
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Positively-charged plasmonic nanostructures for SERS sensing applications
Mariacristina Turinoa,
Nicolas Pazos-Perez a,
Luca Guerrini*a and
Ramon A. Alvarez-Puebla*ab
a,
Luca Guerrini*a and
Ramon A. Alvarez-Puebla*ab
aDepartment of Physical and Inorganic Chemistry – EMaS, Universitat Rovira I Virgili, Carrer de Marcel·lí Domingo s/n, 43007 Tarragona, Spain. E-mail: luca.guerrini@urv.cat; ramon.alvarez@urv.cat
bICREA, Passeig Lluís Companys 23, 08010 Barcelona, Spain
First published on 4th January 2022
Abstract
Surface-enhanced Raman (SERS) spectroscopy has been establishing itself as an ultrasensitive analytical technique with a cross-disciplinary range of applications, which scientific growth is triggered by the continuous improvement in the design of advanced plasmonic materials with enhanced multifunctional abilities and tailorable surface chemistry. In this regard, conventional synthetic procedures yield negatively-charged plasmonic materials which can hamper the adhesion of negatively-charged species. To tackle this issue, metallic surfaces have been modified via diverse procedures with a broad array of surface ligands to impart positive charges. Cationic amines have been preferred because of their ability to retain a positive zeta potential even at alkaline pH as well as due to their wide accessibility in terms of structural features and cost. In this review, we will describe and discuss the different approaches for generating positively-charged plasmonic platforms and their applications in SERS sensing.
1. Introduction
Surface-enhanced Raman (SERS) spectroscopy is an ultrasensitive analytical tool that has been continuously expanding its range of applications in several fields, including biosensing, medicine, environmental monitoring, food safety, and catalysis.1–10 As the magnification of the Raman scattering primarily depends on the excitation of localized surface plasmon resonances (LSPRs) to generate intense electric fields at the metallic surfaces (mostly silver or gold), the design of the plasmonic substrate is central to SERS analysis. Besides the intrinsic optical response of the nanoconstruct, the fine-tuning of its surface physicochemical properties is key to maximizing the sensing performances as well as to guiding, when desired, its potential assembly into more complex architectures.12–14Broadly speaking, SERS sensing strategies are generally classified based on two general principles: direct and indirect strategies. The direct methodologies rely on the acquisition of the intrinsic SERS spectra of the target species to detect its presence and, possibly, quantify its content in the sample.5 Conversely, indirect methods monitor the alteration of the optical signal originated from extrinsic SERS-active molecules whose extent, based on the appropriate design of the sensing platform, can be selectively and quantitatively correlated with the presence of the analyte. Notably, the most common implementation of indirect SERS is by using SERS encoded nanoparticles (also referred to as SERS tags or probes) which function similarly as fluorescent labels but with enhanced multiplexing, photostability, and quantitative response.16,17 Regardless of the sensing scheme, target species or SERS labels and chemosensing elements have to be brought into close contact with the metallic surface to yield intense SERS signals due to the exponential decay of the electric field intensity with distance from the plasmonic material.6 For indirect sensing, SERS labels and chemosensors are typically chosen among those carrying a mercapto group (or, to a lesser extent, a dithiocarbamate group) to promote the covalent attachment onto the nanostructure via the formation of the strong metal–S bond. In this case, then, the surface chemistry is tailored via ligand exchange upon displacement of the surface molecules retained at the plasmonic structure after the synthesis which, thus, makes this approach feasible only when the analyte displays higher affinity for the metal as compared to the native ligands.12 On the other hand, non-covalent interactions commonly takes place through electrostatic binding of the target onto the charged layer adhering onto the metallic surface, such as the well-studied interaction between surface halide anions (Cl−, Br−, I−) and quaternary aryl- and alkyl-ammonium compounds.20,21 Obviously, the chemistry of the metallic surface is directly related to the original synthetic procedures which, by and large, typically result in a negatively-charged interface formed by ligands such as citrate (and its oxidation products) and halide anions that can be found on conventional gold and silver colloids.14 Thus, the adsorption of negatively-charged analytes, especially those unequipped of reactive moieties capable of forming strong covalent bonds with the metal surface, has been traditionally hindered by charge repulsion. To tackle this issue, several strategies have been continuously developing to impart positive charge to the nanomaterial surface, including designing novel synthetic methods to yield intrinsically positive plasmonic materials as well as post-synthetic treatments to retool the pre-formed metal interface with appropriate capping ligands. Regardless of the approach, positively-charged plasmonic nanomaterials were obtained using surface ligand exposing quaternary ammonium groups towards the metal–liquid interface, mostly because of their ability to retain a positive zeta potential even at alkaline pH as well as their wide accessibility in terms of structural features and cost.22
Herein, we intend to provide a systematic overview on the topic by presenting and discussing the current state-of-the-art of the different methodologies of generation of positively-charged SERS substrates and their implementation into sensing platforms for detection of a wide range of analytes of interest. These targets were, for the sake of discussion, arbitrarily classified according to their relative size: from inorganic ions to biologically and environmentally relevant small molecules, from large biomolecules as nucleic acids to pathogenic microorganisms and cells.
2. Synthesis of positively-charged nanomaterials
Synthesis of positively-charged nanoparticles typically relies on two main strategies: the direct reduction of the metal precursor in the presence of positive stabilizing molecules or a post functionalization of previously formed nanoparticles (NPs) with molecules carrying positive groups. Both approaches have their advantages and drawbacks. The direct synthesis is usually easier and faster as it is performed via one-pot approaches. However, the monodispersity of the so-produced nanostructures is often relatively low with the additional problem of a limited tunability of NP size and morphology. On the other hand, post functionalization methods allow to better control the size and shape of the NPs using well-known established protocols for nanoparticle fabrication but frequently require tedious and complicated cleaning steps which can also be the source of undesired colloidal aggregation. Regardless, the surface functional moieties used up to date to impart positive charge to the nanomaterials are amine ligands of very diverse structural features. For the sake of discussion, these amine ligands were arbitrarily classified into the following groups: polymers, cationic surfactants, short polyamines and aminothiolated compounds. In this section, the most used strategies to produce positive NPs will be revised according to the different type of amino surface elements used.2.1 Polymers
In general, the most common and easy way to produce positive NPs is via initial synthesis of conventional negative NPs followed by the subsequent coating with positive polymers, as similarly reported for the well-known layer-by-layer technique. In this case, standard negative citrate NPs can be easily turned into positive NPs by simply adding a solution containing a positive polymer. The most commonly used ones are polyethyleneimine (PEI),23 polyallylamine (PAA),24 poly-L-lysine (PLL),25 polyarginine (PA),26 and chitosan.27,28 This approach can be extended also to non-citrate stabilized NPs that exhibit negative charge on their surface as, for example, Au nanotriangles stabilized with dioctyl sodium sulfosuccinate (AOT)29 or Au nanostars stabilized with polyvinylpyrrolidone (PVP).26 This approach is very easy and effective however such coating process retains, as an intermediate layer between the metallic surface and the external positively-charged shell, the original surfactants. This can pose major limitations on their applicability for surface-dependent techniques like SERS. In addition, this type of coatings can suffer from degradation with time due to polymer detachment. Therefore, great efforts have been performed to achieve the direct synthesis of homogeneous NPs coated and stabilized with these amine ligands. For instance, the direct synthesis of Ag NPs has been achieved using PEI as a stabilizer and ascorbic acid as the reducing agent. The resulting Ag NPs show an average size of around 8.2 nm and the UV-vis extinction spectrum shows a strong plasmon resonance band at 399 nm.30 In a different report, larger Ag NPs of ca. 14 ± 6 nm were produced by simply boiling a AgNO3 solution in the presence of PEI without ascorbic acid.31 Further increase in the size of PEI-coated Ag nanoparticles of around 33 nm, was achieved at room temperature under UV irradiation for 120 min in the presence of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and PEI.32,33Besides PEI, other polymers have been successfully exploited in the direct synthesis of cationic NPs using the same one-pot strategy. For example, the highly biocompatible polypeptide poly-L-lysine (PLL) has been employed to produce Au@PLL the NPs showed in Fig. 1A by simply mixing the gold precursor with PLL while boiling. Fig. 1A also shows the UV-vis absorption spectrum of two different batches of Au@PLL NPs and after eight months. All of them have a LSPR peak at 532 nm without appreciable changes indicating the reproducibility of the synthetic procedure and the long-term stability.34 A similar approach has been used with chitosan, a polysaccharide bioactive polymer with a wide variety of applications due to its functional properties such as antibacterial activity, non-toxicity, ease of modification, and biodegradability. For instance, HAuCl4 and chitosan were combined and heated at ∼95 °C for ∼3 h to generate the corresponding chitosan-coated nanoparticles.35
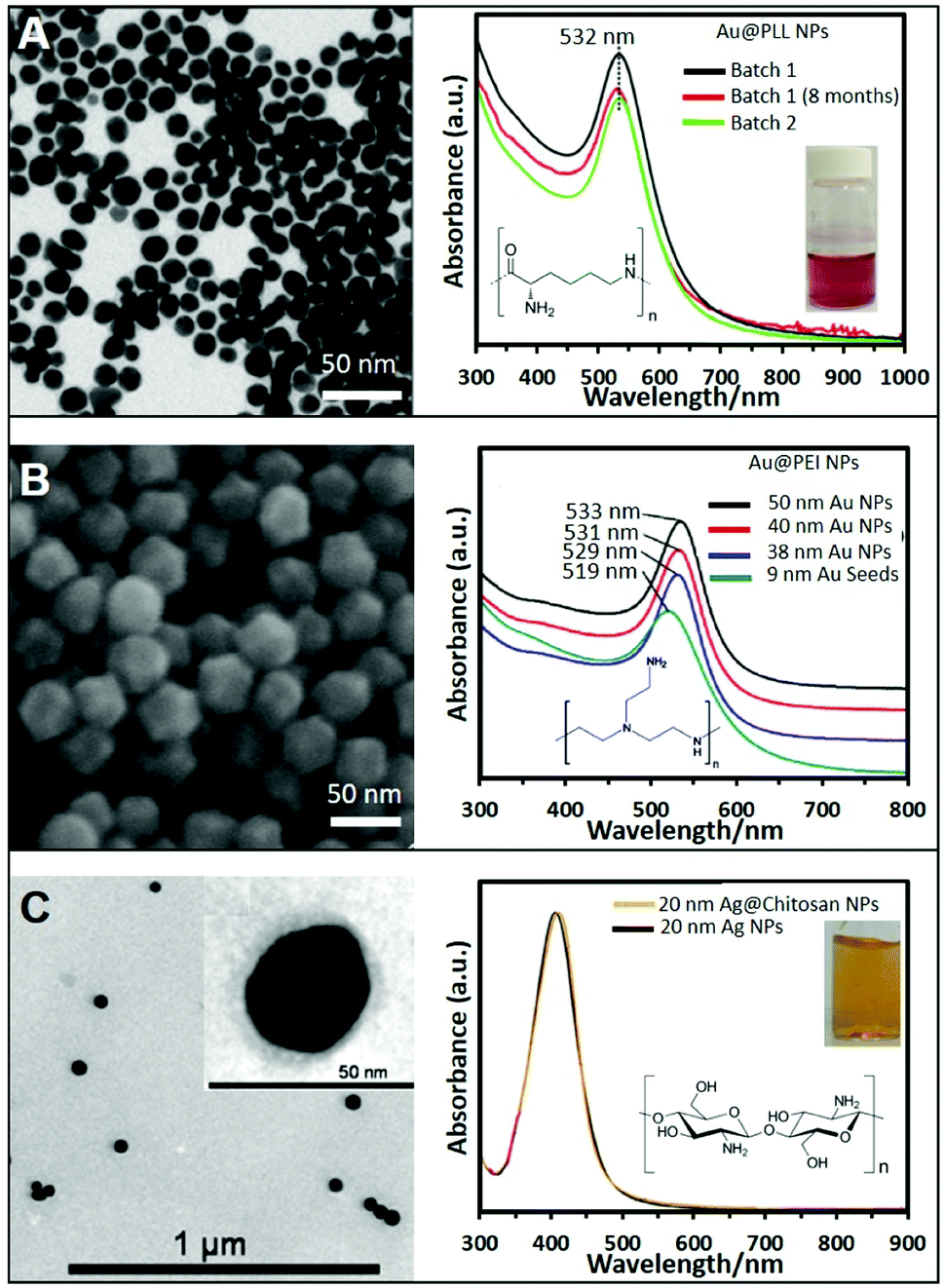 | ||
| Fig. 1 (A) TEM image of gold nanoparticles (Au NPs) stabilized with poly-L-lysine and the corresponding UV-vis spectra showing the reproducibility and stability of the produced NPs; the inset shows a photograph of the colloidal dispersion. Adapted with permission from ref. 34 Copyright 2020, Springer. (B) SEM image of 50 nm Au NPs stabilized with PEI and produced by the seeded growth method. UV-vis absorption spectra of PEI-coated NPs of different sizes (9, 38, 40, and 50 nm). Adapted with permission from ref. 36 Copyright 2018, Elsevier. (C) TEM image of 20 nm chitosan-coated silver nanoparticles (Ag NPs). The chitosan layer can be clearly distinghuised in the inset image. UV-vis spectra of Ag NPs in the presence of chitosan (orange) and without (black). Adapted with permission from ref. 38. Copyright 2011, IOP Publishing Ltd. | ||
