
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA minireview on catalysts for photocatalytic N2 fixation to synthesize ammonia
Ping Qi
a,
Xiaoxu Gaoa,
Jian Wanga,
Huimin Liu
 *a,
Dehua Heb and
Qijian Zhang
*a
*a,
Dehua Heb and
Qijian Zhang
*a
aSchool of Chemical and Environmental Engineering, Liaoning University of Technology, Jinzhou 121001, P. R. China. E-mail: liuhuimin08@tsinghua.org.cn; zhangqijian@tsinghua.org.cn
bInnovative Catalysis Program, Key Lab of Organic Optoelectronics & Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China
First published on 14th January 2022
Abstract
Ammonia (NH3) is an important feedstock in chemical industry. Nowadays NH3 is mainly produced via the industrialized Haber–Bosch process, which requires substantial energy input, since it operates at high temperatures (400–650 °C) and high pressures (20–40 Mpa). From the energy conservation point of view, it is of great significance to explore an alternative avenue to synthesize NH3, which is in line with the concept of sustainable development. Very recently, photocatalytic N2 fixation (PNF) has been discovered as a safe and green approach to synthesize NH3, as it utilizes the inexhaustible solar energy and the abundant N2 in nature to synthesize NH3 under mild conditions. A highly efficient catalyst is the core of PNF. Up to now, extensive studies have been conducted to design efficient catalysts for PNF. Summarizing the catalysts reported for PNF and unraveling their reaction mechanisms could provide guidance for the design of better catalysts. In this review, we will illustrate the development of catalysts for PNF, including semiconductors, plasmonic metal-based catalysts, iron-based catalysts, ruthenium-based catalysts and several other catalysts, point out the remaining challenges and outline the future opportunities, with the aim to contribute to the development of PNF.
 Ping Qi | Ping Qi, lecturer, School of chemistry and environmental engineering, Liaoning University of Technology. 1993–1997, Fushun Petroleum Institute, petrochemical branch, undergraduate. 1997–2004, engineer, isopropanol workshop, Jinzhou Petrochemical Company. 2004–2007, Liaoning University of Technology, master, advisor: Qijian Zhang. 2007-present, lecturer, School of chemistry and environmental engineering, Liaoning University of Technology. |
 Huimin Liu | Huimin Liu received the Ph.D. degree from Tsinghua University, China (2013), and then joined Kansai University (2013–2014), National Institute of Materials Science (NIMS, 2014–2017) and the University of Sydney (2017–2019) as post-doctoral researcher and Lecturer. Now she is working in Liaoning University of Technology as a professor. Her research interests are photochemistry, environmental chemistry and heterogeneous catalyst design. |
 Qijian Zhang | Qijian Zhang received the Ph.D. degree from Tsinghua University, China (2003), and then joined the University of Kitakyushu for cooperative research (2003–2005). From 2003 to now, he is working in Liaoning University of Technology as a professor. His research interests are nano catalysis and energy chemistry, which is dimethyl ether reforming to hydrogen, application of nano Ni based catalyst in CH4 reforming, photo-thermal catalysis, etc. |
Introduction
Ammonia (NH3) is one of the basic raw materials in industrial chemistry and has been widely applied in various fields.1–4 Industrially, NH3 could be used to produce fertilizers, synthetic fibres, nitrile rubber and so on; medically, NH3 is often utilized as a drug to treat dizziness and fainting; militarily, NH3 could serve as a biological disinfectant to disinfect sarin agents. The wide applications of NH3 make it essential in promoting the development of national economies.At present, the Haber-Bosch process is the main approach to synthesize NH3.5–8 However, the Haber-Bosch process is carried out under harsh reaction conditions (the pressure is high up to 20–40 MPa and the temperature is in the range of 400–650 °C), which consumes extensive energy.9,10 From the context of global energy crisis, it is necessary to search for an alternative avenue to synthesize NH3 which is in line with the concept of sustainable development.
Photocatalytic N2 fixation (PNF) is a process which utilizes the inexhaustible solar energy and the abundant N2 in nature to synthesize NH3 under mild conditions.11–18 It is a safe and green approach and has great potential to solve the problems encountered in the industrialized Haber-Bosch process, provided that efficient photocatalysts are adopted.19–31
In photocatalytic N2 fixation reaction, the cleavage of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond is the rate determining step. That is, effective catalysts for PNF should be able to accelerate the NN bond cleavage process.
N bond is the rate determining step. That is, effective catalysts for PNF should be able to accelerate the NN bond cleavage process.
Over the past few years, an enormous amount of research effort has been devoted to explore effective catalysts for PNF. A large number of photocatalysts, such as semiconductors, plasmonic metal-based catalysts, iron-based catalysts, ruthenium-based catalysts and several other catalysts, have been designed for PNF (a schematic illustration is shown in Fig. 1).
 | ||
| Fig. 1 Schematic illustration of catalysts that have been designed for PNF. | ||
For each type of these photocatalysts, a specific photocatalyst for PNF could be simply divided into two different functional units. One is the light harvesting unit (which is used to denote the active sites that could adsorb light) and the other is the thermal-driven active unit (which is referred as the active center that directly involves in the polarization and activation of N2). The tailoring of each of the type of functional unit could contribute to the performance of a catalyst in PNF. In this review, we will summarize the photocatalysts that have been reported active for PNF, generalize the principles for the design of efficient catalysts (tailoring the light harvesting unit or the thermal-driven active unit), unravel their reaction mechanisms, point out the remaining challenges and prospect the future development, with the aim to provide guidance for the rational design of more efficient photocatalysts and contribute to the development of PNF.
Photocatalysts for PNF
In this section, the photocatalysts for PNF are classified into five categories, including semiconductors, plasmonic metal-based catalysts, iron-based catalysts, ruthenium-based catalysts and other catalysts. The progress of each category of photocatalysts is summarized and discussed in the following sub-sections.Semiconductors as photocatalysts for PNF
When semiconductors are adopted to catalyse PNF, suitable energy band levels are required. Generally speaking, semiconductors for PNF are designed according to the following principles, ① the semiconductor could absorb light efficiently and it could be excited by light easily, ② the separation and transfer efficiency of the photogenerated electron hole pairs should be effective, and ③ the energy levels of the semiconductor photocatalyst could meet the standards for the two photo-induced half reactions, referring to the reduction of N2 and the oxidation of H2 or H2O.Some pristine semiconductors could meet the standards to drive PNF, however, their efficiencies are quite low. Introducing vacancies or foreign elements into the semiconductor, functionalizing the pristine semiconductor, constructing heterojunctions/homojunctions or design a semiconductor-based hydrophilic-hydrophobic catalyst, are approaches to extend the light harvesting spectrum, enhance the light harvesting capacity (which means its capacity to absorb more light), facilitate the separation and transfer of photoinduced electron hole pairs and further accelerate PNF.
The activities of pristine semiconductors have been verified experimentally. For example, ultrathin MoS2 could convert N2 into NH3 via a six-electron reduction process (Fig. 2), achieving a NH3 synthesis rate of 325 μmol g−1 h−1 without any sacrificial reagents or co-catalysts.34 The electron-rich property of ultrathin MoS2 as well as the high concentration of localized electrons upon light irradiation accounted for its activity.34 Bismuth monoxide (BiO) quantum dots, synthesized via a facile hydrothermal method, are reported as a highly efficient catalyst for PNF, recording a NH3 generation rate of 1226 μmol g−1 h−1 without the assistance of any sacrificial reagents or co-catalysts.35 It is postulated that all the low valence Bi(II) in BiO were potential active sites for N2 activation.35
 | ||
| Fig. 2 Schematic illustration of trion induced six-electron N2 reduction. This figure has been adapted from ref. 34 with permission from Elsevier, copyright 2017. | ||
The catalytic activity of pristine semiconductor could be improved, in case that a 2-dimensional (2D) material is used as co-catalyst. For instance, P25 itself yielded NH3 at a rate of 2.11 μmol g−1 h−1 under full spectrum light irradiation, while the activity was fivefold increased when Ti3C2 MXene was used as a co-catalyst.36 Similarly, the catalytic performance of CdS in PNF boosted obviously when black phosphorous nanosheet was adopted as a co-catalyst.37 It is reported that 2D co-catalysts facilitated the separation of electron–hole pairs and promoted N2 chemisorption and activation.
Despite that some pristine semiconductors are active for PNF, their efficiencies are generally low. To further improve their efficiencies, multiple approaches are adopted to modify the pristine semiconductors.
(1) The introduction of defective sites engineers the light harvesting unit, such as lowers the conduction band position, engineers the band gap and improves the light harvesting capacity of the semiconductor. Few-layer g-C3N4 (ref. 38) and one-dimensional g-C3N4 (ref. 39) are rich in N defects. The existence of N defective sites lowered the conduction band position and increased its light harvesting capacity, which then contribute to their performance in PNF.38,39 Band gaps and the light harvesting capacities could be consecutively tuned by dedicatedly controlling the content of surface vacancies.40
(2) The defective sites serve as the active sites to enhance the adsorption and activation of N2. N vacancies on nitrides generally activate N2 via a pathway analogous to Mar-van Krevelen mechanism. That is, N2 is firstly activated at the N vacancies and then transformed into NH3 by reacting with H2 or H2O. Wang et al.'s work is a typical example. In their work, a N deficient g-C3N4 catalyst was prepared by the dielectric barrier discharge plasma treatment method, which displayed a NH3 production rate of 161.8 μmol g−1 h−1.41 Mechanism exploration disclosed that N2 was activated via the two-path analogous Mar-van Krevelen mechanism.41
Oxygen vacancies on oxide semiconductors are active centers for N2 adsorption and capable of activating N2.42–44 Theoretical study revealed that the oxygen vacancies on (001) and (100) facets of MoO3−x nanobelts could chemisorb N2 via side-on and end-on models, respectively, which accelerates the sluggish rate determining step (N2 activation) in PNF and boosted its performance.43
Cation defective sites on a semiconductor are electron-rich, which could effectively activate the NN bond and accelerate catalytic activity in PNF. For instance, Zn deficient Zn3In2S6 exhibited a NH3 generation rate of 261.2 μmol g−1 h−1 under visible light irradiation, 15 times higher than the one with poor defects.45
(3) On some defective semiconductors, the defective sites not only improve light harvesting capacity but also facilitate N2 activation. For example, Bi2WO6 hollow microspheres prepared by a solvothermal template-free method are rich in oxygen vacancies.46 The oxygen vacancies induce a sub-band (defect energy level), which not only narrows its band gap and extends its light absorption region (up to 700 nm), but also localizes metastable electrons. These metastable electrons jump to the anti-bonding orbitals of N2 via a manner of non-radiative transfer and activate N2. Overall, the Bi2WO6 hollow microspheres demonstrated a NH3 yield of ∼53 μmol g−1 h−1 under simulated sunlight.46
Similarly, in addition to enhance the light harvesting capacity, the oxygen vacancies on BiOCl,47 TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48–50 and BiOBr51 also serve as the active sites for N2 activation and reduction while the oxygen vacancies on Bi3FeMo2O12 help adsorb and stabilize the N–H intermediate during N2 activation,52 which cooperatively boost NH3 production via PNF (Fig. 3).53
48–50 and BiOBr51 also serve as the active sites for N2 activation and reduction while the oxygen vacancies on Bi3FeMo2O12 help adsorb and stabilize the N–H intermediate during N2 activation,52 which cooperatively boost NH3 production via PNF (Fig. 3).53
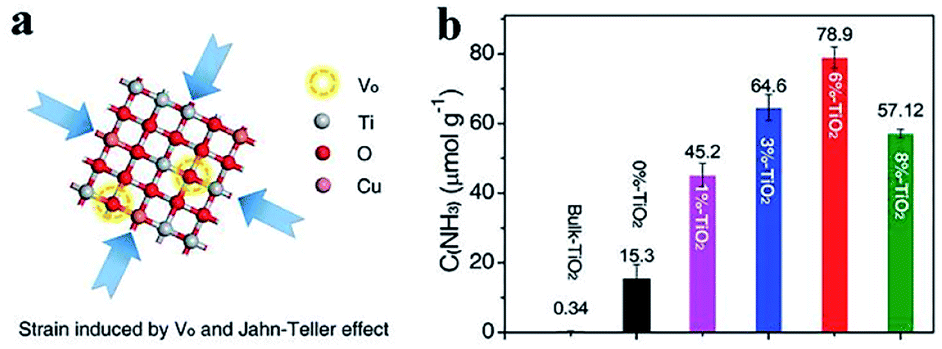 | ||
| Fig. 3 (a) Strain induced by oxygen vacancies and Jahn-Teller effects in TiO2 nanosheet. (b) NH3 yield over different catalysts under ultraviolet (UV)-vis light irradiation for 1 h. This figure has been adapted from ref. 53 with permission from Wiley-VCH, copyright 2019. | ||
Intrinsic strain is occasionally induced by vacancies. Thereby the strain and vacancies cooperatively contribute to PNF. Zhang et al. synthesized an ultrathin TiO2 nanosheet catalyst with abundant oxygen vacancies via a facile copper-doping method, which could absorb visible light high up to 700 nm.53 It was discovered that there was intrinsic compression strain in the as-prepared TiO2 nanosheet. The oxygen vacancies and the strain worked in concert to chemisorb and activate N2 and H2O effectively, leading to a high NH3 production rate (78.9 μmol g−1 h−1).53
(1) Suitable dopant could serve as active sites for promoting N2 activation. For example, Mo1−xWxS2 nanosheets, which could be considered as Mo doped WS2, recorded a NH3 production rate of 111 μmol g−1 h−1 under visible light irradiation, when the concentration of 1T phase was 33.6% and Mo/W = 0.68:0.32.54 The doping of Mo into WS2 resulted in a higher electron density on W 5d orbitals, which polarized the adsorbed N2 and responded for its PNF activity.54
(2) Under most of cases, the introduction of dopant not only engineers the optical properties but also manipulates the active sites for N2 activation.
Doping B into g-C3N4 led to the formation of a new chemical bond B–N–C, which not only effectively enhanced the light harvesting capacity and suppressed the recombination of photoinduced electron–hole pairs, but also served as an active center for N2 chemisorption and activation.55 In the issue, B-g-C3N4 gave a NH3 yield of 313.9 μmol g−1 h−1 under visible light assistance.55 Mn2+ could be doped into W18O49 via partially replacing the W sites.56 The doped Mn2+ played multiple roles in PNF. ① Mn2+ acted as the active sites for the chemisorption of N2 and H2O, ② Mn2+ weakened the NN bond through proton coupling process and ③ the doped Mn2+ facilitated the separation and migration of photoinduced electron–hole pairs. Based on these advantages, the as prepared Mn–W18O49 catalyst exhibited a NH3 production rate of 97.9 μmol g−1 h−1 under full spectrum irradiation of a 300 W Xe lamp.56
When a foreign element is doped into a semiconductor with vacancies sites, the dopants and vacancies sites might synergistically activate N2 and contribute to PNF. For example, in sulfur doped oxygen deficient TiO2 (TiO2−xSy), the oxygen vacancies and sulfur dopant worked in concert to facilitate N2 adsorption and extend its light absorbing capacity to near-infrared region.57 As a result, TiO2−xSy yielded NH3 at a rate of 114.1 μmol g−1 h−1 under full spectrum light irradiation.57 Similar phenomena were also observed over Br doped BiOCl with oxygen vacancies,58 Ni doped vacancy-rich TiO2,59 as well as S doped g-C3N4 with carbon vacancies.60
Doping a foreign element into a semiconductor sometimes leads to the generation of vacancies. Tang et al. fabricated a carbon doped TiO2 nanosheet catalyst (C-TiOx) from Ti3SiC2 via a bottom-up approach (Fig. 4a).29 It was discovered that the doping of carbon led to the generation of oxygen vacancies in TiO2. As charge compensation, controllable Ti3+ sites were produced. The oxygen vacancies broadened its light harvesting region and the electron-rich Ti3+ were active for N2 activation. With Ru/RuO2 as co-catalyst to promote the separation and migration of photoinduced electron–hole pairs, the optimal C-TiOx recorded a 109.3 μmol g−1 h−1 NH3 synthesis rate under visible light irradiation and an apparent quantum yield of 1.1% at 400 nm (Fig. 4b).29 Similarly, oxygen vacancies could also be generated by doping carbon into BiOI.61 Carbon dopant decreased the band gap, extended the light harvesting region, facilitated the separation and migration of electron–hole pairs and consequently hastened PNF, leading to a NH3 generation rate of 311 μmol g−1 h−1 under the illumination of simulated sunlight.61
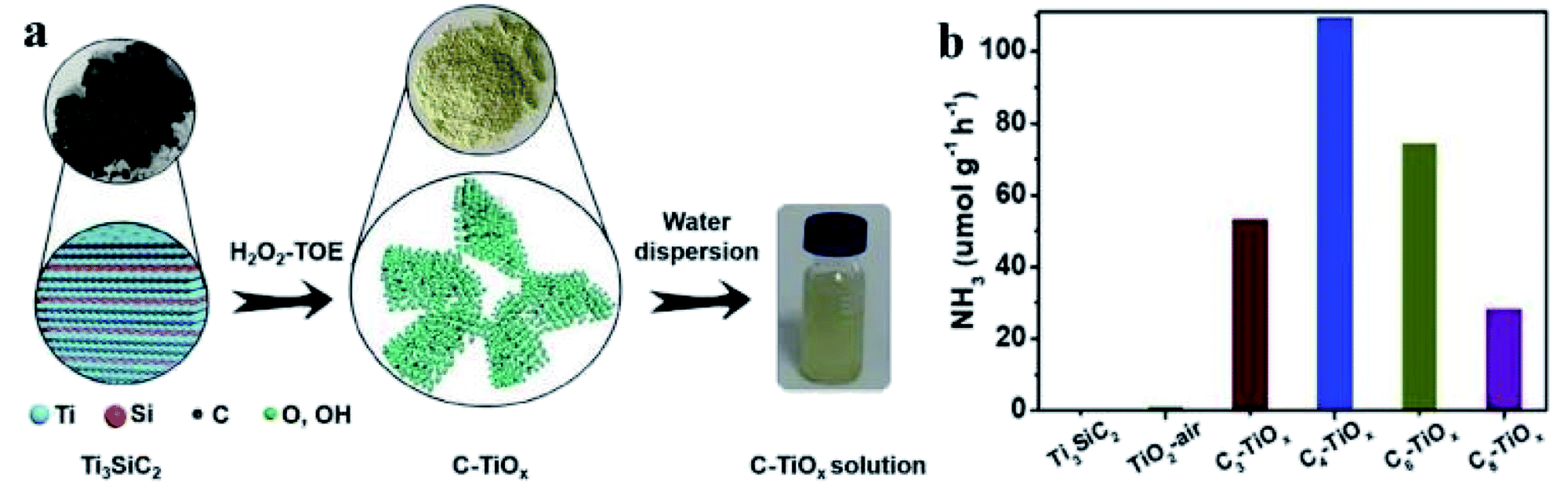 | ||
| Fig. 4 (a) Schematic illustration for the synthesis of C-TiOx. (b) Photocatalytic performance of Cn-TiOx (n denotes the treatment time of Ti3SiC2) and Ti3SiC2 in PNF under visible light irradiation. Ru/RuO2 was used as co-catalyst. This figure has been adapted from ref. 29 with permission from Wiley-VCH, copyright 2021. | ||
As for doping, it is clear that most of the doped semiconductors exhibited a NH3 production rate of ∼100 μmol g−1 h−1, with only two exceptions on B-g-C3N4 and C– BiOI, who gave NH3 yields over 300 μmol g−1 h−1. Even though limited by the number of studies, it might provide a clue that nonmetal element doping could endow the doped semiconductors better performance in PNF.
(1) Functionalized semiconductors with engineered light harvesting unit.
Acid treated semiconductors. Tian et al. reported that salicylic acid (SA) modification could enlarge the Brunauer–Emmett–Teller surface area, improve the optical absorption capacity as well as promote the separation of photoinduced electron–hole pairs in g-C3N4, which resulted in a much enhanced PNF activity over g-C3N4-SA.62
Base treated semiconductors. It has been reported that base treated semiconductors exhibited improved electron–hole separation efficiency. Typical examples are given by Yangjeh et al.63 and Wang et al.,64 who reported that MgO decorated g-C3N4 and KOH treated g-C3N4 gave superior performance in PNF than pristine g-C3N4, and the reduced recombination rate of electron–hole pairs was one of the reasons for their premier activities.
Metal decorated semiconductors. Cu, Fe, Ni and Pd decoration could promote the charge transfer efficiency. Cu, Fe or Ni decorated TiO2, synthesized via microwave assisted hydrothermal method, not only exhibited promoted charge transfer efficiency, but also had larger specific surface area and stronger capacity in harvesting visible light.65 As a result, they exhibited 1.9–6.0 times higher PNF activities than the pristine TiO2 under simulated light irradiation.65 Pd decorated TiO2, prepared by one-pot microwave synthesis techniques, also demonstrated reduced recombination of photoinduced electron–hole pairs and led to a 4 fold higher NH3 production rate than the unmodified one.66
Quantum dots modified semiconductors. Graphene quantum dots modified Bi2WO6, with graphene quantum dots uniformly dispersed on the surface of Bi2WO6, exhibited remarkably enhanced PNF activity than the two single component counterparts. Characterization results implied that the recombination of photoinduced electron–hole pairs was significantly reduced and the junction between graphene quantum dots and Bi2WO6 helped to boost the photocatalytic activity.67
(2) Functionalized semiconductors with tailored active sites.
(3) Functionalized semiconductors with two functional units modified.
Acid treated semiconductors. Wang et al. reported that phosphate acid treated LaFeO3 could catalyze PNF effectively, producing NH3 at a rate of ∼250 μmol g−1 h−1 under simulated light irradiation. Mechanism exploration suggested that phosphate acid served as the Lewis acid center, it worked synergistically with Fe in LaFeO3 to activate N2 via the “push–pull” hypothesis. That is, the electron density is pulled from Fe and pushed into N2 by the adjacent hydrogen bonding sites. In addition, phosphate modification facilitated H2O dissociation.72
(1) Heterojunctions/homojunctions with modified light harvesting unit. Bi2Te3/BiOCl,74 g-C3N4/ZrO2,75 MoO2/BiOCl76 perovskite/attapulgite77 and CdS/LDH (LDH: layered double hydroxide)78 are typical heterojunctions that have been reported active in PNF. Here CdS/LDH is taken as an example for elaboration. In the case that CdS/LDH heterojunction was constructed between (003) or (012) facet of LDH and (002) facet of CdS, a build-in electric field would be induced under light irradiation, which promoted charge transfer from the heterostructure to the reaction media for N2 activation via a favorable configuration and resulted in a better catalytic activity.78
Z scheme is a special case of heterojunctions. Z schemes AgBr/Bi4O5Br2,79 Bi2O3@CoAl-LDHs,80 3,4-dihydroxybenzaldehyde-functionalized Ga2O3/g-C3N4,81 g-C3N4/Mg1.1Al0.3Fe0.2O1.7,82 nano-MOF@defected g-C3N4 (MOF: metal organic frameworks)83 and SiW9Co3/PDA/BWO (PDA: poly-dopamine; BWO: Bi2WO6)84 have been reported active in PNF. In these Z schemes, the separation of photoinduced electron–hole pairs was significantly improved, which played a dispensable role in boosting their catalytic activities.
(2) Heterojunctions/homojunctions with modified light harvesting unit and active sites.
The catalytic activities of heterojunctions in PNF could be further improved by doping foreign elements, introducing defective sites or loading another component to one of the semiconductors of heterojunctions. N deficient g-C3N4/Cu2(OH)2CO3,85 N deficient g-C3N4/Ag2CO3,86 Bi2MoO6/oxygen-vacancy-rich BiOBr,87 MoS2/C–ZnO (C was loaded onto ZnO),88 TiO2@C g-C3N4,89 B doped g-C3N4/Ni2P90 and In2O3/In2S3 (oxygen vacancies are generated in situ)91 are representatives. In this type of heterojunctions, the junctions between the two semiconductors as well as the doped/loaded component or defective sites synergistically contributed to their activities in PNF.
Homojunction catalyst such as ordered/disordered TiO2 exhibited a superior activity in PNF, affording a NH3 formation rate of 432 μmol g−1 h−1 under solar illumination.92 In the homojunction catalyst, ordered TiO2 exhibited a stronger N2 adsorption capacity with a reduced activation barrier while the disordered TiO2 was rich in oxygen vacancies which selectively chemisorbed N2 and enhanced visible light harvesting. The synergistic effect between ordered TiO2 and disordered TiO2, together with the rapid interfacial charge separation, ensured its superior activity (Fig. 5).92
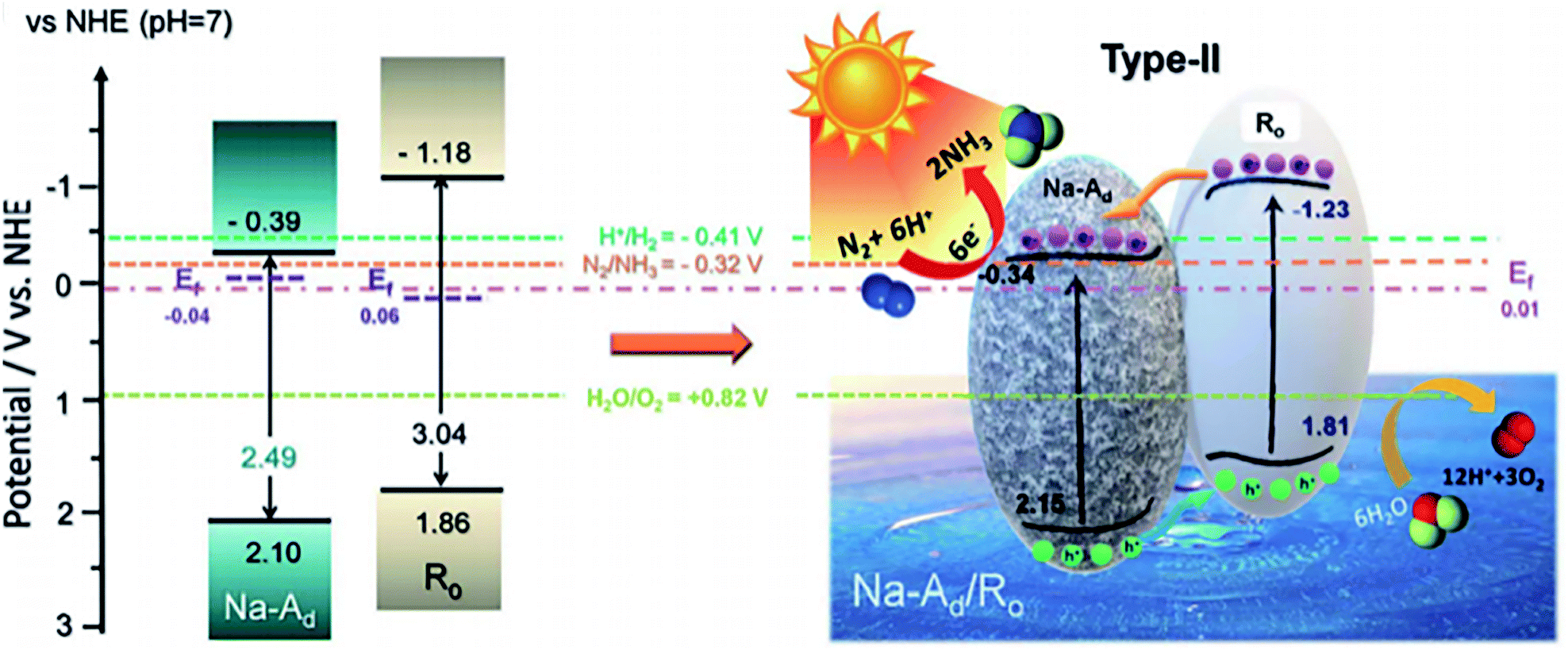 | ||
| Fig. 5 Energy band structure of disordered TiO2 (short for Na-Ad) and ordered TiO2 (represented by Ro) as well as the reaction mechanism. This figure has been adapted from ref. 92 with permission from Royal Society of Chemistry, copyright 2020. | ||
Plasmonic metal-based catalysts for PNF
Plasmonic metals, such as Au and Ag, exhibit localized surface plasmon resonance (LSPR) effect upon light irradiation. The LSPR effects empower the plasmonic metal-based catalysts applicability in PNF. Over most of the plasmonic metal-based catalysts, the light harvesting unit and the active center work synergistically for improved performance. As the particle sizes, morphologies as well as the particle–particle distances of plasmonic metals are crucial for their light harvesting capacities, the tailoring of these properties are generally adopted with the aim to accelerate the reaction rate of plasmonic metal-based catalysts in PNF.For example, Wang et al. encapsulated Au nanoparticles into a MOF membrane (Uio-66) and realized a NH3 production rate of 359.1 μmol g−1 h−1 under visible light irradiation (λ > 400 nm, 100 mW cm−2).94 It was discovered that N2 adsorbed on Au nanoparticles. Upon light irradiation, hot electrons generated on Au nanoparticles. The hot electrons on Au activated N2 via two pathways, ① induced an electromagnetic field to polarize N2 and ② directly injected into the anti-bonding orbitals of N2 molecules (Fig. 6a). Moreover, the gas permeable nature of MOF membrane facilitated the mass transfer of the reactants, which further boosted its photocatalytic activity at the gas (N2)-membrane (Uio-66 encapsulated Au nanoparticles)-solution (H2O) interface (Fig. 6b).94
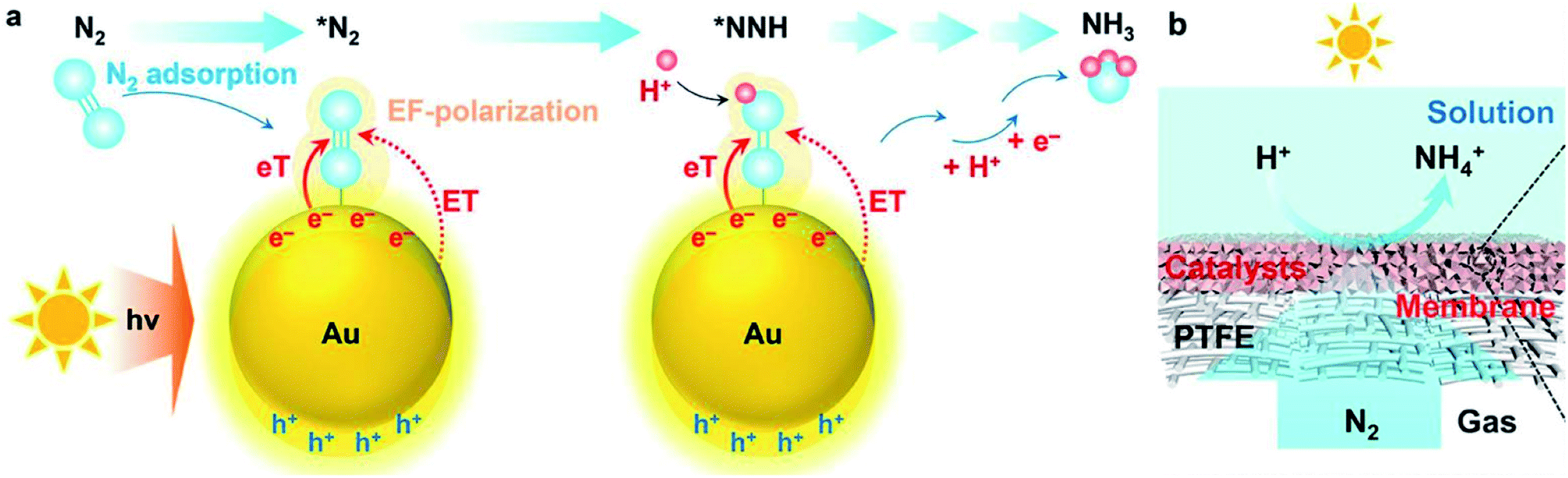 | ||
| Fig. 6 (a) Schematic illustration of PNF over Uio-66 encapsulated Au nanoparticles. (b) Schematic illustration of the interface design for PNF. This figure has been adapted from ref. 94 with permission from Royal Society of Chemistry, copyright 2021. | ||
Yang et al. reported that Au nanoparticles supported on nitrogen deficient g-C3N4 could catalyze PNF, achieving a NH3 production rate of ∼783 μmol g−1 h−1 under visible light irradiation.95 In this catalytic system, the nitrogen vacancy sites adsorbed N2; Au and g-C3N4 harvested visible light and induced electrons upon light irradiation, which then injected into N2 for its activation.95 Liao et al. synthesized small Ag doped g-C3N4 catalyst and unraveled that Ag enhanced N2 adsorption, generated more electrons, facilitated the separation and migration of photogenerated electron–hole pairs and consequently resulted in high activity in PNF.96 Wang et al. supported Au nanocrystals onto Mo doped W18O49 and realized a NH3 synthesis rate of ∼399 μmol g−1 h−1.97 Au not only harvested visible light but also decreased the desorption energy of the product NH3, which accelerated the regeneration of the active sites for next catalytic cycle.97 Introducing a moderate amount of alkali metal cations could further promote N2 activation and then enhance the performance of these plasmonic metal-based catalysts in PNF.98
Iron-based catalysts for PNF
Iron is the most active metal for N2 activation. The strong N2 activation capacity of iron makes it applicable as active center for PNF. However, the light harvesting capacity of iron is very weak. Integrating iron with a material that could harvest light efficiently is an approach to design the photocatalyst for PNF. Based on this principle, doping iron into semiconductors, loading iron onto semiconductors, constructing a Fe-based heterostructure catalyst and loading iron onto other light harvesting materials are promising avenues. Therefore, over most of the iron-based catalysts for PNF, both the light harvesting unit and the active center are regulated. Up to now, various iron-based catalysts have been designed. In this section, we will discuss the types of iron-based catalysts for PNF and emphasize their progress.(1) Fe doped into semiconductors. Doping Fe into a semiconductor could tune the local electronic structure of the catalysts and thereby facilitate N2 activation. For example, when Fe is doped into TiO2, Fe will substitute Ti atoms in TiO2 owing to their similar radii, which creates oxygen vacancies at the neighbor of Fe atoms to meet the local charge balance.99 The Fe atoms and oxygen vacancies work in concert to facilitate N2 adsorption and polarization, which enables N2 hydrogenation via the favorable associative distal pathway and contributes to PNF. As shown in Fig. 7a and b, 5wt% Fe/TiO2 (5-FTNFs) afforded a stable NH3 production rate of ∼64 μmol g−1 h−1 under the full spectrum illumination of a 300 W Xe lamp.99 Isotope labeling experiment as well as 1H-NMR (NMR: Nuclear magnetic resonance, Fig. 7c and d) indicated that the generated NH3 was originated from N2 instead of other contaminants.99 Similarly, Fe-doped BiOBr with oxygen vacancies afforded a NH3 yield of 46.1 μmol g−1 h−1 without any sacrificial reagent under 400 mW cm−2 visible light irradiation.100 The doped Fe and the oxygen vacancies synergistically modulated the band structure, improved charge transfer and thereby enhanced the photocatalytic activities.100
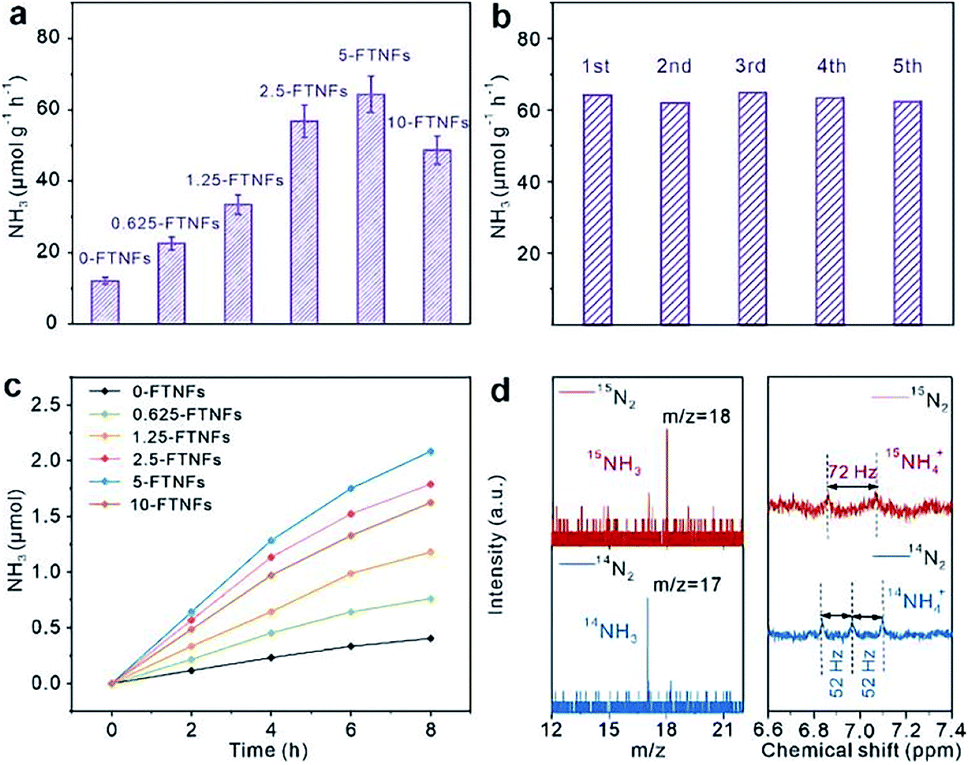 | ||
| Fig. 7 (a) Performance of Fe doped TiO2 (x-FTNFs, where x is the loading of Fe) in PNF. (b) Stability test of 5-FTNFs in PNF. (c) Time-dependent NH3 production over x-FTNFs. (d) Isotope labeling experiment (left panel) and 1H-NMR (right panel) of the products. This figure has been adapted from ref. 99 with permission from Royal Society of Chemistry, copyright 2021. | ||
Doping exerts functions beyond creating oxygen vacancies. For instance, the Fe atoms on the surface of Fe doped SrTiO3 (FexSr1−xTiO3) could not only chemisorb and activate N2, but also promote the electron transfer from FexSr1−xTiO3 to N2, which resulted in high N2 fixation capacity and a NH3 production rate of 30.1 μmol g−1 h−1 over FexSr1−xTiO3 (x = 0.1) under Xe lamp illumination.101 Doping Fe into Mo-based semiconductors could narrow their band gaps, extend their light absorption capacities as well as generate new Fe–Mo active centers, which enables Fe doped Mo-based semiconductor to harvest more solar light and facilitate the electron–hole separation and migration efficiency.102 Chang et al. doped Fe into 2D MoTe2 nanosheets to construct Fe–Mo active centers.103 It is reported that Fe doped into MoTe2 facilitated the separation and transfer of photoinduced electron–hole pairs, prolonged their lifetime and accounted for an obviously boosted NH3 production rate.103 Similarly, Fe doped SrMoO4 (Fe/Sr = 1.6) achieved a NH3 production rate of 93.1 μmol g−1 h−1 under Xe lamp illumination.102
(2) Fe loaded onto semiconductors. Zhang et al. reported that Fe/TiO2−xHy could serve as a dual temperature zone catalyst for PNF.104 Under solar light irradiation, catalyst surface temperature reached 495 °C, with a temperature difference between Fe and TiO2−xHy of 137 °C, owing to the plasmonic local heating effect of Fe. Then Fe acted as the hot zone catalyst to dissociate N2 via its photogenerated hot electrons, while TiO2−xHy accommodated the N atoms from Fe and hydrogenated them into NH3.104 Fe/TiO2−xHy delivered a NH3 concentration of 109.5 μmol g−1 h−1, an order of magnitude higher than the commercial Haber-Bosch Fe catalyst.104
(3) Fe based heterostructure catalysts. Fe2O3/g-C3N4 prepared by thermal treatment method exhibited a NH3 production rate of 7044 μmol g−1 h−1 under the illumination of a 300 W Xe lamp, where the heterostructure facilitated light harvesting and Fe2O3 played key roles in N2 adsorption.105 Oxygen deficient Fe2O3/ZnO could stably produce NH3 at a rate of 80 μmol g−1 h−1 without any sacrificial agent under visible light irradiation.106 Over Fe2O3/ZnO, the synergistic effect between oxygen vacancies and Fe2O3 activated N2, while the heterostructure of Fe2O3/ZnO prohibited the recombination of electron–hole pairs, which accounted for its superior photocatalytic performance.106 Fe-modified palygorskite supported FeS2, synthesized by microwave hydrothermal method, could serve as a Z scheme type photocatalysts for PNF and recorded a NH3 production rate of 147 μmol g−1 h−1 under solar light irradiation.107 Its activity was ascribed to the narrowed band gap, widened light harvesting region of Fe-modified palygorskite as well as the facilitated charge transfer between Fe-modified palygorskite and FeS2.107
(4) Other light responsive materials, such as graphdiyne and MOFs, supported Fe catalysts. For instance, Li et al. reported that the morphology, coordination environment and the valence state of iron oxide could be manipulated by encapsulating versatile shaped Fe3O4 by graphdiyne.108 The encapsulation of graphdiyne endowed Fe3O4@graphdiyne heterojunctions strong light harvesting capacity, a structural evolution during PNF, as well as a NH3 yield of an unprecedented level of ∼1762 μmol g−1 h−1.108 The Fe atoms in Fe-based MOFs (e.g., MIL-101(Fe), MIL-100(Fe), MIL-88(Fe)) have high electron densities, low reaction barriers for the activation of N2 and H2 to N–H bond, and could serve as the catalytic active center for PNF.109 Taking MIL-101(Fe) as an example, it gave a NH3 production rate of 100.7 μmol g−1 h−1 under the illumination of a 300 W Xe lamp.109
Ruthenium-based catalysts for PNF
Ruthenium is another metal capable of activating N2 under thermally-driven conditions. Ruthenium-based catalysts that could harvest solar light effectively have been successfully applied in PNF. Since the light harvesting capacity of ruthenium is weak, the light harvesting unit of ruthenium-based photocatalysts are generally semiconductors or other light responsive materials. Similar as iron-based catalysts, over most of the ruthenium-based catalysts for PNF, both the light harvesting unit and the active center are regulated. Based on the nature of the light responsive components, these ruthenium-based catalysts could be divided into the following categories.(1) Semiconductor, such as TiO2, GaN, C3N4 and CeO2, supported ruthenium catalysts. For example, Ru/TiO2, in which singly dispersed Ru atoms were decorated onto TiO2 nanosheets rich in oxygen vacancies, is active for PNF.110 Over Ru/TiO2, the single Ru atoms were possibly located at the oxygen vacancy sites and stabilized by the vacancies.110 The isolated Ru atoms promoted the chemisorption of N2, boosted the electron–hole separation and overall recorded a NH3 generation rate of 3.3 μmol g−1 h−1 upon irradiation by a 300 W high pressure Xe lamp.110 Ru/P25, prepared by the facile synthetic method, exhibited Ru particle sizes of 2–3 nm. In PNF, Ru/P25 dissociated H2O to hydrogen atoms continuously and then hydrogenated N2 molecules via a distal reaction pathway at the gas (N2)–liquid (H2O) interface.111 The NH3 yield over Ru/P25 was high up to 5.2 μmol g−1 h−1 under the irradiation of a Xe lamp.111 GaN supported Ru catalysts, Ru/GaN, behaved tailorable electronic and morphological properties.112 The interfacial Schottky junction between Ru and GaN facilitated the electron transfer from GaN to Ru, then the electron tank in Ru promoted NN bond dissociation and achieved NH3 synthesis at low temperatures. Notably, 5 wt% Ru/GaN afforded an average NH3 production rate of 120 μmol g−1 h−1 after 2 h UV irradiation at 10 °C (Fig. 8).112
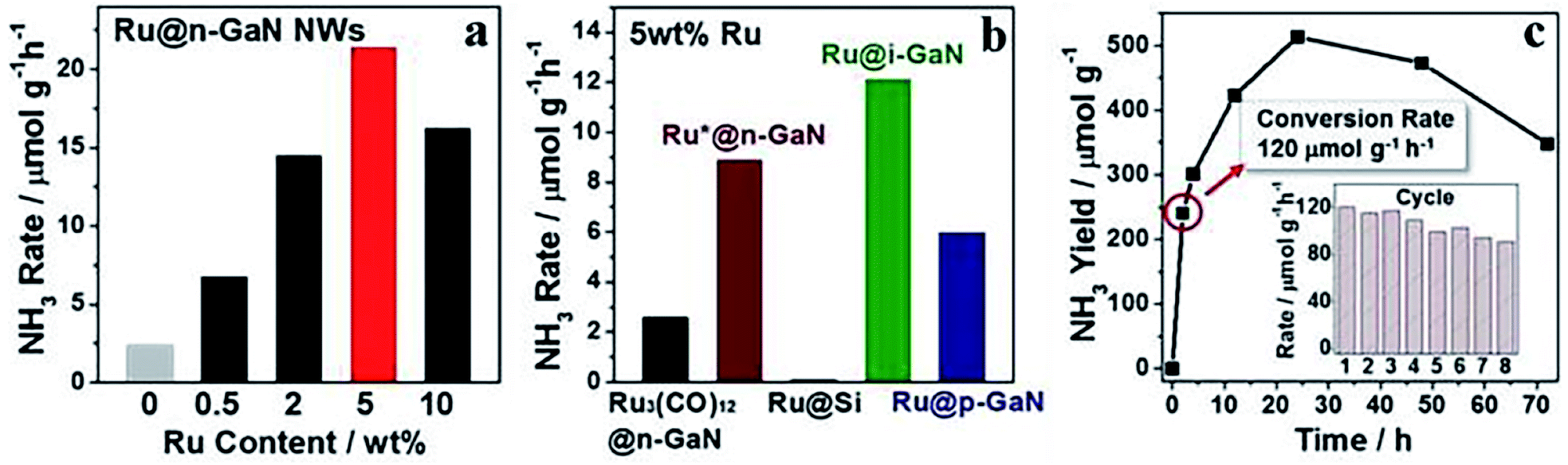 | ||
| Fig. 8 NH3 production rate over (a) Ru/GaN of different Ru loadings, (b) 5 wt% Ru loaded onto various supports, and (c) NH3 production rate as a function of time over 5 wt% Ru/GaN under UV irradiation, with reusability of 5 wt% Ru/GaN as an inset. This figure has been adapted from ref. 112 with permission from American Chemical Society, copyright 2019. | ||
Modifying the semiconductor supported Ru catalysts could speed up NH3 production rate. For instance, modifying Ru/g-C3N4 or Ru/TiO2 by K could enrich the electrons in Ru, enhance the catalyst capacities in activating N2 and consequently improve NH3 generation rate.113,114 Doping Zr4+ into CeO2 could increase the electron densities on Ce and create oxygen vacancies, which strengthened the interactions between Ru nanoparticles and supports.115 The strong interaction upshifted the Ru d-band center relative to Fermi level and enhanced N2 cleavage.115 Ternary heterostructure Ru/RuO2/g-C3N4 catalyst gave an average NH3 production rate of 13.3 μmol g−1 h−1, 6 times higher than Ru/g-C3N4.116 Characterization results indicated that under light irradiation, the electrons transferred to Ru whereas holes migrated to RuO2 to facilitate the reduction and oxidation reactions, respectively, meanwhile, the electron-rich Ru activated N2 effectively.116 Decorating Ru/g-C3N4 catalyst by S-deficient CoSx could construct a bimetallic center at the interface of Ru/CoSx, which facilitated N2 polarization and activation via electron transfer from Ru and Co to N2 upon light irradiation and ultimately gave a NH3 production rate high up to 440 μmol g−1 h−1.117
(2) Other light responsive materials supported Ru catalysts. For example, coal-based carbon nanosheet supported Ru catalyst yielded 55.3 μmol g−1 h−1 NH3 under a 300 W Xe lamp irradiation.118 Graphene oxide/silica could sufficiently disperse Ru and enhance the LSPR effect of Ru species, which excited more electron–hole pairs upon light irradiation and accelerated NH3 generation.119 TiO2–Mxene hybrid nanostructure supported Ru catalysts afforded an ammonia production rate of ∼5.7 μmol g−1 h−1, which was principally stemmed from the synergetic effects among TiO2, Mxene and Ru.120
Other catalysts for PNF
Numerous other catalysts have also been designed for PNF. Phosphorus is one of them. Yu et al. disclosed that the edges of black phosphorus (BP) could absorb and reduce N2. They synthesized an edge-rich BP nanostructure with a flake-like shape via chemical etching exfoliation method. The edge-rich BP is of good dispersibility in H2O, which allows its full contact with the reactants. Owing to the abundant active sites for N2 chemisorption and reduction as well as the efficient contact between the reactants and catalyst, the edge-rich BP delivered a NH3 production rate of 2370 μmol g−1 h−1.121 Lin et al. loaded red phosphorus (RP) onto photoinactive SiO2 via a facile sublimation-deposition method, in which RP was modified by in situ formed carbon.122 The hybrid SiO2/C-RP catalyst was of large specific surface area, strong light harvesting capacity and high charge separation efficiency, which accounted for a NH3 production rate of 36.5 μmol g−1 h−1.122MOFs have also been successfully applied in PNF. Chen et al. unraveled that the Ce species in MOF-76(Ce) was an electron tank, which accepted photoinduced electrons to its 4f orbitals and then donated the electrons to the anti-bonding orbitals of N2.123 As a result, MOF-76(Ce) gave an average NH3 yield of 34 μmol g−1 h−1 under ambient conditions.123 Ye et al. reported that functionalized MIL-125(Ti) could act as photocatalysts for PNF under visible light irradiation without any sacrificial reagent.124 Notably, amine-functionalized NH2-MIL-125(Ti) afforded a NH3 production rate of 12.3 μmol g−1 h−1. The electron transfer from the ligand to metal in MIL-125(Ti) induced Ti3+, which was the active sites for N2 activation. Functionalization extended the light harvesting capacity of MIL-125(Ti) and further enhanced catalyst activity in PNF.124
LDH with oxygen defects and electron-rich metals have been discovered active in PNF, in which vacancies and metal centers synergistically promote N2 adsorption, facilitate the separation of photoinduced electron–hole pairs, and thereby boost activity in PNF.125,126 Zhang et al. uncovered that 0.5% mol Cu modification could impart ZnAl-LDH oxygen vacancies and electron-rich unsaturated Cuδ+ (δ < 2);125 NaOH treatment could also induce vacancies and low-coordinated metal centers in ZnCr-LDH, ZnAl-LDH and NiAl-LDH.126 Taking Cuδ+-ZnAl-LDH as an example, it realized a NH3 production rate of 110 μmol g−1 h−1 under UV-vis light irradiation.125
In addition, Mo-based catalysts (e.g., Mo1/g-C3N4),127 carbon-tungstic-acid hybrids,128 Pr3+:LiNbO3,129 Pt GO/SiO2 (GO: graphene oxide)130 have also been utilized in PNF. These catalysts produce NH3 at the magnitude of μmol g−1 h−1. Meanwhile, the studies on these catalysts are quite limited and the reaction mechanism is not fully understood.
Summary and outlooks
Ideally, PNF is a safe and green approach to synthesize NH3 under ambient conditions, using the inexhaustible solar light as the sole energy input and the abundant N2 as a reactant. Catalyst is the key for PNF. Over the past few years, extensive studies have been conducted to search for efficient catalysts for PNF. Semiconductor, plasmonic metal-based catalysts, iron-based catalysts, ruthenium-based catalysts and several other catalysts, have been reported active in PNF. Table 1 lists the performances of some typical catalysts. This review summarizes the progress of each category of the catalysts designed for PNF, with a special attention on semiconductor-based catalysts. Generally speaking, the catalyst development is still in the infant stage and huge challenges need to be overcome.| Catalysts | Catalytic types | Reaction conditions | NH3 yield (μmol g−1 h−1) | Ref. |
|---|---|---|---|---|
| Ultrathin MoS2 | Pristine semiconductors | Water (200 mL), catalyst (15 mg), reaction temperature (25 °C), N2 bubble, under light irradiation (500 W, λ > 420 nm., Xe lamp) | 325 | 34 |
| BiO quantum dots | Pristine semiconductors | Water (200 mL), catalyst (50 mg), reaction temperature (25 °C), N2 bubble, under light irradiation (500 W, Xe lamp) | 1226 | 35 |
| PFL-g-C3N4 (PFL: porous few-layer) | Defective semiconductors | 20% CH3OH (100 mL), catalyst (20 mg), N2 (30 min), under light irradiation (500 W, AM 1.5G, 100 mW cm−2, Xe lamp) | 8200 | 38 |
| D-CN (1D defective g-C3N4) | Defective semiconductors | 0.1 mol L−1 K2SO4 solution (95 mL) and methanol (5 mL), catalyst (200 mg), reaction temperature (25 °C), N2 bubble (2 h), under light irradiation (600 mW cm−2), reaction time (8 h) | 17.4 | 39 |
| BOC/OV (surface oxygen vacancies modified micro-nanosheet structure Bi2O2CO3) | Defective semiconductors | 0.1 mmol L−1 Na2SO3 solution (50 mL), catalyst (30 mg), N2 (60 mL min−1, 30 min), under light irradiation (300 W, λ > 420 nm, Xe lamp) | 14.7 | 40 |
| A-SmOCl (amorphous SmOCl nanosheets) | Defective semiconductors | Water (20 mL), catalyst (10 mg), N2 (30 mL min−1, 30 min), under light irradiation (250 mW cm−2, 320–780 nm., Xe lamp) | 426 | 42 |
| TiO2 (B) nanotubes | Defective semiconductors | Mixture of water (90 mL) and methanol (10 mL), catalyst (25 mg), ultrasonic oscillation (10 min), reaction time (10 min), N2 (30 min) under simulated sunlight irradiation (300 W, 60 min, AM 1.5G, Xe lamp) | 106 | 48 |
| TiO2-OVs (reduced TiO2) | Defective semiconductors | Mixture of water (90 mL) and methanol (10 mL), catalyst (50 mg), ultrasonic oscillation (15 min), N2 (30 mL min−1), reaction temperature (25 °C), under light irradiation (300 W, Xe lamp) | 324.86 | 49 |
| BCN (B-doped g-C3N4 nanosheets) | Doped semiconductors | Aqueous solution of Na2SO3 (40 mL, 1.0 × 10−3 mol L−1), catalyst (20 mg), N2 (30 mL min−1, 30 min), reaction time (1 h), under light irradiation (250 W, λ > 400 nm, 500 mW cm−2, Xe lamp) | 313.9 | 55 |
| C-BiOI (carbon-doped BiOI) | Doped semiconductors | Mixture of water (90 mL) and ethanol (10 mL), catalyst (50 mg), under light irradiation (300 W, Xe lamp) | 311 | 61 |
| NCN/MgO (g-C3N4 nanosheets decorated with MgO nanoparticles) | Functionalized semiconductors | Water (40 mL) and 40 μL absolute ethanol (0.789 g L−1), catalyst (40 mg), ultrasonic oscillation (6 min), N2 (1 h), reaction temperature (25 °C), under light irradiation (500 W, 100 mW cm−2, λ > 420 nm, Xe lamp) | 4554 | 63 |
| KOH treated g-C3N4 | Functionalized semiconductors | CH3OH (150 mL), catalyst (20 mg), reaction temperature (25 °C), under light irradiation (300 W, 100 mW cm−2, Xe lamp) | 3632 | 64 |
| Cu/TiO2 (transition metal modified TiO2) | Functionalized semiconductors | 5.0 vol% glycerol aqueous solution (200 mL), catalyst (100 mg), under simulating solar light irradiation (300 W, AM 1.5 filter, Xe lamp) | 6780 | 65 |
| P-LFO (phosphate modified LaFeO3) | Functionalized semiconductors | Water (40 mL), catalyst (20 mg), N2 (1 h), under light irradiation (500 W, λ > 420 nm, Xe lamp) | 250 | 72 |
| Bi@BiOBr | Functionalized semiconductors | Water (100 mL), catalyst (10 mg), N2 (80 mL min−1, 30 min), reaction temperature (15 °C), under simulating solar light irradiation (300 W, 1.63 W cm−2, Xe lamp) | 1350 | 73 |
| MOF@DF-C3N4 (nano-MOF@defected thin film C3N4) | Heterojunctions and homojunctions | Mixture of water (48 mL) and methanol (2 mL), catalyst (10 mg), ultrasonic oscillation (15 min), reaction temperature (25 °C) under light irradiation (300 W, λ > 400 nm, Xe lamp) | 2320 | 83 |
| MoS2/C–ZnO | Heterojunctions and homojunctions | Mixture of water (190 mL) and ethanol (10 mL), catalyst (100 mg), reaction time (5 h), air, under light irradiation (300 W, λ > 420 nm, Xe lamp) | 49.1 | 88 |
| TiO2@C/g-C3N4 | Heterojunctions and homojunctions | 20 vol% CH3OH (100 mL), catalyst (50 mg), N2 (60 mL min−1), under light irradiation (300 W, λ < 420 nm, Xe lamp) | 250.6 | 89 |
| Na-Ad/Ro (Na treatment of P25-TiO2, Ad: Disordered anatase, RO: disordered rutile) | Heterojunctions and homojunctions | Water (50 mL), catalyst (50 mg), isopropyl alcohol (7 mL), N2 (0.3 L min−1) under simulated AM 1.5G sunlight irradiation (1000 W, Xe lamp) in a double-layered jacket with cooling water circulating line | 432 | 92 |
| Bi4O5Br2/ZIF-8 | Semiconductor-based hydrophilic-hydrophobic catalyst | Water (50 mL), catalyst (50 mg), N2 (80 mL min−1) under simulated sunlight irradiation (300 W, 200–800 nm, Xe lamp) | 16.4 | 93 |
| Au@UiO-66 | Plasmonic metal-based catalysts | Aqueous solution of K2SO4 (50 mL, 0.5 mol L−1), catalyst (15 mg), N2 (80 mL min−1, 30 min), reaction temperature (25 °C) under light irradiation (300 W, λ > 400 nm, 100mW cm−2, Xe lamp) in a conventional solution-based (i.e., PiS) protocol | 359.1 | 94 |
| Au/HCNS-NV (HCNS: Hollow mesoporous carbon nitride sphere, NV: nitrogen vacancies) | Plasmonic metal-based catalysts | Mixture of water (80 mL) and methanol (20 mL), catalyst (50 mg), ultrasonic oscillation (10 min), reaction temperature (room temperature), N2 (100 mL min−1, 30 min), under light irradiation (300 W, Xe lamp) | 783.4 | 95 |
| Au/P25-K+ | Plasmonic metal-based catalysts | Water (50 mL), catalyst (5 mg), N2 (60 mL min−1) under light irradiation (300 W, λ > 400 nm, 100mW cm−2, Xe lamp) | 430 | 98 |
| Fe–MoTe2 | Iron-based catalysts | Milli-Q (80 mL), catalyst (10 mg), reaction temperature (25 °C), N2 (50 sccm, 60 min), under light irradiation (300 W, λ < 420 nm, 400 mW cm−2, h, Xe lamp) | 129.08 | 103 |
| MIL-101(Fe) | Iron-based catalysts | Water (100 mL), catalyst (50 mg), ultrasonic oscillation (10 min), reaction temperature (room temperature), N2 (80 mL min−1, 30 min), under simulated light irradiation (300 W, 1 h, Xe lamp) | 100.7 | 109 |
| Ru–TiO2 | Ruthenium-based catalysts | 20% ethanol solution (100 mL), catalyst (40 mg), reaction temperature (25 °C), N2 (1 h), light irradiation (300 W, Xe lamp), reaction time (4 h) | 3.3 | 110 |
| Ru-Vs-CoS/CN (Ru/CoSx with S-vacancy on graphitic carbon nitride nanosheets) | Ruthenium-based catalysts | 10% methanol solution (50 mL), catalyst (25 mg), N2 (30 min), light irradiation (300 W, 200 mW cm−2, Xe lamp) | 438 | 117 |
| SiO2/C-RP (RP: red phosphorous) | Other catalysts | Water (40 mL), catalyst (20 mg), ultrasonic oscillation, N2, reaction temperature (25 °C) under light irradiation (300 W, 320 mW cm−2, Xe lamp) | 36.5 | 122 |
| Mo–PCN SACs (PNC: polymeric carbon nitride, SACs: single-atom catalysts) | Other catalysts | Water (6 mL), catalyst (3 mg), ultrasonic oscillation (60 min), pH = 5, N2, reaction temperature (room temperature), reaction time (12 h), light irradiation (300 W, Xe lamp) | 830 | 127 |
| Pr3+: LiNbO3 | Other catalysts | Aqueous solution (100 mL), catalyst (40 mg), N2 (30 min), reaction temperature (30 °C), under light irradiation (300 W, λ > 420 nm, Xe lamp) | 38.4 | 129 |
(1) NH3 production rates are still low. Over most of the catalysts, NH3 production rates are in the magnitude of mmol g−1 h−1 or even μmol g−1 h−1. It is far away from the industrial applications. Meanwhile, other chemicals (e.g., N2H4) are occasionally generated as byproducts. Therefore, persistent efforts should be devoted to design catalysts that could drive PNF efficiently and selectively to the desired product NH3. Adopting novel materials as catalysts for PNF might benefit this research area.
(2) There is a long way to make clear the reaction mechanism. In spite that some studies carried out mechanism explorations, little progress have been made in understanding the fundamental mechanism. The physicochemical properties of the photocatalysts under working states remain unclear; N2 chemisorption, activation and reduction pathway on the active sites are not clarified; the electron–hole transfer and migration routes need to be understood; in-depth understanding on the structure/property–performance correlations in PNF needs to be unraveled. Theoretical studies together with in situ characterization techniques might offer potential approaches to make clear the reaction mechanism.
In summary, despite that the development of catalysts for PNF is still in the primary stage, progress has been made. PNF has been proved as a promising avenue to replace the industrialized Haber-Bosch process to produce NH3. Numerous opportunities exist to move the research field forwards.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work received financial support from the National Natural Science Foundation of China (21902116), and Liaoning Revitalization Talents Program (XLYC1902070).References
- H. Koch, M. A. H. J. van Kessel and S. Lucker, Appl. Microbiol. Biotechnol., 2019, 103, 177–189 CrossRef CAS PubMed.
- H. Cheng, P. Cui, F. Wang, L.-X. Ding and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 15541–15547 CrossRef CAS PubMed.
- J. Xiong, P. Song, J. Di and H. Li, Chem. Eng. J., 2020, 402, 126208 CrossRef CAS.
- R. Su, Z. Liu, H. N. Abbasi, J. Wei and H. Wang, Materials, 2020, 13, 4559 CrossRef CAS PubMed.
- K. H. R. Rouwenhorst, A. G. J. Van der Ham and L. Lefferts, Int. J. Hydrogen Energy, 2021, 46, 21566–21579 CrossRef CAS.
- C. G. Balesdent, J. L. Crossland and D. R. Tyler, 242nd ACS National Meeting Denver, 2011 Search PubMed.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schloegl, Angew. Chem., Int. Ed., 2013, 52, 12723–12726 CrossRef CAS PubMed.
- B. A. Rohr, A. R. Singh and J. K. Norskov, J. Catal., 2019, 372, 33–38 CrossRef CAS.
- X. Wang, W. Wang, M. Qiao, G. Wu, W. Chen, T. Yuan, Q. Xu, M. Chen, Y. Zhang, X. Wang, J. Wang, J. Ge, X. Hong, Y. Li, Y. Wu and Y. Li, Sci. Bull., 2018, 63, 1246–1253 CrossRef CAS.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S.-X. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- W. Wang, H. Zhang, S. Zhang, Y. Liu, G. Wang, C. Sun and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16644–16650 CrossRef CAS PubMed.
- X. Chen, N. Li, Z. Kong, W.-J. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- X. Xue, R. Chen, C. Yan, P. Zhao, Y. Hu, W. Zhang, S. Yang and Z. Jin, Nano Res., 2019, 12, 1229–1249 CrossRef CAS.
- M. Li, H. Huang, J. Low, C. Goo, R. Long and Y. Xiong, Small Methods, 2019, 3, 1800388 CrossRef.
- D. Yan, H. Li, C. Chen, Y. Zou and S. Wang, Small Methods, 2019, 3, 1800331 CrossRef.
- Z. Li, Z. Gao, B. Li, L. Zhang, R. Fu, Y. Li, X. Mu and L. Li, Appl. Catal., B, 2020, 262, 118276 CrossRef CAS.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- Q. Hao, C. Liu, G. Jia, Y. Wang, H. Arandiyan, W. Wei and B.-J. Ni, Mater. Horiz., 2020, 7, 1014–1029 RSC.
- S. Chen, D. Liu and T. Peng, Sol. RRL, 2021, 5, 2000487 CrossRef CAS.
- V. Manh-Hiep, M. Sakar and D. Trong-On, Catalysts, 2018, 8, 621 CrossRef.
- G. Zhang, C. D. Sewell, P. Zhang, H. Mi and Z. Lin, Nano Energy, 2020, 71, 104645 CrossRef CAS.
- M.-H. Vu, M. Sakar, S. A. Hassanzadeh-Tabrizi and T.-O. Do, Adv. Mater. Interfaces, 2019, 6, 1900091 CrossRef.
- S. Zhang, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, J. Energy Chem., 2019, 1, 100013 Search PubMed.
- D. Ziegenbalg, J. Zander and R. Marschall, Chemphotochem, 2021, 5, 792 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, Indian Chem. Eng., Sect. A, 1996, 35, 443–453 Search PubMed.
- Q. Han, H. Jiao, L. Xiong and J. Tang, Adv. Mater., 2021, 2, 564 RSC.
- J. Yang, J. Chem., 2018, 2018, 3286782 Search PubMed.
- Q. Han, C. Wu, H. Jiao, R. Xu, Y. Wang, J. Xie, Q. Guo and J. Tang, Adv. Mater., 2021, 33, 2008180 CrossRef CAS PubMed.
- G. Zhang, Y. Li, C. He, X. Ren, P. Zhang and H. Mi, Adv. Energy Mater., 2021, 11, 2003294 CrossRef CAS.
- M. Nazemi and M. A. El-Sayed, Nano Energy, 2019, 63, 103886 CrossRef CAS.
- B. M. Comer and A. J. Medford, ACS Sustainable Chem. Eng., 2018, 6, 4648–4660 CrossRef CAS.
- G. Zhang, Y. Meng, B. Xie, Z. Ni, H. Lu and S. Xia, Appl. Catal., B, 2021, 296, 120379 CrossRef CAS.
- S. Sun, X. Li, W. Wang, L. Zhang and X. Sun, Appl. Catal., B, 2017, 200, 323–329 CrossRef CAS.
- S. Sun, Q. An, W. Wang, L. Zhang, J. Liu and W. A. Goddard III, J. Mater. Chem. A, 2017, 5, 201–209 RSC.
- Y. Liao, J. Qian, G. Xie, Q. Han, W. Dang, Y. Wang, L. Lv, S. Zhao, L. Luo, W. Zhang, H.-Y. Jiang and J. Tang, Appl. Catal., B, 2020, 273, 119054 CrossRef CAS.
- Z.-K. Shen, Y.-J. Yuan, P. Wang, W. Bai, L. Pei, S. Wu, Z.-T. Yu and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 17343–17352 CrossRef CAS PubMed.
- Y. Xue, X. Kong, Y. Guo, Z. Liang, H. Cui and J. Tian, J. Materiomics, 2020, 6, 128–137 CrossRef.
- D. Hao, C. Liu, X. Xu, M. Kianinia, I. Aharonovich, X. Bai, X. Liu, Z. Chen, W. Wei, G. Jia and B.-J. Ni, New J. Chem., 2020, 44, 20651–20658 RSC.
- Y. Feng, Z. Zhang, K. Zhao, S. Lin, H. Li and X. Gao, J. Colloid Interface Sci., 2021, 583, 499–509 CrossRef CAS PubMed.
- Y. Zhao, E. Wang and R. Jin, Diamond Relat. Mater., 2019, 94, 146–154 CrossRef CAS.
- T. Hou, R. Guo, L. Chen, Y. Xie, J. Guo, W. Zhang, X. Zheng, W. Zhu, X. Tan and L. Wang, Nano Energy, 2019, 65, 104003 CrossRef CAS.
- Y. Li, X. Chen, M. Zhang, Y. Zhu, W. Ren, Z. Mei, M. Gu and F. Pan, Catal. Sci. Technol., 2019, 9, 803–810 RSC.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed.
- H. Han, Y. Yang, J. Liu, X. Zheng, X. Wang, S. Meng, S. Zhang, X. Fu and S. Chen, ACS Appl. Energy Mater., 2020, 3, 11275–11284 CrossRef CAS.
- T. Wang, C. Feng, J. Liu, D. Wang, H. Hu, J. Hu, Z. Chen and G. Xue, Chem. Eng. J., 2021, 414, 128827 CrossRef CAS.
- Y. Shiraishi, M. Hashimoto, K. Chishiro, K. Moriyama, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2020, 142, 7574–7583 CrossRef CAS PubMed.
- S. Wu, X. Tan, K. Liu, J. Lei, L. Wang and J. Zhang, Catal. Today, 2019, 335, 214–220 CrossRef CAS.
- G. Zhang, X. Yang, C. He, P. Zhang and H. Mi, J. Mater. Chem. A, 2020, 8, 334–341 RSC.
- Y. Zhang, X. Chen, S. Zhang, L. Yin and Y. Yang, Chem. Eng. J., 2020, 401, 126033 CrossRef CAS.
- X. Xue, R. Chen, H. Chen, Y. Hu, Q. Ding, Z. Liu, L. Ma, G. Zhu, W. Zhang, Q. Yu, J. Liu, J. Ma and Z. Jin, Nano Lett., 2018, 18, 7372–7377 CrossRef CAS PubMed.
- B. Liu, A. S. Yasin, T. Musho, J. Bright, H. Tang, L. Huang and N. Wu, J. Electrochem. Soc., 2019, 166, H3091–H3096 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed.
- J. Qin, W. Zhao, X. Hu, J. Li, P. Ndokoye and B. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 7127–7134 CrossRef CAS PubMed.
- W. Wang, H. Zhou, Y. Liu, S. Zhang, Y. Zhang, G. Wang, H. Zhang and H. Zhao, Small, 2020, 16, 1906880 CrossRef CAS PubMed.
- Z. Ying, S. Chen, S. Zhang, T. Peng and R. Li, Appl. Catal., B, 2019, 254, 351–359 CrossRef CAS.
- X. Xue, H. Chen, Y. Xiong, R. Chen, M. Jiang, G. Fu, Z. Xi, X. L. Zhang, J. Ma, W. Fang and Z. Jin, ACS Appl. Mater. Interfaces, 2021, 13, 4975–4983 CrossRef CAS PubMed.
- D. Wu, R. Wang, C. Yang, Y. An, H. Lu, H. Wang, K. Cao, Z. Gao, W. Zhang, F. Xu and K. Jiang, J. Colloid Interface Sci., 2019, 556, 111–119 CrossRef CAS PubMed.
- J. Li, D. Wang, R. Guan, Y. Zhang, Z. Zhao, H. Zhai and Z. Sun, ACS Sustainable Chem. Eng., 2020, 8, 18258–18265 CrossRef CAS.
- S. Cao, B. Fan, Y. Feng, H. Chen, F. Jiang and X. Wang, Chem. Eng. J., 2018, 353, 147–156 CrossRef CAS.
- L. Zeng, F. Zhe, Y. Wang, Q. Zhang, X. Zhao, X. Hu, Y. Wu and Y. He, J. Colloid Interface Sci., 2019, 539, 563–574 CrossRef CAS PubMed.
- P. Qiu, Z. Liang, X. Liu, X. Qian, H. Cui and J. Tian, J. Colloid Interface Sci., 2020, 571, 318–325 CrossRef CAS PubMed.
- E. Vesali-Kermani, A. Habibi-Yangjeh and S. Ghosh, J. Ind. Eng. Chem., 2020, 84, 185–195 CrossRef CAS.
- X. Li, X. Sun, L. Zhang, S. Sun and W. Wang, J. Mater. Chem. A, 2018, 6, 3005–3011 RSC.
- Y. Liu, Z. Yu, S. Guo, L. Yao, R. Sun, X. Huang and W. Zhao, New J. Chem., 2020, 44, 19924–19932 RSC.
- J. M. Walls, J. S. Sagu and K. G. U. Wijayantha, RSC Adv., 2019, 9, 6387–6394 RSC.
- T. Fei, L. Yu, Z. Liu, Y. Song, F. Xu, Z. Mo, C. Liu, J. Deng, H. Ji, M. Cheng, Y. Lei, H. Xu and H. Li, J. Colloid Interface Sci., 2019, 557, 498–505 CrossRef CAS PubMed.
- Y. Hao, X. Dong, S. Zhai, H. Ma, X. Wang and X. Zhang, Chem.–Eur. J., 2016, 22, 18722–18728 CrossRef CAS PubMed.
- S. Cao, H. Chen, F. Jiang and X. Wang, Appl. Catal., B, 2018, 224, 222–229 CrossRef CAS.
- Y. Kong, C. Lv, C. Zhang and G. Chen, Appl. Surf. Sci., 2020, 515, 146009 CrossRef CAS.
- Y. Bi, Y. Wang, X. Dong, N. Zheng, H. Ma and X. Zhang, RSC Adv., 2018, 8, 21871–21878 RSC.
- X. Sun, D. Jiang, L. Zhang, S. Sun and W. Wang, ACS Sustainable Chem. Eng., 2017, 5, 9965–9971 CrossRef CAS.
- M. Lan, N. Zheng, X. Dong, H. Ma and X. Zhang, Colloids Surf., A, 2021, 623, 126744 CrossRef CAS.
- X. Rong, Y. Mao, J. Xu, X. Zhang, L. Zhang, X. Zhou, F. Qiu and Z. Wu, Catal. Commun., 2018, 116, 16–19 CrossRef CAS.
- H. Mou, J. Wang, D. Yu, D. Zhang, W. Chen, Y. Wang, D. Wang and T. Mu, ACS Appl. Mater. Interfaces, 2019, 11, 44360–44365 CrossRef CAS PubMed.
- C. Xiao, H. Wang, L. Zhang, S. Sun and W. Wang, Chemcatchem, 2019, 11, 6467–6472 CrossRef CAS.
- H. Zhang, X. Li, H. Su, X. Chen, S. Zuo, X. Yan, W. Liu and C. Yao, J. Sol-Gel Sci. Technol., 2019, 92, 154–162 CrossRef CAS.
- G. Zhang, T. Dai, Y. Wang, Y. Meng, B. Xie, Z. Ni and S. Xia, Appl. Catal., B, 2021, 288, 119990 CrossRef CAS.
- Y. Chen, C. Zhao, S. Ma, P. Xing, X. Hu, Y. Wu and Y. He, Inorg. Chem. Front., 2019, 6, 3083–3092 RSC.
- S. Xia, G. Zhang, Z. Gao, Y. Meng, B. Xie, H. Lu and Z. Ni, J. Colloid Interface Sci., 2021, 604, 798–809 CrossRef CAS PubMed.
- S. Cao, N. Zhou, F. Gao, H. Chen and F. Jiang, Appl. Catal., B, 2017, 218, 600–610 CrossRef CAS.
- Y. Wang, W. Wei, M. Li, S. Hu, J. Zhang and R. Feng, RSC Adv., 2017, 7, 18099–18107 RSC.
- Z. Ding, S. Wang, X. Chang, D.-H. Wang and T. Zhang, RSC Adv., 2020, 10, 26246–26255 RSC.
- T. Wang, J. Liu, P. Wu, C. Feng, D. Wang, H. Hu and G. Xue, J. Mater. Chem. A, 2020, 8, 16590–16598 RSC.
- H. Liang, L. Fang and S. Hu, New J. Chem., 2019, 43, 12094–12102 RSC.
- W. Guang, Y. Lihua, L. Yifu, Z. Jiaming, H. Zheng and G. Gui, Appl. Surf. Sci., 2019, 481, 649–660 CrossRef.
- X. Xue, R. Chen, C. Yan, Y. Hu, W. Zhang, S. Yang, L. Ma, G. Zhu and Z. Jin, Nanoscale, 2019, 11, 10439–10445 RSC.
- P. Xing, P. Chen, Z. Chen, X. Hu, H. Lin, Y. Wu, L. Zhao and Y. He, ACS Sustainable Chem. Eng., 2018, 6, 14866–14879 CrossRef CAS.
- Q. Liu, L. Ai and J. Jiang, J. Mater. Chem. A, 2018, 6, 4102–4110 RSC.
- Y. Shiraishi, K. Chishiro, S. Tanaka and T. Hirai, Langmuir, 2020, 36, 734–741 CrossRef CAS PubMed.
- H. Xu, Y. Wang, X. Dong, N. Zheng, H. Ma and X. Zhang, Appl. Catal., B, 2019, 257, 117932 CrossRef CAS.
- J. Lee, X. Liu, A. Kumar, Y. Hwang, E. Lee, J. Yu, Y. D. Kim and H. Lee, Chem. Sci., 2021, 12, 9619–9629 RSC.
- J. Liu, R. Li, X. Zu, X. Zhang, Y. Wang, Y. Wang and C. Fan, Chem. Eng. J., 2019, 371, 796–803 CrossRef CAS.
- L.-W. Chen, Y.-C. Hao, Y. Guo, Q. Zhang, J. Li, W.-Y. Gao, L. Ren, X. Su, L. Hu, N. Zhang, S. Li, X. Feng, L. Gu, Y.-W. Zhang, A.-X. Yin and B. Wang, J. Am. Chem. Soc., 2021, 143, 5727–5736 CrossRef CAS PubMed.
- Y. Guo, J. Yang, D. Wu, H. Bai, Z. Yang, J. Wang and B. Yang, J. Mater. Chem. A, 2020, 8, 16218–16231 RSC.
- Y. Wang, R. Zhao, F. Wang, Y. Liu, X. Yu, L. Chen, Y. Yao, S. Lu and X. Liao, Catal. Sci. Technol., 2020, 10, 7652–7660 RSC.
- P. Qiu, C. Huang, G. Dong, F. Chen, F. Zhao, Y. Yu, X. Liu, Z. Li and Y. Wang, J. Mater. Chem. A, 2021, 9, 14459–14465 RSC.
- T.-A. Bu, Y.-C. Hao, W.-Y. Gao, X. Su, L.-W. Chen, N. Zhang and A.-X. Yin, Nanoscale, 2019, 11, 10072–10079 RSC.
- Y. Bo, H. Wang, Y. Lin, T. Yang, R. Ye, Y. Li, C. Hu, P. Du, Y. Hu, Z. Liu, R. Long, C. Gao, B. Ye, L. Song, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 16085–16092 CrossRef CAS PubMed.
- X. Chen, X. Zhang, Y.-H. Li, M.-Y. Qi, J.-Y. Li, Z.-R. Tang, Z. Zhou and Y.-J. Xu, Appl. Catal., B, 2021, 281, 119516 CrossRef CAS.
- Z. Ying, S. Chen, T. Peng, R. Li and J. Zhang, Eur. J. Inorg. Chem., 2019, 2019, 2182–2192 CrossRef CAS.
- J. Luo, X. Bai, Q. Li, X. Yu, C. Li, Z. Wang, W. Wu, Y. Liang, Z. Zhao and H. Liu, Nano Energy, 2019, 66, 104187 CrossRef CAS.
- H. Li, S. Gu, Z. Sun, F. Guo, Y. Xie, B. Tao, X. He, W. Zhang and H. Chang, J. Mater. Chem. A, 2020, 8, 13038–13048 RSC.
- C. Mao, H. Li, H. Gu, J. Wang, Y. Zou, G. Qi, J. Xu, F. Deng, W. Shen, J. Li, S. Liu, J. Zhao and L. Zhang, Chem, 2019, 5, 2702–2717 CAS.
- S. Liu, S. Wang, Y. Jiang, Z. Zhao, G. Jiang and Z. Sun, Chem. Eng. J., 2019, 373, 572–579 CrossRef CAS.
- Q. Chen, Y. Zhou, J.-X. Zhu, T.-T. Liang, R.-B. Huang and A.-M. Chen, Chin. J. Inorg. Chem., 2020, 36, 426–434 CAS.
- X. Ye, X. Li, X. Chu, Z. Wang, S. Zuo, T. Wang and C. Yao, J. Alloys Compd., 2021, 871, 159542 CrossRef CAS.
- Y. Fang, Y. Xue, L. Hui, H. Yu and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3170–3174 CrossRef CAS PubMed.
- G. Li, F. Li, J. Liu and C. Fan, J. Solid State Chem., 2020, 285, 121245 CrossRef CAS.
- S. Liu, Y. Wang, S. Wang, M. You, S. Hong, T.-S. Wu, Y.-L. Soo, Z. Zhao, G. Jiang, J. Qiu, B. Wang and Z. Sun, ACS Sustainable Chem. Eng., 2019, 7, 6813–6820 CrossRef CAS.
- Y. Liao, J. Lin, B. Cui, G. Xie and S. Hu, J. Photochem. Photobiol., A, 2020, 387, 112100 CrossRef CAS.
- L. Li, Y. Wang, S. Vanka, X. Mu, Z. Mi and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 8701–8705 CrossRef CAS PubMed.
- H. Liu, P. Wu, H. Li, Z. Chen, L. Wang, X. Zeng, Y. Zhu, Y. Jiang, X. Liao, B. S. Haynes, J. Ye, C. Stampfl and J. Huang, Appl. Catal., B, 2019, 259, 118026 CrossRef CAS.
- C. Mao, L. Yu, J. Li, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 224, 612–620 CrossRef CAS.
- Z. Ma, X. Xiong, C. Song, B. Hu and W. Zhang, RSC Adv., 2016, 6, 51106–51110 RSC.
- H. Wang, X. Li, Q. Ruan and J. Tang, Nanoscale, 2020, 12, 12329–12335 RSC.
- Y. Jili, Y. Xuanying, T. Yanhong, L. Meijun and L. Chengbin, Adv. Funct. Mater., 2020, 30, 1906983 CrossRef.
- A. Awati, H. Maimaiti, S. Wang and B. Xu, Sci. Total Environ., 2019, 695, 133865 CrossRef CAS PubMed.
- H. Maimaiti, S. Wang, A. Awati and B. Xu, J. Nanopart. Res., 2020, 22, 101 CrossRef CAS.
- C. Hao, Y. Liao, Y. Wu, Y. An, J. Lin, Z. Gu, M. Jiang, S. Hu and X. Wang, J. Phys. Chem. Solids, 2020, 136, 109141 CrossRef CAS.
- S. Bian, M. Wen, J. Wang, N. Yang, P. K. Chu and X.-F. Yu, J. Phys. Chem. Lett., 2020, 11, 1052–1058 CrossRef CAS PubMed.
- L. Lin, Q. Zhu, A. Cheng and L. Ma, Catal. Sci. Technol., 2020, 10, 4119–4125 RSC.
- C. Zhang, Y. Xu, C. Lv, X. Zhou, Y. Wang, W. Xing, Q. Meng, Y. Kong and G. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 29917–29923 CrossRef CAS PubMed.
- H. Huang, X.-S. Wang, D. Philo, F. Ichihara, H. Song, Y. Li, D. Li, T. Qiu, S. Wang and J. Ye, Appl. Catal., B, 2020, 267, 118686 CrossRef CAS.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2020, 10, 1901973 CrossRef CAS.
- Y. Zhao, L. Zheng, R. Shi, S. Zhang, X. Bian, F. Wu, X. Cao, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2020, 10, 2002199 CrossRef CAS.
- X.-W. Guo, S.-M. Chen, H.-J. Wang, Z.-M. Zhang, H. Lin, L. Song and T.-B. Lu, J. Mater. Chem. A, 2019, 7, 19831–19837 RSC.
- X. Li, W. Wang, D. Jiang, S. Sun, L. Zhang and X. Sun, Chem.–Eur. J., 2016, 22, 13819–13822 CrossRef CAS PubMed.
- L. Sun, X. Chu, C. He, S. Zuo, X. Li and C. Yao, Appl. Phys. A: Mater. Sci. Process., 2021, 127, 35 CrossRef CAS.
- B. Xu, H. Maimaiti, S. Wang, P. Zhai and H. Zhang, React. Kinet., Mech. Catal., 2020, 130, 1155–1170 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |