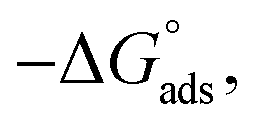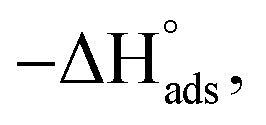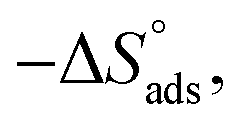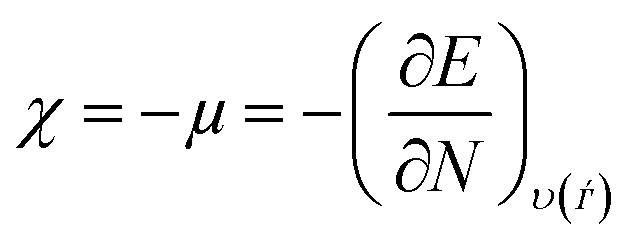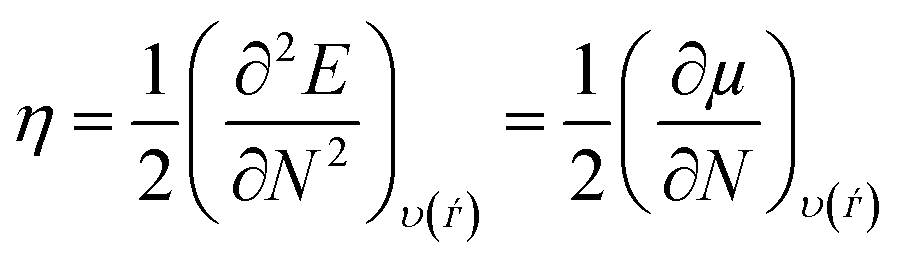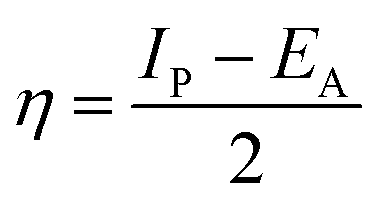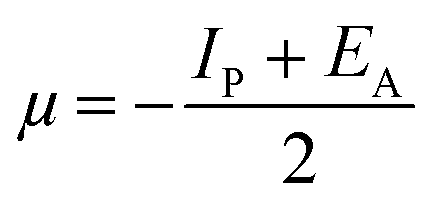DOI:
10.1039/D1RA07983B
(Paper)
RSC Adv., 2022,
12, 3253-3273
Electrochemical and quantum chemical studies on the corrosion inhibition of 1037 carbon steel by different types of surfactants
Received
30th October 2021
, Accepted 6th January 2022
First published on 24th January 2022
Abstract
In this work, three different types of surfactants, namely, dodecyl trimethyl ammonium chloride (DTAC, C12H25N (CH3)3Cl)−, octyl phenol poly(ethylene glycol ether)x (TX-100, C34H62O11 for x = 10) and dioctyl sodium sulfosuccinate (AOT-100, C20H37O7NaS) with corrosion restraint were utilized as corrosion inhibitors for 1037 CS in 0.5 M HCl. The protection efficacy (% IE) was indicated by weight loss and electrochemical measurements. Polarization curves showed that the investigated compounds are mixed-type inhibitors. The protection efficacy (% IE) increases with the increase in the surfactant concentration and reached 64.42–86.46% at 8 × 10−4 M and 30 °C. Adsorption of these utilized surfactants (DTAC, TX-100, and AOT) onto the CS surface concurred with the Langmuir adsorption isotherm. Impedance data revealed that by increasing the surfactant concentration, the charge transfer resistance (Rct) increases and vice versa for the capacitance of double layer (Cdl). Surface morphological investigations such as scanning electron microscopy (SEM) combined with EDX and atomic force microscopy (AFM) were used to further investigate the inhibitors' protective abilities. Monte Carlo simulations showed the great interaction between the tested surfactants and the metal surface of the CS. The theoretical results (density functional theory, DFT) were in good agreement with experimental measurements. The restraint efficiencies of anionic, neutral, and cationic surfactants regarded a certain dating to HSAB precept and Fukui indices.
1. Introduction
The hydrochloric acid solution was utilized as a pickling agent in oil fields because it is the most inexpensive manner to interrupt down calcium carbonate, CaCO3, scale interior the pipelines beneath maximum conditions. Appropriately, corrosion surfactants (regularly surfactants) should be injected with the hydrochloric acid solution to avoid the adverse impact of acid on the surface of the pipelines.1 CS has been extensively applied as a production material for pipe paintings inside the oil and fuel lines, which include down hollow tubular, float lines, and transmission pipelines.2 Surfactants are compounds that may be determined in a huge variety of business settings to analyze laboratories and are part of our everyday lives. The surfactants have severe focal factors including excessive inhibition efficiency, low price, low toxicity, and clean manufacturing.3 Moreover, research on surfactants adsorbed onto steel surfaces is extraordinarily critical for electrochemical properties such as corrosion inhibition, adhesion, lubrication, and detergency.4 “The effective type of corrosion surfactant for these applications is film-forming surfactant. Nowadays, surfactants are widely used, and find a wide range of applications in the petroleum industry. This is attributed to their remarkable ability to influence the properties of surfaces and interfaces. Recently, one of the most important applications of surfactants is effective corrosion inhibition in the oil industry. It interacts with anodic or cathodic reaction sites and retards corrosive oxidation and reduction reactions. An increment in surfactant action was observed when the concentration within the destructive arrangement approaches the basic critical micelle concentration (CMC). Above this value, there was no further increment in the efficiency that remains constant for further increment in surfactant concentration. Within the nonappearance of a charged head group, the driving force of micellization is the hydrophobic drive and van der Waals attractions. The strong interaction between water particles repulses the hydrocarbon chain from the water bulk phase. This drives the surfactants to form aggregates where the hydrophilic head groups conceal the hydrocarbon chains. It was observed that the adsorption of these surfactants depends on the physico-chemical properties of the useful groups and the electron density at the donor atoms. The adsorption happens due to the interaction of the lone pair and/or π-orbitals of surfactants with the d-orbitals of the metal surface atoms, which brings out more prominent adsorption of the surfactant particles onto the surface, leading to the formation of a corrosion protection film.5–7 The adsorption is additionally affected by the structure and the charge of the metal surface, and the type of testing electrolyte.8–10 Recently, quantum chemical strategies have been demonstrated to be exceptionally valuable in deciding the atomic structure as well as elucidating the electronic structure and reactivity.11 In this way, it has gotten to be a common practice to carry out quantum chemical calculations in corrosion hindrance studies. The concept of evaluating the productivity of a corrosion surfactant by computational chemistry is to search for compounds with desirable properties using chemical intuition and experience into a mathematically quantified and computerized form. Once a relationship between the structure and movement or property is found, any number of compounds, counting those not however synthesized, can be promptly screened by a computational technique12 and a set of numerical conditions, which can speak to the chemical phenomenon beneath study.13,14 Surfactants, because of their remarkable ability to influence the properties of surfaces and interfaces, have been effectively exploited as corrosion surfactants in an acidic medium.15–25 Surfactants are amphiphilic molecules containing one hydrophilic (head) and other hydrophobic (tail) parts; this favors the adsorption process at metallic surfaces”. The hydrophilic part of the surfactant can be positive, negative, impartial or zwitterionic and the hydrophobic component includes one or more hydrocarbon chains, commonly with 6–22 carbon atoms.
The aim of this paper is to get data on the level of corrosion activity in a system, utilizing the chemical and electrochemical procedures. This information is utilized to assess the corrosion surfactant capability. Another objective of this work is to calculate the more related molecular properties based on its activity as corrosion inhibitors. The nearby reactivity was analyzed using Fukui indices, since they indicate the reactive regions within the shape of the nucleophilic and electrophilic behavior of each atom in the molecule. These surfactants are not used as corrosion inhibitors for CS in HCl media in the literature.
2. Experimental
2.1 Chemicals and materials
Hydrochloric acid (37% wt), “ethyl alcohol and acetone were acquired from Al-Gomhoria Company (Egypt). Surfactants (dodecyl trimethyl ammonium chloride: DTAC (C12H25N (CH3)3 –Cl); octyl phenol poly(ethylene glycol ether)x: TX-100 (C34H62O11 for x = 10); dioctyl sodium sulfosuccinate: AOT-100 (C20H37O7NaS)) were obtained from Aldrich Chemical Company. The molecular structures of DTAC, TX-100 and AOT-100 are shown in Scheme 1. Refined water was utilized for preparing test solutions for all measurements. The corrosion tests were performed on 1037 carbon steel. Rectangular specimens with dimensions 2.00 cm × 2.00 cm × 0.20 cm were utilized for weight loss estimations”. For electrochemical estimations, the exposed surface area of carbon steel was 1.00 cm2. Scheme 1 reports the structure, names and molecular formulas of the utilized surfactants.
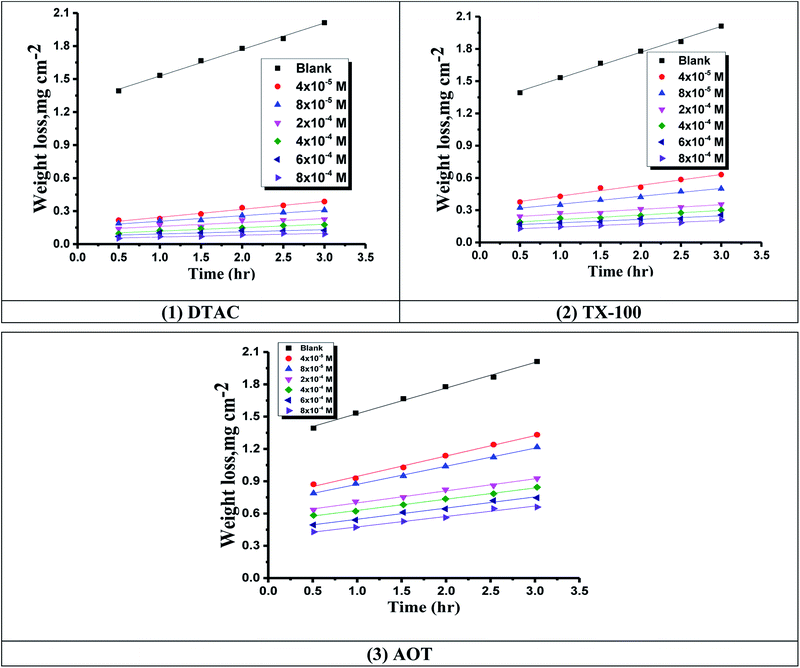 |
| | Fig. 1 Time-WL bends of CS in half molar HCl in the existence and lack of various doses of (1) DTAC, (2) TX-100 and (3) AOT at 30 °C. | |
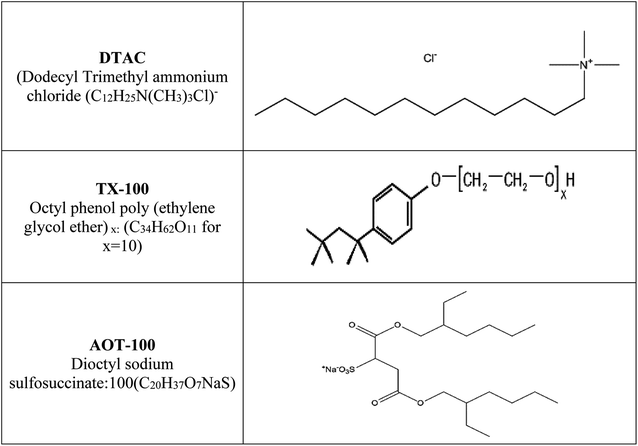 |
| | Scheme 1 The structure, names and molecular formulas of the utilized surfactants. | |
2.2 Methods
2.2.1 Weight loss (WL) estimations.
The WL tests were carried out in accordance with the American Society for Testing and Materials (ASTM) standard protocol. CS specimens were mechanically cleaned with 80 to 1200 grit sizes of emery papers and degreased with acetone, washed two times with distilled water and finally dried using filter papers. After precisely weighing, the specimens were dipped in 100 ml of 0.5 M HCl with and without altered concentrations of surfactants at 30 °C. After a distinctive submersion time period (30, 60, 90, 120, 150 and 180 min), the CS coins were taken out, washed with distilled water, dried and then weighted precisely. The tests were performed in triplicate and the average value of the WL was considered.
2.3. Electrochemical measurement
Three electrochemical techniques, “specifically potentiodynamic polarization (PDP), electrochemical impedance spectroscopy (EIS), and electrochemical frequency modulation (EFM), were utilized to study the corrosion behavior. All tests were conducted in an ordinary three cathodes glass cell. A Pt electrode as the counter electrode and a saturated calomel electrode as the reference electrode were utilized in this. The CS specimen was machined into a rectangular shape (1.00 cm × 1.00 cm × 0.30 cm) and fixed with epoxy resin taking off a working zone of 1.00 cm2, and the specimens were cleaned”, degreased, and washed as portrayed in WL estimations.
2.3.1. PDP measurements.
These were carried out using a Volta Lab “PGZ 100 system connected to a personal computer using the Volta Master 4 version 7.08 software for calculation. All the tests were performed at a temperature of 30 °C. The balance time leading to the consistent state of the specimens was 20 min and the open circuit potential (OCP) was noted”. The potentiodynamic bends were recorded from −900 to −200 mV at a scan rate 1 mV s−1.
2.3.2. EIS and EFM measurements.
These were carried out using a Gamry Instrument Series “G 750™ Potentiostat/Galvanostat/ZRA equipped with a Gamry framework system based on ESA400. Gamry applications include software EIS300 for EIS measurements, and EFM140 to calculate the corrosion current density and the Tafel constants for EFM measurements. A computer was utilized for collecting information. The Echem Analyst 5.5 software was utilized for plotting, graphing and fitting information. EIS estimations were carried out in the frequency range of 105 to 10 Hz with an amplitude of 10 mV peak to peak using ac signals at a respective corrosion potential. EFM was carried out utilizing two frequencies 0.2 and 0.5 Hz”. The base frequency became 0.1 Hz, so the waveform repeats after 0.1 s. In this study, we make use of a perturbation sign with an amplitude of 10 mV for each perturbation frequency of 20 and 50 m Hz.
2.4 Quantum chemical calculation
HOMO energy “(highest occupied molecular orbital), LUMO energy (lowest unoccupied molecular orbital) and Fukui index estimations were performed using Materials Studio DMol3 version 4.4.0,26,27 a high-quality quantum mechanics computer program (available from Accelrys Inc., San Diego, CA). These estimations employed an ab initio, gradient-corrected functional (GGA) method with a double numeric plus polarization (DNP) basis set and a Becke One Parameter (BOP) functional. It is well known that the phenomena of electrochemical corrosion appear in an aqueous phase. For this reason, it is essential to incorporate the solvent effect in the computational calculations. In a comparable way, it is imperative to consider the impacts that can appear as much in the geometric properties as within the electrical ones. DMol3 incorporates certain COSMO”28 controls, which permit for the behavior of solvation impacts.
3. Results and discussion
3.1 Weight loss (WL) measurements
The WL values were used to calculate the corrosion rate (CR) in milli-meter consistent with year (mm per year) using the eqn (1):
where K = 8.75 × 104.
The protection efficiency (% IE) and the degree of surface coverage (θ) were calculated using the following relation eqn (2) and (3):
| | 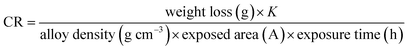 | (1) |
| | 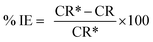 | (2) |
| | 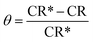 | (3) |
where CR* and CR are the corrosion rates of C-carbon steel with and without surfactants, respectively.
Fig. 1 reports the WL-time bends for CS in 0.5 M HCl at various doses of the three surfactants at 30 °C.
Table 1 shows the dependence of “% IE on the altered concentrations of surfactants (AOT, TX-100 and DTAC) in the range from 4.0 × 10−5 mol l−1 to 8.0 × 10−4 mol l−1. From the data in this table, the following conclusions can be made”:
Table 1 WL for CS in 0.5 M HCl solution in the presence and absence of altered doses of surfactants at 30 °C
| Compound |
Conc., M |
CR (mm per year) |
θ
|
% IE |
| Blank |
0.5 |
2.68 ± 0.0173 |
— |
— |
| AOT |
4.0 × 10−5 |
2.13 ± 0.0145 |
0.205 |
20.5 |
| 8.0 × 10−5 |
1.94 ± 0.0145 |
0.276 |
27.6 |
| 2.0 × 10−4 |
1.27 ± 0.0230 |
0.526 |
52.6 |
| 4.0 × 10−4 |
1.17 ± 0.0155 |
0.563 |
56.3 |
| 6.0 × 10−4 |
1.16 ± 0.0239 |
0.567 |
56.7 |
| 8.0 × 10−4 |
1.08 ± 0.0153 |
0.597 |
59.7 |
| TX-100 |
4.0 × 10−5 |
1.10 ± 0.0203 |
0.590 |
59.0 |
| 8.0 × 10−5 |
0.82 ± 0.0014 |
0.694 |
69.4 |
| 2.0 × 10−4 |
0.48 ± 0.0015 |
0.820 |
82.0 |
| 4.0 × 10−4 |
0.46 ± 0.0023 |
0.828 |
82.8 |
| 6.0 × 10−4 |
0.36 ± 0.0015 |
0.866 |
86.6 |
| 8.0 × 10−4 |
0.341 ± 0.0023 |
0.873 |
87.3 |
| DTAC |
4.0 × 10−5 |
0.80 ± 0.0015 |
0.700 |
70.0 |
| 8.0 × 10−5 |
0.56 ± 0.0014 |
0.791 |
79.1 |
| 2.0 × 10−4 |
0.39 ± 0.0017 |
0.854 |
85.4 |
| 4.0 × 10−4 |
0.35 ± 0.0015 |
0.869 |
86.9 |
| 6.0 × 10−4 |
0.22 ± 0.0016 |
0.918 |
91.8 |
| 8.0 × 10−4 |
0.19 ± 0.0012 |
0.929 |
92.9 |
(1) At a constant temperature, the % PE rises with improvement in the surfactant concentration.
(2) The lowest CR was obtained by DTAC and the highest by AOT-100; therefore, IE tends to decrease in the subsequent order: DTAC > TX-100 > AOT.
(3) The reticence action of the surfactants in “HCl can be simply considered as electrostatic adsorption29 and covalent bonding chemisorption. This action was attributed to the effect of chloride ions of DATC surfactants, chloride ions of acid solution and chemisorption of C12H25N+ (CH3)3, C34H62O11 and C20H37O7S−. In addition, other factors such as CMC and structure of surfactant might be affecting the inhibition activity”.