While these approaches offer very straightforward routes to produce positive nanoparticles, they typically yield relatively small colloids with subpar SERS-enhancing performances. Therefore, great efforts have been devoted to the fabrication of larger and homogeneous plasmonic NPs via, among others, seed-mediated approaches. For instance, the direct synthesis of well-defined polyhedral gold nanoparticles with various sizes (38, 40, and 50 nm) using PEI as a capping molecule has been successfully achieved using preformed PEI-coated gold seeds of 9 nm. Size tuning was achieved by combining different amounts of seed to a growth solution containing HAuCl4 and PEI (ascorbic acid was employed as the reducing agent). Fig. 1B shows a SEM image of the 50 nm nanoparticles, demonstrating the monodispersity of the produced structures, as well as the red-shift of the LSPR maximum with the nanoparticle size (Fig. 1B).36,37
Another two-step growth method has been proposed to produce chitosan-coated Ag NPs. In the first step, a stock of negatively-charged silver seeds was synthesized via reduction of silver nitrate with sodium borohydride in the presence of sodium citrate. In a second step, an aqueous solution of trisodium citrate, ascorbic acid, chitosan, and seeds was prepared. To this mixture, AgNO3 was added dropwise under continuous magnetic stirring at 0 °C. Fig. 1C shows a TEM image of the so prepared Ag spherical particles with an average diameter of 20 nm. The inset shows a higher magnification TEM image of the stained NPs showing the presence of the thin chitosan outer layer. The optical spectrum of the chitosan-coated NPs shows a LSPR centered at around 410 nm, approximately 5 nm more than the chitosan-free colloids (Fig. 1C), which been associated to the change in the local refractive index of nanoparticles resulting from the polymer coating.38 Following these protocols, it is also possible to produce Ag/Au bimetallic NPs by first synthesizing Ag nanoparticles and then induce a controlled galvanic replacement reaction by the stepwise addition of HAuCl4 while boiling under constant stirring.39 In addition, cationic Ag layers have been also grown on colloidal templates like for instance poly(glycidyl methacrylate) (PGMA) nanoparticles. To this end, PGMA NPs were previously coated with PEI to modify the nanoparticle charge to further grow Ag nanoparticles on the surface by adding a AgNO3 solution and NaBH4.40
2.2 Cationic surfactants
Cationic surfactants like cetyltrimethylammonium bromide (CTAB), cetyltrimethylammonium chloride (CTAC), and, to a lesser extent, benzyldimethyldodecylammonium chloride (BDAC) are widely used to produce gold structures of different geometries and sizes. Seed mediated protocols using these surfactants allow the control over the size and morphology of the produced NPs with a high degree of tunability. For instance, spherical Au NPs from sizes ranging from 50 nm to 1 μm can be produced by preparing a seed solution by reducing HAuCl4 in the presence of sodium citrate with NaBH4. To synthesize Au NPs of different sizes, a specific amount of the as-prepared seeds was added to a growth solution containing CTAB, HAuCl4, potassium iodide, and ascorbic acid. Fig. 2A shows SEM images of the produced NPs and the trend corresponding extinction spectra. As it can be seen, 50 nm NPs show the characteristic dipolar plasmon mode around 532 nm. However, as particle size increases, the band is noticeably red-shifted and broadened. For NPs above ca. 113 nm, a shoulder appears at lower wavelengths which corresponds to the quadrupolar resonance. From particles sizes above 330 nm, scattering effects dominate the spectra.41 This synthetic protocol has been also reproduced replacing CTAB with CTAC, which avoids the use of iodide.42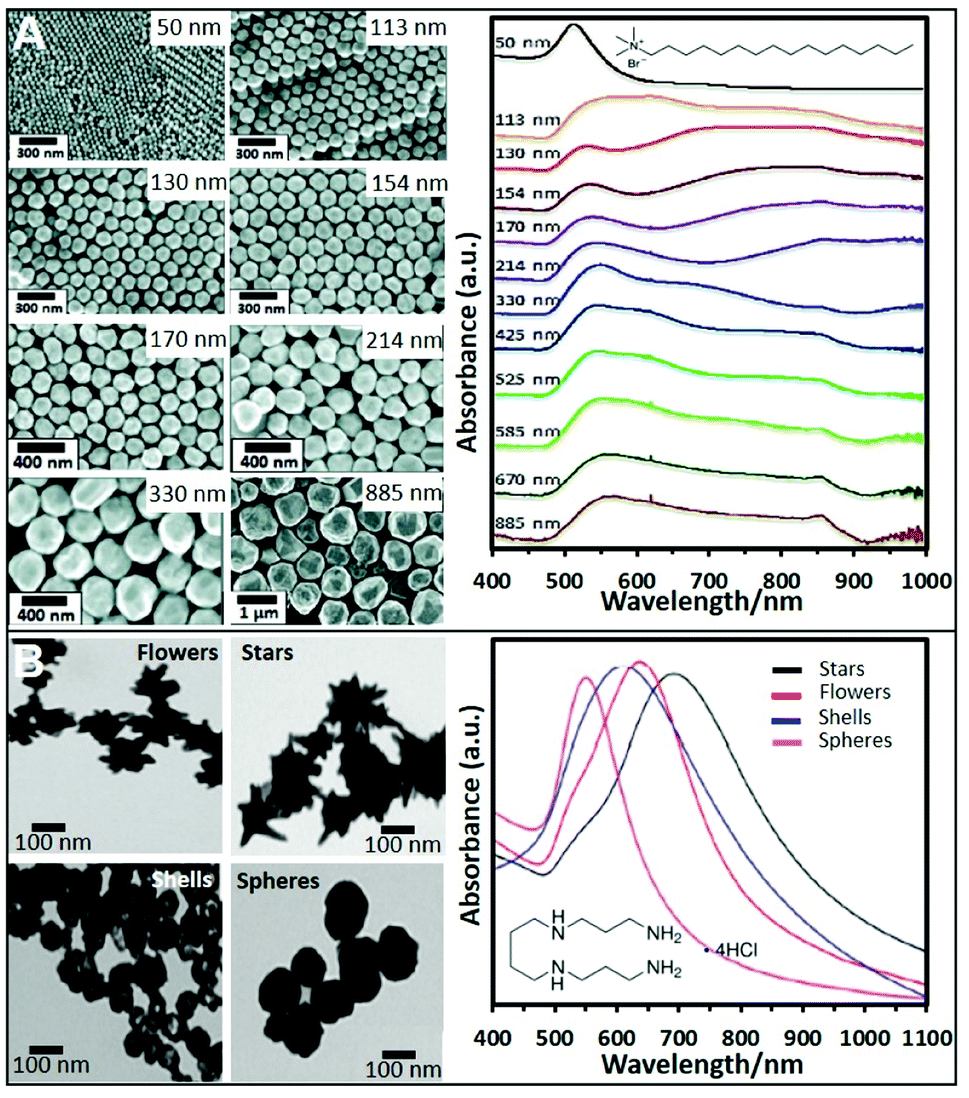 | ||
| Fig. 2 (A) SEM images of CTAB-stabilized Au NPs from 50 to 885 nm diameter size and the corresponding UV-vis spectra. Adapted with permission from ref. 41 Copyright 2012, American Chemical Society. (B) TEM images and corresponding UV-vis spectra of four different nanoparticle morphologies (flowers, stars, shells, and spheres) produced using a seedless and surfactant-free method and subsequently coated with spermine molecules. Adapted with permission from ref. 60. Copyright 2020, American Chemical Society. | ||
CTAB, CTAC and BDAC were also employed in the production of anisotropic NPs such as gold nanorods (NRs),21,43 bipyramids,44 or nanostars (NSt).45 By preparing the seeds in the presence of these surfactants rather than citrate and replacing the iodide with AgNO3 to break the symmetry of the seeds, Au NRs can be obtained.30,31,46 The use of seeds formed in the presence of citrate and CTAC, and aged for 90 min at 80 °C, enabled the formation of bipyramids.44 The production of positive Ag NPs with tunable sizes is also possible using CTAB with a similar approach as for Au. The main difference is that a base like NH4OH has to be added to initiate the reduction process.47–51
Such a class of surfactants forms a bilayer around the NPs which imparts colloidal stability while providing a net positive surface charge. However, nanoparticle aggregation is prevented only for surfactant content above the critical micellar concentration (i.e., when a large content of unbound surfactant molecules is still present in the solution).44 Furthermore, the surfactant bilayer can significantly hinder the ability of target molecules to approximate the metallic surface (the CTAB bilayer has been reported to be around 3–5 nm thick).52
2.3 Small molecules
Short polyamine molecules (e.g., spermine; spermidine, and ethylenedioxydiethylamine) were also used to fabricate positively-charged plasmonic colloids. Among them, spermine (Sp) tetrahydrochloride has been the most widely employed chemical. Sp molecules adhere onto the NP surfaces via electrostatic binding and adopting a flat orientation, which ideally minimizes the separation between the plasmonic materials and the media. This type of NPs is generally prepared by adding NaBH4 to a solution containing AgNO3 and spermine. By performing this approach and adding different amounts of NaBH4 different sizes of Ag NPs can be produced.53–56 For instance, spermine coated Ag NPs of 20, 40, and 60 nm could be obtained. Their corresponding surface plasmon resonance bands showed a redshift with increasing Ag NP size, 394 nm, 400 nm, and 413 nm respectively.57 The corresponding zeta potentials reported in a different work for similar sizes were 13, 8.4, and 21 mV.19Nonetheless, even when reported, the possibility to produce different NPs sizes using spermine remains elusive. Therefore, other strategies using preformed NPs have been also proposed. For instance, citrate-capped Ag colloids were cleaned by centrifugation and mixed with an equal volume of spermine solution, adopting a similar post-modification strategy as for polymer coatings.58 Using this approach, spermine-coated gold nanoparticles of different morphologies (nanostars, nanoflowers, or nanoshells, see Fig. 2B) were obtained.59,60
2.4 Aminothiolated compounds
Besides electrostatic anchoring of the surface ligands onto the metallic nanomaterials, formation of direct covalent bonds has been exploited by using amino molecules equipped with an addition thiol or, to a lesser extent, dithiocarbamate group.Several approaches have been proposed for their direct synthesis. Gold nanoparticles have been produced by the dropwise addition of sodium borohydride to an aqueous solution of tetrachloroauric(III) acid under magnetic stirring at room temperature. Once the solution became red, indicating the NPs formation, a solution of cysteamine hydrochloride was added as stabilizing agent.61,62 The same approach has been used to produce Ag NPs that were further stabilized by the addition of L-cysteine. In this case, sodium borohydride was added dropwise to a silver nitrate solution under stirring.63 Alternatively, positively-charged Au NPs were obtained by mixing first the thiolated molecule 2-aminoethanethiol with the metal precursor (HAuCl4) followed by the addition of NaBH4.64 However, nanoparticles obtained using NaBH4 as a reducing agent are typically small (ca. 12 nm). Larger NPs (ca. 51 nm) were then produced by replacing NaBH4 with heating (cysteamine was added to a boiling solution of HAuCl4 under magnetic stirring). However, in the reported protocol, thiolated polyethylene glycol (SH-mPEG) was added to improve the stability of the synthesized Au NPs.65
Nevertheless, the control over the size and shape of the plasmonic nanoparticles remains very limited by the direct synthesis in the presence of thiols. Thus, the post-functionalization of preformed plasmonic nanoparticles with thio–amino molecules has been often reported to overcome this limitation. In this regard, citrate-capped nanoparticles were largely used as colloidal materials to generate positively-charged colloids by simple mixing, under constant agitation, with aminothiolated molecules such as cysteamine hydrochloride,66–69 diethyldithiocarbamate (DDTC),70 thiocholine,22 or L-cysteine methyl ester hydrochloride.71 Fig. 3A shows an example where Ag was grown over preformed Au NPs and further functionalized with cysteamine. The same protocol with small modifications can be applied to NPs stabilized whit different surfactants or polymers like PVP or CTAB. For instance, PVP–Au NPs need to be centrifuged twice to remove excess PVP before functionalization with the thioamine solution (cysteamine).72 In the case of CTAB, CTAC, or BDAC stabilized NPs, the nanomaterials must be submitted to carefully tuned washing cycles to remove the excess of the surfactants while retaining the colloidal stability. Subsequently, the nanoparticles, such as Au NRs,73 or bipyramids,44 were combined with a cysteamine hydrochloride solution under magnetic stirring at 40 °C. The corresponding SEM and TEM images with their optical characterization are shown in Fig. 3B and C. In the same way, Au NSts prepared with a mixture of CTAB/BDAC were functionalized L-cysteine.45
 | ||
| Fig. 3 (A) High-resolution TEM image of Au@Ag NPs functionalized with cysteamine and their extinction spectrum (those of the AuNP core and non-modified Au@AgNPs were also included). Adapted with permission from ref. 69 Copyright 2020, Elsevier. (B) SEM image and extinction spectra of Au nanorods (NRs) functionalized with cysteamine. Adapted with permission from ref. 73 Copyright 2020, Elsevier. (C) TEM image of Au bipyramids and the monitoring of the ligand exchange process from CTAB to cysteamine via UV-vis spectroscopy. Adapted with permission from ref. 44 Copyright 2021, American Chemical Society. | ||
In Table 1 the most common strategies for the direct synthesis of spherical positive plasmonic nanoparticles are summarized.
| Stabilizing | Metal | Reductant | NP size (nm) |
|---|---|---|---|
| PEI32,33 | Au | NaBH4 | 9 |
| Spermine57 | Ag | NaBH4 | 20–60 |
| Cysteamine61,62 | Au | NaBH4 | 12 |
| L-Cysteine63 | Ag | NaBH4 | 22 |
| PEI31 | Ag | T (90 °C) | 14 |
| Chitosan35 | Au | T (95 °C) | 17 |
| Cysteamine65 | Au | T (100 °C) | 51 |
| PEI30 | Ag | Asc. Ac + T (90 °C) | 8 |
| PEI28,29 | Ag | UV irradiation | 33 |
| PEI32,33 | Au | Asc. Ac + seeds | 37–100 |
| Chitosan38 | Ag | Citrate + Asc. Ac + seeds | 20 |
| CTAB/CTAC41,42 | Au/Ag | Asc. Ac + seeds | 10–100 |
To summarize, post-coating of pre-formed colloids with polymers such as PEI offers a straightforward route to obtain positive nanoparticles with resistance against aggregation. However, the relatively large thickness of the polymeric layer can limit the suitability of the substrates to SERS applications by acting as an efficient spacer from the plasmonic surface, as we will further discuss in the next section. Post-functionalizations with smaller-sized surface ligands such as short amino molecules (e.g.; spermine), which are electrostatically adhering onto the colloids similarly as the polymeric species, or covalently-bound amino-thiolated molecules, facilitate the closer contact of the plasmonic surface with the negatively-charged analyte. Although, it is frequently accompanied by a reduction of the colloidal stability. On the other hand, direct fabrication of positively-charged nanoparticles is typically limited to spherical nanoparticles with, often, relatively small diameters and broad size distributions, except for cationic surfactant-based synthesis (e.g., CTAB). However, in the latter case, the residual surfactant bilayer at the plasmonic surface can, once again, limit the analyte accessibility to the nanomaterial while imparting colloidal stability only for relatively high surfactant concentration in the media (i.e., above the critical micellar concentration). Since there is no ideal positively-charged nanomaterial to be used as universal SERS substrates, each specific application with its distinct requirements will determine the substrate of choice.
Finally, it is also worth stressing that the positively-charged nanomaterials can be integrated into hybrid composites comprising additional building units (e.g., magnetic particles, inert supports, quantum dots, microfluidic chips, etc.) to generate multifunctional materials with synergistic properties. These types of multifunctional nanostructures are generally produced in multistep processes where the individual components are separately synthesized and subsequently assembled.2,68,74,75
3. SERS sensing applications
SERS-sensing with positively-charged nanostructures typically relies on electrostatic binding of the target species with the amino groups of the surface ligand to promote its adhesion onto the plasmonic surface while the detection is mostly achieved via the acquisition of the intrinsic SERS spectrum of the analyte (i.e., direct SERS). However, we will also discuss indirect sensing strategies using positively-charged nanoparticles as SERS-encoded nanoparticles or SERS tags. For the sake of discussion, the studies reported in the literature were arbitrarily classified and discussed according to the size range of the target molecules.3.1 Inorganic anions
Integration of positively-charged plasmonic nanomaterials into SERS sensing schemes for the quantification of inorganic anions of human and environmental concern22,32,33,56,70,76–80 has been extensively investigated in the literature. For instance, Hao et al.79 fabricated a Ag nanofilm on Cu foils which, subsequently, was immersed into a cysteamine (Cys) solution to generate a Cys-self-assembled monolayer (SAM) for the direct SERS detection of perchlorate (ClO4−), a ubiquitous environmental contaminant. The SERS spectrum of Cys (Fig. 4A) displays, among others, an intense band at ca. 635 cm−1, assigned to the C–S stretching of Cys gauche-conformation and a weaker feature at ca. 715 cm−1 ascribed to the Cys trans-conformation, which suggests that the gauche conformer is predominant in the SAM. Immersion into a 50 μM perchlorate solution leads to the appearance of the characteristic ClO4− band at ca. 930 cm−1 (Fig. 4B) which intensity decreases in the presence of an increasing concentration of co-existing anions, such as bicarbonate (Fig. 4B), indicating the following order of selectivity: ClO4− > SO42− > HCO3−, NO3− > Cl− > H2PO4− (Fig. 4C). Shi et al.80 fabricated a positively-charged silver nanowire membrane via post-functionalization of the PVP-capped wire with diethyldithiocarbamate (DDTC, Fig. 4D). The SERS-active, flexible membrane has been used to recover typical oxidizers contained in explosives (perchlorates, ClO4−; chlorates, ClO3−; and nitrates, NO3−) present at the nanogram levels on surfaces via simple surface swab. This enables the rapid on-site SERS detection using a portable Raman spectrometer via the collection of the characteristic bands of ClO4−/ClO3− at 930 cm−1 and NO3− at 1041 cm−1. Contrarily, unmodified-Ag nanowire membranes did not allow the detection of the target analytes.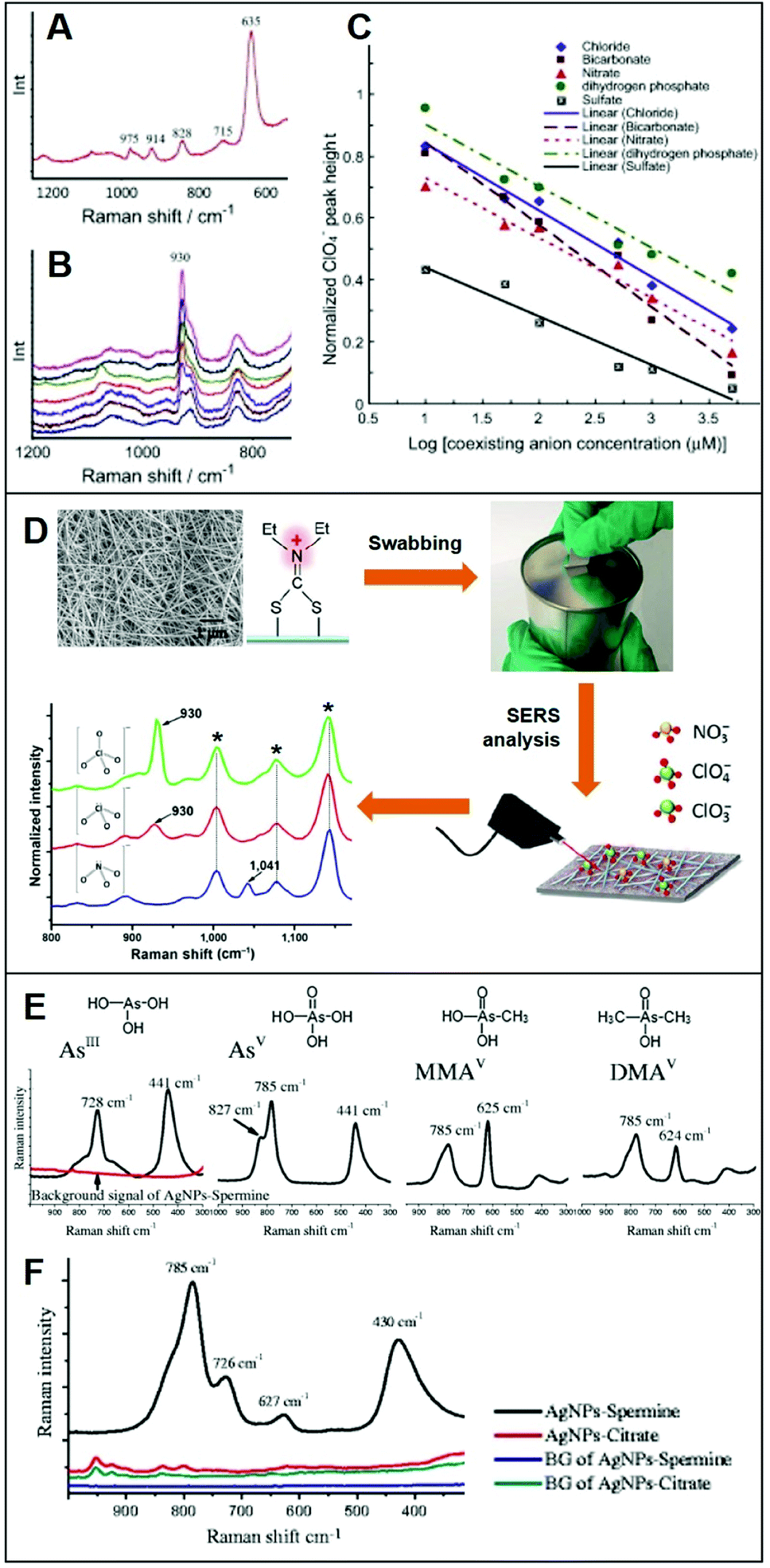 | ||
| Fig. 4 Direct SERS detection of inorganic anions. (A) Background spectrum of the Cys on Ag nanofilm/Cu foil in deionized water. (B) SERS spectra of a 50 μM ClO4− solution in the presence of different concentrations of HCO3− (0, 10, 50, 100, 500, 1000 and 5000 μM, from top to bottom). (C) Normalized ClO4− peak height plotted against concentration of coexisting anions (Cl−, NO3−, SO42−, HCO3− and H2PO4−) in logarithmic scale (log μM). Adapted with permission from ref. 79 Copyright 2015, Elsevier. (D) SEM image of the Ag nanowire membrane and surface modification with diethyldithiocarbamate (DDTC) which is used as a swab to collect trace amounts of ClO4−, ClO3− and NO3− anions onto a surface. SERS analysis of the swabbed membrane onto surfaces with 2.0 μg of ClO4−, 1.7 μg of ClO3− and 1.2 μg of NO3− (asterisks: DDTC bands). Adapted with permission from ref. 80 Copyright 2016, Springer. (E) Individual SERS spectra of fours arsenic species (4 ppm) on spermine-coated silver nanoparticles (AgSp). (F) SERS spectra of a mixture of the four arsenic species (200 ppb) in the cell lysate on negatively-charged citrate silver colloids (AgCit) and AgSp. The background signals AgCit and AgSp were also included. Adapted with permission from ref. 56 Copyright 2017, Springer. | ||
On the other hand, Yang et al.56 exploited spermine-coated silver colloids (AgSp) to detect toxic arsenic species such as arsenite (AsIII), arsenate (AsV), and two biologically relevant metabolic products of arsenic (monomethylarsonic acid, MMAV; and dimethylarsinic acid, DMAV) in cell lysates. Arsenic speciation in biological samples is important to better discern the impact of arsenic toxicity and its mode of action. Fig. 4D shows the SERS spectra of all arsenic species on AgSp colloids, together with the molecular structures of each analyte. For comparison, the authors also tested negatively-charged citrate silver colloids (AgCit) of similar size, showing that arsenic speciation in cell lysate can only be achieved on positively-charged colloids, while AgCit do not yield distinguishable SERS signals under the applied experimental conditions (Fig. 4E illustrates the SERS spectra obtained by a mixture of the arsenic species combined with the two colloids).
3.2 Small organic molecules
In addition to inorganic anions, a large set of small negatively-charged organic molecules such as explosives,22,48,68,70,71,81–83 pollutants,28,66,73,84,85 and pigments25,31,44 has been targeted by SERS-sensing approaches using positively-charged plasmonic nanomaterials.As previously discussed, CTAB is a surfactant commonly used as a shape-directing agent to yield asymmetric nanoparticles with tunable and improved plasmonic responses as compared to the traditional spherical counterparts. While imparting an overall positive charge, the CTAB bilayer at the metallic surface also negatively impacts the ability of analytes to closely approach the plasmonic nanostructures and, thus, the possibility of acquiring intense SERS spectra of the molecular target. In this regard, an illustrative example is provided by the work of Amin et al.,44 who first synthesized CTAB-capped gold nano-bipyramids (BP–CTAB) and then submitted to ligand exchange with cysteamine (Cys) to generate the corresponding BP–Cys materials (Fig. 5A). The addition of negatively-charged pigments (e.g., acid blue) to the colloidal suspensions causes the electrostatic-induced aggregation of the nanoparticles and the formation of interparticle hot-spots that boost the SERS signal (Fig. 5B). While characteristic acid blue bands are observed on both colloids, their intensities are much larger for BP–Cys (Fig. 5C and D) which is consistent with the thinner spacing layer determined by the self-assembling of small Cys molecules on the gold surface.
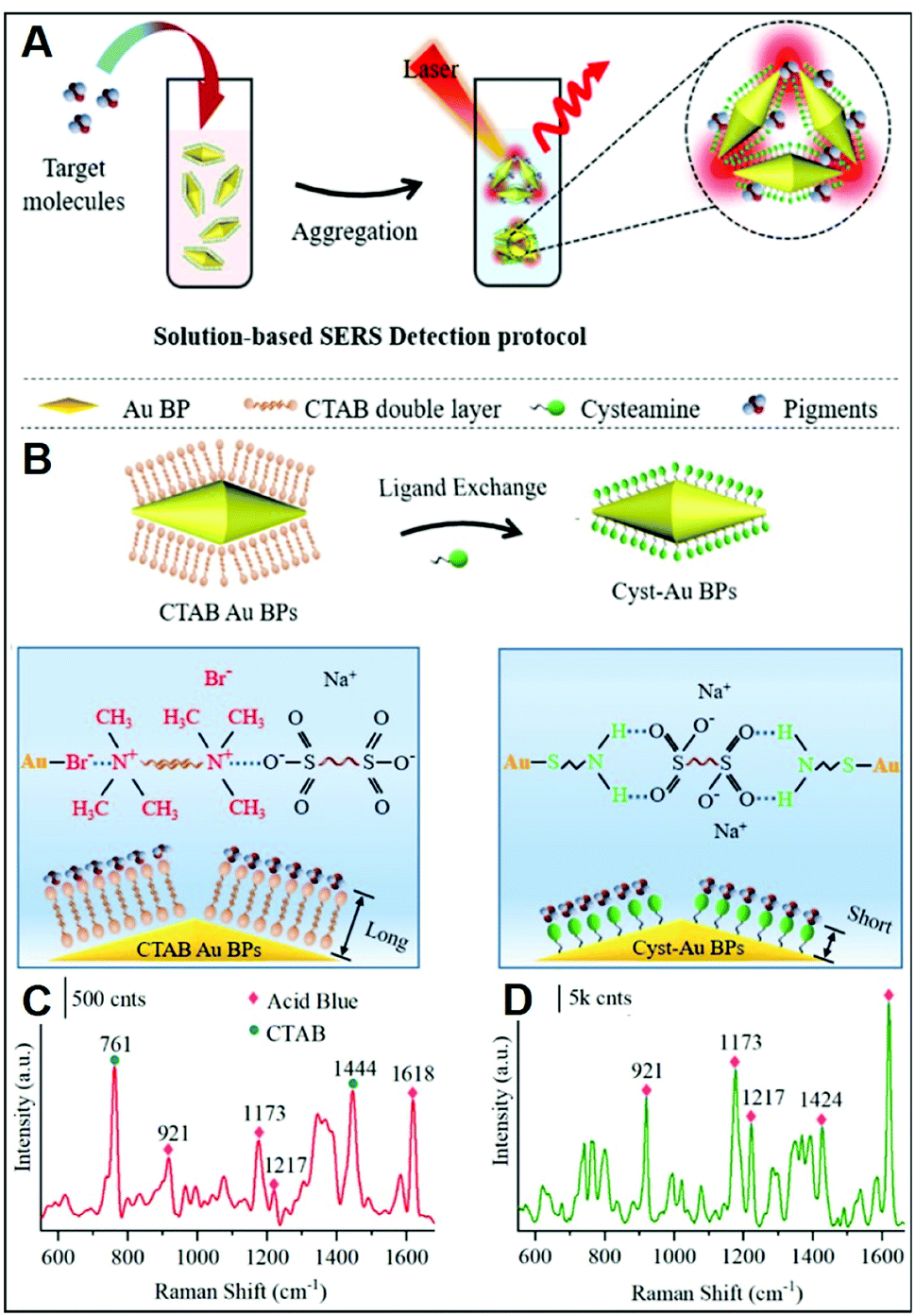 | ||
| Fig. 5 Direct SERS detection of pigments. (A) Outline of the sensing scheme for pigment detection using positively-charged gold nano-bipyramids (AuBPs). (B) Surface modification of CTAB-capped AuBPs with cysteamine. (C and D) SERS spectra of acid blue (1 ppm) using CTAB-capped (C) and cys-Au BPs (D) together with an illustration of the electrostatic interaction with their respective surface molecules. Adapted with permission from ref. 44 Copyright 2021, American Chemical Society. | ||
Adopting an analogous approach, Weng et al.73 replaced the CTAB bilayer on gold nanorods (NRs) with Cys for the SERS detection of the toxic pesticide acephate in rice samples over a broad dynamic range of concentration and down to 0.5 mg L−1. In this case, however, NR–Cys colloids were combined with a solution extracted from acephate-contaminated rice and dropped on a silicon chip. SERS spectra were then continuously acquired during the drying process, which can be broadly separated into three distinct stages: wet state, critical state, and dry state (Fig. 6A). Maximum SERS intensities, for 785 nm excitation laser, are observed during the second stage, which has been attributed to the peculiar geometrical organization of the NR–Cys into a 3D long-range-ordered array that optimizes the generation of closely-spaced interparticle gaps. SERS spectra acquired during the critical state display, among others, characteristic features of acephate centered at ca. 358, 400, 560, and 692 cm−1. Multivariant methods in machine or deep learning were finally exploited by the authors to establish regression models for fast automatic analysis (ca. 5 min) of acephate levels in rice samples (Fig. 6A). In a recent study, Gao et al.68 generated core-satellite structures via electrostatic assembling of cysteamine-modified gold nanoparticles (core) with negatively-charged mercaptopropionic acid-capped CdTe quantum dots (satellites, QDs), which causes the QD photoluminescence quenching due to energy transfer (Fig. 6B). The addition of the trinitrotoluene (TNT) explosive selectively promotes the detachment of the satellites from the plasmonic core as a result of the formation of the strong cysteamine–TNT complex. Such disassembly is reflected in the TNT concentration dependent changes of the extinction spectra and fluorescent intensity of QDs (Fig. 6C and D), thereby facilitating the naked-eye detection of the explosive. On the other hand, the formation of the cysteamine–TNT complex triggers gold nanoparticle aggregation and the consequent enhancement of the SERS signal (Fig. 6E), which also scales linearly with the target concentration. By combining the trimodal responses, it is possible to achieve an outstanding linear dynamic range of concentration (from 10 fM to 200 μM) with a limit of detection (LOD) of 3.2 fM. The viability of the multichannel sensing platform was validated by TNT detection in environmental and biological specimens such as liquor, fruit, clothing, and soil.
 | ||
| Fig. 6 Direct SERS detection of small hazardous molecules. (A) Outline of the SERS detection of acephate in rice using dynamic SERS analysis in combination multivariant methods and cysteamine-modified gold nanorods as SERS substrates. Adapted with permission from ref. 73 Copyright 2020, Elsevier. (B) Depiction of the tri-modal sensing platform (colorimetric, fluorescence, and SERS) consisting of negatively-charged satellites (mercaptopropionic acid-capped CdTe quantum dots, QDs) assembled on a positively-charged plasmonic core (cysteamine-modified gold nanoparticles, GNP) for TNT analysis: extinction (C), photoluminescence (D) and SERS (E) spectra of GNPs-QDs assemblies in the presence of increasing concentrations of TNT (0–200 μM, 0–6000 nM and 0–100 nM, respectively). Adapted with permission from ref. 68 Copyright 2021, Elsevier. | ||
3.3 Large biomolecules
The use of positively-charged nanoparticles as SERS platforms is particularly suited for the analysis of nucleic acids as the negatively-charged phosphate backbone of the biomolecule acts as an electrostatic glue promoting nanoparticle clustering. Such an interaction selectively locates the target sequences at the interparticle gaps (i.e., hot-spots) to maximize their SERS response while simultaneously generating, under optimized conditions, stable clusters in suspension (Fig. 7A). This also avoids the need of using external aggregating agents, as in the case of conventional negatively-charged colloids, which negatively affects the reliability of the SERS outcome mostly due to the poorly controlled and time-dependent colloidal aggregation causing signal fluctuations.5 In a seminal work, van Lierop et al. prepared silver nanoparticles coated with a wide range of positively-charged ligands (including PLL, PEI, CTAB, spermine, spermidine, and ethylenedioxydiethylamine) and tested their performance in the SERS detection of 5-carboxyfluorescein labeled single-stranded DNA. Among different substrates, spermine-coated nanoparticles (AgSp) allowed the acquisition of much more intense SERS signals with the lowest LOD (1 nM), which has been attributed to, on the one hand, the smaller molecular size as compared to polymers and CTAB layers (i.e., shorter interparticle gaps with larger EM enhancements) and, on the other hand, to the higher affinity for DNA binding as compared to other small amines. On these bases, AgSp were subsequently used as plasmonic substrates in the label-free SERS analysis of nucleic acids, demonstrating their ability to yield very intense and well-defined SERS spectra with an unprecedented level of batch-to-batch reproducibility at the nanogram per mL level. As emphasized in the comparison of the SERS spectra of a single and double-stranded DNA (ssDNA and dsDNA, respectively) reported in Fig. 7A, the obtained spectra provide an accurate and reliable structural fingerprint of the analyzed sample which allows for effective discrimination and classification between nucleic acids (a vibrational band assignment can be found in ref. 5). Our group pioneered this approach11,18,86–93 and implemented it in a variety of studies focusing, among others, on the discrimination and quantification of hybridization events and nucleobase modifications,11,91 structural classification of diverse classes of RNAs,88 quantification of nucleobase content,89 characterization of DNA interaction with exogenous molecules,92 etc. In particular, we demonstrated the viability of this method for being integrated into clinical applications differentiating clinically relevant point mutations in 141-nt K-Ras oncogene segments (Fig. 7B).18 In combination with partial least-squares discriminant analysis (PLS-DA), a well-established statistical classification method, we were able to discriminate diverse patterns for SERS spectra of large ssDNA strand with single-point mutations. Differently to short sequences which linearly extends over the metallic surface, AgSp nanoparticles function as compaction agents for long strands stimulating their folding into different structural forms (A- and B-forms, and a combination of thereof) depending on the specific nucleobase sequence and composition, as consistently suggested by the spectral changes of diagnostic backbone conformation bands. This study was conducted in buffered solutions while, in clinical samples (e.g., blood, urine, saliva), nucleic acids typically coexist, at very low concentrations, with a complex mixture of other molecules that compete for adsorption onto the plasmonic surface. For this reason, upstream amplification processes (e.g., polymerase chain reaction, PCR) are necessary to selectively enrich the target sequences while reducing their lengths and separate it from the biological media.19,57,60,94 Trau and co-workers implemented such an approach by utilizing multiplex reverse transcription-recombinase polymerase amplification (RT-RPA) to enrich prostate cancer (PCa) RNA biomarkers (T1E4 and RN7SL1) from urine samples.19 As outlined in Fig. 7C, the target RNA sequences are amplified as ca. 200 bp dsDNA amplicons, which are separately interrogated by SERS using AgSp colloids. Multivariate statistical analysis of the data allowed the risk stratification of PCa in 43 patient urinary samples with high specificity, sensitivity, and accuracy. In a second study,57 the authors successfully extended such a method beyond the proof-of-concept stage to validate the clinical performance of the nanodiagnostic tool by evaluating the clinical metrics with a well-suited cancer risk stratification scoring model. Similarly, Liu et al.60 implemented polymerase chain reaction (PCR), direct SERS interrogation and principal component analysis-linear discriminant analysis (PCA-LDA) to identify and classify BRAF wild type (WT) and V600E mutant genes, firstly, from both genome DNA (gDNA) and cell-free DNA (cfDNA) collected from cell culture media and, subsequently in plasma samples. In this case, however, the authors tested the SERS enhancing performances of different spermine-coated plasmonic nanoparticles (i.e., including gold or silver nanospheres, nanoshells, nanoflowers, and nanostars). Under the reported experimental conditions, gold/silver nanostars showed the highest SERS activity for DNA detection.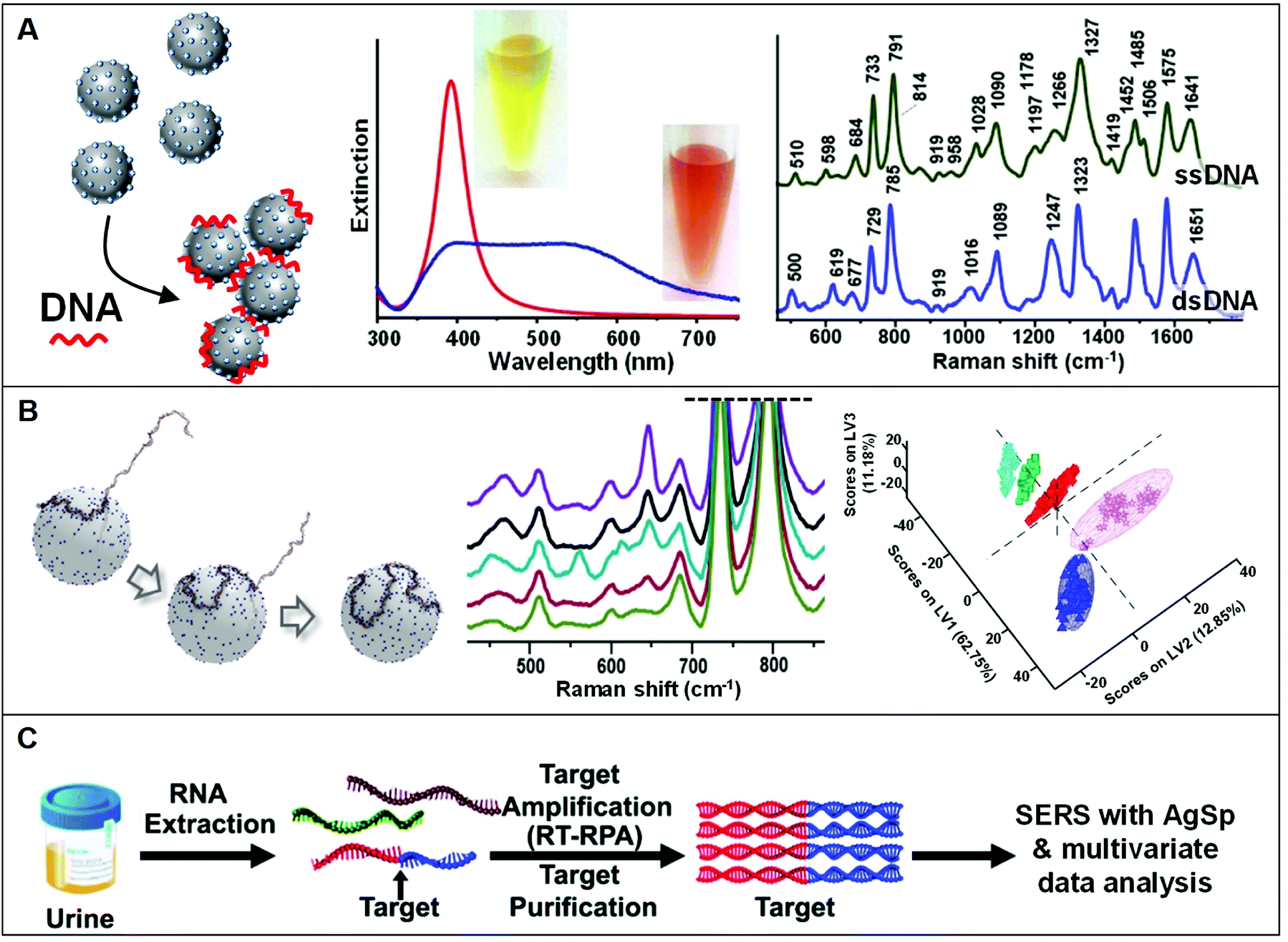 | ||
| Fig. 7 Direct SERS detection of nucleic acids. (A) Schematic of the DNA-mediated assembly of spermine-coated silver colloids (AgSp) into stable clusters in suspension. Adapted with permission from ref. 11 Copyright 2015, Wiley-VCH. Examples of extinction spectra of AgSp before (red curve) and after (blue curve) the addition of a 21-base pair dsDNA (optical images of the colloids are also included). SERS spectra of the ssDNA = CATCGCAGGTACCTGTAAGAG, and the dsDNA consisting of the mentioned ssDNA and its complementary strand. Adapted with permission from ref. 15. Copyright 2021, MDPI. (B) Molecular dynamics simulation of a 141-nt ssDNA adsorption and wrapping around a AgSp nanoparticle at low salt concentration. SERS spectra of 141 nt ssDNAs with single and double-point mutations, and corresponding partial least-squares discriminant analysis (PLS-DA). Adapted with permission from ref. 18 Copyright 2017 WILEY-VCH. (C) Outline of the coupling of a nucleic acid amplification method to direct SERS analysis with AgSp colloids and chemometrics. In this case, total RNA content was initially extracted from urinary samples followed by reverse transcription-recombinase polymerase amplification (RT-RPA) of specific RNA biomarkers to yield ∼200 bp dsDNA amplicons, which were discriminated via SERS using spermine-coated silver colloids in combination with multivariate statistical analysis. Adapted with permission from ref. 19 Copyright 2017 Royal Society of Chemistry. | ||
In addition to nucleic acids, Liu et al.53 also employed AgSp colloids for monitoring protein kinase A (PKA) activity in cell extracts. PKA-induced phosphorylation selectively alters the net charge state of a dye-labelled peptide (TAMRA-LRRASLG) which promotes the electrostatic interaction with silver nanoparticles and their aggregation. The intensity of the SERS spectra of the TAMRA dye quantitatively correlates with PKA concentration in a wide range of 0.0001–0.5 U μL−1 with a LOD equals to 0.00003 U μL−1.
It is also worth noting that the structural and functional plasticity of dsDNA to simultaneously yield stable clusters of AgSp nanoparticles in suspension and non-covalently interacting with small aromatic molecules via intercalative binding was exploited in the simultaneous quantification of Fe(III) and Al(III) in tap water.95 Specifically, in this study, dsDNA complexation with the anthraquinone derivative alizarin red S (ARS) prevents the direct interaction of ARS with the plasmonic surface while locating it close enough to the silver surface to yield intense SERS signals. In this scenario, the ARS chelating properties towards iron and aluminum ions are retained and well-revealed in large changes in the SERS spectral profiles, which can be quantitatively correlated with the ion concentration. In the absence of dsDNA, ARS molecules directly adhere onto the AgSp surfaces through the same coordination sites for metal ion binding, thereby hindering their use as SERS chemosensors.
3.4 Pathogens and vesicles
Conventional methods for microorganism detection (e.g., culture-based approaches) are laborious and very time-consuming, while other molecular diagnostic tools (e.g., ELISA, PCR) suffer from high cost and/or low sensibility. Thus, SERS has been explored as an analytical tool for the rapid, low-cost, and sensitive detection of pathogens (e.g.; bacteria, fungi), adopting diverse indirect96,97 and direct sensing strategies, along with positively-charged nanoparticles.27,34,38–40,46,47,50,51,75 A representative workflow to carry out direct SERS analysis of pathogens using AgNPs is outlined in Fig. 8A. In this work, Chen et al.51 use CTAB-coated silver nanoparticles for the identification of methicillin-resistant Staphylococcus aureus (MRSA) as well as discrimination of 6 microorganisms (S. aureus 29213, S. aureus 25923, C. albicans, B. cereus, E. coli, and P. aeruginosa). A pathogen suspension prepared with ultrapure water was incubated for 30 min with positively charged nanoparticles, which self-assemble on the microorganism membrane via electrostatic adhesion (conversely, citrate-capped silver nanoparticles poorly accumulate at the pathogen surface). Samples were then dried on a silica chip to be investigated by SERS, yielding the corresponding spectra which contain an ensemble of features ascribed proteins, lipids and DNA, mostly arising from membrane components. Due to the spectral similarity between pathogens, chemometric methods (e.g., partial least squares discriminant analysis, PLS-DA) are appropriately implemented for data processing and accurate spectroscopic discrimination of different microorganisms. Adopting a similar sensing scheme, two different species of Cryptococcus (Cryptococcus neoformans and Cryptococcus gattii) with different pathogenicity, diagnosis, and treatment were accurately discriminated using CTAB-coated silver nanoparticles and direct SERS analysis in combination with orthogonal partial least-squares discriminant analysis.47 In another study, discrimination of three bacteria (Escherichia coli, Salmonella typhimurium, and Bacillus subtilis) was achieved with excellent reproducibility by using PEI-coated bimetallic Ag/Au nanoparticles. Here, bacteria were drop-cast onto a slide and then exposed to the nanoparticles to yield intense SERS signals.39 Interestingly, chitosan-coated silver nanoparticles mixed with two strains of Gram-positive Staphylococcus aureus have been shown to exhibit a synergistic antibacterial activity.38 Direct SERS analysis is then used to monitor the resulting biochemical changes in the bacterial cell wall.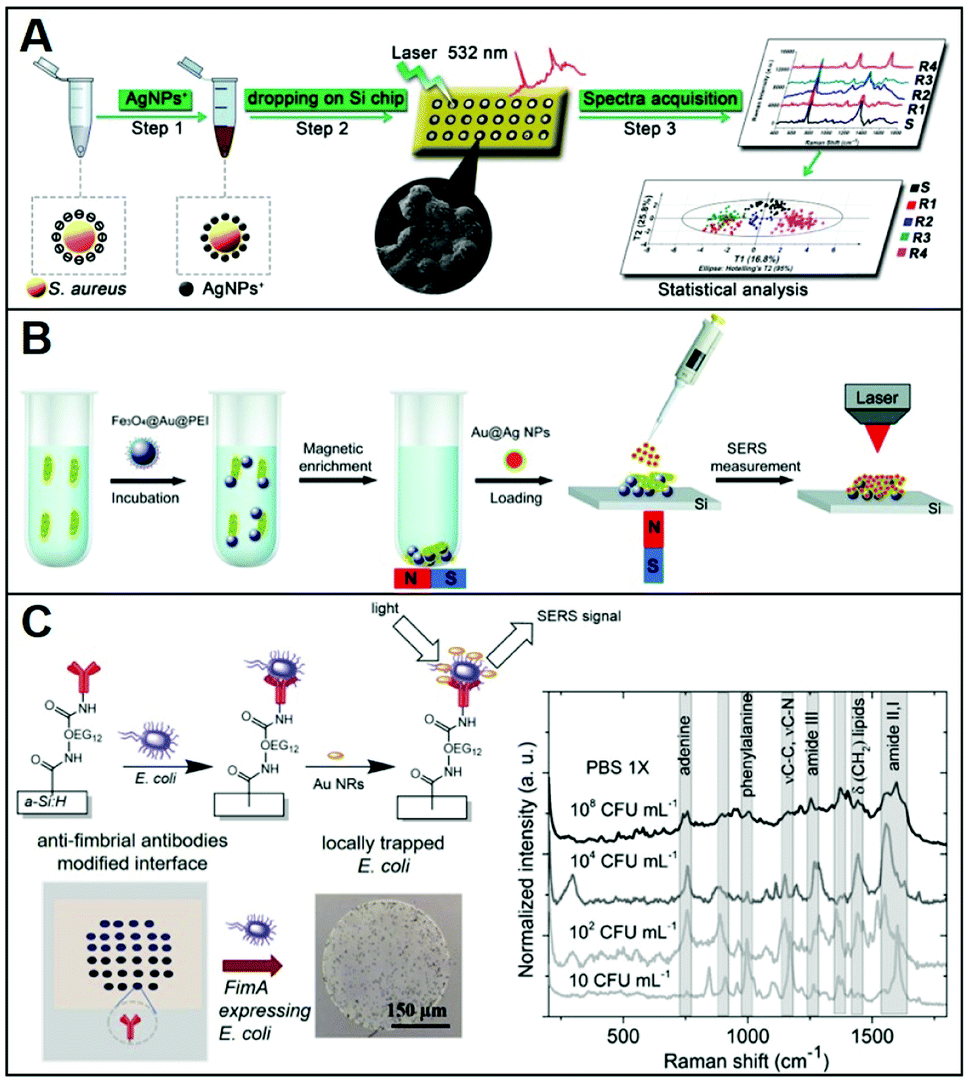 | ||
| Fig. 8 Direct SERS identification of pathogens. (A) Schematic of the direct SERS analysis of pathogens using positively-charged plasmonic nanoparticles which involves the electrostatic accumulation of the nanoparticles at the cell membrane, the deposition of the sample onto a solid support, and subsequent SERS interrogation. Chemometric methods are used for data analysis. Adapted with permission from ref. 51 Copyright 2019, Springer. (B) Outline of the magnetic/SERS sensing strategy for bacteria detection. Step 1: Fe3O4@Au@PEI are used to capture and concentrate pathogens from the media via electrostatic interactions. Step 2: acquisition of the pathogen SERS spectra via deposition of a compact layer of citrate-capped Au@Ag nanoparticles on dried Fe3O4@Au@PEI–bacteria samples. Adapted with permission from ref. 75 Copyright 2016, Royal Society of Chemistry. (C) Bacteria selective capturing at hydrogenated amorphous silicon surfaces modified with anti-fimbrial antibodies and SERS detection via electrostatic adhesion of CTAB-caped gold nanorods. The illustration also includes optical images of the anti-fimbriae modified array and the spots after interaction with bacteria. Averaged and normalized SERS spectra of captured E. coli K12 MG1655 extracted from SERS mapping of the trapped bacteria at different E. coli concentrations (108, 104, 102 and 10 CFU mL−1) in PBS 1×. Adapted with permission from ref. 46 Copyright 2020, Elsevier. | ||
Magnetic functionalities were also integrated into the SERS sensing schemes to facilitate the separation and accumulation of the pathogens at a specific point of the slide while removing the need for time-consuming and complex procedures.50,75 Notably, amine-functionalized magnetic particles have shown to be used as effective capture agents for microrganisms such as bacteria over a wide pH range (5.0–8.0).98 For instance, PEI-coated magnetic particles (Fe3O4@PEI) were used to electrostatically capture Candida species causing fungal infections (Candida albicans, Candida tropicalis, and Candida krusei) from serum with high efficiency (step 1) and, once separated from the media, mixed with CTAB-coated silver nanoparticles and, finally, deposited on a silicon wafer for SERS analysis (step 2).50 Multivariate data analysis was used to accurately discriminate among the Candida species; the whole test can be completed within 40 minutes.
Alternatively, Wang et al.75 afforded plasmonic properties to the magnetic microbeads by integrating an external layer of gold nanoparticles prior to the final PEI-coating (i.e., Fe3O4@Au@PEI). These substrates simultaneously allow for the rapid separation and accumulation of bacteria from the media and their SERS detection (Fig. 8B). However, the obtained LODs were relatively high (ca. 1 × 105 cells per mL for E. coli) thus an additional step was integrated to enhance the sensitivity of the platform. Specifically, a concentrated suspension of citrate-capped Au@Ag nanoparticles dissolved in ethanol was deposited on the dried sample (i.e., Fe3O4@Au@PEI–E. coli complexes). The increased Au@Ag nanoparticles content and the use of ethanol as a solvent ease the formation of a densely packed layer of nanoparticles around the beads thanks to the rapid evaporation of the solvent. With such an increased density of hot-spots around the captured bacteria, Gram-positive bacterium E. coli and Gram-positive bacterium S. aureus can be detected at LOD as low as 103 cells per mL. Validity of the method was also demonstrated in tap water and milk spiked with bacteria, while the optimization of the experimental conditions allowed achieving rapid bacterial detection within 10 minutes (principal component analysis, PCA, was applied in this work).
Otherwise, for selective capturing of a specific class of bacteria from biological samples, appropriate antibodies, when available, can be used. In this regard, Andrei et al.46 fabricated a biochip consisting of a glass substrate coated with a thin film of hydrogenated amorphous silicon, which has been conjugated with anti-fimbrial E. coli antibodies for the specific trapping of E. coli strains containing type-1 fimbriae. The capturing surface was also modified with CH3O–PEG750, an oligoethylene glycol molecule that imparts anti-fouling character (Fig. 8C), preventing unspecific adhesion. In a fluid cell, the bacteria containing suspension is put into contact with the biochip surface through cycles of flow and static contact stages to maximize bacteria trapping. Subsequently, CTAB-capped gold nanorods are deposited on the support and a SERS mapping is performed on the dried sample. The bacterial vibrational fingerprint of E. coli is preserved in the biochip configuration. As shown in the SERS spectra in Fig. 8C, characteristic features ascribed to amide bands, phenylalanine stretching deformation of lipidic C–H bonds, and adenine vibrations are clearly discernible. The applicability of the method was demonstrated in artificial urine spiked with E. coli K12, where reproducible SERS fingerprints of the bacteria have been acquired down to 10 colony-forming units (CFU) per mL, far below the clinical threshold, in less than 3 h.
The electrostatic interactions of positively-charged nanostructures and negatively-charged biological membranes have been also exploited in the direct SERS analysis of exosomes in combination with multivariate analysis.2,59,99–101 Exosomes are heterogeneous single-membrane vesicles with diameters between 30 and 150 nm and secreted by almost all cells, which are emerging as one of the most compelling biomarkers in liquid biopsies for early cancer diagnosis, prognosis, and treatment monitoring.2 Among others, Shin et al.99 functionalized a compact film of gold nanoparticles with cysteamine to favor the accumulation of exosome suspension at the SERS mapping areas. In addition to the spectral differentiation between normal (HPAEC) and lung cancer (H1299, PC9) cell lines, the molecular origin of the Raman markers at the basis of such discrimination was elucidated, indicating the membrane protein EGFR as the primary source of exosomal differentiation. Analogously, Rojalin et al.101 used cysteamine to impart a positive charge on silver nanoparticles integrated onto microscale biosilicate materials for SERS analysis of ovarian and endometrial cancer exosomes. On the other, Liu et al.59 employed spermine-coated gold nanostars to acquire the SERS fingerprint of vesicles from different isolation protocols, batches, and cell origins, demonstrating the viability of such an approach to correctly classify the vesicles obtained using three different isolation methods.
3.5 Cell SERS imaging
The molecular specificity, sensitivity, and high special resolution of SERS are particularly suited for living cell imaging and the dynamic monitoring of biological processes at the cellular and subcellular level through the use of appropriately designed biocompatible plasmonic nanoparticles.102 To this end, nanoparticle surface properties are, among others, tuned to afford long-term stability in cell media and efficient cellular internalization, a feature that is strictly related to nanomaterial surface charge.24,26 As a representative example, we will discuss the work performed by Liz-Marzan and co-workers,26 in which high-resolution 3D SERS imaging of the intracellular pH was performed in living MCF7 breast cancer cells. The pH-responsive plasmonic nanosensor comprised gold nanostars (AuNSt) functionalized with 4-mercaptobenzonic acid (4-MBA) as a pH-sensitive molecule and subsequently wrapped with an external layer of the cationic poly-L-arginine hydrochloride (PA) polymer via electrostatic interactions (Fig. 9A). The cationic polymer shell equips the pH-SERS sensitive probes with high colloidal stability and biocompatibility as well as a higher degree of cellular internalization as compared to other classes of protective layers yielding neutral or negative charge due to the ability of PA to adhere onto the cell membrane via electrostatic interactions and facilitate endocytosis and translocation in cells.103 Fig. 9B shows the pH-dependent MBA SERS profiles of the nanosensors in cell culture media. Most notably, protonation and deprotonation of the carboxylic group make COO− stretching modes at 1415/1430 cm−1 particularly sensitive to changes in pH, contrarily to the intense 1081 cm−1 feature, mainly associated with ring breathing of the phenyl moiety. Thus, the intensity ratio between the sum of the 1415 and 1429 cm−1 contributions and the 1081 cm−1 band was chosen as the spectral marker of the proton concentration and plotted in Fig. 9C as a function of pH. Interestingly, the PA coating also improves the quality of the pH response curve (i.e., milder slope in the 4–8 pH range), which has been tentatively ascribed to the effect of the PA permeability on the diffusion of ions as well as the structural impact that the PA layer may have on the underlying 4MBA molecules. Then, MCF7 cells were exposed to the nanoparticles for 2 or 8 hours and 3D imaging was carried out at 2 μm separated x–y planes along the orthogonal z-axis. Fig. 9D shows the corresponding stacked 2D images of SERS intensities which localize the 3D distribution of the nanoparticles at the levels 3–5, in agreement with the volumetric height of a live MCF7 cell (ca. 6–8 μm). Nanoparticle cellular distribution is visualized in Fig. 9D by plotting the number of pixels with distinguishable SERS intensity at 1081 cm−1 at the different x–y planes. Furthermore, the pH value is also extracted from the SERS spectra as previously discussed. Direct comparison of results at t = 2 h and 8 h suggests a progressive motion of the nanoparticles from the cell membrane toward the perinuclear area which is accompanied by a drop of the environmental pH from 6–7 during the first stage of cellular endocytosis (endosomal compartment) to 4.5–5, when nanoparticles are entrapped into mature lysosomes.104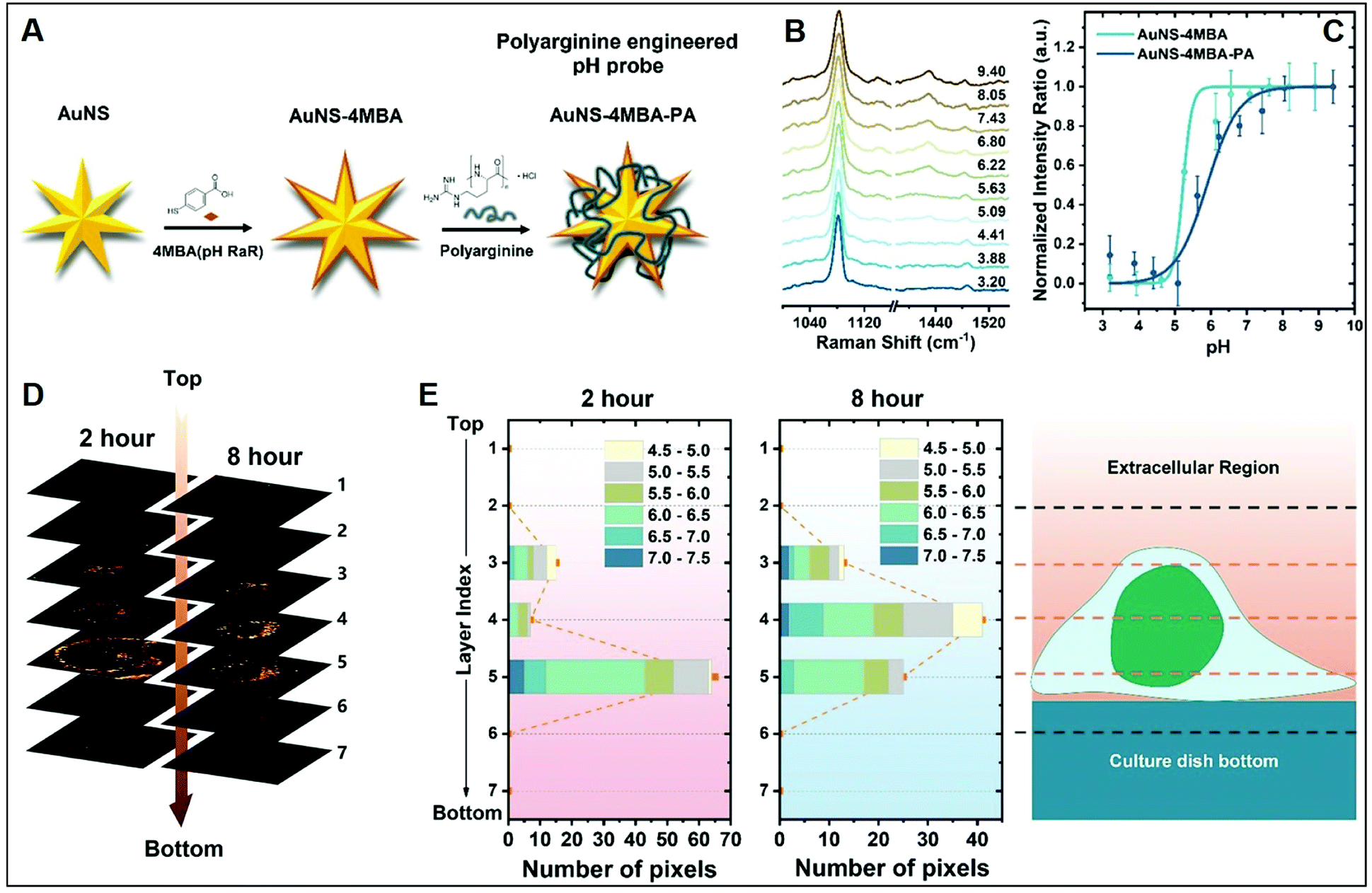 | ||
| Fig. 9 Cell SERS imaging. (A) Fabrication scheme of the preparation of pH-sensitive plasmonic sensor comprising gold nanostars (AuNS) functionalized with 4MBA (4-mercaptobenzoic acid) as the pH-sensitive molecular reporter and wrapped with polyarginine. (B) pH-dependent SERS spectra of the probes in complete cell culture media. (C) SERS intensity ratios (I1425 + I1429)/I1081 vs. pH of the cell culture media for MBA-functionalized AuNSt with and without PA coating. (D) Reconstruction of 3D SERS imaging of the internalization of the nanoparticles into a single MCF7 cell at different x–y planes separated by 2 μm along the orthogonal z-axis upon 2 and 8 hours incubation. The brightest areas correspond to larger nanoparticle accumulations. (E) pH range distribution at the different x–y planes as a function of pixel number. Adapted with permission from ref. 26 Copyright, American Chemical Society, 2020. | ||
On the other hand, Sujai et al.24 showed that, while imparting a positive charge to nanoparticles enhances their cellular internalization, it depresses their ability to penetrate within multilayered cell assemblies. In this work, the authors synthesized SERS-encoded gold nanoparticles coated either with thiolated-polyethylene glycol (HS-PEG) or polyallylamine (PAA) to afford colloidally stable negative and positive materials, respectively. Uptake efficiency was monitored via SERS in monolayer (2D) cells and multi-layered spheroids (3D), as a model system for living tissues. Positively-charged NPs demonstrated their faster internalization via an endocytic pathway in monolayer cells but ca. a 9 times lower penetration capability in cell spheroids as compared to negatively-charged SERS-encoded NPs. This has been attributed to the electrostatic repulsion between negatively-charged cell membranes (and other biological macromolecules) and negatively charged SERS-encoded NPs, which would propel the diffusion of the particles through the interstitial spaces and, eventually, their internalization into deep-seated cells. The results clearly highlight the importance of the rational design of the nanomaterials to be used as efficient nanotheranostic agents. In fact, positively-charged NPs appear to be the optimal candidates for rapid uptake by small sized tumors, while negatively-charged NPs are expected to penetrate deeper into the core of larger tumors.
4. Conclusions
In this review, we summarized and discuss the main procedures to prepare positively-charged plasmonic nanomaterials via surface functionalization with amine ligands of very different nature and size, using different synthetic strategies. Successively, a comprehensive list of studies integrating such substrates for SERS sensing was also examined and liberally classified according to the size of the target analyte which structural properties outline, by and large, the class of nanomaterial and the respective type of amino surface ligands that are more suited for the specific purpose. Future challenges regarding the engineering and exploitation of this sub-class of nanomaterials largely overlap those of all substrates designed for analytical SERS applications, most notably (i) the affordable production of reliable, robust, and effective plasmonic platforms obtained via, ideally, simple standardized protocols, and (ii) the integration into multifunctional systems (e.g., microfluidics, magnetic, multi-optical read-outs for enhanced sensing, chemometrics, etc.) to overcome the intrinsic limitations of SERS as stand-alone technique and enable automatization/standardization of the measuring procedures. On a more specific note, we emphasize a certain technological delay in the development and design of positively-charged nanomaterials as compared to the state-of-the-art nanofabrication research, which it may be ascribed to the more recent implementation of this sub-class of nanostructures to relevant applications. Similarly, mono and polyamine molecules exploited as surface ligands to fabricate positively-charged nanomaterials have been selected so far only among those commercially available. As demonstrated, even minor structural changes in the amine ligands can play a central role at determining the SERS analytical performances of the material. Thus, we expect that à la carte synthesis of different amine molecules, such as thiolated-spermine analogs, could significantly improve the quality of the sensing platform, also in terms of robustness and reusability, with particular focus on paving the way for their application in complex biological media for SERS bio-sensing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Spanish Ministerio de Economia y Competitividad (RYC-2015-19107, RYC-2016-20331, PID2020-120306RB-I00 and PDC2021-121787-I00), the Generalitat de Cataluña (2017SGR883), and the Universitat Rovira I Virgili (2021PFR-URV-B2-02). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 713679 and from the Universitat Rovira i Virgili (URV).Notes and references
- L. Guerrini and R. A. Alvarez-Puebla, ACS Omega, 2021, 6, 1054–1063 CrossRef CAS
.
- L. Guerrini, E. Garcia-Rico, A. O'Loghlen, V. Giannini and R. A. Alvarez-Puebla, Cancers, 2021, 13, 2179 CrossRef CAS
- J. M. Romo-Herrera, K. Juarez-Moreno, L. Guerrini, Y. Kang, N. Feliu, W. J. Parak and R. A. Alvarez-Puebla, Mater. Today Bio, 2021, 11, 100125 CrossRef CAS
- L. Guerrini and R. A. Alvarez-Puebla, Cancers, 2019, 11, 748 CrossRef CAS
- E. Garcia-Rico, R. A. Alvarez-Puebla and L. Guerrini, Chem. Soc. Rev., 2018, 47, 4909–4923 RSC
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef
- Z. D. Li and D. Kurouski, Acc. Chem. Res., 2021, 54, 2477–2487 CrossRef CAS
- Y. C. Zhang, S. He, W. X. Guo, Y. Hu, J. W. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS
- L. Jiang, M. M. Hassan, S. Ali, H. Li, R. Sheng and Q. Chen, Trends Food Sci. Technol., 2021, 112, 225–240 CrossRef CAS
- J. Morla-Folch, H.-n. Xie, P. Gisbert-Quilis, S. Gómez-de Pedro, N. Pazos-Perez, R. A. Alvarez-Puebla and L. Guerrini, Angew. Chem., Int. Ed., 2015, 54, 13650–13654 CrossRef CAS
- B. D. Howes, L. Guerrini, S. Sanchez-Cortes, M. P. Marzocchi, J. V. Garcia-Ramos and G. Smulevich, J. Raman Spectrosc., 2007, 38, 859–864 CrossRef CAS
- L. Guerrini and D. Graham, Chem. Soc. Rev., 2012, 41, 7085–7107 RSC
- L. Guerrini, R. Alvarez-Puebla and N. Pazos-Perez, Materials, 2018, 11, 1154 CrossRef
- I. Calderon, L. Guerrini and R. A. Alvarez-Puebla, Biosensors, 2021, 11, 230 CrossRef CAS
- L. Fabris, ChemNanoMat, 2016, 2, 249–258 CrossRef CAS
- L. Guerrini, N. Pazos-Perez, E. Garcia-Rico and R. Alvarez-Puebla, Cancer Nanotechnol., 2017, 8, 5 CrossRef
- J. Morla-Folch, P. Gisbert-Quilis, M. Masetti, E. Garcia-Rico, R. A. Alvarez-Puebla and L. Guerrini, Angew. Chem., Int. Ed., 2017, 56, 2381–2385 CrossRef CAS
- J. Wang, K. M. Koo, E. J. H. Wee, Y. Wang and M. Trau, Nanoscale, 2017, 9, 3496–3503 RSC
- L. Guerrini, I. Izquierdo-Lorenzo, J. V. Garcia-Ramos, C. Domingo and S. Sanchez-Cortes, Phys. Chem. Chem. Phys., 2009, 11, 7363–7371 RSC
- S. Jessl, M. Tebbe, L. Guerrini, A. Fery, R. A. Alvarez-Puebla and N. Pazos-Perez, Small, 2018, 14, 1703879 CrossRef
- A. Stewart, S. Murray and S. E. J. Bell, Analyst, 2015, 140, 2988–2994 RSC
- S. Sanchez-Cortes, R. M. Berenguel, A. Madejón and M. Pérez-Méndez, Biomacromolecules, 2002, 3, 655–660 CrossRef CAS
- P. T. Sujai, M. M. Joseph, G. Saranya, J. B. Nair, V. P. Murali and K. K. Maiti, Nanoscale, 2020, 12, 6971–6975 RSC
- L. Marsich, A. Bonifacio, S. Mandal, S. Krol, C. Beleites and V. Sergo, Langmuir, 2012, 28, 13166–13171 CrossRef CAS
- Y. Z. Zhang, D. J. de Aberasturi, M. Henriksen-Lacey, J. Langer and L. M. Liz-Marzan, ACS Sens., 2020, 5, 3194–3206 CrossRef CAS
- E. Efeoglu and M. Culha, Appl. Spectrosc., 2013, 67, 498–505 CrossRef CAS
- C. Y. Wang, Y. Zeng, A. G. Shen and J. M. Hu, Anal. Methods, 2018, 10, 5622–5628 RSC
- F. Liebig, R. M. Sarhan, C. Prietzel, A. F. Thünemann, M. Bargheer and J. Koetz, Langmuir, 2018, 34, 4584–4594 CrossRef CAS
- A. Shahzad, M. Chung, T. Yu and W. S. Kim, Chem.–Asian J., 2015, 10, 2512–2517 CrossRef CAS
- K. Kim, J. W. Lee and K. S. Shin, ACS Appl. Mater. Interfaces, 2012, 4, 5498–5504 CrossRef CAS
- S. Tan, M. Erol, A. Attygalle, H. Du and S. Sukhishvili, Langmuir, 2007, 23, 9836–9843 CrossRef CAS
- S. Tan, M. Erol, S. Sukhishvili and H. Du, Langmuir, 2008, 24, 4765–4771 CrossRef CAS
- R. Manisekaran, E. J. C. Amieva, C. M. Valdemar-Aguilar and L. M. Lopez-Marin, Gold Bull., 2020, 53, 135–140 CrossRef CAS
- E. O. Ganbold, J. H. Park, U. Dembereldorj, K. S. Ock and S. W. Joo, J. Raman Spectrosc., 2011, 42, 1614–1619 CrossRef CAS
- B. Ankudze, A. Philip and T. T. Pakkanen, Sens. Actuators, B, 2018, 265, 668–674 CrossRef CAS
- A. Philip, B. Ankudze and T. T. Pakkanen, Appl. Surf. Sci., 2018, 444, 243–252 CrossRef CAS
- M. Potara, E. Jakab, A. Damert, O. Popescu, V. Canpean and S. Astilean, Nanotechnology, 2011, 22, 135101 CrossRef
- O. Prakash, S. Sil, T. Verma and S. Umapathy, J. Phys. Chem. C, 2020, 124, 861–869 CrossRef CAS
- H. Gong, K. Zhang, C. Dicko, L. Bülow and L. Ye, ACS Appl. Nano Mater., 2019, 2, 1655–1663 CrossRef CAS
- N. Pazos-Perez, F. J. Garcia de Abajo, A. Fery and R. A. Alvarez-Puebla, Langmuir, 2012, 28, 8909–8914 CrossRef CAS
- R. Zhou, Z. W. Wu, Z. H. Sun and X. F. Su, Nanosci. Nanotechnol. Lett., 2015, 7, 801–805 CrossRef
- B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368–6374 CrossRef CAS
- M. U. Amin, R. Y. Zhang, L. W. Li, H. J. You and J. X. Fang, Anal. Chem., 2021, 93, 7657–7664 CrossRef CAS
- P. Ndokoye, J. Ke, J. Liu, Q. Zhao and X. Li, Langmuir, 2014, 30, 13491–13497 CrossRef CAS
- C. C. Andrei, A. Moraillon, S. Lau, N. Felidj, N. Yamakawa, J. Bouckaert, E. Larquet, R. Boukherroub, F. Ozanam, S. Szunerits and A. C. Gouget-Laemmel, Talanta, 2020, 219, 121174 CrossRef CAS
- S. Hu, F. Gu, M. Chen, C. W. Wang, J. Li, J. Yang, G. Y. Wang, Z. Zhou and Y. Yang, Sci. Rep., 2020, 10, 12480 CrossRef CAS
- Y. Li, R. Lu, J. Y. Shen, W. Q. Han, X. Y. Sun, J. S. Li and L. J. Wang, Analyst, 2017, 142, 4756–4764 RSC
- J. Hedberg, M. Lundin, T. Lowe, E. Blomberg, S. Wold and I. O. Wallinder, J. Colloid Interface Sci., 2012, 369, 193–201 CrossRef CAS
- S. Hu, H. Q. Kang, F. Gu, C. W. Wang, S. Y. Cheng, W. J. Gong, L. P. Wang, B. Gu and Y. Yang, Int. J. Nanomed., 2021, 16, 941–950 CrossRef
- X. Chen, M. Tang, Y. Liu, J. Huang, Z. Liu, H. Tian, Y. Zheng, M. L. de la Chapelle, Y. Zhang and W. Fu, Microchim. Acta, 2019, 186, 102 CrossRef
- M. Blanco-Formoso, N. Pazos-Perez and R. A. Alvarez-Puebla, ACS Omega, 2020, 5, 25485–25492 CrossRef CAS
- R. Y. Liu, C. G. Xie, Y. H. Yan, L. Hu, S. H. Wang, K. A. Alamry, H. M. Marwani and L. J. Chen, Nanomaterials, 2020, 10, 575 CrossRef CAS
- D. van Lierop, Z. Krpetic, L. Guerrini, I. A. Larmour, J. A. Dougan, K. Faulds and D. Graham, Chem. Commun., 2012, 48, 8192–8194 RSC
- X. Li, T. Yang, C. S. Li, Y. Song, D. Wang, L. Jin, H. Lou and W. Li, Theranostics, 2020, 10, 898–909 CrossRef CAS
- M. Yang, S. Matulis, L. H. Boise, A. J. McGoron and Y. Cai, Anal. Bioanal. Chem., 2017, 409, 4683–4695 CrossRef CAS
- K. M. Koo, J. Wang, R. S. Richards, A. Farrell, J. W. Yaxley, H. Samaratunga, P. E. Teloken, M. J. Roberts, G. D. Coughlin, M. F. Lavin, P. N. Mainwaring, Y. Wang, R. A. Gardiner and M. Trau, ACS Nano, 2018, 12, 8362–8371 CrossRef CAS
- Y. Nie, Y. Teng, P. Li, W. Liu, Q. Shi and Y. Zhang, Spectrochim. Acta, Part A, 2018, 191, 271–276 CrossRef CAS
- Y. Liu, W. Zhang, T. H. Phan, W. Chrzanowski, A. Rodger and Y. Wang, Anal. Methods, 2020, 12, 5908–5915 RSC
- Y. Liu, N. Lyu, V. K. Rajendran, J. Piper, A. Rodger and Y. Wang, Anal. Chem., 2020, 92, 5708–5716 CrossRef CAS
- M. Ocwieja, J. Maciejewska-Pronczuk, Z. Adamczyk and M. Roman, J. Colloid Interface Sci., 2017, 501, 192–201 CrossRef CAS
- D. Lupa, M. Oćwieja, N. Piergies, A. Baliś, C. Paluszkiewicz and Z. Adamczyk, Colloid Interface Sci. Commun., 2019, 33, 100219 CrossRef CAS
- M. Oćwieja, A. Barbasz, S. Walas, M. Roman and C. Paluszkiewicz, Colloids Surf., B, 2017, 160, 429–437 CrossRef
- Z. Guo, J. Hwang, B. Zhao, J. H. Chung, S. G. Cho, S.-J. Baek and J. Choo, Analyst, 2014, 139, 807–812 RSC
- X. Bai, S. Xu, G. Hu and L. Wang, Sens. Actuators, B, 2016, 237, 224–229 CrossRef CAS
- A. Hussain, H. Pu and D.-W. Sun, J. Food Meas. Charact, 2020, 14, 2021–2029 CrossRef
- Q. Tu, T. Yang, Y. Qu, S. Gao, Z. Zhang, Q. Zhang, Y. Wang, J. Wang and L. He, Analyst, 2019, 144, 2017–2025 RSC
- R. Gao, D. Li, Q. M. Zhang, S. Q. Zheng, X. G. Ren and W. Deng, Sens. Actuators, B, 2021, 328, 128960 CrossRef CAS
- A. Hussain, H. B. Pu and D. W. Sun, Spectrochim. Acta, Part A, 2020, 229, 117994 CrossRef CAS
- J. Chen, Y. E. Shi, M. Zhang and J. H. Zhan, RSC Adv., 2016, 6, 51823–51829 RSC
- Z. Xu, J. Hao, W. Braida, D. Strickland, F. Li and X. Meng, Langmuir, 2011, 27, 13773–13779 CrossRef CAS
- H. He, P. Li, X. Tang, D. Lin, A. Xie, Y. Shen and L. Yang, Spectrochim. Acta, Part A, 2019, 212, 293–299 CrossRef CAS
- S. Z. Weng, W. X. Zhu, P. Li, H. C. Yuan, X. Y. Zhang, L. Zheng, J. L. Zhao, L. S. Huang and P. Han, Food Chem., 2020, 310, 125855 CrossRef CAS
- Y. Pang, C. Wang, L. Lu, C. Wang, Z. Sun and R. Xiao, Biosens. Bioelectron., 2019, 130, 204–213 CrossRef CAS
- C. Wang, J. Wang, M. Li, X. Qu, K. Zhang, Z. Rong, R. Xiao and S. Wang, Analyst, 2016, 141, 6226–6238 RSC
- S. Zhu, X. L. Zhang, J. C. Cui, Y. E. Shi, X. H. Jiang, Z. Liu and J. H. Zhan, Analyst, 2015, 140, 2815–2822 RSC
- J. Hao, M.-J. Han, J. Li and X. Meng, J. Colloid Interface Sci., 2012, 377, 51–57 CrossRef CAS
- C. M. Ruan, W. Wang and A. H. Gu, Anal. Chim. Acta, 2006, 567, 114–120 CrossRef CAS
- J. Hao, M.-J. Han, X. Meng, W. Weimer and Q. K. Wang, Spectrochim. Acta, Part A, 2015, 136, 1593–1599 CrossRef CAS
- Y. E. Shi, W. S. Wang and J. H. Zhan, Nano Res., 2016, 9, 2487–2497 CrossRef CAS
- A. K. M. Jamil, E. L. Izake, A. Sivanesan, R. Agoston and G. A. Ayoko, Anal. Methods, 2015, 7, 3863–3868 RSC
- A. K. M. Jamil, A. Sivanesan, E. L. Izake, G. A. Ayoko and P. M. Fredericks, Sens. Actuators, B, 2015, 221, 273–280 CrossRef CAS
- A. K. M. Jamil, E. L. Izake, A. Sivanesan and P. M. Fredericks, Talanta, 2015, 134, 732–738 CrossRef CAS
- L. Zhang, L. Zhou, W. Ji, W. Song and S. Zhao, Food Anal. Methods, 2017, 10, 1940–1947 CrossRef
- X. Jiang, M. Yang, Y. Meng, W. Jiang and J. Zhan, ACS Appl. Mater. Interfaces, 2013, 5, 6902–6908 CrossRef CAS
- L. Guerrini and R. A. Alvarez-Puebla, Nanomaterials, 2021, 11, 326 CrossRef CAS
- P. Gisbert-Quilis, M. Masetti, J. Morla-Folch, J. M. Fitzgerald, N. Pazos-Perez, E. Garcia-Rico, V. Giannini, R. A. Alvarez-Puebla and L. Guerrini, Adv. Mater. Interfaces, 2017, 4, 1700724 CrossRef
- J. Morla-Folch, H.-N. Xie, R. A. Alvarez-Puebla and L. Guerrini, ACS Nano, 2016, 10, 2834–2842 CrossRef CAS
- J. Morla-Folch, R. A. Alvarez-Puebla and L. Guerrini, J. Phys. Chem. Lett., 2016, 7, 3037–3041 CrossRef CAS
- A. Torres-Nunez, K. Faulds, D. Graham, R. A. Alvarez-Puebla and L. Guerrini, Analyst, 2016, 141, 5170–5180 RSC
- L. Guerrini, Ž. Krpetić, D. van Lierop, R. A. Alvarez-Puebla and D. Graham, Angew. Chem., Int. Ed., 2015, 54, 1144–1148 CrossRef CAS
- M. Masetti, H.-n. Xie, Ž. Krpetić, M. Recanatini, R. A. Alvarez-Puebla and L. Guerrini, J. Am. Chem. Soc., 2015, 137, 469–476 CrossRef CAS
- L. Guerrini and R. A. Alvarez-Puebla, Analyst, 2019, 144, 6862–6865 RSC
- X. Z. Li, T. Y. Yang, C. S. Li, Y. T. Song, H. Lou, D. G. Guan and L. L. Jin, Theranostics, 2018, 8, 1678–1689 CrossRef CAS
- L. Guerrini and R. A. Alvarez-Puebla, Anal. Chem., 2019, 91, 11778–11784 CrossRef CAS
- C. Catala, B. Mir-Simon, X. Feng, C. Cardozo, N. Pazos-Perez, E. Pazos, S. Gómez-de Pedro, L. Guerrini, A. Soriano, J. Vila, F. Marco, E. Garcia-Rico and R. A. Alvarez-Puebla, Adv. Mater. Technol., 2016, 1, 1600163 CrossRef
- N. Pazos-Perez, E. Pazos, C. Catala, B. Mir-Simon, S. Gomez-de Pedro, J. Sagales, C. Villanueva, J. Vila, A. Soriano, F. Javier Garcia de Abajo and R. A. Alvarez-Puebla, Sci. Rep., 2016, 6, 29014 CrossRef CAS
- Y.-F. Huang, Y.-F. Wang and X.-P. Yan, Environ. Sci. Technol., 2010, 44, 7908–7913 CrossRef CAS
- H. Shin, H. Jeong, J. Park, S. Hong and Y. Choi, ACS Sens., 2018, 3, 2637–2643 CrossRef CAS
- J. C. Fraire, S. Stremersch, D. Bouckaert, T. Monteyne, T. De Beer, P. Wuytens, R. De Rycke, A. G. Skirtach, K. Raemdonck, S. De Smedt and K. Braeckmans, ACS Appl. Mater. Interfaces, 2019, 11, 39424–39435 CrossRef CAS
- T. Rojalin, H. J. Koster, J. Liu, R. R. Mizenko, D. Tran, S. Wachsmann-Hogiu and R. P. Carney, ACS Sens., 2020, 5, 2820–2833 CrossRef CAS
- Y. T. Shen, J. Yue, W. Q. Xu and S. P. Xu, Theranostics, 2021, 11, 4872–4893 CrossRef CAS
- D. J. Mitchell, L. Steinman, D. T. Kim, C. G. Fathman and J. B. Rothbard, J. Pept. Res., 2000, 56, 318–325 CrossRef CAS
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Chem. Soc. Rev., 2011, 40, 233–245 RSC
| This journal is © The Royal Society of Chemistry 2022 |