme 40). The reaction proceeds at elevated temperatures, leading to the splitting of ammonium iodide into HI and NH3. HI reduces the reaction inducers, DMSO and benzenesulfonyl hydrazide. The electrophilic intermediate obtained from DMSO easily reacts with nucleophilic flavone and subsequently attacks I and NH3 to give the final thioether product from DMSO. Similarly, the benzenesulfonyl hydrazide counterpart undergoes dehydration followed by reductive dehydration supported by HI to give thiodiazonium. The thiodiazonium regioselectively attacks the nucleophilic flavone, yielding the corresponding thioether. The regioselectivity of the reaction was studied by blocking the C-3 position of flavone, yielding no desired product, which implicates the regioselectivity of the reaction.92
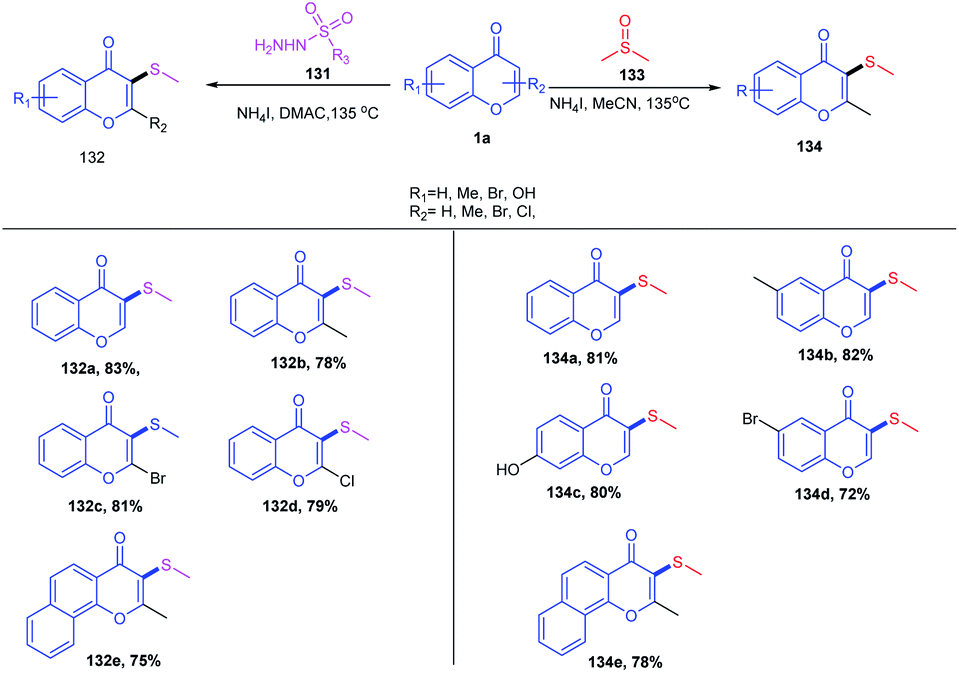 | ||
| Scheme 40 Ammonium iodide-induced sulfenylation at the C-3 position of flavones. | ||
T. Guo et al. 2020 synthesised a C-3-functionalized chromone by the C-3 vulcanisation of imidazoheterocycle (Scheme 41). The dual C–H sulfenylation was achieved by odourless elemental sulphur under an additive and catalyst-free reaction condition. The C-3-functionalized asymmetric sulphides were obtained in good yields with C–H activation of both chromones and imidazoheterocycles using DMSO as both solvent and internal oxidant. The method acts as an efficient methodology for the synthesis of different organosulfur compounds using commercially available S8.93
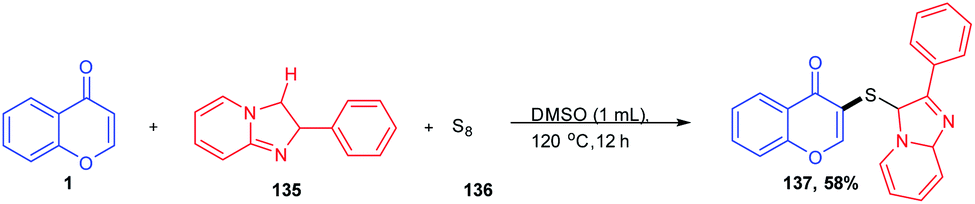 | ||
| Scheme 41 Catalyst and additive-free sulfenylation at the C-3 position of chromones. | ||
4. C-2 activation of chromones
4.1. Transition metal-catalyzed C-2 activation of chromones
K. H. Kim et al. in 2012 carried out the synthesis of C-2 arylated chromones by palladium-catalyzed oxidative arylation in benzene (Scheme 42). The reaction proceeds with regioselective C-2 arylation of chromones via a double C–H activation process. The reaction yields C-2 arylated chromones in moderate yields through regioselective palladation on the C-2 position of chromones via a CMD process and a subsequent C–H bond activation.52 Similarly, M. Min et al. 2012 put forward the regiocontrolled C–H activation at the C-2 position of chromones. The method enables the palladium-catalyzed oxidative cross coupling reaction of nonactivated arenes with the C-2 position of chromones. The reaction offers a direct route to the C-2 arylation under mild conditions with a wide substrate scope.94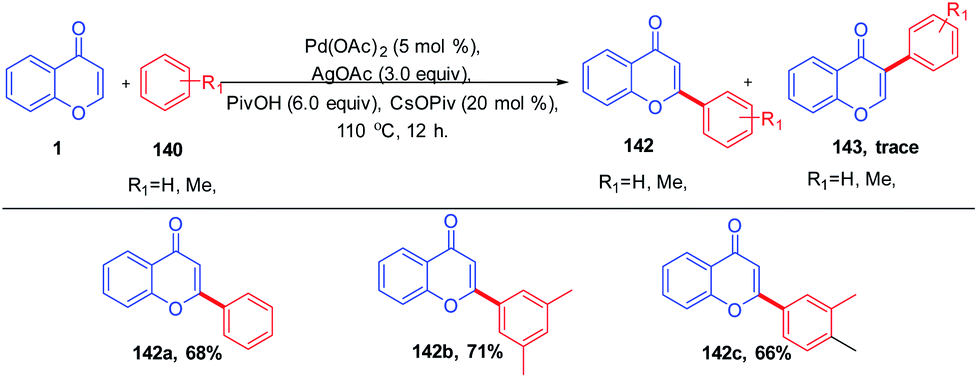 | ||
| Scheme 42 Palladium-catalyzed oxidative arylation of chromones. | ||
The mechanism proceeds via regioselective palladation at the C-2 position of chromones via a CMD process with 138 for the generation of intermediate 139. A subsequent C–H bond activation of benzene could occur via CMD mechanism or an electrophilic palladation (SEAr) process to generate the intermediate 141. The intermediate 141 on the removal of palladium (0) yields the final C-2 arylated product. Furthermore, Pd(0) was oxidized to Pd(II) by the action of AgOAc for the continuation of the catalytic cycle82 (Scheme 43).