3.1.1 Effect of temperature.
The effect of temperature, “in the range of 30–60 °C with an increment of 10 °C on both the corrosion rate and the % IE of different surfactants in 0.5 M HCl, was studied by WL measurements and was given in Fig. 2. In Fig. 2, we can see that increasing the temperature leads to an increase in the corrosion rate of CS both in free acid and inhibited acid solutions and a decrease in the % IE of surfactants, which suggested that corrosion inhibition of CS by the investigated surfactants is caused by the adsorption of surfactant molecules onto the CS surface, while at higher temperatures, the desorption of the investigated surfactants from the CS surface occurred”.30
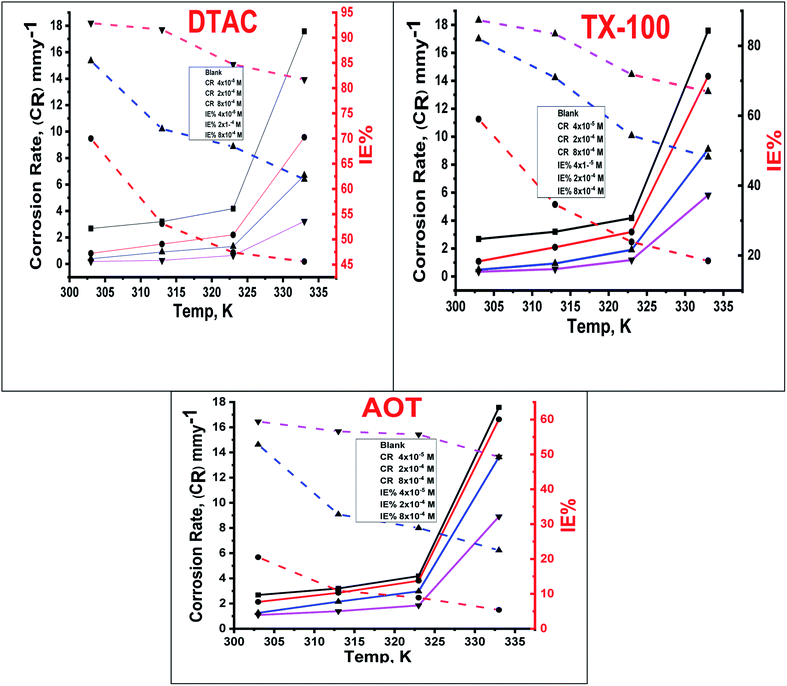 |
| | Fig. 2 Variation of % IE and (CR) at different doses of utilized surfactants in 0.5 M HCl at different temperatures. | |
3.1.2 Kinetic parameters of the used surfactants on the CS surface.
The inhibition properties of the used surfactants can be explained by means of kinetic model. The activation parameters were calculated from Arrhenius eqn (4) and transition state eqn (5):| | 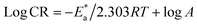 | (4) |
where A represents the pre-exponential factor.| | CR = RT/Nh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(ΔS*/R)exp(−ΔH*/RT) exp(ΔS*/R)exp(−ΔH*/RT) | (5) |
where “h is Planck's constant, N is the Avogadro number, ΔS* is the entropy of activation, and ΔH* is the enthalpy of activation. The values of CR (in g m−2 h−1) were obtained at different temperatures, which is a linear function with 1/T and permits the calculation of the Arrhenius activation energy, 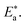 The regression between logkcorr. and 1/T was calculated, and Arrhenius plots of logkcorr.vs. 1/T for the blank and different concentrations of surfactants are shown in Fig. 3. All parameters are given in Table 2. The values of
The regression between logkcorr. and 1/T was calculated, and Arrhenius plots of logkcorr.vs. 1/T for the blank and different concentrations of surfactants are shown in Fig. 3. All parameters are given in Table 2. The values of 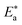 in the presence of surfactants are higher than those in the uninhibited acid solution. The results agreed with that of previous study reported by Fouda et al.31 and Popova et al.32 for CS in 1 M HCl. The addition of surfactants to the solution increases the activation energy
in the presence of surfactants are higher than those in the uninhibited acid solution. The results agreed with that of previous study reported by Fouda et al.31 and Popova et al.32 for CS in 1 M HCl. The addition of surfactants to the solution increases the activation energy 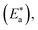 as shown in Table 2 and the extent of the increase is proportional to the IE of the surfactant, indicating that the energy barrier for the corrosion reaction increases in the presence of these surfactants. This means that by addition of the surfactant in the acid solution, the corrosion reaction will be further pushed to surface sites that are characterized by higher values of activation energy
as shown in Table 2 and the extent of the increase is proportional to the IE of the surfactant, indicating that the energy barrier for the corrosion reaction increases in the presence of these surfactants. This means that by addition of the surfactant in the acid solution, the corrosion reaction will be further pushed to surface sites that are characterized by higher values of activation energy 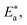 indicating that CS corrosion occurs at the uncovered part of the surface. Thus, adsorption of the surfactant was assumed to occur on the higher energy sites,33 and the presence of surfactant, which results in the blocking of the active sites, must be associated with an increase in the activation energy,
indicating that CS corrosion occurs at the uncovered part of the surface. Thus, adsorption of the surfactant was assumed to occur on the higher energy sites,33 and the presence of surfactant, which results in the blocking of the active sites, must be associated with an increase in the activation energy, 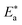 which indicates the physical adsorption or weak chemical bonding between the surfactant molecules and the CS surface.34 The increase in
which indicates the physical adsorption or weak chemical bonding between the surfactant molecules and the CS surface.34 The increase in 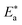 in the presence of the surfactants indicates the physical adsorption that occurs in first stage,35,36 weak chemical bonding between the surfactants and the steel surface37 or due to the decrease in the adsorption of these surfactants with the increase in temperature.38 Generally, one can say that the nature and concentration of electrolytes greatly affect the activation energy for the corrosion process. A plot of log(kcorr./T) vs. (1/T) gives a straight line with a slope of [−ΔH*/2.303R] and an intercept of [log(R/Nh) + ΔS*/2.303R]. The plots are shown in Fig. 4. The calculated values of ΔH* and ΔS* are given in Table 2. The values of ΔH* are positive indicating that the corrosion process is endothermic one. The values of
in the presence of the surfactants indicates the physical adsorption that occurs in first stage,35,36 weak chemical bonding between the surfactants and the steel surface37 or due to the decrease in the adsorption of these surfactants with the increase in temperature.38 Generally, one can say that the nature and concentration of electrolytes greatly affect the activation energy for the corrosion process. A plot of log(kcorr./T) vs. (1/T) gives a straight line with a slope of [−ΔH*/2.303R] and an intercept of [log(R/Nh) + ΔS*/2.303R]. The plots are shown in Fig. 4. The calculated values of ΔH* and ΔS* are given in Table 2. The values of ΔH* are positive indicating that the corrosion process is endothermic one. The values of 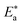 are larger than the analogous values of ΔH*, indicating that the corrosion process must involve a gaseous reaction, simply the hydrogen evolution reaction, associated with a decrease in the total reaction volume”. These data verified the known thermodynamic relation among
are larger than the analogous values of ΔH*, indicating that the corrosion process must involve a gaseous reaction, simply the hydrogen evolution reaction, associated with a decrease in the total reaction volume”. These data verified the known thermodynamic relation among 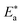 and ΔH*39 which is as follows eqn (6):
and ΔH*39 which is as follows eqn (6):| | 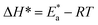 | (6) |
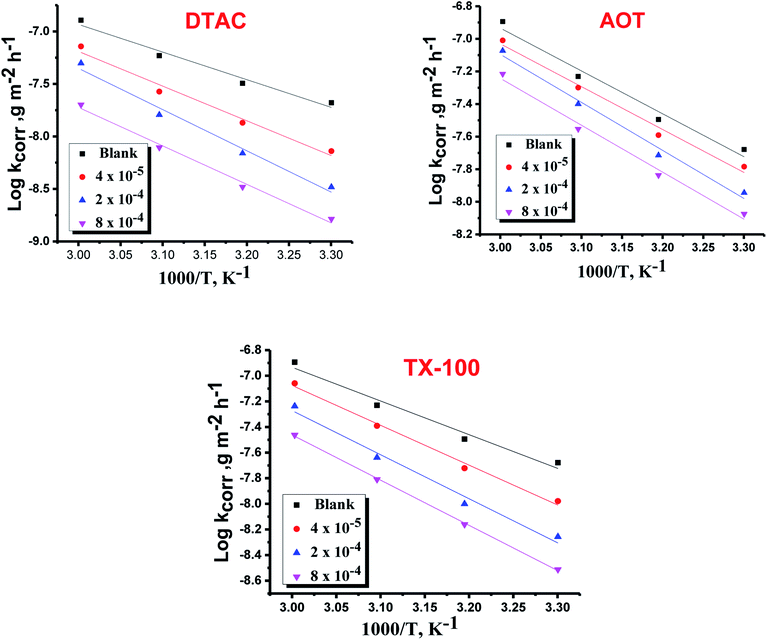 |
| | Fig. 3 Arrhenius bends of variation of logkcorr.vs. 1/T for the dissolution of CS in half molar HCl in the presence and absence of different doses of DTAC, AOT and TX-100. | |
Table 2 Kinetic parameters for CS in 0.5 M HCl in the absence and presence of different doses of surfactants
| Comp. |
Conc., M |
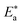
kJ mol−1 |
ΔH*, kJ mol−1 |
−ΔS*, J mol−1 K−1 |
| Blank |
0.5 |
50.4 ± 0.2309 |
47.7 ± 0.1453 |
236.3 ± 0.1453 |
| AOT |
4 × 10−5 |
50.4 ± 0.2028 |
49.3 ± 0.2333 |
233.3 ± 0.2729 |
| 2 × 10−4 |
56.4 ± 0.20278 |
53.8 ± 0.2404 |
221.2 ± 0.1764 |
| 8 × 10−4 |
56.8 ± 0.1732 |
54.2 ± 0.2028 |
218.4 ± 0.1528 |
| TA-100 |
4 × 10−5 |
59.7 ± 0.2333 |
57.1 ± 0.2333 |
199.8 ± 0.1732 |
| 2 × 10−4 |
71.2 ± 0.2603 |
68.5 ± 0.2309 |
178.2 ± 0.1764 |
| 8 × 10−4 |
74.8 ± 0.2646 |
72.2 ± 0.2603 |
172.7 ± 0.1856 |
| DTAC |
4 × 10−5 |
63.3 ± 0.1528 |
60.7 ± 0.2333 |
202.4 ± 0.1764 |
| 2 × 10−4 |
75.2 ± 0.2028 |
72.5 ± 0.1764 |
169.8 ± 0.1528 |
| 8 × 10−4 |
78.2 ± 0.2048 |
76.2 ± 0.2404 |
159.5 ± 0.1453 |
 |
| | Fig. 4 Variation of logkcorr./T vs. 1/T for the dissolution of CS in 0.5 M HCl in the presence and absence of various doses of DTAC, AOT and TX-100. | |
The entropy of activation (ΔS*) is negative in each in the presence and absence of components, implying that the activated complex represented the rate determining step with respect to the association in place of the dissociation step. It means that a lower disorder took place whilst intending from reactants to the activated complex.40 In addition, the much less negative values of ΔS* in the presence of additives created a near-equilibrium corrosion system state.41
3.2 Electrochemical measurements
3.2.1 PDP measurements.
All electrochemical measurements were carried out using Volta Master 4, which calculates and displays Ecorr. (mV), icorr. (mA cm−2), βa (mV), βc (mV) and the corrosion rate (CR) in mm per year. This CR was calculated using the following relation eqn (7):42| | 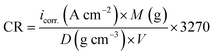 | (7) |
where “icorr. is the corrosion current density, D is the density of CS, M is the atomic mass of Fe and V is the valence entered in the Tafel dialogue box. With 3270 = 10 × [1 year (in seconds)/96497.8] and 96497.8 = 1 Faraday in Coulombs”. The % IE was calculated as follows eqn (8):| | 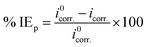 | (8) |
where i0corr. and icorr. are the corrosion current densities of uninhibited and inhibited solutions, respectively.
The PDP curves for CS in 0.5 M HCl with and without “DTAC, TX-100 and AOT are shown in Fig. 5. It can be observed from these Figures that the presence of surfactants in 0.5 M HCl solution shifts the corrosion potential (Ecorr.) to more positive values, while the polarization curves were shifted to lower current regions. These phenomena certify the inhibitory effect of these surfactants by increasing their concentrations in HCl and both cathodic and anodic reactions were inhibited by increasing the surfactant concentration. Moreover, the cathodic curves give approximately parallel lines, suggesting that the hydrogen discharge reaction slows down, its activation being controlled43 by surfactant addition in a HCl medium. The selected surfactants act as mixed-type inhibitors i.e., promoting retardation of both anodic dissolution of CS and cathodic hydrogen discharge reactions. This was confirmed by the slow displacement of Ecorr. (less than 85 mV). By inspecting the data in Table 3, it can be observed that: (a) with the increase in surfactant concentration, icorr. values decrease gradually, (b) the anodic and cathodic Tafel slopes (βc & βa)) are slightly changed after surfactant addition, and (c) this indicates that surfactants influence both anodic and cathodic processes;44 the IE increases reaching a maximum value of 86.46% at 8 × 10−4 M. The irregular trends of βa and βc values indicate the participation of more than one type of species adsorbed onto the metal surface”. The maximum inhibiting impact is achieved with the surfactant DTAC, and % IE values decrease as follows (Table 3): DTAC > TX-100 > AOT.
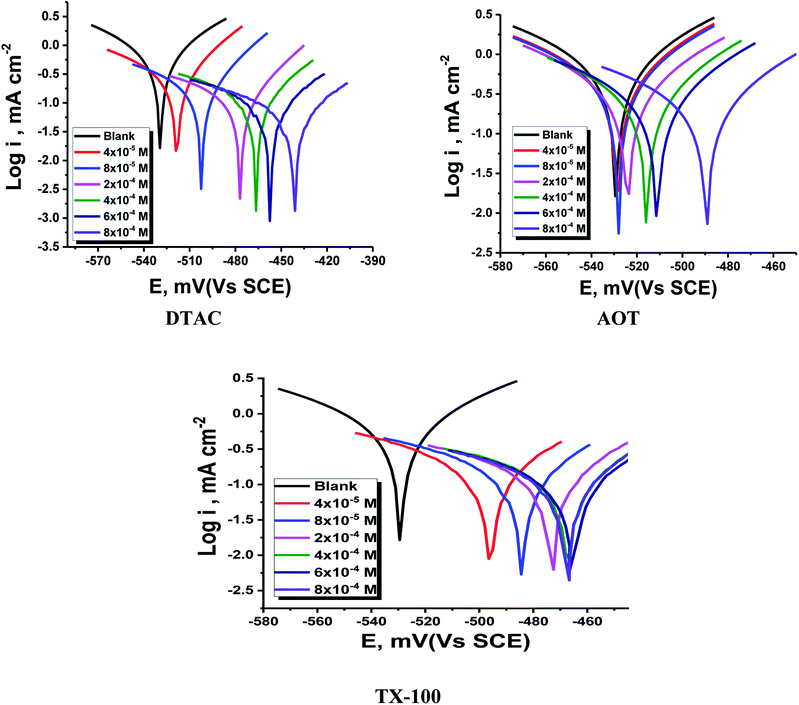 |
| | Fig. 5 PDP for CS dissolution in half-molar HCl existence and absence of altered doses of DTAC, TX-100 and AOT at 30 °C. | |
Table 3 PDP data of CS in half molar HCl and in the existence of different doses of surfactants at 30 °C
| Comp. |
Conc., M |
−Ecorr., mV vs. SCE |
i
corr., μA cm−2 |
−βc, mV dec−1 |
β
a, mV dec−1 |
θ
|
% IE |
CR, μm per year |
| Blank |
0.5 |
529.2 ± 0.1455 |
515.6 ± 0.1155 |
69.1 ± 0.2309 |
54.2 ± 0.1175 |
— |
— |
5976 |
| AOT |
4.0 × 10−5 |
528.72 ± 0.2333 |
405.4 ± 0.1732 |
72.4 ± 0.1453 |
51.3 ± 0.2028 |
0.2137 |
21.37 |
4641 |
| 8.0 × 10−5 |
528.2 ± 0.1453 |
387.8 ± 0.2028 |
72.7 ± 0.1653 |
50.9 ± 0.3528 |
0.2479 |
24.79 |
4495 |
| 2.0 × 10−4 |
516.6 ± 0.17634 |
283.9 ± 0.2603 |
68.1 ± 0.1553 |
52.6 ± 0.2028 |
0.4485 |
44.85 |
2427 |
| 4.0 × 10−4 |
515.7 ± 0.1453 |
201.7 ± 0.1764 |
64.5 ± 0.1732 |
46.3 ± 0.1453 |
0.6088 |
60.88 |
2338 |
| 6.0 × 10−4 |
511.1 ± 0.1732 |
190.9 ± 0.2028 |
67.9 ± 0.2309 |
48.6 ± 0.2906 |
06298 |
62.98 |
2212 |
| 8.0 × 10−4 |
489.5 ± 0.1732 |
181.5 ± 0.1732 |
67.9 ± 0.2028 |
50.3 ± 0.1732 |
0.6480 |
64.80 |
2103 |
| TX-100 |
4.0 × 10−5 |
507.1 ± 0.1453 |
224.7 ± 0.2028 |
69.0 ± 0.2028 |
49.6 ± 0.2028 |
0.5642 |
56.42 |
2604 |
| 8.0 × 10−5 |
496.7 ± 0.2333 |
179.4 ± 0.2309 |
71.3 ± 0.1732 |
51.9 ± 0.1732 |
0.6520 |
65.20 |
2051 |
| 2.0 × 10−4 |
487.1 ± 0.1453 |
160.5 ± 0.1732 |
70.6 ± 0.2309 |
53.0 ± 0.2082 |
0.6887 |
68.87 |
1860 |
| 4.0 × 10−4 |
484.5 ± 0.1000 |
140.0 ± 0.1453 |
72.1 ± 0.1453 |
49.5 ± 0.1732 |
0.7285 |
72.85 |
1622 |
| 6.0 × 10−4 |
483.7 ± 0.1202 |
114.9 ± 0.1523 |
79.3 ± 0.2603 |
52.5 ± 0.2028 |
0.7772 |
77.72 |
1332 |
| 8.0 × 10−4 |
479.3 ± 0.1732 |
101.7 ± 0.2603 |
81.5 ± 0.2309 |
49.6 ± 0.1764 |
0.8028 |
80.28 |
1179 |
| DTAC |
4.0 × 10−5 |
518.4 ± 0.1732 |
213.3 ± 0.1453 |
74.6 ± 0.2028 |
41.7 ± 0.2028 |
0.5863 |
58.63 |
2472 |
| 8.0 × 10−5 |
502.3 ± 0.2309 |
130.9 ± 0.2333 |
77.5 ± 0.1202 |
38.6 ± 0.2128 |
0.7461 |
74.61 |
1517 |
| 2.0 × 10−4 |
477.4 ± 0.2028 |
87.2 ± 0.1202 |
82.0 ± 0.1732 |
39.0 ± 0.2231 |
0.8309 |
83.09 |
1010 |
| 4.0 × 10−4 |
471.5 ± 0.2028 |
85.1 ± 0.2309 |
78.5 ± 0.1752 |
45.9 ± 0.2124 |
0.8349 |
83.49 |
993 |
| 6.0 × 10−4 |
452.2 ± 0.2028 |
82.3 ± 0.2028 |
89.2 ± 0.1522 |
45.1 ± 0.2227 |
0.8404 |
84.04 |
954 |
| 8.0 × 10-4 |
445.5 ± 0.1732 |
69.8 ± 0.2028 |
80.4 ± 0.1202 |
45.4 ± 0.2325 |
0.8646 |
86.46 |
809 |
3.2.2 EIS measurements.
The EIS provides protective layer stability, mechanistic and kinetic information for an electrochemical system under investigation. “The Nyquist impedance plots obtained for the CS electrode at respective corrosion potentials after 15 min immersion in 0.5 M HCl with and without altered concentrations of surfactants are shown in Fig. 6. These diagrams exhibit one capacitive loop appearing as a semicircle in Nyquist plots, being more pronounced with the increase in surfactant concentration. Bode plots given in Fig. 7 show that a single maximum of the phase angle corresponds to each capacitive loop in Nyquist plots. From Bode graphs plotted as phase vs. logF, it may observed that the phase angle does not exceed 90° (like for pure capacitive impedance).45 Consequently, logZ vs. log frequency Bode plots (Fig. 5) show two horizontal plateaus at the highest frequencies, which is attributed to logRs (solution resistance), at the lowest frequencies that represent log(Rs + Rct). Furthermore, the Nyquist plots do not yield perfect semicircles as expected from the theory of EIS, the impedance loops measured are depressed semicircles with their centers below the real axis, where the kind of phenomenon is known as the “dispersing effect” as a result of frequency dispersion46 and mass transport resistance47 as well as electrode surface heterogeneity, resulting from surface roughness, impurities, dislocations, grain boundaries, adsorption of surfactants, formation of porous layers,48–51etc., so one constant phase element (CPE) is substituted for the capacitive element, to explain the depression of the capacitance semi-circle, to give a more accurate fit. Impedance data are analyzed using the circuit in Fig. 8, in which Rs represents the electrolyte resistance, Rct represents the charge-transfer resistance of the constant phase element (CPE). According to Hsu and Mansfeld,52 the correction of capacity to its real data is as follows eqn (9):where “Yo is the CPE coefficient, ωmax is the frequency at which the imaginary part of impedance (−Zi) has a maximum53 and n is the CPE exponent (phase shift). The data obtained from fitted spectra are listed in Table 4”. The value of (θ) was calculated from the EIS data using the following relation eqn (10):| | 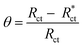 | (10) |
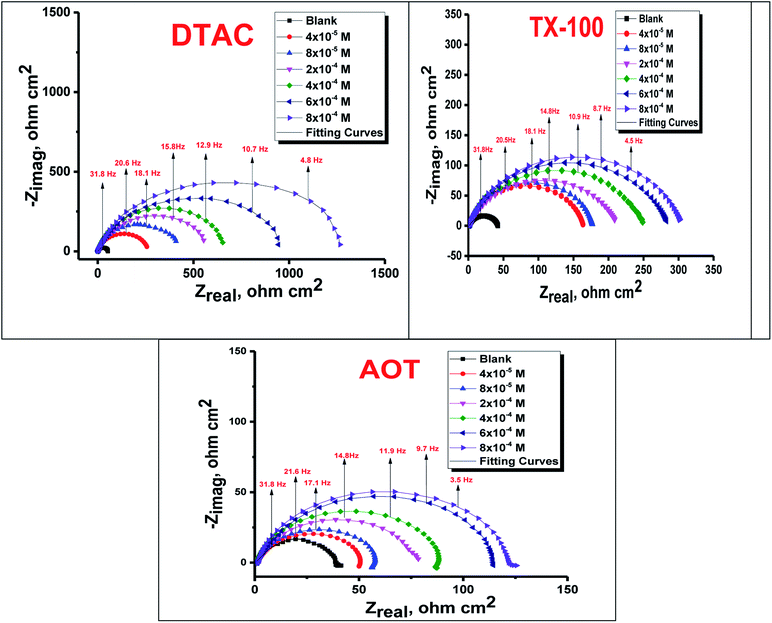 |
| | Fig. 6 Nyquist bends for CS in half molar HCl in the absence and presence of DTAC, TX-100 and AOT surfactants at 30 °C. | |
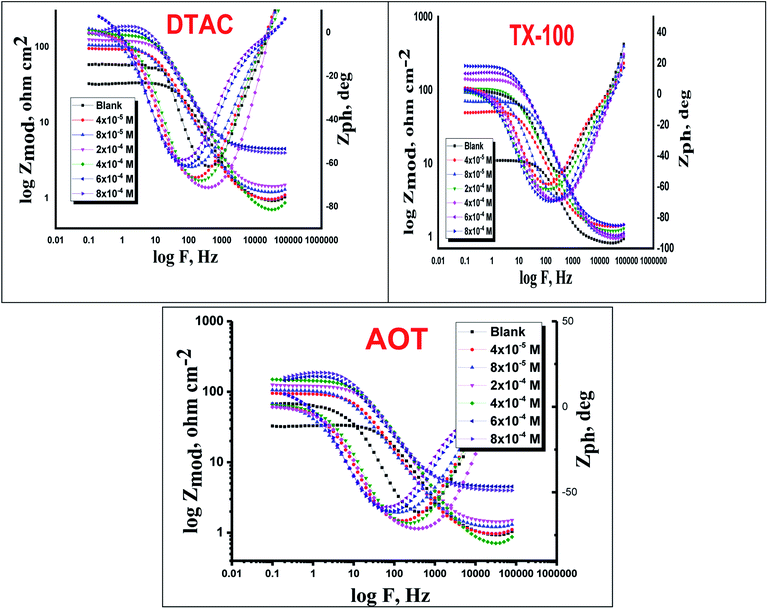 |
| | Fig. 7 Bode bends for CS in half molar HCl in the absence and presence of DTAC, TX-100 and AOT at 30 °C. | |
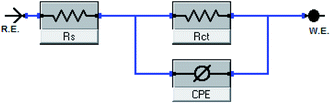 |
| | Fig. 8 Equivalent circuit model utilized to fit EIS data. | |
Table 4 EIS data of CS in half molar HCl in the absence and presence of different doses of surfactants at 30 °C
| Comp. |
Conc., M |
R
S, Ω cm2 |
Y
o, μΩ−1 sn cm−2 |
n
|
R
ct, Ω cm2 |
C
dl, μF cm−2 |
θ
|
% IE |
| Blank |
0.5 |
1.946 ± 0.0173 |
542 |
0.865 |
32.1 ± 0.1453 |
437.0 ± 0.2333 |
— |
— |
| AOT |
4.0 × 10−5 |
2.165 ± 0.0145 |
381 |
0.867 |
47.09 ± 0.1764 |
236.5 ± 0.1453 |
0.318 |
31.8 |
| 8.0 × 10−5 |
2.184 ± 0.0233 |
331 |
0.872 |
56.43 ± 0.2309 |
202.6 ± 0.1732 |
0.432 |
43.2 |
| 2.0 × 10−4 |
1.814 ± 0.0145 |
259 |
0.876 |
74.11 ± 0.1732 |
183.1 ± 0.1732 |
0.567 |
56.7 |
| 4.0 × 10−4 |
2.344 ± 0.0230 |
165 |
0.890 |
84.91 ± 0.2028 |
120.6 ± 0.1562 |
0.622 |
62.2 |
| 6.0 × 10−4 |
3.284 ± 0.0155 |
137 |
0.893 |
110 ± 0.2128 |
99.2 ± 0.1322 |
0.708 |
70.8 |
| 8.0 × 10−4 |
3.103 ± 0.0239 |
129 |
0.895 |
119.9 ± 0.1732 |
93.3 ± 0.1202 |
0.732 |
73.2 |
| TX-100 |
4.0 × 10−5 |
1.102 ± 0.0153 |
133 |
0.856 |
175.0 ± 0.1453 |
93.1 ± 0.1732 |
0.817 |
81.7 |
| 8.0 × 10−5 |
1.757 ± 0.0203 |
78 |
0.895 |
190.2 ± 0.1623 |
58.9 ± 0.1202 |
0.831 |
83.1 |
| 2.0 × 10−4 |
1.835 ± 0.0155 |
73 |
0.897 |
217.9 ± 0.1403 |
55.52 ± 0.2309 |
0.853 |
85.3 |
| 4.0 × 10−4 |
2.433 ± 0.0145 |
63 |
0.898 |
268.2 ± 0.2028 |
42.5 ± 0.1553 |
0.88 |
88 |
| 6.0 × 10−4 |
2.527 ± 0.0172 |
59 |
0.929 |
303.2 ± 0.2309 |
38.9 ± 0.1651 |
0.894 |
89.4 |
| 8.0 × 10−4 |
2.706 ± 0.0161 |
56 |
0.946 |
316.8 ± 0.1523 |
38.7 ± 0.2028 |
0.899 |
89.9 |
| DTAC |
4.0 × 10−5 |
1.711 ± 0.0155 |
113 |
0.834 |
187.5 ± 0.1851 |
79.4 ± 0.1553 |
0.0829 |
82.9 |
| 8.0 × 10−5 |
1.835 ± 0.0203 |
73 |
0.839 |
382.0 ± 0.1233 |
48.9 ± 0.1732 |
0.916 |
91.6 |
| 2.0 × 10−4 |
1.894 ± 0.0153 |
57 |
0.840 |
535.3 ± 0.1553 |
40.2 ± 0.2309 |
0.94 |
94 |
| 4.0 × 10−4 |
1.926 ± 0.0239 |
55 |
0.844 |
637.0 ± 0.1163 |
37.2 ± 0.2028 |
0.95 |
95 |
| 6.0 × 10−4 |
2.426 ± 0.0155 |
53 |
0.857 |
867.5 ± 0.2028 |
36.7 ± 0.1453 |
0.963 |
96.3 |
| 8.0 × 10−4 |
2.673 ± 0.0153 |
52 |
0.869 |
1135 ± 0.2309 |
36.6 ± 0.1553 |
0.972 |
97.2 |
The % IE was calculated as follows eqn (11):
| | 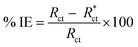 | (11) |
where
Rct and
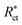
are the resistance in the presence and absence of surfactants, respectively.
The data given in Table 4 demonstrate that the Rs data are slightly associated with the Rct values. “By increasing the surfactant concentrations, the Rct values increase and the calculated Cdl values decrease, which causes an increase in θ and IE. The high Rct values are generally associated with a slower corroding system.54 The decrease in Cdl suggests that the surfactant molecule function by adsorption at the metal/solution interface and can result in a decrease in the local dielectric constant and/or an increase in the thickness of the electrical double layer.55 It was also observed from the Table that the (n) value varies directly with the surfactant concentration, while the reverse is the case with Yo, the (n) value being a measure of the roughness of the surface of the working electrode resulting from the adsorption of the surfactant molecules”.56
3.2.3 EFM measurements.
EFM is an electrochemical technique in which two sinusoidal potential signals are summed and applied to a corrosion sample via a Potentiostat. “The great strength of the EFM is the causality factor which serves as an internal check on the validity of the EFM measurement.57 With the causality factors, the experimental EFM data can be verified. EFM is a nondestructive corrosion measurement like EIS; it is a small signal ac technique. Unlike EIS, however, two sine waves (at different frequencies) are applied to the cell simultaneously. The results of EFM experiments are a spectrum of current response as a function of frequency. The spectrum is called the intermodulation spectrum. The spectra contain current responses assigned for harmonica and intermodulation current peaks. The larger peaks were used to calculate the corrosion current density (icorr.), the Tafel slopes (βc and βa) and the causality factors (CF-2 and CF-3). Intermodulation spectra obtained from EFM measurements are presented in Fig. 9 for 0.5 M HCl in the absence and presence of 8 × 10−4 M of AOT, TX-100 and DTAC, respectively. Similar curves were obtained for other concentrations of surfactants (not shown). As can be seen from Table 5, the corrosion current densities decrease by increasing the concentrations of the studied surfactants. The % IEEFM calculated from eqn (8) increase by increasing the studied surfactant concentrations. The causality factors in Table 5 are very close to theoretical values, which according to the EFM theory58 should guarantee the validity of Tafel slopes and corrosion current densities. Fig. 9 shows the % IE recorded for DTAC, TX-100 and AOT at a concentration of 8 × 10−4 M using the four different techniques, namely, WL, PP, EIS and EFM”. The calculated % IE found is in excellent agreement.
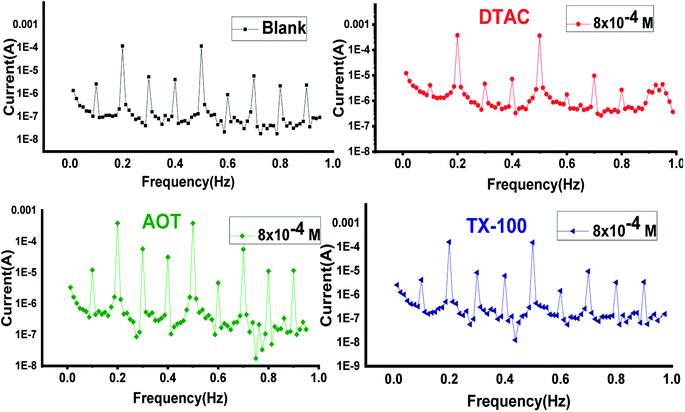 |
| | Fig. 9 EFM bends for CS in half molar HCl in the absence and presence of 8 × 10−4 M dose of DTAC, AOT and TX-100 at 30 °C. | |
Table 5 EFM parameters obtained for CS in the presence and absence of different doses of surfactants in 0.5 M HCl at 30 °C
| Comp. |
Conc., M |
i
corr., μA cm−2 |
β
c, mV dec−1 |
β
a, mV dec−1 |
CF-2 |
CF-3 |
θ
|
% IE |
CR , μm per year |
| Blank |
0.00 |
488.9 ± 0.2028 |
126 ± 0.1453 |
106 ± 0.2906 |
1.86 |
2.95 |
— |
— |
5763 |
| AOT |
4 × 10−5 |
394.3 ± 0.1553 |
96 ± 0.1732 |
88 ± 0.2027 |
1.67 |
2.98 |
0.193 |
19.3 |
4575 |
| 8 × 10−5 |
321.9 ± 0.1653 |
107 ± 0.2309 |
88 ± 0.1763 |
1.84 |
2.92 |
0.342 |
34.2 |
3735 |
| 2 × 10−4 |
272.8 ± 0.1453 |
102 ± 0.2309 |
87 ± 0.2028 |
1.80 |
3.21 |
0.4420 |
44.2 |
3165 |
| 4 × 10−4 |
238.6 ± 0.2431 |
92 ± 0.1732 |
86 ± 0.2082 |
1.33 |
2.82 |
0.512 |
51.2 |
2768 |
| 6 × 10−4 |
208.7 ± 0.2055 |
97 ± 0.2102 |
88 ± 0.2028 |
1.74 |
2.73 |
0.573 |
57.3 |
2422 |
| 8 × 10−4 |
117.4 ± 0.1452 |
95 ± 0.1732 |
83 ± 0.2123 |
1.87 |
2.87 |
0.759 |
75.9 |
1362 |
| TX-100 |
4 × 10−5 |
88.3 ± 0.1742 |
98 ± 0.2101 |
93 ± 0.2234 |
1.44 |
2.77 |
0.819 |
81.9 |
1025 |
| 8 × 10−5 |
87.1 ± 0.2102 |
95 ± 0.2423 |
89 ± 0.2131 |
1.46 |
3.01 |
0.822 |
82.2 |
1011 |
| 2 × 10−4 |
83.1 ± 0.2209 |
97 ± 0.2512 |
91 ± 0.2028 |
1.56 |
2.77 |
0.830 |
83 |
965 |
| 4 × 10−4 |
70.0 ± 0.2010 |
107 ± 0.1202 |
93 ± 0.1732 |
1.72 |
2.64 |
0.857 |
85.7 |
812 |
| 6 × 10−4 |
56.2 ± 0.1453 |
98 ± 0.2333 |
85 ± 0.2309 |
1.86 |
3.32 |
0.885 |
88.5 |
652 |
| 8 × 10−4 |
50.88 ± 0.1353 |
85 ± 0.1453 |
73 ± 0.1453 |
1.99 |
5.684 |
0.896 |
89.6 |
590 |
| DTAC |
4 × 10−5 |
79.1 ± 0.1725 |
114 ± 0.2027 |
105 ± 0.2028 |
1.67 |
3.01 |
0.838 |
83.8 |
691 |
| 8 × 10−5 |
42.4 ± 0.2333 |
114 ± 0.2022 |
106 ± 0.1453 |
1.55 |
3.13 |
0.913 |
91.3 |
493 |
| 2 × 10−4 |
41.3 ± 0.2121 |
132 ± 0.1027 |
113 ± 0.1732 |
1.91 |
2.69 |
0.916 |
91.6 |
476 |
| 4 × 10−4 |
30.7 ± 0.2028 |
130 ± 0.1121 |
115 ± 0.1453 |
2.01 |
2.77 |
0.937 |
93.7 |
357 |
| 6 × 10−4 |
28.6 ± 0.2228 |
118 ± 0.1327 |
113 ± 0.2028 |
1.63 |
2.67 |
0.941 |
94.1 |
333 |
| 8 × 10−4 |
27.8 ± 0.2108 |
149 ± 0.1125 |
134 ± 0.2027 |
1.91 |
2.62 |
0.943 |
94.3 |
323 |
The order of the % IE found for the Rct data is DTAC > TX-100 > AOT (Fig. 10). “The % IE, calculated from EIS results, show the same trend as those obtained from PP measurements. The difference in % IE from two methods may be attributed to the different surface status of the electrode in two measurements. EIS was performed at the rest potential, while in polarization measurements, the electrode potential was polarized to high overpotential and non-uniform current distributions, resulting from cell geometry, solution conductivity, counter and reference electrode placement, etc., which will lead to the difference between the electrode area undergoing polarization and the total area”.59
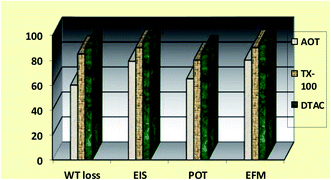 |
| | Fig. 10 Comparison of “% IE (recorded using EFM, EIS, PP and WL measurements) obtained for the three surfactants during CS corrosion in 0.5 M HCl solutions containing 8 × 10−4 M of these three surfactants at 30 °C”, respectively. | |
3.3 Adsorption of surfactants
Adsorption of surfactants on solid surfaces can regulate their hydrophobicity, surface charge, and different key properties such as corrosion protection that govern the interfacial process. “In general, adsorption is governed by several forces such as covalent bonding, electrostatic attraction, hydrogen bonding or non-polar interactions between the adsorbed species, lateral associative interaction, solvation, and desolvation.60 The total adsorption is usually the cumulative result of some or all the above-mentioned forces.61 The standard free energy of adsorption can be written as follows” eqn (12):62| | 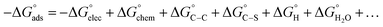 | (12) |
where “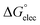 is the electrostatic interaction term,
is the electrostatic interaction term, 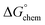 the chemical term due to covalent bonding,
the chemical term due to covalent bonding, 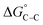 the free energy gained upon association of methyl groups in the hydrocarbon chain,
the free energy gained upon association of methyl groups in the hydrocarbon chain, 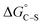 the free energy due to interactions between the hydrocarbon chains and hydrophobic sites on the solid,
the free energy due to interactions between the hydrocarbon chains and hydrophobic sites on the solid, 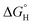 the hydrogen bonding term and
the hydrogen bonding term and 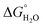 the term owing to dissolution or solvation of the adsorbate species, or any species displaced from the interface due to adsorption”. In the following, the major forces involved in surfactant adsorption are discussed:
the term owing to dissolution or solvation of the adsorbate species, or any species displaced from the interface due to adsorption”. In the following, the major forces involved in surfactant adsorption are discussed:
3.3.1 Driving forces for surfactant adsorption.
For each surfactant–solid system, several of the above-mentioned terms can be operative depending on the solid and the surfactant type, surfactant concentration, electrolyte, pH, temperature, etc.:
(i) Electrostatic interactions 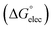 : in systems where the ionic surfactants and the solid particles are charged, electrostatic interactions play a governing role in the adsorption process.
: in systems where the ionic surfactants and the solid particles are charged, electrostatic interactions play a governing role in the adsorption process.
(ii) Chemical interactions 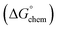 : chemical interaction is another important driving force for adsorption of surfactants on the solid particles. Compared to other driving forces, this interaction is specific to certain systems where covalent bonding can occur between the surfactant and the solid.
: chemical interaction is another important driving force for adsorption of surfactants on the solid particles. Compared to other driving forces, this interaction is specific to certain systems where covalent bonding can occur between the surfactant and the solid.
(iii) Hydrophobic lateral interactions 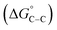 : “At a concentration above a threshold value, analogous to the aggregation in the bulk, surfactant molecules tend to form two dimensional aggregates at the solid/liquid interface, causing an abrupt increase in the adsorption density. These aggregates have been called “hemi-micelles”63 or, in general, “colloids” for surface colloids.64 The driving force for adsorption
: “At a concentration above a threshold value, analogous to the aggregation in the bulk, surfactant molecules tend to form two dimensional aggregates at the solid/liquid interface, causing an abrupt increase in the adsorption density. These aggregates have been called “hemi-micelles”63 or, in general, “colloids” for surface colloids.64 The driving force for adsorption 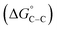 results from the free energy of transferring the hydrocarbon chains from the aqueous environment into the hydrophobic interior of the aggregates.
results from the free energy of transferring the hydrocarbon chains from the aqueous environment into the hydrophobic interior of the aggregates. 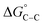 can be represented as a linear function of the energy gained per –CH2 group”.65
can be represented as a linear function of the energy gained per –CH2 group”.65
(iv) Hydrophobic interaction between the hydrocarbon chains and hydrophobic sites on the solid 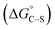 : “the hydrophobic interaction
: “the hydrophobic interaction 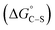 between the alkyl chain of a surfactant and the hydrophobic sites on the solid becomes a significant factor for surfactant adsorption on fully or partially hydrophobic surfaces. In this case, the surfactant molecules attach to the hydrophobic sites with the hydrocarbon chains aligned parallel to the surface at low concentrations and normal to the surface at higher concentrations. Such an adsorption process often results in a two-step isotherm”.
between the alkyl chain of a surfactant and the hydrophobic sites on the solid becomes a significant factor for surfactant adsorption on fully or partially hydrophobic surfaces. In this case, the surfactant molecules attach to the hydrophobic sites with the hydrocarbon chains aligned parallel to the surface at low concentrations and normal to the surface at higher concentrations. Such an adsorption process often results in a two-step isotherm”.
(v) Hydrogen bonding 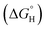 : “Hydrogen bonding between surfactant species and the solid surface species could occur in systems containing hydroxyl, phenolic, carboxylic and amine groups on the surfactant. For instance, adsorption of a nonionic surfactant such as ethoxylated alcohol and sugar-based alkyl glucoside on oxides has been proposed to involve hydrogen bonding.66,67 It should be noted that for adsorption due to hydrogen bonding to take place, the bond formed between the surfactant functional groups and mineral surfaces should be stronger than that formed between the mineral and interfacial water molecules”.
: “Hydrogen bonding between surfactant species and the solid surface species could occur in systems containing hydroxyl, phenolic, carboxylic and amine groups on the surfactant. For instance, adsorption of a nonionic surfactant such as ethoxylated alcohol and sugar-based alkyl glucoside on oxides has been proposed to involve hydrogen bonding.66,67 It should be noted that for adsorption due to hydrogen bonding to take place, the bond formed between the surfactant functional groups and mineral surfaces should be stronger than that formed between the mineral and interfacial water molecules”.
(vi) Desolvation energy 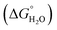 : “When a hydrated head group of the surfactant transfers from the bulk to the mineral–solution interfacial region, partial removal of water molecules from the secondary solvation shell around the surfactant head groups can occur. In contrast to other driving forces, desolvation energy due to such a process is unfavorable for the adsorption process”.
: “When a hydrated head group of the surfactant transfers from the bulk to the mineral–solution interfacial region, partial removal of water molecules from the secondary solvation shell around the surfactant head groups can occur. In contrast to other driving forces, desolvation energy due to such a process is unfavorable for the adsorption process”.
3.4 Mechanism of adsorption68
The adsorption of ionic surfactants on oppositely charged surface could be taking the following path: (i) “At low surfactant concentrations, the adsorption is due to electrostatic interaction between individual isolated charged monomeric species and the oppositely charged solid surface. (ii) Surfactant species begin to form surface aggregates, colloids (surface colloids), including hemi-micelles and ad-micelles, due to lateral interactions between hydrocarbon chains. Thus, the additional driving force resulting from the lateral association with the electrostatic interaction is still active. (iii) When the solid surface is electrically neutralized by the adsorbed surfactant ions, the electrostatic attraction is no longer operative, and adsorption takes place due to lateral attraction alone with a reduced slope. (iv) When the surfactant concentration reaches critical micelle concentration, the surfactant monomer activity becomes constant and any further increase in the concentration contributes only to the micellization in solutions and it does not change the adsorption density. The adsorption in this region is mainly through lateral hydrophobic interaction between the hydrocarbon chains. In steps (iii) and (iv), surfactant molecules adsorb with a reversed orientation (head groups facing the bulk solution), resulting in a decrease in the hydrophobicity of the particles in this region. The pH plays a very significant role in controlling the adsorption of ionic surfactants. Thus, the adsorption of anionic surfactants is higher on positively charged surfaces (pH below isoelectric point (IEP)) than on negatively charged surfaces, while the cationic surfactants adsorb more on negatively charged surfaces.69,70 The molecular structure of surfactants does influence its adsorption behavior markedly. Most nonionic surfactants contain polar groups that form hydrogen bonds with the hydroxyl groups on the solid surface. Since the hydrogen bonding is weaker than the electrostatic interaction, the adsorption of the nonionic surfactant to most solids is less than that of ionic surfactants. Nonionic surfactants exhibit adsorption like those of cationic surfactants, except for a sharp increase in step III of the adsorption mechanism because of the absence of electrostatic interactions. The adsorption of the nonionic surfactants also depends on the pH and number of the hydrophilic groups and the hydrocarbon chain length”. Several adsorption isotherms have been assessed, and the Langmuir adsorption isotherm become determined to be the excellent description of the adsorption conduct of the applied surfactants, which obey the subsequent eqn (13):| | | Cinh/θ =1/Kads + Cinh | (13) |
| | 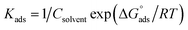 | (14) |
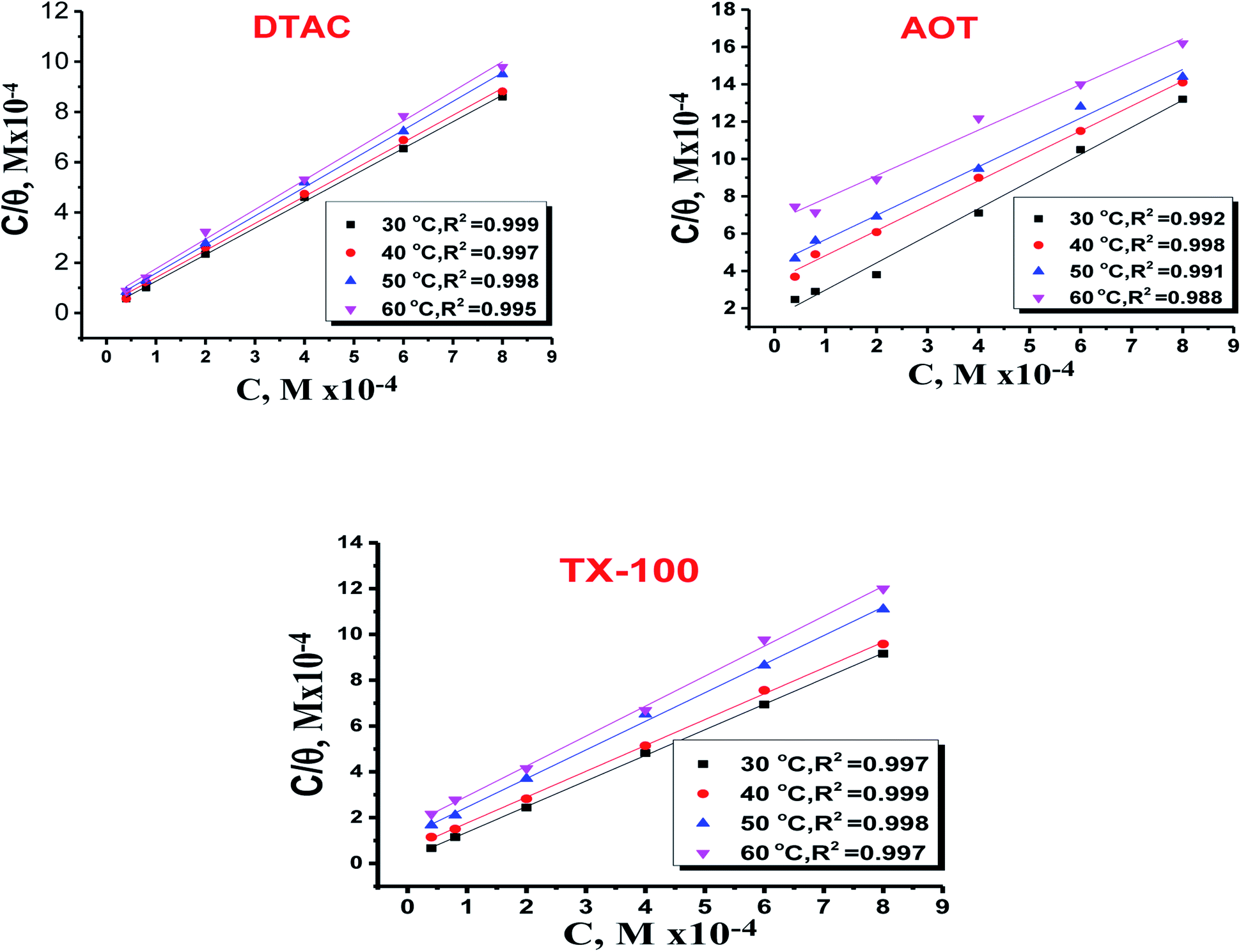
where “Cinh is the surfactant concentration, θ is the fraction of the surface coverage, Kads is the modified adsorption equilibrium constant, which can be related to the free energy of adsorption 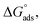 and Csolvent is the molar concentration of solvent, which in the case of water is 55.5 mol l−1. The
and Csolvent is the molar concentration of solvent, which in the case of water is 55.5 mol l−1. The 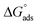 value was calculated according to eqn (14). Fig. 11 shows the dependence of the fraction of the surface coverage (C/θ) as a function of the concentration (C) of AOT, TX-100 and DTAC. The degrees of surface coverage (θ) were evaluated from EIS measurements using eqn (10) and are given in Table 6. The
value was calculated according to eqn (14). Fig. 11 shows the dependence of the fraction of the surface coverage (C/θ) as a function of the concentration (C) of AOT, TX-100 and DTAC. The degrees of surface coverage (θ) were evaluated from EIS measurements using eqn (10) and are given in Table 6. The 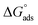 values are negative, suggesting the spontaneity of the adsorption process. The regression coefficient is R2 = 0.999. It is well known that values of
values are negative, suggesting the spontaneity of the adsorption process. The regression coefficient is R2 = 0.999. It is well known that values of 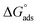 in the order of 20 kJ mol−1 or lower indicate physisorption, while those in the order of 40 kJ mol−1 or higher involve charge sharing or charge transfer from the surfactant molecules to the metal surface to form a coordinate type of bond.71,72 The calculated values of
in the order of 20 kJ mol−1 or lower indicate physisorption, while those in the order of 40 kJ mol−1 or higher involve charge sharing or charge transfer from the surfactant molecules to the metal surface to form a coordinate type of bond.71,72 The calculated values of 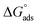 for DTAC are around −38.1 to 37.3 kJ mol−1, for TX-100 approximately −36.8 to 35.2 kJ mol−1 and for AOT-100 approximately −32.2 to 31.4 kJ mol−1, so physical and chemical adsorption (mixed type) may proposed.
for DTAC are around −38.1 to 37.3 kJ mol−1, for TX-100 approximately −36.8 to 35.2 kJ mol−1 and for AOT-100 approximately −32.2 to 31.4 kJ mol−1, so physical and chemical adsorption (mixed type) may proposed.
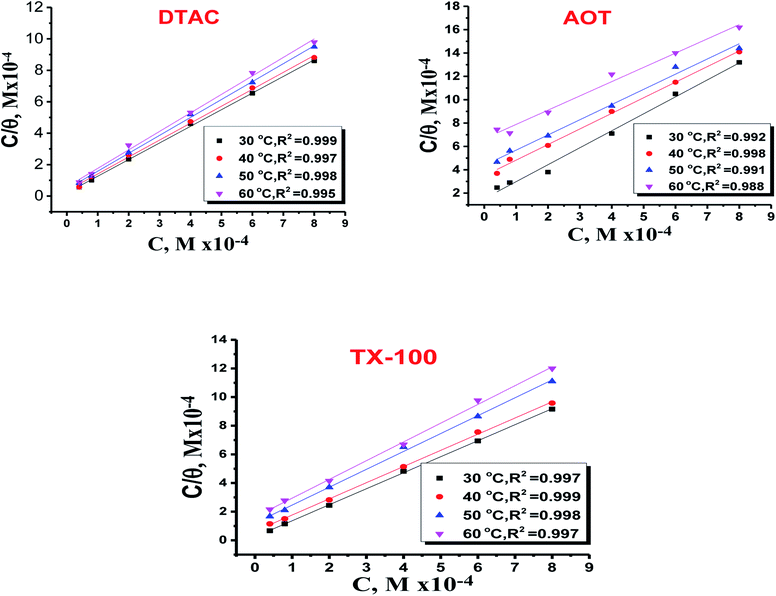 |
| | Fig. 11 Langmuir bends for the adsorption of DTAC, AOT and TX-100 on CS in half molar HCl at different temperatures. | |
Table 6 Parameters for the adsorption of surfactants in half molar HCl on the CS at different temperatures
| Surfactant |
Temp. |
K
ads ×10−4 M−1 |
Slope |
R
2
|
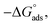
kJ mol−1 |

kJ mol−1 |
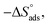
J mol−1 K−1 |
| AOT |
303 |
0.65 |
1.3 |
0.991 |
32.2 ± 0.1732 |
37.77 ± 0.2082 |
38.68 ± 0.2333 |
| 313 |
0.28 |
1.3 |
0.993 |
31.1 ± 0.2028 |
| 323 |
0.23 |
1.2 |
0.998 |
31.6 ± 0.1453 |
| 333 |
0.15 |
1.2 |
0.976 |
31.4 ± 0.2028 |
| TX-100 |
303 |
4.03 |
1.1 |
0.999 |
36.8 ± 0.1028 |
52.15 ± 0.1732 |
70.82 ± 0.1453 |
| 313 |
1.57 |
1.1 |
0.999 |
35.6 ± 0.1732 |
| 323 |
0.83 |
1.2 |
0.998 |
35.0 ± 0.2028 |
| 333 |
0.60 |
1.3 |
0.998 |
35.2 ± 0.1453 |
| DTAC |
303 |
4.88 |
1.0 |
0.999 |
37.3 ± 0.1732 |
27.47 ± 0.2333 |
12.10 ± 0.2028 |
| 313 |
1.93 |
1.0 |
0.994 |
36.5 ± 0.2028 |
| 323 |
1.39 |
1.0 |
0.995 |
37.4 ± 0.1732 |
| 333 |
1.08 |
1.1 |
0.998 |
38.1 ± 0.1001 |
3.5 Surface analyses results
Surface analysis was conducted by both electron scanning microscopy (SEM) and atomic electron microscopy (AFM), as depicted in Fig. 12. “SEM examination was used to acquire a thorough knowledge of the corrosion morphology prior to and after immersion in 1 M HCl solution with and without DTAC, TX-100, and AOT at 30 °C. Fig. 12(a) shows a polished CS surface with no fractures or pits save for a few polished abrasions. Fig. 12(b) depicts the CS specimen following immersion in 1 M HCl without surfactants, revealing a badly degraded CS surface with a larger number of fractures and pits. Fig. 12(c–e) shows a CS specimen following immersion in 1 M HCl with DTAC, TX-100, and AOT, which showed a substantial improvement with a lower number of cracks and pits as compared to the CS surface without surfactants, due to the inhibitive layer development on the CS surface. The fact that the DTAC-inhibited surface is smoother than the TX-100 and AOT-inhibited surfaces indicates that DTAC is a superior inhibitor than TX-100 and AOT. Fig. 12(a–e) shows 3D micrographs of the CS surface obtained from AFM with and without surfactants. Fig. 12(a) depicts a polished CS surface with an average roughness of 39.63 nm. The CS surface is shown in Fig. 12(b) after being immersed in a 1 M HCl solution without DTAC, TX-100, or AOT; the average roughness is 233.5 nm. Fig. 12(c–e) show the CS surface after submersion in 1 M HCl solution containing DTAC, TX-100, and AOT, with average roughness values of 63.5, 72.0, and 79.0 nm, respectively”. DTAC has a lower average roughness than that of TX-100, whereas AOT has better adsorption than that of DTAC.
 |
| | Fig. 12 (a–e) SEM and AFM images for blank (free sample), sample + 1 M HCl, sample + 1 M HCl + 8 × 10−4 DTAC, sample + 1 M HCl + 8 × 10−4 TX-100, sample + 1 M HCl + 8 × 10−4 AOT. | |
3.6 Computational study and corrosion mechanism
Lower values of ionization potential “IP (−EHOMO) are likely to indicate a tendency of the molecule to donate electrons to appropriate acceptor molecules with low energy or empty electron orbital. The higher the values of electron affinity EA (−ELUMO), the stronger the electron accepting abilities of the molecules. However, the hydrophobic properties of the long hydrocarbon tail could be associated with the formation of a protective film that drastically reduces the corrosion process. Pearson introduced the quantities of electronic hardness (η) and softness (σ) in his hard–soft-acid–base principle73 (HSAB) in the early stage of the reactivity theory. The species are classified as soft (hard) if their valence electrons are easy (hard) to polarize or to remove and the relationship between hardness or softness and the chemical reactivity was given by the HSAB principle. A soft base will interact favorably with a soft acid, sharing electrons, to form bonds of covalent character. Hard acids prefer hard bases and form bonds dominated by electrostatic forces, or ionic character. The concepts of electronegativity (χ) and global hardness (η)”74 are given as follows eqn (15) and (16):| | 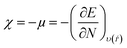 | (15) |
| | 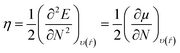 | (16) |
where “μ is the chemical potential, E is the total energy, N is the number of electrons, and ν(ѓ) is the external potential of the system. The global hardness (η), softness (σ), and chemical potential (μ) were calculated in terms of IP and EA”75 as follows eqn (17) and (18):| | 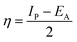 | (17) |
| | 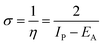 | (18) |
| | 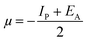 | (19) |
In Table 7, certain quantum-chemical parameters related to the molecular electronic structure are presented, “which were obtained by means of the application of POP/DNP basis set in each one of the interest systems. The results for the above-mentioned calculations in a gaseous phase as well as in a liquid phase are presented. These results show the effect of applying a calculation in the liquid phase. From these results, TX-100 exhibits the lowest value of global hardness (high softness) in liquid phases compared with the cationic part of DTAC and the anionic part of AOT. This suggested a higher tendency of covalent adsorption of TX-100 to occur than other used surfactants. It means that this one has a higher reactivity. The role of the counter ions on the adsorption of ionic surfactants is an important factor. High hardness of Cl− (counter ion effect) and cationic part of DTAC suggested higher tendency of electrostatic adsorption of DTAC to occur (cooperative effect) than other used surfactants that lead to a high % IE. The feasible adsorption of organic cations in the presence of the halide ions is due to the formation of intermediate bridge, the negative ends of the halide metal dipoles being oriented towards the solution. The dipoles of the surface compound formed are oriented with their negative ends towards the solution, whereby setting up an additional potential difference between the metal and the solution. This will shift the zero-charge potential positively. This shift will make the charge on the metal surface more negative and facilitates the adsorption of positively charged quaternary ammonium compound [C12H25N+(CH3)3] by the formation of ionic bonds. Cl− ions act as adsorption mediators for bonding the two positive partners, the metal surface and the positively charged ammonium compound. This gives rise to the formation of an adsorption composite film, in which the chloride ions (Cl−) are sandwiched between the metal and positively charged part of the surfactant.76 This film acts as a barrier facing the corrosion process, as shown in Fig. 13. Anionic part of AOT is a softer base than Cl− thus it attached with metals (softer) via covalent adsorption. The anionic part of AOT suggested a competitive effect with Cl− ions of the HCl solution. In this case (showing the anodic process of iron corrosion), Cl− ion-catalyzed iron dissolution occurs”. The anionic part of AOT can retard the corrosion process by adsorption onto a positively charged steel surface.
Table 7 Quantum-chemical descriptors for surfactants found with DFT meted
| Quantum-chemical descriptors |
| |
|
|
I
P, eV |
E
A, eV |
ΔE, eV |
η, eV |
μ, eV |
σ, eV |
X, eV |
−E
tot. |
| AOT |
Anionic part |
Gas phase |
0.55 |
2.848 |
3.403 |
1.70 |
0.588 |
1.15 |
−0.58 |
1709.2 |
| Liquid phase |
5.342 |
0.982 |
4.36 |
2.18 |
0.459 |
−3.162 |
3.162 |
1709.3 |
| Na+ counter |
Gas phase |
36.23 |
6.99 |
29.24 |
9.62 |
0.104 |
−21.61 |
21.61
|
162.19 |
| Liquid phase |
29.81 |
0.756 |
29.05 |
14.53 |
0.069 |
−15.28 |
15.28
|
162.2 |
| TX-100 |
Cationic part |
Gas phase |
5.276 |
0.838 |
4.438 |
2.219 |
0.451 |
−3.057 |
3.057 |
2160.1 |
| Liquid phase |
4.74 |
0.41 |
4.33 |
3.748 |
0.461 |
−2.58 |
2.58 |
2160.0 |
| DTAC |
Gas phase |
8.158 |
2.934 |
5.224 |
2.612 |
0.383 |
−5.546 |
5.546 |
646.3 |
| Liquid phase |
6.700 |
0.795 |
7.495 |
2.17 |
0.267 |
−2.952 |
2.952 |
646.4 |
| Cl− counter |
Gas phase |
−2.33 |
19.08 |
16.75 |
8.375 |
0.119 |
10.705 |
−5.35 |
460.23 |
| Liquid phase |
4.53 |
12.22 |
16.75 |
8.375 |
0.119 |
3.85 |
−3.85 |
460.35 |
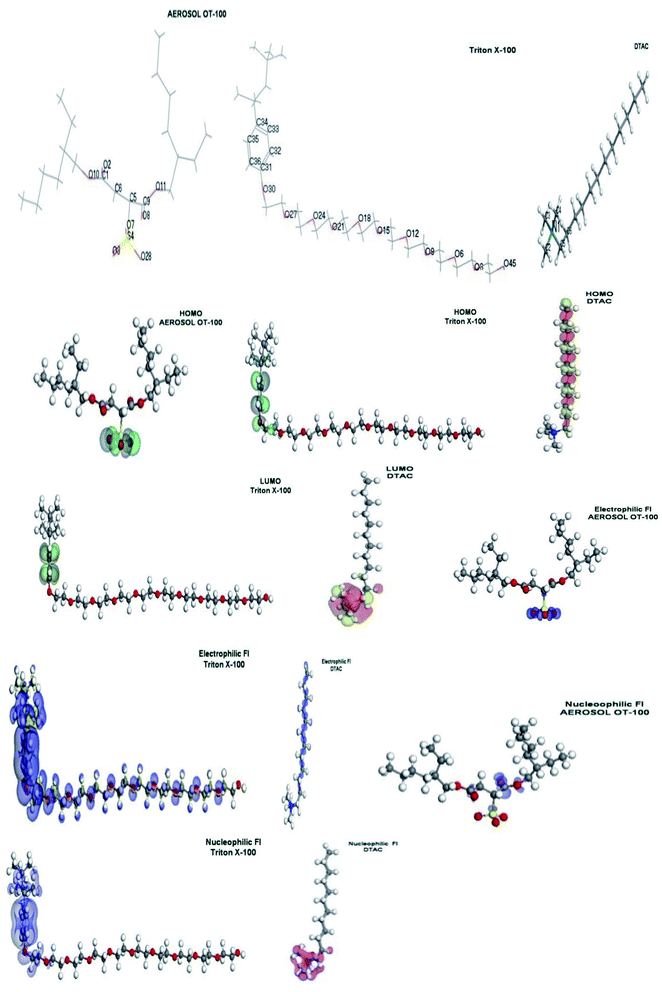 |
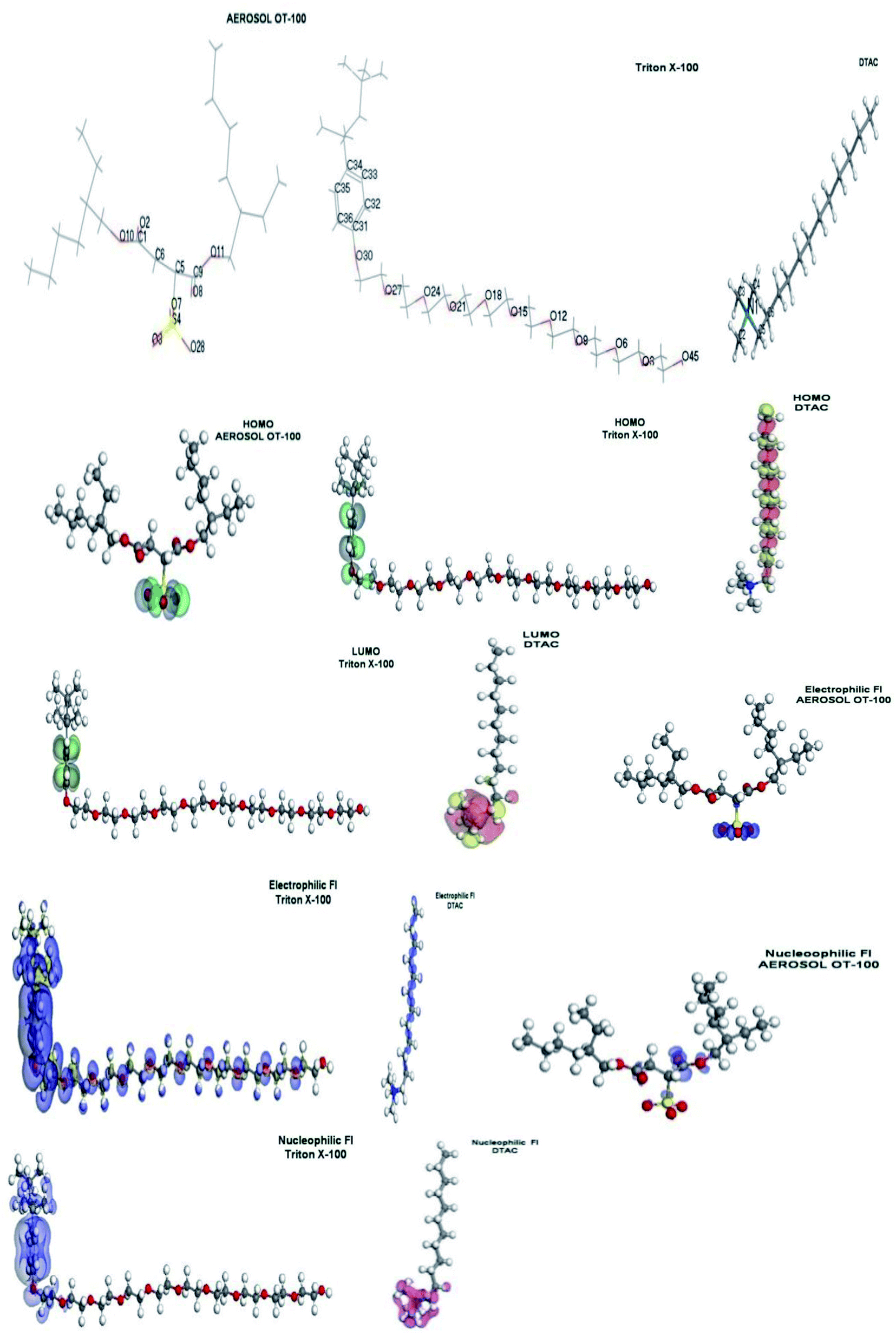
| | Fig. 13 Molecular orbital plots as well as the active sites for electrophilic and nucleophilic attack for AOT, TX-100 and DTAC, respectively. | |
The anodic process of iron corrosion eqn (20)–(22):
| | | FeClads− → Fe Cl+ + 2e | (21) |
From Fig. 13, we can observe that:
The HOMO location in the cationic part of DTAC surfactants is mostly distributed on the hydrophobic part. “The LUMO location in the cationic part of DTAC is mostly distributed on the nitrogen cation that the preferred sites for the nucleophilic attack through metallic negative centers. The position of the surfactant (lying vertically) could be the reason for the high surfactant efficiency. The HOMO location in the nonionic surfactant is mostly distributed on the benzene ring (hydrophobic part) and oxygen atom attached to it. The LUMO location in the nonionic surfactant is mostly distributed on the benzene ring (hydrophobic part). However, the molecular structure of TX-100 suggests that they are able to adsorb onto the metal surface through two lone pair of electrons of the polar hydrophilic head groups by forming hydrogen bonds. Oxygen atoms can form a protective layer (which blocs the active sites of corrosion on the metal surface). The position of the surfactant (lying flat) could be the reason for the high surfactant efficiency. The HOMO location in anionic surfactants is mostly distributed on oxygen atoms attached to the sulfur atom, indicating that the preferred sites for the electrophilic attack through metallic positive center are located on these oxygen atoms. The LUMO location in the anionic surfactant is mostly distributed on oxygen atoms of the nearest acetate group to the sulfur atom, indicating that the preferred sites for the nucleophilic attack through metallic negative centers are located on the oxygen atoms. From the above, it is mentioned that a hydrophilic metal surface attracts a large hydrophilic head group of chosen surfactants. Table 7 shows that the values of ΔE dropped in the following order: AOT+ > TX-100 > DTAC, showing that DTAC inhibits more effectively than TX-100 and AOT. From this table, because DTAC has a greater σ and lower η value, it can be inferred that this molecule has a higher potential to be adsorbed on steel surfaces. It should also be noticed that DTAC has the maximum number of electrons, indicating that this chemical has a strong theoretical capacity to suppress steel corrosion. These findings indicate that the active centers on the steel surface are extremely well adsorbent of this chemical. Among the theoretical models proposed to compute local reactivity indices are Fukui functions that make it possible to rationalize the reactivity of individual molecular orbital contributions, thus to account for the response of the whole molecular spectrum and not only of the frontier orbitals. Frontier orbital electron densities on atoms provide a useful means for the detailed characterization of donor–acceptor interactions. In the case of a donor molecule, f−(r) electrophilic electron density corresponds to reactivity with respect to electrophilic attack or when the molecule loss electrons and in the case of an acceptor molecule, f+(r) nucleophilic electron density corresponds to the reactivity with respect to nucleophilic attack. However, frontier electron densities can strictly be used only to describe the reactivity of different atoms in the same molecule. The highest FI values are presented in Table 8. The most susceptible sites for electrophilic attack are located on C10, C11, C12 and C13 atoms in case of DTAC, oxygen atoms and carbon atoms of benzene ring in case of TX-100 and O3, O7, O28, and S4 in case of AOT”. In addition, susceptible sites are experiential to be confronted by anions or nucleophilic attack, positioned on C2, C3, C4, C5 and N1 of DTAC, C32, C33, C35, and C36 in case of TX-100 and O10 and O12 in case of AOT (Fig. 11).
Table 8 The highest FI values
| “The highest Fukui indices values for the three surfactants by Hirshfeld methods in liquid phase calculated with BOP/DNP basis set” |
| AOT |
TX-100 |
DTAC |
| |
Liquid phase |
|
Liquid phase |
|
Liquid phase |
|
f
−
(r) |
f
+
(r) |
f
−
(r) |
f
+
(r) |
f
−
(r) |
f
+
(r) |
| C1 |
0.003 |
0.048 |
O3 |
0.010 |
−0.001 |
N1 |
0.000 |
0.034 |
| O2 |
0.016 |
0.054 |
O6 |
0.010 |
−0.001 |
C2 |
0.001 |
0.077 |
| O3 |
0.226 |
0.050 |
O9 |
0.011 |
−0.001 |
C3 |
0.000 |
0.054 |
| S4 |
0.105 |
0.039 |
O12 |
0.011 |
−0.001 |
C4 |
0.001 |
0.062 |
| C5 |
0.020 |
−0.026 |
O15 |
0.011 |
−0.001 |
C5 |
0.007 |
0.059 |
| C6 |
0.007 |
−0.039 |
O18 |
0.011 |
−0.001 |
C6 |
0.012 |
0.018 |
| O7 |
0.245 |
0.026 |
O21 |
0.011 |
−0.001 |
C7 |
0.020 |
0.009 |
| O8 |
0.027 |
0.019 |
O24 |
0.012 |
0.000 |
C8 |
0.032 |
0.005 |
| C9 |
0.006 |
0.054 |
O27 |
0.018 |
0.004 |
C9 |
0.041 |
0.002 |
| O10 |
0.007 |
0.179 |
O30 |
0.091 |
0.031 |
C10 |
0.052 |
0.001 |
| O11 |
0.011 |
0.162 |
C31 |
0.070 |
0.065 |
C11 |
0.054 |
0.000 |
| C12 |
0.004 |
0.024 |
C32 |
0.063 |
0.116 |
C12 |
0.057 |
0.000 |
| O28 |
0.244 |
0.038 |
C33 |
0.051 |
0.107 |
C13 |
0.051 |
0.000 |
| |
|
|
C34 |
0.077 |
0.061 |
C14 |
0.046 |
0.000 |
| |
|
|
C35 |
0.051 |
0.117 |
C15 |
0.035 |
0.000 |
| |
|
|
C36 |
0.054 |
0.111 |
C16 |
0.029 |
0.000 |
| |
|
|
O45 |
0.002 |
0.002 |
|
|
|
3.7 Monte Carlo (MC) simulation
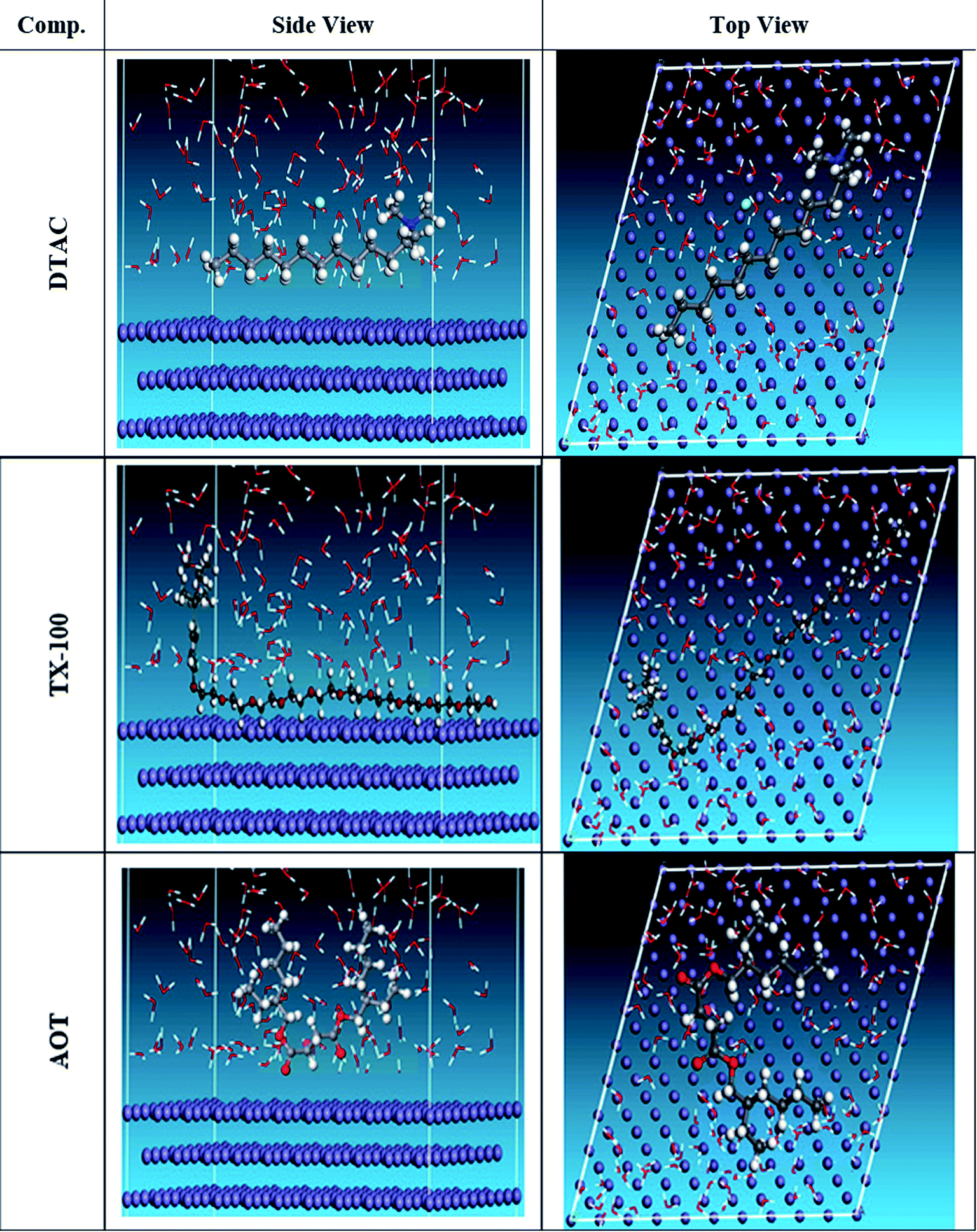
The MC simulation was performed to study the adsorption behavior of the three inhibitors on the Fe (110) surface. Fig. 14 shows the most suitable nonionic surfactant configurations simulated in water solution adsorbed on the Fe (110) substrate attained by an adsorption locator module. According to the equilibrium configurations of the three inhibitors adsorbed onto the Fe (110) surface, we can draw a conclusion that surfactants can be absorbed onto the Fe surface through their oxygen atoms. In this way, the covering with surfactants, consequently preventing the surface from contact with water, can reduce the exposure of the Fe surface. Therefore, the corrosion inhibition is achieved by this factor. It can be inferred that the inhibitor molecules will form a water-proof film on the Fe surface after addition to the solution. The Adsorption Locator Study Table 9 output contain the following columns: structure: the configurations of the adsorbate components on the Fe surface. Total energy: the total energy, in kcal mol−1, of the Fe surface–non ionic surfactant–water configuration. The Fe surface energy is taken as zero. The total energy is defined as the sum of the energies of the adsorbate components, the rigid adsorption energy, and the deformation energy. Adsorption energy: the energy, in kcal mol−1, released (or required) when the relaxed adsorbate components are adsorbed onto the Fe surface. This is the sum of the rigid adsorption energy and the deformation energy for the adsorbate components. Rigid adsorption energy: the energy, in kcal mol−1, released (or required) when the unrelaxed adsorbate components (before the geometry optimization step) are adsorbed onto the Fe surface. Deformation energy: the energy, in kcal mol−1, released when the adsorbed components are relaxed on the Fe surface. dEad/dNi: the energy, in kcal mol−1, of Fe surface-adsorbate configurations where one of the adsorbate components has been removed. Table 9 shows that the DTAC compound has the maximum binding energy, followed by TX-100, while AOT was the least effective compound in the simulation experiment. The inhibitory trend is still changing, with the adsorption energy increasing in the order of DTAC > TX-100 > AOT. Hence, the better stability formed complicated happened with DTAC and accordingly will increase its inhibition efficiency and that is steady with experimental results. In addition, the simulation was conducted in the presence of water species, with the same compound inhibition order. This demonstrates that DTAC inhibits most of the other chemicals in all of the systems studied. The results given in Table 9 reflect that the average value of adsorption energies is higher of order DTAC-water > AOT-water > TX-100-water system. In addition, the average energy needs to release DTAC are higher than those of other surfactants.
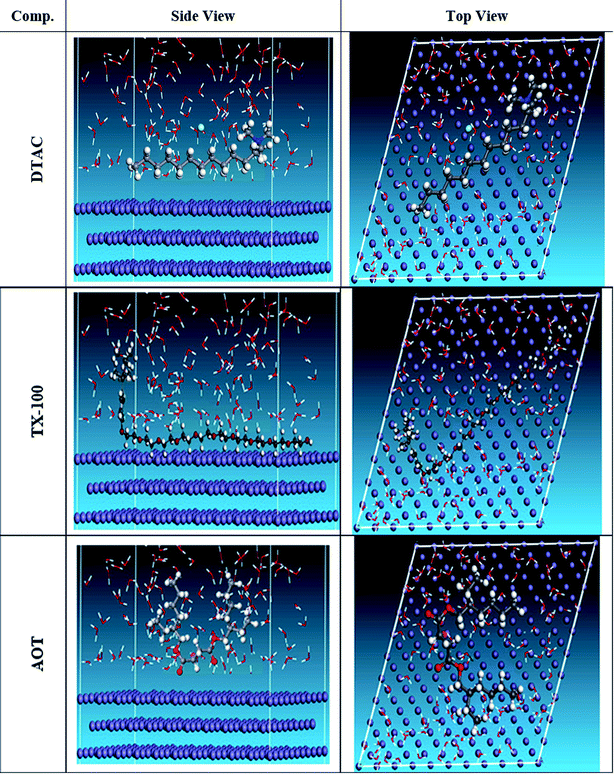 |
| | Fig. 14 The most appropriate configuration for the adsorption of the surfactant molecules on the Fe (110) substrate received by an adsorption locator module. | |
Table 9 Outputs and descriptors estimated by the Monte Carlo simulation for the adsorption of surfactants on iron (110)
| Surf., |
Total energy |
Adsorption energy |
Rigid adsorption energy |
Deformation energy |
Compound dEad/dNi |
water dEad/dNi |
| DTAC |
−2057.9 |
−2195.2 |
−2127.18 |
−67.825 |
−84.8 |
−11.271 |
| TX-100 |
−1260.0 |
−1370.0 |
−1330.00 |
−40.00 |
−20.00 |
−9.70 |
| AOT |
−1260.0 |
−1375.0 |
−1330.00 |
−45.38 |
−32.50 |
−8.25 |
4. Conclusions
DTAC, AOT and TX-100 showed good performance as corrosion inhibitors in hydrochloric acid media. The inhibition is because of the adsorption of the surfactant molecules onto the CS surface and blocking off of its active sites. Adsorption of the surfactants suits the Langmuir isotherm model. The results obtained from WL, PDP, EIS, and EFM tests are in reasonably excellent agreement and display improved surfactant performance with the growing surfactant concentration. PDP records display that the applied surfactants act as mixed-kind inhibitors in half molar HCl. The theoretical examination of molecules in each gaseous section and liquid section indicated the difference among AOT, TX-100, and DTAC, consistent with the HSAB principle. SEM combined with EDX and AFM analyses of the morphology of the CS surface revealed the presence of a persistent and insoluble adherent layer that prevents the electrolyte from reaching the metal's surface. Monte Carlo simulations reveal a strong interaction between the surfactants studied and the CS surface.
Author contributions
A. S. Fouda, designated the experiments, carried out the experiments and data analysis, wrote the manuscript, discussion and edits the manuscript.
Conflicts of interest
The author declares no competing interests.
References
- J. E. Oddo and M. B. Tomson, J. Pet. Technol., 1982, 1583–1590 CrossRef CAS.
-
B. Ridd, T. J. Blakset, D. Queen, Corrosion, NACE, ( 1998). p. 78 Search PubMed.
- G. Banerjee and S. N. Malhotra, Corrosion, 1992, 48, 10–15 CrossRef CAS.
- M. El Azhar, B. Mernari, M. Traisnel, F. Bentiss and M. Lagrenee, Corros. Sci., 2001, 43, 2227–2238 CrossRef.
- O. Olivares, N. V. Likhanova, B. Gomez, J. Navarrete, E. A. Llanos-Serrano and J. M. Hallen, Appl. Surf. Sci., 2006, 252, 2894–2909 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1992, 37, 2137–2144 CrossRef CAS.
- A. Popova, E. Sokolova, S. Raicheva and M. Christov, Corros. Sci., 2003, 45, 33–58 CrossRef.
- M. Lagrenee, B. Mernari, M. Bouanis, M. Traisnel and F. Bentiss, Corros. Sci., 2002, 44, 573–588 CrossRef CAS.
- S. Tamil Selvi, V. Raman and N. Rajendran, J. Appl. Electrochem., 2003, 33, 1175–1182 CrossRef CAS.
- M. Kissi, M. Bouklah, B. Hammouti and M. Benkaddour, Appl. Surf. Sci., 2006, 252, 4190–4197 CrossRef CAS.
- E. Kraka and D. Cremer, J. Am. Chem. Soc., 2000, 122, 8245–8264 CrossRef CAS.
- M. Karelson and V. Lobanov, Chem. Rev., 1996, 96, 1027–1044 CrossRef CAS PubMed.
-
A. Hinchliffe, Chemical Modelling from Atoms to Liquids, John Wiley & Sons, New York, 1999 Search PubMed.
- F. Dabosi, Y. Derbali, M. Etman, A. Srhiri and A. de, J. Savignac, Appl. Electrochem., 1991, 21, 225–232 Search PubMed.
- M. El Achouri, M. S. Hajji, S. Kertit, M. Essassi, M. Salem and R. Coudert, Corros. Sci., 1995, 37, 381–389 CrossRef CAS.
- T. Vasudevan, S. Muralidharan, S. Alwarappan and S. V. K. Iyer, Corros. Sci., 1995, 37, 1235–1244 CrossRef CAS.
- M. El Achouri, M. S. Hajji, M. Salem, S. Kertit, J. Aride, R. Coudert and E. M. Essassi, Corrosion, 1996, 52, 103–108 CrossRef CAS.
- M. A. Migahed and A. M. Al-Sabagh, Chem. Eng. Commun., 2009, 196, 1054–1075 CrossRef CAS.
- M. L. Free, Corrosion, 2002, 58, 1025–1030 CrossRef CAS.
- M. Saleh and A. A. Atia, J. Appl. Electrochem., 2006, 36, 899–905 CrossRef CAS.
- W. L. Wang and M. L. Free, Corros. Sci., 2004, 46, 2601–2611 CrossRef CAS.
- A. A. Atia and M. M. Saleh, J. Appl. Electrochem., 2003, 33, 171–177 CrossRef CAS.
- T. Y. Soror and M. A. El-Ziady, Mater. Chem. Phys., 2002, 77, 697–703 CrossRef.
- X. Li, L. Tang., H. lie, G. Mu and G. Lie, Mater. Lett., 2008, 62, 2321–2324 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1995, 23, 1833–1840 CrossRef.
- H. Ma, S. Chen, B. Yin, S. Zhao and X. Liu, Corros. Sci., 2003, 45, 867–882 CrossRef CAS.
-
Volt Master 4 Manual, 2000 Search PubMed.
- C. N. Cao, Chem. Ind. Eng., 2004, 27(3), 325–340 Search PubMed.
- A. S. Fouda, E. S. El-Gharkawy, H. Ramadan and A. El-Hossiany, Biointerface Res. Appl. Chem., 2021, 11, 9786–9803 CAS.
- A. Popova, E. Sokolova, S. Raicheva and M. Christov, Corros. Sci., 2003, 45, 33–58 CrossRef.
- A. S. Fouda, E. Abdel-Latif, H. M. Helal and A. El-Hossiany, Russ. J. Electrochem., 2021, 57, 159–171 CrossRef CAS.
- X. H. Li and G. N. Mu, Appl. Surf. Sci., 2005, 252, 1254–1265 CrossRef CAS.
- A. S. Fouda, H. Ibrahim, S. Rashwaan, A. El-Hossiany and R. M. Ahmed, Int. J. Electrochem. Sci., 2018, 13, 6327–6346 CrossRef CAS.
- L. Larabi, O. Benali and Y. Harek, Mater. Lett., 2007, 61, 3287–3291 CrossRef CAS.
- A. S. Fouda, M. Eissa and A. El-Hossiany, Int. J. Electrochem. Sci., 2018, 13, 11096–11112 CrossRef CAS.
- O. L. Riggs and R. M. Hurd, Corrosion, 1967, 23, 252–260 CrossRef CAS.
- A. S. Fouda, R. E. Ahmed and A. El-Hossiany, Prot. Met. Phys. Chem. Surf., 2021, 57, 398–411 CrossRef CAS.
- A. Y. Musa, A. A. H. Kadhum, A. B. Mohamad, A. R. daud, M. S. Takriff and S. K. Kamarudin, Corros. Sci., 2009, 51(10), 2393–2399 CrossRef CAS.
- M. A. Khaled, M. A. Ismail, A. A. El-Hossiany and A. S. Fouda, RSC Adv., 2021, 11, 25314–25333 RSC.
- A. S. Fouda, S. A. Abd El-Maksoud, A. El-Hossiany and A. Ibrahim, Int. J. Electrochem. Sci., 2019, 14, 2187–2207 CrossRef CAS.
- A. S. Fouda, M. A. A. El-Ghaffar, M. H. Sherif, A. T. El-Habab and A. El-Hossiany, Prot. Met. Phys. Chem. Surf., 2020, 56, 189–201 CrossRef CAS.
- K. F. Khaled, Electrochim. Acta, 2003, 48, 2493–2503 CrossRef CAS.
- A. S. Fouda, S. A. El-Maksoud, A. El-Hossiany and A. Ibrahim, Int. J. Electrochem. Sci., 2019, 14, 6045–6064 CrossRef CAS.
- F. B. Growcock and J. H. Jasinski, J. Electrochem. Soc., 1989, 136, 2310–2314 CrossRef CAS.
- A. S. Fouda, M. Abdel Azeem, S. A. Mohamed, A. El-Hossiany and E. El-Desouky, Int. J. Electrochem. Sci., 2019, 14, 3932–3948 CrossRef CAS.
- A. H. Mehaute and G. Grepy, Solid State Ionics, 1983, 9–10, 17–30 Search PubMed.
- M. M. Motawea, A. El-Hossiany and A. S. Fouda, Int. J. Electrochem. Sci., 2019, 14, 1372–1387 CrossRef CAS.
- C. H. Hsu and F. Mansfeld, Corrosion, 2001, 57, 747–748 CrossRef CAS.
- A. S. Fouda, F. I. El-Dossoki, A. El-Hossiany and E. A. Sello, Surf. Eng. Appl. Electrochem., 2020, 56, 491–500 CrossRef.
-
Gamry Echem Analyst Manual, 2003 Search PubMed.
- R. W. Bosch, J. Hubrecht, W. F. Bogaerts and B. C. Syrett, Corrosion, 2001, 57, 60–70 CrossRef CAS.
- A. S. Fouda, S. A. Abd El-Maksoud, A. M. Belal, A. El-Hossiany and A. Ibrahium, Int. J. Electrochem. Sci., 2018, 13, 9826–9846 CrossRef CAS.
- P. Somasundaran and R. B. Grieves, AIChE Symp. Ser., 1975, 71, 124 Search PubMed.
- A. S. Fouda, A. M. El-Wakeel, K. Shalabi and A. El-Hossiany, Elixir Corros. Day, 2015, 83, 33086–33094 Search PubMed.
-
D. W. Fuerstenau, The chemistry of bio surfaces, Marcel Dekker, New York, 1971. p. 143 Search PubMed.
- D. W. Fuerstenau, T. W. Healy and P. Somasundaran, Trans. AIME, 1964, 202, 958 Search PubMed.
- B. Zerga, A. Attayibat, M. Sfaira, M. Taleb, B. Hammouti, M. Ebn Touhami, S. Radi and Z. Rais, J. Appl. Electrochem., 2010, 40, 1575–1582 CrossRef CAS.
- P. Somasundaran and J. T. Kunjappu, Colloids Surf., 1989, 37, 245–268 CrossRef.
- I. J. Lin and P. Somasundaran, J. Colloid Interface Sci., 1971, 37(4), 731–743 CrossRef CAS.
- L. Zhang, P. Somasundaran, J. Mielczarski and E. Mielczarski, J. Colloid Interface Sci., 2002, 256, 16–22 CrossRef CAS.
- R. O. James and T. W. Healy, J. Colloid Interface Sci., 1972, 40(1), 65–81 CrossRef CAS.
- R. Zhang and P. Somasundaran, Adv. Colloid Interface Sci., 2006, 215, 123–126 Search PubMed.
- L. K. Koopal, E. M. Lee and M. R. Böhmer, J. Colloid Interface Sci., 1995, 170, 85–97 CrossRef CAS.
- A.-X. Fan, P. Somasundaran and N. J. Turro, Langmuir, 1997, 13, 506–510 CrossRef CAS.
- F. M. Donahue and K. Nobe, J. Electrochem. Soc., 1965, 112, 886–890 CrossRef CAS.
- E. Kamis, F. Belluci, R. M. Latanision and E. S. H. El-Ashry, Corrosion, 1991, 47, 677–686 CrossRef.
- A. S. Fouda, S. Rashwan, A. El-Hossiany and F. E. El-Morsy, J. Chem., Biol. Phys. Sci., 2019, 9, 1–24 CAS.
- D. A. Ló pez, S. N. Simison and S. R. de Sánchez, Corros. Sci., 2005, 47, 735–755 CrossRef.
- K. Shalabi, E. Abdel-Galil, A. H. El-Askalany and Y. M. Abdallah, J. Mol. Liq., 2022, 348, 118420 CrossRef CAS.
- O. A. Elgyar, A. M. Ouf, A. El-Hossiany and A. S. Fouda, Biointerface Res. Appl. Chem., 2021, 11, 14358–14344 Search PubMed.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- V. S. Sastri and J. R. Perumareddi, Corros. Sci., 1997, 53, 617–622 CrossRef CAS.
- T. Y. Soror and M. A. El-Ziady, Mater. Chem. Phys., 2002, 77, 697–703 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Ameena M.
Al-bonayan
b,
Mohamed
Eissa
cd and
Dalia M.
Eid
e
*a,
Ameena M.
Al-bonayan
b,
Mohamed
Eissa
cd and
Dalia M.
Eid
e

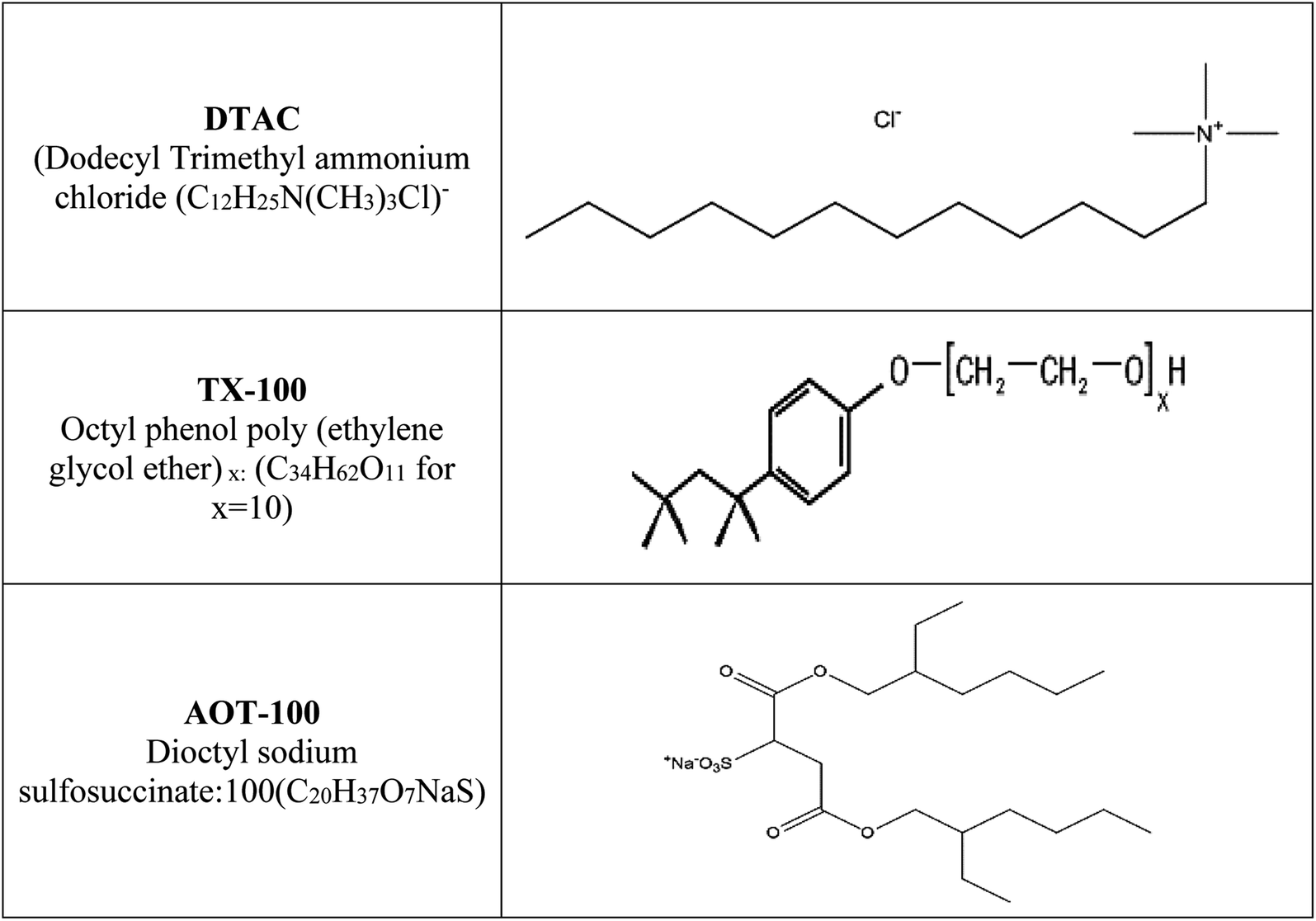
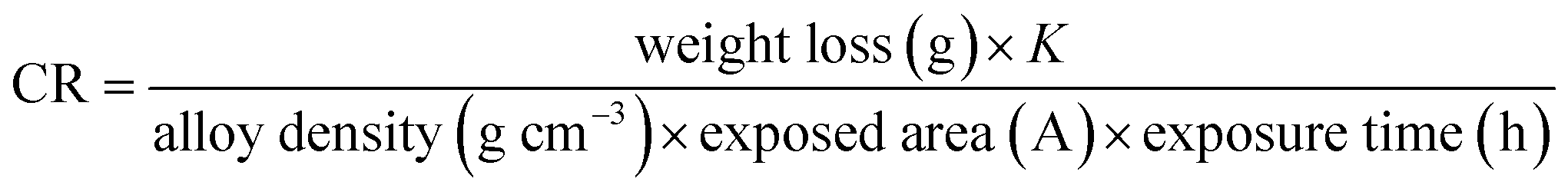
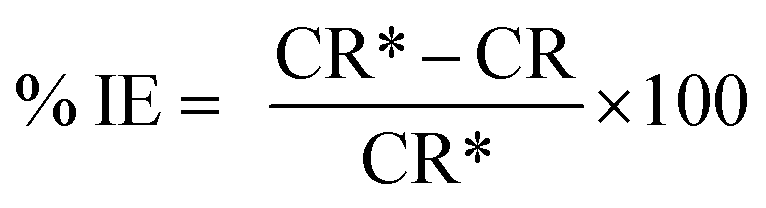
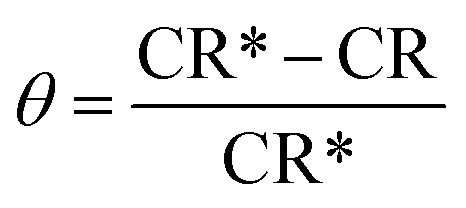
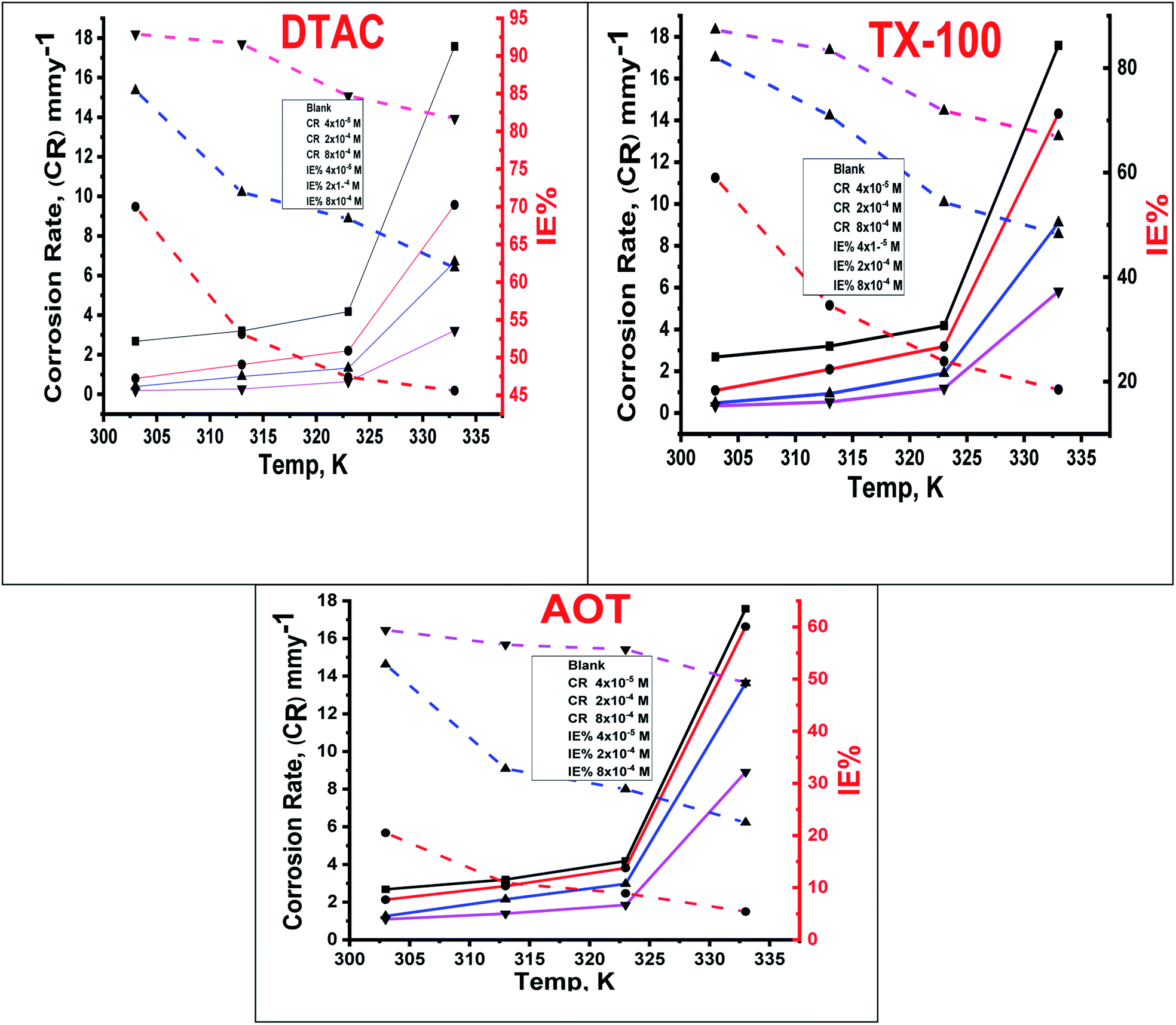

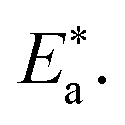 The regression between log
The regression between log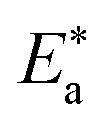 in the presence of surfactants are higher than those in the uninhibited acid solution. The results agreed with that of previous study reported by Fouda et al.31 and Popova et al.32 for CS in 1 M HCl. The addition of surfactants to the solution increases the activation energy
in the presence of surfactants are higher than those in the uninhibited acid solution. The results agreed with that of previous study reported by Fouda et al.31 and Popova et al.32 for CS in 1 M HCl. The addition of surfactants to the solution increases the activation energy 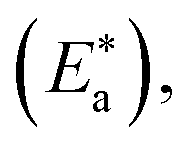 as shown in Table 2 and the extent of the increase is proportional to the IE of the surfactant, indicating that the energy barrier for the corrosion reaction increases in the presence of these surfactants. This means that by addition of the surfactant in the acid solution, the corrosion reaction will be further pushed to surface sites that are characterized by higher values of activation energy
as shown in Table 2 and the extent of the increase is proportional to the IE of the surfactant, indicating that the energy barrier for the corrosion reaction increases in the presence of these surfactants. This means that by addition of the surfactant in the acid solution, the corrosion reaction will be further pushed to surface sites that are characterized by higher values of activation energy 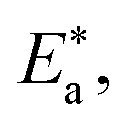 indicating that CS corrosion occurs at the uncovered part of the surface. Thus, adsorption of the surfactant was assumed to occur on the higher energy sites,33 and the presence of surfactant, which results in the blocking of the active sites, must be associated with an increase in the activation energy,
indicating that CS corrosion occurs at the uncovered part of the surface. Thus, adsorption of the surfactant was assumed to occur on the higher energy sites,33 and the presence of surfactant, which results in the blocking of the active sites, must be associated with an increase in the activation energy, 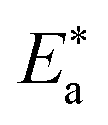 which indicates the physical adsorption or weak chemical bonding between the surfactant molecules and the CS surface.34 The increase in
which indicates the physical adsorption or weak chemical bonding between the surfactant molecules and the CS surface.34 The increase in 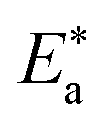 in the presence of the surfactants indicates the physical adsorption that occurs in first stage,35,36 weak chemical bonding between the surfactants and the steel surface37 or due to the decrease in the adsorption of these surfactants with the increase in temperature.38 Generally, one can say that the nature and concentration of electrolytes greatly affect the activation energy for the corrosion process. A plot of log(kcorr./T) vs. (1/T) gives a straight line with a slope of [−ΔH*/2.303R] and an intercept of [log(R/Nh) + ΔS*/2.303R]. The plots are shown in Fig. 4. The calculated values of ΔH* and ΔS* are given in Table 2. The values of ΔH* are positive indicating that the corrosion process is endothermic one. The values of
in the presence of the surfactants indicates the physical adsorption that occurs in first stage,35,36 weak chemical bonding between the surfactants and the steel surface37 or due to the decrease in the adsorption of these surfactants with the increase in temperature.38 Generally, one can say that the nature and concentration of electrolytes greatly affect the activation energy for the corrosion process. A plot of log(kcorr./T) vs. (1/T) gives a straight line with a slope of [−ΔH*/2.303R] and an intercept of [log(R/Nh) + ΔS*/2.303R]. The plots are shown in Fig. 4. The calculated values of ΔH* and ΔS* are given in Table 2. The values of ΔH* are positive indicating that the corrosion process is endothermic one. The values of 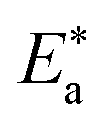 are larger than the analogous values of ΔH*, indicating that the corrosion process must involve a gaseous reaction, simply the hydrogen evolution reaction, associated with a decrease in the total reaction volume”. These data verified the known thermodynamic relation among
are larger than the analogous values of ΔH*, indicating that the corrosion process must involve a gaseous reaction, simply the hydrogen evolution reaction, associated with a decrease in the total reaction volume”. These data verified the known thermodynamic relation among 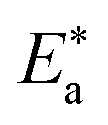 and ΔH*39 which is as follows eqn (6):
and ΔH*39 which is as follows eqn (6):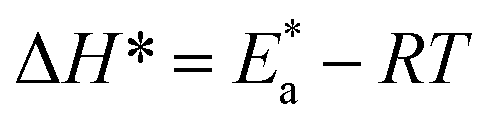
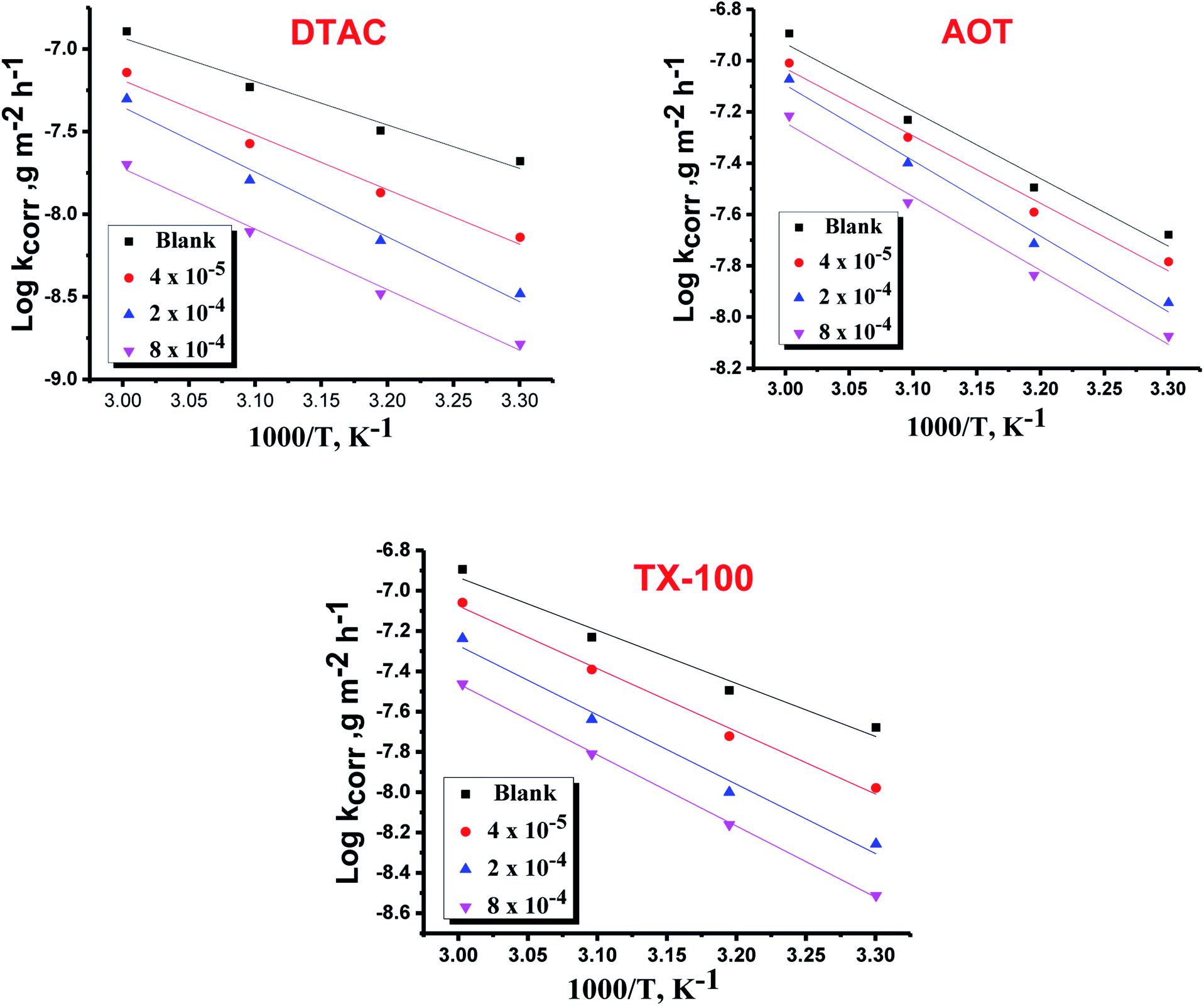
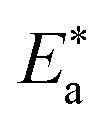
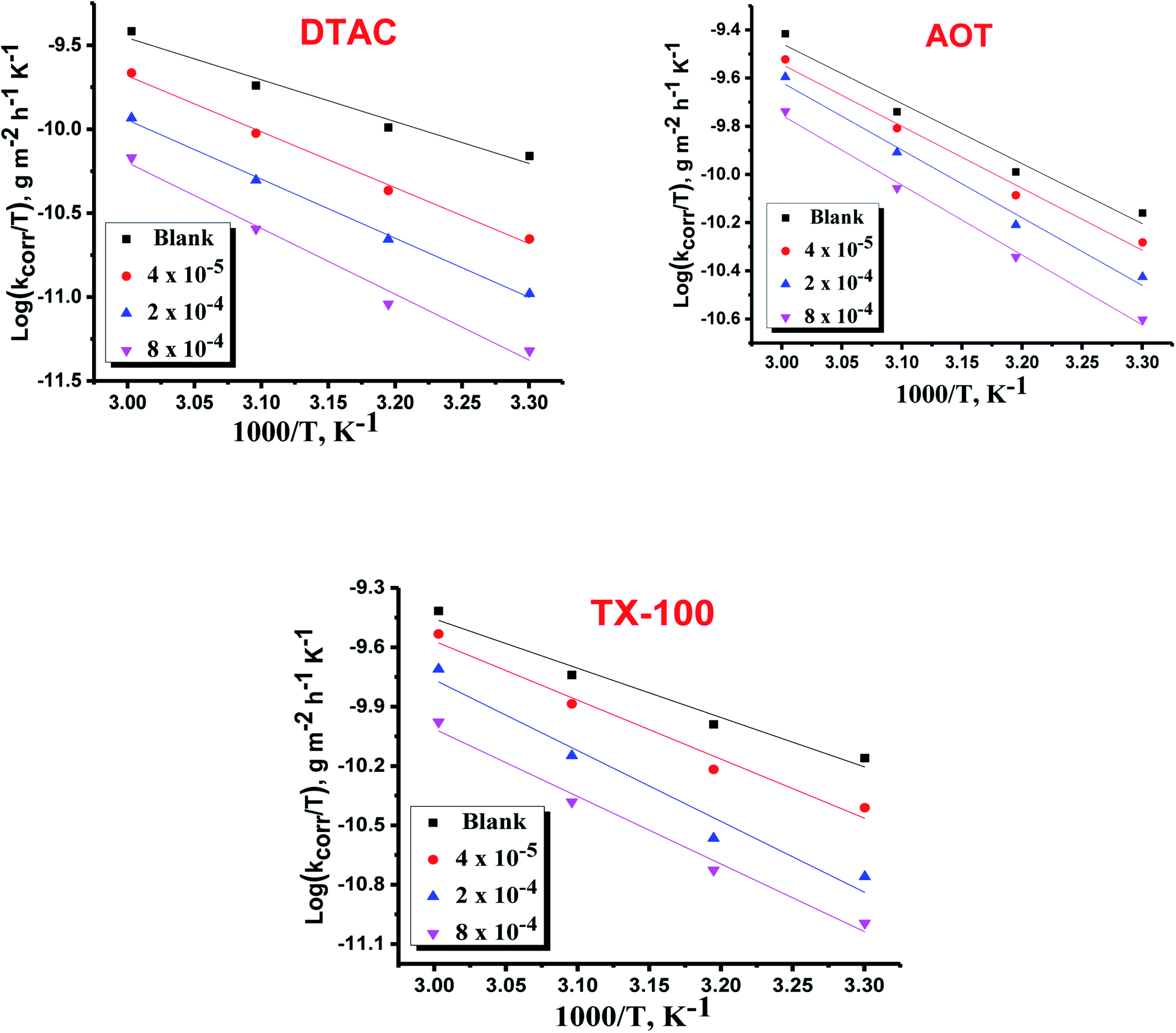
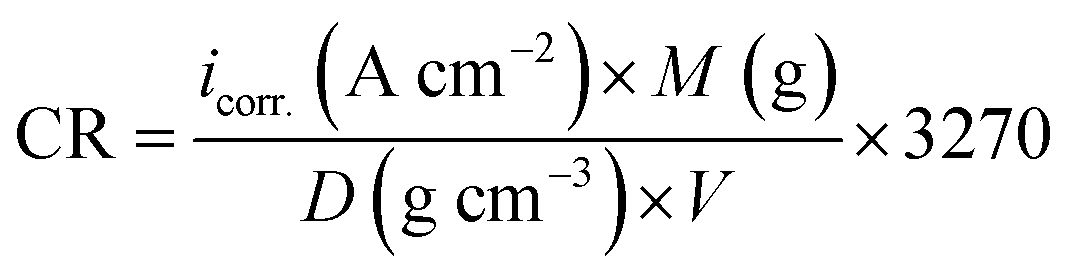
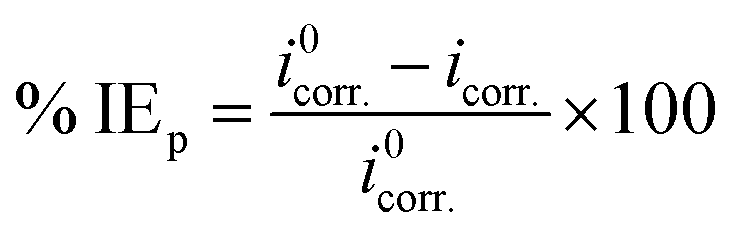
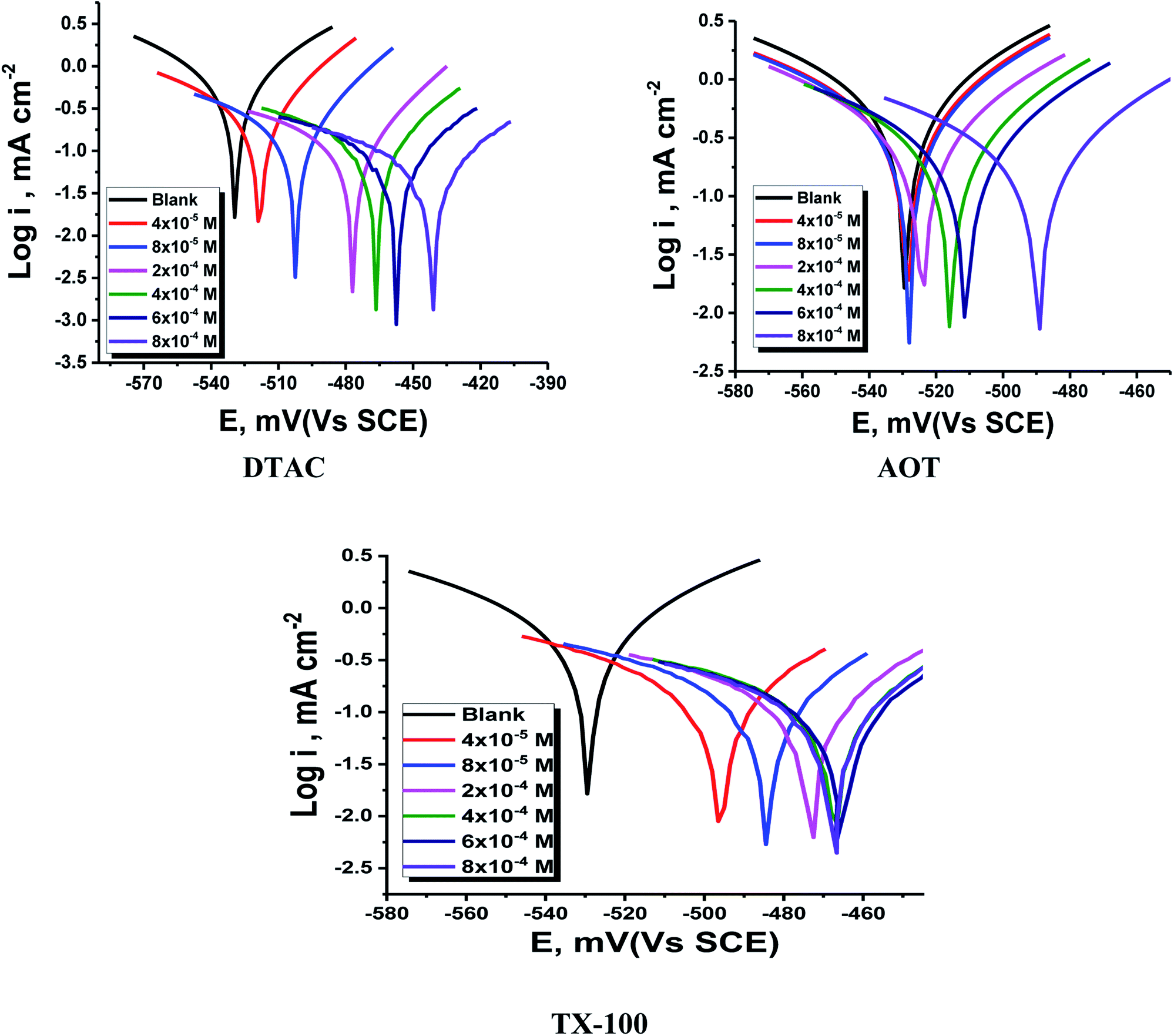
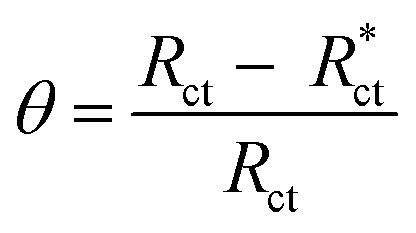
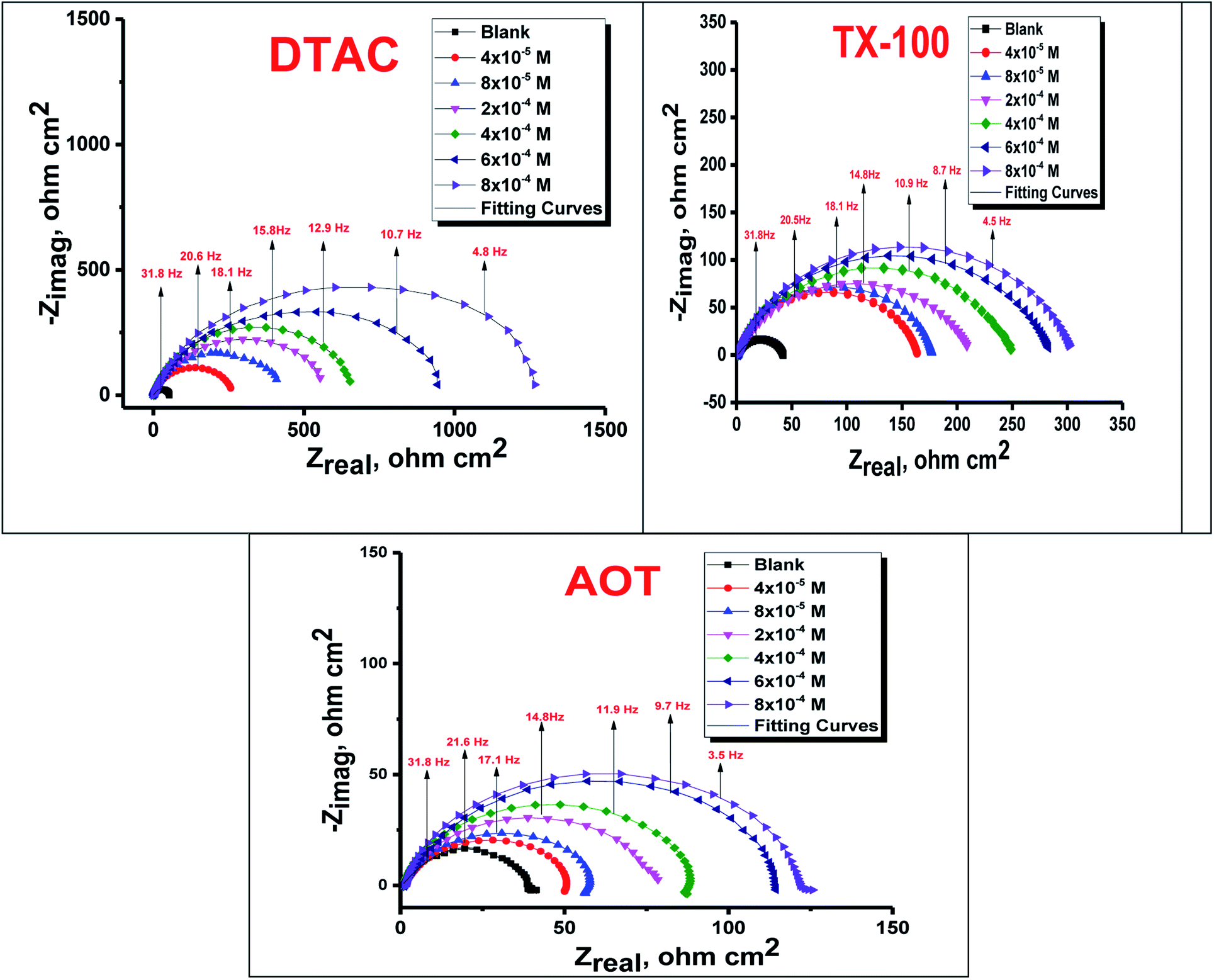
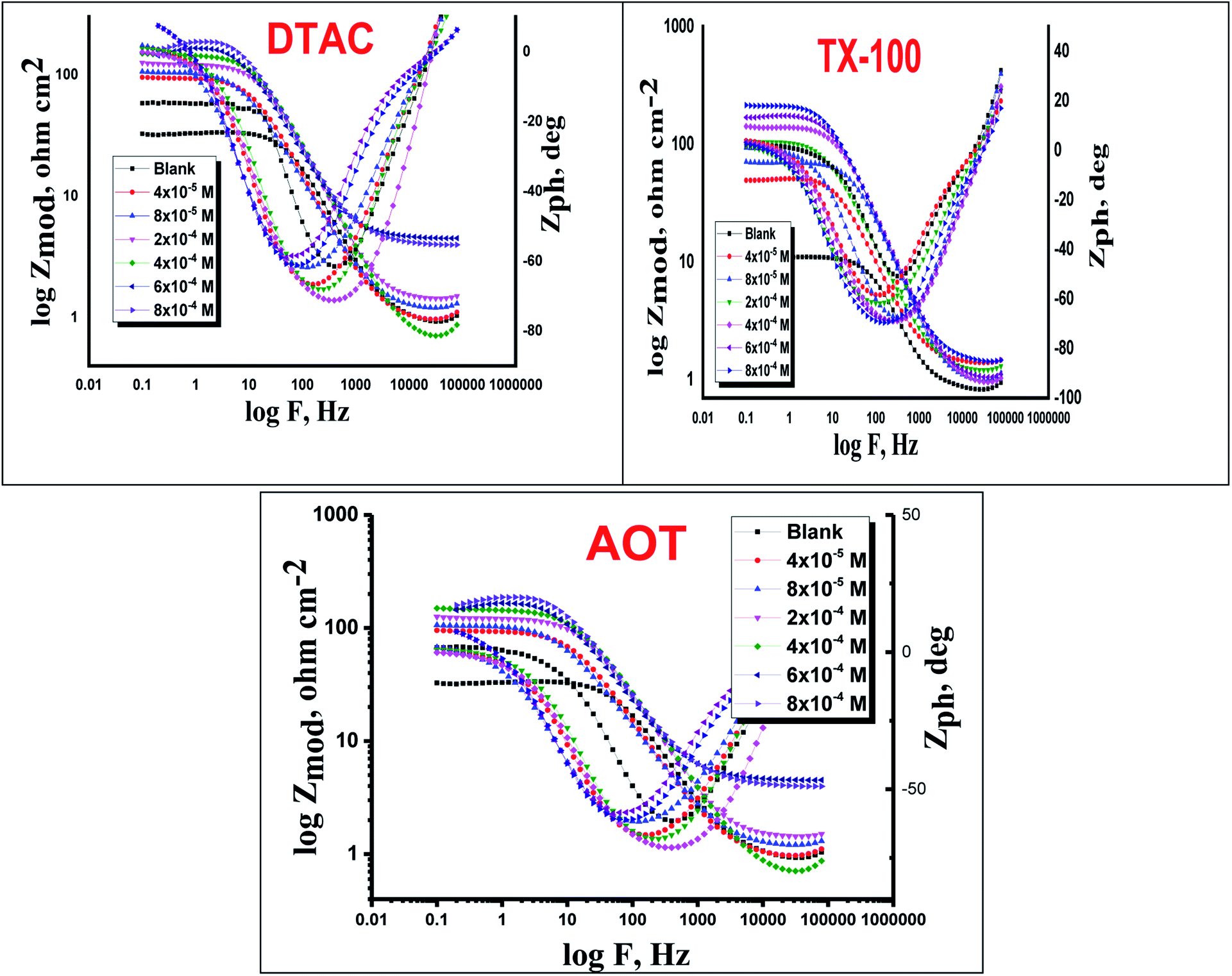
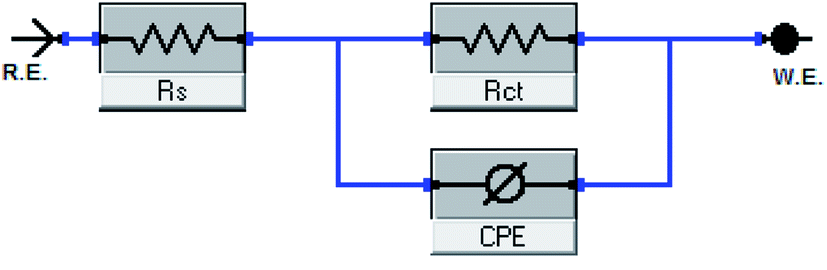
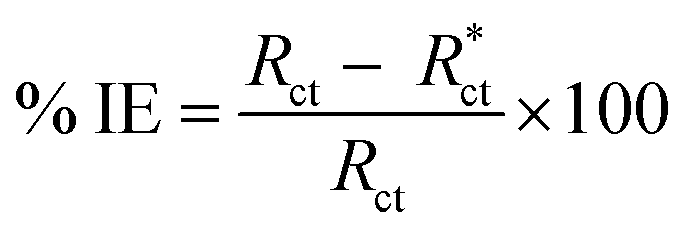
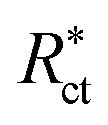 are the resistance in the presence and absence of surfactants, respectively.
are the resistance in the presence and absence of surfactants, respectively.
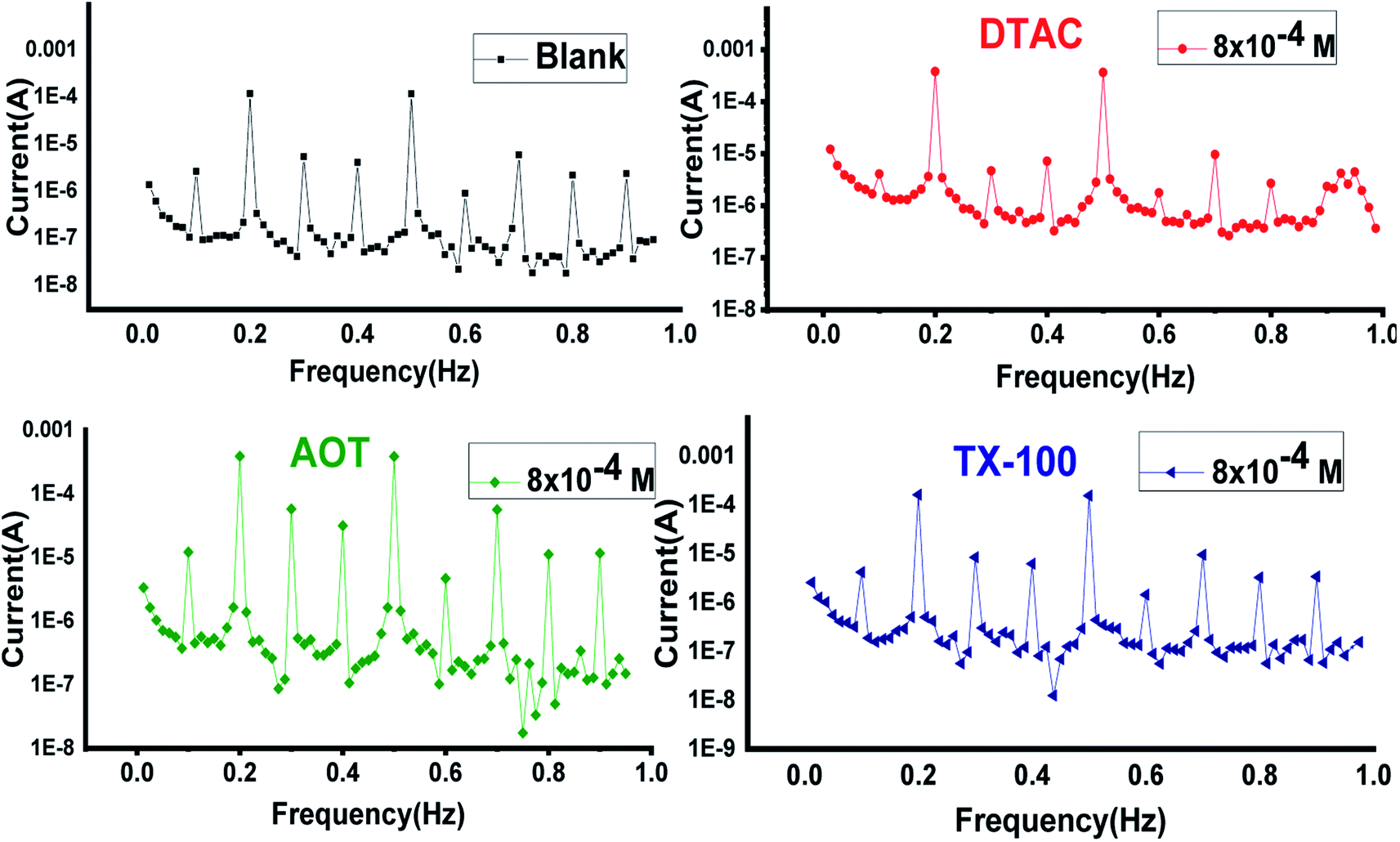
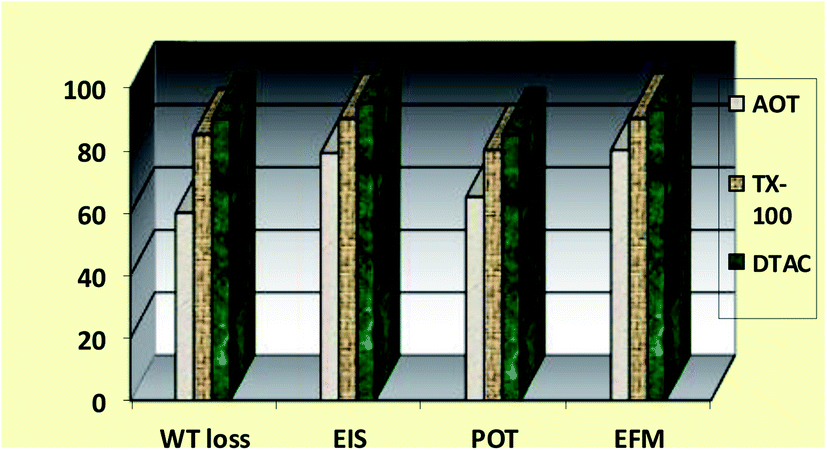
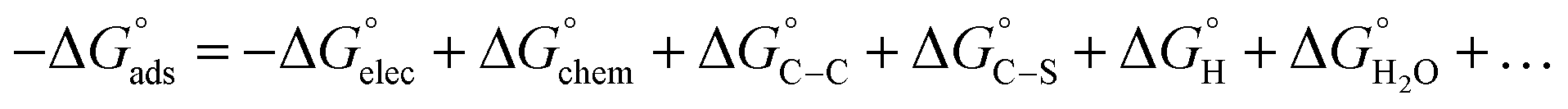
 is the electrostatic interaction term,
is the electrostatic interaction term, 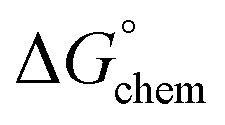 the chemical term due to covalent bonding,
the chemical term due to covalent bonding, 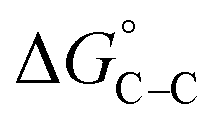 the free energy gained upon association of methyl groups in the hydrocarbon chain,
the free energy gained upon association of methyl groups in the hydrocarbon chain, 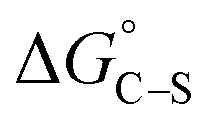 the free energy due to interactions between the hydrocarbon chains and hydrophobic sites on the solid,
the free energy due to interactions between the hydrocarbon chains and hydrophobic sites on the solid,  the hydrogen bonding term and
the hydrogen bonding term and 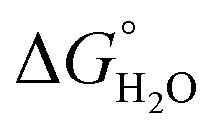 the term owing to dissolution or solvation of the adsorbate species, or any species displaced from the interface due to adsorption”. In the following, the major forces involved in surfactant adsorption are discussed:
the term owing to dissolution or solvation of the adsorbate species, or any species displaced from the interface due to adsorption”. In the following, the major forces involved in surfactant adsorption are discussed:
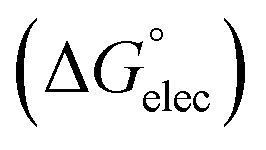 : in systems where the ionic surfactants and the solid particles are charged, electrostatic interactions play a governing role in the adsorption process.
: in systems where the ionic surfactants and the solid particles are charged, electrostatic interactions play a governing role in the adsorption process.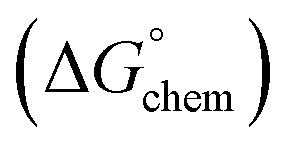 : chemical interaction is another important driving force for adsorption of surfactants on the solid particles. Compared to other driving forces, this interaction is specific to certain systems where covalent bonding can occur between the surfactant and the solid.
: chemical interaction is another important driving force for adsorption of surfactants on the solid particles. Compared to other driving forces, this interaction is specific to certain systems where covalent bonding can occur between the surfactant and the solid.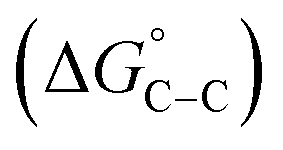 : “At a concentration above a threshold value, analogous to the aggregation in the bulk, surfactant molecules tend to form two dimensional aggregates at the solid/liquid interface, causing an abrupt increase in the adsorption density. These aggregates have been called “hemi-micelles”63 or, in general, “colloids” for surface colloids.64 The driving force for adsorption
: “At a concentration above a threshold value, analogous to the aggregation in the bulk, surfactant molecules tend to form two dimensional aggregates at the solid/liquid interface, causing an abrupt increase in the adsorption density. These aggregates have been called “hemi-micelles”63 or, in general, “colloids” for surface colloids.64 The driving force for adsorption 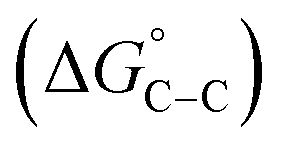 results from the free energy of transferring the hydrocarbon chains from the aqueous environment into the hydrophobic interior of the aggregates.
results from the free energy of transferring the hydrocarbon chains from the aqueous environment into the hydrophobic interior of the aggregates.  can be represented as a linear function of the energy gained per –CH2 group”.65
can be represented as a linear function of the energy gained per –CH2 group”.65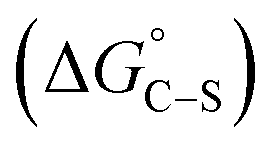 : “the hydrophobic interaction
: “the hydrophobic interaction 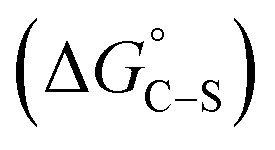 between the alkyl chain of a surfactant and the hydrophobic sites on the solid becomes a significant factor for surfactant adsorption on fully or partially hydrophobic surfaces. In this case, the surfactant molecules attach to the hydrophobic sites with the hydrocarbon chains aligned parallel to the surface at low concentrations and normal to the surface at higher concentrations. Such an adsorption process often results in a two-step isotherm”.
between the alkyl chain of a surfactant and the hydrophobic sites on the solid becomes a significant factor for surfactant adsorption on fully or partially hydrophobic surfaces. In this case, the surfactant molecules attach to the hydrophobic sites with the hydrocarbon chains aligned parallel to the surface at low concentrations and normal to the surface at higher concentrations. Such an adsorption process often results in a two-step isotherm”.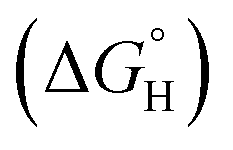 : “Hydrogen bonding between surfactant species and the solid surface species could occur in systems containing hydroxyl, phenolic, carboxylic and amine groups on the surfactant. For instance, adsorption of a nonionic surfactant such as ethoxylated alcohol and sugar-based alkyl glucoside on oxides has been proposed to involve hydrogen bonding.66,67 It should be noted that for adsorption due to hydrogen bonding to take place, the bond formed between the surfactant functional groups and mineral surfaces should be stronger than that formed between the mineral and interfacial water molecules”.
: “Hydrogen bonding between surfactant species and the solid surface species could occur in systems containing hydroxyl, phenolic, carboxylic and amine groups on the surfactant. For instance, adsorption of a nonionic surfactant such as ethoxylated alcohol and sugar-based alkyl glucoside on oxides has been proposed to involve hydrogen bonding.66,67 It should be noted that for adsorption due to hydrogen bonding to take place, the bond formed between the surfactant functional groups and mineral surfaces should be stronger than that formed between the mineral and interfacial water molecules”.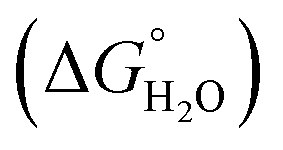 : “When a hydrated head group of the surfactant transfers from the bulk to the mineral–solution interfacial region, partial removal of water molecules from the secondary solvation shell around the surfactant head groups can occur. In contrast to other driving forces, desolvation energy due to such a process is unfavorable for the adsorption process”.
: “When a hydrated head group of the surfactant transfers from the bulk to the mineral–solution interfacial region, partial removal of water molecules from the secondary solvation shell around the surfactant head groups can occur. In contrast to other driving forces, desolvation energy due to such a process is unfavorable for the adsorption process”.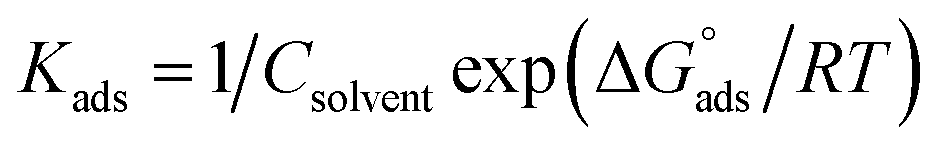
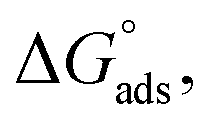 and Csolvent is the molar concentration of solvent, which in the case of water is 55.5 mol l−1. The
and Csolvent is the molar concentration of solvent, which in the case of water is 55.5 mol l−1. The 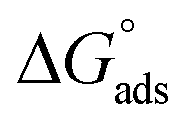 value was calculated according to eqn (14). Fig. 11 shows the dependence of the fraction of the surface coverage (C/θ) as a function of the concentration (C) of AOT, TX-100 and DTAC. The degrees of surface coverage (θ) were evaluated from EIS measurements using eqn (10) and are given in Table 6. The
value was calculated according to eqn (14). Fig. 11 shows the dependence of the fraction of the surface coverage (C/θ) as a function of the concentration (C) of AOT, TX-100 and DTAC. The degrees of surface coverage (θ) were evaluated from EIS measurements using eqn (10) and are given in Table 6. The 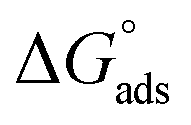 values are negative, suggesting the spontaneity of the adsorption process. The regression coefficient is R2 = 0.999. It is well known that values of
values are negative, suggesting the spontaneity of the adsorption process. The regression coefficient is R2 = 0.999. It is well known that values of 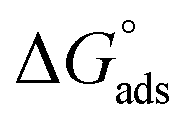 in the order of 20 kJ mol−1 or lower indicate physisorption, while those in the order of 40 kJ mol−1 or higher involve charge sharing or charge transfer from the surfactant molecules to the metal surface to form a coordinate type of bond.71,72 The calculated values of
in the order of 20 kJ mol−1 or lower indicate physisorption, while those in the order of 40 kJ mol−1 or higher involve charge sharing or charge transfer from the surfactant molecules to the metal surface to form a coordinate type of bond.71,72 The calculated values of 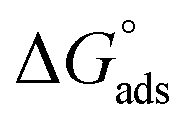 for DTAC are around −38.1 to 37.3 kJ mol−1, for TX-100 approximately −36.8 to 35.2 kJ mol−1 and for AOT-100 approximately −32.2 to 31.4 kJ mol−1, so physical and chemical adsorption (mixed type) may proposed.
for DTAC are around −38.1 to 37.3 kJ mol−1, for TX-100 approximately −36.8 to 35.2 kJ mol−1 and for AOT-100 approximately −32.2 to 31.4 kJ mol−1, so physical and chemical adsorption (mixed type) may proposed.