
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSamarium(II) iodide-mediated reactions applied to natural product total synthesis
Majid. M. Heravi * and
Azadeh Nazari
* and
Azadeh Nazari
Department of Chemistry, School of Science, Alzahra University, PO Box 1993891176, Vanak, Tehran, Iran. E-mail: mmh1331@yahoo.com; mmheravi@alzahra.ac.ir; Fax: +98 21 88041344; Tel: +98 21 88044051
First published on 30th March 2022
Abstract
Natural product synthesis remains a field in which new synthetic methods and reagents are continually being evaluated. Due to the demanding structures and complex functionality of many natural products, only powerful and selective methods and reagents will be highlighted in this proceeding. Since its introduction by Henri Kagan, samarium(II) iodide (SmI2, Kagan's reagent) has found increasing use in chemical synthesis. Over the years, many reviews have been published on the application of SmI2 in numerous reductive coupling procedures as well as in natural product total synthesis. This review highlights recent advances in SmI2-mediated synthetic strategies, as applied in the total synthesis of natural products since 2004.
1. Introduction
Natural product total synthesis remains a challenging field, especially if the structures of natural products are uncertain, and even more so if the target molecules or the late-stage synthetic precursors are highly unstable. As a result, this area of research allows for testing various synthetic methodologies and strategies, together with new chemical reagents according to their versatility, reactivity, reliable selectivities, and compatibility with functional groups, to develop a new route for a total synthesis or to find an achievable pathway for the first time.Samarium diiodide (SmI2) also known as Kagan's reagent, was reported by Kagan in 1980.1
It has been considered among the most valuable reagents and utilized in the development of a broad variety of reactions, e.g., reduction of different functional groups comprising sulfones and sulfoxides, alkyl and aryl halides, epoxides, phosphine oxides, carbonyls, and conjugated double bonds, in addition to C–C bond-construction, and cascade or sequential reactions.
Over the years, several reviews have been published on the application of Kagan's reagent and also its application in the natural product total synthesis.2,3 In this review, we try to emphasize the crucial role of samarium diiodide in the total synthesis of natural products from 2004 to date.2 An introduction to the reagent and different classes of SmI2-mediated transformations is also included; however, previous reviews must be considered for a detailed discussion of this field. Samarium diiodide (SmI2) was first introduced by Matignon in 1906.4 Over seventy years later, in 1977 Kagan et al. used this reagent in organic chemistry,1 and since then this mild and selective single-electron reducing agent has been used in the development of numerous reactions and has evolved into a prominent reagent for the modification of natural products and the natural products total syntheses. Due to the high versatility of samarium diiodide, the SmI2-mediated reactions are broadly utilized as key steps in total syntheses, delivering extraordinary outcomes that cannot be attributed to other reagents and the large reduction potential of SmI2 (up to −2.05 V in the presence of HMPA)5 enables access to a large number of reactive intermediates, e.g. Accordingly, SmI2 demonstrates a broad application from the total synthesis of natural and bioactive products to fields of materials and polymer science.3s
Due to the high oxophilicity of samarium and the ability to coordinate at several Lewis basic centers simultaneously, SmI2-mediated reactions often proceed through well-defined transition states and produce products with excellent stereoselectivity. SmI2 acts as a one-electron transfer reagent, and therefore, radical or anionic reaction mechanisms can come into play (Scheme 1). The SmI2-mediated reduction of alkyl halides occurs through a single-electron transfer to produce a radical species or via two successive single-electron transfers to generate organosamarium reactive intermediates (Scheme 1).
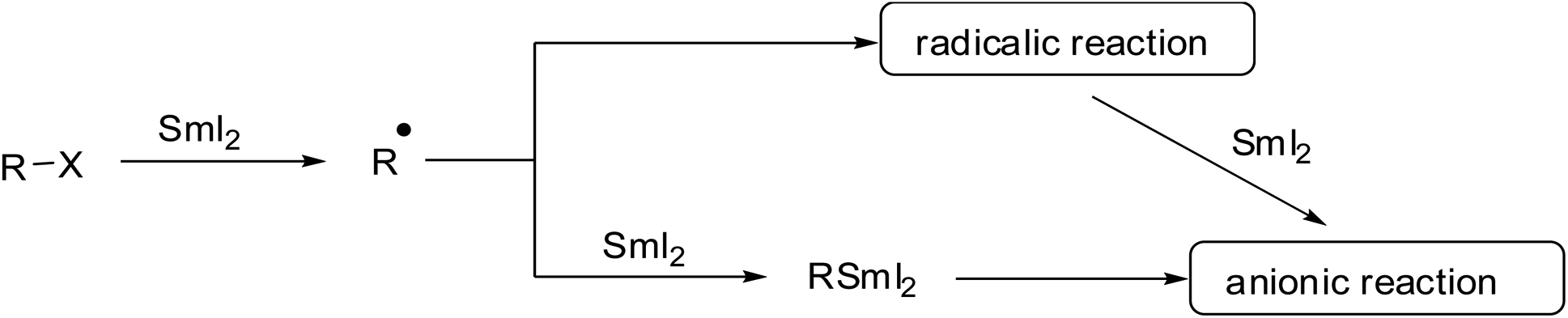 | ||
| Scheme 1 Possible pathways for SmI2-mediated reactions. | ||
Comparably, a SmI2-mediated reduction of carbonyl compounds results in either ketyl radicals or reactive carbanions. The resulting radical or organosamarium anionic intermediate can trigger various reductions including many variations of C–C bond-forming processes and cascade or sequential reactions under mild conditions. Generally, SmI2 is prepared as a 0.1 M solution in THF via the addition of molecular iodine or 1,2-diiodoethane to samarium powder. Kagan used diiodoethane as a source of iodine.1 Imamoto introduced a more atom-efficient method using elemental iodine.6a Further procedures utilized alternative sources of iodine such as diiodomethane6b or a combination of TMSCl and NaI.6c There are also methods for faster preparation such as microwave irradiation or ultrasound, and several other methods7 (Scheme 2).
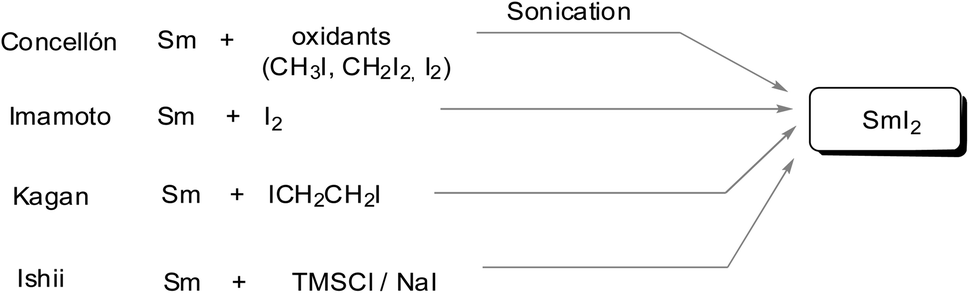 | ||
| Scheme 2 Preparation of samarium iodide. | ||
The strong reactivity of SmI2 can be determined very specifically by carefully optimizing the reaction conditions. The high versatility of samarium diiodide makes a variety of radical and/or ionic reactions selectively accessible. The effective combination of useful reactivity and tunable selectivity of SmI2 makes it one of the most important one-electron reducing agents in synthetic chemistry. One of the extraordinary aspects of SmI2-induced reactions is the power to manipulate the outcome of reactions and to control the rate of reductions using selective cosolvents or additives.5c The difference in additives can stimulate the chemo- or stereoselectivity of the SmI2-mediated transformations, can change the reactivity, and even affect the selectivity of one functional group among others, which makes this user-friendly reagent the most intelligent electron transfer (ET) reagent available to the synthetic chemist. The real reduction potential of SmI2 and the chemo- or stereoselectivity of the reactions mediated by SmI2 can differ based on the temperature of the reaction, the solvent, or any additives, used in the reaction.8
Additives, which are generally used to fine-tune the reactivity of samarium diiodide can be categorized into three major groups: (a) Lewis bases, such as hexamethylphosphoramide (HMPA), ethers, and other electron donor ligands, (b) proton sources mainly including alcohols and water, and (c) inorganic additives, such as metal ion salts or complexes such as LiCl, FeCl3, NiI2, and LiBr. The solvent atmosphere can also alter the coordination sphere of the samarium metal, and consequently, changing the reactivity of the Sm(II) reducing agent. HMPA is still a prominent additive that significantly increases the potency of SmI2 as an electron-transfer reagent via coordination to Sm(II) and stabilizing the Sm(III) oxidation state.5a,8b,c
SmI2-mediated reactions can be classified into two major classes: (1) functional group reduction and (2) reductive C–C bond formation. SmI2 is an extremely chemoselective reagent and its chemoselectivity and reactivity can be determined by the presence or absence of additives and different reaction conditions. This reagent reduces various functional groups such as alkyl and aryl halides, carbonyls, oxides, epoxides, sulfoxides, sulfones, and phosphine, as well as conjugated double bonds.3f (Scheme 3). Recent advances demonstrate the power and extraordinary potential of Kagan's reagent for the mild and selective electron-transfer reduction of functional groups such as carboxylic acids, esters, amides, and nitriles which have limitations and require violent conditions to be transformed.3
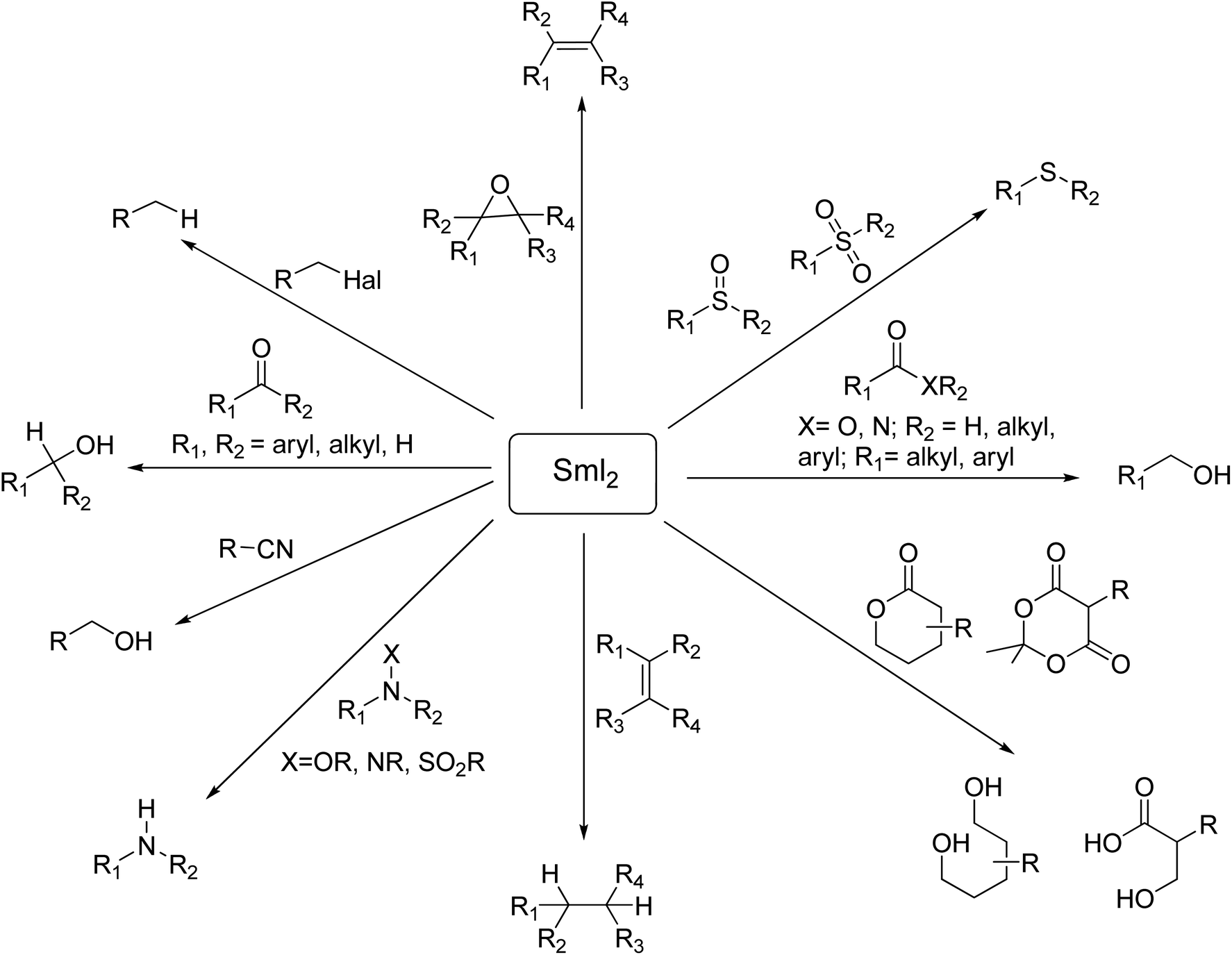 | ||
| Scheme 3 Selective SmI2-mediated functional group transformations. | ||
The various applications of SmI2-mediated reactions to construct carbon–carbon bonds have been utilized for the development of synthetic strategies in natural product total synthesis due to the remarkable combination of the high reducing ability of this reagent with an extraordinary control of stereochemistry (Scheme 4).
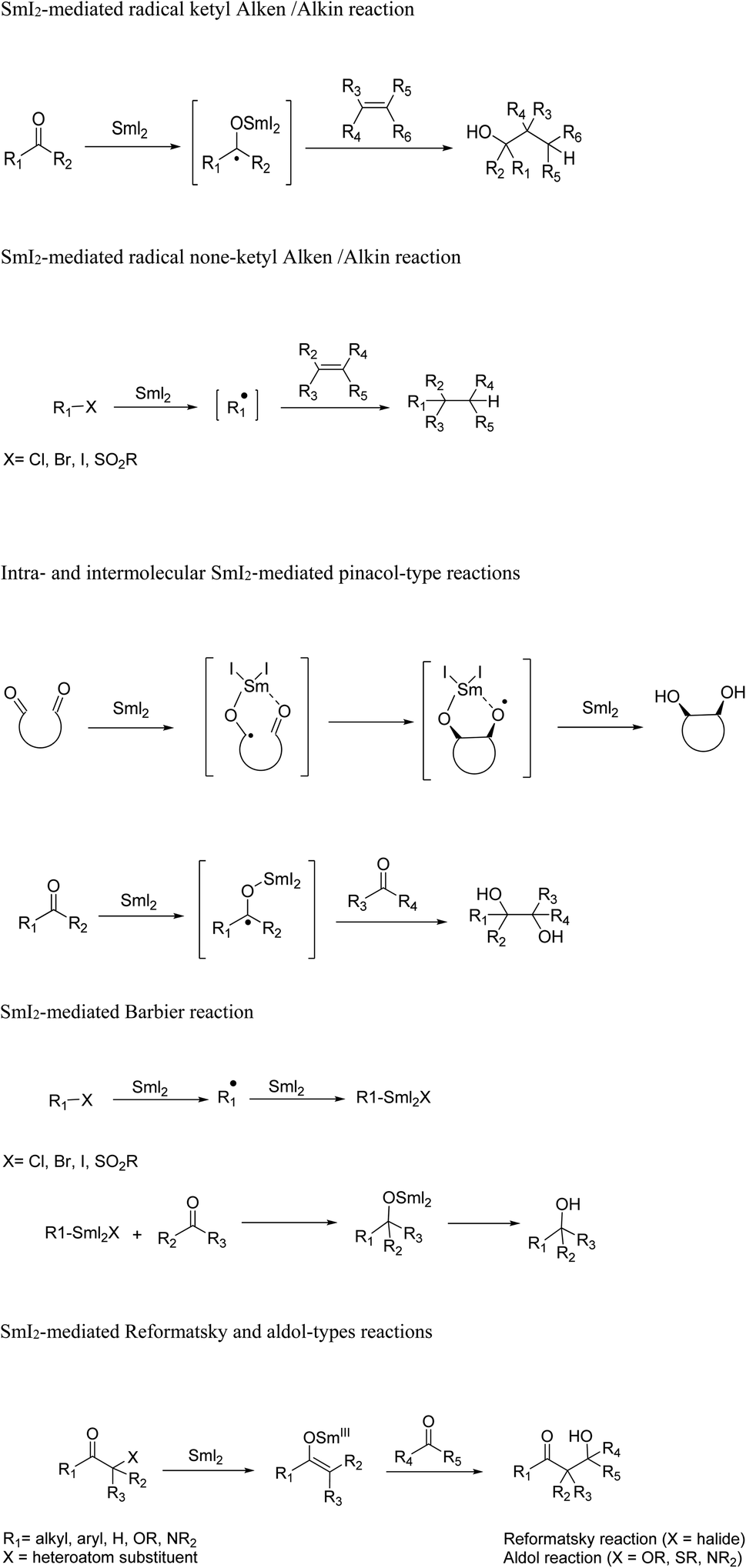 | ||
| Scheme 4 Selective SmI2-mediated reactions. | ||
Samarium diiodide (SmI2) catalyzed intramolecular reductive coupling reactions of ketyl radicals with unsaturated compounds like alkenes, alkynes, and nitriles develop functionalized carbocycles comprising tertiary alcohols. Intramolecular ketyl–olefin coupling reactions produce carbocycles in great yield and favorable diastereoselectivity. The ketyl–allene coupling reaction catalyzed by SmI2 delivers stereocontrolled, highly functionalized carbocycles and heterocycles. The reaction conditions play an important role in the regioselectivity of free-radical additions to allenes ranging from complete terminal attack to restricted central carbon addition.9–11 Molander12 reported also SmI2-mediated-ketyl cyclizations with allenes to synthesize highly functionalized carbocycles or heterocycles in acceptable to excellent yield and great stereochemical control (Scheme 5).
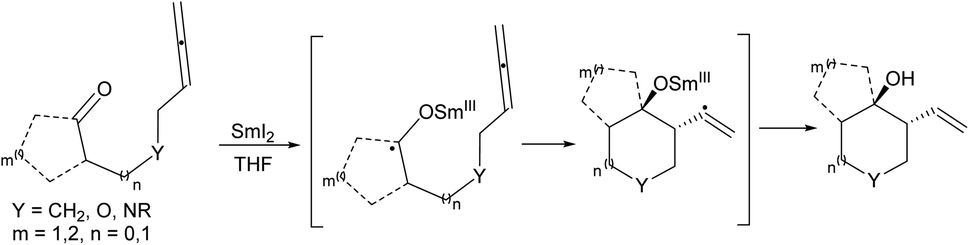 | ||
| Scheme 5 SmI2-mediated ketyl–allene cyclizations. | ||
Molander introduced a pathway that provided increased selectivity for addition to the proximal sp2 center, preventing regio- and stereoisomeric product mixtures, and the prospect of leading to sequential reactions (Scheme 6, pathway a). By using HMPA as an additive, the reactions improve sufficiently and when the toxicity of HMPA is a problem, DMPU can be used instead albeit requiring longer reaction times.
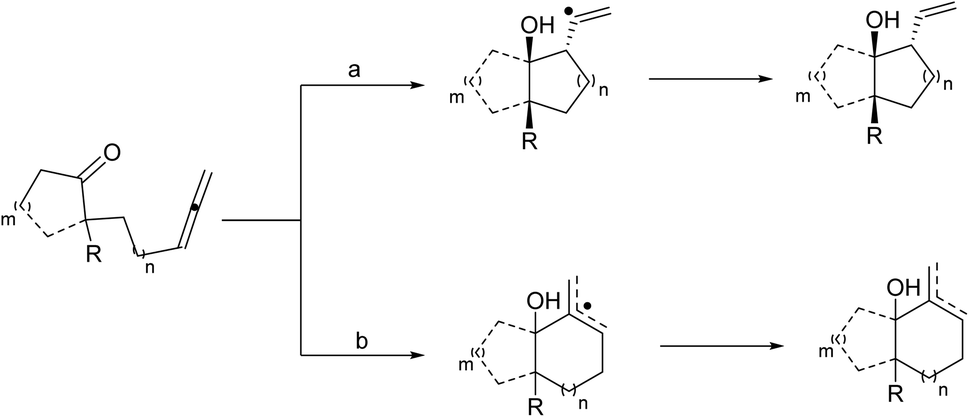 | ||
| Scheme 6 Reaction pathways of SmI2-mediated ketyl–allene cyclizations. | ||
The reactions underwent a chair-like transition state regulated by the samarium alkoxide in the pseudoequatorial position to obtain high diastereoselectivity. The reaction generated two neighboring stereocenters, and the resulting hydroxyl group and alkene moiety can be utilized for additional development. The ketyl–allene coupling can be extended to SmI2-mediated cascade reactions. In the absence of a hydrogen donor, vinyl radicals are relatively rigid, making them favorable for sequential reactions. The developed SmI2-catalyzed reactions often desired superstoichiometric amounts of a metal co-reductant to restore the Sm(II) catalyst. Procter13 recently developed a SmI2-catalyzed intermolecular radical coupling of aryl cyclopropyl ketones and alkynes to give cyclopentenes using 15–25 mol% of SmI2 in THF at 45 °C in good to excellent yields without requiring a superstoichiometric co-reductant to regenerate SmI2. The reactions underwent a reversible single-electron transfer from SmI2 to the ketone, opening the cyclopropyl ring, intermolecular coupling with the alkan closing of the cyclopentene ring resulting in a ketyl radical. The ketyl radical regenerated the Sm(II) catalyst by transferring an electron back to Sm(III) (Scheme 7).
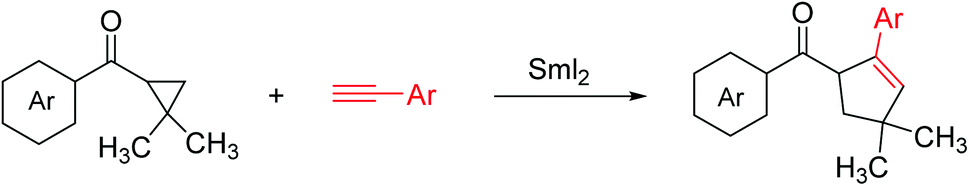 | ||
| Scheme 7 SmI2-catalyzed intermolecular coupling of cyclopropyl ketones and alkynes. | ||
SmI2-mediated cascade reactions are remarkable synthetic tools in the total synthesis of natural products and other complex molecules affording ring formation and/or constructing multiple stereocenters. Due to extraordinary properties of samarium(II) iodide such as the excellent functional group tolerance, high Lewis acidity, fine-tuning the reactivity, the thermodynamic control of single-electron processes, mild reaction conditions, and particularly the ability of this reagent to combine radical and ionic transformations to promote the challenging transformations in any combination or order, it has given rise to this reagent as a crucial tool to achieve the polycyclic complex structures bearing multiple stereocenters from simple, linear synthetic precursors via one-pot, multistep sequences. Over the past decade in the synthesis of complex natural products, various SmI2-catalyzed radical cyclizations to develop different radical/anionic sequential strategies have been utilized for the selective synthesis of complex carbocycles bearing quaternary stereocenters. The ability of SmI2 to mediate both individual reactions in a remarkably particular way and tandem reactions in an equally productive manner has permitted different tandem reactions such as sequential radical reactions, transformations initiated by radical sequences followed by anionic reactions, anionic processes followed by radical reactions and tandem anionic processes to be possible. Several SmI2 asymmetric reactions in presence of a chiral auxiliary to develop the enantioenriched compounds have also been reported, and are also expected to be a common method for high selectivity. Several C–C bond formations using samarium diiodide as electron-donor in the presence of chiral auxiliary also exist.3q In SmI2-mediated asymmetric reactions, samarium enolates can generate in situ which then undergo asymmetric protonation.
The Reformatsky-aldol and the pinacol coupling can be suitable methods to develop asymmetric synthesis considering reasonable synthetic manipulation. Due to the strong oxophilicity of the samarium ion, high stereoselectivities will be achieved using samarium(II) complexes. The intelligent choice of a chiral ligand is important. The suitable ligand should possess many oxygen or nitrogen atoms to coordinate with the samarium and should not be reducible. The superstoichiometric amounts of SmI2 and the ligand also has to be considered. The ease of preparation and recyclability of the ligand should also be evaluated in the case of green chemistry and the development of asymmetric catalysis.3q Samarium diiodide can be a useful reducing agent in asymmetric synthesis for the mild cleavage of the chiral auxiliaries or certain protecting groups.
In continuation of our interest in total synthesis of natural products,14 in this reviews we try to underscore the applications of SmI2 in the total synthesis of natural products covering the literature from 2004 to date.
2. Applications of SmI2-mediated reactions in natural products total synthesis
2.1. SmI2-mediated functional groups transformations
Mycinolide IV (6),15 the aglycon of mycinamicin IV, is a 16-membered macrolide antibiotic and was isolated from Micromonospora griseorubida sp. nov.15 In 2005, Mukaiyama and colleagues16 reported the enantioselective synthesis of the C11–C17 fragment of mycinolide IV using an asymmetric samarium(II) iodide-mediated aldol reaction by installing a chiral oxazolidinone on the unsaturated epoxide. The key steps contain the preparation of samarium enolates by epoxide-fragmentation of, γ.δ-oxiranyl-α,β-unsaturated esters with two moles of SmI2 and a stereoselective synthesis of δ,β′-dihydroxy-β,γ-unsaturated esters by aldol reaction of aldehydes with the samarium enolates. This type of aldol reaction was effectively utilized in the enantioselective synthesis of the C11–C17 segment of mycinolide IV containing two chiral centers and a trans double bond, and in the stereochemical assignment of this compound.The enantioselective synthesis of 5 (C11–C17 segment of mycinolide IV (6)) started with the reaction of 2,4-pentadienoic acid ethyl ester 1 to produce γ.δ-oxiranyl-α,β-unsaturated ester 2 in 2 steps in 52%. Next, an asymmetric SmI2-mediated aldol reaction of 2 and propanal 3 with 2 equiv. of SmI2 at 78 °C provided the aldol adduct 4 in 85% yield and high diastereoselectivity (dr = 8.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Compound 4 was then converted to compound 5 in 2 steps and 87% yield (Scheme 8).
:1). Compound 4 was then converted to compound 5 in 2 steps and 87% yield (Scheme 8).
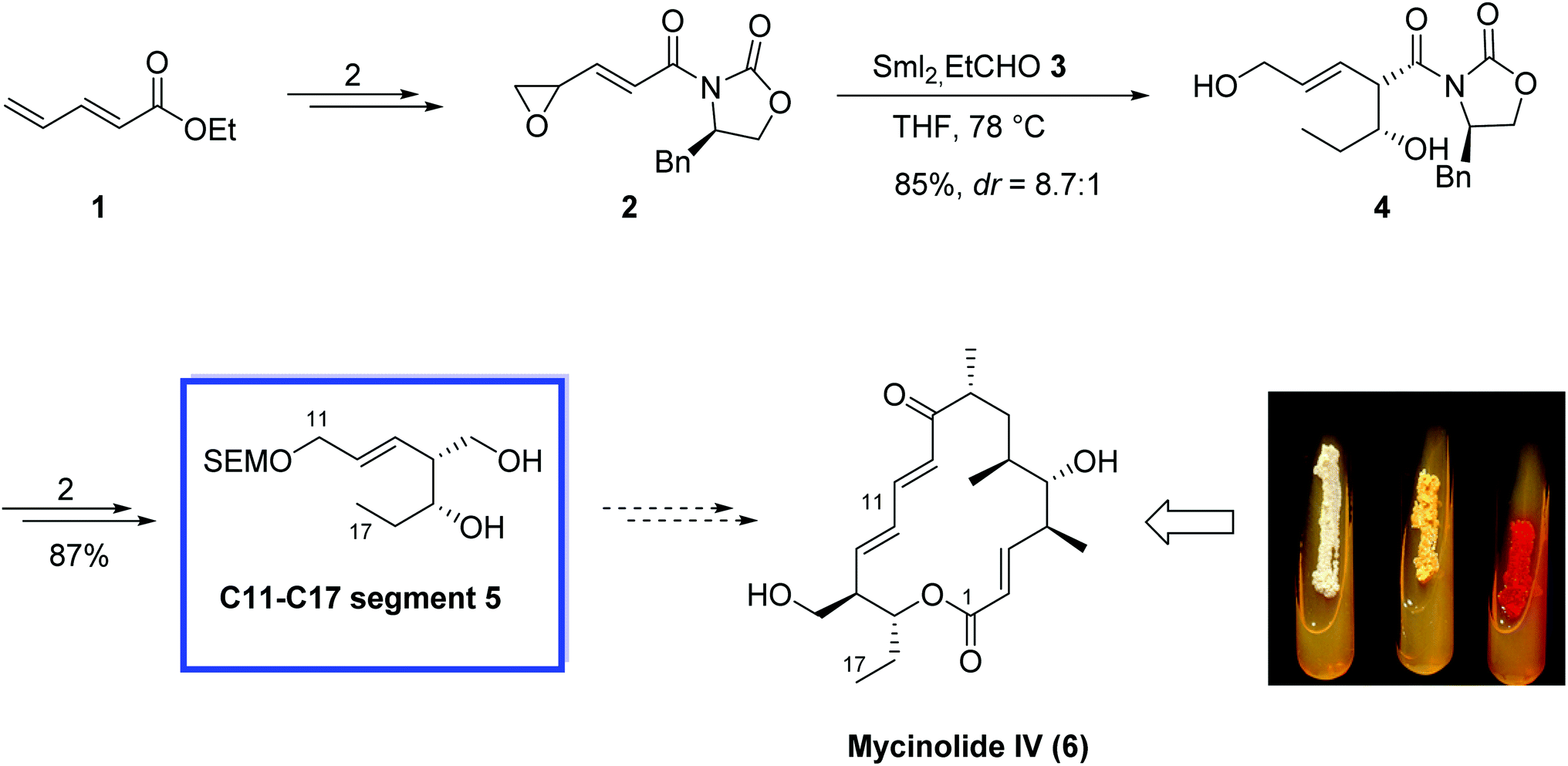 | ||
| Scheme 8 Enantioselective synthesis of 5 (C11–C17 segment of mycinolide IV (6)). | ||
The complex macrolide (+)-acutiphycin 12 was originally isolated from the blue-green alga Oscillatoria acutissima; however, this alga no longer produces this metabolite. (+)-Acutiphycin 12 shows an antineoplastic effect against murine cancer cells and also exhibits cytotoxicity in contact with KB and NIH/3T3 cell lines.17
In 2006, Jamison and colleagues18 reported a highly convergent synthesis of (+)-acutiphycin 12 in 18 steps and an overall yield of 4.0% from commercial starting materials. Both the relative and absolute stereochemistry of the synthetic (+)-acutiphycin 12 were in good agreement with those reported for the natural product. They used an alkynyl ether for the first time as a macrolactone precursor in total synthesis. They also used an intermolecular, SmI2-mediated Reformatsky reaction as a fragment coupling procedure for the first time. The authors claimed that the developed synthetic route is a standard and compatible synthetic pathway that facilitated the immediate and organized analysis of the structure–activity relationships of (+)-acutiphycin 12.
The total synthesis of (+)-acutiphycin 12 began with the coupling of alkyne 7 and aldehyde 8 to provide the required α-bromoketone 9 in 5 steps and quantitative yield. While activated zinc failed to generate the desired enolate, a SmI2-mediated Reformatsky reaction of α-bromoketone 9 and aldehyde 10 afforded the β-hydroxy ketone 11 as a mixture of diastereomers in 90% yield. (+)-Acutiphycin 12 was then synthesized from 11 in 10 steps (Scheme 9).
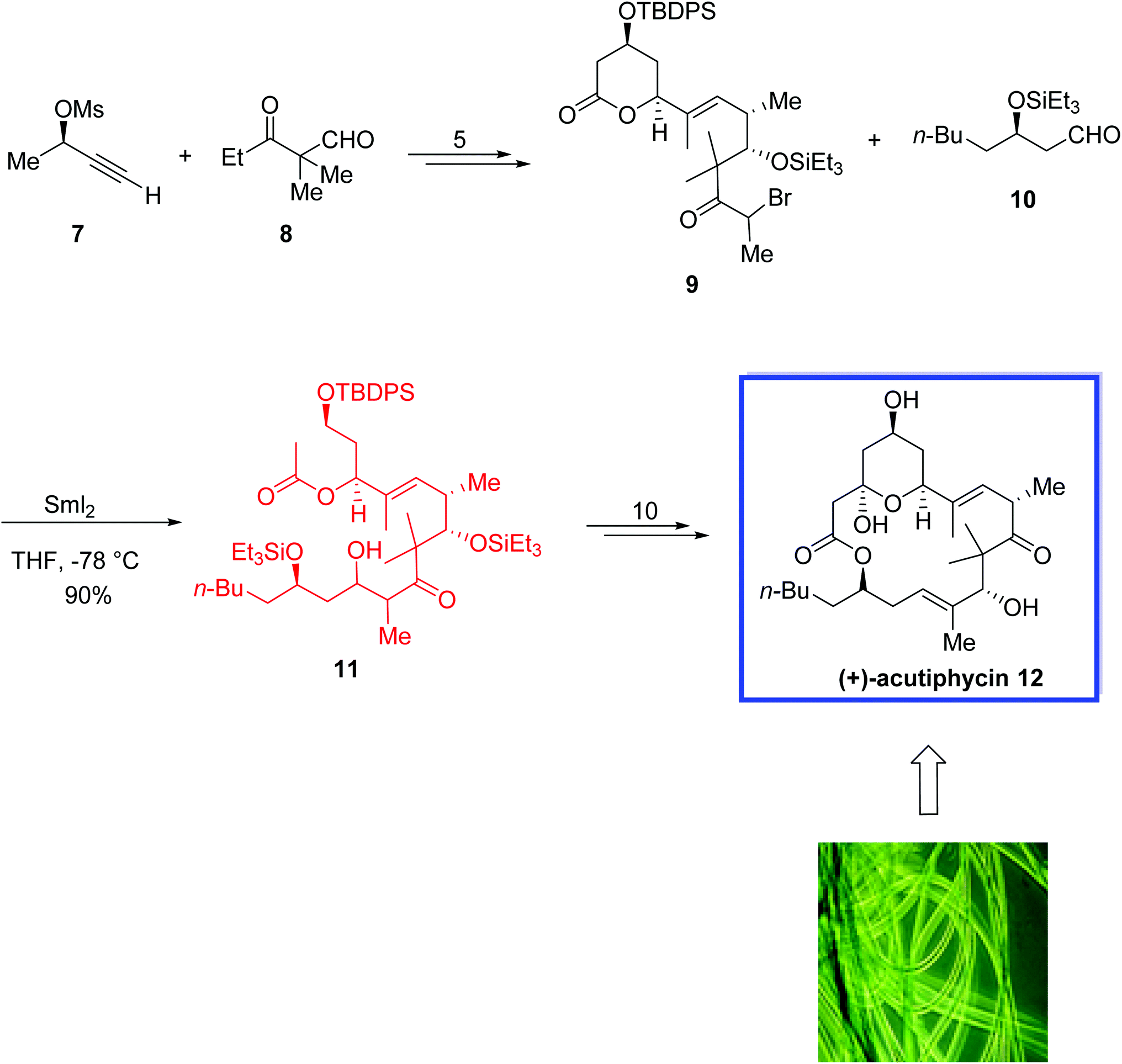 | ||
| Scheme 9 Total synthesis of (+)-acutiphycin 12. | ||
(±)-Subincanadine F 17, isolated from the barks of the Brazilian medicinal plant Aspidosperma subincanum Mart.,19 is a bioactive indole alkaloid with cytotoxic activity in vitro toward murine lymphoma L1210 cells and human epidermoid carcinoma KB cells. Due to remarkable structural characteristics and extraordinary pharmacological activities of the subincanadine family of alkaloids, they have drawn much interest from synthetic chemists.
In 2006, Zhai and coworkers20 developed the first total synthesis of (±)-subincanadine F 17, possessing a 1-azabicyclo[4.3.1]-decane core. The six-step total synthesis of indole alkaloid (±)-subincanadine F 17 began with 1-(para methoxybenzyl)tryptamine 13, prepared from commercially available tryptamine in one step. Key steps contain a SmI2-mediated ring opening and an acid-mediated Mannich reaction to construct the bridge-containing tetracyclic scaffold of subincanadine F 17. They also developed the key intermediate tetracyclic ketoester 15 in one step from α,β-diketoester 14. According to the researchers, the developed strategy to (±)-subincanadine F 17 as well as the synthesized key intermediate tetracyclic ketoester 15 can be potentially useful to synthesize various indole alkaloids with related structures.
A reaction of 13 with α,β-diketoester 14 in acetonitrile at room temperature for 8 h afforded the tetracyclic ketoester 15 in a 75% yield. The tetracyclic core of subincanadine F 17 was easily constructed via a samarium diiodide mediated ring-opening of 15 in THF, at r.t. for 3 h and the 6/5/9 tricycle 16 was obtained in 86% yield. Finally (±)-subincanadine F 17 was synthesized from 16 in 3 steps and 28% yield (Scheme 10).
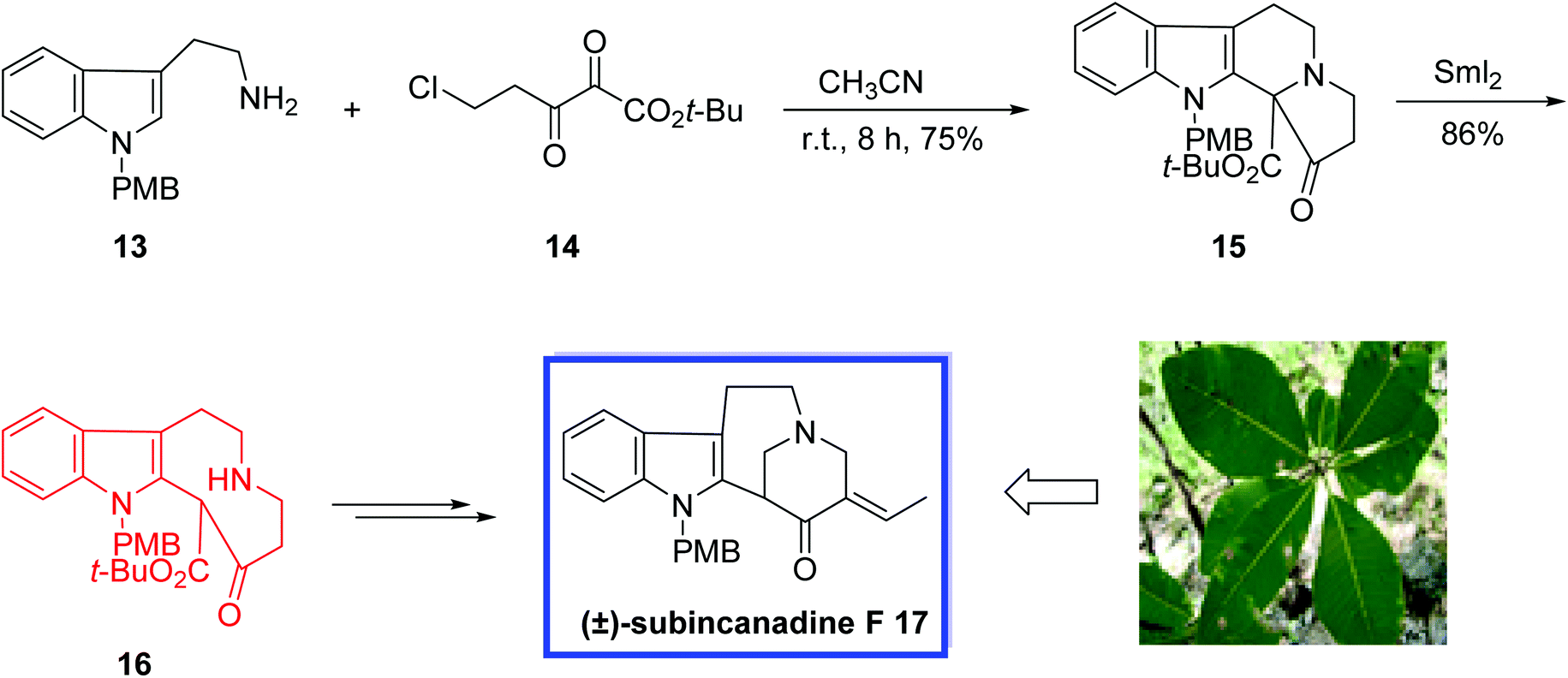 | ||
| Scheme 10 Total synthesis of (±)-subincanadine F 17. | ||
Lactone-containing natural products (6R)-6-[(1E,4R,6R)-4,6-dihydroxy-10-phenyldec-1-en-1-yl]-5,6-dihydro-2H-pyran-2-one 23, (6S)-5,6-dihydro-6-[(2R)-2-hydroxy-6-phenylhexyl]-2H-pyran-2-one 24, isolated from Ravensara crassifolia,21 belong to the group of 5,6-dihydro-α-pyrone-containing natural products with a substituted arylalkyl side chain at C6. These compounds continue to be of great interest because the β-unsaturated α-pyrones can function as Michael-acceptors for the amino acid residues of receptors. α,β-Unsaturated α-pyrones exhibit various biological activities such as antibacterial, anticancer, antifungal, antifeedant, HIV protease inhibition, and antitumor activities. Compounds 23 and 24 show antifungal activities against the phytopathogenic fungus Cladosporium cucumarinum.22
In 2014, Venkateswarlu and colleagues23 reported an efficient stereoselective total synthesis of the 6-alkylated pyranones (6R)-6-[(1E, 4R, 6R)-4,6-dihydroxy-10-phenyldec-1-en-1-yl]-5,6 dihydro-2H-pyran-2-one 23 and (6S)-5,6-dihydro-6-[(2R)-2-hydroxy-6-phenylhexyl]-2H-pyran-2 one 24 from commercially available 5-phenylpentan-1-ol 18 and l-aspartic acid 21. The key steps include a SmI2-mediated reduction of alkoxy ketone, a Crimmins aldol reaction, and a Grubbs-II-catalyzed olefin cross-metathesis. The spectral data and optical rotation of the synthetic compounds 23 and 24 were in good agreement with those of the natural product. The synthesis of 23 started from 5-phenylpentan-1-ol 18 to provide the ketone 19 in 5 steps and 90% yield. Treatment of 19 with SmI2 in THF and MeOH for 12 h resulted in a stereoselective reduction of the oxo group to the desired anti-1,3-diastereoisomer 20 as the major product in 76% yield. Compound 20 was then reacted with 5,6-dihydro-6-vinyl-α-pyrone 22, which was prepared from l-aspartic acid 21 in 6 steps and 80% yield, to afford 23 in 2 steps and 78% yield. The natural product 24 was also prepared in 7 steps starting from 5-phenylpentan-1-ol 18 and resulted in a separable mixture of the natural product 24 and bicyclic lactone 25 (7:3) in 78% yield (Scheme 11).
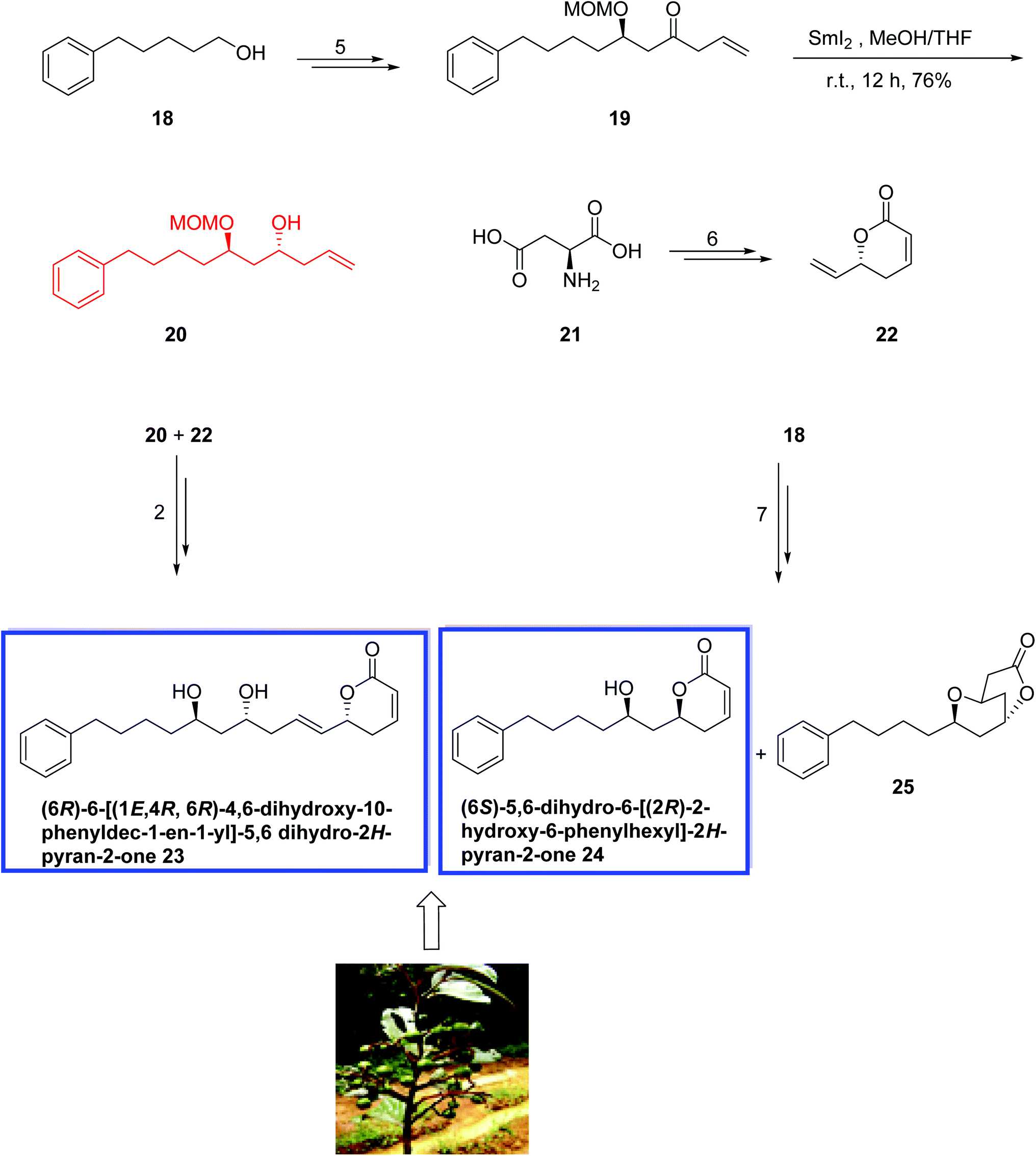 | ||
| Scheme 11 Total synthesis of (6R)-6-[(1E, 4R, 6R)-4,6-dihydroxy-10-phenyldec-1-en-1-yl]-5,6 dihydro-2H-pyran-2-one 23 and (6S)-5,6-dihydro-6-[(2R)-2-hydroxy-6-phenylhexyl]-2H-pyran-2 one 24. | ||
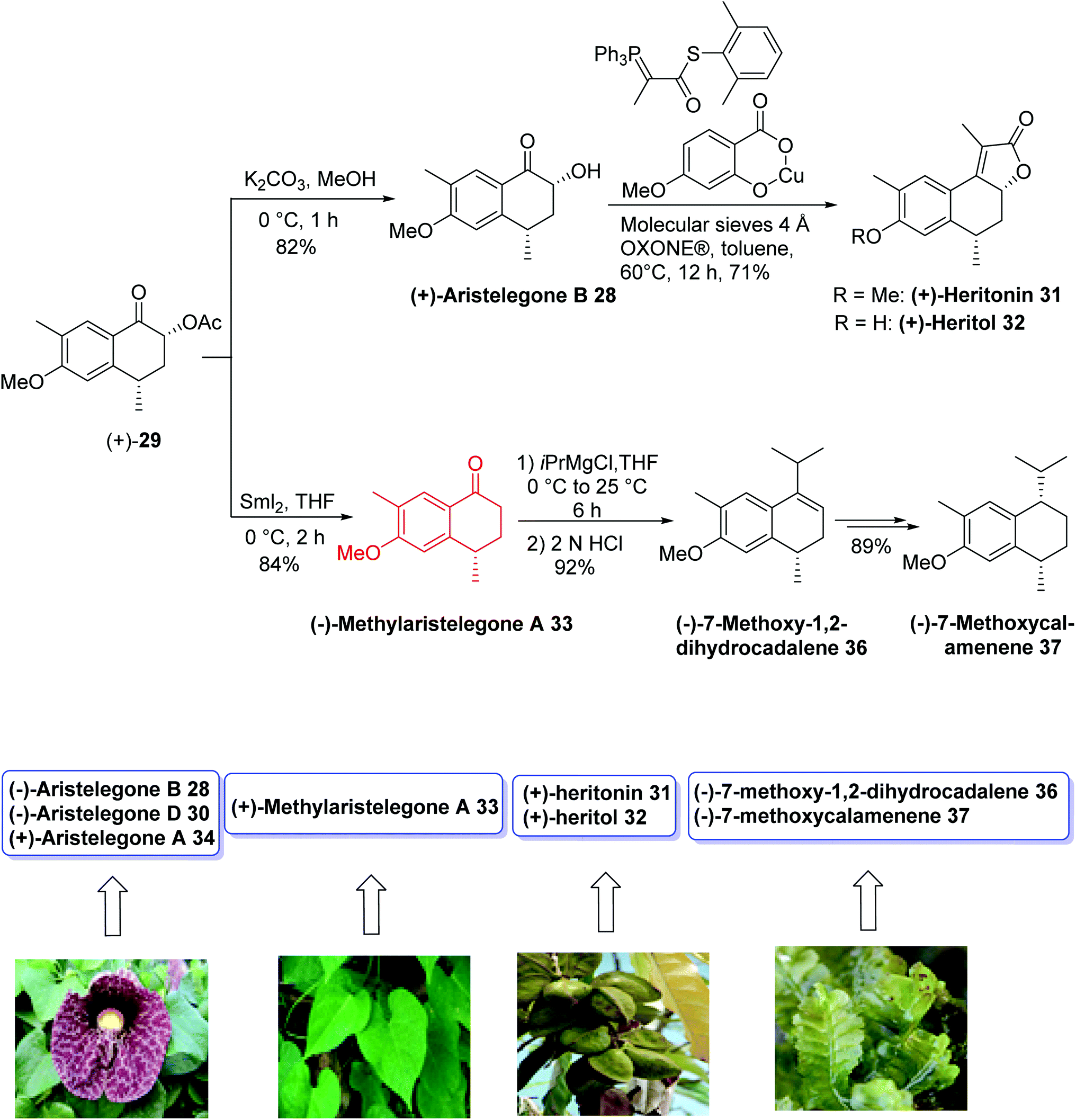
Hydroxy(methyl)tetrahydronaphthalene and methoxy(methyl)tetrahydronaphthalene are known classes of natural products with various biological activities.24 The (+)-aristelegone A 34, (−)-aristelegone B 28, and (−)-aristelegone D 30, isolated from Aristolochia elegans; (+)-methylaristelegone A 33 (antispasmodic), isolated from Aristolochia constructa;25; (+)-heritonin 31 and (+)-heritol 32, isolated from Heritiera littoralis; (+) mutisianthol 35 (antitumor), isolated from Mutisia homoeantha;26 and (−)-7-methoxy-1,2-dihydrocadalene 36 and (−)-7-methoxycalamenene 37, isolated from a Heteroscyphus planus culture27 belong to the hydroxy(methyl)tetrahydronaphthalene class of natural products.
In 2015, Argade and coworkers28 developed the chemoenzymatic total synthesis of nine bioactive tetrahydronaphthalene-based natural products using antipode (−)/(+)-aristelegone B 28 as a single common precursor. Both (+)/(−)-isomers as potential building blocks were prepared through the late-stage efficient enzymatic resolution. It was planned to employ (±)/(+)/(−)-28/29 as key building blocks bearing well-positioned substituents and necessary functional groups to achieve the stereoselective collective synthesis of target compounds. The key steps were SmI2-mediated deoxygenations, the syn-selective installation of hydroxyl groups at the α-position of the ketone component, a sequence of acylation-Wittig reactions, regioselective benzylic oxidations, enantiomerically pure enzymatic resolutions, and stereoselective reductions. The analytical and spectral data, obtained for the synthetic product, were in good agreement with those reported for the natural product (−)-28.
The total synthesis of all assigned target compounds began with the reaction of 2-methylanisole 26 and succinic anhydride 27 to obtain (−)-aristelegone B 28 (54% yield, 94% ee) and (+)-acylaristelegone B 29 (46% yield, 96% ee) in 8 steps. The natural product (−)-aristelegone D 30 was obtained from (−)-aristelegone B 28 within two steps in 84% yield; therefore, the first chemoenzymatic synthesis of (−)-aristelegone D 30 was achieved in ten steps with an 8% overall yield. Tandem acylation-Wittig reaction of (−)-aristelegone B 28 provided (−)-heritonin 31 in a 74% yield (93% ee) and 8% overall yield in nine steps without any racemization. Treatment of (−)-heritonin 31 with AlCl3 provided (−)-heritol 32 in 80% yield. Cleavage of the hydroxyl group of (−)-aristelegone B 28 with samarium iodide provided (+)-methylaristelegone A 33 in a 65% yield. Further treatment of (+)-methylaristelegone A 33 with BBr3 gave (+)-aristelegone A 34 in a 74% yield, from which (+)-mutisianthol 35 was then synthesized by a known procedure in 5 additional steps. To synthesize (−)-7-methoxy-1,2-dihydrocadalene 36, (−)-7 methoxycalamenene 37 and (+)-heritonin 31, the second enantiomerically pure building block (+)-acylaristelegone B 29 was selected, which was achieved via enzymatic resolution. Cleavage of the acetoxy group of (+)-acylaristelegone B 29 by samarium iodide furnished (−)-methylaristelegone A 33 in an 84% yield. The reaction of isopropylmagnesium bromide with (−)-methylaristelegone A 33 followed by acid-catalyzed in situ dehydration of the formed intermediate tertiary alcohol yielded another natural product, (−)-7-methoxy-1,2-dihydrocadalene 36, in a 92% yield; therefore, the first chemoenzymatic synthesis of (−)-7-methoxy-1,2-dihydrocadalene 36 was accomplished in ten steps with an 8% overall yield. An enantioselective reduction of the naturally-isolated natural product (−)-36 gave (−)-7-methoxycalamenene 37 via known procedure. Base-induced deacylation of compound (+)-29 to establish (+)-aristelegone B 28, followed by a similarly performed tandem acylation-Wittig reaction provided the natural product (+)-heritonin 31 in a 71% yield. The diastereoselective synthesis of the common precursor (±)-aristelegone B 28 and its efficient enzymatic resolution allowed for the syntheses shown below in Schemes 12 and 13.
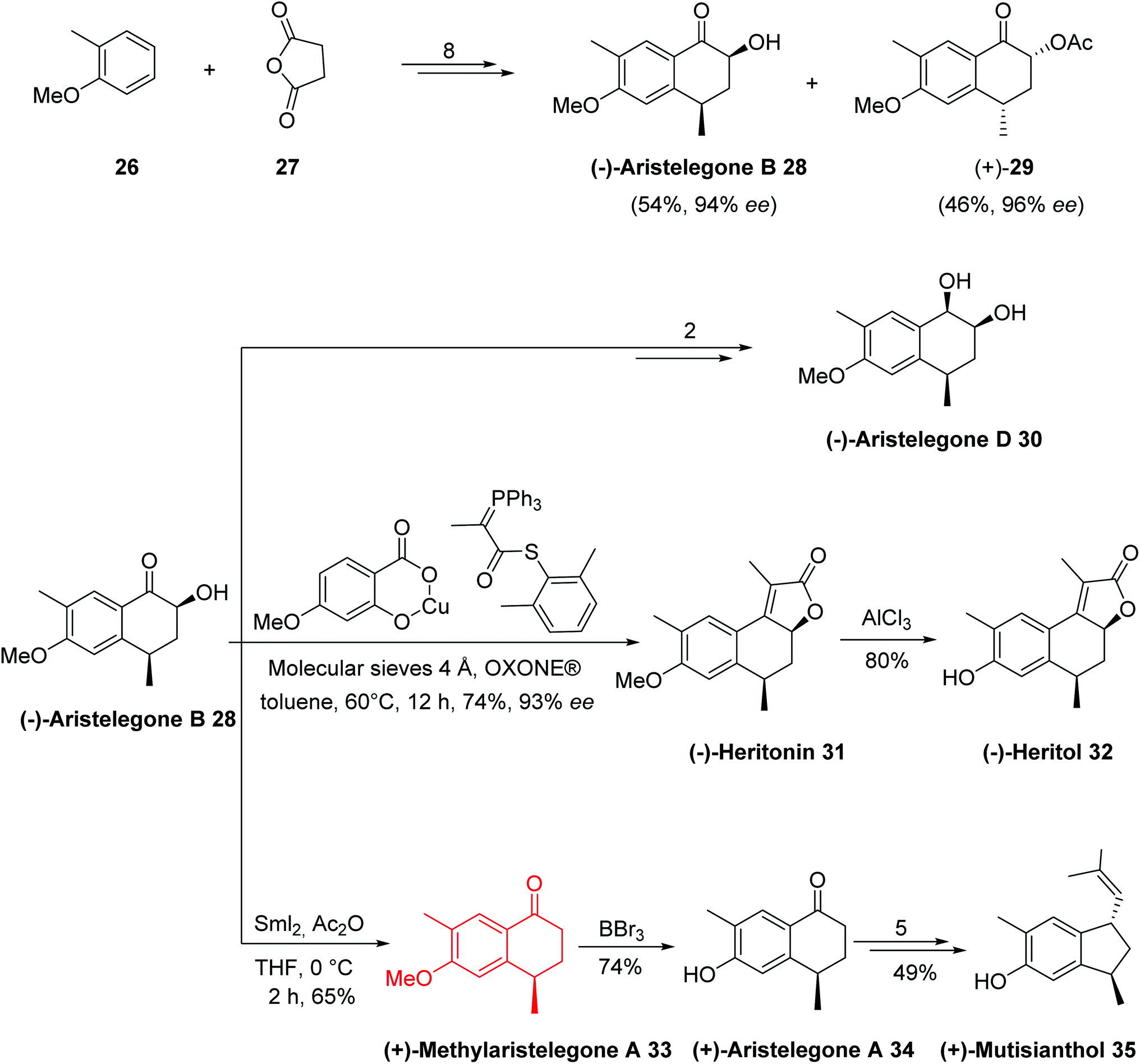 | ||
| Scheme 12 Total synthesis of tetrahydronaphthalene-based natural products 28–35. | ||
 | ||
| Scheme 13 Total synthesis of tetrahydronaphthalene-based natural products 28–37. | ||
(−)-Securinine 43 and (−)-14,15-dihydrosecurinine 4429 were isolated from the roots of Securinega suffructicosa,30 and belong to the Securinega alkaloids31 with a wide range of pharmacological activities. They have received considerable attention from the synthetic community. Most securinine-type alkaloids are characterized by possessing a unique tricyclic skeleton with an α,β-unsaturated-γ-lactone ring that exists only in the Euphorbiaceae in species Phyllantus and Securinega.32 (−)-Securinine 43 is the major component of the alkaloid fraction in S. suffruticosa and is a potential central nervous system (CNS) stimulant that shows antimalarial, antibacterial, and cytotoxic activities.33 (−)-14,15-Dihydrosecurinine 44 is also a potent CNS stimulant and an acute poison,30 and has potent inhibition activities against [3H]GABA34 and AChE.35
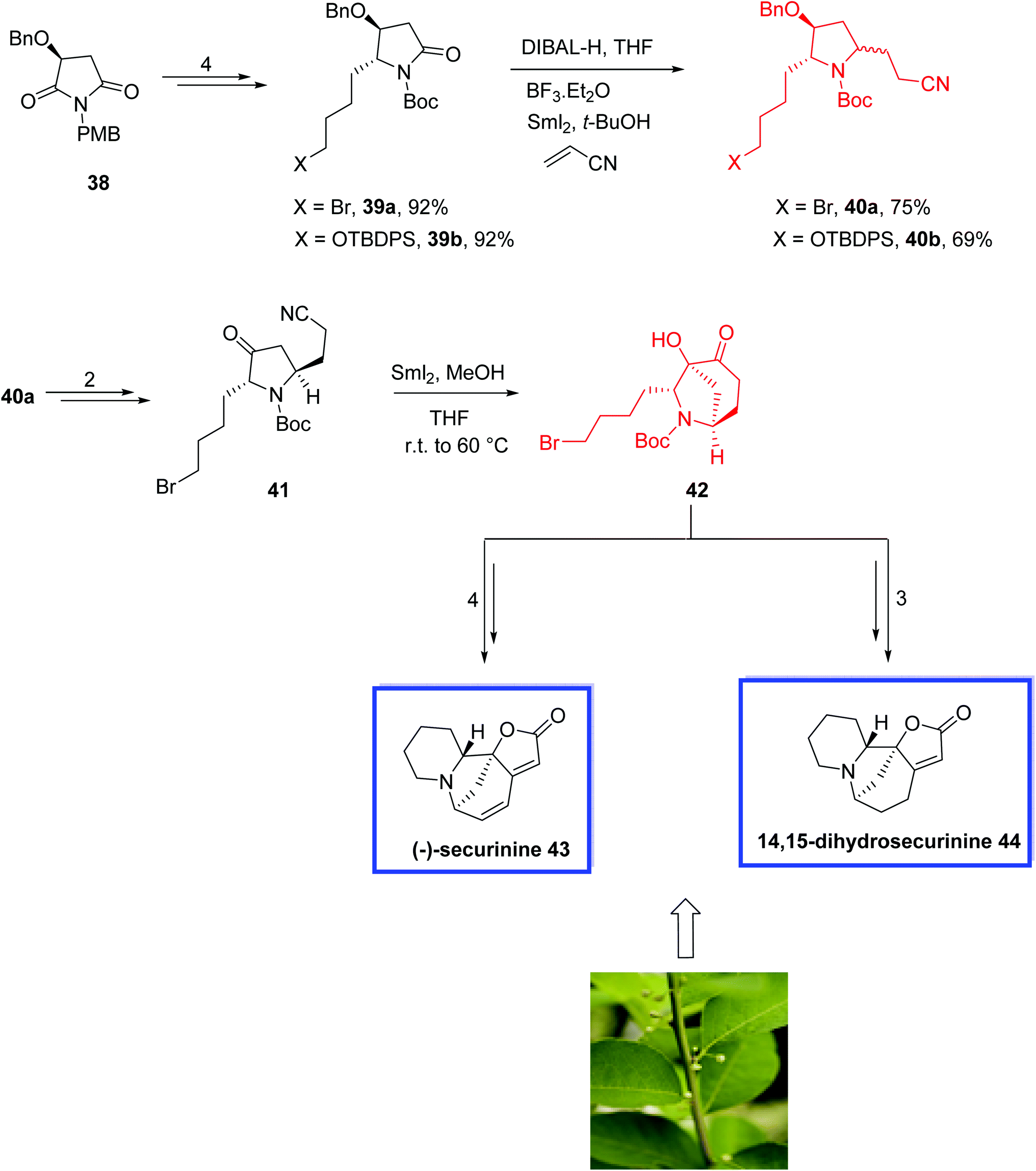
In 2015, Zheng and coworkers36 developed the total synthesis of (−)-14,15-dihydrosecurinine 44 in 12 steps in an overall yield of 14.4%. They also reported the total synthesis of (−)-securinine 43 in 10 steps in an overall yield of 20.2% from the intermediate 42. The key steps contain a six-step procedure to construct the piperidine ring A, a SmI2-induced reductive coupling reaction for the diastereoselective construction of α-hydroxy-6-azabicyclo[3.2.1]octanone and an intramolecular olefination reaction to form the butenolide ring D. The spectral data of 43 and 44 were in good agreement with those previously reported and the enantiopurity of the synthetic 14,15-dihydrosecurinine 44 was greater than 99% ee.
The total synthesis of (−)-14,15-dihydrosecurinine 44 and (−)-securinine 43 started from optically pure maleimide 38 to obtain imides 39a and 39b in 4 steps and 92% yield.
Reduction of 39a/39b with DIBAL-H, followed by SmI2-induced reductive radical coupling with acrylonitrile provided the desired stereoisomer 40 as the major product in a diastereomeric mixture of 40a/40b (d.r. = 75:25 for 40a, d.r. = 88:12 for 40b). Of note, SmI2-mediated debromination of 39a occurred if the reaction occurred at a temperature above −40 °C. Compound 40a was then converted into ketonitrile 41 in 2 steps. Treatment of compound 41 with excess SmI2 in methanol afforded the cyclized product 42 as a single diastereoisomer in 75% yield, from which (−)-securinine 43 and 14,15-dihydrosecurinine 44 were synthesized in 4 and 3 steps, respectively (Scheme 14).
 | ||
| Scheme 14 Total synthesis of (−)-securinine 43 and (−)-14,15-dihydrosecurinine 44. | ||
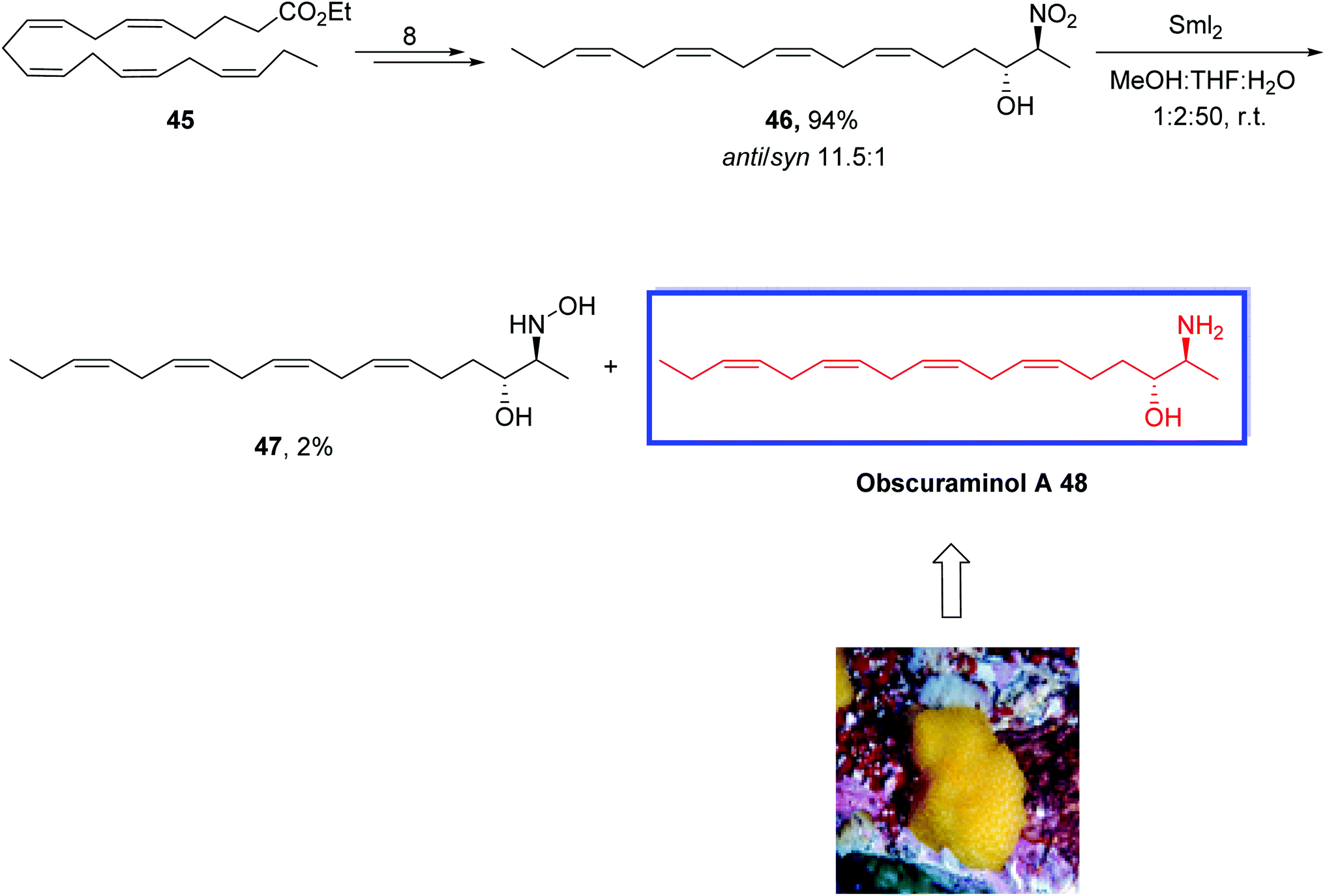
The vicinal amino alcohol motif occurs in various biologically active natural products, and several bioactive long-chain vicinal amino alcohols such as obscuraminol A 48 have been isolated from marine sources. The sphingolipid class of natural products obtained from long-chain vicinal amino alcohols with the 1-deoxygenated sphingosine core occurs in marine invertebrates, particularly in marine ascidian.37 Obscuraminol A 48 is a vicinal anti-amino alcohol that belongs to the class of sphingoids possessing a polyunsaturated Z-skipped side chain in their structure. Obscuraminol A 48 retains the same (2S,3R)-configuration as spisulosine and was isolated from the chloroform extracts of the marine ascidian Pseudodistoma obscurum.38 This class of substances exhibits several bioactivities such as cytotoxicity, antimicrobial, and antifungal effects.39
In 2016, Hansen and colleagues40 reported the first stereoselective synthesis of the polyunsaturated amino alcohol natural product obscuraminol A 48 in 6% overall yield over 11 steps from the ethyl ester of eicosapentaenoic acid 45. The key steps were an anti- and enantioselective organocatalyzed Henry reaction and a challenging chemoselective SmI2-mediated reduction of a nitro-group in the Henry product. The configuration of the four Z-skipped double bonds of synthesized obscuraminol A 48 was maintained by the starting material, i.e., the ethyl ester of (all-Z)-eicosa-5,8,11,14,17-pentaenoic acid. The synthesized obscuraminol A 48 was in complete agreement with the natural product.
The stereoselective synthesis of obscuraminol A 48 started with the ethyl ester of (all-Z)-eicosa-5,8,11,14,17-pentaenoic acid 45 to obtain the (2S,3R)-nitroaldol 46 in 94% yield and good diastereoselectivity (anti/syn 11.5:1) in 8 steps. Reduction of vicinal nitroaldols 46 into related amino alcohols with common methods was not successful; therefore, they tried the previously reported SmI2-mediated reduction method on the nitroaldol product 46. Treatment of 46 with SmI2 in MeOH/THF/H2O gave the desired amino-alcohol 48 in 60% yield together with the hydroxylamine 47 as a by-product. Increasing the reaction time and molar equivalents of SmI2 to 10 equivalents did not affect the full conversion to the target molecule 48. The synthesized obscuraminol A 48 validated the assigned structure of the natural product 48. With lower diastereoselectivity than anticipated from the starting material defined to be 5.2:1 in favor of the desired anti-isomer and the absolute configuration of the stereogenic centers in 48 to be 2S,3R as initially allocated, this provided evidence for the initial assignment of the relative configuration of 46 also as anti. Hence, the absolute configuration of the stereogenic centers in 48 is indeed 2S,3R as originally assigned (Scheme 15).
 | ||
| Scheme 15 Total synthesis of obscuraminol A 48. | ||
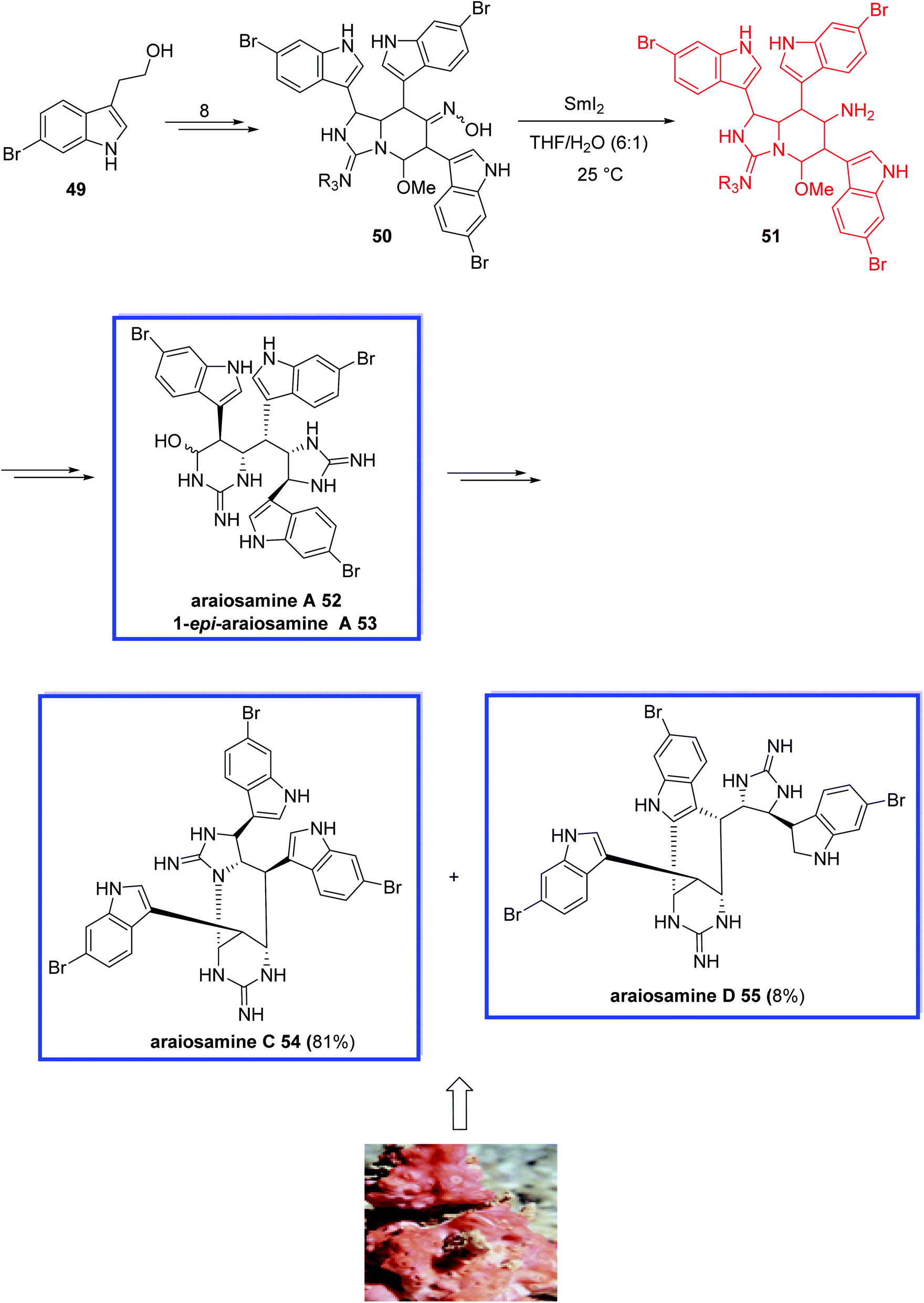
Araiosamines A–D were isolated from the methanol extract of a marine sponge, Clathria (Thalysias) araiosa.41 They relate to a small family of unusual marine alkaloids with extraordinary structures with two guanidine units, three bromoindole heterocycles and six contiguous stereocenters.
In 2016, Baran and coworkers42 reported the total synthesis of araiosamine A 52 and araiosamines C–D 54–55 in 11 steps. Key steps featured the installation of guanidine through a new chemoselective reagent, a SmI2-mediated stereoselective reduction, chemo- and stereoselective C–H functionalization, and a simple final step to synthesize the biosynthetic intermediate. The absolute configuration of these natural araiosamines was not assigned; therefore, they synthesized (+)-araiosamine C 54, which was found to be the natural enantiomer, in reference to the optical rotation. They show that the synthetic araiosamines A,C, and D, 52, 54, and 55, respectively, exhibit significant antibacterial activities against both Gram-positive and -negative bacteria.
The total synthesis of araiosamines A, C, and D (52, 54, 55) began with the commercially available 6-bromotryptophol 49 to obtain oxime 50 in 8 steps. The reduction of the oxime in 50 was a challenging step due to the sterically placed oxime moiety and the existence of other functional groups being prone to be reduced. Using lithium aluminum hydride and sodium or different combinations of sodium borohydride and metal salts caused debromination. Alone, the combination of SmI2 and H2O reduced oxime 50 to primary amine 51 stereoselectively, from which araiosamine A 52 and 1-epi-araiosamine A 53 were then obtained in several steps. Heating the mixture of 52 and 53 resulted in araiosamines C 54 and D 55 (Scheme 16).
 | ||
| Scheme 16 Total synthesis of araiosamine A 52, araiosamine C 54, and araiosamine D 55. | ||
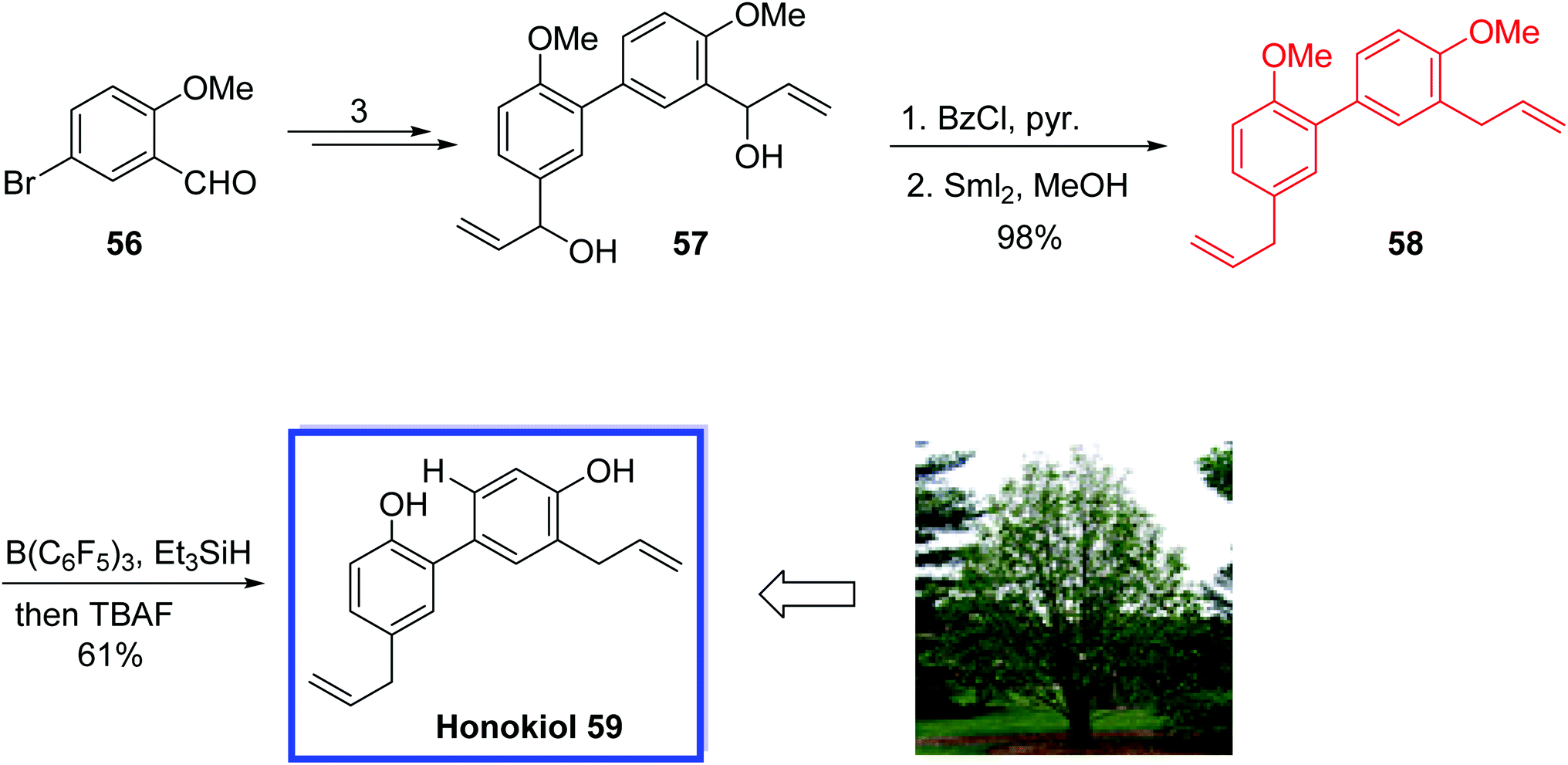
Honokiol 59, isolated from the bark of Magnolia officinalis, is a neolignan, which exhibits a significant range of promising biological activities such as antitumor43 and anti-angiogenic44 activities. Honokiol 59 was isolated together with its isomeric compound magnolol45 with similar physical properties, but with different bioactivities.
In 2016, O'Neil and colleagues46 reported the total synthesis of the honokiol 59 in 4 steps and 42% overall yield. The key steps were a Suzuki coupling to construct the biphenyl core and a samarium-mediated bis-benzoyl ester reduction to install both allyl groups at the same time at the last stage to avoid problems related to allylic deprotonation or isomerization.
This total synthesis demonstrates the capability of this samarium diiodide for organic synthesis, establishing sensitive olefins without any unwanted isomerization. It was anticipated that the procedure could be scaled up for more biological investigations.
The total synthesis of honokiol 59 began with the commercially available aldehyde 56 which was utilized to obtain intermediate diol 57 in 3 steps. Acylation of 57 with benzoyl chloride and reduction of both benzoyl esters with SmI2/MeOH provided dimethylhonokiol 58 in 98% yield and >10:1 selectivity for both nonconjugated allyls substituents (Scheme 17).
 | ||
| Scheme 17 Total synthesis of honokiol 59. | ||
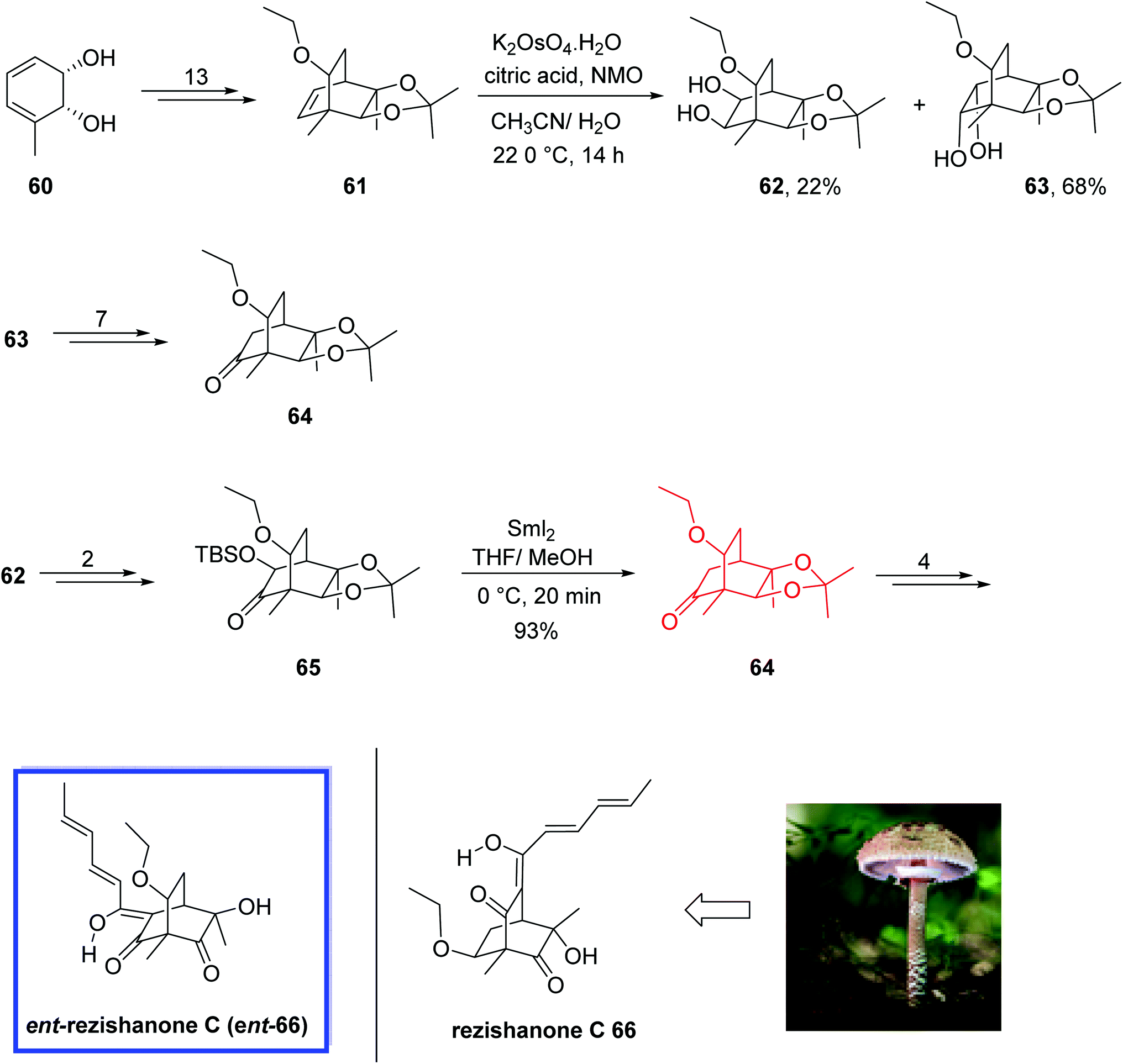
The sorbicillins belong to the class of polypeptide metabolites derived from fungi.47 They exhibit considerable structural variety and biological activities.48 Rezishanone C 66 relates to a hybrid sub-type48a of sorbicillins, which is generated via a Diels–Alder reaction between sorbicillinol and several non-sorbicillinoid-derived compounds with a dienophilic residue. In the case of rezishanone C 66, it has been proposed that an ethyl vinyl ether group is the dienophile included.
In 2017, Banwell and colleagues49 reported the total synthesis of target compound ent-66, the enantiomer of the true structure of the sorbicillinoid rezishanone C 66 (sorbivinetone), from a homochiral cis-1,2-dihydrocatechol 60. All the spectral data of compound ent-66 were in complete agreement with the assigned structure and the specific rotation was also comparable but opposite sign to the reported data for rezishanone C 66. Therefore, they reported the total synthesis of ent-rezishanone C (ent-66) and instead of the originally illustrated structure, compound 66 was demonstrated as the correct structure for rezishanone C 66. They performed dispersion-corrected DFT calculations to approximate the transition state energies that correlate with the different possible modes of Diels–Alder cycloaddition occurring between sorbicillinol and ethyl vinyl ether. These calculations proposed that the most energetically favorable reaction path was directly to compound 66 instead of the already proposed structures.
The total synthesis of rezishanone C 66 commenced from homochiral cis-1,2-dihydrocatechol 60 to convert into the desired ethyl ether 61 in 13 steps, which included several key structural components of the scaffold of ent-rezishanone C 66.
Compound 61 was then subjected to dihydroxylation to obtain a mixture of diols 62 (22%) and 63 (68%), which were separated by chromatography. Diol 63 was then converted into the desired ketone 64 in 7 steps (79%) from which the target ent-66 was synthesized in 3 steps and 52% yield. Diol 62 was also converted into ketone 64 by first converting to ketone 65 in 89% yield over 2 steps. Then treatment of ketone 65 with samarium diiodide in THF provided ketone 64 in 93% yield. An additional four steps led to ent-rezishanone C 66 (Scheme 18).
 | ||
| Scheme 18 Total synthesis of ent-rezishanone C (ent-66). | ||
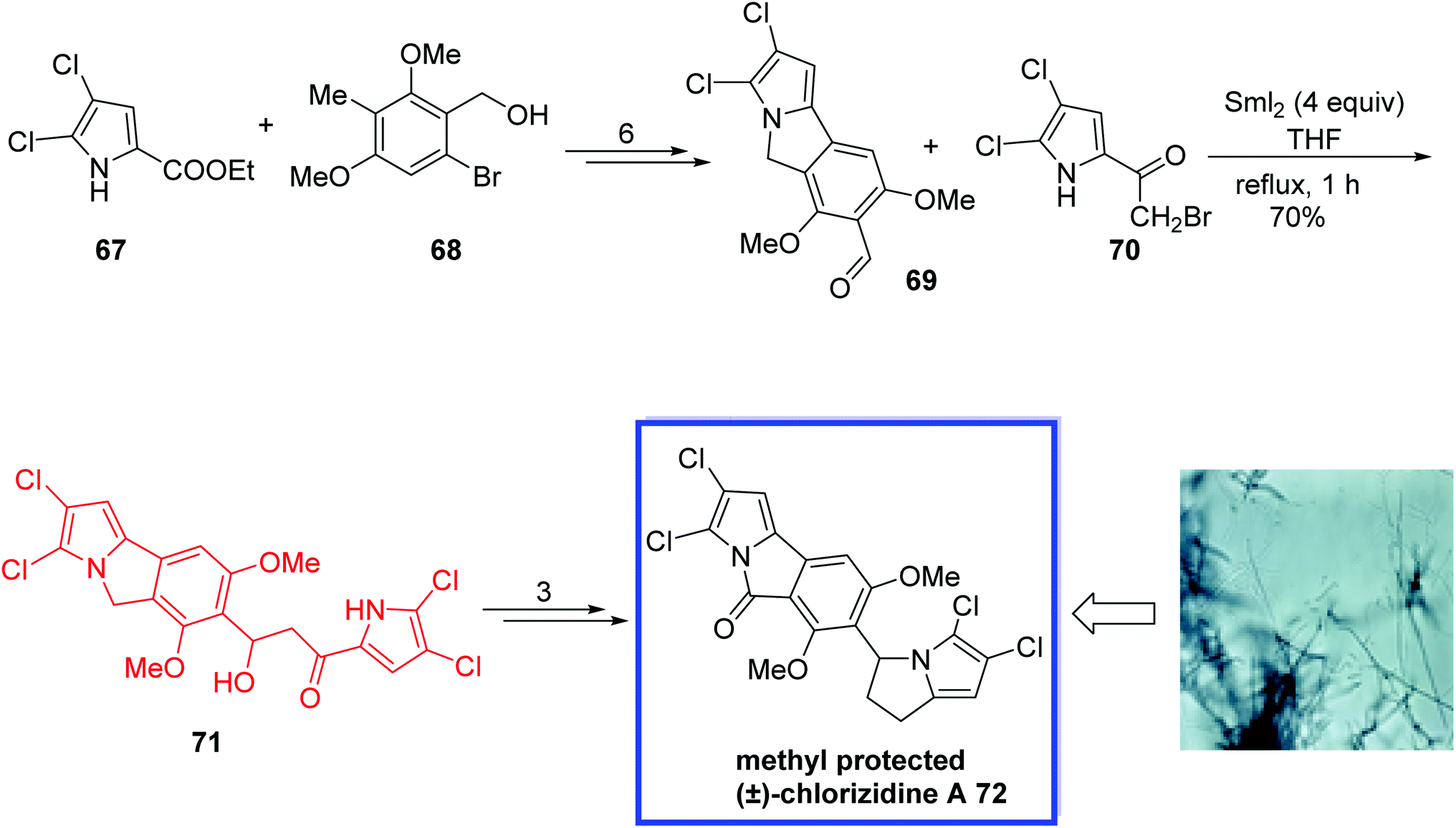
Pyrrole-containing natural products, isolated from terrestrial and marine origins,50 show a various range of applications and diverse interesting biological activities such as antimalarial, antifungal, antibacterial, anticancer, antiprotozoal, etc.50 Chlorizidine A 72, a new pyrrole-containing natural product, was isolated from Streptomyces sp. strain CNH-287 and exhibits significant cytotoxicity against HCT-116 adenocarcinoma cell line.50g Chlorizidine A 72 retains the 5H-pyrrolo[2,1-a]isoindol-5-one scaffold, which was found for the first time in the field of natural products and is perhaps responsible for the cytotoxicity.
In 2017, Mhaske and coworkers51 developed the first total synthesis of the methyl-protected (±) chlorizidine A 72 in 10 steps. The key steps were a Pd-catalyzed decarboxylative coupling Mitsunobu reaction and a samarium iodide-mediated Reformatsky reaction to synthesize the unique 5H-pyrrolo[2,1-a]isoindol-5-one ring and 2,3-dihydropyrrolizine scaffold. Chlorizidine A 72 is highly prone to degradation; hence, the methyl group of (±)-chlorizidine A 72 was protected. The spectral and analytical data of the synthesized methyl-protected chlorizidine A 72 were in complete agreement with the reported data, which confirms the structure of the isolated natural product chlorizidine A 72. The designed synthetic route is suitable to synthesize other potential analogues for structure–activity relationship analyses.
The total synthesis of the methyl-protected (±)-chlorizidine A 72 began with the reaction of dichloro ester 67 and alcohol 68, which were coupled and transformed to the desired aldehyde 69 within 6 steps. With aldehyde 69 in hand, they installed the 2,3-dihydropyrrolizine ring system through the effective use of a samarium iodide-mediated Reformatsky reaction with bromoketone 70 in THF under reflux to synthesize the β-hydroxy ketone 71 in 70% yield. The methyl-protected (±)-chlorizidine A 72 was then synthesized in 3 steps (Scheme 19).
 | ||
| Scheme 19 Total synthesis of methyl-protected (±)-chlorizidine A 72. | ||
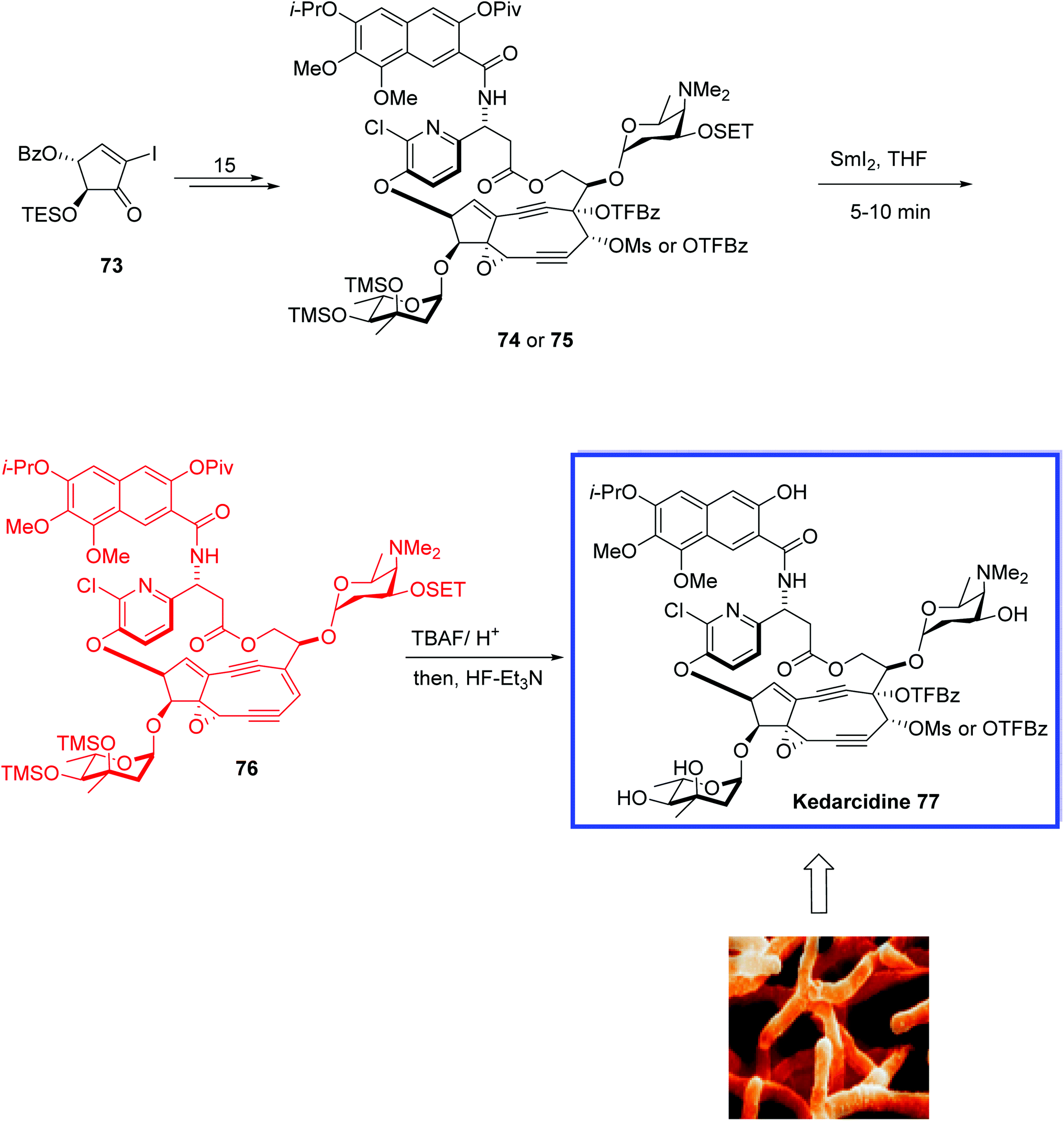
Kedarcidin 77, isolated from Streptoalloteichus sp. ATCC 53650 (not Saccharothrix),52 is a nine-membered cyclic enediyne, in which an alkene directly is attached to two alkynyl appendages. The kedarcidin chromophore is a highly unstable natural product. It consists of an ansa-bridged enediyne chromophore and an apoprotein that stabilizes the toxin in the Actinomycete.52 Kedarcidin 77 is a chromoprotein antitumor antibiotic and shows selective in vivo antitumor activity against P388 leukemia and B16 melanoma cells.52
In 2019, Lear and coworkers53 reported the total synthesis of Myers' structure of the kedarcidin chromophore 77 in 17 steps from gram-scale intermediates. Key steps contain a stereoselective epoxyalkyne formation, an α-selective glycosylation, a Mitsunobu aryl etherification to establish a steric 1,2-cis-configuration, a Sonogashira–Shiina cyclization sequence, a Ohfune-mediated amidation, a Ce(III)-mediated nine-membered enediyne cyclization, and a SmI2-mediated reductive olefination. HRMS data of the synthetic natural product 77 was achieved for the first time.
The total synthesis of kedarcidin 77 began with the iodocyclopentenone 73 as its C10-OSET silyl ether to establish the nine-membered enediyne cyclisation and to prepare C4–O-trifluorobenzoate (TFBz) ester 74 or 75 in 15 steps. Deoxygenation of 74 or 75 by SmI2 in THF at −20 °C for 5–10 min gave olefin 76. Finally, deprotection of 76 under buffered hydrogen fluoride conditions resulted in kedarcidin chromophore 77 (Scheme 20).
 | ||
| Scheme 20 Total synthesis of kedarcidin 77. | ||
Rumphellclovanes E 82, a rare marine natural product and family member of clovane-type sesquiterpenoids, was isolated from the gorgonian coral Rumphella antipathies.54 Clovane-type sesquiterpenoids comprise a tricyclo[6.3.1.01,5]dodecane ring system including three to five stereocenters, of which at least two of them are quaternary centers.
Clovane-type sesquiterpenoids exhibit several biological properties such as inhibition of superoxide anion and elastane, produced by human neutrophils,54 and inhibition of embryonic hippocampal and cortical neurons enlargement.55
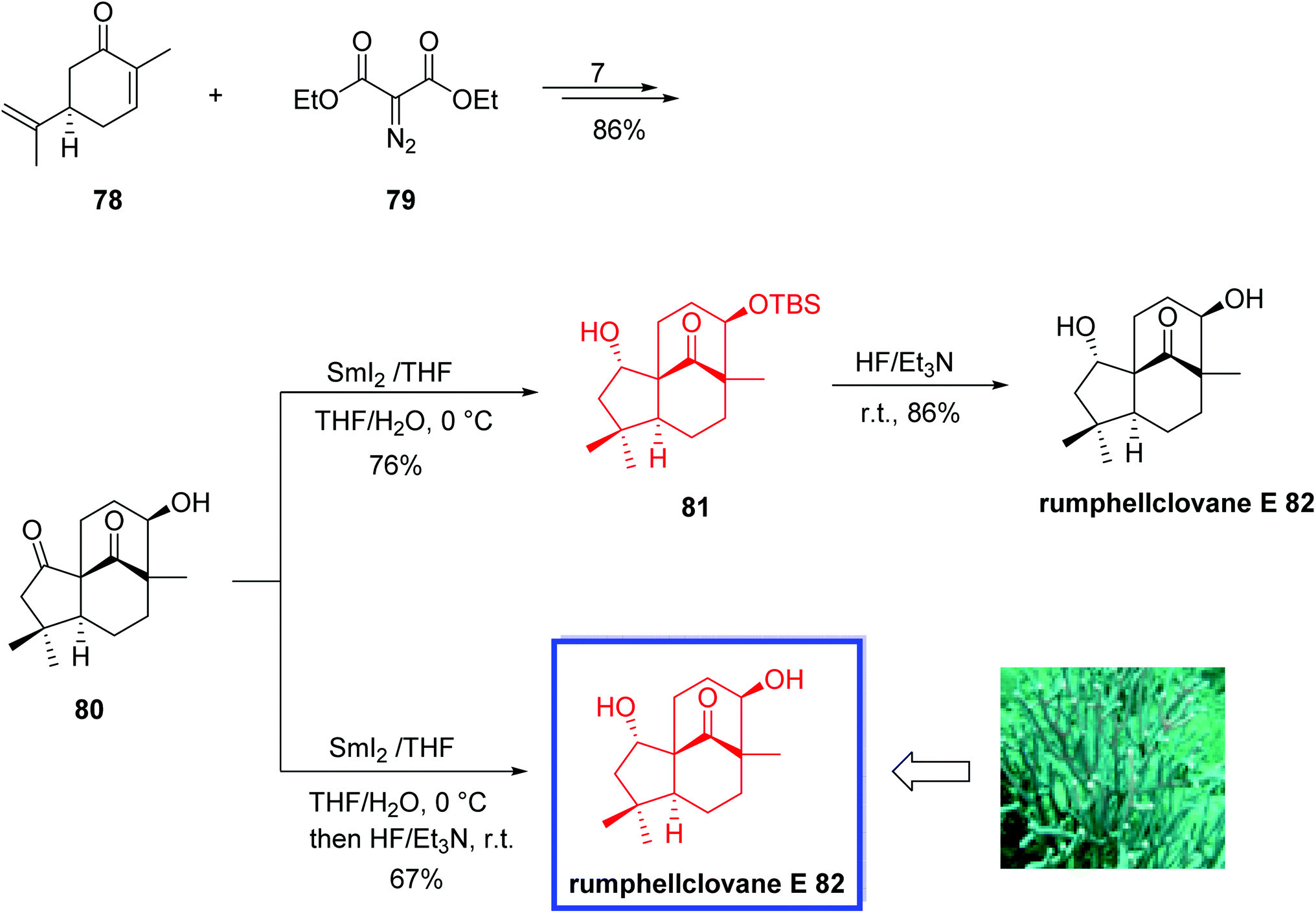
In 2021, Liu and coworkers56 reported the first asymmetric total synthesis of rumphellclovane E 82, in eight steps from commercially available (R)-carvone 78 via a B–AB–ABC sequence. Key steps included a SmI2-mediated chemo- and diastereoselective reduction of the cyclopentanone to install the desired stereocenter at C2, an iron-catalyzed intramolecular reductive aldol reaction to construct ring C, and a Rh-catalyzed cyclopropanation to construct ring A. The structure of rumphellclovane E was confirmed by X-ray crystallographic analysis. 1H and 13C NMR data of synthetic rumphellclovane E 82 were in good agreement with those of the natural product. However, the optical rotation of the synthetic 82 was different from that of natural product probably because of the insufficient quantity attained from nature. According to the authors, the developed total synthesis will be a general strategy to synthesize the clovane-type sesquiterpenoids.
The total synthesis of rumphellclovane E 82 began with the reaction of (R)-carvone 78 and the diazo compound 79 to afford ketone 80 in 7 steps and 86% yield. Treatment of 80 with SmI2 in THF/H2O (10:1) at 0 °C provided the reduction product 81 in 76% yield. The slow addition of diluted SmI2 solution (0.1 mol L−1 in THF) in this step was especially important for sufficient yield. Finally, TBS deprotection of the hydroxyl at C9 in 81 using HF·Et3N (10:1, v/v) in THF at room temperature delivered rumphellclovane E 82 in 86% yield. Rumphellclovane E 82 was also synthesized in 100 mg-scale in one pot from 80 in 67% yield (Scheme 21).
 | ||
| Scheme 21 Total synthesis of rumphellclovanes E 82. | ||
2.2. SmI2-mediated cyclization reactions
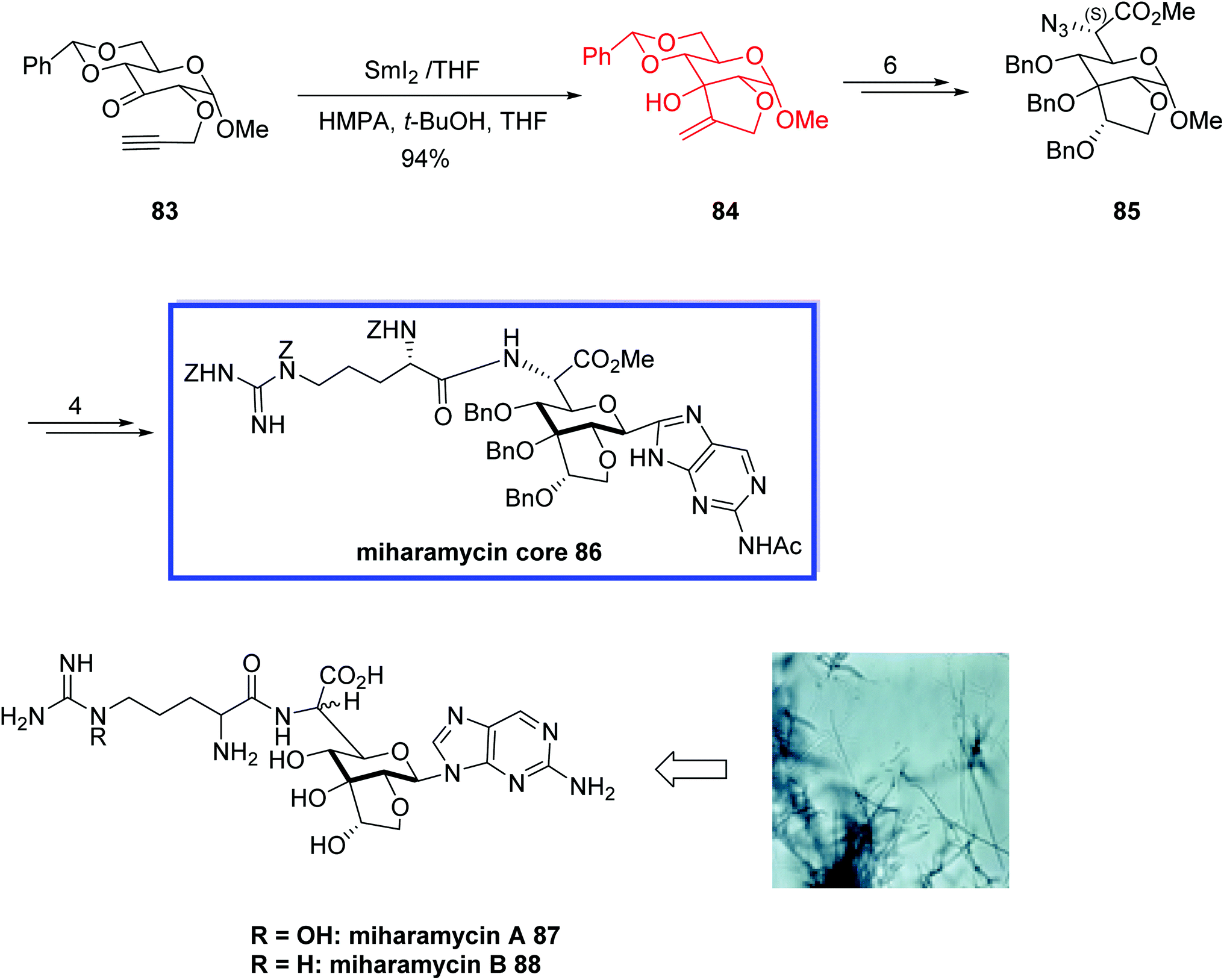
Miharamycins A 87 and B 88, isolated from Streptomyces miharaensis SF-489, are novel 2-aminopurinyl peptidyl nucleoside antibiotics (PNAs) with complex nine-carbon core saccharides and N5-hydroxyarginine. They show significant inhibition effects for the rice blast disease caused by Pyricularia oryzae.57In 2008, Blériot and colleagues58 reported the relative configuration at C-6′ of miharamycin A 87 and the structure of miharamycin A 87 was confirmed. They also attempted to develop the first total synthesis of miharamycin B 88. They synthesized the remarkable core structure of miharamycin B 88, though efforts to deprotect the core structure 86 failed to provide the miharamycin B 88. The key steps contained a SmI2-mediated keto–alkyne coupling to construct the bicyclic sugar moiety and the regio- and stereoselective N-glycosylation to form the nucleosidic part of miharamycin B 88. According to the researchers, the reported synthetic route could be applied to synthesize structurally similar nucleoside natural products, like amipurimycin. First, the relative configuration at C-6′ of miharamycin A 87 was determined. Since both antibiotics have been isolated from the same microorganism and most of the peptidyl nucleoside family of antibiotics retain S-configured natural amino acids, they proposed the S- configuration at C-6′ of both miharamycins A 87 and B 88.
The total synthesis of miharamycins B 88 began with the SmI2-mediated 5-exo-dig ketyl–alkyne cyclization of α-propargyloxy ketone 83 in the presence of hexamethylphosphoramide (HMPA) and tBuOH at room temperature to give the desired exoalkene 84 in 94% yield. The desired C-6 (S)-azido ester 85 was then synthesized from 84 in 6 steps and 82% yield. Compound 85 was then converted into N-benzyloxycarbonyl-protected miharamycin B core 86 in 4 steps and 70% yield (Scheme 22).
 | ||
| Scheme 22 Total synthesis of miharamycin B core 86. | ||
(−)-GB 13 9659 and (+)-GB 16 97,60 isolated from the bark of Galbulimima belgraveana, belong to the gabulimima alkaloid natural products. GB alkaloids have held the attention of the organic synthesis community due to the extraordinary structures of these alkaloids, but also in the pharmaceutical industry, particularly because the Galbulimima belgraveana bark has been used as a medicine and some of the members of these alkaloids show potent muscarinic or thrombin receptor antagonist activities.61 Vorapaxar (SCH 530348), based on the natural product himbacine, is a thrombin receptor antagonist, and is used for preventing heart attacks in patients with acute coronary syndrome.
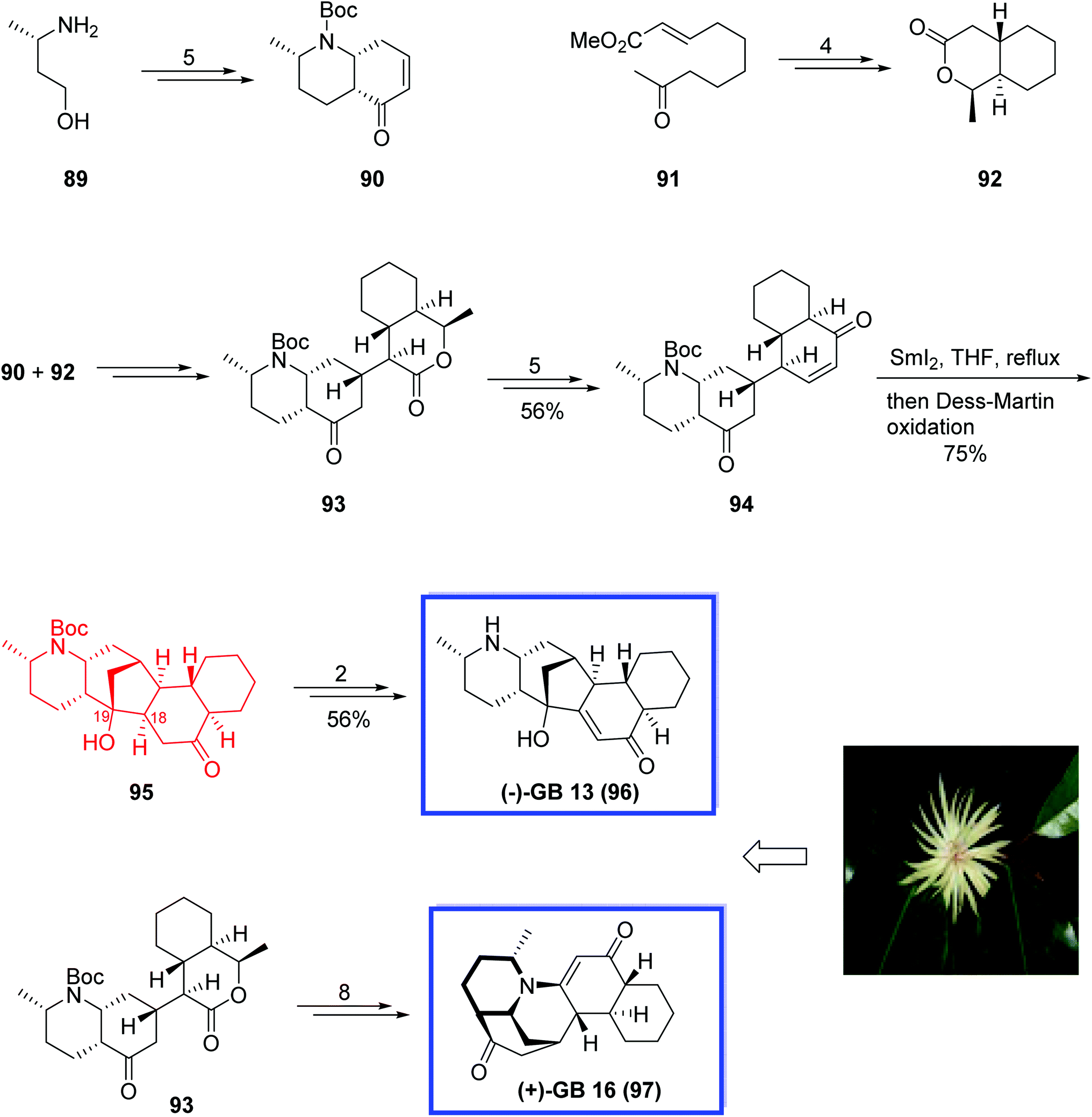
In 2010, Ma and coworkers62 developed a convergent strategy for the asymmetric total synthesis of the alkaloid (−)-GB 13 96 in 19 linear steps and an overall yield of 6.1% from the commercially available starting material. The key steps contain a Mukaiyama–Michael addition for connecting two bicyclic fractions and a SmI2-mediated carbonyl–alkene reaction to construct the C ring. They also developed the first total synthesis of (+)-GB 16 97 using an advanced intermediate from the (−)-GB 13 96 total synthesis as a starting material. The analytical data of synthetic 96 and 97 were in good agreement with those reported for the natural products. X-ray crystal structure analysis of (+)-GB 16 97 was also reported.
Total synthesis of the alkaloid (−)-GB 13 96 began with the preparation of the two needed bicyclic fractions 90 and 92 for the Mukaiyama–Michael addition. The desired enone 90 was prepared using (S)-3-aminobutan-1-ol 89 in 5 steps and 79% yield. In a parallel procedure, the lactone 92 was prepared from olefin 91 in 4 steps and 89% yield. The addition of lactone 92 onto the enone 90 provided ketone 93 in 80% yield. Further reaction of 93 generated the desired enone 94 in 5 steps and 74% yield. SmI2-mediated ketyl–alkene reductive coupling of enone 94 by slowly adding the enone 94 to a solution of SmI2 in THF at reflux without any additives, followed by the oxidation with Dess–Martin periodinane constructed C ring and resulted in the desired reductive coupling product 95 in 75% yield. Finally, (−)-GB 13 96, was synthesized from 95 in 2 steps and 71% yield. (+)-GB 16 97 was synthesized from lactone 93 in 8 steps and 87% yield (Scheme 23).
 | ||
| Scheme 23 Total synthesis of (−)-GB 13 (96) and (+)-GB 16 (97). | ||
Sieboldine A 105, isolated from club moss Lycopodium sieboldii, and alopecuridine, isolated from L. alopecuroides (foxtail club moss), are two members of the fawcettimine-type Lycopodium alkaloids with unusual polycyclic structures and a broad range of biological activities.63 The Lycopodium alkaloids are a growing family of structurally various and complex natural products. To date, more than 250 Lycopodium alkaloids have been isolated and characterized. Due to their exciting molecular structures and significant biological activities, the Lycopodium alkaloids are challenging targets for their total synthesis and have attracted broad attention from biogenetic and biological points of view.
Sieboldine A 105 is a significant acetylcholinesterase (AChE) inhibitor and shows significant cytotoxic activity against murine lymphoma L1210 cells.63,64 Both alkaloids comprise a unique tetracyclic skeleton and two contiguous quaternary stereocenters, additionally, sieboldine A 105 possesses an N-hydroxyazacyclononane ring bridged to a tetrahydrofuran ring.
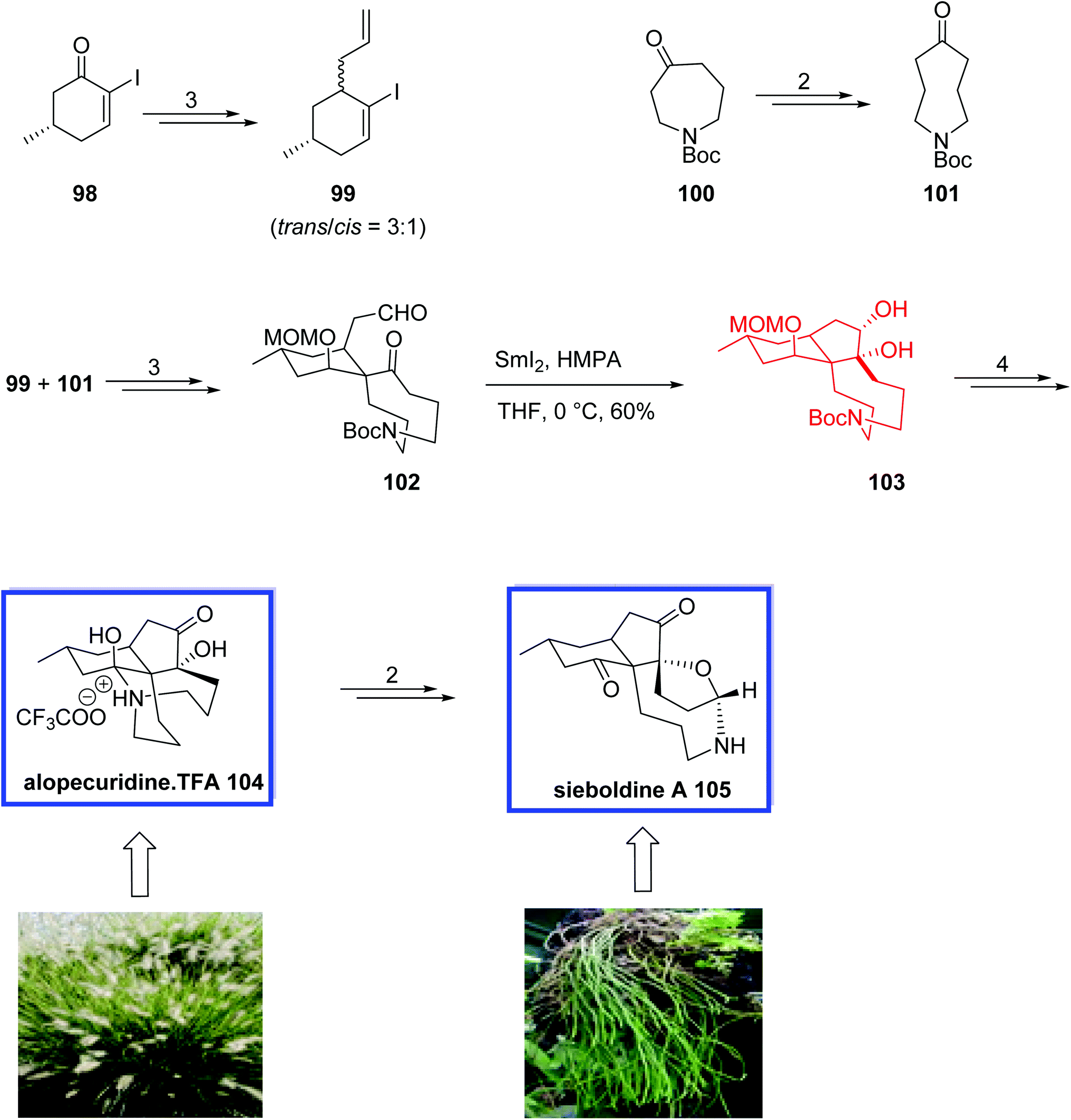
In 2011, Tu and coworkers65 reported the first total synthesis of (±)-alopecuridine·TFA 104 and a biomimetic synthesis of (±)-sieboldine A 105 through the known iodide 98 in 13 and 15 steps, respectively. Key steps contain a semipinacol rearrangement of a medium-sized ring and a SmI2-mediated intramolecular pinacol coupling. The spectroscopic data for synthetic alopecuridine·TFA 104 and sieboldine A 105 were identical to those reported for the natural product.
The total synthesis of (±)-alopecuridine·TFA 104 and its biomimetic conversion into (±)-sieboldine A 105 began with the preparation of two coupling fragments 99 and 101. Iodoalkene 99 was prepared from known iodide 98 in 3 steps and 75% yield. Ketone 101 was prepared from commercially available azepine 100 in 2 steps and 75% yield. The two fragments, 99 and 101, were then coupled to give aldehyde 102 in 3 steps and 86% overall yield. The key tricyclic core and two contiguous quaternary carbon atoms of alopecuridine·TFA 104 were constructed through the SmI2-mediated stereoselective intramolecular pinacol coupling of aldehyde 102 in THF at 0 °C and resulted in cis-diol 103. Finally, alopecuridinium trifluoroacetate 104 was synthesized from diol 103 in 4 steps and 96% yield. The biomimetic transformation of (±)-alopecuridine·TFA 104 to sieboldine A 105 occurred over two steps in 60% yield (Scheme 24).
 | ||
| Scheme 24 The total synthesis of (±)-alopecuridine·TFA 104 and (±)-sieboldine A 105. | ||
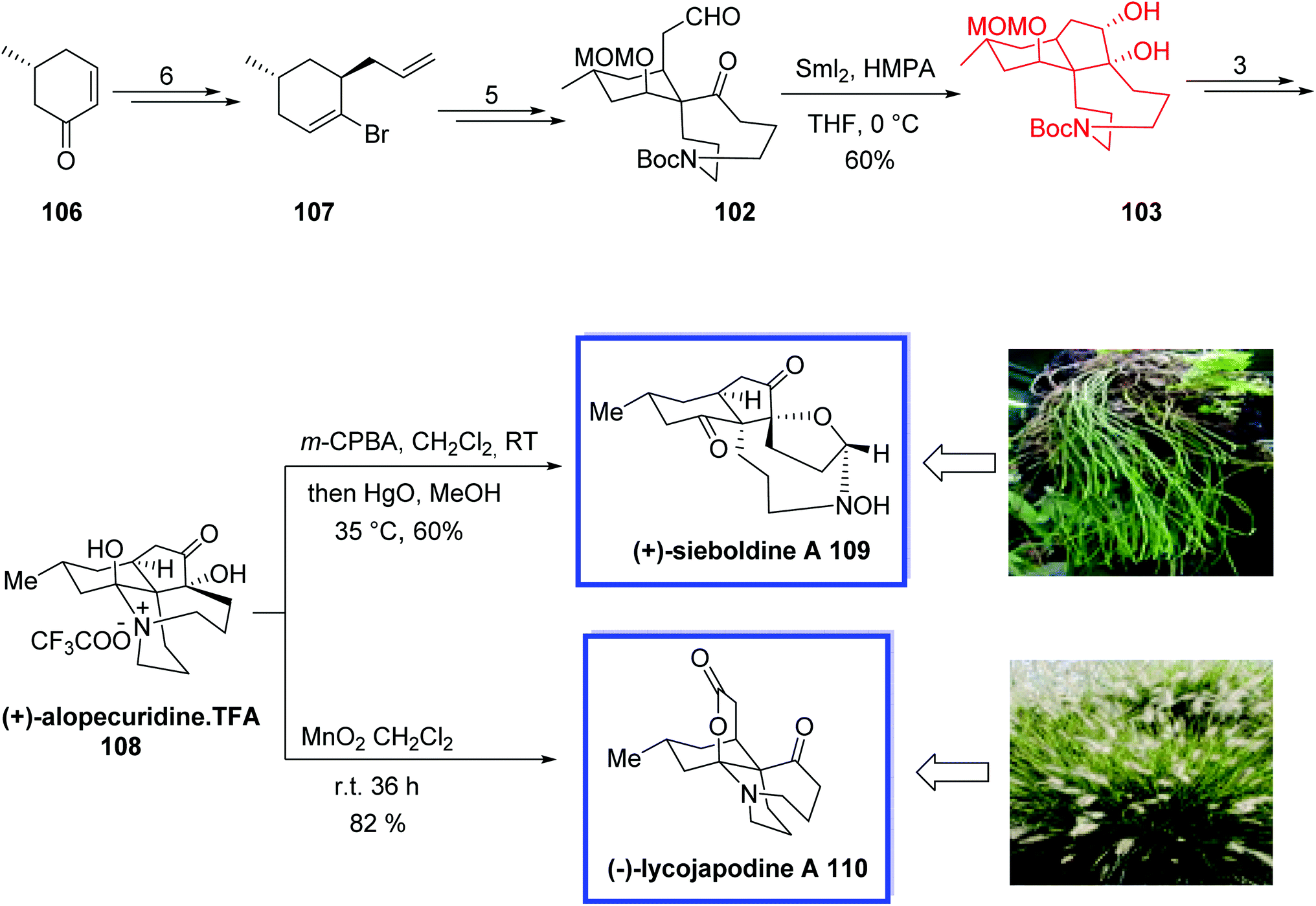
In 2012, Wang and coworkers66 developed an efficient strategy for the asymmetric total synthesis of (+)-alopecuridine 108, (+)-sieboldine A 109, and (−)-lycojapodine A 110,67 in 15, 16, and 16 steps, respectively, starting from chiral enone 106. All three alkaloids are structurally similar fawcettimine-type alkaloids and possess a unique tetracyclic skeleton and two contiguous quaternary stereocenters. (−)-Lycojapodine A 110, isolated from the club moss Lycopodium japonicum, retains anti-HIV-1 activity. Both (−)-lycojapodine A 110 and (+)-sieboldine A 109, are also acetylcholinesterase (AChE) inhibitors.
The key steps for the total synthesis of (+)-alopecuridine·TFA 108 contain the construction of the spiro 6,9-azacarbocycles with an all-carbon quaternary center via a semipinacol rearrangement of a hydroxyl epoxide, and the construction of the 5-membered ring through a late-stage SmI2-induced intramolecular pinacol coupling reaction. The biogenetic relationship of these three alkaloids was also confirmed through the biomimetic synthesis of (+)-sieboldine A 109 and (−)-lycojapodine A 110. An N-oxidation/nitrone formation reaction of (+)-alopecuridine·TFA 108 resulted in the synthesis of (+)-sieboldine A 109. The biogenetic synthesis of (−)-lycojapodine A 110 from (+)-alopecuridine·TFA 108 through diol formation/diol cleavage strategy was developed for the first time. The spectral data of the synthetic products were in good agreement with the reported values and their rotations were identical to those of the natural products and confirmed the absolute configurations.
The asymmetric total synthesis of (+)-alopecuridine·TFA 108 and sieboldine A 109 began with the known enone 106 to provide chiral bromoalkene 107 in 6 steps, from which aldehyde 102 was prepared in 5 steps and 86% yield. SmI2-induced intramolecular pinacol coupling of aldehyde 102 provided tricyclic compound 103. Finally, (+)-alopecuridine·TFA 108 was obtained from 103 in 3 steps and 96% yield. The one-pot synthesis of (+)-sieboldine A 109 occurred through the biomimetic oxidation of (+)-alopecuridine·TFA 108 using m-CPBA in CH2Cl2 followed by addition of HgO in MeOH to generate 109 in 60% yield. Lacojapodine A 110 was synthesized in 82% yield through the biomimetic oxidation of (+)-alopecuridine·TFA 108 using MnO2 in CH2Cl2 at room temperature (Scheme 25).
 | ||
| Scheme 25 The total synthesis of (+)-alopecuridine·TFA 108, (+)-sieboldine A 109 and lacojapodine A 110. | ||
The fawcettimine-type Lycopodium alkaloids, containing alopecuridine 108,63a,b sieboldine A 109,64d and lycojapodine A 110,66 (+)-fawcettidine 119 and (+)-fawcettimine 12067 have continued to be of great interest to synthetic chemists. The serratinine-type Lycopodium alkaloids, such as (−)-8-deoxyserratinine 121,67d exemplify another important class of Lycopodium alkaloids possessing a 6/5/6/5 tetracyclic framework with two contiguous quaternary stereogenic centers. They are structurally comparable to the fawcettimine-type alkaloids. The fawcettimine-type alkaloids are different from other classes of Lycopodium alkaloids because of the extensive ring–chain tautomerism, which is important to their biogenesis.
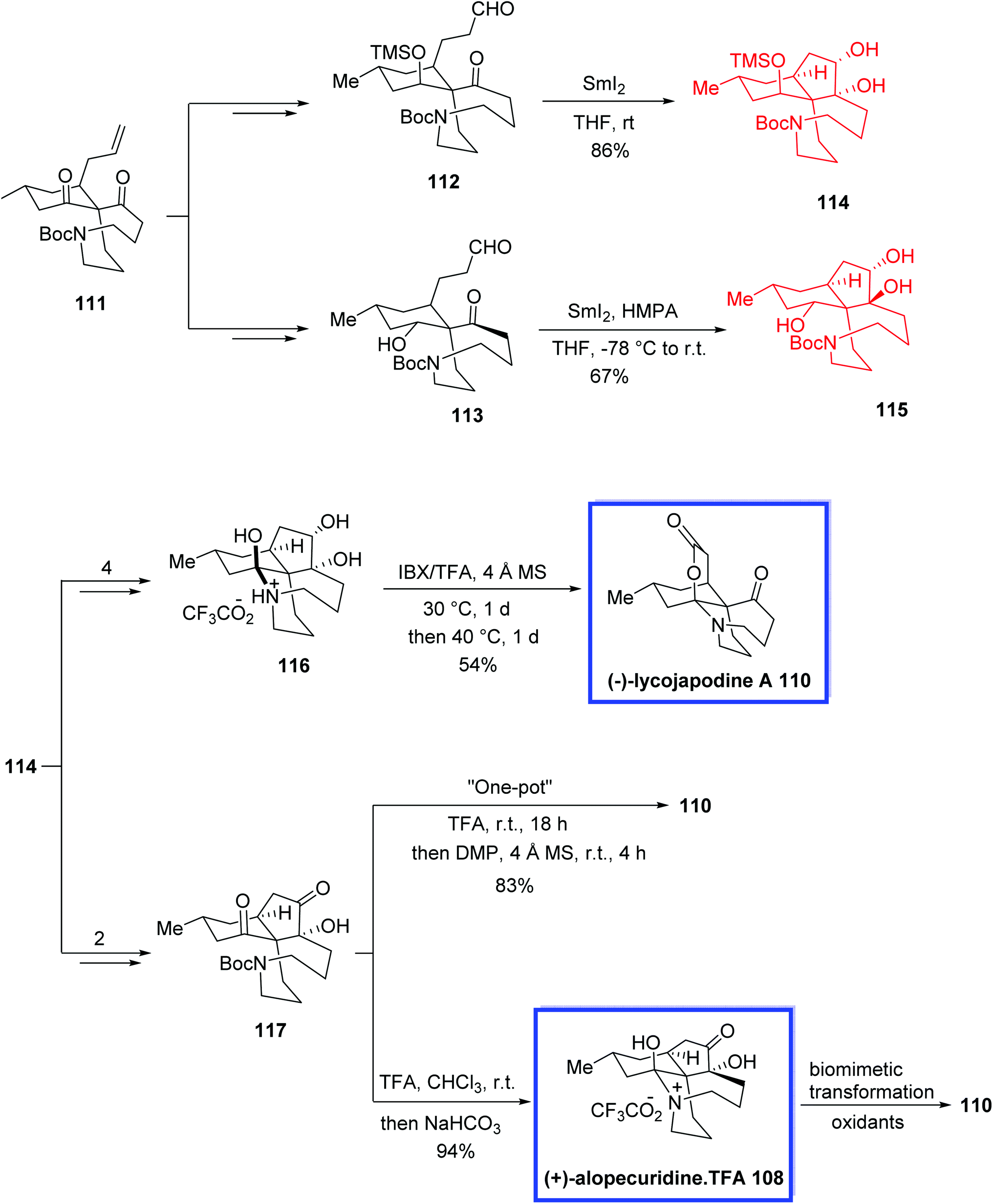
In 2013, Lei and colleagues68 reported the total synthesis of both fawcettimine- and serratinine-type Lycopodium alkaloids (+)-alopecuridine 108, (−)-lycojapodine A 110, (+)-fawcettidine 119, (+)-fawcettimine 120 and (−)-8-deoxyserratinine 121, from a known precursor 111 in either 12 or 13 steps employing a very compact pathway stimulated by the suggested biosynthesis of the fawcettimine- and serratinine-type alkaloids. The key steps contain an intramolecular C-alkylation to install the spiro quaternary carbon center and the aza-cyclononane ring, SmI2-mediated pinacol couplings to synthesize the tricyclic skeleton and also to confirm the relative stereochemistry of the oxa-quaternary center, and a sequential transannular N-alkylation to achieve the desired tetracyclic core. The total synthesis of (−)-lycojapodine A 110 was achieved either via a late-stage IBX/TFA-mediated tandem oxidation reaction or through a one-pot biogenetic pathway from (+)-alopecurdine 108. The spectroscopic data of the synthetic 119, 120, and 121 were in good agreement with those already reported.
The total synthesis of 108, 110 and 119-121 started from the common intermediate 111, to obtain aldehydes 112 and 113. Pinacol coupling of 112 using SmI2 in THF at room temperature resulted in diol 114 as the sole product in 86% yield. A pinacol coupling reaction of aldehyde 113 using SmI2 in THF in the presence of HMPA at −78 °C to room temperature resulted in the 4,5-trans diol 115 as the sole product in 67% yield. (−)-Lycojapodine A 110 was synthesized via a late-stage IBX/TFA-mediated tandem oxidation reaction of tautomer-locked diol 116, which was synthesized from diol 114 in 3 steps and 54% yield. (−)-Lycojapodine A 110 was also synthesized via a one-pot process using α-ketol 117, which was obtained from diol 114 in 2 steps and 50% yield. Removal of the Boc group from α-ketol 117 with TFA and neutralization with NaHCO3 afforded (+)-alopecuridine·TFA 108 in 94% yield. Additionally, the successive biomimetic transformation of (+)-alopecuridine·TFA 108 also resulted in (−)-lycojapodine A 110. The absolute configuration of 108 was also confirmed (Scheme 26).
 | ||
| Scheme 26 Total synthesis of (+)-alopecuridine 108, (−)-lycojapodine A 110. | ||
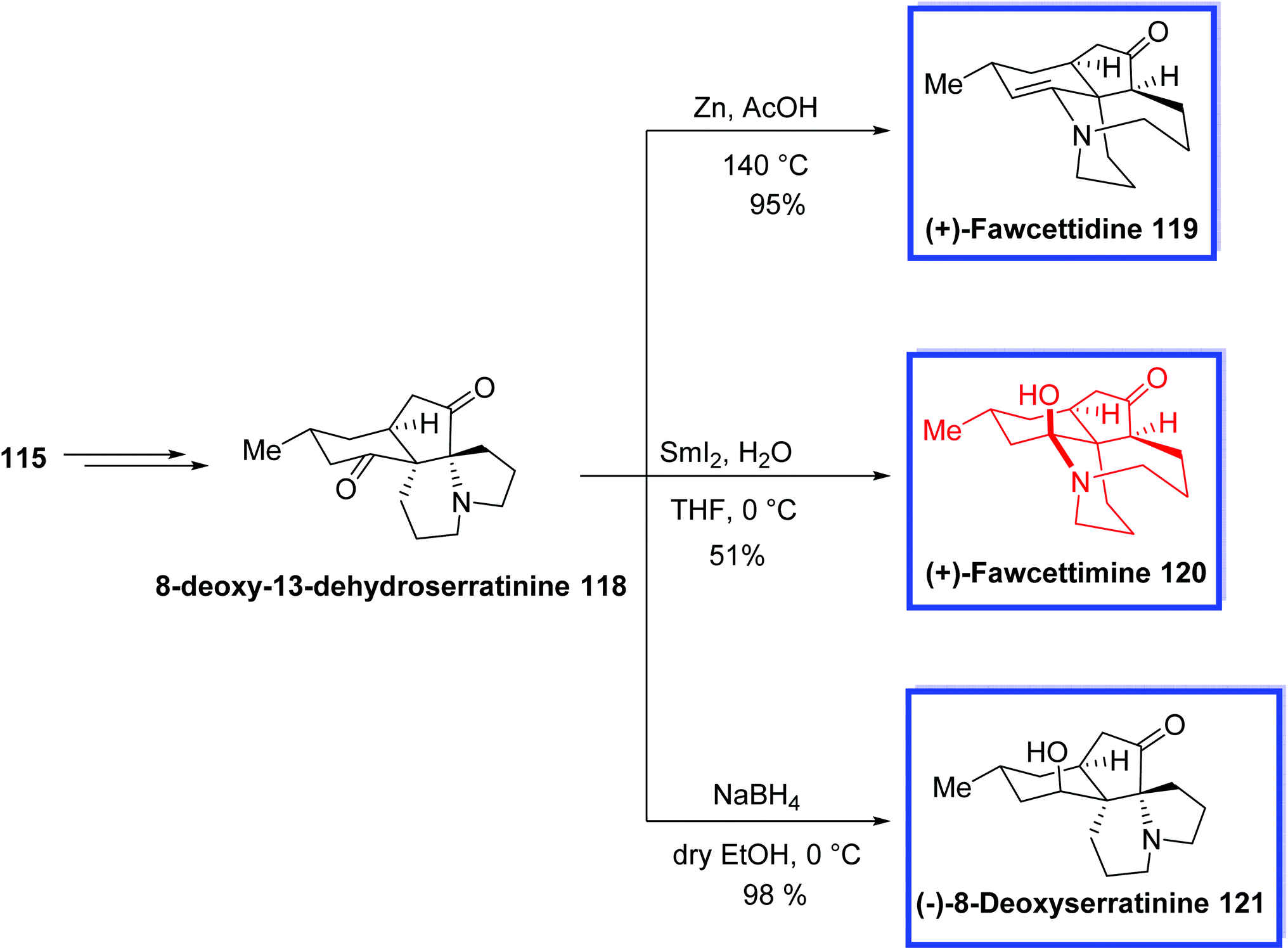
The total syntheses of (+)-fawcettimine 119, (+)-fawcettidine 120, and (−)-8-deoxyserratinine 121 were achieved using 8-deoxy-13-dehydroserratinine 118, which retained the key aza-quaternary carbon center at C4 and was prepared from diol 115. Treatment of 118 with NaBH4 at 0 °C afforded (−)-8-deoxyserratinine 121 in 98% yield. The reductive reaction of 118 with Zn/AcOH at 140 °C resulted in (+)-fawcettidine 119 in 95%. Finally, (+)-fawcettimine 120 was attained by treatment of 118 with SmI2 in THF/H2O at 0 °C in 51% yield (Scheme 27).
 | ||
| Scheme 27 Total synthesis of (+)-fawcettidine 119, (+)-fawcettimine 120 and (−)-8-deoxyserratinine 121. | ||
(−)-Actinophyllic acid 129, isolated from the leaves of the tree Alstonia actinophylla,69 is a biologically active indole alkaloid with five contiguous stereocenters and a potent inhibitor of the zinc-dependent carboxypeptidase U (CPU). Inhibitors of CPU promote fibrinolysis and inhibit blood clot formation which is a cause of several cardiovascular disorders.70,71
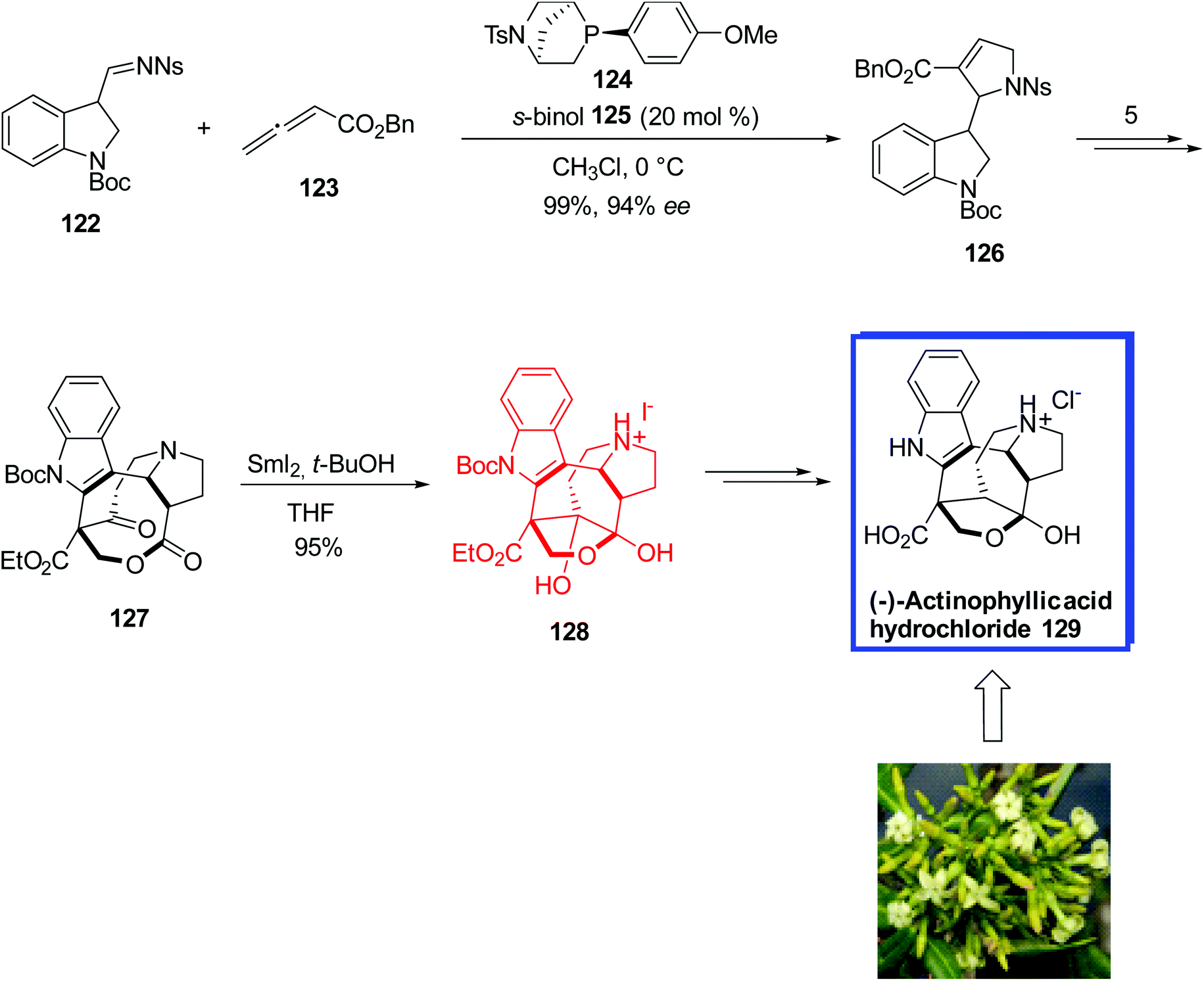
In 2016, Kwon and coworkers72 reported the first enantioselective total synthesis of (−)-actinophyllic acid 129 in 13 steps from a known aldehyde in 12.4% yield. They used the same starting materials for the synthesis of (−)-actinophyllic acid 129 as the racemic synthesis of actinophyllic acid. The key steps were a chiral phosphine-catalyzed [3 + 2] annulation of an allenoate and an indole imine to prepare a proline intermediate, a CuI-catalyzed coupling to form the tetrahydroazocine ring, an intramolecular alkylative lactonization, a SmI2-mediated intramolecular pinacol coupling between a ketone and a lactone to construct the caged framework of (−)-actinophyllic acid 129, and a remarkable regioselective dehydroxylation. The spectral data of the synthesized structures were in complete agreement with those reported in the literature.
The asymmetric total synthesis of (−)-actinophyllic acid 129 began with the [3 + 2] annulation between N-nosylimine 122 and benzyl allenoate 123 using phosphine B 124 and 20 mol% s-BINOL 125 and as the additive gave the desired (S)-enantiomer 126 with the enantioselectivity of 94% ee in 99% yield. With the desired (S)-enantiomer 126 in hand, the lactone 127 was synthesized in 35−48% yield in 5 steps. To synthesize the desired coupling product 128 from the lactone 127 via a pinacol reaction, Kwon and colleagues evaluated several single-electron-transfer reagents, comprising Ti3+, Li, Na, and Sm2+. Only the reaction of lactone 127 with SmI2 and 10 equiv. of t-BuOH in THF furnished the desired hemiketal 128 in quantitative yield, from which (−)-actinophyllic acid hydrochloride 129 was synthesized over 2 steps in 90% yield (Scheme 28).
 | ||
| Scheme 28 The total synthesis of (−)-actinophyllic acid 129. | ||
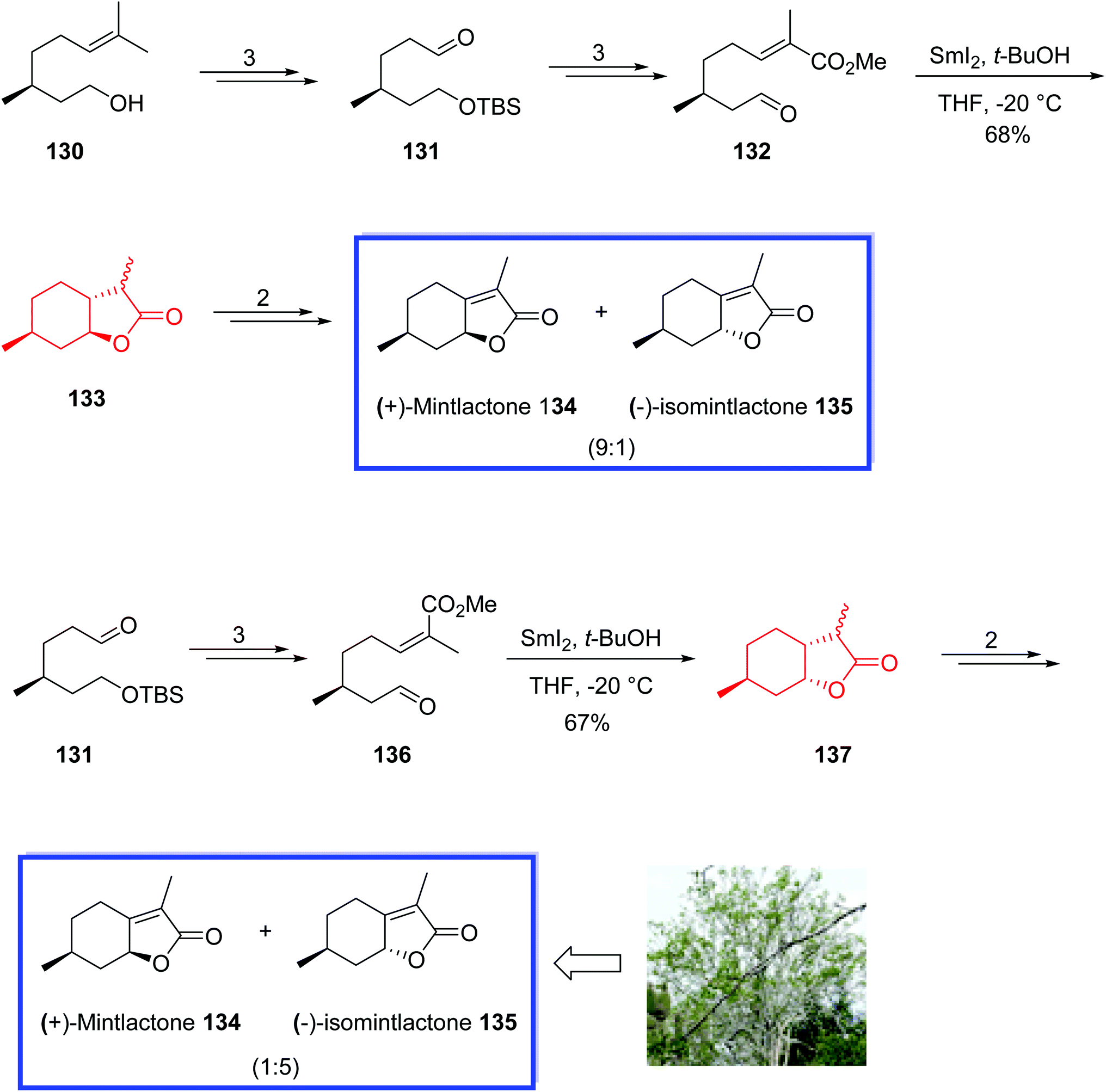
(+)-Mintlactone 134 and (−)-isomintlactone 135, isolated from Bursera graveolens, are bicyclic monoterpene natural products with two stereocenters.73 These compounds typically have a sweet aroma and are used as food additives, tobacco additives, and intermediates for the synthesis of dihydrogen mint and menthofuran.73
In 2017, Wang and coworkers74 introduced the divergent procedure for the total synthesis of (+)-mintlactone 134 and (−)-isomintlactone 135 through a SmI2-mediated intramolecular radical cyclization to construct two rings and a stereocenter in one step.
The stereochemistry of the final natural products was controlled by the stereocenter of (−)-citronellol 130 and facilitated by the coordination of the oxygen atoms of the aldehyde and ester with the samarium atom in the transition state. This strategy would be used as a general route for the synthesis of complex natural products with a butenolide moiety. The total synthesis of (+)-mintlactone 134 and (−)-isomintlactone 135 began with (−)-citronellol 130 to obtain aldehyde 131 in 3 steps, from which α,β-unsaturated ester 132 was prepared in 3 steps. SmI2-induced radical cyclization of the key substrate 132 with SmI2 in anhydrous THF and t-BuOH at −20 °C under an argon atmosphere provided bicyclic product 133 in 68% yield, from which a mixture of 134 and 135 (9:1) was synthesized in 2 steps and 68% yield.
(−)-Isomintlactone 135 was prepared using aldehyde 131, which was converted to Z-α,β-unsaturated ester 136 in 3 steps. Treatment of compound 136 with SmI2 in anhydrous THF and t-BuOH at −20 °C under an argon atmosphere provided lactone 137 with an opposite chirality on C8 as expected. With the key substrate 137 in hand, a mixture of 134 and 135 (1:5) was synthesized in 2 steps and 51% yield (Scheme 29).
 | ||
| Scheme 29 The total synthesis of (+)-mintlactone 134 and (−)-isomintlactone 135. | ||
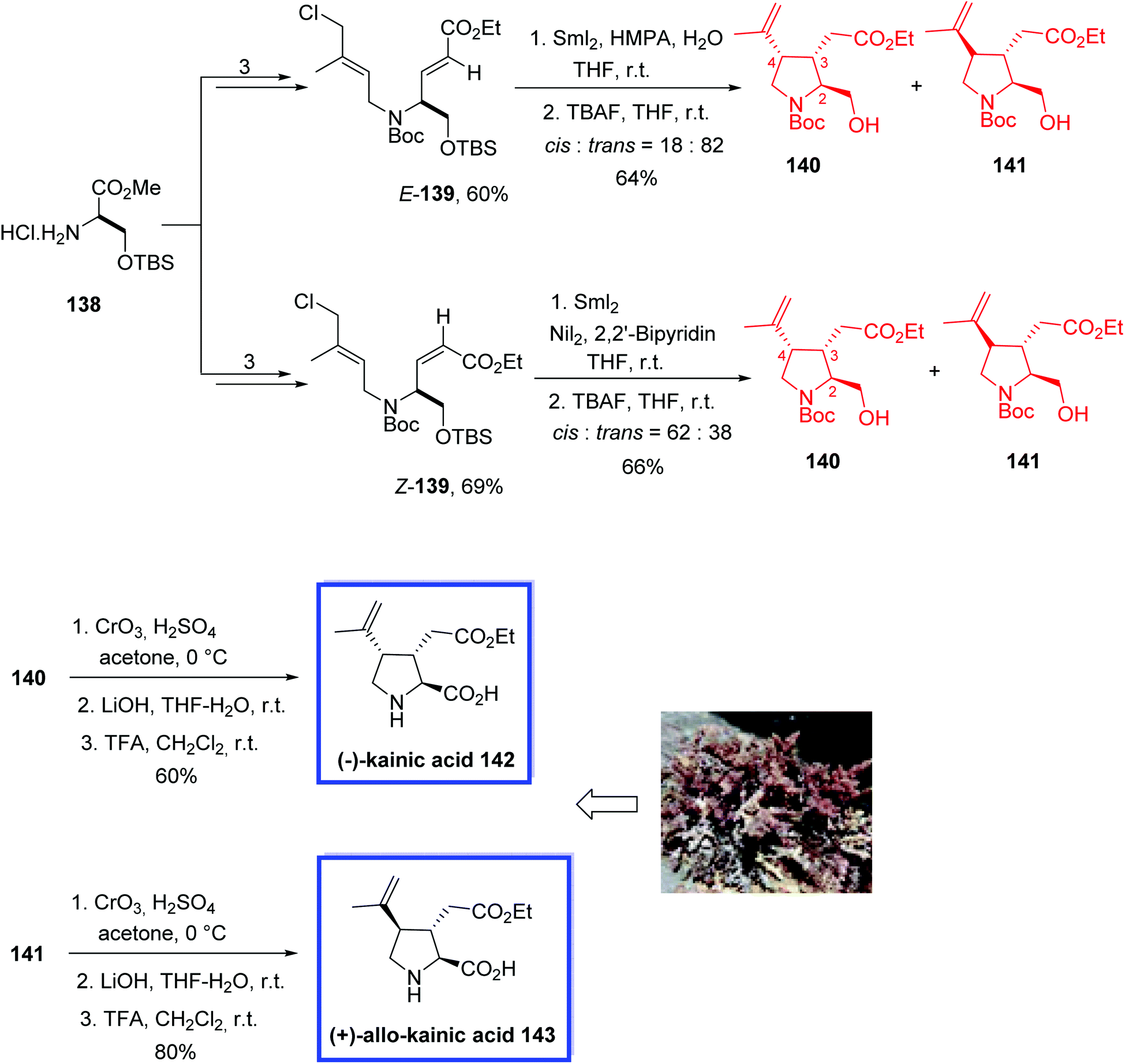
(−)-Kainic acid 142 and its C4-epimer, (+)-allo-kainic acid 143, are pyrrolidine alkaloids with three contiguous stereocenters, and were isolated initially from the Japanese marine alga Digenea simplex (Japanese name, kainin-sou).75 (−)-Kainic acid 142 promoted a strong activation of neurons and was evaluated as a KA-type ionotropic receptor. Therefore, it was used in neuroscience and neuropharmacology as an effective molecular probe.76
In 2017, Matsuda and coworkers77 developed the total syntheses of (−)-kainic acid 142 and (+)-allo-kainic acid 143 in 12 steps from a commercially available starting material. The key step was a SmI2-mediated reductive 3,4-disubstituted pyrrolidine cyclization. They found that the preference of the 3,4-cis- or 3,4-trans-selective cyclization can be achieved by changing the additives from NiI2 to HMPA during the SmI2-promoted pyrrolidine cyclization. Spectroscopic data (1H and 13C NMR, and specific rotation) of synthesized 142 and 143 were in good agreement with those reported from previous total syntheses.
The total syntheses of (−)-kainic acid 142 and (+)-allo-kainic acid 143 began with the amine hydrochloride 138 to obtain E-139 and Z-139 in 3 steps in 60% and 69% yield, respectively. The SmI2-mediated cyclization of E-139 or Z-139 and removal of the TBS group with TBAF resulted in a mixture of only two of the four possible diastereomeric pyrrolidines, 140 and 141. Using HMPA and H2O, 3,4-trans-pyrrolidine 141 was the major diastereomer (cis:trans = 18:82) for the cyclization of E-139. Then 3,4-cis-pyrrolidine 140 was the major diastereomer (cis:trans = 62:38) for the cyclization of Z-139 in the presence of NiI2 and 2,2′-bipyridine. Finally, Jones oxidation of alcohol 140 or 141, hydrolysis of the ester with LiOH in THF-H2O, and removal of the Boc group with TFA provided (−)-kainic acid 142 or (+)-allo-kainic acid 143 in 60% or 80% yield (3 steps), respectively (Scheme 30).
 | ||
| Scheme 30 The total synthesis of (−)-kainic acid 142 and (+)-allo-kainic acid 143. | ||
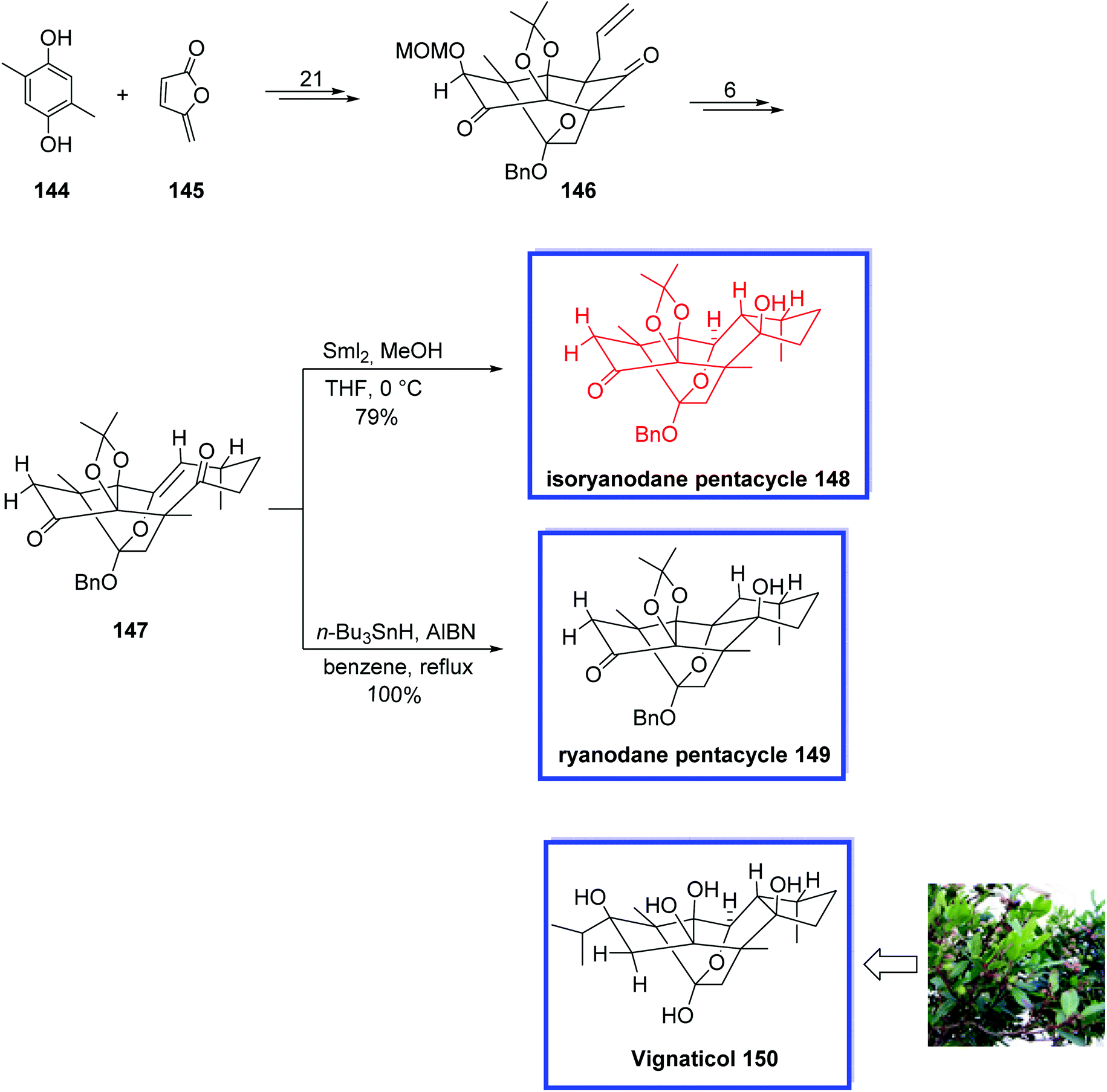
Isoryanodane diterpenoids contain a rare diterpenoid skeleton. They have been proven to show anti-complementary and antifeedant activities.78 Vignaticol 150, isolated from Persea indica (Lauraceae), is an isoryanodane diterpenoid and contains a densely oxygenated and complex fused pentacycle (ABCDE-ring) with 10 contiguous stereogenic centers that are structurally different in the substitution structures of their hydroxyl groups. These compounds demonstrate several important biological activities such as anti-COX-2, anti-complement, and immunosuppressive.79 In 2018, Inoue and coworkers80 reported a new procedure for establishing the isoryanodane ABCDE-ring structure of 148 for the first time. They synthesized the 6/5/7-membered isoryanodane BCD-ring system of 148 from the 5/6-membered ryanodane BD-ring system of 146 in 7 steps.
Compound 148 differs from vignaticol 150 only at the C2,3-positions and has 9 of the 10 contiguous stereogenic centers of vignaticol 150, and thus, it may likely lend itself to be a useful scaffold for the total synthesis of vignaticol 150 and other densely oxygenated and complex fused terpenoids. The key steps were the installation of the C8-vinyl group at the C6-position, an oxy-Cope rearrangement, and SmI2-mediated transannular carbonyl-ene reaction establishing the C6-, C10-, and C11-stereogenic centers. The DFT calculation confirmed that the SmI2 reaction pertained to an intramolecular carbonyl-ene reaction and indicated that the samarium reagent functioned as both a Lewis acid and reluctantly proceeded through a regio- and stereoselective cyclization without incurring any cation-caused rearrangement or E1cB elimination. The stereochemistry of 147 and its C9-(R)-stereocenter was determined by X-ray crystallographic analysis.
The total synthesis of isoryanodane pentacycle 148 began with the reaction of 2,5-dimethylbenzene-1,4-diol 144 and maleic anhydride 145 to give the common enantiopure ryanodane tetracycle 146 within 21 steps. The 9-membered ring of 147 was then synthesized from 146 in 6 steps and 60% yield. Transannular combination of the oxy-Cope rearrangement and transannular carbonyl-ene reaction of 147 using SmI2 in the presence of MeOH gave isoryanodane pentacycle 148 in 79% yield. Alternatively, the transannular cyclization of compound 147 in the presence of n-Bu3SnH provided ryanodane pentacycle 149 in 100% yield (Scheme 31).
 | ||
| Scheme 31 The total synthesis of isoryanodane pentacycle 148. | ||
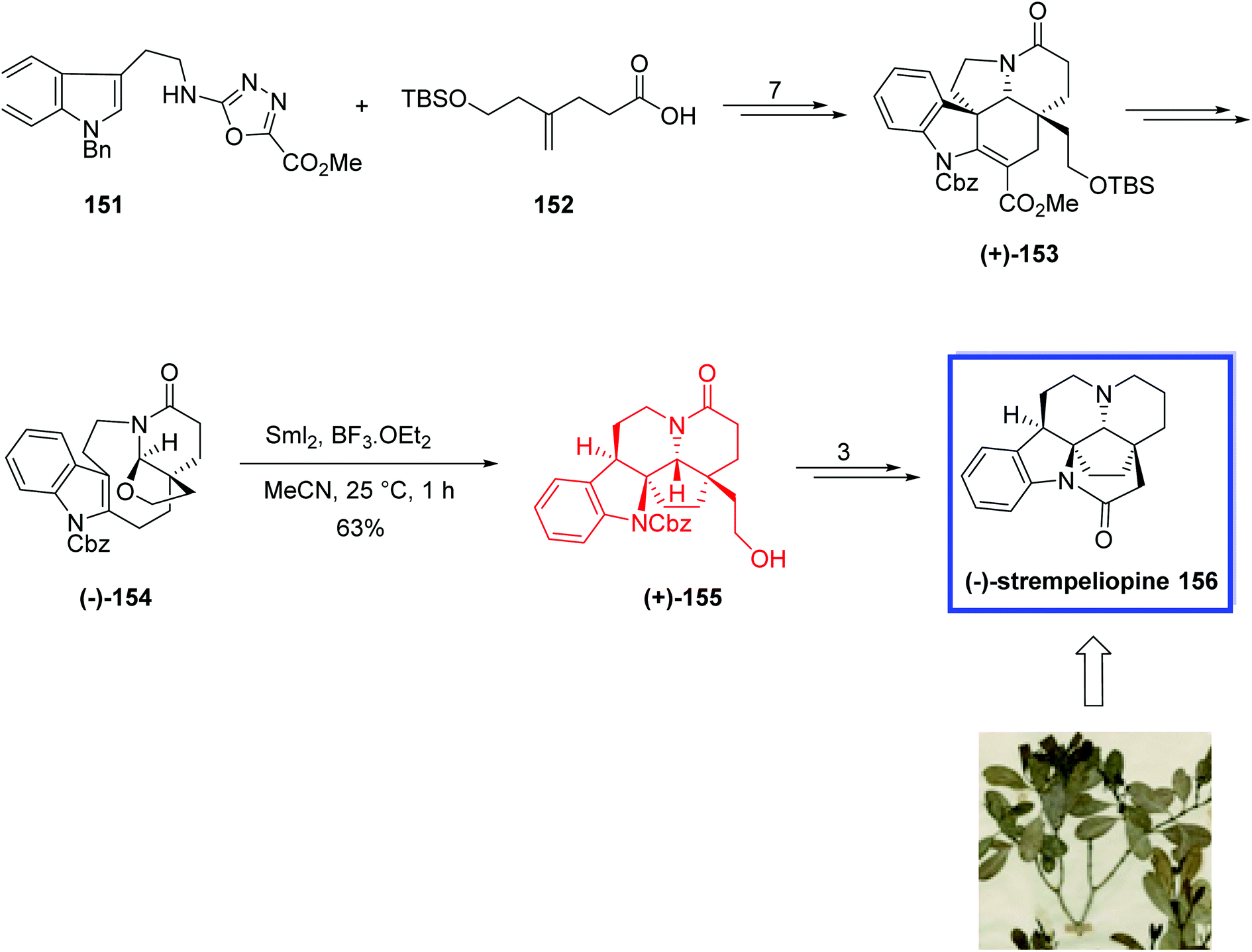
(−)-Strempeliopine 156, isolated from the roots of Strempeliopsis strempelioides K. Scrum, is the parent base of the schizozygane alkaloids. The schizozygane alkaloids are biogenetically presumed to be correlated to the Aspidosperma alkaloids.81 The complex structure of (−)-strempeliopine 156, containing a highly fused hexacyclic core with an installed transannular ethane bridge, has attracted considerable attention of synthetic chemists.
In 2021, Boger and coworkers82 developed the total synthesis of (−)-strempeliopine 156 from the intermediate (+)-153. Key step was a combined Lewis acid/SmI2-mediated dearomative transannular radical cyclization onto an indole to construct the C19–C2 bond of (−)-strempeliopine 156. They selected CH3CN as solvent for the SmI2-mediated transannular radical cyclization to prevent the effective complexation of BF3·OEt2 with the N-acyl hemiaminal, and to reduce its power to facilitate the formation of the iminium ion intermediate.
Total synthesis of (−)-strempeliopine 156 started with the previously reported intermediate (+)-153, which was prepared by the reaction of N-1,3,4-oxadiazole 151 and 4-(2-tert-butyldimethylsilyloxy)pent-4-enoic acid 152 in 7 steps. (+)-153 was then converted into the carbamate (−)-154 in 73% yield. Treatment of 154 with BF3·OEt2 to produce the N-acyliminium ion and the following SmI2-mediated radical cyclization in CH3CN at 25 °C afforded (+)-155 regioselectively (8:1) and diastereoselectively (>30:1) in 63% yield. Finally, (−)-strempeliopine 156 was synthesized from (+)-155 in 3 steps and 48% yield (Scheme 32).
 | ||
| Scheme 32 Total synthesis of (−)-strempeliopine 156. | ||
Kopsia indole alkaloids,83 isolated from several Kopsia (Apocynaceae) species, exhibit various remarkable biological activities, such as cholinergic, anti-rheumatism, and anti-inflammation antimicrobial, anticancer, anti-inflammatory, and antitussive properties.84 Based on the scaffold of the kopsine family there are four subtypes: kopsine, isokopsine, chanofruticosine, and fruticosine. Kopsinine 160, isolated initially from Kopsia longiflora Merr.,85 belongs to the Aspidosperma alkaloids family of natural products and shows in vitro anticholinergic activity.84j
In 2013, Boger and coworkers86 reported the total synthesis of kopsinine 160. The key steps included the intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles to form the pentacyclic core and an extraordinary SmI2-induced transannular cyclization reaction to construct the hexacyclic ring system of kopsinine from the bicyclo[2.2.2]octane core, perhaps ensuing from a radical-mediated cyclization with subsequent kinetic protonation of the extra reduced conjugate addition ester enolate from the less overly involved convex face. This hexacyclic ring system can directly correlate to the structures of the related Aspidosperma alkaloids.
The total synthesis of kopsinine 160 began with the cycloaddition adduct 157 to provide the key intermediate methyldithiocarbonate 158 in 6 steps and 86% yield. Treatment of 158 with SmI2 in THF/HMPA (10:1) provided 159 in 75% yield as a single diastereomer. Finally, kopsinine 160 was synthesized from 159 in 2 steps (Scheme 33).
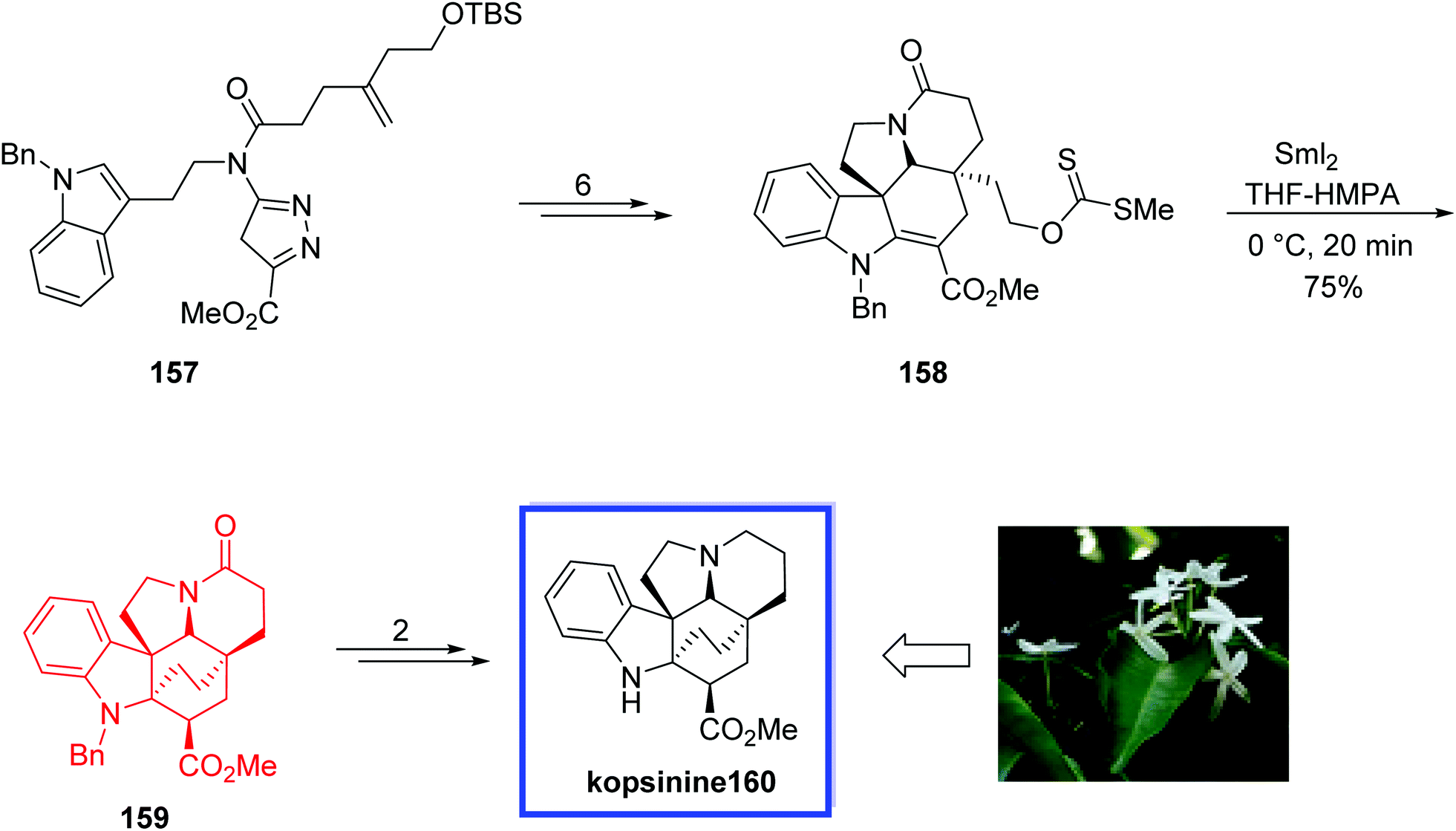 | ||
| Scheme 33 Total synthesis of kopsinine 160. | ||
Later in 2015, Boger and colleague87 developed the asymmetric total synthesis of (−)-kopsinine 165 and its unnatural enantiomer from the common Aspidosperma-like pentacyclic intermediate 161. The key steps contain a tandem intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles to provide the stereochemically rich C21-functionalized pentacyclic core precursor 161 of the natural product, and the C21–C2 bond formation via a late-stage SmI2-promoted transannular free radical conjugate addition reaction constructing the bicyclo[2.2.2]octane core. The spectral data of synthetic (−)-kopsinine 165 were in good agreement with those reported for the natural product.
The total synthesis of (−)-kopsinine 165 began with the Aspidosperma-like pentacyclic intermediate 161 to provide the natural (+)-162 and ent-(−)-162 enantiomers in 3 steps and 97% yield. The natural enantiomer (+)-162 was then converted into the primary iodide (−)-163 in 4 steps and 81% yield. The key transannular cyclization of (−)-163 with SmI2 in THF/HMPA (10:1) at 0 °C for 1 h smoothly gave (−)-164 in excellent yield (85%) along with <5% of the C3 diastereomer. This generation of essentially a single diastereomer (>17:1). The natural (−)-kopsinine 165 was synthesized from (−)-164 in 2 steps and 82% yield. The unnatural enantiomer of kopsinine, ent-(+)-165, was also synthesized in a similar pathway from ent-(−)-162 (Scheme 34).
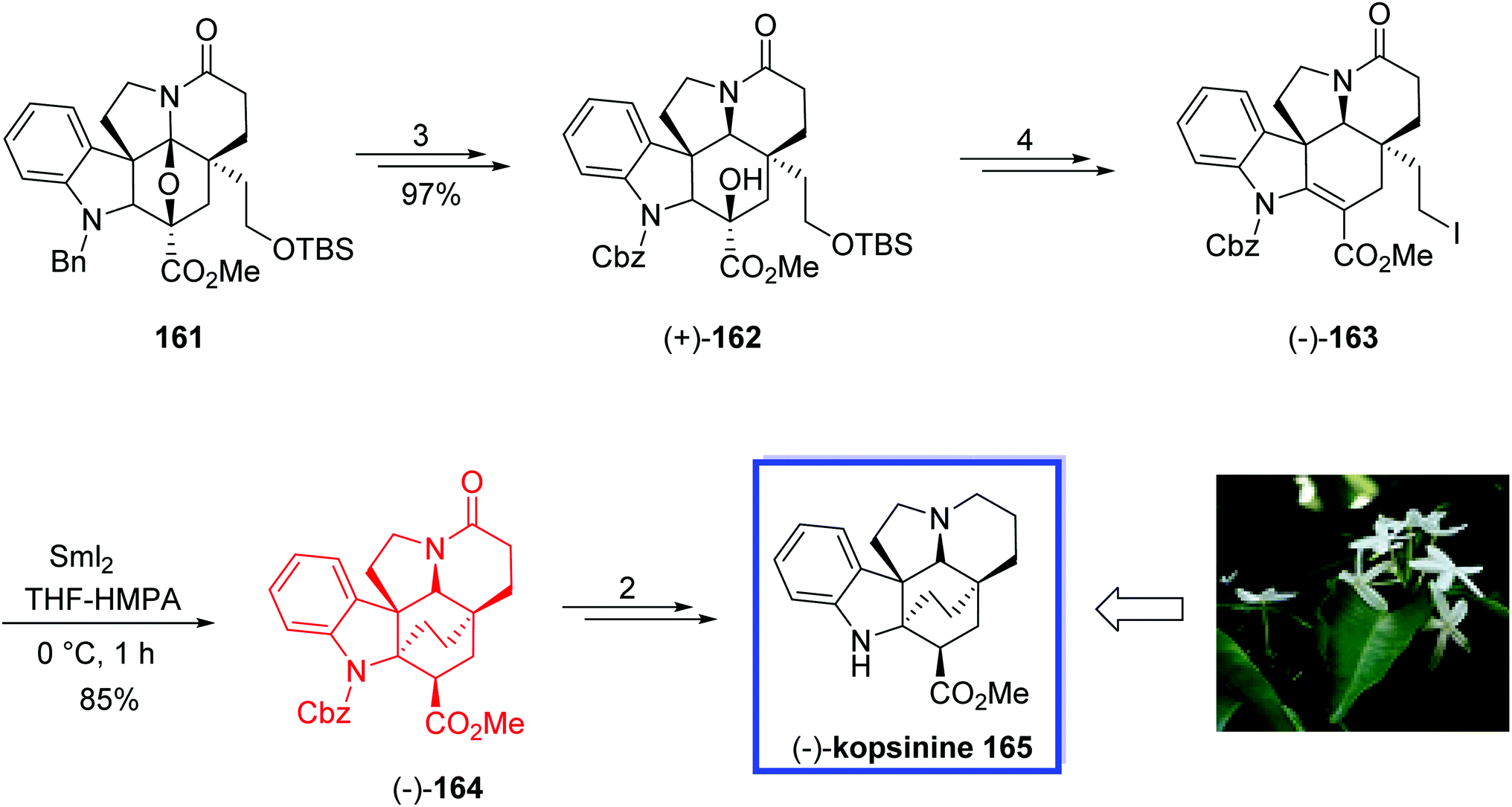 | ||
| Scheme 34 The total synthesis of (−)-kopsinine 165. | ||
(−)-Fruticosine 173 and (−)-kopsine 175, isolated from Kopsia fruticosa, (−)-kopsanone 176, isolated from Aspidosperma macrocarpon Mart., and (+)-methylchanofruticosinate 172, isolated from K. arborea, belong to the subfamilies of kopsia indole alkaloids.83 These alkaloids show several impressive types of biological activity, including cholinergic, antirheumatic, and anti-inflammation effects.84
In 2017, Qin and colleagues88 reported the asymmetric total syntheses of all four subfamilies of kopsia indole alkaloids, i.e., (−)-kopsine 175, (−)-isokopsine 171, (+)-methyl chanofruticosinate 172, (−)-fruticosine 173, and (−)-kopsanone 176. The key steps contain an asymmetric Tsuji–Trost rearrangement to install the first quaternary carbon center at C20, an intramolecular cyclopropanation by diazo decomposition to generate the second and third quaternary carbon centers at C2 and C7, respectively, and a SmI2-mediated acyloin reaction to form the isokopsine core. They prepared and applied two key intermediates, N-decarbomethoxyisokopsine 169 and N-decarbomethoxykopsine 170, to synthesize Kopsia alkaloids with different subtype core structures.
The total synthesis of (−)-isokopsine 171, (+)-methyl chanofruticosinate 172, (−)-fruticosine 173, (−)-kopsine 175 and (−)-kopsanone 176 began with the commercially available tetrahydrocarbazolone 166 to obtain ketone 167 in 15 steps. SmI2-mediated acyloin condensation of 167 in THF at room temperature provided nitrile intermediate 168 in 74% yield, generating the C16–C22 bond. N-decarbomethoxyisokopsine 169 was then synthesized from 168 in 3 steps together with N-decarbomethoxykopsine 170 in 36% and 30% yield, respectively. (−)-Isokopsine, (−)-171, was obtained from 169 in 64% yield over two steps. (−)-Fruticosine, (−)-173, was synthesized from (−)-isokopsine, (−)-171, in 55% overall yield within 3 steps, and treatment of (−)-171 with Pb(OAc)4 caused the cleavage of the C16–C22 bond and provided (+)-methyl chanofruticosinate, (+)-172, in 65% yield. (−)-Kopsine, (−)-175, was synthesized from 170 in 42% yield together with 174 in 47% yield within 2 steps. (−)-Kopsanone, (−)-176, was also synthesized from 170 in two steps and 77% yield (Scheme 35).
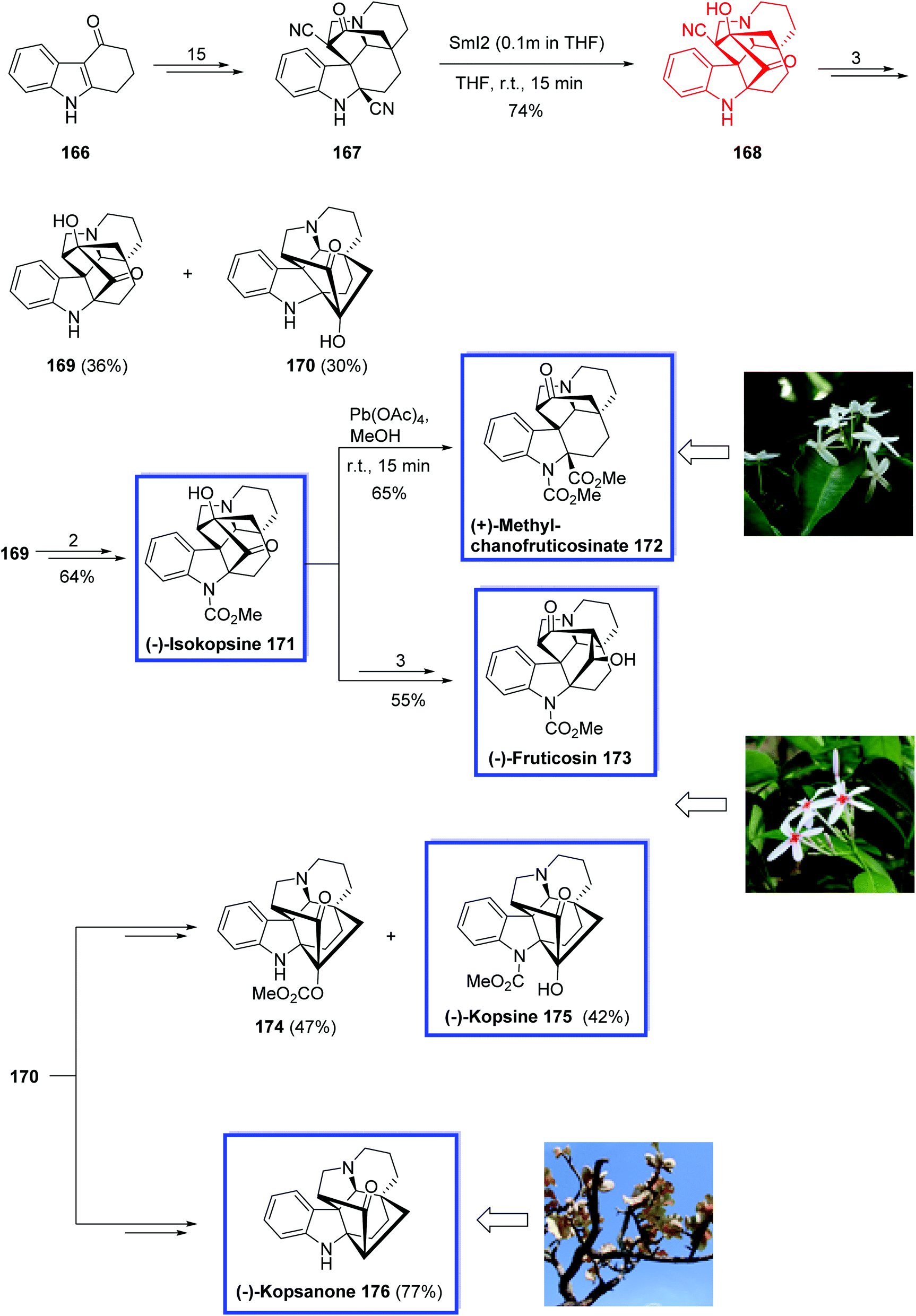 | ||
| Scheme 35 Total synthesis of (−)-isokopsine 171, (+)-methyl chanofruticosinate 172, (−)-fruticosine 173, (−)-kopsine 175 and (−)-kopsanone 176. | ||
In 2018, Ma and coworkers89 developed a modified thiourea catalyst for the synthesis of 3,3-disubstituted carbazolones via the Michael addition of carbazolones to 2-chloroacrylonitrile 182 with excellent enantioselectivity. 3,3-Disubstituted carbazolones have been used as the valuable building blocks for developing Aspidosperma and Kopsia indole alkaloids during the past few decades. The key steps for the total syntheses of Kopsia alkaloids 5,22-dioxokopsane 190, isolated from Kopsia pauciflora, kopsinidine C 194 and demethoxycarbonylkopsin 193, isolated from Kopsia officinalis, involved the MnIII-mediated oxidative cyclization to install the caged ring system and a SmI2-mediated reductive coupling.
The total synthesis of 5,22-dioxokopsane 190, kopsinidine C 194, and demethoxycarbonylkopsin 193 began with the reaction of tetracarbazolone 179 and 2-naphthyl isocyanate 180 in the presence of KHMDS in THF to provide β-keto amide 181 in 53% yield. Michael addition of β-ketoamide 181 to chloroacrylonitrile 182 on gram scale with 5 mol% of the n-pentyl-substituted catalyst 178 at −10 °C and then treatment with zinc powder at room temperature provided 3,3-disubstituted carbazolone 183 in >95% yield and 96% ee. The thiourea catalyst 178 was prepared from commercially available tert-butyl ((1R, 2R)-2-aminocyclohexyl)carbamate 177 in 4 steps and 86% yield. Compound 183 was then converted into aldehyde 184 in 7 steps and 79% yield. A SmI2-mediated Reformatsky-type cyclization of 184, followed by the Dess–Martin oxidation provided ketoamide 185 in 53% yield. Ketoamide 185 was then converted into two cyclization products 186 and 187 using Mn(OAc)3 as the single-electron oxidant and Cu(OAc)2 as the co-oxidant in methanol in 76% and 10% yield, respectively. Next, aminonitrile 188 was synthesized from amino ketal 186 in two steps and 92% yield. A SmI2-mediated reductive coupling between the keto and nitrile groups of amino nitrile 188 at ambient temperature gave isokopsine lactam 189 in 79% yield, from which 5,22-dioxokopsane 190 was synthesized in 3 steps and 44% yield (Scheme 36). In a parallel process, amino nitrile 191 was synthesized from hemiketal 187 in 2 steps and 91% yield. A SmI2-mediated reductive coupling of 197 in THF at ambient temperature gave hydroxy ketone 198 in 75% yield. Subsequent NaOH-mediated ketol rearrangement of hydroxy ketone 192 in NaOH/dioxane (1:1) at room temperature gave demethoxycarbonylkopsin 193 in 80% yield. Further reduction of the ketone with LiAlH4 delivered kopsinidine C 194 and 22-epi-kopsinidine C 195 in 40% and 30% yield (Scheme 37).
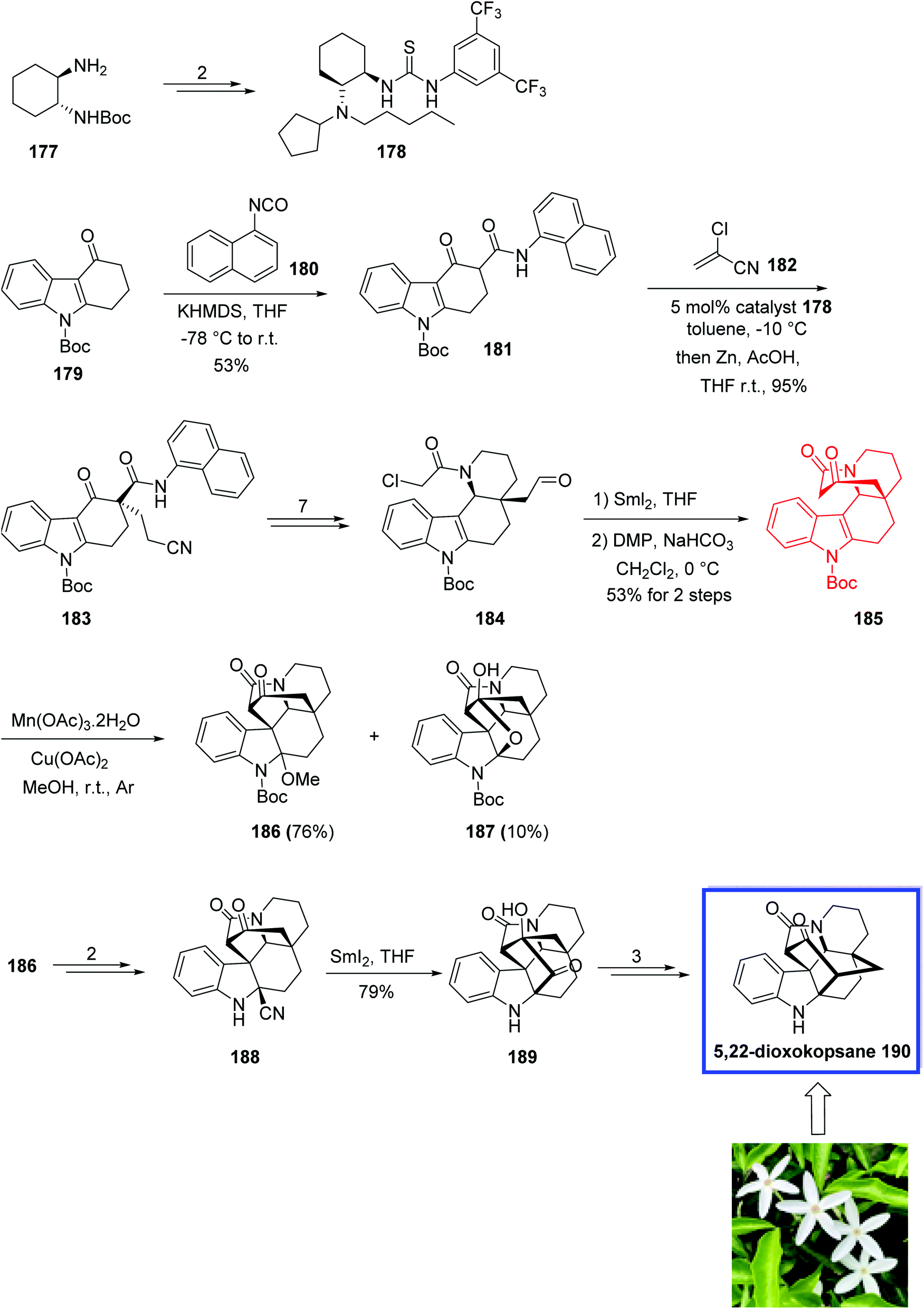 | ||
| Scheme 36 Total synthesis of 5,22-dioxokopsane 190. | ||
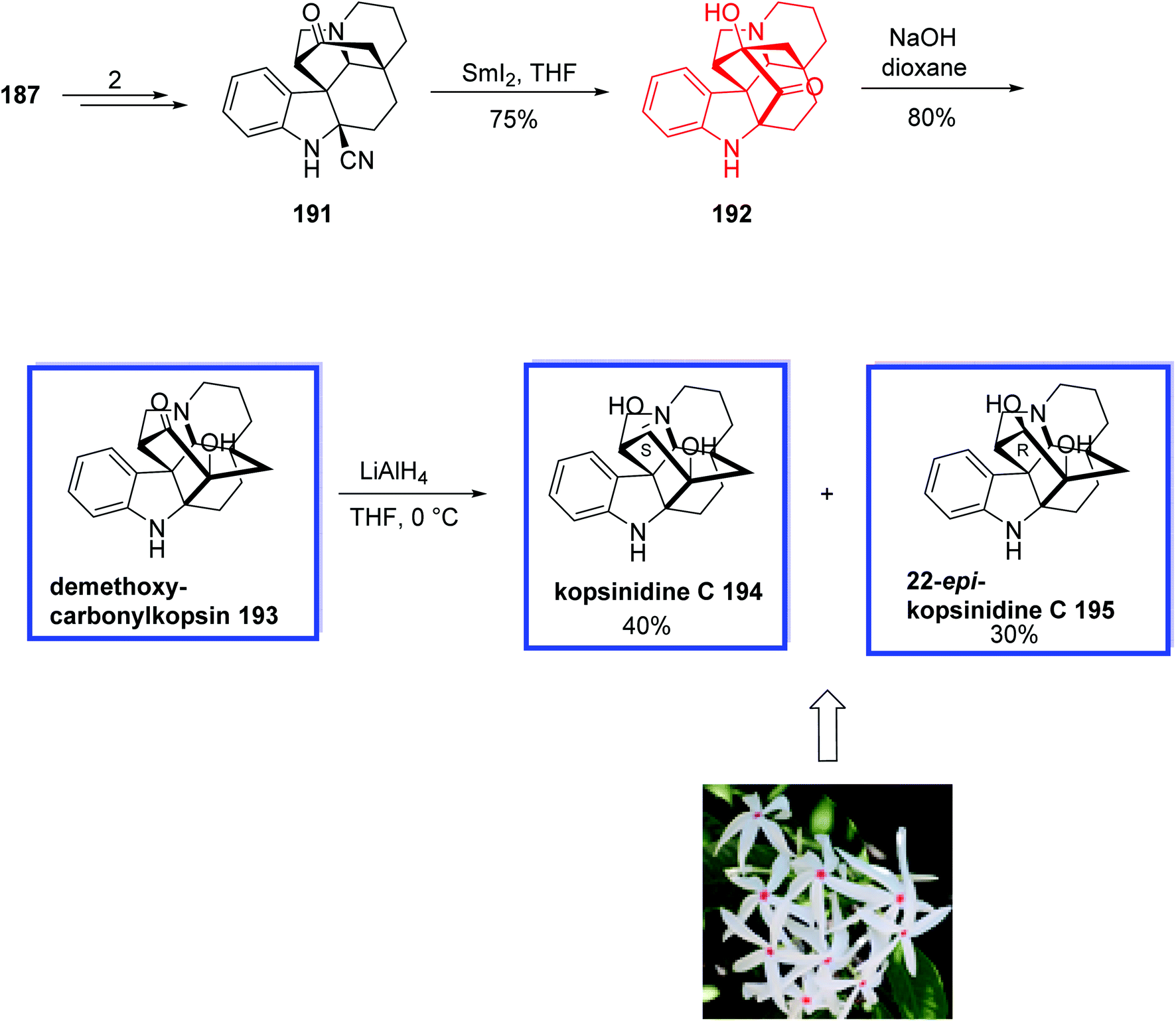 | ||
| Scheme 37 The total synthesis of demethoxycarbonylkopsin 193, kopsinidine C 194 and 22-epi-kopsinidine C 195. | ||
(−)-Histrionicotoxin 283A (HTX-283A, 206) belongs to azaspirocyclic histrionicotoxin alkaloids and was first isolated from skin extracts of the Colombian poison arrow frog Dendrobates histrionicus. A mixture of six alkaloids including 206 and other members have been isolated and identified so far. (−)-Histrionicotoxin 283A 206 shows highly selective inhibition of nicotinic acetylcholine receptors.90a
(−)-Histrionicotoxin 235A (HTX-235A, 205) was isolated from another poison frog Dendrobates auratus.90b It was used towards important investigations into neurophysiology.
In 2018, Morimoto and coworkers91 developed the formal total synthesis of histrionicotoxin alkaloids. The key steps were a Hg(OTf)2-catalyzed cycloisomerization and a SmI2-mediated ring expansion reaction.
The SmI2-mediated ring expansion was used as a key step to synthesize 1-azaspiro[5.5]undecane skeleton 204 in 15 steps and 15% overall yield, based on the known silyl ether 196, in comparison with Tokuyama's method (5% overall yield and 14 steps based on the ketodiester and benzylamine). This synthetic approach was the first SmI2-mediated radical ring expansion applied to the synthesis of such a complex system as a 1-azaspiro[5.5]undecane skeleton of histrionicotoxins.
The total synthesis of 205 and 206 started from the known silyl ether 196. Alkylation of a lithium acetylide of silyl ether 196 provided sulfone 197 in 2 steps. Reacrion of 191 with known pyrrolidinone 198 followed by SmI2-mediated desulfonylation provided the desired cyclization precursor 199. The Hg(OTf)2-catalyzed cycloisomerization reaction of precursor 199 using 20 mol% catalyst in −30 °C provided the desired spirocyclic product 200 in 77% yield. The reaction of 200 with SmI2 in the presence of H2O and triethylamine provided the desired equatorial-alcohol 201 as a major diastereomer in 58% yield. The eq-alcohol 201 was then converted into silyl ether 202 in 8 steps. Compound 202 was further converted into 1-azaspiro[5.5]undecane 204 in high yield via the key SmI2-mediated ring expansion reaction using HMPA and pivalic acid 203. The key intermediate 204 was transformed to (−)-HTX-235A 205 in two steps, and then further converted into HTX-283A, 206 in 5 steps (Scheme 38).
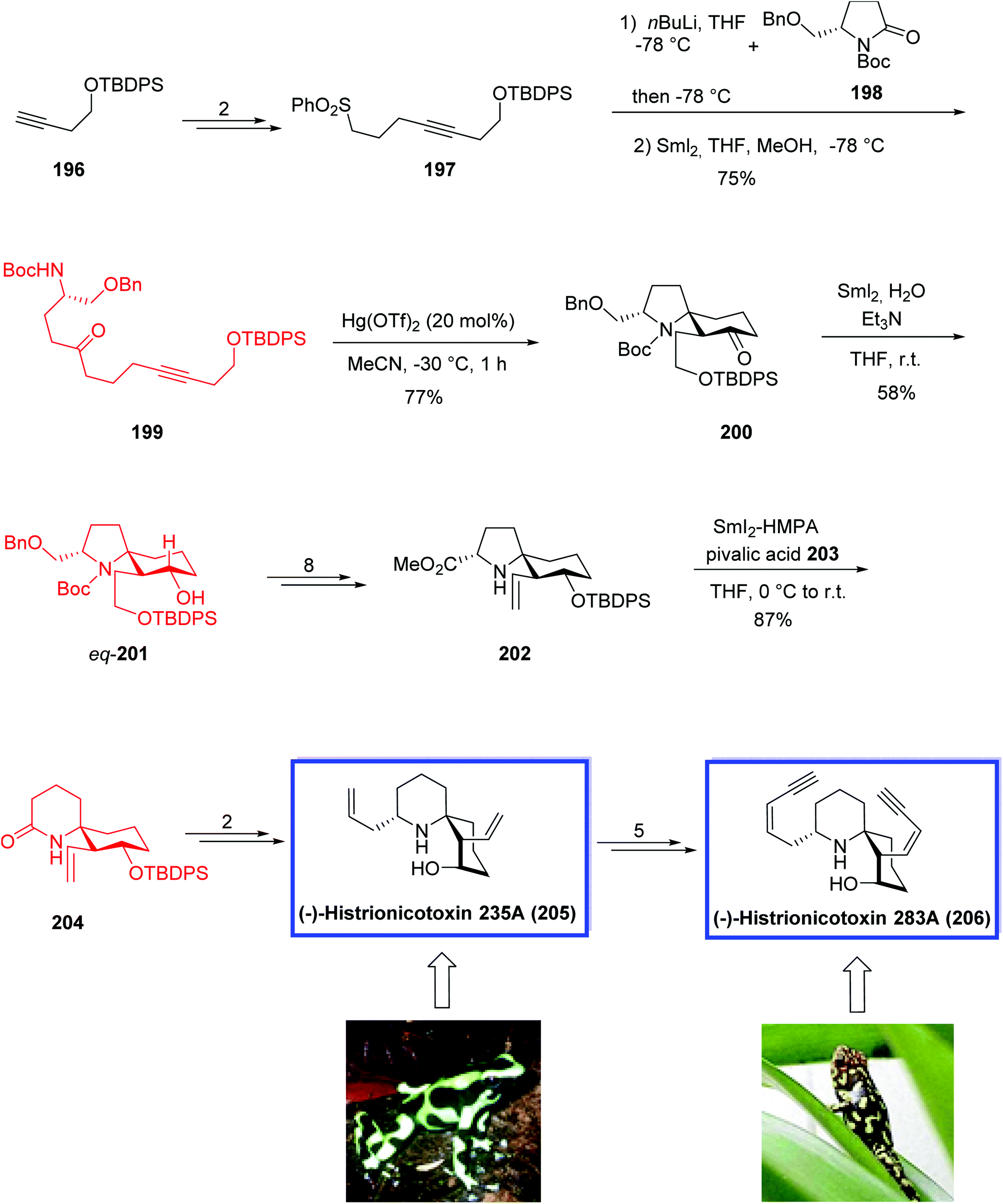 | ||
| Scheme 38 The total synthesis of (−)-histrionicotoxin 235A (HTX-235A, 205) and (−)-histrionicotoxin 283A (HTX-283A, 206). | ||
Wickerols A 211 and B 212 are diterpene natural products and were isolated from the fungus Trichoderma atroviride FKI-3849,92 and more recently, from cultures of the marine red alga-epiphytic fungus Trichoderma koningiopsis Y10-2.93 They are similar to the types of molecules with potential activity against the influenza virus.94
Wickerol B 212, possesing a hydroxyl group at C8, is less active than Wickerol A 212 whereas wickerol A 211 shows antiviral activity against the H1N1 type A influenza virus.92–94 Therefore, different strategies for the synthesis of wickerols and their analogues would extremely promote an extensive investigation of the structure–activity relationships.
In 2020, Gui and coworkers95 reported a divergent total synthesis of wickerols A 211 and B 212 in 16 and 15 steps, respectively, from sitolactone 207.
The key steps included a SmI2-induced intramolecular reductive cyclization, a Claisen rearrangement, and an intramolecular alkylation/aldol reaction to develop the tetracyclic core stereoselectively, and the absolute configurations of wickerols A 211 and B 212 were assigned. Under optimized conditions, they observed that the key SmI2-induced intramolecular reductive cyclization can be performed on a gram scale with similar yields. All the spectroscopic data and for synthetic wickerol A 211 and wickerol B 212 were in good agreement with those reported for the natural products, and they had absolute configurations the same as the natural isolates.
The total synthesis of wickerol A 211 and wickerol B 212 began with the commercially available sitolactone 207 to obtain ketone 208 in 3 steps and 95% yield. SmI2-mediated intramolecular ketone–allylic acetate reductive cyclization of ketone 208 using hexamethylphosphoramide (HMPA) in THF under the optimized conditions afforded desired compound 209 in 52% yield, from which ketone 210 was then synthesized in 5 steps and 70% yield. The terminal olefin 210 was then used to complete the synthesis of wickerols A 211 and B 212 in 7 and 6 steps and 91% and 55% yields, respectively (Scheme 39).
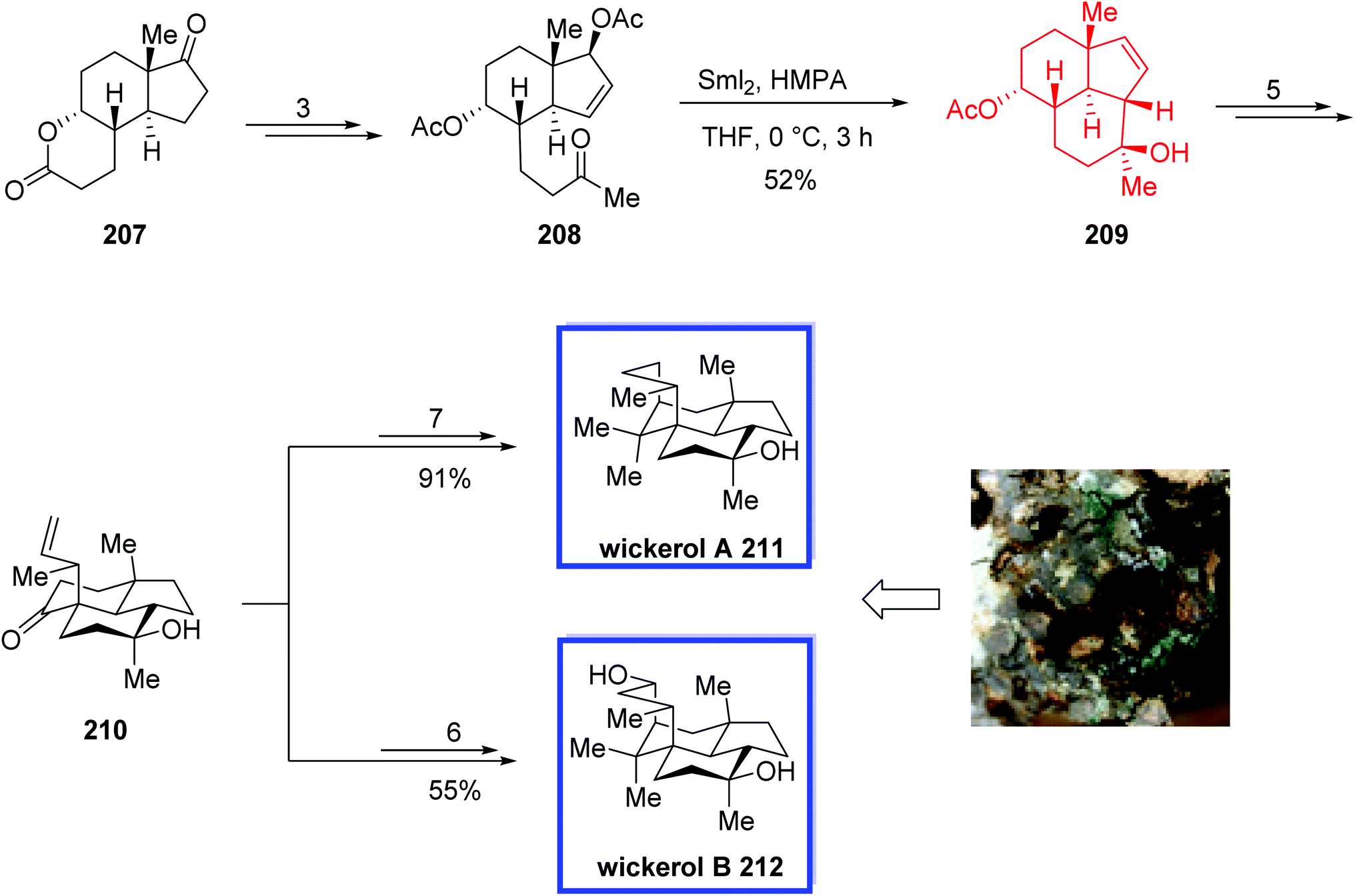 | ||
| Scheme 39 The total synthesis of wickerol A 211 and wickerol B 212. | ||
Gambierol 221, a neurotoxin isolated from the cultured cells of the ciguatera causative dinoflagellate Gambierdiscus toxicus, belongs to the fused polycyclic ether marine natural products, is a potent blocker of voltage-gated potassium channels, and also is responsible for ciguatera fish poisoning.96 Due to the molecular complexity of structures of this class of marine natural products and their potent and multiple biological activities, they continue to be of great interest to synthetic chemists. Gambierol 221 contains a trans-fused octacyclic ring system with 18 stereogenic centers, a partly conjugated triene side chain, and two 1,3-diaxial dimethyl-substituted tetrahydropyranyl rings.
In 2002, Sasaki and colleague97 developed the first total synthesis of (−)-gambierol 221. Key steps contain B-alkyl Suzuki–Miyaura cross-coupling of the ABC and EFGH ring fragments, 214 and 220, respectively, to construct the octacyclic polyether scaffold, a SmI2-mediated reductive cyclization to improve the synthetic method to construct the F and H rings (the EFGH ring fragment 220), and a late-stage installation of the sensitive triene side chain via Pd(PPh3)4/CuCl/LiCl-induced Stille coupling. This very efficient route enabled the synthesis of 214 and 220 in multigram quantities and easy access to a variety of structural analogues of gambierol 221. The spectral data of synthetic gambierol 221 was in good agreement with that of the natural product. The absolute configuration of gambierol 221 was also confirmed.
The total synthesis of (−)-gambierol 221 began with the preparation of the ABC and EFGH ring fragments, 214 and 220, respectively. The ABC ring exocyclic enol ether 214 was synthesized from the known olefin 213 in 36 steps and 18% overall yield.
Synthesis of the EFGH ring fragment 220 started from the known ester 215 to provide aldehyde 216 in 6 steps and 91% yield. Stereoselective reductive cyclization of 216 using SmI2 in the presence of methanol/THF and at room temperature afforded the desired γ-lactone 217 in 70% yield, which was then converted to β-alkoxy acrylate 218 in 18 steps and almost quantitative yield. Stereoselective ring closure of the F ring containing 1,3-diaxial angular methyl groups occurred through the treatment of 218 with SmI2 in methanol/THF at 0 °C and gave the FGH ring system 219 in 87% yield as a single stereoisomer. The EFGH ring ketene acetal phosphate 219 was then achieved in 7 steps. Accordingly, the EFGH ring fragment 220 was synthesized in 33 steps and 22% overall yield from 215. With the coupling of two key intermediates 214 and 220, and construction of the octacyclic polyether core, the total synthesis of (−)-gambierol 221 was then completed in 24 steps and 43% yield (Scheme 40).
 | ||
| Scheme 40 Total synthesis of (−)-gambierol 221. | ||
The ent-kaurene diterpenoids, one special classification of the diterpenoid family, have gained increasing attention due to their structural variety and complexity, together with comprehensive bioactivity outlines. The Isodon species of plants produce an enormous collection of terpene natural products that are biosynthetically originated from the familiar ent-kaurene nucleus. Among these ent-kaurene diterpenoids, maoecrystal P 225, isolated from the Chinese medicinal plant Isodon eriocalyx, exhibits promising cytotoxicity against human tumor cells.98
In 2018, Luo and co-workers99 introduced a new strategy for the total synthesis of maoecrystal P 225 with 27 steps from commercially available 2-bromocyclohex-2-one 222. The total synthesis of maoecrystal P 225 included a highly regio- and diastereoselective intermolecular Diels–Alder cycloaddition, redox manipulations, a SmI2-mediated carbonyl–alkene cyclization, and isomerization reactions. This novel strategy could be used for the synthesis of other highly oxidized ent-kauranoids for biological evaluation.
The total synthesis of maoecrystal P 225 began with ketone 222 and delivered the cyclization precursor 223 in 11 steps. The reaction of 223 with excess SmI2 in toluene at 0 °C provided ketol 224 as a single diastereomer in the modest yield of 50–60% with the completion of the ent-kauranoid skeleton. The modest yield of 224 can be justified due to the difficulty of building a hindered quaternary center and a forced ring construction. Finally, maoecrystal P 225 was synthesized from 224 in 14 steps and 55% yield, completing the ent-kauranoid skeleton with a hindered quaternary center and a strained ring system (Scheme 41).
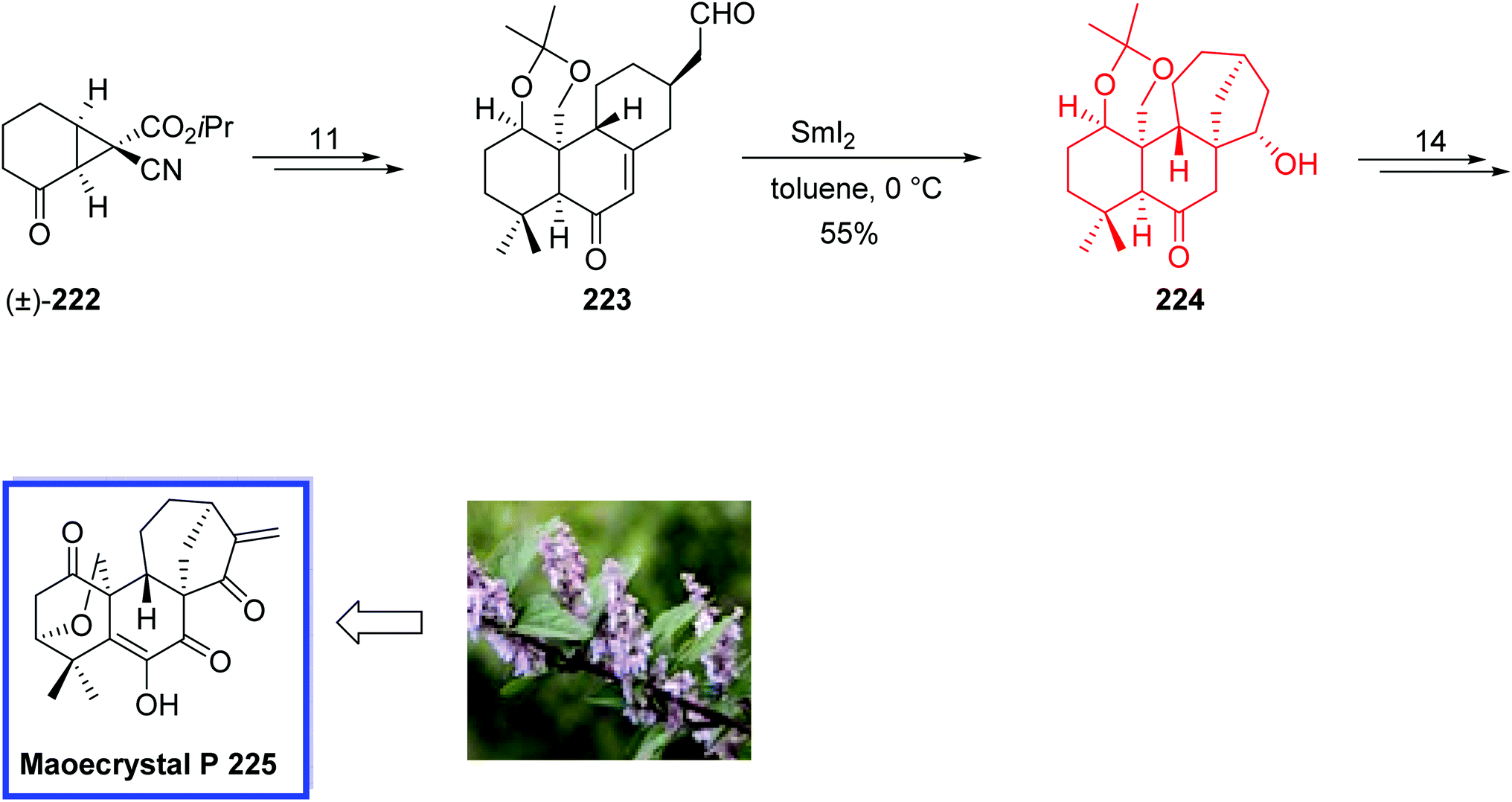 | ||
| Scheme 41 Total synthesis of maoecrystal P 225. | ||
Grayanane diterpenoids (grayanoids), with unique carbon skeletons, were isolated from plants of the Ericaceae family. Grayanoids show various biological properties such as anticancer, antiviral, antifeedant, antinociceptive, analgesic, insecticidal, toxicity, and protein tyrosine phosphatase 1B (PTP1B) properties. Principinol D 233, isolated from Rhododendron principis, belongs to the grayanane diterpenoid natural products with a broad range of biological activities.100 Grayananes are a wider class of rearranged kaurane natural products. More than one thousand kaurane diterpenoids have been isolated; however, only a few of them have been synthesized. Structurally, principinol D 233 contains a highly oxidized tetracyclic core, consisting of a bicyclo[3.2.1]octane ring system with five stereocenters. It inhibits protein tyrosine phosphatase 1B (PTP1B) in vitro, which is a potential target for obesity and types 2 diabetes therapy.101
In 2019, Newhouse and colleagues102 reported the first asymmetric total synthesis of principinol D 233 in 19-steps. The key steps contain a SmI2-mediated reductive cyclization to construct the central seven-membered ring of the grayanane skeleton, a convergent fragment coupling protocol through a 1,2-addition reaction between a bicyclo[3.2.1]octane fragment 229 and a cyclopentyl fragment 230, and Ni-catalyzed α-vinylation reaction to establish the bicyclo[3.2.1]octane. Reductive strategies involved a diastereoselective SmI2-induced ketone reduction of a bicyclic ketone and a new procedure for selective ester reduction in the presence of ketones. The NMR spectra of the product were in good agreement with those reported for the isolated natural product. According to the researchers, the reported fragment-based strategy is expected to be utilized for SAR-investigations and the synthesis of various grayanane derivatives.
The synthesis of bicyclo[3.2.1]octane fragment 229 began with cyclohexenone 226 to afford ketone 227 in 5 steps. Diastereoselective reduction of ketone 227 with SmI2 in the presence of HMPA and PhSH provided the desired alcohol 228 in an excellent yield (95%, >20:1 d.r.). From alcohol 228, the bicyclo[3.2.1]octane 229 was obtained in 2 steps and 87% yield. The racemic bicyclo[3.2.1]octane 229 was then coupled with the enantioenriched cyclopentyl aldehyde 230 (er = 93:7) to give enone-aldehyde 231 in 5 steps. Closure of the seven-membered ring occurred through treatment of 231 with SmI2 in the presence of water to produce the desired tetracycle 232 in 63% yield as a single diastereomer, from which principinol D 233 was synthesized in 5 steps and 78% yield (Scheme 42).
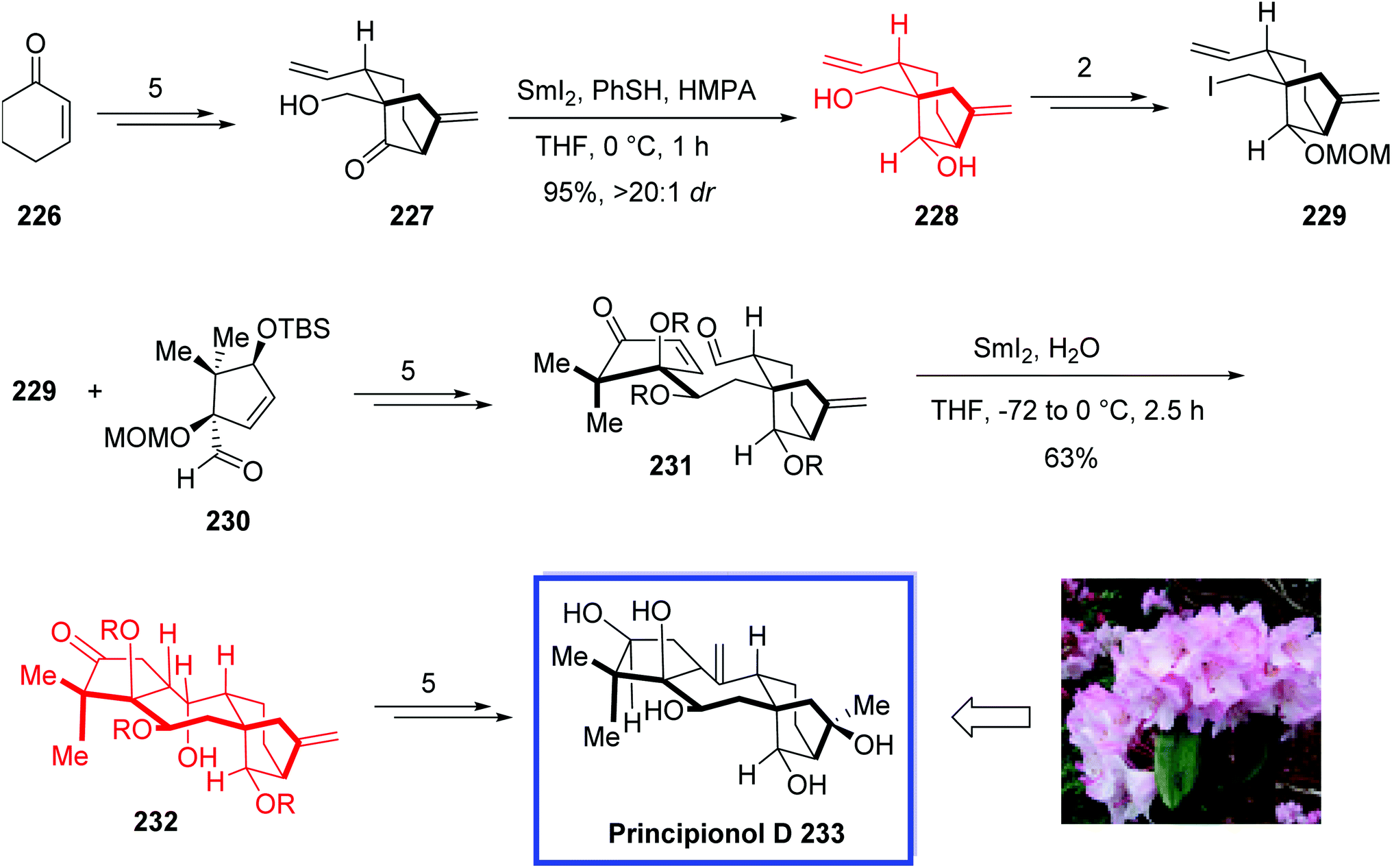 | ||
| Scheme 42 Total synthesis of principinol D 233. | ||
(+)-Pleuromutilin 237, isolated from the fungus Clitopilus passeckerianus,103 is a diterpene natural product with antibiotic activity. This antibiotic prevents protein synthesis, by which it connects to the peptidyl transferase center of bacterial ribosomes.104
In 2018, Reisman and colleagues105 developed the total synthesis of the antibiotics (+)-pleuromutilin 237 and (+)-12-epi-pleuromutilin 12-epi-237 each in 18 steps from (+)-transdihydrocarvone. The key steps included the construction of the eight-membered ring through a highly stereoselective SmI2-promoted cyclization and the installation of the C10 stereocenter via a stereospecific transannular [1,5]-hydrogen atom transfer. According to researchers the shortness and modularity of the reported strategy will facilitate the design and synthesis of new mutilin antibiotics.
The total syntheses of (+)-pleuromutilin 237 and (+)-12-epi-pleuromutilin 12-epi-237 began with the enone 234. Aldehydes 235 and 12-epi-235 were then synthesized from enone 234 each in 12 steps. It was found that dropwise addition of SmI2 (3 equiv.) to 235 with 6 equivalents of H2O in THF at 0 °C in the absence of oxygen, followed by quenching with trimethylsilyl chloride (TMSCl) and water provided tricycle 236 as a separable mixture of diastereomers (23:1) in 93% yield. H2O played a significant role to attain high diastereoselectivity. (+)-Pleuromutilin 237 was then synthesized from 236 in 4 steps and 80% yield. SmI2 cyclization of 12-epi-235 under optimal conditions provided 12-epi-236 in 77% yield (17:1 d.r.), and 12-epi-237 was synthesized from 12-epi-236 in 4 steps (Scheme 43).
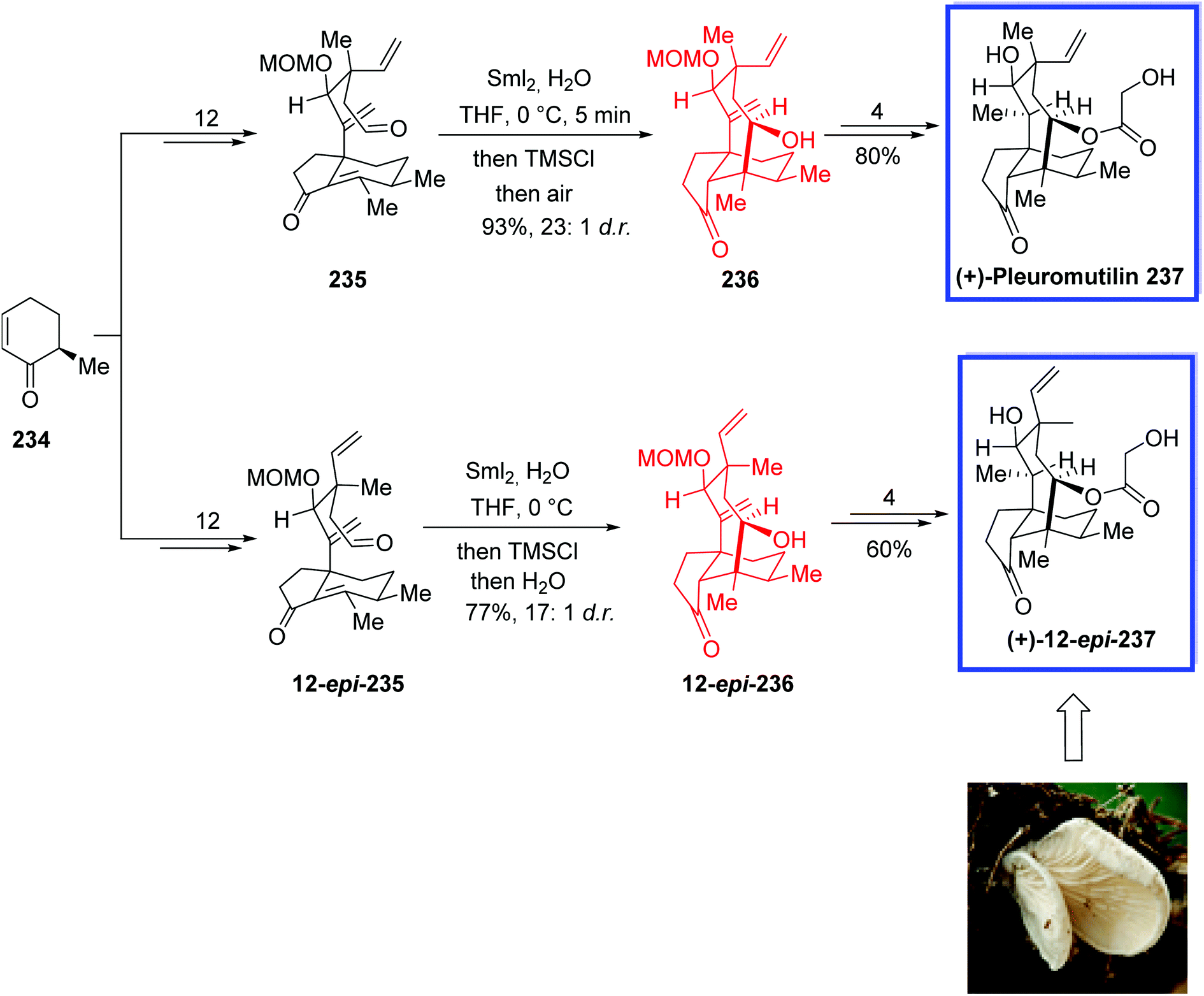 | ||
| Scheme 43 Total synthesis of (+)-pleuromutilin 237 and (+)-12-epi-pleuromutilin 237. | ||
Paucidirinine 244, isolated from the stem bark of Kopsia pauciflora, is a member of the aspidofractinine alkaloid family containing a constricted five-membered lactam ring. The aspidofractinine alkaloids are a class of structurally complex alkaloids isolated from the leaves of Pleiocarpa tubicana and Aspidosperma refractum.106 They display considerable medicinal properties and biological activity, and several members of this family can reverse multidrug resistance in human cancer.107
In 2019, She and coworkers108 developed the first total synthesis of paucidirinine 244 in 10 steps and 8% overall yield from tryptamine hydrochloride salt. Key steps of this potentially scalable strategy contain a highly stereocontrolled intramolecular [4 + 2] Diels–Alder reaction, an acid-catalyzed lactamization, and a challenging late-stage SmI2-induced radical cyclization to construct the bicyclic[2.2.2]octane core. The relative configuration of this potentially bioactive alkaloid was also confirmed through X-ray diffraction analysis.
The total synthesis of paucidirinine 244 began with 1,4-butanediol 238 to obtain aldehyde 239 in 3 steps and 61% yield. The key indole precursor 241 was prepared in 3 steps and 98% yield using tryptamine hydrochloride salt 240 as the starting material. Indole 241 was then coupled with aldehyde 239 to obtain alkyl iodide 242 in 5 steps. The SmI2-mediated cyclization of 242 in THF/HMPA (10:1) for 1 h at 25 °C provided the desired cycloadduct 243 in 2 steps and 67% yield. Finally, removal of the p-methoxybenzyl group with TFA provided paucidirinine 244 in 76% yield (Scheme 44).
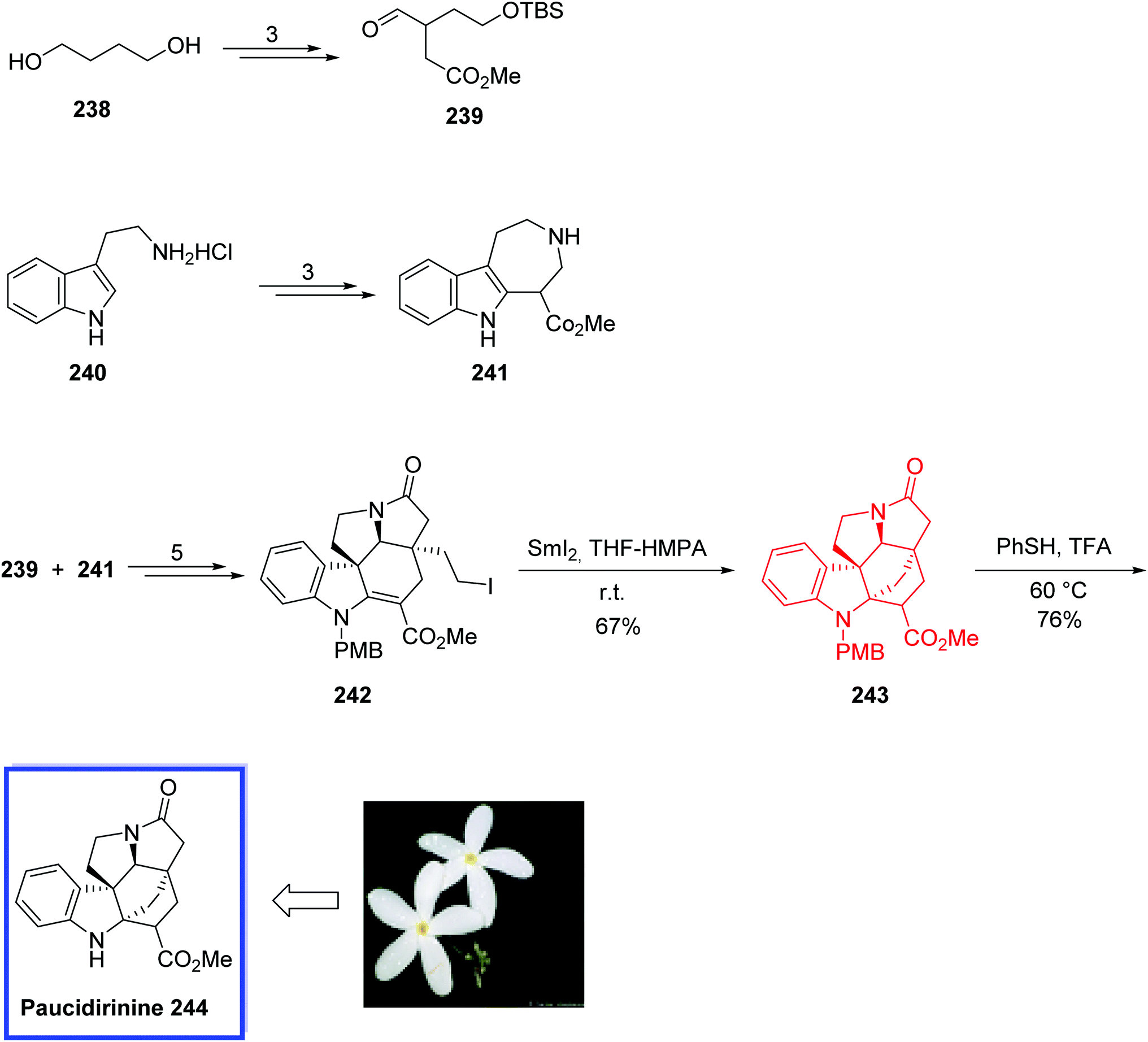 | ||
| Scheme 44 Total synthesis of paucidirinine 244. | ||
Kopsinitarine E 248, isolated from K. teoi L. Allorge,109 is a new member of the Kopsia species. Kopsinitarines A–E are Kopsia alkaloids, comprising a cage-like system that contains two five-membered rings and three six-membered rings.
In 2020, Ma and colleagues110 reported the first total synthesis of kopsinitarine E 248 in 20 steps from commercially available carbazolone 245. The key steps comprised a remarkable SmI2-mediated radical cyclization cascade, followed by semi-pinacol rearrangement to install the 5,7-fused ring system, a chemoselective hydrosilyl amide reduction to establish the hemiaminal ether bridge, and an intramolecular Mannich reaction for closure of the highly strained cage system. According to the authors, the SmI2-key procedure would be useful for developing other Kopsia alkaloids containing this key skeleton. The spectral data of synthetic kopsinitarine E 248 were in good agreement with those reported for natural kopsinitarine E.
The total synthesis of kopsinitarine E 248 commenced with a known Boc-protected carbazolone 245 to provide the bromide 246 in 8 steps and 63% yield. A SmI2-mediated Dieckmann-like condensation of 246 gave alcohol 247 in 64% yield. Finally, kopsinitarine E 248 was synthesized from 247 in 10 steps and 64% yield (Scheme 45).
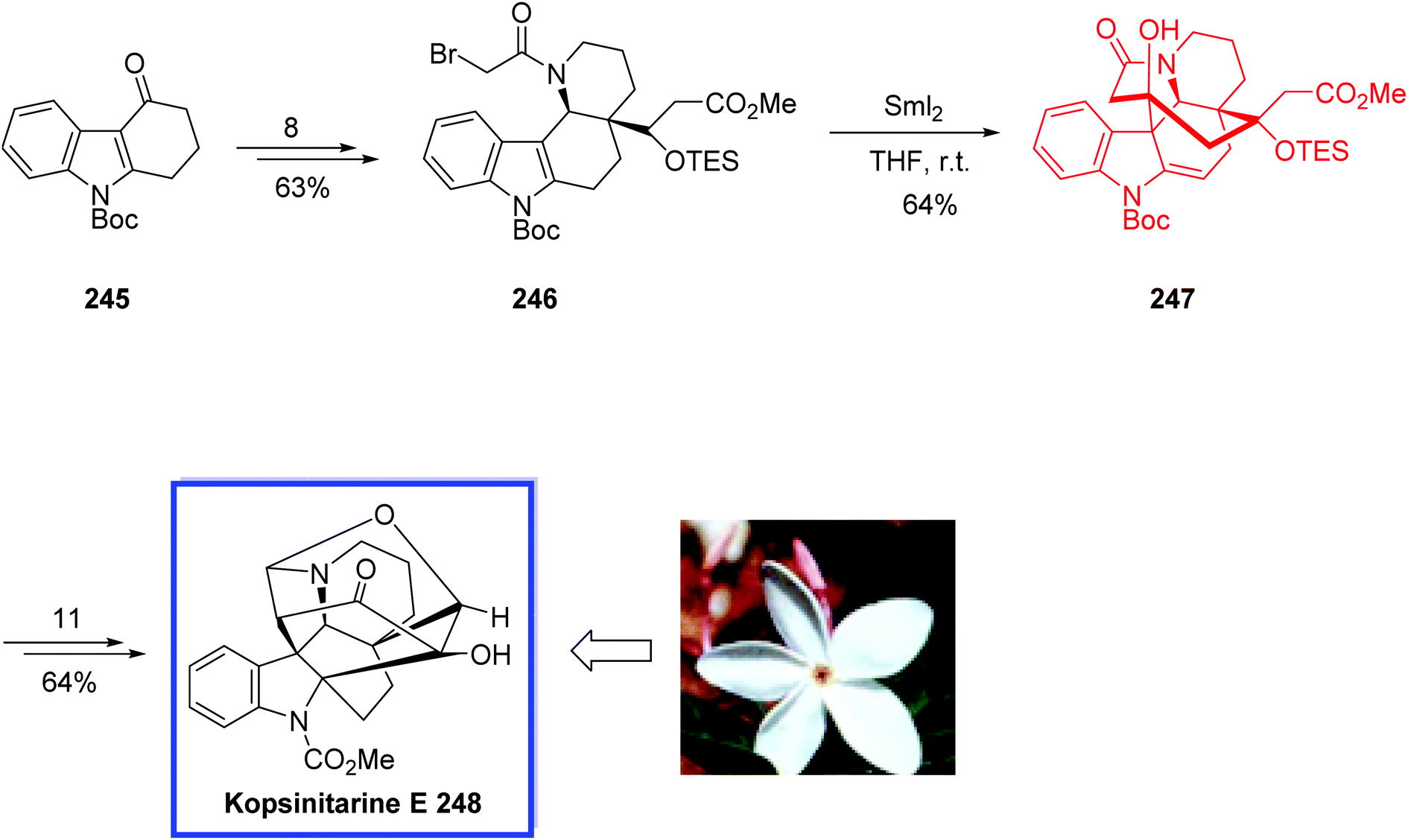 | ||
| Scheme 45 Total synthesis of kopsinitarine E 248. | ||
Q-1047H-A-A 253 and Q-1047H-R-A 254, isolated from a culture of Pseudonocardia sp. Q-1047 (type strain deposited under FERM-2331),111 belong to a class of macrolactam bacterial metabolites and are members of the macrolactam natural products, which exhibit various biological and pharmacological activities.112
Q-1047H-A-A 253 and Q-1047H-R-A 254 contain 13-membered lactams with four stereogenic centers. The general structural scaffold of Q-1047H-A-A 253 makes it a key intermediate to access other natural products.
In 2013, Yang and colleagues113 reported the first total synthesis of Ansamacrolactams (+)-Q-1047H-A-A 255 and (+)-Q-1047H-R-A 256 in 17 steps. The key steps in the synthetic work included an asymmetric chelation-controlled vinylogous Mukaiyama aldol reaction for the stereoselective synthesis of the syn-aldol adduct and an intramolecular SmI2-mediated Reformatsky reaction for the formation of the macrocyclic lactam 252. The spectral data for the synthetic Q-1047H-A-A 255 were in good agreement with those reported for the natural product, but with the opposite sign of its optical rotation. Therefore, it was found that the synthetic ansamacrolactams (+)-Q-1047H-A-A 253 and (+)-Q-1047H-R-A 254 were the enantiomers of the compounds reported as the natural products, and the relative stereochemistry and absolute configuration of the natural products (+)-Q-1047H-A-A 253 and (+)-Q-1047H-R-A 254 were reassigned as compounds 255 and 256. According to researchers, the developed synthetic strategy should be used for the syntheses and SAR analysis of other structurally and biologically related products.
The total synthesis of (+)-Q-1047H-A-A 253 and (+)-Q-1047H-R-A 254 began with the reaction of N,O-acetal 249 with benzaldehyde 250 to afford aldehyde 251 in 11 steps and 60% overall yield. The key SmI2-promoted Reformatsky reaction of 251 occurred via the addition of 251 to the solution of SmI2 in THF under reflux, and the desired cyclization product 252 was obtained in 84% yield as a mixture of two pairs of diastereomers (3.9:1:4.0:3.3) related to the C2 and C3 carbon centers. Q-1047H-A-A 253 was then synthesized from the mixture of 252 in 2 steps and 51% yield. Finally, the reduction of Q-1047H-A-A 253 with Na2S2O4 afforded Q-1047H-R-A 254 in 64% yield (Scheme 46).
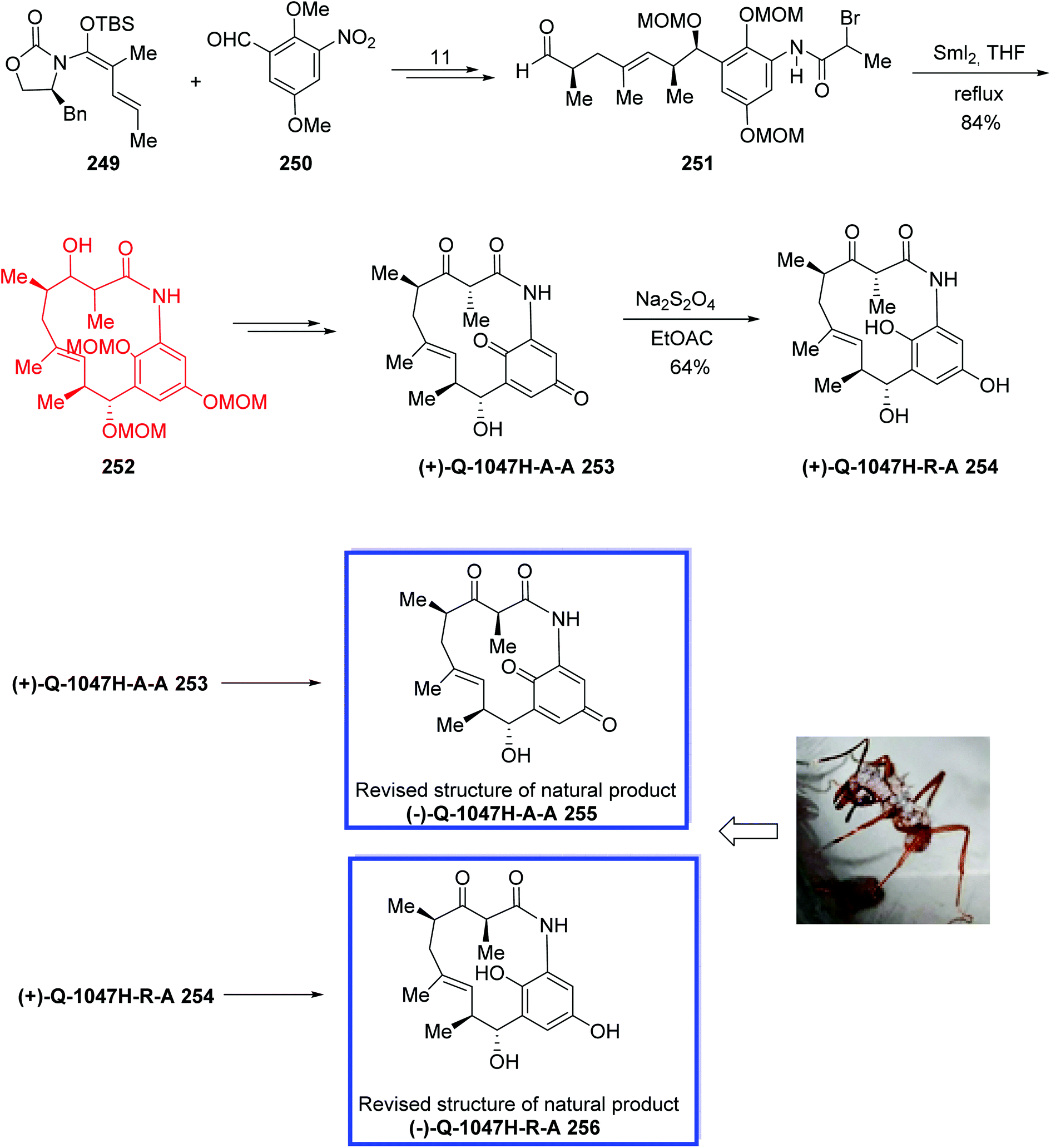 | ||
| Scheme 46 Total syntheses of (+)-Q-1047H-A-A 253 and (+)-Q-1047H-R-A 254. | ||
2.3. SmI2-mediated sequential reactions
Strychnine 263, isolated from the seeds of the Strychnos nux-vomica tree belong to the family of Strychnos alkaloids,114 which are one of the most fascinating classes of natural products. Strychnine 263 is the most complex natural product of this size, and its elevated level of complexity has continued to persuade synthetic chemists. Its structure retains seven fused rings and six asymmetric centers; hence, it is one of the most complex natural products of this size.In 2010, Reissig and coworkers115 developed the shortest formal total synthesis of strychnine 263. In this strategy, they show the power of SmI2-promoted cascade reaction to construct two new rings, three stereocenters, and a quaternary carbon atom in one step from a commercially available indole derivative. The key strychnine precursor 259 was accomplished from commercially available indolylacetonitrile in only five steps in an overall yield of 33%, and its incorrectly reported configuration at C-13a was also revised. By examination of the scope of this SmI2-induced ketyl–aryl coupling/acylation cascade reaction, it was accepted that this method could be applied to various highly functionalized tetracyclic indole derivatives stereoselectively.
The total synthesis of strychnine 263 began with a rapid SmI2-promoted cascade reaction of indole derivative 257. Treatment of 257 with 2.4 equivalents of samarium diiodide and hexamethylphosphoramide (HMPA) provided diastereomerically pure tetracycle 258 as the major product in 70–75%. The tetracyclic building block 258 was then converted to the desired key building block 259 as the major product in good selectivity (5:1:1 ratio). Finally, carbamate 259 was converted into the pentacyclic strychnine precursor 262, known as Rawals key building block, to complete the formal total synthesis of strychnine 263 (Scheme 47).
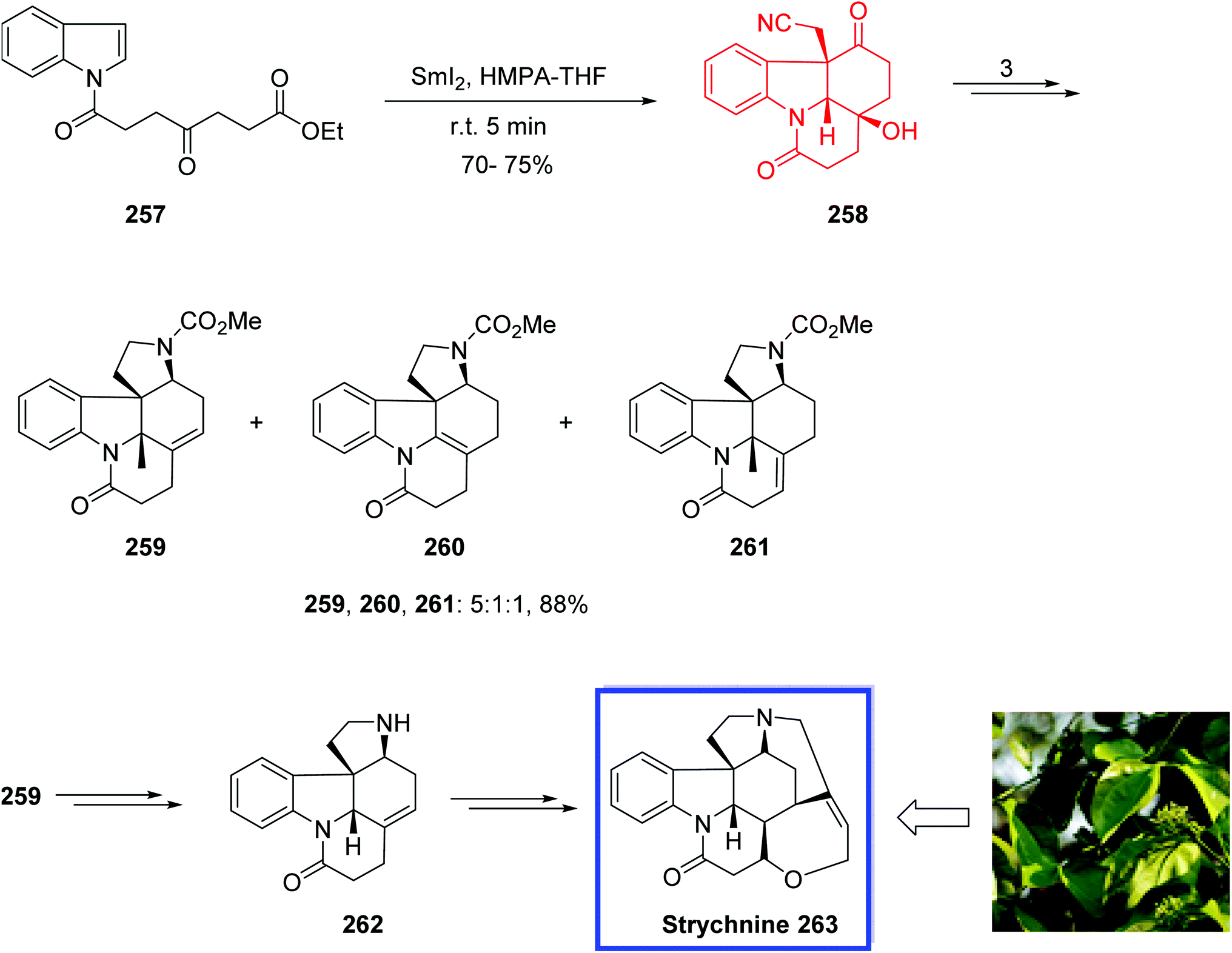 | ||
| Scheme 47 Total synthesis of strychnine 263. | ||
(+)-Crotogoudin 268, derived from the Madagascan Croton plant,116 belongs to the unusual 3,4-seco atisane family of diterpenoid natural products. Species of the Croton genus are a rich source of secondary metabolites with a broad spectrum of biological activities.116,117 (+)-Crotogoudin 268 exhibits antiproliferative activity in vitro against different human cancer cell lines.116,117 Structurally, (+)-crotogoudin 268 consists of a tetracyclic ring system with four contiguous stereocenters and with a six-membered ring fused to a bicyclo[2.2.2]octane core.
In 2013, Carreira and coworkers118 reported the first total synthesis of ent-crotogoudin 268. The key step involved a radical cyclopropane-opening/annulation/elimination cascade for the diastereoselective synthesis of the tetracyclic carbon core. They also used an efficient pathway to synthesize 1-substituted bicyclo[2.2.2]octa-2,6-diones. The absolute configuration of the (+)-crotogoudin natural product was also assigned. Spectral data for synthetic (+)-crotogoudin 268 were in good agreement with those reported for the natural product. Comparison of the optical rotation shows identical signs for both synthetic and natural material, but with significantly different magnitude. Therefore, the absolute configuration of the natural product (+)-crotogoudin (ent-268) was assigned to 5R, 10R, belonging to the ent-atisane family.
The total synthesis of (+)-crotogoudin 268 began with the reaction of ketoester 264 with enal 265 to afford allylic pivaloate 266 in 12 steps. Tetracyclic compound 267 was synthesized in 80% yield and high diastereoselectivity (d.r. = 7.7:1) via a SmI2-mediated cascade reaction of allylic pivaloate 266 under optimized reaction conditions (THF/1,3-dimethyl-2-oxohexahydropyrimidine 9:1, 0 °C to r.t.) and due to steric hindrance of the allylic leaving group of 266. The reaction was found to be sensitive to the method of preparation of SmI2. Finally, methyl carboxylate 267 was converted into (+)-crotogoudin 268 in 54% and 4 steps (Scheme 48).
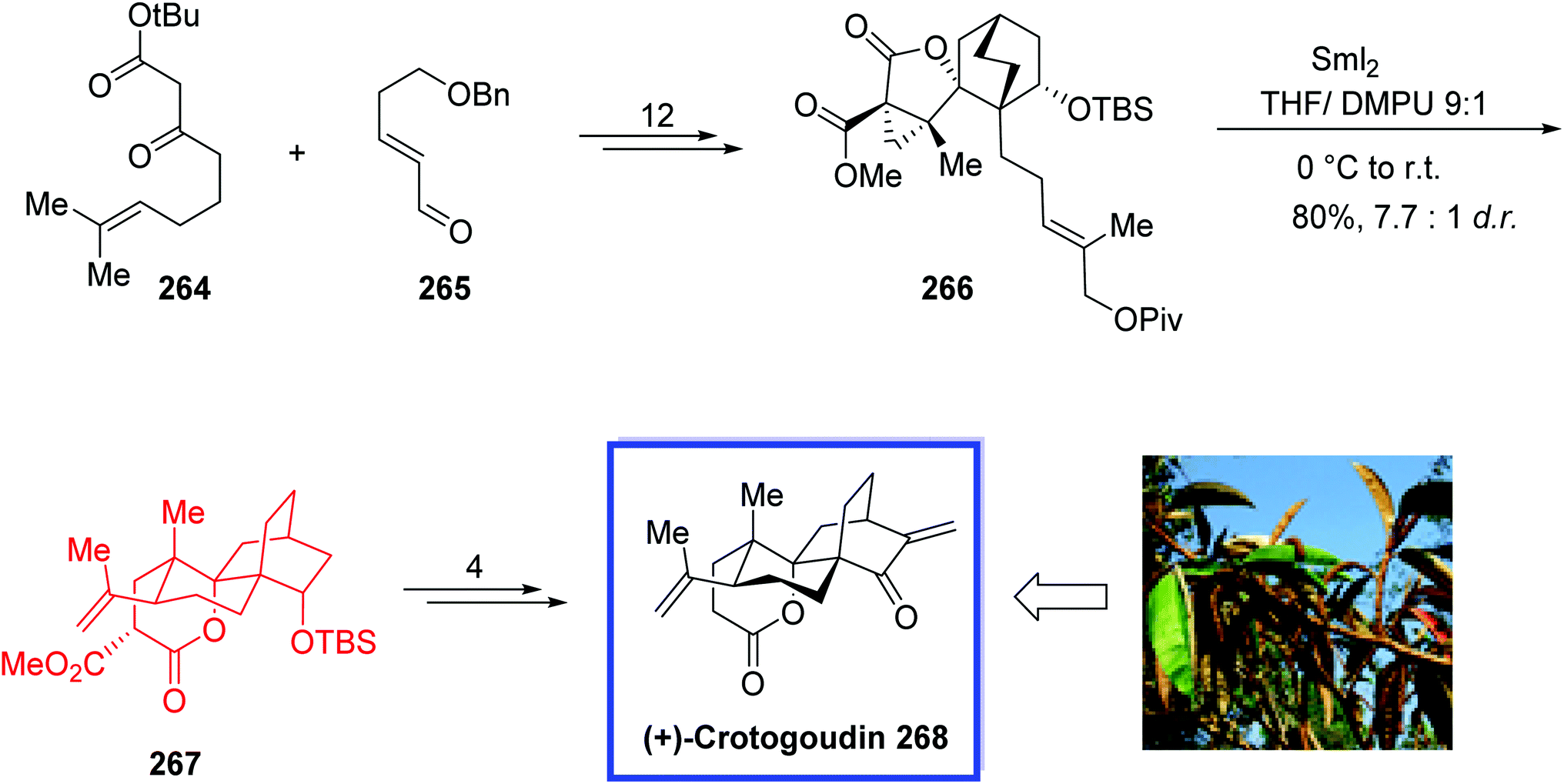 | ||
| Scheme 48 Total synthesis of (+)-crotogoudin 268. | ||
Aplykurodinone-1 272, isolated from the sea hare Syphonota geographica, belongs to a family of highly degraded marine steroids with cytotoxic activities against the various human cancer cell lines.119 Structurally, the aplykurodines have gained much interest in the synthetic community. Aplykurodinone-1 272 includes six contiguous stereocenters, a quaternary carbon center, and an unusual cis-fused C–D ring with epimeric C8 and unsaturated side chain.
In 2014, Li and coworkers120 developed an extraordinary method for the total synthesis of aplykurodinone-1 272 in 8 steps from known enone 269 without applying any protecting group. The key steps contain a SmI2-mediated reductive cyclization cascade reaction to synthesize an advanced intermediate and a direct cuprate-induced 1,4-addition for the direct installation of the C11 side chain. Their work was the first SmI2-promoted intramolecular cascade cyclization reaction between halide, alkene, and aldehyde functional groups.
The total synthesis of aplykurodinone-1 272 began with the easily available enone 269 to prepare the haloacetal-aldehyde 270, as an inseparable 1:1 mixture of diastereomeric haloacetal 271 in 3 steps. The SmI2-mediated reductive cascade cyclization reaction of precursor 270 with 2.0 equiv. of SmI2 in THF in the presence of 2.0 equiv. of HMPA easily constructed the tricyclic core and afforded the desired cyclization product 271a (C9-diastereoisomers 3.7:1 d.r.) in 42% yield together with the monocyclization byproduct 271b in 17% yield. The total synthesis of aplykurodinone-1 272 was completed from 271a in 3 steps installing the C11 side chain to afford 272, C13-epi-aplykurodinone-1 273 and undesired facial isomer C11-epi-aplykurodinone-1 274 each in 32% yield (Scheme 49).
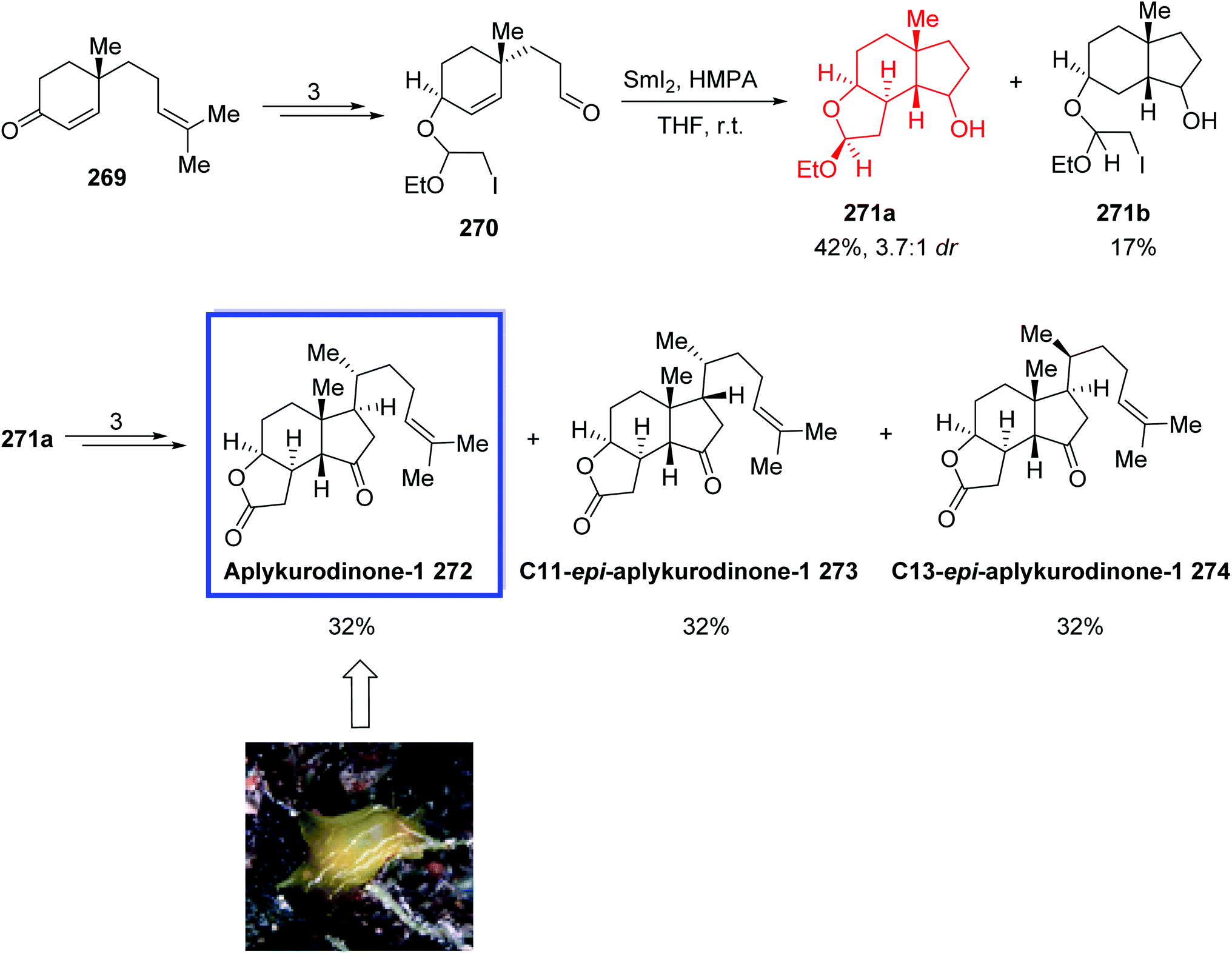 | ||
| Scheme 49 Total synthesis of aplykurodinone-1 272. | ||
Longeracemine 280, isolated from the fruits of Daphniphyllum longeracemosum, is a member of the Daphniphyllum alkaloids121 with 35 unique carbon frameworks to date, and over 320 members that have been isolated from evergreen shrubs and trees from southeast Asia. These alkaloids with diverse structures and plenty of polycyclic fused structures display various biological activities, such as antioxidant and anti-HIV activities, cytotoxicity against various cancer cell lines, insecticidal, and also known platelet aggregation inhibitors.121d,e Longeracemine retains a 5/6/5/5/6/5 skeleton, which comprises a highly functionalized 2-azabicyclo[2.2.1]heptane core and three contiguous quaternary carbons.
In 2020, Wood and coworkers122 reported the total synthesis of the 2-azabicyclo[2.2.1]heptane core of longeracemine 280. The key steps contained a stereoselective SmI2-mediated cascade reaction to synthesize the 2-azabicyclo[2.2.1]heptene from azabicyclo[2.2.1]heptadiene. The key SmI2-induced spirocyclization/rearrangement cascade created two adjacent all-carbon quaternary centers and provided the C-15 hydroxyl stereochemistry essential to synthesize 280. The spectral analysis shows the strong NOE correlation between the C15 hydrogen and the equatorial C1 hydrogen in both spirocycles 278a and 278b.
The total synthesis of 2-azabicyclo[2.2.1]heptanes 278a und 278b began with the reaction of N-Boc pyrrole 275 and ethyl 3-bromopropiolate 276 to obtain aldehyde 277 in 8 steps and 50% yield as a 3:2 mixture of diastereomers. The key SmI2-mediated spirocyclization/rearrangement cascade of 277 occurred under very mild conditions using three molar equivalents of SmI2 in THF at 23 °C for 1 h and stereoselectively provided the desired 2-azabicyclo[2.2.1]heptenes 278a (41%) and 278b (29%) in good yield with excellent selectivity. C15 hydroxyl protection of 278a as a triethylsilyl (TES) ether and removal of the Boc group resulted in secondary amine 279, which could establish the stage for additional development of 279 to 280 (Scheme 50).
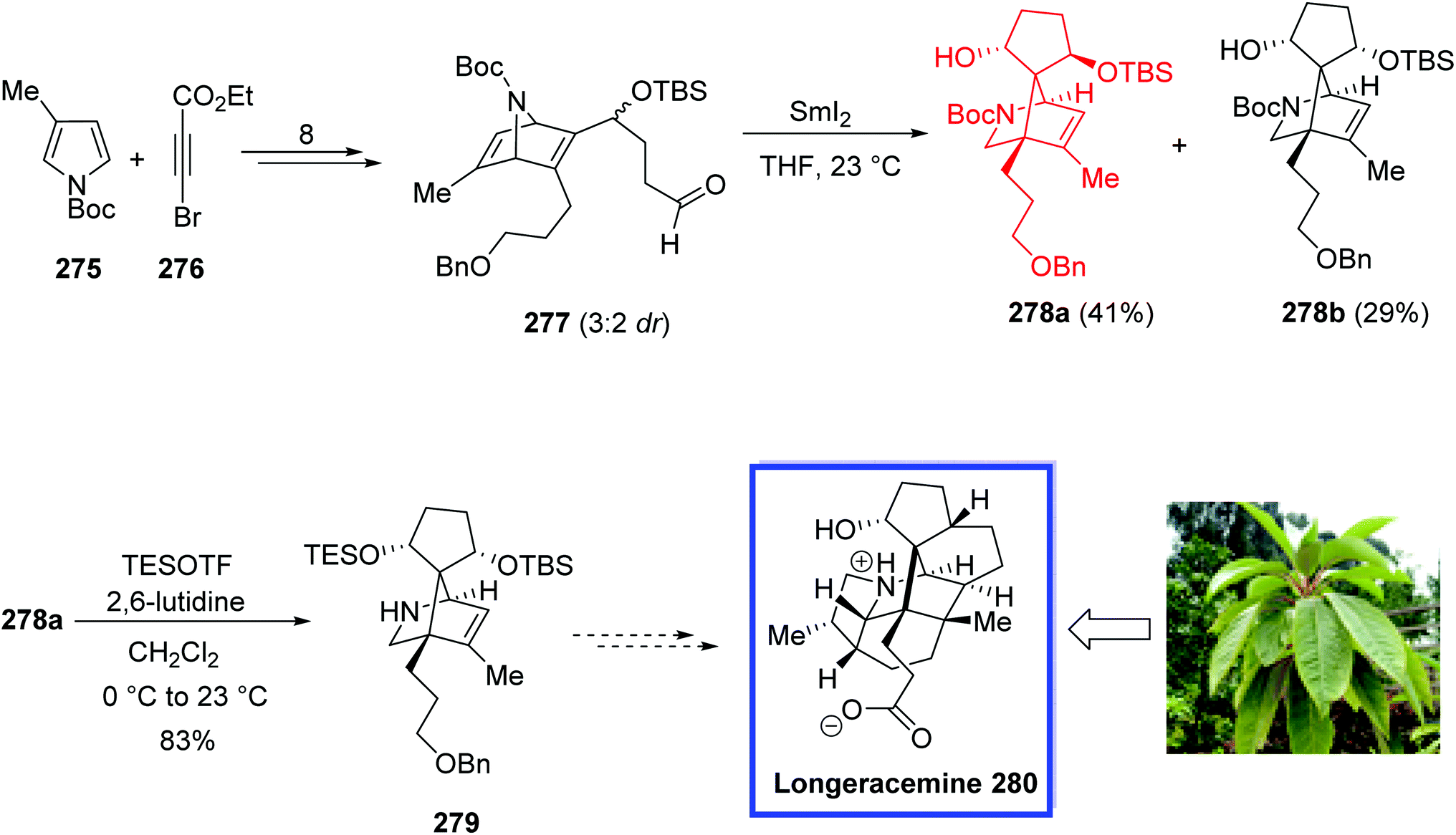 | ||
| Scheme 50 Total synthesis of 2-azabicyclo[2.2.1] heptane core of longeracemine 280. | ||
3. Conclusion
Manipulation and fine-tuning of the reduction potential, reaction rate, yield, high chemoselectivity, and stereoselectivity of SmI2 can be affected by varying the different additives and cosolvents of a reaction mixture. The very extensive reactivity window of this reagent and the remarkable ability of SmI2 to mediate both anionic and radical processes promote this highly chemoselective reagent a wide spectrum of synthetic strategies varying from functional group modifications to carbon–carbon bond-forming reactions, and in the case of natural products total synthesis, facilitating an otherwise impossible reaction or clean switching of reaction procedure.Knowing the enormous potential of SmI2-mediated reactions in modifications of functional groups, a survey of new reaction types and mechanistic routes and developing more general and powerful strategies with non-toxic additives are crucial for the application to large-scale synthesis. Additionally, to expand the use of the SmI2-mediated reactions in the future synthesis of complex, biologically active, and chiral target compounds in asymmetric syntheses and natural product total syntheses, it would be expected that this area is still very much considered a work in progress.
Conflicts of interest
There are no conflicts to declare.References
- (a) J. L. Namy, P. Girard and H. B. Kagan, New J. Chem., 1977, 1, 5–7 CAS; (b) P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS.
- D. J. Edmonds, D. Johnston and D. J. Procter, Chem. Rev., 2004, 104(7), 3371–3403 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48(39), 7140–7165 CrossRef CAS PubMed , and references therein;; (b) T. Nakata, Chem. Rec., 2010, 10(3), 159–172 CrossRef CAS PubMed; (c) T. Nakata, Chem. Soc. Rev., 2010, 39(6), 1955–1972 RSC; (d) E. Benedetti, C. Bressy, M. Smietana and S. Arseniyadis, Efficiency in Natural Product Total Synthesis, 2018, Chapter 6, Recent Applications of Kagan's Reagent (SmI2) in Natural Product Synthesis—First Edition, ed. P.-Q. Huang, Z. J. Yao and R. P. Hsung, John Wiley & Sons, 2018, and references therein Search PubMed; (e) K. Gopalaiah and H. B. Kagan, Chem. Rec., 2013, 13, 187–208 CrossRef CAS; (f) S. E. Dibrell, Y. Tao and S. E. Reisman, Acc. Chem. Res., 2021, 54(6), 1360–1373 CrossRef CAS; (g) S. Maity and S. Hoz, Eur. J. Org. Chem., 2021, 2021(7), 1103–1112 CrossRef CAS; (h) C. Beemelmanns and H.-U. Reissig, Pure Appl. Chem., 2011, 83(3), 507–518 CAS; (i) C. Beemelmanns and H.-U. Reissig, Pure Appl. Chem., 2011, 40(5), 2199–2210 CAS; (j) T. Honda, Heterocycles, 2010, 81(12), 2719–2747 CrossRef CAS; (k) T. Honda, Heterocycles, 2011, 83(1), 1–46 CrossRef CAS; (l) D. V. Sadasivam, K. A. Choquette and R. A. Flowers, J. Visualized Exp., 2013, 72, 4323–4327 Search PubMed; (m) T. Motoo and S. Masakazu, Heterocycles, 2014, 89(6), 1369–1391 CrossRef; (n) M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed; (o) K. Gopalaiah and H. B. Kagan, New J. Chem., 2008, 32(4), 607–637 RSC; (p) S. Shi and M. Szostak, Molecules, 2017, 22, 2018 CrossRef PubMed; (q) C. D. J. Aretz, Doctoral thesis, 2018; (r) C. T. Valeska, Doctoral thesis, 2017; (s) D. J. Procter, R. A. Flowers II and T. Skrydstrup, Organic Synthesis Using Samarium Diiodide: A Practical Guide, Published by the Royal Society of Chemistry, Cambridge, 2009 Search PubMed; (t) H. Pellissier, Beilstein J. Org. Chem., 2018, 14, 325–344 CrossRef CAS PubMed; (u) M. Sinast, M. Zuccolo, J. Wischnat, T. Sube, F. Hasnik, A. Baro, S. Dallavalle and S. Laschat, J. Org. Chem., 2019, 84, 10050–10064 CrossRef CAS PubMed.
- C. Matignon and E. Crazes, Ann. Chim., 1906, 8, 417–426 CAS.
- (a) M. Shabangi and R. A. Flowers II, Tetrahedron Lett., 1997, 38, 1137–1140 CrossRef CAS; (b) M. Shabangi, M. L. Kuhlman and R. A. Flowers II, Org. Lett., 1999, 1, 2133–2135 CrossRef CAS; (c) R. A. Flowers II, Synlett, 2008, 10, 1427–1439 CrossRef.
- (a) T. Imamoto and M. Ono, Chem. Lett., 1987, 16, 501–502 CrossRef; (b) G. A. Molander and C. Kenny, J. Org. Chem., 1991, 56, 1439–1445 CrossRef CAS; (c) N. Akane, T. Hatano, H. Kusui, Y. Nishiyama and Y. Ishii, J. Org. Chem., 1994, 59, 7902–7907 CrossRef CAS.
- (a) J. A. Teprovich, P. K. S. Antharjanam, E. Prasad, E. N. Pesciotta and R. A. Flowers, Eur. J. Inorg. Chem., 2008, 5015–5019 CrossRef CAS; (b) A. Dahlén and G. Hilmersson, Eur. J. Inorg. Chem., 2004, 3020 CrossRef; (c) J. M. Concellón, H. Rodríguez-Solla and E. B. M. Huerta, Eur. J. Org. Chem., 2003, 9, 1775–1778 CrossRef; (d) N. Akane, Y. Kanagawa, Y. Nishiyama and Y. Ishii, Chem. Lett., 1992, 21(12), 2431–2434 CrossRef; (e) M. Szostak, M. Spain and D. J. Procter, J. Org. Chem., 2012, 77, 3049–3059 CrossRef CAS; (f) J.-L. Namy, M. Colombo and H. B. Kagan, Tetrahedron Lett., 1994, 35(11), 1723–1726 CrossRef CAS; (g) S. M. Ruder, Tetrahedron Lett., 1992, 33(19), 2621–2624 CrossRef CAS; (h) M. Kunishima, K. Hioki, T. Ohara and S. J. Tani, J. Chem. Soc., Chem. Commun., 1992, 3, 219–220 RSC.
- (a) M. L. Kuhlman and R. A. Flowers II, Tetrahedron Lett., 2000, 41(42), 8049–8052 CrossRef CAS; (b) H. Farran and S. Hoz, Org. Lett., 2008, 10(5), 865–867 CrossRef CAS PubMed; (c) R. J. Enemærke, T. Hertz, T. Skrydstrup and k. Daasbjerg, Chem.–Eur. J., 2000, 6(20), 3747–3754 CrossRef.
- T. Gillmann, Tetrahedron Lett., 1993, 34, 607–610 CrossRef CAS.
- (a) K. Griesbaum, Angew. Chem., Int. Ed. Engl., 1966, 5, 933–946 CrossRef CAS; (b) H. G. Kuivila, W. Rahman and R. H. Fish, J. Am. Chem. Soc., 1965, 87, 2835–2840 CrossRef CAS; (c) D. A. Pasto and G. J. L'Hermine, Org. Chem., 1990, 55, 685–694 CrossRef CAS.
- A. Hölemann and H.-U. Reissig, Org. Lett., 2003, 5, 1463–1466 CrossRef PubMed.
- G. A. Molander and E. P. Cormier, J. Org. Chem., 2005, 70, 2622–2626 CrossRef CAS PubMed.
- S. Agasti, N. A. Beattie, J. Joseph, W. McDouall and D. J. Procter, J. Am. Chem. Soc., 2021, 143(9), 3655–3661 CrossRef CAS PubMed.
- A. Nazari, M. M. Heravi and V. Zadsirjan, J. Organomet. Chem., 2021, 932, 121629 CrossRef CAS; M. M. Heravi, V. Zadsirjan, M. Daraie and M. Ghanbarian, ChemistrySelect, 2020, 5, 9654–9690 CrossRef; M. M. Heravi, M. Ghanbarian, V. Zadsirjan and B. Alimadadi Jani, Monatsh. Chem., 2019, 150, 1365–1407 CrossRef; M. M. Heravi, T. B. Lashaki, B. Fattahi and V. Zadsirjan, RSC Adv., 2018, 8, 6634–6659 RSC; M. M. Heravi, V. Zadsirjan, M. Esfandyari and T. B. Lashaki, Tetrahedron: Asymmetry, 2017, 28(8), 987–1043 CrossRef; M. M. Heravi, S. Rohani, V. Zadsirjan and N. Zahedi, RSC Adv., 2017, 7(83), 52852–52887 RSC; M. M. Heravi, T. B. Lashaki and N. Poorahmad, Tetrahedron: Asymmetry, 2015, 26(8–9), 405–495 CrossRef; M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24, 1149–1188 CrossRef; M. M. Heravi and V. F. Vavsari, RSC Adv., 2015, 5, 50890–50912 RSC; M. M. Heravi and N. Nazari, Curr. Org. Chem., 2015, 19, 2358–2408 CrossRef; M. M. Heravi, V. Zadsirjan and Z. Bozorgpour Savadjani, Curr. Org. Chem., 2014, 18, 2857–2891 CrossRef; M. M. Heravi, A. Bakhtiari and Z. Faghihi, Curr. Org. Synth., 2014, 11, 787–823 CrossRef; M. M. Heravi, E. Hashemi and N. Nazari, Mol. Diversity, 2014, 18, 441–472 CrossRef PubMed; M. M. Heravi, E. Hashemi and F. Azimian, Tetrahedron, 2014, 1, 7–21 CrossRef; M. M. Heravi, E. Hashemi and N. Ghobadi, Curr. Org. Chem., 2013, 17, 2192–2224 CrossRef; M. M. Heravi, S. Asadi and B. M. Lashkariani, Mol. Diversity, 2013, 17, 389–407 CrossRef PubMed.
- (a) M. Hayashi, M. Ohno, K. Kinoshita, S. Satoi, M. Suzuki and K. Harada, J. Antibiot., 1981, 34, 346–349 CrossRef CAS PubMed; (b) M. Hayashi, H. OHara, M. Ohno, H. Sakakibara, S. Satoi, K. Harada and M. Suzuki, J. Antibiot., 1981, 34, 1075–1077 CrossRef CAS PubMed; (c) B. Herlé, G. Späth, L. Schreyer and A. Fürstner, Angew. Chem., Int. Ed., 2021, 60, 7893–7899 CrossRef PubMed; (d) S. Satoi, N. Muto, M. Hayashi, T. Fujii and M. Otani, Jpn. J. Antibiot., 1980, 33, 364–376 CrossRef CAS PubMed.
- Y. Ogawa, K. Kuroda and T. Mukaiyama, Chem. Lett., 2005, 34(5), 698–699 CrossRef CAS.
- J. J. Barchi, R. E. Moore and G. M. L. Patterson, J. Am. Chem. Soc., 1984, 106, 8193–8197 CrossRef CAS.
- R. M. Moslin and T. F. Jamison, J. Am. Chem. Soc., 2006, 128, 15106–15107 CrossRef CAS PubMed.
- (a) J. Kobayashi, M. Sekiguchi, S. Shimamoto, H. Shigemori, H. Ishiyama and A. J. Ohsaki, Org. Chem., 2002, 67, 6449–6455 CrossRef CAS PubMed; (b) H. Ishiyama, M. Matsumoto, M. Sekiguchi, H. Shigemori, A. Ohsaki and J. Kobayashi, Heterocycles, 2005, 66, 651–658 CrossRef CAS.
- P. Gao, Y. Liu, L. Zhang, P.-F. Xu, S. Wang, Y. Lu, M. He and H. J. Zhai, Org. Chem., 2006, 71, 9495–9498 CrossRef CAS PubMed.
- (a) G. E. Raoelison, C. Terreaux, E. F. Queiroz, F. Zsila, M. Simonyi, S. Antus, A. Randriantsova and K. Hostetmann, Helv. Chim. Acta, 2001, 84, 3470 CrossRef CAS; (b) L. D. Juliawaty, M. Kitajima, H. Takayama, S. A. Achmad and N. Aimi, Phytochemistry, 2000, 54, 989 CrossRef CAS PubMed.
- (a) K. Yasui, Y. Tamura, T. Nakatani, K. Kawada and M. Ohtani, J. Org. Chem., 1995, 60, 7567 CrossRef CAS; (b) X.-P. Fang, J. E. Anderson, C.-J. Chang, J. L. McLaughlin and P. E. Fanwick, J. Nat. Prod., 1991, 54, 1034 CrossRef CAS PubMed; (c) A. D. Argoudelis and A. D. Zieserl, Tetrahedron Lett., 1966, 7, 1969 CrossRef; (d) K. R. Romines and R. A. Chrusciel, Curr. Med. Chem., 1995, 2, 825 CrossRef CAS; (e) S. H. Inayat-Hussain, A. B. Osman, L. B. Din and N. Taniguchi, Toxicol. Lett., 2009, 131, 153 CrossRef; (f) H. Kikuchi, K. Sasaki, J. Sekiya, Y. Maeda, A. Amagai, Y. Kubohara and Y. Ohsima, Bioorg. Med. Chem., 2004, 12, 3203 CrossRef CAS PubMed; (g) A. D. Fátima, L. K. Kohn, M. A. Antônio, J. E. de Carvalho and D. R. A. Pilli, Bioorg. Med. Chem., 2005, 13, 2927 CrossRef PubMed.
- S. Rajaram, D. Ramesh, U. Ramulu, P. Prabhakar and Y. Venkateswarlu, Helv. Chim. Acta, 2014, 97, 112–121 CrossRef CAS.
- (a) J.-X. Pu, X.-M. Gao, C. Lei, W.-L. Xiao, R.-R. Wang, L.-B. Yang, Y. Zhao, L.-M. Li, S.-X. Huang, Y.-T. Zheng and H.-D. Sun, Chem. Pharm. Bull., 2008, 56, 1143 CrossRef CAS PubMed; (b) C. F. Chyu, M. R. Ke, Y. S. Chang, S. C. Chien and Y. H. Kuo, Helv. Chim. Acta, 2007, 90, 1514 CrossRef CAS; (c) C.-H. Chao, J.-C. Cheng, D.-Y. Shen and T.-S. Wu, J. Nat. Prod., 2014, 77, 22 CrossRef CAS PubMed; (d) L. S. Long, k. Lee, H.-B. Chai, P. Rasoanaivo, Q. Gao, H. Navarro, M. E. Wall, M. C. Wani, N. R. Farnsworth, G. A. Cordell, J. M. Pezzuto and A. D. Kinghorn, Tetrahedron, 1997, 53, 15663 CrossRef CAS; (e) D. H. Miles, V. Chittawong, D.-S. Lho, A. M. Payne, A. A. De la Cruz, E. D. Gomez, J. A. Weeks and J. L. Atwood, J. Nat. Prod., 1991, 54, 286 CrossRef CAS.
- (a) T.-S. Wu, Y.-L. Tsai, A. G. Damu, P.-C. Kuo and P.-L. Wu, J. Nat. Prod., 2002, 65, 1522 CrossRef CAS PubMed; (b) Z. Gang, PhD dissertation, Chiba University, Japan, 2010.
- (a) D. H. Miles, A. M. Ly, V. Chittawong, A. A. De la Cruz and E. D. Gomez, J. Nat. Prod., 1989, 52, 896 CrossRef CAS PubMed; (b) D. H. Miles, D. S. Lho, A. A. De la Cruz, E. D. Gomez, J. A. Weeks and J. L. Atwood, J. Org. Chem., 1987, 52, 2930 CrossRef CAS; (c) F. Bohlmann, C. Zdero and N. Le Van, Phytochemistry, 1979, 18, 99 CrossRef CAS.
- K. Nabeta, K. Katayama, S. Nakagawara and K. Katoh, Phytochemistry, 1993, 32, 117 CrossRef.
- R. U. Batwal and N. P. Argade, Org. Biomol. Chem., 2015, 13, 11331–11340 RSC.
- (a) S. Saito, K. Kotera, N. Shigematsu, A. Ide, N. Sugimoto, Z. Horii, M. Hanaoka, Y. Yamawaki and Y. Tamura, Tetrahedron, 1963, 19, 2085–2099 CrossRef CAS PubMed; (b) I. Satoda, M. Murayama, J. Tsuji and E. Yoshii, Tetrahedron Lett., 1962, 3, 1199–1206 CrossRef; (c) S. Saito, K. Kotera, N. Sugimoto, Z. Horii and Y. Tamura, Chem. Ind., 1962, 1652–1653 CAS.
- S. Saito, N. Shigematsu and Z.-I. Horii, J. Pharm. Soc. Jpn., 1963, 83, 800–801 CrossRef CAS.
- V. I. Murav’eva and A. I. Ban’kovskii, Dokl. Akad. Nauk SSSR, 1956, 110, 998–1000 Search PubMed.
- Z.-I. Horii, T. Tanaka, Y. Tamura, S. Saito, C. Matsumura and N. J. Sugimoto, J. Pharm. Soc. Jpn., 1963, 83, 602–605 CrossRef CAS.
- D. Rajand and M. Luczkiewicz, Fitoterapia, 2008, 79, 419–427 CrossRef PubMed.
- J. A. Beutler, E. W. Karbon, A. N. Brubaker, R. Malik, D. R. Curtis and S. J. Enna, Brain Res., 1985, 330, 135–140 CrossRef CAS PubMed.
- C. H. Jang, J. S. Eun, H. W. Park, S. M. Seo, J. H. Yang, K. H. Leem, S. H. Oh, C. H. Oh, N. I. Baek and D. K. Kim, Korean J. Pharmacogn., 2003, 34, 14–17 CAS.
- X. Zheng, J. Liu, C.-X. Ye, A. Wang, A.-E. Wang and P.-Q. Huang, J. Org. Chem., 2015, 80, 1034–1041 CrossRef CAS PubMed.
- (a) N. Bartke and Y. A. Hannun, J. Lipid Res., 2009, 50, 91–96 CrossRef PubMed; (b) S. T. Pruett, A. Bushnev, K. Hagedorn, M. Adiga, C. A. Haynes and A. H. Merrill, J. Lipid Res., 2008, 49, 1621–1639 CrossRef CAS PubMed; (c) T. H. Won, M. You, S.-H. Lee, B. J. Rho, D.-C. Oh, K.-B. Oh and J. Shin, Mar. Drugs, 2014, 12, 3754–3769 CrossRef CAS PubMed.
- L. Garrido, E. Zubia, M. J. Ortega, N. Santiago and J. Salva, Tetrahedron, 2001, 57, 4579–4588 CrossRef CAS.
- (a) C. Jimenez and P. Crews, J. Nat. Prod., 1990, 53, 978–982 CrossRef CAS PubMed; (b) R. J. Clark, M. J. Garson and J. N. Hooper, J. Nat. Prod., 2001, 64, 1568–1571 CrossRef CAS PubMed; (c) L. Garrido, E. Zubia, M. J. Ortega, N. Santiago and J. Salva, Tetrahedron, 2001, 57, 4579–4588 CrossRef CAS; (d) E. A. Jares-Erijman, C. P. Bapat, A. Lithgow-Bertelloni, K. L. Rinehart and R. Sakai, J. Org. Chem., 1993, 58, 5732–5737 CrossRef CAS; (e) M. L. Giavatta, E. Manzo, G. Nuzzo and G. Villani, Tetrahedron, 2010, 66, 7533–7538 CrossRef; (f) A. Aiello, E. Fattorruso, A. Giordano, M. Menna, C. Navarrete and E. Munoz, Bioorg. Med. Chem., 2007, 15, 2920–2926 CrossRef CAS PubMed.
- L. Filippova, S. Antonsen, Y. Stenstrøm and T. V. Hansen, Tetrahedron, 2016, 72, 6572–6577 CrossRef CAS.
- For some reviews on marine alkaloids, see: (a) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2016, 33, 382 RSC , and references therein; For some reviews on guanidine alkaloid syntheses, see:; (b) R. G. S. Berlinck and S. Romminger, Nat. Prod. Rep., 2016, 33, 456 RSC , and references therein;; (c) X. Wei, N. M. Henriksen, J. J. Skalicky, M. K. Harper, T. E. Cheatham, III, C. M. Ireland and R. M. J. Van Wagoner, Org. Chem., 2011, 76, 5515 CrossRef CAS PubMed.
- M. Tian, M. Yan and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 14234–14237 CrossRef CAS PubMed.
- (a) L. Li, W. Han, Y. Gu, S. Qiu, Q. Lu, J. Jin, J. Luo and X. Hu, Cancer Res., 2007, 67, 4894–4903 CrossRef CAS PubMed; (b) H.-C. Pan, D.-W. Lai, K.-H. Lan, C.-C. Shen, S. M. Wu, C.-S. Chiu, K.-B. Wang and M.-L. Sheu, Carcinogenesis, 2013, 34, 2568–2579 CrossRef CAS PubMed; (c) G. D. Kim, J. Oh, H.-J. Park, K. Bae and S. K. Lee, Int. J. Oncol., 2013, 43, 600–610 CrossRef CAS PubMed.
- (a) X. Bai, F. Cerimele, M. Ushiko-Fukai, M. Waqas, P. M. Campbell, B. Govindarajan, C. J. Der, T. Battle, D. A. Frank, K. Ye, E. Murad, W. Dubiel, G. Soff and J. L. Arbiser, J. Biol. Chem., 2003, 278, 3551–3557 Search PubMed; (b) L. E. Fried and J. L. Arbiser, Antioxid. Redox Signaling, 2009, 11, 1139–1148 CrossRef CAS PubMed; (c) L. Ma, J. Chen, X. Wang, X. Liang, Y. Luo, W. Zhu, T. Wang, M. Peng, S. Li, S. Jie, A. Peng, Y. Wei and L. J. Chen, Med. Chem., 2011, 54, 6469–6481 CrossRef CAS PubMed.
- Y. J. Lee, Y. M. Lee, C. K. Lee, J. K. Jung, S. B. Han and J. T. Hogn, Pharmacol. Ther., 2011, 130, 157–176 CrossRef CAS PubMed.
- A. M. Wright and G. W. O'Neil, Tetrahedron Lett., 2016, 57, 3441–3443 CrossRef CAS.
- (a) T. El-Elimat, H. A. Raja, M. Figueroa, S. M. Swanson, J. O. Falkinham, III, D. M. Lucas, M. R. Grever, M. C. Wani, C. J. Pearce and N. H. Oberlies, J. Antibiot., 2015, 68, 191 CrossRef CAS PubMed; (b) M.-M. Zhai, F.-M. Qi, J. Li, C.-X. Jiang, Y. Hou, Y.-P. Shi, D.-L. Di, J.-W. Zhang and Q.-X. Wu, J. Agric. Food Chem., 2016, 64, 2298 CrossRef CAS PubMed.
- (a) A. M. Harned and K. A. Volp, Nat. Prod. Rep., 2011, 28, 1790 RSC; (b) J. Meng, X. Wang, D. Xu, X. Fu, X. Zhang, D. Lai, L. Zhou and G. Zhang, Molecules, 2016, 21, 715 CrossRef PubMed.
- Q. Yan, M. G. Banwell, M. L. Coote, R. Lee and A. C. Willis, Chem.–Asian J., 2017, 12, 1480–1484 CrossRef CAS PubMed.
- (a) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801–1839 RSC; (b) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2010, 39, 4402–4421 RSC; (c) Ch. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep., 2006, 23, 517–531 RSC; (d) J. T. Gupton, Top. Heterocycl. Chem., 2006, 2, 53–92 CAS; (e) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC; (f) M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed; (g) X. Álvarez-Mico, P. R. Jensen, W. Fenical and C. C. Hughes, Org. Lett., 2013, 15(5), 988–991 CrossRef PubMed.
- J. P. Mahajan and S. B. Mhaske, Org. Lett., 2017, 19, 2774–2776 CrossRef CAS PubMed.
- (a) K. S. Lam, G. A. Hesler, D. R. Gustavson, A. R. Crosswell, J. M. Veitch, S. Forenza and K. Tomita, J. Antibiot., 1991, 44, 472–478 CrossRef CAS PubMed; (b) S. J. Hofstend, J. A. Matson, A. R. Malacko and H. Marquardt, J. Antibiot., 1992, 45, 1250–1254 CrossRef PubMed; (c) J. R. Lohman, S. X. Huang, G. P. Horsman, P. E. Dilfer, T. Huang, Y. Chen, E. Wendt-Pienkowski and B. Shen, Mol. BioSyst., 2013, 9, 478–491 RSC; (d) J. E. Leet, D. R. Schroeder, S. J. Hofstead, J. Gorik, K. L. Colson, S. Huang, S. E. Klohr, T. W. Doyle and T. W. Matson, J. Am. Chem. Soc., 1992, 114, 7946–7948 CrossRef CAS; (e) J. E. Leet, et al., J. Am. Chem. Soc., 1993, 115, 8432–8443 CrossRef CAS; (f) K. L. Constantine, K. L. Colson, M. Wittekind, M. S. Friedrichs, N. Zein, J. Tuttle, D. R. Langley, J. E. Leet, D. R. Schroeder, K. S. Lam, B. T. Farmer II, W. J. Metzler, R. E. Bruccoleri and L. Mueller, Biochemistry, 1994, 33, 11438–11452 CrossRef CAS PubMed; (g) N. Zein, A. M. Casazza, T. W. Doyle, J. E. Leet, D. R. Schroeder, W. Solomon and S. G. Nadler, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8009–8012 CrossRef CAS PubMed; (h) K. Iida and M. Hirama, J. Am. Chem. Soc., 1995, 117, 8875–8876 CrossRef CAS; (i) M. Hirama, Pure Appl. Chem., 1997, 69, 525–530 CrossRef CAS.
- M. J. Lear, K. Hirai, K. Ogawa, S. Yamashita and M. Hirama, J. Antibiot., 2019, 72, 350–363 CrossRef CAS PubMed.
- H.-M. Chung, J.-H. Su, T.-L. Hwang, J.-J. Li, J.-J. Chen, Y.-H. Chen, Y.-C. Chang, Y.-D. Su, Y.-H. Chen, L.-S. Fang, J.-H. Sheu, W.-H. Wang and P.-J. Sung, Tetrahedron, 2013, 69, 2740–2744 CrossRef CAS.
- (a) Z. Khaing, D. Kang, A. M. Camelio, C. E. Schmidt and D. Siegel, Bioorg. Med. Chem. Lett., 2011, 21, 4808–4812 CrossRef CAS PubMed; (b) X. Cheng, N. Harzdorf, Z. Khaing, D. Kang, A. M. Camelio, T. Shaw, C. E. Schmidt and D. Siegel, Org. Biomol. Chem., 2012, 10, 383–393 RSC.
- G. Liu, Z. Zhang, S. Fu and Bo. Liu, Org. Lett., 2021, 23, 290–295 CrossRef CAS PubMed.
- (a) T. Tsuruoka, H. Yumoto, N. Ezaki and T. Niida, Sci. Rep., 1967, 9, 1–4 Search PubMed; (b) T. Noguchi, Y. Yasuda, T. Niida and T. Shomura, Ann. Phytopathol. Soc. Jpn., 1968, 34, 323–327 CrossRef CAS; (c) H. Seto, M. Koyama, H. Ogino, T. Tsuruoka, S. Inouye and N. Otake, Tetrahedron Lett., 1983, 24, 1805–1808 CrossRef CAS.
- F. Marcelo, J. Jiménez-Barbero, J. Marrot, A. P. Rauter, P. Sinaÿ and Y. Blériot, Chem.–Eur. J., 2008, 14, 10066–10073 CrossRef CAS PubMed.
- (a) S. V. Binns, P. J. Dunstan, G. B. Guise, G. M. Holder, A. F. Hollis, R. S. McCredie, J. T. Pinhey, R. H. Prager, M. Rasmussen, E. Ritchie and W. C. Taylor, Aust. J. Chem., 1965, 18, 569 CrossRef CAS; (b) L. N. Mander, R. H. Prager, M. Rasmussen, E. Ritchie and W. C. Taylor, Aust. J. Chem., 1967, 20, 1473 CrossRef CAS; (c) L. N. Mander, R. H. Prager, M. Rasmussen, E. Ritchie and W. C. Taylor, Aust. J. Chem., 1967, 20, 1705 CrossRef CAS; (d) L. N. Mander and M. M. McLachlan, J. Am. Chem. Soc., 2003, 125, 2400 CrossRef CAS PubMed; (e) M. Movassaghi, D. K. Hunt and M. Tjandra, J. Am. Chem. Soc., 2006, 128, 8126 CrossRef CAS PubMed; (f) U. Shah, S. Chackalamannil, A. K. Ganguly, M. Chelliah, S. Kolotuchin, A. Buevich and A. McPhail, J. Am. Chem. Soc., 2006, 128, 12654 CrossRef CAS PubMed.
- L. N. Mander, A. C. Willis, A. J. Herlt and W. C. Taylor, Tetrahedron Lett., 2009, 50, 7089 CrossRef CAS.
- (a) S. Chackalamannil, Y. Wang, W. J. Greenlee, Z. Hu, Y. Xia, H.-S. Ahn, G. Boykow, Y. Hsieh, J. Palamanda, J. Agans-Fantuzzi, S. Kurowski, M. Graziano and M. Chintala, J. Med. Chem., 2008, 51, 3061 CrossRef CAS PubMed; (b) M. J. Malaska, A. H. Fauq, A. P. Kozikowski, P. J. Aagaard and M. McKinney, Bioorg. Med. Chem. Lett., 1995, 5, 61 CrossRef CAS.
- W. Zi, S. Yu and D. Ma, Angew. Chem., Int. Ed., 2010, 49, 5887–5890 CrossRef CAS PubMed.
- (a) W. A. Ayer, B. Altenkirk, S. Valverde-Lopez, B. Douglas, R. F. Raffauf and J. A. Weisbach, Can. J. Chem., 1968, 46, 15 CAS; (b) W. A. Ayer, B. Altenkirk and Y. Fukazawa, Tetrahedron, 1974, 30, 4213 CrossRef CAS; (c) W. A. Ayer and L. S. Trifonov, in The Alkaloids, ed. G. A. Cordell and A. Brossi, Academic Press, New York, 1994, vol. 45, p. 233 Search PubMed.
- (a) Y. Hirasawa, H. Morita, M. Shiro and J. Kobayashi, J. Org. Lett., 2003, 5, 3991 CrossRef CAS PubMed; (b) Y. Hirasawa, J. Kobayashi and H. Morita, Heterocycles, 2009, 77, 679–729 CrossRef CAS; (c) J. Kobayashi and H. Morita, in The Alkaloids, ed. G. A. Cordell, Academic Press, New York, 2005, vol. 61, pp. 1–57 Search PubMed; (d) X. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752–772 RSC; (e) M. Kitajima and H. Takayama, Isolation and Asymmetric Synthesis in Topics in Current Chemistry, ed. H. J. Knölker, Springer, Berlin, 2011, vol. 309, p. 1 Search PubMed; (f) Y. Hirasawa, H. Morita, M. Shiro and J. Kobayashi, Org. Lett., 2003, 5, 3991 CrossRef CAS PubMed; (g) J. He, X.-Q. Chen, M.-M. Li, Y. Zhao, G. Xu, X. Cheng, L.-Y. Peng, M.-J. Xie, Y.-T. Zheng, Y.-P. Wang and Q.-S. Zhao, Org. Lett., 2009, 11, 1397 CrossRef CAS PubMed.
- X.-M. Zhang, Y.-Q. Tu, F.-M. Zhang and S.-H. Wang, Angew. Chem., Int. Ed., 2011, 50, 3916–3919 CrossRef CAS PubMed.
- X.-M. Zhang, H. Shao, Y.-Q. Tu, F.-M. Zhang and S.-H. Wang, J. Org. Chem., 2012, 77, 8174–8181 CrossRef CAS PubMed.
- (a) J. He, X.-Q. Chen, M.-M. Li, Y. Zhao, G. Xu, X. Cheng, L.-Y. Peng, M.-J. Xie, Y.-T. Zheng, Y.-P. Wang and Q.-S. Zhao, Org. Lett., 2009, 11, 1397 CrossRef CAS PubMed; (b) R. H. Burnell, J. Chem. Soc., 1959, 3091 CAS; (c) R. H. Burnell, C. G. Chin, B. S. Mootoo and D. R. Taylor, Can. J. Chem., 1963, 41, 3091 CrossRef CAS; (d) Y. Inubushi, H. Ishii, B. Yasui, Y. Harayama, M. Hosokawa, R. Nishino and Y. Nakahara, Yakugaku Zasshi, 1967, 87, 1394 CrossRef CAS PubMed.
- H. Li, X. Wang, B. Hong and X. Lei, J. Org. Chem., 2013, 78, 800–821 CrossRef CAS PubMed.
- (a) A. R. Carroll, E. Hyde, J. Smith, R. J. Quinn, G. Guymer and P. I. Forster, J. Org. Chem., 2005, 70, 1096 CrossRef CAS PubMed , later, Morita et al.;; (b) K. Koyama, Y. Hirasawa, T. Hosoya, T. Hoe, K. Chan and H. Morita, Bioorg. Med. Chem., 2010, 18, 4415 CrossRef CAS PubMed , in 2005, Luo et al.;; (c) X. Qin, Y. Zhao, P. Lunga, X. Yang, C. Song, G. Cheng, L. Liu, Y. Chen, Y. Liu and X. Luo, Tetrahedron, 2015, 71, 4372 CrossRef CAS.
- (a) J. Leurs, V. Nerme, Y. Sim and D. J. J. Hendriks, Thromb. Haemostasis, 2004, 2, 416 CrossRef CAS PubMed; (b) M. O. Polla, L. Tottie, C. Norden, M. Linschoten, M. Musil, S. Trumpp-Kallmeyer, I. R. Aurkurst, R. Ringom, K. H. Holm, S. M. Neset, M. Sandberg, J. Thurmond, P. Yu, G. Hategan and H. Anderson, Bioorg. Med. Chem., 2004, 12, 1151 CrossRef CAS PubMed.
- (a) J. Leurs and D. Hendriks, Thromb. Haemostasis, 2005, 94, 417 Search PubMed; (b) V. J. Marder, W. C. Aird, J. S. Bennett, S. Schulman and G. C. White, Hemostasis and Thrombosis: Basic Principles and Clinical Practice, Lippincott Williams & Wilkins, Philadelphia, 6th edn, 2013 Search PubMed.
- L. Cai, K. Zhang and O. Kwon, J. Am. Chem. Soc., 2016, 138, 3298–3301 CrossRef CAS PubMed.
- (a) H. Iwabuchi, Abstracts of Papers: 41st Symposium on the Chemistry of Terpenes, Essential Oils, and Aromatics, Morioka, Japan, 1997 Search PubMed; (b) K. Takahashi, T. Someya, S. Muraki and T. Yoshida, Agric. Biol. Chem., 1980, 44, 1535 CAS; (c) S. K. Tsuboi, K. Shimozuma and A. Takeda, J. Org. Chem., 1980, 45, 1517 CrossRef CAS; (d) J. A. Hirsch and R. H. Eastman, J. Org. Chem., 1967, 32, 2915 CrossRef CAS.
- X. Wang, H. Wanga, X. Wu, T. Yua, W. Gaoa, T. Shi, X. Peng, D. He and Z. Wang, Synlett, 2017, 28, 1660–1662 CrossRef CAS.
- (a) S. Murakami, T. Takemoto and Z. Shimizu, J. Pharm. Soc. Jpn., 1953, 73, 1026–1029 CrossRef CAS; (b) I. Nitta, H. Watase and Y. Tomiie, Nature, 1958, 181, 761–762 CrossRef CAS PubMed; (c) I. Nitta, H. Watase and Y. Tomiie, Bull. Chem. Soc. Jpn., 1958, 31, 714–725 CrossRef; (d) H. Watase, Bull. Chem. Soc. Jpn., 1958, 31, 932–940 CrossRef CAS; (e) W. Oppolzer and K. Thirring, J. Am. Chem. Soc., 1982, 104, 4978–4979 CrossRef CAS.
- (a) P. R. Simon, Excitatory Amino Acids, ed. Thieme Medical, New York, 1992 Search PubMed; (b) P. J. Roberts, J. Storm-Mathisen and G. A. R. Johnston, Glutamate: Transmitter in the Central Nervous System, John Wiley, Chichester, UK, 1981, pp. 89–115 Search PubMed; (c) R. L. Johnson and J. F. Koerner, J. Med. Chem., 1988, 31, 2057–2066 CrossRef CAS PubMed; (d) E. G. McGeer, J. W. Olney and P. L. McGeer. Kainic Acid as a Tool in Neurobiology, Raven, New York, 1978 Search PubMed; (e) H. Shinozaki and S. Konishi, Brain Res., 1970, 24, 368–371 CrossRef CAS PubMed.
- J. Suzuki, N. Miyano, S. Yashiro, T. Umezawa and F. Matsuda, Org. Biomol. Chem., 2017, 15, 6557 RSC.
- (a) T. Nohara, Y. Kashiwada and T. Tomimatsu, Tetrahedron Lett., 1980, 21, 2647–2648 CrossRef CAS; (b) M. F. Braulio, G. C. Azucena, G. Carmen and T. David, J. Nat. Prod., 1997, 60, 880–888 CrossRef.
- (a) A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111 CrossRef CAS PubMed; (b) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef CAS PubMed; (c) T. K. Allred, F. Manoni and P. G. Harran, Chem. Rev., 2017, 117, 11994 CrossRef CAS PubMed.
- M. Koshimizu, M. Nagatomo and M. Inoue, Tetrahedron, 2018, 74, 3384–3390 CrossRef CAS.
- (a) A. Laguna, L. Novotny, L. Dolejs and M. Bude sinsky, Planta Med., 1984, 50, 285–288 CrossRef CAS PubMed; (b) G. Bentham and J. D. Hooker, Gen. Pl., 1876, 2, 702 Search PubMed; (c) J. Hajicek, J. Taimr and M. Budesinsky, Tetrahedron Lett., 1998, 39, 505–508 CrossRef CAS; (d) R. M. Kariba, P. J. Houghton and A. Yenesew, J. Nat. Prod., 2002, 65, 566–569 CrossRef CAS PubMed; (e) Y. Atilaw, M. Heydenreich, A. Ndakala, H. M. Akala, E. Kamau and A. Yenesew, Phytochem. Lett., 2014, 10, 28–31 CrossRef CAS; (f) J. Hajicek and J. Trojanek, Collect. Czech. Chem. Commun., 1986, 51, 1731–1742 CrossRef CAS.
- X. Zeng and D. L. Boger, J. Am. Chem. Soc., 2021, 143, 12412–12417 CrossRef CAS PubMed.
- T.-S. Kam and K.-H. Lim, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Elsevier, New York, 2008, vol. 66, pp. 1–111 Search PubMed.
- (a) K. Awang, O. Thoison, A. H. Hadi, M. Pais and T. Sevenet, Nat. Prod. Lett., 1993, 3(4), 283–289 CrossRef CAS; (b) T. S. Kam and K. Yoganathan, Nat. Prod. Lett., 1997, 10(1), 69–74 CrossRef CAS; (c) C. Kann, J. R. Deverre, T. Sevenet, J. C. Quirion and H. P. Husson, Nat. Prod. Lett., 1995, 7(4), 275–281 CrossRef; (d) K. Husain, I. Jantan, N. Kamaruddin, I. M. Said, N. Aimi and H. Takayama, Phytochemistry, 2001, 57(4), 603–606 CrossRef CAS PubMed; (e) K. Husain, I. Jantan, I. M. Said, N. Aimi and H. Takayama, J. Asian Nat. Prod. Res., 2003, 5(1), 63–67 CrossRef CAS PubMed; (f) D. J. Middleton, Harv. Pap. Bot., 2004, 9(1), 89–142 Search PubMed; (g) D. J. Middleton, Harv. Pap. Bot., 2000, 9, 89 Search PubMed; (h) K.-H. Lim, O. Hiraku, K. Komiyama, T. Koyano, M. Hayashi and T.-S. Kam, J. Nat. Prod., 2007, 70, 1302 CrossRef CAS PubMed; (i) M. J. Tan, C. Yin, C. P. Tang, C. Q. Ke, G. Lin and Y. Ye, Planta Med., 2011, 77(9), 939–944 CrossRef CAS PubMed; (j) H. Naaz, S. Singh, V. P. Pandey, P. Singh and U. N. Dwivedi, Indian J. Biochem. Biophys., 2013, 50(2), 120–125 CAS.
- (a) W. D. Crow and M. Michael, Aust. J. Chem., 1955, 8, 129 CrossRef CAS; (b) W. D. Crow and M. Michael, Aust. J. Chem., 1962, 15, 130 CrossRef CAS; (c) W. G. Kump and H. Schmid, Helv. Chim. Acta, 1961, 44, 1503 CrossRef; (d) W. G. Kump, D. J. L. Count, A. R. Battersby and H. Schmid, Helv. Chim. Acta, 1962, 45, 854 CrossRef CAS; (e) W. G. Kump, M. B. Patel, J. M. Rowson and H. Schmid, Helv. Chim. Acta, 1964, 47, 1497 CrossRef CAS.
- J. Xie, A. L. Wolfe and D. L. Boger, Org. Lett., 2013, 15(4), 868–870 CrossRef CAS PubMed.
- K. Lee and D. L. Boger, Tetrahedron, 2015, 71, 3741–3746 CrossRef CAS PubMed.
- L. Leng, X. Zhou, Q. Liao, F. Wang, H. Song, D. Zhang, X.-Y. Liu and Y. Qin, Angew. Chem., Int. Ed., 2017, 56, 3703–3707 CrossRef CAS PubMed.
- D. Ni, Y. Wei and D. Ma, Angew. Chem., Int. Ed., 2018, 57, 10207–10211 CrossRef CAS PubMed.
- (a) J. W. Daly, I. Karle, C. W. Myers, T. Tokuyama, J. W. Waters and B. Witkop, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1870–1875 CrossRef CAS PubMed; (b) T. F. Spande, H. M. Garraffo, J. W. Daly, T. Tokuyama and A. Shimada, Tetrahedron, 1992, 48, 1823–1836 CrossRef CAS.
- K. Matsumura, K. Nishikawa, H. Yoshida, M. Doe and Y. Morimoto, RSC Adv., 2018, 8, 11296–11303 RSC.
- T. Yamamoto, N. Izumi, H. Ui, A. Sueki, R. Masuma, K. Nonaka, T. Hirose, T. Sunazuka, T. Nagai, H. Yamada, S. Ōmura and K. Shiomi, Tetrahedron, 2012, 68, 9267–9271 CrossRef CAS.
- Z. Shi, PhD thesis, University of Chinese Academy of Sciences, 2018.
- (a) A. S. Monto, Clin. Infect. Dis., 2009, 48, 397–399 CrossRef CAS PubMed; (b) M. Hussain, H. D. Galvin, T. Y. Haw, A. N. Nutsford and M. Husain, Drug Resist., 2017, 10, 121–134 CrossRef CAS PubMed.
- J. Deng, Y. Ning, H. Tian and J. Gui, J. Am. Chem. Soc., 2020, 142, 4690–4695 CrossRef CAS PubMed.
- (a) T. Yasumoto and M. Murata, Chem. Rev., 1993, 93, 1897 CrossRef CAS; (b) P. J. Scheuer, Tetrahedron, 1994, 50, 3 CrossRef CAS; (c) M. Murata and T. Yasumoto, Nat. Prod. Rep., 2000, 17, 293 RSC; (d) T. Yasumoto, Chem. Rec., 2001, 1, 228 CrossRef CAS PubMed; (e) M. Satake, M. Murata and T. J. Yasumoto, J. Am. Chem. Soc., 1993, 115, 361 CrossRef CAS; (f) A. Morohashi, M. Satake and T. Yasumoto, Tetrahedron Lett., 1999, 40, 97 CrossRef CAS.
- H. Fuwa, N. Kainuma, K. Tachibana and M. Sasaki, J. Am. Chem. Soc., 2002, 124, 14983–14992 CrossRef CAS PubMed.
- (a) H.-D. Sun, S.-X. Huang and Q.-B. Han, Nat. Prod. Rep., 2006, 23, 673–698 RSC; (b) P. A. Garc&a, A. B. De Oliveira and R. Batista, Molecules, 2007, 12, 455–483 Search PubMed; (c) M. Liu, W.-G. Wang, H.-D. Sun and J.-X. Pu, Nat. Prod. Rep., 2017, 34, 1090–1140 RSC; (d) J. Wang, Z. Lin, Q. Zhao and H. Sun, Phytochemistry, 1998, 47, 307–309 CrossRef CAS; (e) X.-M. Niu, S.-H. Li, S.-X. Mei, Z. Na, Q.-S. Zhao, Z.-W. Lin and H.-D. Sun, J. Nat. Prod., 2002, 65, 1892–1896 CrossRef CAS PubMed; (f) W.-G. Wang, X.-N. Li, X. Du, H.-Y. Wu, X. Liu, J. Su, Y. Li, J.-X. Pu and H.-D. Sun, J. Nat. Prod., 2012, 75, 1102–1107 CrossRef CAS PubMed.
- F. Su, Y. Lu, L. Kong, J. Liu and T. Luo, Angew. Chem., Int. Ed., 2018, 57, 760–764 CrossRef CAS PubMed.
- (a) A. F. Koca and I. Koca, Food Chem. Toxicol., 2007, 45, 1315–1318 CrossRef PubMed; (b) A. Gunduz, S. Turedi, R. M. Russell and F. A. Ayaz, Clin. Toxicol., 2008, 46, 437–442 CrossRef CAS PubMed; (c) S. A. Jansen, I. Kleerekooper, Z. L. Hofman, I. F. P. Kappen, A. Stary-Weinzinger and M. A. G. van der Heyen, Cardiovasc. Toxicol., 2012, 12, 208–215 CrossRef CAS PubMed.
- (a) S. Durdagi, G. Scozzafava, D. Vullo, H. Sahin, S. Kolayli and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2014, 29, 469–475 CrossRef CAS PubMed; (b) C.-H. Li, J.-Y. Zhang, X.-Y. Zhang, S.-H. Li and J.-M. Gao, Eur. J. Med. Chem., 2019, 166, 400–416 CrossRef CAS PubMed; (c) M. Stevens, S. Peigneur and J. Tytgat, Front. Pharmacol., 2011, 2, 1–13 Search PubMed; (d) S. K. Bagal, A. D. Brown, P. J. Cox, K. Omoto, R. M. Owen, D. C. Pryde, B. Sidders, S. E. Skerratt, E. B. Stevens, R. I. Storer and N. A. Swain, J. Med. Chem., 2013, 56, 593–624 CrossRef CAS PubMed; (e) A. Nardi, N. Damann, T. Hertrampf and A. Kless, ChemMedChem, 2012, 7, 1712–1740 CrossRef CAS PubMed; (f) M. de Lera Ruiz and R. L. Kraus, J. Med. Chem., 2015, 58, 7093–7118 CrossRef CAS PubMed; (g) S. Zhang and Z. Y. Zhang, Drug Discovery Today, 2007, 12, 373–381 CrossRef CAS PubMed; (h) M. Stuible, K. M. Doody and M. L. Tremblay, Cancer Metastasis Rev., 2008, 27, 215–230 CrossRef CAS PubMed; (i) L. Lessard, M. Stuible and M. L. Tremblay, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 613–619 CrossRef CAS PubMed; (j) C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168 CrossRef CAS PubMed.
- A. Turlik, Y. Chen, A. C. Scruse and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 8088–8092 CrossRef CAS PubMed.
- (a) F. Kavanagh, A. Hervey and W. Robbins, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 570 CrossRef CAS PubMed; (b) A. J. Birch, C. W. Holzapfel and R. W. Rickards, Tetrahedron, 1966, 22(suppl. 8), 359 CrossRef; (c) B. Gierczyk, A. Kujawa, A. Szczepkowski, T. Ślusarczyk, T. Pachlewski, P. Chachuła and G. Domian, Acta Mycol., 2019, 54(2), 1124 Search PubMed.
- S. M. Poulsen, M. Karlsson, L. B. Johansson and B. Vester, Mol. Microbiol., 2001, 41, 1091–1099 CrossRef CAS PubMed.
- E. P. Farney, S. S. Feng, F. Schafers and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 1267–1270 CrossRef CAS PubMed.
- (a) T. S. Kam, K. Yoganathan and C. H. Chuah, Tetrahedron Lett., 1994, 35, 4457–4460 CrossRef CAS; (b) G. H. Tan, T. M. Lim and T. S. Kam, Tetrahedron Lett., 1995, 36, 1327–1330 CrossRef CAS; (c) T. S. Kam, K. Yoganathan, T. Koyano and K. Komiyama, Tetrahedron Lett., 1996, 37, 5765–5768 CrossRef CAS.
- (a) T. S. Kam, in The Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon, Amsterdam, 1999, vol. 14, pp. 285–435 Search PubMed; (b) T. S. Kam and Y. M. Choo, The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Academic Press, Amsterdam, 2006, vol. 63, pp. 181−363 Search PubMed; (c) T. S. Kam and K. H. Lim, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Academic Press, The Netherlands, 2008, vol. 66, pp. 1–111 Search PubMed.
- W. Zhang, S. Lin, Ch. Du, S. Feng, Z. Liu, J. Zhang, X. Xie, X. Wang, H. Li and X. She, J. Org. Chem., 2019, 84, 1111–1116 CrossRef CAS PubMed.
- (a) S.-H. Lim, K.-M. Sim, Z. Abdullah, O. Hiraku, M. Hayashi, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2007, 70, 1380 CrossRef CAS PubMed; (b) D. J. Middleton, Harv. Pap. Bot., 2004, 9, 89–142 Search PubMed.
- K. Nagaraju, D. Ni and D. Ma, Angew. Chem., Int. Ed., 2020, 59, 22039–22042 CrossRef CAS PubMed.
- (a) H. Yazawa, H. Imai, K. Suzuki, S. Kadota and T. Saito, US Pat. 4912215, 1990 Search PubMed; (b) Y. Imai, S. Yazawa and T. Saito, Japanese Patent, JP01168671, 1989; (c) Y. Imai, S. Yazawa, K. Suzuki, Y. Yamaguchi, M. Shibazaki and T. Saito, Japanese Patent, JP01106884, 1989.
- (a) H. G. Floss and T.-W. Yu, Chem. Rev., 2005, 105, 621 CrossRef CAS PubMed; (b) M. Hara, K. Asano, I. Kawamoto, T. Takiguchi, S. Katsumata, K. Takahashi and H. Nakano, J. Antibiot., 1989, 42, 1768 CrossRef CAS PubMed; (c) C. Deboer, P. A. Meulman, R. J. Wnuk and D. H. Peterson, J. Antibiot., 1970, 23, 442 CrossRef CAS PubMed.
- S. Yang, Y. Xi, R. Zhu, L. Wang, J. Chen and Z. Yang, Org. Lett., 2013, 15(4), 2013 Search PubMed.
- (a) R. Huisgen, Angew. Chem., 1950, 62, 527–534 CrossRef; (b) J. Bosch, J. Bonjoch and M. Amat, Alkaloids, ed. G. A. Gordell, Academic Press, New York, 1996, vol. 48, pp. 75–189 Search PubMed; (c) J. Buckingham, Bitter Nemesis—The Intimate History of Strychnine, CRC, New York, 2008 Search PubMed; (d) K. C. Nicolaou and T. Montagnon, Molecules that Changed the World, Wiley-VCH, Weinheim. 2008, pp. 91–96 Search PubMed.
- C. Beemelmanns and H.-U. Reissig, Angew. Chem., Int. Ed., 2010, 49, 8021–8025 CrossRef CAS PubMed.
- (a) O. L. Rakotonandrasana, F. H. Raharinjato, M. Rajaonarivelo, V. Dumontet, M.-T. Martin, J. Bignon and P. Rasoanaivo, J. Nat. Prod., 2010, 73, 1730–1733 CrossRef CAS PubMed; (b) P. E. B. K. Kainulainen and B. W. van Ee, A Nomenclator of Croton (Euphorbiaceae) in Madagascar, the Comoros Archipelago, and the Mascarene Islands PhytoKeys, 2017, vol. 90, pp. 1–87 Search PubMed.
- (a) D. Goldsmith, The Total Synthesis of Natural Products, ed. J. ApSimon, Wiley, New York, 1992, vol. 8, pp. 1–243 Search PubMed; (b) A. Abad, C. Agull, A. C. CuÇat, I. deAlfonso Marzal, I. Navarro and A. Gris, Tetrahedron, 2006, 62, 3266–3283 CrossRef CAS , and references therein..
- S. Breitler and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 11168–11171 CrossRef CAS PubMed.
- (a) M. J. Ortega, E. Zubia and J. Salva, J. Nat. Prod., 1997, 60, 488–489 CrossRef CAS PubMed; (b) A. Spinella, M. Gavagnin, A. Crispino, G. Cimino, E. Martinez, J. Ortea and G. Sodano, J. Nat. Prod., 1992, 55, 989–993 CrossRef CAS PubMed; (c) T. Miyamoto, R. Higuchi and T. Komori, Tetrahedron Lett., 1986, 27, 1153–1156 CrossRef CAS; (d) R. A. Hill, D. N. Kirk, H. L. J. Makin and G. M. Murphy, Dictionary of Steroids, Chapman and Hall, London, 1991, pp. xiv–xxix CrossRef; (e) M. Gavagnin, M. Carbone, M. Nappo, E. Mollo, V. Roussis and G. Cimino, Tetrahedron, 2005, 61, 617–621 CrossRef CAS.
- G. Liu, G. Mei, R. Chen, H. Yuan, Z. Yang and C. c. Li, Org. Lett., 2014, 16, 4380–4383 CrossRef CAS PubMed.
- (a) Q. Zhang, Y. Zhang, T.-Q. Yang, Y.-T. Di and X.-J. Hao, RSC Adv., 2013, 3, 9658–9661 RSC; (b) K. Kubitzki, Daphniphyllaceae flowering plants eudicots, Springer, Berlin, Heidelberg, 2007, pp. 127–128 CrossRef; (c) S. Yagi, Kyoto-furitsu Ika Daigaku Zasshi, 1909, 6, 208–222 Search PubMed; (d) J. Kobayashi and T. Kubota, Nat. Prod. Rep., 2009, 26, 936–962 RSC; (e) H. Wu, X. Zhang, L. Ding, S. Chen, J. Yang and X. Xu, Planta Med., 2013, 79, 1589–1598 CrossRef CAS PubMed; (f) A. K. Chattopadhyay and S. Hanessian, Chem. Rev., 2017, 117, 4104–4146 CrossRef CAS PubMed.
- K. Komine, K. M. Lambert, Q. R. Savage, J. B. Cox and J. L. Wood, Chem. Sci., 2020, 11, 9488–9493 RSC.
| This journal is © The Royal Society of Chemistry 2022 |