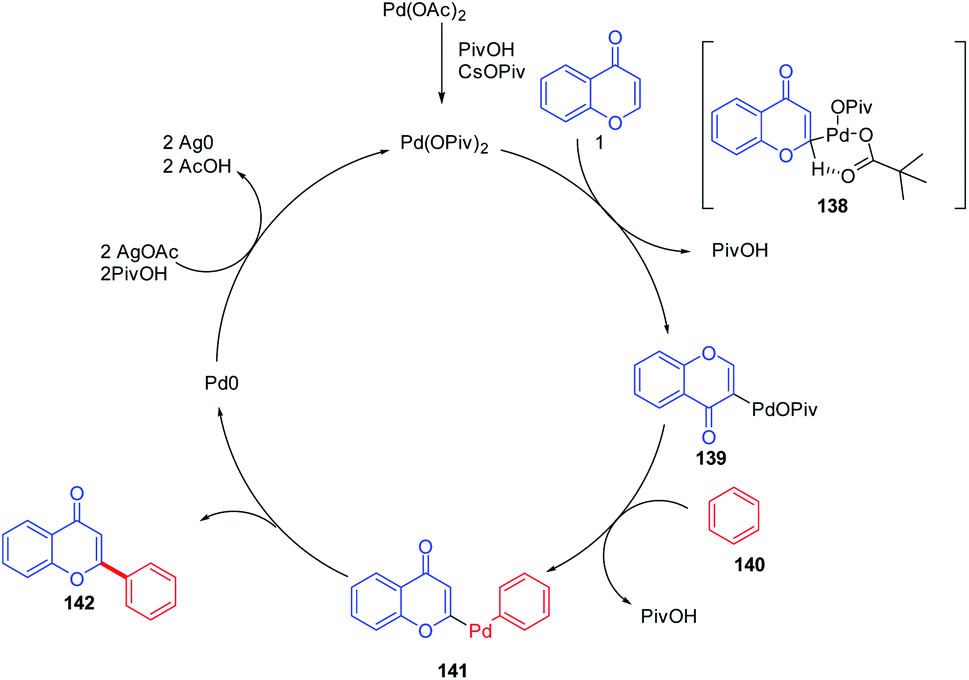 | ||
| Scheme 43 Proposed mechanism for palladium-catalyzed oxidative arylation of chromones. | ||
D. Kim et al. in 2012 reported the C-2 functionalization of chromones via 1,4-conjugate addition of aryl boronic acid with substituted chromones. The reaction yielded a mixture of flavones and flavanones in the presence of the catalytic amount of Pd(OAc)2 and Lewis acid, whereas the addition of the catalytic amount of oxidants DDQ and KNO2 to the reactions solely yielded flavones (Scheme 44). The scope of the reaction was fairly well giving good yields for a wide range of flavanones and flavones.95
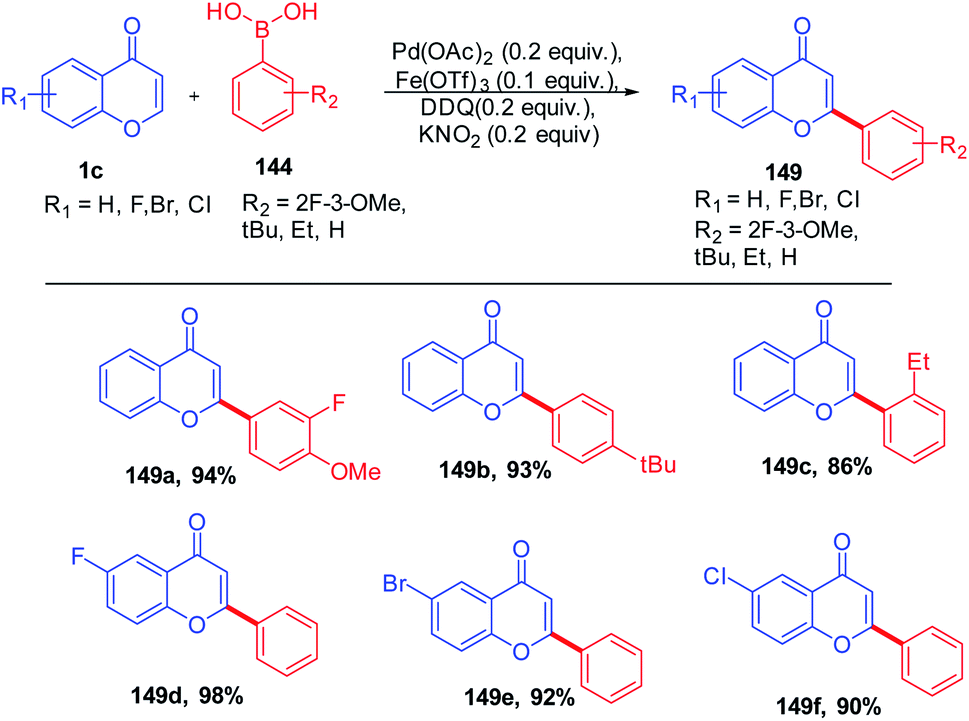 | ||
| Scheme 44 Ligand-free conjugate addition of aryl boronic acids on chromones by a palladium(II) catalyst. | ||
The plausible mechanism for the direct arylation of chromones at the C-2 position by boronic acid is depicted in Scheme 45. Initially, Fe(OTf3) coordinates with the carbonyl oxygen of the chromone moiety, thereby activating the electrophilicity of enolone of chromones. The phenylpalladium formed in situ via transmetalation of the aryl group of supplied boronic acid adds to the C-2 position of chromones forming alkylpalladium adduct 146 and enolate 147. Further protonolysis of the formed enolate yields the conjugate addition product flavanone. Similarly, β-elimination of the palladium adduct 146 generates a Heck-type product flavone as a minor product. The usage of the oxidant system DDQ/KNO2 provides flavone exclusively via oxidation of the resulting flavanone. Finally, the reoxidation of Pd(0) to Pd(II) completes the catalytic cycle.
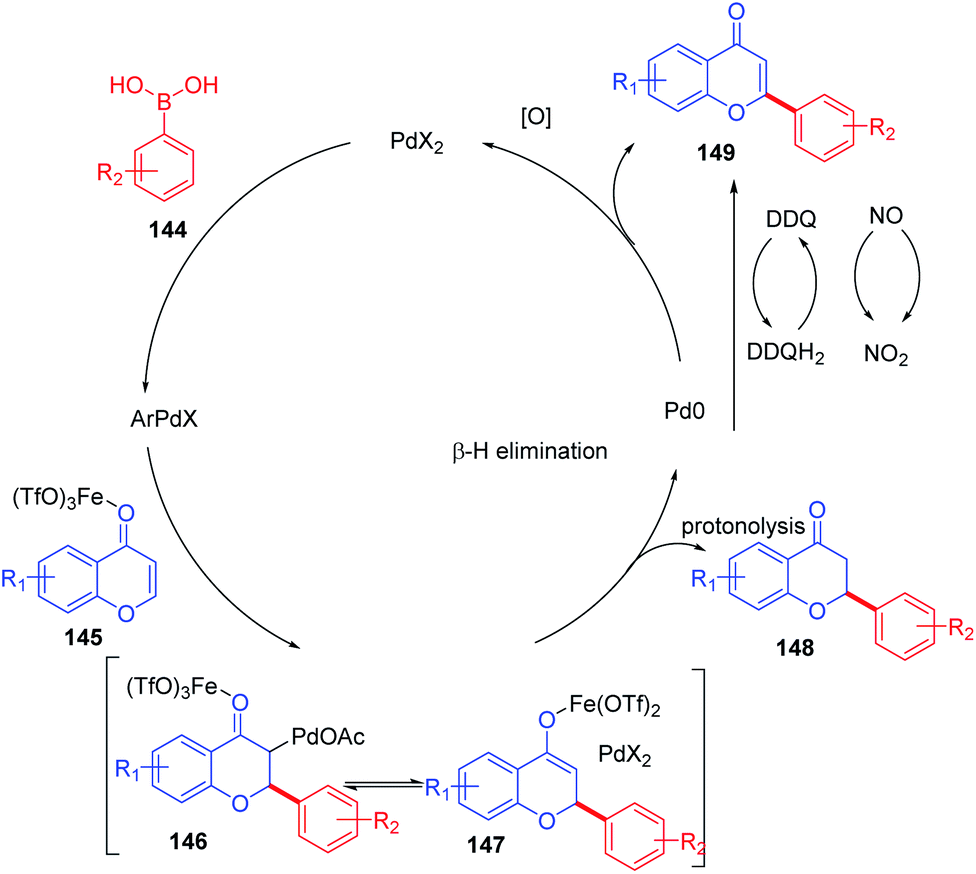 | ||
| Scheme 45 Proposed mechanism for ligand-free palladium(II)-catalyzed conjugate addition of aryl boronic acids to chromones. | ||
B. Niu et al. 2015 introduced copper-catalyzed dehydrogenative coupling of coumarins and flavones with various ethers yielding new substituted ethers (Scheme 46). The reaction proceeds in the presence of a catalytic amount of copper oxide in combination with tert-butylhydrogen peroxide (TBHP) and 1,4-diazabicyclo[2.2.2]octane (DABCO) in good yields with excellent functional group tolerance.96
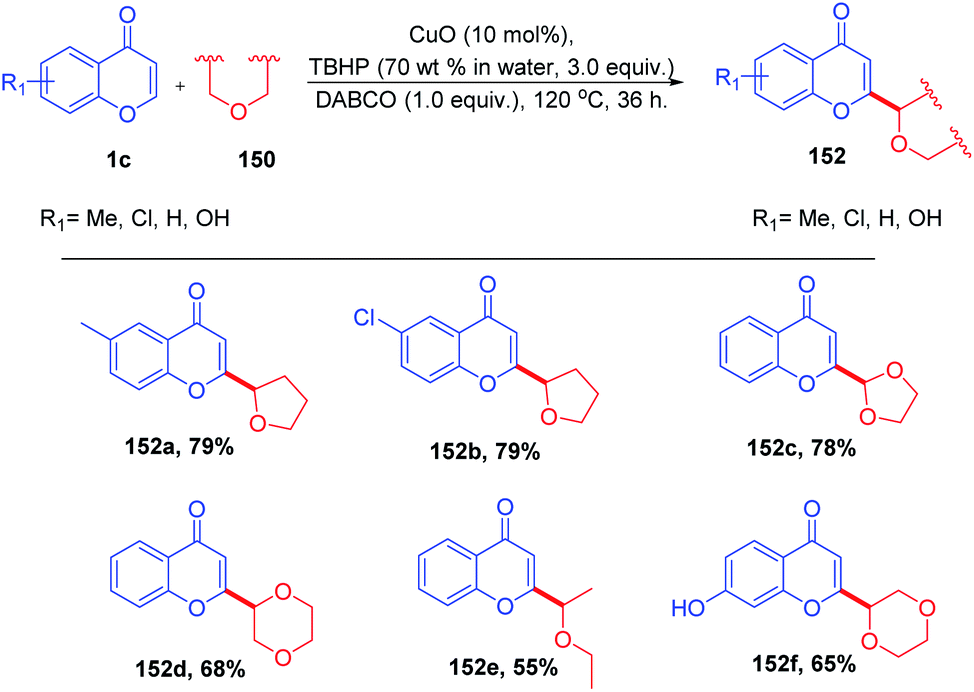 | ||
| Scheme 46 Copper-catalyzed dehydrogenative coupling of flavones with various ethers. | ||
The cross-dehydrogenative coupling of chromones with ethers initiates the formation of two radicals under thermal conditions (Scheme 47). The radicals abstract hydrogen from the α position of supplied ether. The relatively electron-rich ether radical 150 thus formed attacks the electron-deficient C-2 position of flavones. The radical species 151 thus generated upon oxidation with a copper catalyst yields C-2-functionalized products.
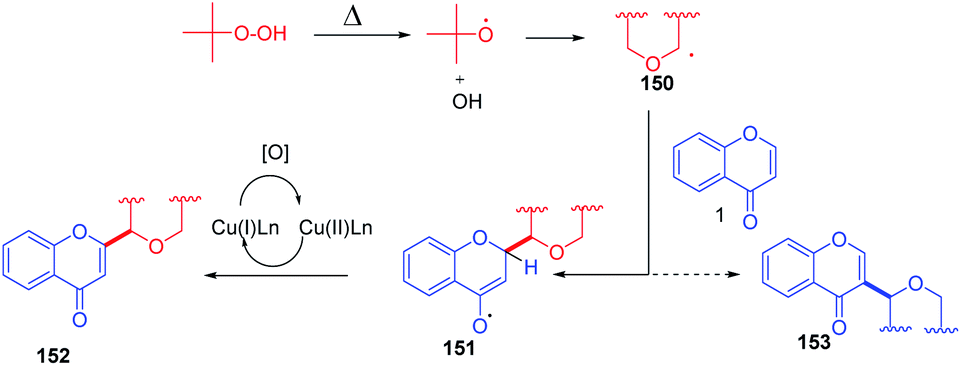 | ||
| Scheme 47 Proposed mechanism for copper-catalyzed cross-dehydrogenative coupling of flavones with ethers. | ||
B. M. Trost et al. 2019 reported the enantio- and diastereoselective 1,4-addition of butenolides to chromones (Scheme 48). The zinc-prophenol catalyst was compatible for both α,β- and β,γ-butenolide nucleophiles without any preactivation. The reaction proceeds with moderate to good yields for electrophiles including chromones, thiochromones and quinolones.97
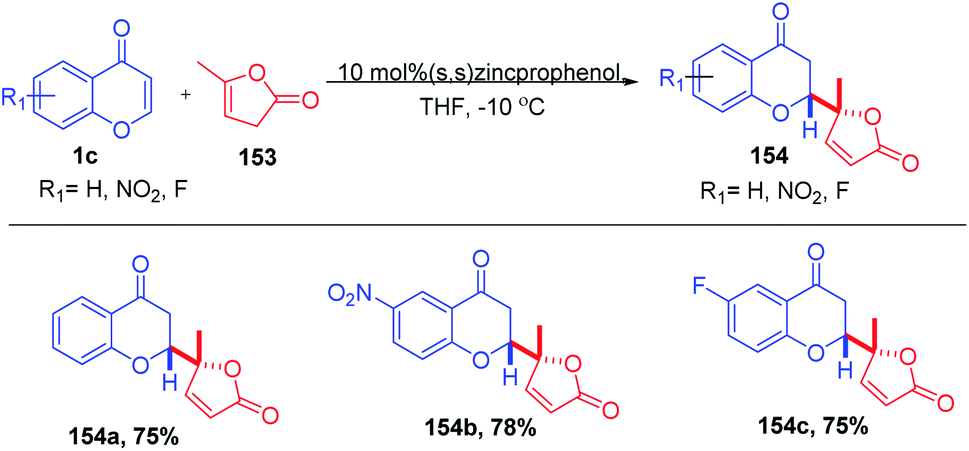 | ||
| Scheme 48 Zinc prophenol-catalyzed vinylogous addition of butenolides to chromones. | ||
4.2. Metal-free C-2 activation of chromones
R. Narayan et al. in 2014 reported hypervalent iodine-mediated selective oxidative functionalization of chromones by aliphatic alkanes proceeds well in both cyclic and acyclic alkanes (Scheme 49). They suggested that iodine(III) supplied in the reaction generates alkyl radicals in situ, which will attack the most electrophilic C-2 position of chromones. The presence of the catalytic amount of PhI(O2CCF3)2 and NaN3 under room temperature conditions yields C-2 alkylated chromones.98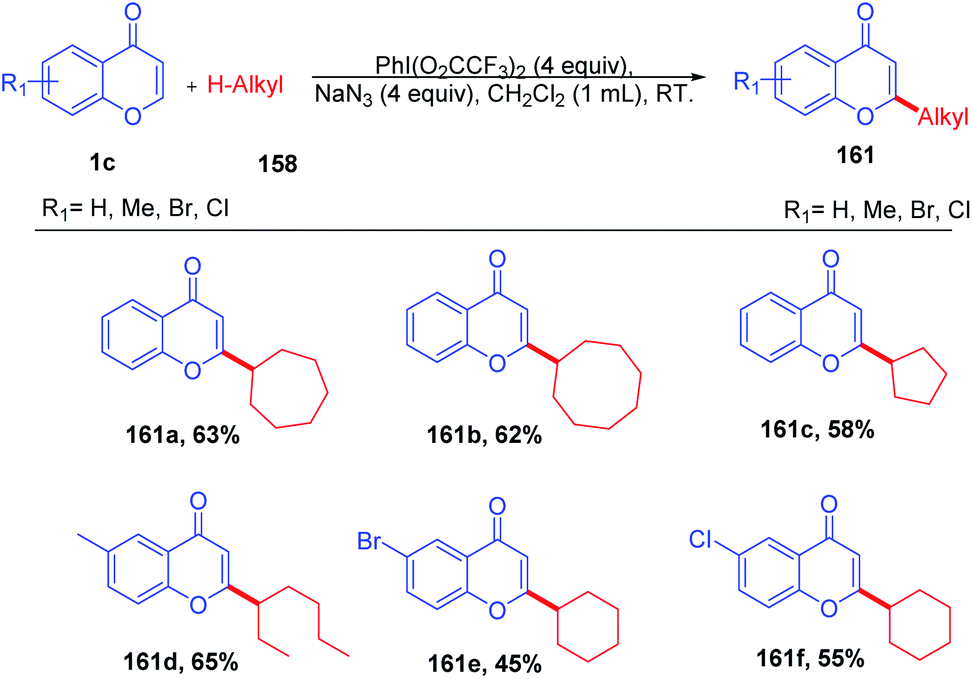 | ||
| Scheme 49 Hypervalent iodine-mediated cross coupling of chromones with alkanes. | ||
The reaction mechanism was proposed as a radical pathway and supported by the suppression of the reaction in the presence of radical scavengers (Scheme 50). Initially, trifluoroacetate of PhI(O2CCF3)2 was exchanged by azides of sodium azide by a ligand exchange method to form 156, and thermolysis of 156 generates 160 and an azide radical. The azide radical abstracts H from the alkane to afford alkyl radical 157, which upon reaction with the electrophilic C-2 position of chromones and further oxidation gives C-2 alkylated chromones.
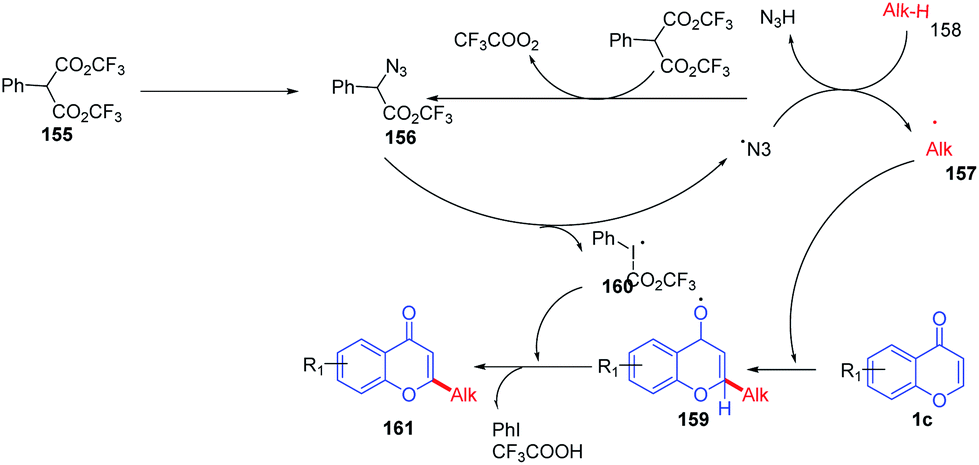 | ||
| Scheme 50 Proposed mechanism for hypervalent iodine-mediated cross coupling of chromones with alkanes. | ||
R. Samanta et al. in 2015 introduced transition metal-free oxidative, regioselective cross-coupling of chromones with non-functionalized azoles for the generation of C-2-functionalized chromones with a new C–N bond (Scheme 51). The reaction is having prior importance due to the formation of the C–N bond, which is ubiquitous in natural products and bioactive molecules.99
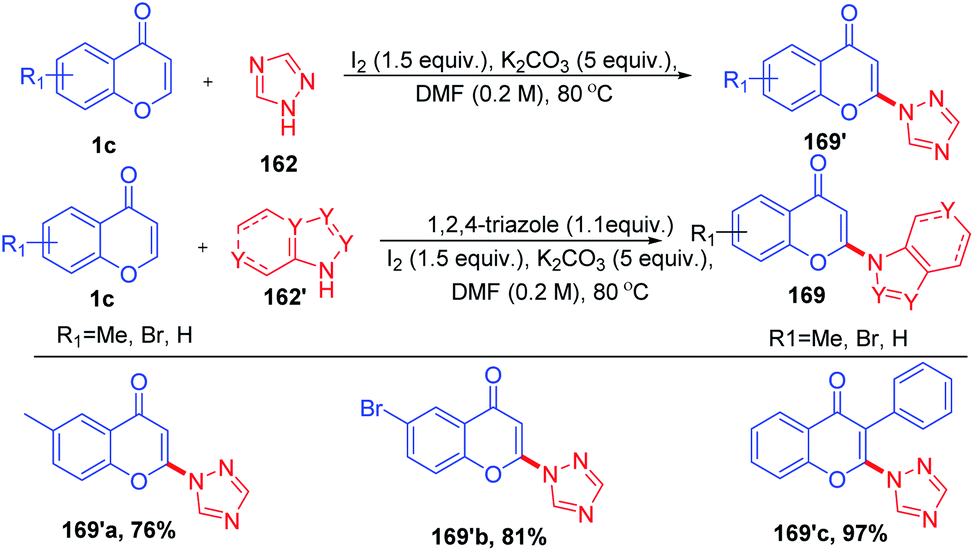 | ||
| Scheme 51 Oxidative regioselective amination of chromones. | ||
Initially, 162 reacts with molecular iodine forming an electrophilic iodine source 163 (Scheme 52). The reaction of 164 with 1c gives an oxonium ion. The oxonium ion 164 thus formed exists in equilibrium with the corresponding iodonium ion 165. Subsequently, 164 or 165 gets trapped by iodide to form product 166. The compound 166, which is not stable under basic conditions, thus undergoes a base-mediated dehydroiodination to generate 3-iodochromone 167.
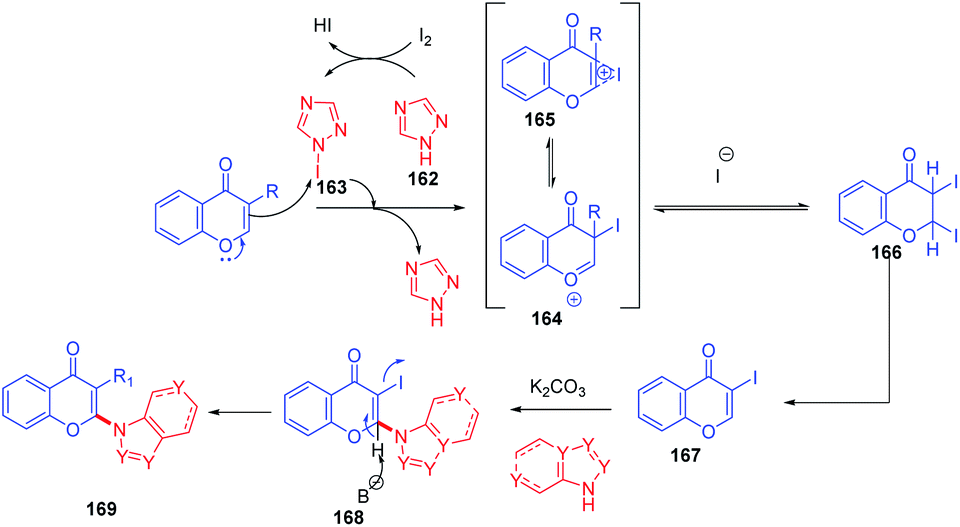 | ||
| Scheme 52 Proposed mechanism for oxidative regioselective amination of chromones. | ||
Yi-Han Chang et al. 2021 synthesised 2-(1H-1,2,4-triazol-1-yl)-4H-chromen-4-one by dehydroiodination (Scheme 53). The method used 1,2,4-triazole 162 as a catalyst for the generation of 3-iodochromones by reaction of molecular iodine with chromones at the C-3 position. Dehydroiodination on 3-iodochromone yields 2-(1H-1,2,4-triazol-1-yl)-4H-chromen-4-one 170a. Yi-Han Chang et al. used the method for the generation of biologically relevant compounds with phenols and 3-fluorothiol substitution at the C-2 position. The anti-inflammatory effect of phenols and 3-fluorothiol-substituted chromones against superoxide anion generation was studied. The tetrazole group acts as the leaving group in the reaction of chromones with phenols and thiophenols.100
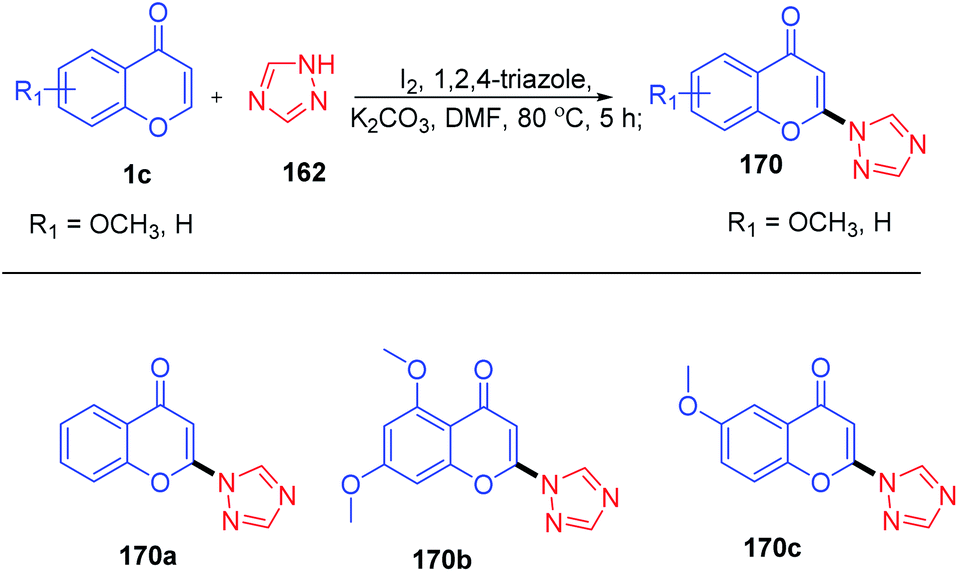 | ||
| Scheme 53 Synthesis of 2-(1H-1,2,4-triazol-1-yl)-4H-chromen-4-one by C-2 H activation. | ||
5 Conclusion
To summarize, the review covered the site-selective functionalisation of non-prefunctionalized chromones at the C-5, C-3 and C-2 positions. All the functionalization reactions either catalyzed by a transition metal or occurring under metal-free conditions were mentioned in the review. A detailed review of all the reaction transformations by hydrogen at the active positions of chromone into different functional groups was presented. The review classifies the reactions into two categories to enhance the understanding. They are transition metal-catalysed reactions in which the reaction proceeds by the formation of a metallacycle intermediate and the other one as a metal-free reaction transformation. The second topic is a wider one where the reaction can be nitration, a base-catalysed allylation, a nucleophilic or an electrophilic reaction, and a catalyst- and ligand-free reaction. The C-2 position of chromones is more electron deficient and it may go for reactions with nucleophilic coupling partners or alkyl radicals. Similarly, the C-3 position is electron rich and it undergoes reactions with electrophilic coupling partners. There are also reports for catalyst and additive-free sulfenylation at the C-3 position. Therefore, to include all the functionalization reactions in the review, we summarise all these reactions under metal-free functionalization.The non-pre-functionalized reactions are having greater importance compared to conventional prefunctionalized reactions. The reaction occurs in fewer steps without the liberation of halogen wastes when compared to conventional reaction methods. Despite these immense advancements in the site-selective sp2 C–H activation of chromones, the methodologies suffer from serious drawbacks. It is difficult to attain site-selective substitution on the C-3 site of the chromone. Most of the reactions depend on the pre-functionalization of the C-2 sites before the functionalization at the C-3 position. On studying the yield of the functionalization reactions, we can see that the yield of these reactions is comparatively low. The lower yield of these site-selective products needed to be addressed in future. Similarly, all the C–H functionalization reactions on the arene ring of chromone take place at the C-5 position alone. This is because of the inherent direction of the carbonyl group on the chromone moiety. Methodologies focusing on these drawbacks will facilitate the future advancement of these reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Anjitha Theres Benny thanks VIT, Vellore for research fellowships respectively. The authors are also thankful to VIT Vellore for financial support in the form of a seed grant.References
- A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 9, 4960–4992 CrossRef PubMed.
- S. Kumar and A. K. Pandey, Sci. World J., 2013, 16, 162750 Search PubMed.
- M. Neelgundmath, K. R. Dinesh, C. D. Mohan, F. Li, X. Dai, K. S. Siveen, S. Paricharak, D. J. Mason, J. E. Fuchs, G. Sethi, A. Bender, K. S. Rangappa, O. Kotresh and S. Basappa, Bioorg. Med. Chem. Lett., 2015, 25, 893–897 CrossRef CAS PubMed.
- D. H. Nam, K. Y. Lee, C. S. Moon and Y. S. Lee, Eur. J. Med. Chem., 2010, 45, 4288–4292 CrossRef CAS PubMed.
- D. Donnelly, R. Geoghegan, C. O’Brien, E. Philbin and T. S. Wheeler, J. Med. Chem., 1965, 8, 872–875 CrossRef CAS PubMed.
- Z. N. Siddiqui, T. N. M. Musthafa and S. Praveen, Med. Chem. Res., 2013, 22, 127–133 CrossRef CAS.
- D. Ashok, K. Rangu, V. Hanumantha Rao, S. Gundu, B. Srilata and M. Vijjulatha, Med. Chem. Res., 2016, 25, 501–514 CrossRef CAS.
- M. Hiruy, D. Bisrat, A. Mazumder and K. Asres, Nat. Prod. Res., 2019, 35, 1052–1056 CrossRef PubMed.
- T. E. S. Ali and M. A. Ibrahim, J. Braz. Chem. Soc., 2010, 21, 1007–1016 CrossRef.
- Z. X. Zou, P. S. Xu, C. R. Wu, W. X. Zhu, G. Z. Zhu, X. A. He, G. G. Zhang, J. Z. Hu, S. Liu, W. Zeng, K. P. Xu and G. S. Tan, Fitoterapia, 2016, 111, 124–129 CrossRef CAS PubMed.
- V. S. Honmore, A. D. Kandhare, P. P. Kadam, V. M. Khedkar, D. Sarkar, S. L. Bodhankar, A. A. Zanwar, S. R. Rojatkar and A. D. Natu, Int. Immunopharmacol., 2016, 33, 8–17 CrossRef CAS PubMed.
- P. S. Mahajan, M. D. Nikam, V. M. Khedkar, P. C. Jha, D. Sarkar and C. H. Gill, Res. Chem. Intermed., 2016, 42, 2707–2729 CrossRef CAS.
- C. F. M. Silva, D. C. G. A. Pinto and A. M. S. Silva, ChemMedChem, 2016, 11, 2252–2260 CrossRef CAS PubMed.
- S. Choodej, D. Sommit and K. Pudhom, Bioorg. Med. Chem. Lett., 2013, 23, 3896–3900 CrossRef CAS PubMed.
- C. Proença, H. M. T. Albuquerque, D. Ribeiro, M. Freitas, C. M. M. Santos, A. M. S. Silva and E. Fernandes, Eur. J. Med. Chem., 2016, 115, 381–392 CrossRef PubMed.
- N. Phosrithong, W. Samee, P. Nunthanavanit and J. Ungwitayatorn, Chem. Biol. Drug Des., 2012, 79, 981–989 CrossRef CAS PubMed.
- C. Demetgül and N. Beyazit, Carbohydr. Polym., 2018, 181, 812–817 CrossRef PubMed.
- Y. M. A. Mohamed, A. Vik, T. Hofer, J. H. Andersen and T. V. Hansen, Chem. Phys. Lipids, 2013, 170, 41–45 CrossRef PubMed.
- M. I. Fernández-Bachiller, C. Pérez, L. Monjas, J. Rademann and M. I. Rodríguez-Franco, J. Med. Chem., 2012, 55, 1303–1317 CrossRef PubMed.
- Q. Liu, X. Qiang, Y. Li, Z. Sang, Y. Li, Z. Tan and Y. Deng, Bioorg. Med. Chem., 2015, 23, 911–923 CrossRef CAS PubMed.
- C. F. M. Silva, D. C. G. A. Pinto and A. M. Silva, Expert Opin. Drug Discovery, 2018, 13, 1141–1151 CrossRef CAS PubMed.
- P. Baruah, M. A. Rohman, S. O. Yesylevskyy and S. Mitra, BioImpacts, 2019, 9, 79 CrossRef CAS PubMed.
- M. Ceylan-Ünlüsoy, E. J. Verspohl and R. Ertan, J. Enzyme Inhib. Med. Chem., 2010, 25, 784–789 CrossRef PubMed.
- S. Nazreen, M. S. Alam, H. Hamid, M. S. Yar, A. Dhulap, P. Alam, M. A. Q. Pasha, S. Bano, M. M. Alam, S. Haider, C. Kharbanda, Y. Ali and K. K. Pillai, Bioorg. Med. Chem. Lett., 2014, 24, 3034–3042 CrossRef CAS PubMed.
- O. Bozdag-Dündar, M. Ceylan-Ünlüsoy, E. J. Verspohl and R. Ertan, Arzneimittelforschung, 2011, 57, 532–536 CrossRef PubMed.
- J. E. Philip, M. Shahid, M. R. Prathapachandra Kurup and M. P. Velayudhan, J. Photochem. Photobiol., B, 2007, 175, 178–191 CrossRef PubMed.
- Y. Chen, Y. Gao, Y. He, G. Zhang, H. Wen, Y. Wang, Q. P. Wu and H. Cui, J. Med. Chem., 2020, 64, 1001–1017, DOI:10.1021/acs.jmedchem.0c01508.
- N. Yadav, R. Kumar, A. K. Singh, S. Mohiyuddin and P. Gopinath, Spectrochim. Acta, Part A, 2020, 235, 1386–1425 CrossRef PubMed.
- W. Lu, J. Chen, J. Shi, L. Xu, S. Yang and B. Gao, J. Biol. Inorg Chem., 2021, 26, 57–66 CrossRef CAS PubMed.
- L. Fan, J. C. Qin, T. R. Li, B. D. Wang and Z. Y. Yang, Sens. Actuators, B, 2014, 203, 550–556 CrossRef CAS.
- H. Kim, R. Manivannan and Y. A. Son, J. Nanosci. Nanotechnol., 2020, 20, 2840–2846 CrossRef CAS PubMed.
- S. Bhardwaj, N. Maurya and A. K. Singh, Sens. Actuators, B, 2018, 260, 753–762 CrossRef CAS.
- D. Şahin Gül, H. Ogutcu and Z. Hayvalı, J. Mol. Struct., 2020, 1204, 127569 CrossRef.
- C. Li, J. Qin, B. Wang, L. Fan, J. Yan and Z. Yang, J. Fluoresc., 2015, 26, 345–353 CrossRef PubMed.
- E. W. F. W. Alton and A. A. Norris, J. Allergy Clin. Immunol., 1996, 98, S102–S106 CrossRef CAS.
- M. Isaka, M. Sappan, P. Auncharoen and P. Srikitikulchai, Phytochem. Lett., 2010, 3, 152–155 CrossRef CAS.
- S. Khadem and R. Marles, Molecules, 2011, 17, 191–206 CrossRef PubMed.
- Y. W. Sun, G. M. Liu, H. Huang and P. Z. Yu, Phytochemistry, 2012, 75, 169–176 CrossRef CAS PubMed.
- D. Ashok, K. Rangu, V. H. Rao, S. Gundu, B. Srilata and M. Vijjulatha, Med. Chem. Res., 2016, 25, 501–514 CrossRef CAS.
- G. Singh, A. Sharma, H. Kaur and M. P. S. Ishar, Chem. Biol. Drug Des., 2016, 87, 213–223 CrossRef CAS PubMed.
- J. Wang, Y. T. Liu, L. Xiao, L. Zhu, Q. Wang and T. Yan, Inflammation, 2014, 37, 2085–2090 CrossRef CAS PubMed.
- J. Morris, D. G. Wishka, W. R. Humphrey, A. H. Lin, A. L. Wiltse, C. W. Benjamin, R. R. Gorman and R. J. Shebuski, Bioorg. Med. Chem. Lett., 1994, 4, 2621–2626 CrossRef CAS.
- S. P. Pawar, D. D. Kondhare and P. K. Zubaidha, Med. Chem. Res., 2013, 22, 753–757 CrossRef CAS.
- X. Zhang, J. Wang, C. Hong, W. Luo and C. Wang, Acta Pharm. Sin. B, 2015, 5, 67–73 CrossRef PubMed.
- R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed.
- J. A. Labinger, Chem. Rev., 2017, 117, 8483–8496 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- X. S. Xue, P. Ji, B. Zhou and J. P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- C. G. Newton, S. G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed.
- C. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144–6147 CrossRef CAS PubMed.
- R. Samanta, R. Narayan and A. P. Antonchick, Org. Lett., 2012, 14, 6108–6111 CrossRef CAS PubMed.
- Catalysis Series, ed. C. C. C. Johansson Seechurn, A. DeAngelis, T. J. Colacot, and T. Colacot, R Soc Chem., 2014, pp. 1–19 Search PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- J. W. Lampe, J. Nutr., 2003, 133, 956S–964S CrossRef CAS PubMed.
- L. Fu, Z. Xu, J.-P. Wan and Y. Liu, Org. Lett., 2020, 22, 9518–9523 CrossRef CAS PubMed.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS.
- D. Kang, K. Ahn and S. Hong, Asian J. Org. Chem., 2018, 7, 1136–1150 CrossRef CAS.
- S. Kathiravan and I. A. Nicholls, Eur. J. Org. Chem., 2014, 2014, 7211–7219 CrossRef CAS.
- M. Min, D. Kim and S. Hong, Chem. Commun., 2014, 50, 8028–8031 RSC.
- K. Kim, H. Choe, Y. Jeong, J. H. Lee and S. Hong, Org. Lett., 2015, 17, 2550–2553 CrossRef CAS PubMed.
- S. H. Han, S. Kim, U. De, N. K. Mishra, J. Park, S. Sharma, J. H. Kwak, S. Han, H. S. Kim and I. S. Kim, J. Org. Chem., 2016, 81, 12416–12425 CrossRef CAS.
- R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2016, 138, 10132–10135 CrossRef CAS PubMed.
- P. Y. Lee, P. Liang and W. Y. Yu, Org. Lett., 2017, 19, 2082–2085 CrossRef CAS PubMed.
- X. Li, G. Wu, X. Liu, Z. Zhu, Y. Huo and H. Jiang, J. Org. Chem., 2017, 82, 13003–13011 CrossRef CAS PubMed.
- K. Shin, Y. Park, M. H. Baik and S. Chang, Nat. Chem., 2018, 10, 218–224 CrossRef CAS PubMed.
- G. Tan, Q. You and J. You, ACS Catal., 2018, 8, 8709–8714 CrossRef CAS.
- S. Debbarma, M. R. Sk, B. Modak and M. S. Maji, J. Org. Chem., 2019, 84, 6207–6216 CrossRef CAS PubMed.
- G. Ncube and M. P. Huestis, Organometallics, 2019, 38, 76–80 CrossRef CAS.
- M. R. Sk and M. S. Maji, Org. Chem. Front., 2019, 7, 19–24 RSC.
- Y. Zhou, H. Liang, Y. Sheng, S. Wang, Y. Gao, L. Zhan, Z. Zheng, M. Yang, G. Liang, J. Zhou, J. Deng and Z. Song, J. Org. Chem., 2020, 85, 9230–9243 CrossRef CAS PubMed.
- M. Recanatini, R. W. Hartmann, A. Bisi, A. Cavalli, F. Belluti, S. Gobbi, A. Rampa, P. Valenti, M. Palzer and A. Palusczak, J. Med. Chem., 2001, 44, 672–680 CrossRef CAS PubMed.
- T. Ishihara, H. Kakuta, H. Moritani, T. Ugawa and I. Yanagisawa, Bioorg. Med. Chem., 2004, 12, 5899–5908 CrossRef CAS PubMed.
- M. Jeganmohan and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 8520–8524 CrossRef CAS PubMed.
- D. Kim and S. Hong, Org. Lett., 2011, 13, 4466–4469 CrossRef CAS PubMed.
- K. H. Kim, H. S. Lee, S. H. Kim and J. N. Kim, Tetrahedron Lett., 2012, 53, 2761–2764 CrossRef CAS.
- F. Chen, Z. Feng, C. Y. He, H. Y. Wang, Y. L. Guo and X. Zhang, Org. Lett., 2012, 14, 1176–1179 CrossRef CAS PubMed.
- B. A. Mir, A. Banerjee, S. K. Santra, S. Rajamanickam and B. K. Patel, Adv. Synth. Catal., 2016, 358, 3471–3476 CrossRef CAS.
- H. Choi, M. Min, Q. Peng, D. Kang, R. S. Paton and S. Hong, Chem. Sci., 2016, 7, 3900–3909 RSC.
- S. Mkrtchyan and V. O. Iaroshenko, Eur. J. Org. Chem., 2018, 2018, 6867–6875 CrossRef CAS.
- Z. Lv, C. Sheng, Y. Zhang, T. Wang, J. Feng, H. Sun, H. Zhong, M. Zhang, H. Chen and K. Li, Bioorg. Med. Chem. Lett., 2010, 20, 7106–7109 CrossRef CAS PubMed.
- L. Klier, T. Bresser, T. A. Nigst, K. Karaghiosoff and P. Knochel, J. Am. Chem. Soc., 2012, 134, 13584–13587 CrossRef CAS PubMed.
- L. Klier, D. S. Ziegler, R. Rahimoff, M. Mosrin and P. Knochel, Org. Process Res. Dev., 2017, 21, 660–663 CrossRef CAS.
- Y. Ding, W. Wu, W. Zhao, Y. Li, P. Xie, Y. Huang, Y. Liu and A. Zhou, Org. Biomol. Chem., 2016, 14, 1428–1431 RSC.
- W. Zhao, P. Xie, Z. Bian, A. Zhou, H. Ge, B. Niu and Y. Ding, RSC Adv., 2015, 5(74), 59861–59864 RSC.
- W. Zhao, P. Xie, Z. Bian, A. Zhou, H. Ge, M. Zhang, Y. Ding and L. Zheng, J. Org. Chem., 2015, 80, 9167–9175 CrossRef CAS PubMed.
- T. Guo, X. N. Wei, M. Zhang, Y. Liu, L. M. Zhu and Y. H. Zhao, Chem. Commun., 2020, 56, 5751–5754 RSC.
- M. Min, H. Choe and S. Hong, Asian J. Org. Chem., 2012, 1, 47–50 CrossRef CAS.
- D. Kim, K. Ham and S. Hong, Org. Biomol. Chem., 2012, 10, 7305–7312 RSC.
- B. Niu, W. Zhao, Y. Ding, Z. Bian, C. U. Pittman, A. Zhou and H. Ge, J. Org. Chem., 2015, 80, 7251–7257 CrossRef CAS PubMed.
- B. M. Trost, E. Gnanamani, C. A. Kalnmals, C. I. J. Hung and J. S. Tracy, J. Am. Chem. Soc., 2019, 141, 1489–1493 CrossRef CAS PubMed.
- R. Narayan and A. P. Antonchick, Chem.–Eur. J., 2014, 20, 4568–4572 CrossRef CAS PubMed.
- R. Samanta, R. Narayan, J. O. Bauer, C. Strohmann, S. Sievers and A. P. Antonchick, Chem. Commun., 2015, 51, 925–928 RSC.
- Y. H. Chang, F. Shu-Yen, H. Y. Lai, T. L. Hwang and H. Y. Hung, Bioorg. Med. Chem. Lett., 2021, 36, 127822 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |