
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of the cross-linker length of thiophene units on photocatalytic hydrogen production of triazine-based conjugated microporous polymers†
Xiao Hana,
Yan Zhanga,
YunYun Dong a,
Jinsheng Zhao*ab,
Shouli Ming*a and
Junhong Zhang*a
a,
Jinsheng Zhao*ab,
Shouli Ming*a and
Junhong Zhang*a
aCollege of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng, 252059, P. R. China. E-mail: j.s.zhao@163.com; mingshouli@126.com; zhangjunhong@lcu.edu.cn
bShandong Provincial Key Laboratory of Chemical Energy Storage and Novel Cell Technology, Liaocheng University, Liaocheng, 252059, PR China
First published on 23rd December 2021
Abstract
Conjugated microporous polymers (CMPs) have been investigated in the field of photocatalytic hydrogen production because of their extended π-conjugation, tunable chemical structure and excellent thermal stability. Herein, we construct three CMPs based on thiophenes and triazine, and prove the effect of cross-linker length on photocatalytic activity of CMPs. BTPT-CMP1 exhibits blue-shifted optical absorption compared to BTPT-CMP2 and BTPT-CMP3 with long cross-linkers, however, possesses higher photocurrent because of the large specific surface area and small interface charge transfer resistance of BTPT-CMP1. It was found that BTPT-CMP1 (5561.87 μmol g−1 h−1) with short cross-linkers exhibits better photocatalytic performance compared to BTPT-CMP2 (1840.86 μmol g−1 h−1) and BTPT-CMP3 (1600.48 μmol g−1 h−1). Also, BTPT-CMP1 possesses a higher hydrogen evolution rate than most reported 1,3,5-triazine based conjugated polymers. These results demonstrate that the cross-linker length has great influence on the photocatalytic properties of conjugated microporous polymers, which offers theoretical direction for designing high-performance CMPs.
1. Introduction
Environmental pollution and the fossil energy crisis have become the two most concerning issues in the 21st century.1,2 Hydrogen energy, as a recognized clean energy with no pollution, has attracted increasing attention from society. Photocatalytic hydrogen evolution was firstly proposed by Honda and Fujishima in 1972 based on the potential applications of hydrogen, which has become a research hotspot in the academic field.3,4 In the last few decades, research mainly focused on metal-based inorganic semiconductor photocatalysts including TiO2, ZnO, CdS, GaP, and SiC.5,6 However, metal-based inorganic semiconductor photocatalysts face many challenges, such as low photocatalytic activity, poor charge separation ability, high toxicity and the limitation of rare metals reserve.7,8 Compared to inorganic semiconductors, organic polymers have many advantages, such as easily-controllable molecular structure and performance, rich raw material reserves, light weight, etc.9–11 The research on organic polymers used as photocatalysts began in the 1980s. After that, various types of organic polymers for photocatalytic hydrogen evolution have been widely studied, such as conjugated microporous polymers (CMPs), linear conjugated polymers and covalent organic framework materials, etc.12–14 Among them, CMPs is emerging class of organic porous materials with large π-conjugated framework and many nanoscale pores.15–17 More importantly, the energy band structure, optical absorption and pore size of CMPs can be fine-tuned by changing the chemical structure. These distinguished traits make CMPs have large potential applications in the field of photocatalysis.17–20In order to obtain high-performance CMPs used in photocatalytic hydrogen production, various strategies have been proposed, such as extending conjugated block, modifying side chain structure, constructing donor–acceptor (D–A) type molecular structure, etc.21,22 For example, Wang et al.23 reported the dibenzothiophene dioxide-containing CMPs with cross-linkers from benzene to biphenyl, and to p-terphenyl, and investigated the effect of cross-linker length on photocatalytic property. Lin et al.24 reported a truxene-based conjugated polymer with hydrophilic amino side chains, which dramatically modified the wettability and hydrogen evolution rate of polymer than that with hydrophobic octyl chains. Shu et al.25 studied the influence of molecular geometry on photocatalytic property. They constructed two bisulphone-containing conjugated polymers with 1D and 3D molecular structures, which suggest that the linear polymers exhibit increased photocatalytic performance related to that with 3D molecular structure due to fast charge transport in 1D molecular skeleton. Lan et al.26 prepared a series of D–A type conjugated polymers based on dibenzothiophene-S,S-dioxide (FSO) as the acceptor unit and pyrene as the donor unit. They found that the introduction of FSO can widen the optical absorption region of polymer and improve the exciton-separation efficiency. Zhao et al.27 also obtained two kinds of FSO based CMPs photocatalysts and investigated the effect of linking sites on the photocatalytic hydrogen evolution. To further widen the optical absorption, Xiao et al.28 used diketopyrrolopyrrole as acceptor unit to construct D–A type CMPs photocatalyst. These results suggest that D–A type conjugated polymers not only regulate the optical absorption, but also are helpful for exciton separation under visible light. The conjugated D–A type CMPs could enable efficient exciton separation, however, the dissociated electron and hole often recombine simultaneously. In order to suppress electron and hole recombination, Guo et al. constructed a series of D–A1–A2 type CMPs comprising two kind of acceptor units with different energy levels.29 In short, various strategies have been proposed to construct CMPs with high hydrogen evolution efficiency, however, the photocatalytic parameters of CMPs can't meet the standards of commercial application. Therefore, identifying the key factors affecting the hydrogen evolution efficiency of CMPs is of great significance for the design of high-performance materials.30,31
Recent research indicate that triazine is an appropriate acceptor unit for the construction of D–A type CMPs applied in photocatalytic hydrogen evolution due to the hydrophilic nature of nitrogen atoms in triazine unit.32,33 In this study, we synthesized three thiophene-triazine based D–A type CMPs by Stille coupling reaction, as shown in Fig. 1. The difference of three CMPs is the cross-linker from monothiophene to bithiophene and p-terthiophene. As we known that polythiophene and its derivatives could be n-doped by ions under negative potential window, which might be helpful to charge transport.34 It is speculated that thiophene-triazine based D–A type CMPs possess high charge transport rate, further produce more photocurrent for photocatalytic hydrogen evolution.
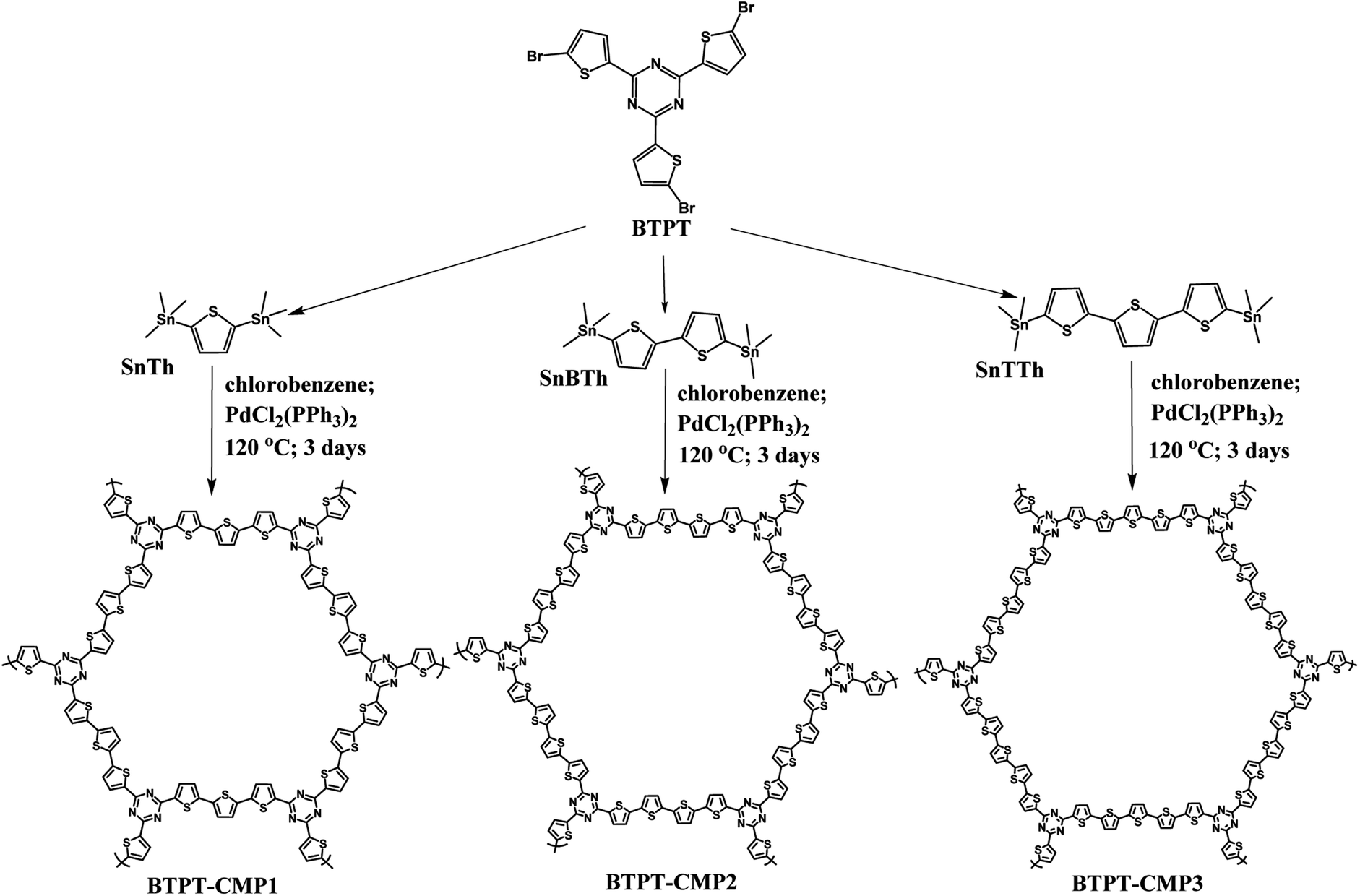 | ||
| Fig. 1 Synthesis routes of BTPT-CMP1, BTPT-CMP2 and BTPT-CMP3. | ||
Herein, the optical and electrochemical properties of the three CMPs were investigated comparatively. BTPT-CMP1 possesses much higher photocurrent relative to BTPT-CMP2 and BTPT-CMP3 due to fast transport rate and less recombination of photogenerated charges in BTPT-CMP1. The results indicate that the hydrogen production performance of the prepared CMPs decrease gradually with increase of the cross-linker length. BTPT-CMP1 with monothiophene cross-linker displays the hydrogen-producing rate of 5561.87 μmol h−1 g−1 under visible light with 3 wt% Pt loaded. Also, BTPT-CMP1 keeps good stability over 15 h without obvious photocatalytic efficiency loss. These results demonstrate that BTPT-CMP1 possess large application potential in photocatalytic hydrogen evolution.
2. Experimental section
2.1. Materials and the characterization methods
The chemical agents except 2,4,6-tris(5-bromothiophene-2-yl)-1,3,5-triazine (BTPT) are obtained from the commercial channels, and BTPT was synthesized according to our previous report.35 All of the detailed information is given in the ESI.† And also, characterization methods for the polymer materials and the related apparatus are given in the ESI.† The detailed information on the hydrogen evolution activity of the polymers are also given in the ESI.†2.2. Synthesis of BTPT-CMP1
BTPT-CMP1 was prepared by the Stille coupling reaction under nitrogen atmosphere (Fig. 1). Typically, 200 mg of BTPT (354.52 μmol) and 217.90 mg of SnTh (531.79 μmol) were dispersed into chlorobenzene under stirring condition. Then, 18.7 mg of PdCl2(PPh3)2 (26.9 μmol) was added to the Schlenk tube. The mixed solution was heated under nitrogen at 120 °C for 72 h. After reaction, the red powder precipitate was obtained, followed by washed with ethanol and acetone, finally vacuum dried at 70 °C for 12 h. The product mass: 132.50 mg, yield: 83%.2.3. Synthesis of BTPT-CMP2
200.00 mg of BTPT (354.52 μmol) and SnBTh (261.57 mg, 531.79 μmol) were firstly dispersed into chlorobenzene (35 mL). Then 18.7 mg of PdCl2(PPh3)2 (26.9 μmol) was added as the catalyst to the Schlenk tube under a N2 atmosphere. Then, the reaction solution was stirred for 3 days at 120 °C. After reaction, the red powder precipitate was obtained, followed by washed with ethanol and acetone, finally vacuum dried at 70 °C for 12 h. The product mass: 164.70 mg, yield: 81%.2.4. Synthesis of BTPT-CMP3
BTPT-CMP3 was synthesized according to the preparation method of BTPT-CMP2, except that SnTTh was used as the organotin compound for the Stille reaction. The product mass: 197.60 mg, yield: 80%.3. Results and discussion
3.1. Synthesis and characterization of CMPs
The triazine compound (BTPT) was synthesized according to previous literature.35 The three thiophene based CMPs photocatalysts were prepared at satisfactory yields by the Stille coupling reaction. These polymers exhibit poor solubility in common organic solvents because of their rigid backbones and the cross-linked networks.19 The structures of the polymers were proved by FT-IR and solid-state 13C CP-MAS NMR. As shown in Fig. 2, a strong peak at 1493 cm−1 is found, which can be ascribed to the aromatic C–N stretching vibration of the triazine ring.36,37 The characteristic peak located at 1420 cm−1 and 1637 cm−1 originate from the skeleton vibrations of aromatic rings and thiophene rings. Besides, the characteristic vibration of the C–H in-plane bending (1035 cm−1) and C–H out-of-plane bending (787 cm−1) on the thiophene groups could be clearly observed. In addition, the peak at 1366 cm−1 is attributed to the O–H of physisorbed water, which is also proved by the broad band in the range of 3132–3669 cm−1. The solid-state 13C CP-MAS NMR was employed to further confirm their structures (Fig. S1†). The chemical shift at about 166 ppm is assigned to C atom on the triazine ring. The signals at 123–143 ppm are ascribed to C atoms on the thiophene units. Further structural details about polymers were investigated by X-ray photoelectron spectroscopy (XPS), as shown in Fig. 3. XPS spectra indicate there are carbon, nitrogen and sulfur in the three polymers, which is also proved by element mapping (Fig. S2 and S3†). For BTPT-CMP1, the C 1s spectrum exhibit two obvious peaks at 284.5 and 286.8 eV, which are assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–S in the thiophene rings, respectively. The peak located at 285.1 eV is ascribed to the C–N in triazine groups, which is also proved by the peak (398.6 eV) in N 1s spectrum. The S 2p spectrum is divided into two main peaks at 164.1 eV and 165.2 eV, corresponding to the S 2p3/2 and S 2p1/2 binding energies, respectively. As BTPT-CMP1, BTPT-CMP2 and BTPT-CMP3 exhibit similar XPS spectra.
C and C–S in the thiophene rings, respectively. The peak located at 285.1 eV is ascribed to the C–N in triazine groups, which is also proved by the peak (398.6 eV) in N 1s spectrum. The S 2p spectrum is divided into two main peaks at 164.1 eV and 165.2 eV, corresponding to the S 2p3/2 and S 2p1/2 binding energies, respectively. As BTPT-CMP1, BTPT-CMP2 and BTPT-CMP3 exhibit similar XPS spectra.
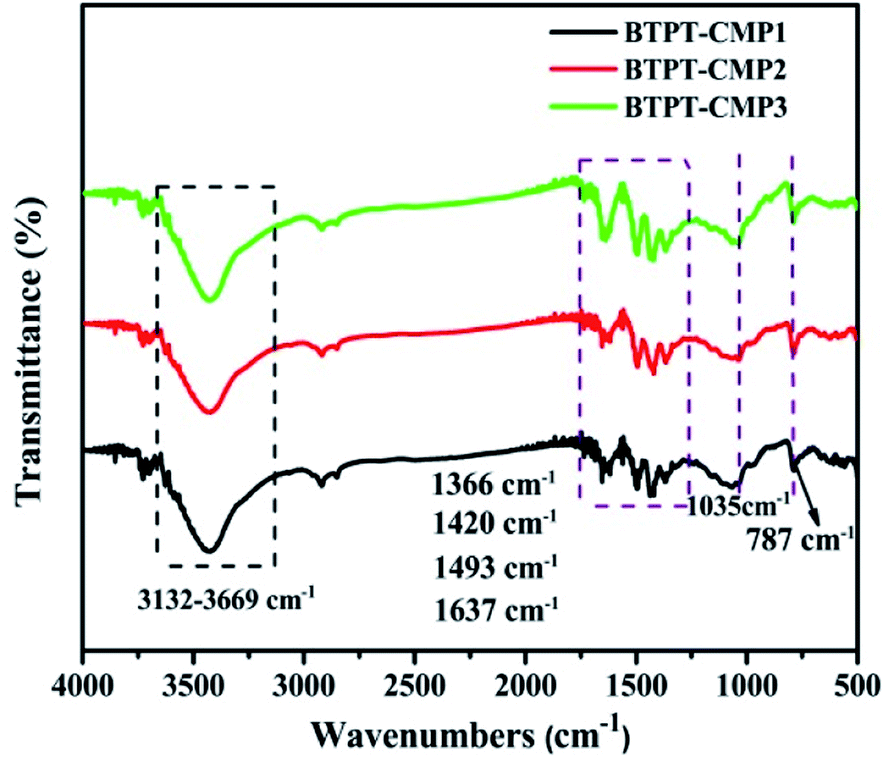 | ||
| Fig. 2 FT-IR spectra of the polymers. | ||
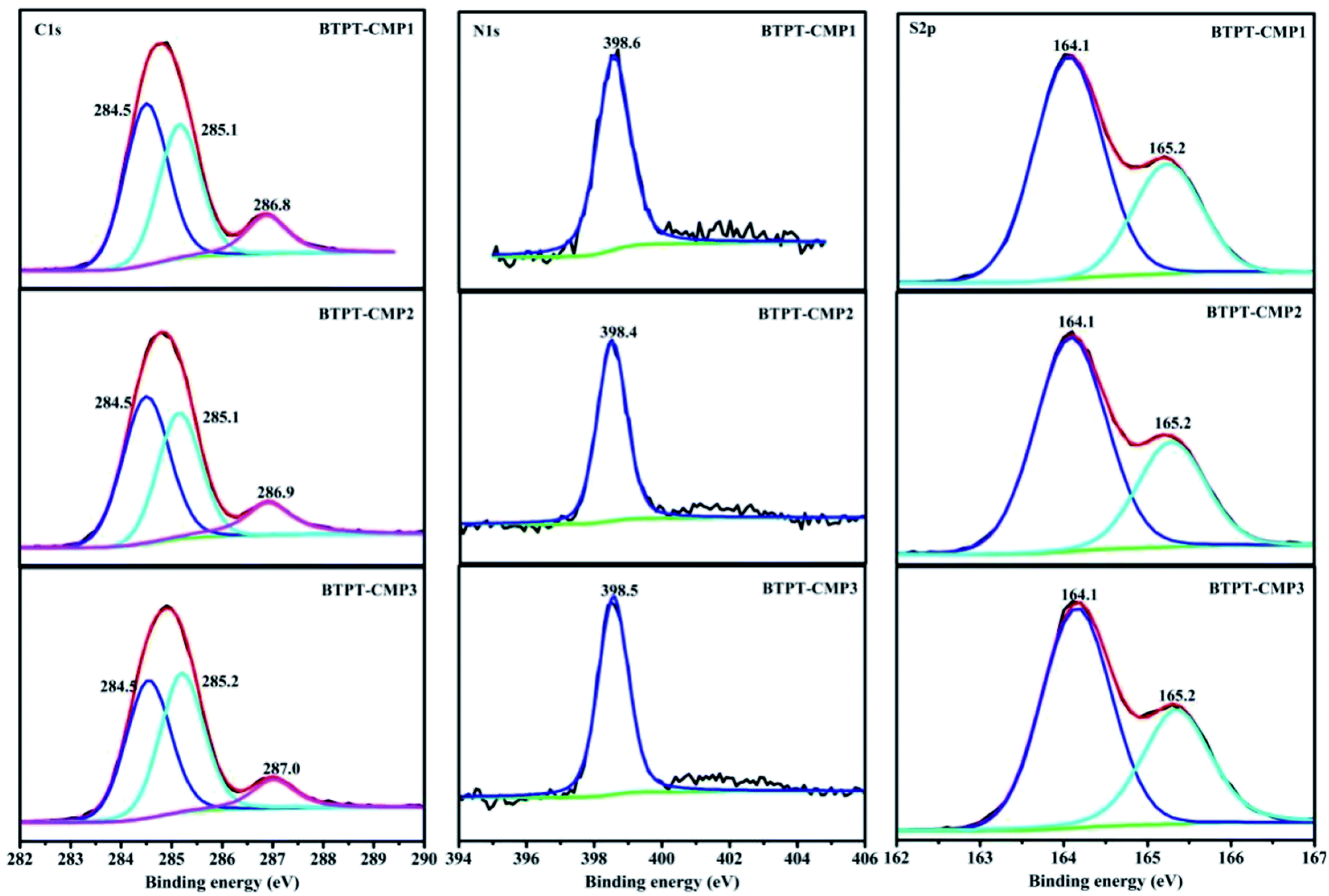 | ||
| Fig. 3 High-resolution XPS spectra of the polymers. | ||
The crystalline structure and aggregation state of polymer could be illustrated with powder X-ray diffraction (PXRD). The broad diffraction peaks were observed in the PXRD as shown in Fig. 4a, which revealed the amorphous structure of the polymers. Also, the thermal stability of the three polymers were measured in nitrogen atmosphere. Thermal gravimetric analysis (TGA) indicate that the initial decomposition temperature (Td) for BTPT-CMP1, BTPT-CMP2 and BTPT-CMP3 are 353 °C, 320 °C and 127 °C, respectively (Fig. 4b). It is observed that BTPT-CMP1 and BTPT-CMP2 exhibit better thermal stability than BTPT-CMP3.
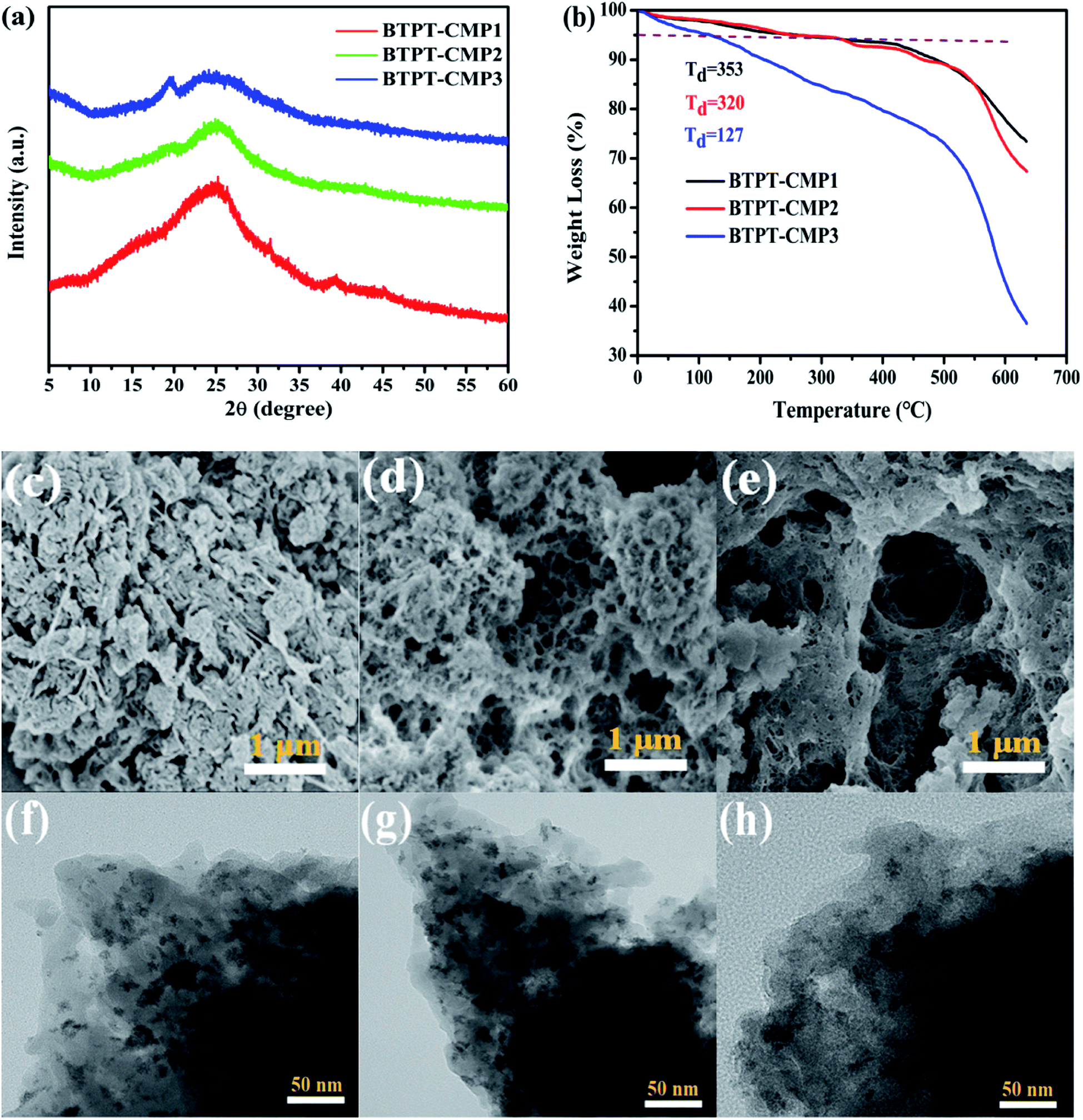 | ||
| Fig. 4 (a) PXRD patterns of the polymers; (b) TGA curves of polymers; SEM images of BTPT-CMP1 (c), BTPT-CMP2 (d) and BTPT-CMP3 (e); TEM images of BTPT-CMP1 (f), BTPT-CMP2 (g) and BTPT-CMP3 (h). | ||
The morphology of the polymers were investigated by scanning electron microscope (SEM) and transmission electron microscopy (TEM). The three polymers have sponge-like structure with obvious macropores and mesopores, as shown in Fig. 4c–e and f–h. The porous structure make polymer possess more specific surface area, which could provide more catalytic sites. The porosity of polymers were investigated by N2 adsorption–desorption experiment at 77 K (Fig. 5). The three polymers display faint N2-adsorption behavior at low relative pressure. The rapid nitrogen adsorption at very high relative pressure could be observed, which suggest that the three polymers possess obvious meso- and macroporous structures.38 The result is consistent with the findings of SEM. Also, the Brunauer–Emmett–Teller (BET) surface area of BTPT-CMP1, BTPT-CMP2 and BTPT-CMP3 were evaluated to be 138.4, 62.1 and 58.8 m2 g−1, respectively. Significantly, the specific surface area of BTPT-CMP1 is beyond 2 times higher than that of BTPT-CMP2 and BTPT-CMP3. The specific surface area difference might be related to polymeric cross-linker length. The phenomenon is usually observed in reported polymers containing different length cross-linkers.39,40 Also, the pore-size analysis of the three polymers was carried out, as shown in the inset of Fig. 5. All the three polymers possess wide pore-size distribution and the pore diameter concentrate into 45.25 nm for BTPT-CMP1, 15.17 nm for BTPT-CMP2 and 30.74 nm for BTPT-CMP3. The wide pore-size distribution of BTPT-CMP1 could be explained that derived from the interparticle porosities or voids.41 The large specific surface area and wide pore-size distribution are beneficial to provide more active sites for BTPT-CMP1, which is conducive to the increase of photocatalytic hydrogen production rate.
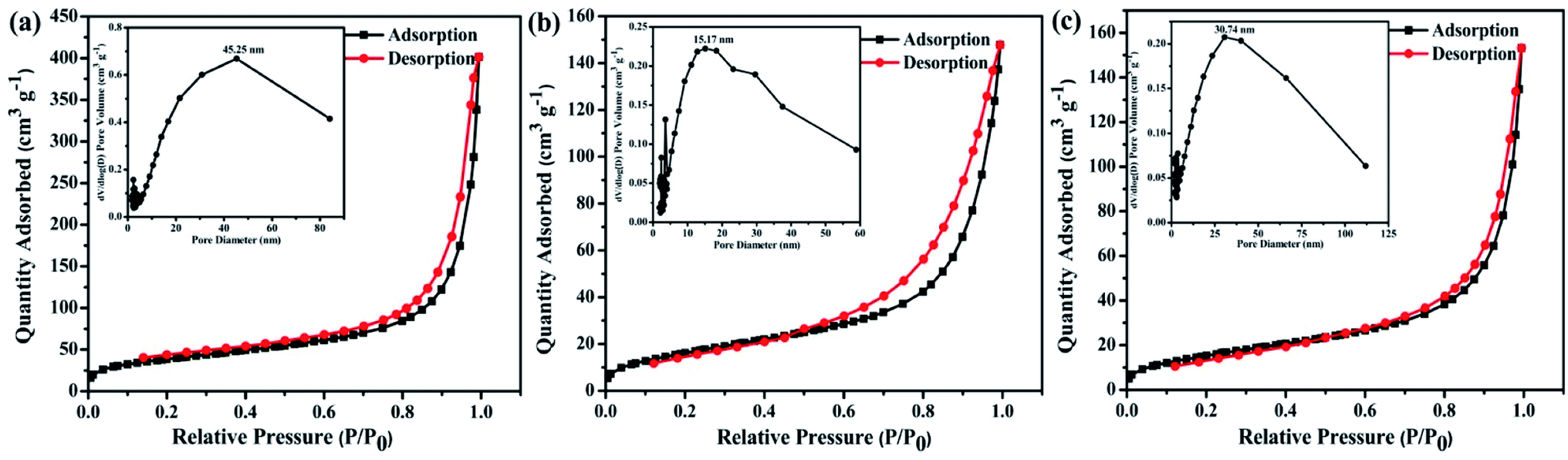 | ||
| Fig. 5 N2 adsorption–desorption isotherms of (a) BTPT-CMP1; (b) BTPT-CMP2; (c) BTPT-CMP3 at 77 K. Pore size distribution are shown in the insets. | ||
3.2. The optical and electric properties of CMPs
The UV-Vis diffuse reflectance spectra (DRS) of the three polymers are displayed in Fig. 6a, which reveal broad absorption from 200 nm to 650 nm. It is observed that BTPT-CMP2 and BTPT-CMP3 exhibit red-shifted absorption bands compared to BTPT-CMP1, which result from BTPT-CMP2 and BTPT-CMP3 possess larger conjugated molecular skeleton than BTPT-CMP1.22 In theory, BTPT-CMP3 possess larger conjugated molecular skeleton relative to BTPT-CMP2, however, no obvious optical absorption shift could be observed. The phenomenon might be ascribed to the poor solubility of BTPT-CMP3 in polymerization system restrict the formation of polymer with large conjugated molecular skeleton, which lead to BTPT-CMP2 and BTPT-CMP3 possess similar conjugation degree. Based on the absorption spectra of the three polymers, the band gaps are calculated to be 2.06 eV for BTPT-CMP1, 1.90 eV for BTPT-CMP2 and BTPT-CMP3 (Fig. 6b). In short, all the three polymers reveal broad absorption and narrow ban gap due to the electron push–pull effect of the D–A structure for polymer.42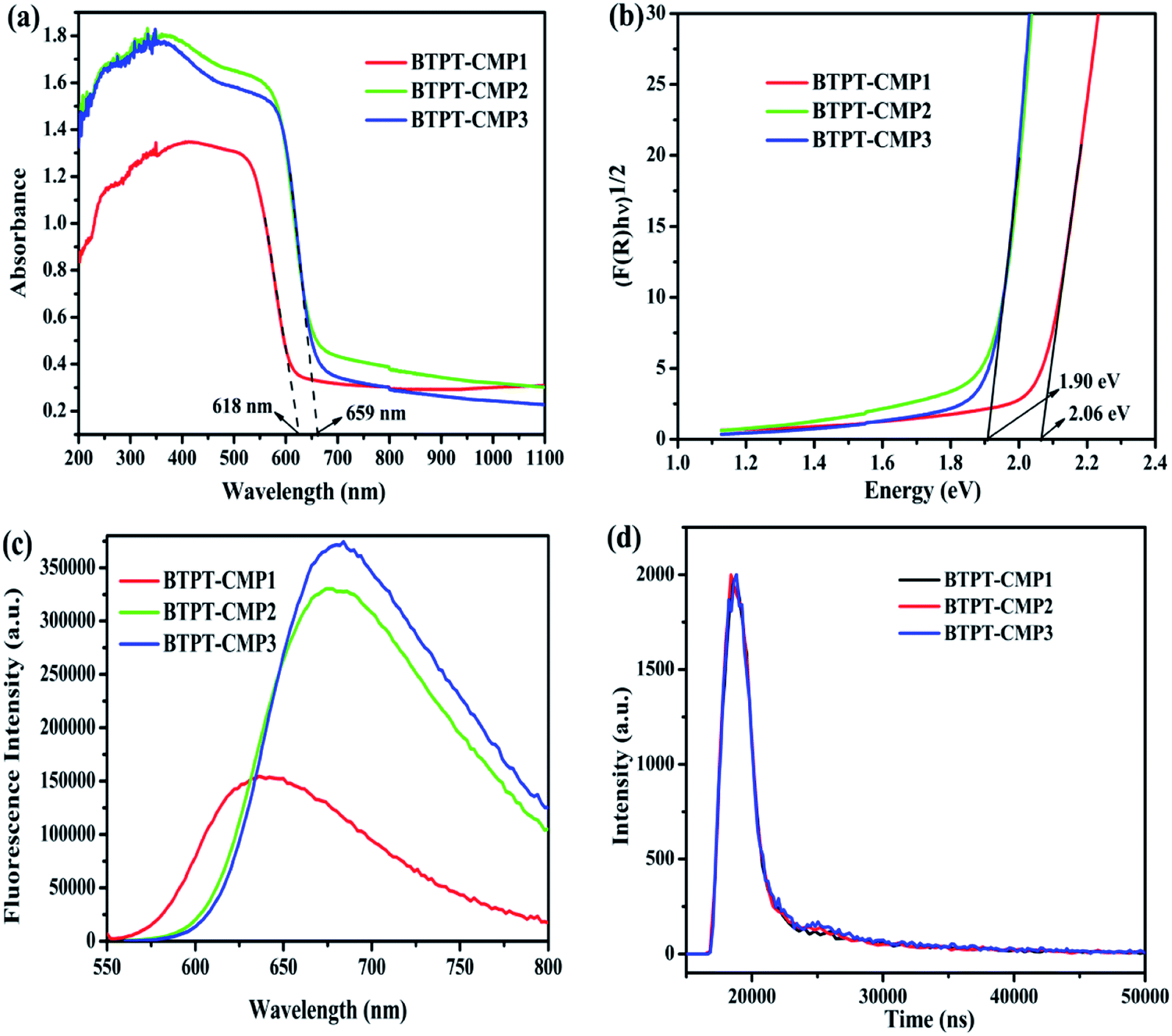 | ||
| Fig. 6 UV-Vis diffuse reflectance spectra (a) and band gaps (b) of polymers; photoluminescence spectra (c) and time-resolved photoluminescence spectra (d) of polymers at the excitation wavelength of 544 nm. | ||
As is well known, photoluminescence (PL) spectrum is one of the effective methods to evaluate the separation/recombination efficiency of photogenerated carriers. Therefore, the photoluminescence spectra of the three polymers were measured comparatively (Fig. 6c). As expected, BTPT-CMP1 (640 nm) exhibits blue-shifted emission peak relative to BTPT-CMP2 (680 nm) and BTPT-CMP3 (684 nm). However, it is observed that BTPT-CMP1 possesses much weaker emission intensity compared with BTPT-CMP2 and BTPT-CMP3, indicating less recombination of photogenerated electron and hole in BTPT-CMP1.43,44 Also, the time-resolved fluorescence decay spectra of the three polymers were studied to measure photogenerated electron lifetime. As shown in Fig. 6d, according to the fitting results, the emission lifetimes of BTPT-CMP1 (τ1 = 0.93 μs, τ2 = 8.68 μs) are much shorter than those of BTPT-CMP2 (τ1 = 0.97 μs, τ2 = 8.88 μs) and BTPT-CMP3 (τ1 = 0.98 μs, τ2 = 9.01 μs). Based on the decay fitting data, the average PL lifetime (τavg) was calculated using the equation:
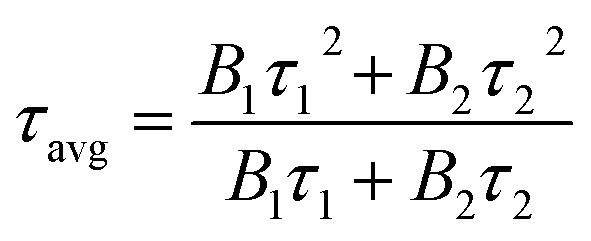
As a result, BTPT-CMP1 shows a τavg (7.63 μs), which is lower than that of BTPT-CMP2 (7.87 μs) and BTPT-CMP3 (8.16 μs), suggesting the higher activity of photogenerated electron in BTPT-CMP1. According to these results, it could be inferred that BTPT-CMP1 might possess better photocatalytic activity than BTPT-CMP2 and BTPT-CMP3.
The separation efficiency and transport properties of photo-generated carriers in polymers were also characterized by photoelectrochemical measurement. As shown in Fig. 7a, the transient photocurrents under light irradiation are 0.87 μA cm−2 for BTPT-CMP1, 0.54 μA cm−2 for BTPT-CMP2 and 0.49 μA cm−2 for BTPT-CMP3, respectively. BTPT-CMP1 possesses much higher photocurrent than BTPT-CMP2 and BTPT-CMP3, implying that BTPT-CMP1 could produce more photo-generated electrons for photocatalytic reaction,45,46 which is consistent with the result of PL spectra. The larger specific surface area may be an critical reason to enhance the photocurrent of BTPT-CMP1 (Fig. 5).47,48
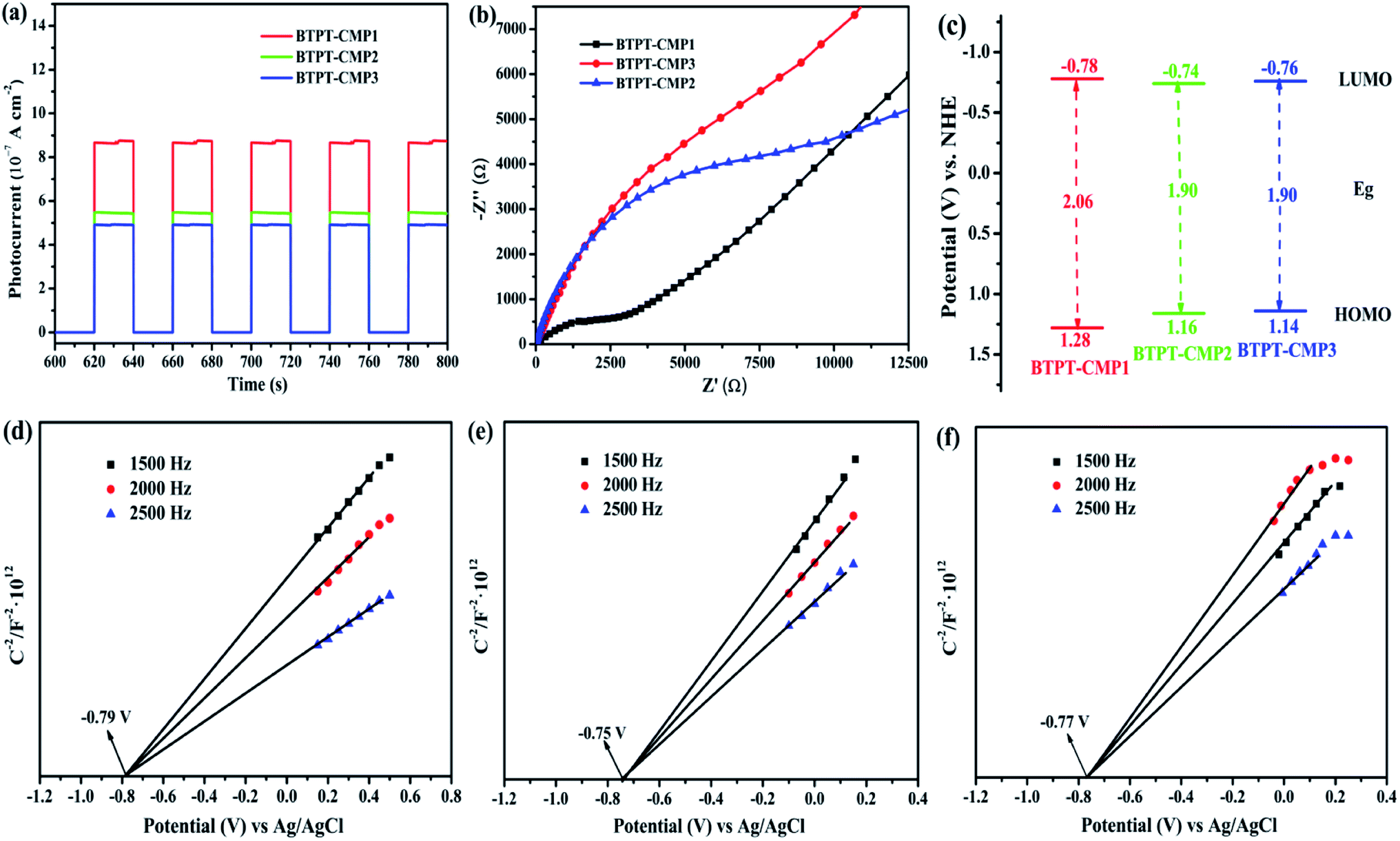 | ||
| Fig. 7 (a) Transient photocurrent and (b) Nyquist impedance plots of polymers measured in 0.5 M Na2SO4; (c) the band structure diagram of polymers; Mott–Schottky plots of BTPT-CMP1 (d), BTPT-CMP2 (e) and BTPT-CMP3 (f) at different frequency in an aqueous solution of Na2SO4 (0.5 M). | ||
The three prepared polymers possess similar molecular structures, however, PL spectra and photoelectrochemical measurement indicate that BTPT-CMP1 could produce more photo-generated electrons relative to BTPT-CMP2 and BTPT-CMP3. To investigate the reasons for the differences, electrochemical impedance spectroscopy (EIS) measurement of the three polymers was performed from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz to 0.01 Hz. BTPT-CMP1 depicts smaller semicircle arc compared with BTPT-CMP2 and BTPT-CMP3 (Fig. 7b), which suggests little interface charge transfer resistance of BTPT-CMP1. As we known that little interface charge transfer resistance of polymer is beneficial to photo-generated electrons transport, further to activate reaction substrates.49
000 Hz to 0.01 Hz. BTPT-CMP1 depicts smaller semicircle arc compared with BTPT-CMP2 and BTPT-CMP3 (Fig. 7b), which suggests little interface charge transfer resistance of BTPT-CMP1. As we known that little interface charge transfer resistance of polymer is beneficial to photo-generated electrons transport, further to activate reaction substrates.49
One important factor to determine the photocatalytic ability is the energy levels of conjugated polymers. Herein, the energy levels of the three polymers were characterized with the help of the cyclic voltammetry curves (Fig. S4†) and the analysis on Mott–Schottky plots. The HOMO and LUMO energy levels of the polymers can be calculated by employing the following equations:
| LUMO = −e(Eonset,re + 4.8 − 0.50) | (1) |
| HOMO = LUMO − Eg | (2) |
| Polymer | SBETa (m2 g−1) | λonsetb (nm) | HOMOc (V) | LUMOc (V) | Egd (eV) | HERe (μmol g−1 h−1) |
|---|---|---|---|---|---|---|
| a Specific surface area calculated from the N2 sorption-desorption isotherm.b Initial optical absorption.c Energy level vs. NHE calculated from CV curve.d The optical bandgap obtained by the plot of (αhν)2 against hν in UV-Vis reflectance spectroscopy.e 50 mg sample with 3 wt% Pt under visible light (λ > 420 nm). | ||||||
| BTPT-CMP1 | 138.4 | 618 | 1.28 | −0.78 | 2.06 | 5561.87 |
| BTPT-CMP2 | 62.1 | 659 | 1.16 | −0.74 | 1.90 | 1840.86 |
| BTPT-CMP3 | 58.8 | 659 | 1.14 | −0.76 | 1.90 | 1600.48 |
3.3. The photocatalytic performance of CMPs
The photocatalytic H2-production activities was evaluated by using 50 mg samples with 3 wt% Pt in a 5:1 (v/v) mixture of H2O/TEOA under visible light. Fig. 8a records the H2 production activity of BTPT-CMP1, BTPT-CMP2 and BTPT-CMP3 within 3 h under the same conditions. The amount of photocatalytic H2 evolution are 16685.6 μmol g−1 for BTPT-CMP1, 5522.5 μmol g−1 for BTPT-CMP2 and 4801.4 μmol g−1 BTPT-CMP3, respectively. The H2 production rate of BTPT-CMP1 is 3.02 times higher than that of BTPT-CMP2 and 3.47 times higher than that of BTPT-CMP3. The corresponding H2 evolution rates of the samples are presented in Fig. 8b. The corresponding H2 evolution rates of BTPT-CMP1, BTPT-CMP2 and BTPT-CMP3 are 5561.87 μmol g−1 h−1, 1840.86 μmol g−1 h−1 and 1600.48 μmol g−1 h−1, respectively. It could be seen that the H2 evolution rates of polymers decreased gradually with the increase of molecular cross-linker length. The better H2 evolution efficiency of BTPT-CMP1 could be ascribed to the following reasons: (1) the larger specific surface area and wider pore-size distribution are beneficial to provide more active sites for BTPT-CMP1; (2) little interface charge transfer resistance of BTPT-CMP1 help photo-generated electrons transport, and further activate more reaction substrates.
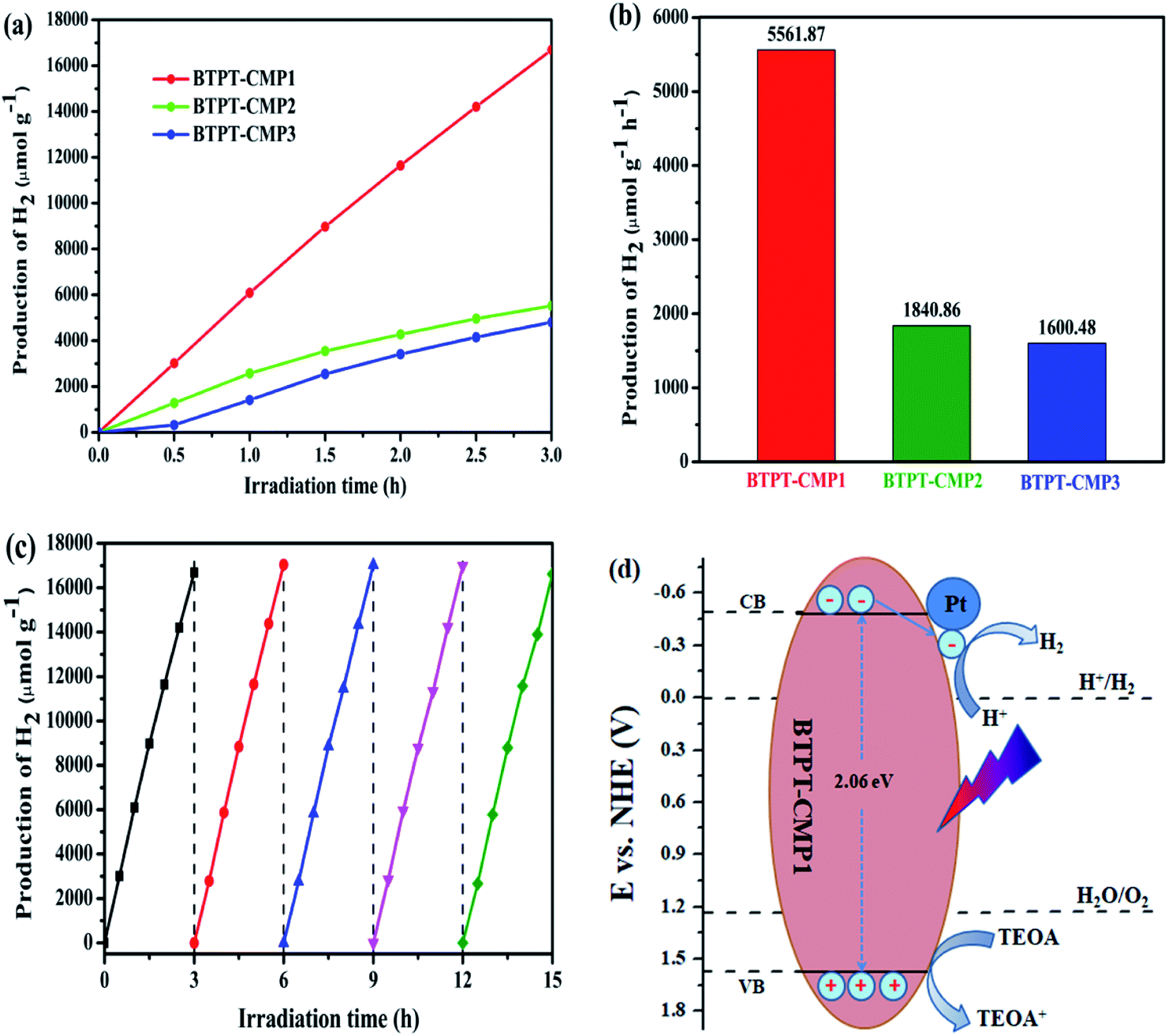 | ||
| Fig. 8 (a) Hydrogen evolution of the three polymers with 3 wt% Pt in a 5:1 mixture of H2O/TEOA under visible light (λ > 420 nm); (b) the H2 production rates of the three polymers; (c) cycling stability of BTPT-CMP1; (d) photocatalytic mechanism of BTPT-CMP1. | ||
Significantly, 5561.87 μmol g−1 h−1 H2 production rate of BTPT-CMP1 is one of the most optimum values among the conjugated polymers. To assess the photocatalytic hydrogen production efficiency of BTPT-CMP1 comprehensively, triazine based conjugated polymers have been list in Table 2. BTPT-CMP1 exhibits higher hydrogen evolution rate than the most 1,3,5-triazine based conjugated polymers, which suggest the great application prospect of BTPT-CMP1 in photocatalysis field.
| Photocatalyst | Cocatalyst content | HER (μmol g−1 h−1) | Reference |
|---|---|---|---|
| BTPT-CMP1 | 3 wt% Pt | 5561 | This work |
| Cl-ECF | 3 wt% Pt | 1296 | 50 |
| CTF-2 | 3 wt% Pt | 296 | 51 |
| THT-TA | 3 wt% Pt | 246 | 52 |
| P1 | 3 wt% Pt | 1000 | 53 |
| PCTF-1 | 1 wt% Pt | 475 | 54 |
| ZrTTA-6SH-ZnTFPP | 3 wt% Pt | 175 | 55 |
| CTFS10 | 3 wt% Pt | 2000 | 56 |
| CTF-1_10 min | 3 wt% Pt | 1072 | 57 |
| CTF-N | 2.11 wt% Pt | 10760 |
58 |
| TFA-COF | 3 wt% Pt | 80 | 59 |
Considering the potential application prospect of BTPT-CMP1, its long-term photocatalytic stability experiment is carried out under same condition. As presented in Fig. 8c, a linearly increasing amount of H2 evolved during 15 h is obtained. No obvious deactivation is observed after five cycles, demonstrating that BTPT-CMP1 as photocatalyst keeps good stability during hydrogen production procedure. The apparent quantum yield (AQY) of polymers were further investigated by using various band-pass filter. As shown in Fig. S5,† the AQY of BTPT-CMP1 recorded at 405, 420, 450, 500, 550 and 630 nm were 3.6%, 3.8%, 3.5%, 3.1%, 2.5% and 0.9%, respectively. The maximum AQY of 3.8% was obtained at 420 nm, which matches well with the UV/Vis absorption spectrum of BTPT-CMP1. The phenomenon implies that optical absorption ability of BTPT-CMP1 has a big influence on the photocatalytic reaction for hydrogen generation. For BTPT-CMP2 and BTPT-CMP3, the AQY values show downward trend as the incident light wavelength increasing. In addition, the AQY values of BTPT-CMP2 and BTPT-CMP3 are lower than that of BTPT-CMP1, which is in agreement with photocatalytic performance change. Also, XRD, FT-IR, UV-Vis spectra, SEM and TEM of BTPT-CMP1 before and after irradiation were characterized, as shown in Fig. S6–S9.† By comparing, there is no obvious structure difference for BTPT-CMP1 before and after irradiation, demonstrating its good structure stability.
3.4. The photocatalytic mechanism of CMPs
The photocatalytic reaction mechanism of BTPT-CMP1 is proposed according to these results, as displayed in Fig. 8d. During the photocatalytic reaction process, the conjugated microporous polymer absorb photons to generate excitons (electron and hole pairs). Then, the photo-induced electrons and holes transfer to CB and VB of BTPT-CMP1, respectively. Owing to the presence of heterojunctions between BTPT-CMP1 and Pt co-catalyst with lower Fermi energy level25,60 electron and hole pairs are directly split into free charges. The free electrons could transfer to Pt co-catalysts, and then react with H+ to evolve H2 directly. At the same time, the free holes on VB of the polymer react with TEOA to form oxidization product, TEOA+.4. Conclusion
In conclusion, we have constructed three D–A type conjugated microporous polymers (CMPs) by using 1,3,5-triazine as the acceptor unit and thiophenes as the donor units, respectively. Due to the small conjugated skeleton, BTPT-CMP1 exhibits blue-shifted optical absorption than BTPT-CMP2 and BTPT-CMP3 with long cross-linkers. However, BTPT-CMP1 possess higher photocurrent than BTPT-CMP2 and BTPT-CMP3 due to the large specific surface area and little interface charge transfer resistance of BTPT-CMP1. An impressive hydrogen production rate of 5561.87 μmol g−1 h−1 was obtained by BTPT-CMP1, which is 3.02 times higher than that of BTPT-CMP2 and 3.47 times higher than that of BTPT-CMP3. Among the reported 1,3,5-triazine based conjugated polymers, BTPT-CMP1 exhibits higher hydrogen evolution rate than the most 1,3,5-triazine based conjugated polymers, which suggest the great application prospect of BTPT-CMP1 in photocatalysis field.Data availability
All related data can be available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (22172069), and the Natural Science Foundation of Shandong Province (ZR2021ME071).References
- H. R. Liu, C. Z. Li, H. Li, Y. Q. Ren, J. Chen, J. T. Tang and Q. H. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 20354–20365 CrossRef CAS PubMed.
- T. M. Su, Q. Shao, Z. Z. Qin, Z. H. Guo and Z. L. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- D. Prusty, L. Paramanik and K. Parida, Energy Fuels, 2021, 35, 4670–4686 CrossRef CAS.
- S. J. Wang, J. Q. Zhang, B. Li, H. Q. Sun and S. B. Wang, Energy Fuels, 2021, 35, 6504–6526 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- M. G. Mohamed, M. H. Elsayed, A. M. Elewa, A. F. M. EL-Mahdy, C. H. Yang, A. A. K. Mohammed, H. H. Chou and S. W. Kuo, Catal. Sci. Technol., 2021, 11, 2229–2241 RSC.
- Z. J. Wang, N. Mao, Y. B. Zhao, T. J. Yang, F. Wang and J. X. Jiang, Polym. Bull., 2019, 76, 3195–3206 CrossRef CAS.
- J. Xiao, X. L. Liu, L. Pan, C. X. Shi, X. W. Zhang and J. J. Zou, ACS Catal., 2020, 10, 12256–12283 CrossRef CAS.
- Z. P. Yu, K. R. Yan, W. Ullah, H. Z. Chen and C. Z. Li, ACS Appl. Polym. Mater., 2021, 3, 60–92 CrossRef CAS.
- H. L. Nguyen and A. Alzamly, ACS Catal., 2021, 11, 9809–9824 CrossRef CAS.
- S. T. Han, T. Huang, Y. Pan, J. W. Zhao, H. Lin, H. X. Lin, Z. X. Ding, H. L. Xi and J. L. Long, Catal. Sci. Technol., 2021, 11, 4021–4025 RSC.
- F. Lan, Q. Wang, H. Chen, Y. Chen, Y. Y. Zhang, B. W. Huang, H. B. Liu, J. Liu and R. Li, ACS Catal., 2020, 10, 12976–12986 CrossRef CAS.
- A. A. Guilbert, Y. Bai, C. M. Aitchison, R. S. Sprick and M. Zbiri, ACS Appl. Polym. Mater., 2021, 3, 765–776 CrossRef CAS PubMed.
- M. G. Mohamed, E. C. Atayde Jr, B. M. Matsagar, J. Na, Y. Yamauchi, K. C. W. Wu and S. W. Kuo, J. Taiwan Inst. Chem. Eng., 2020, 112, 180–192 CrossRef CAS.
- V. R. Battula, H. Singh, S. Kumar, I. Bala, S. K. Pal and K. Kailasam, ACS Catal., 2018, 8, 6751–6759 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- W. J. Zhang, J. T. Tang, W. G. Yu, Q. Huang, Y. Fu, G. C. Kuang, C. Y. Pan and G. P. Yu, ACS Catal., 2018, 8, 8084–8091 CrossRef CAS.
- R. Gaur and P. Jeevanandam, J. Nanopart. Res., 2015, 17, 1–13 CrossRef CAS.
- J. Y. Lee, Y. J. Kwon, J. W. Woo and D. K. Moon, J. Ind. Eng. Chem., 2008, 14, 810–817 CrossRef CAS.
- Y. F. Zhi, Z. J. Yao, W. B. Jiang, H. Xia, Z. Shi, Y. Mu and X. M. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 37578–37585 CrossRef CAS PubMed.
- S. H. Luo, Z. T. Zeng, H. Wang, W. P. Xiong, B. Song, C. Y. Zhou, A. Duan, X. F. Tan, Q. Y. He, G. M. Zeng, Z. F. Liu and R. Xiao, Prog. Polym. Sci., 2021, 115, 101374 CrossRef CAS.
- H. Li, L. P. Wang and G. Yu, Nano Today, 2021, 40, 101247 CrossRef CAS.
- Z. J. Wang, X. Y. Yang, T. J. Yang, Y. B. Zhao, F. Wang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J. X. Jiang, ACS Catal., 2018, 8, 8590–8596 CrossRef CAS.
- K. W. Lin, Z. F. Wang, Z. C. Hu, P. Luo, X. Y. Yang, X. Zhang, M. Rafiq, F. Huang and Y. Cao, J. Mater. Chem. A, 2019, 7, 19087–19093 RSC.
- C. Shu, Y. B. Zhao, C. Zhang, X. M. Gao, W. Y. Ma, S. B. Ren, F. Wang, Y. Chen, J. H. Zeng and J. X. Jiang, ChemSusChem, 2020, 13, 369–375 CrossRef CAS PubMed.
- Z. A. Lan, W. Ren, X. Chen, Y. F. Zhang and X. C. Wang, Appl. Catal., B, 2019, 245, 596–603 CrossRef CAS.
- Y. B. Zhao, W. Y. Ma, Y. F. Xu, C. Zhang, Q. Wang, T. J. Yang, X. M. Gao, F. Wang, C. Yan and J. X. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- W. J. Xiao, Y. Wang, W. R. Wang, J. Li, J. D. Wang, Z. W. Xu, J. J. Li, J. H. Yao and W. S. Li, Macromolecules, 2020, 53, 2454–2463 CrossRef CAS.
- L. P. Guo, Y. L. Niu, S. Razzaque, B. Tan and S. B. Jin, ACS Catal., 2019, 9, 9438–9445 CrossRef CAS.
- S. Ghosh, A. Nakada, M. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, J. Am. Chem. Soc., 2020, 142, 9752–9762 CAS.
- S. Wang, M. K. Wang, Y. C. Liu, X. Y. Meng, Y. Ye, X. W. Song and Z. Q. Liang, Sens. Actuators, B, 2020, 326, 128808 CrossRef.
- D. Kong, X. Y. Han, J. J. Xie, Q. S. Ruan, C. D. Windle, S. Gadipelli, K. Shen, Z. M. Bai, Z. X. Guo and J. W. Tang, ACS Catal., 2019, 9, 7697–7707 CrossRef CAS PubMed.
- N. Z. Xu, Y. B. Liu, W. J. Yang, J. Tang, B. W. Cai, Q. Li, J. W. Sun, K. Q. Wang, B. L. Xu, Q. T. Zhang and Y. Fan, ACS Appl. Energy Mater., 2020, 3, 11939–11946 CrossRef CAS.
- H. Bohra and M. F. Wang, ACS Appl. Polym. Mater., 2019, 1, 1697–1706 CrossRef CAS.
- X. Xue, J. M. Luo, L. Q. Kong, J. S. Zhao, Y. Zhang, H. M. Du, S. Chen and Y. Xie, RSC Adv., 2021, 11, 10688–10698 RSC.
- R. Gomes and A. Bhaumik, RSC Adv., 2016, 6, 28047–28054 RSC.
- C. Yang, W. Huang, L. C. Silva, K. A. Zhang and X. C. Wang, Chem.–Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- J. L. Wang, G. C. Ouyang, D. W. Wang, J. Li, J. H. Yao, W. S. Li and H. X. Li, Macromolecules, 2021, 54, 2661–2666 CrossRef CAS.
- S. J. Ren, R. Dawson, D. J. Adams and A. I. Cooper, Polym. Chem., 2013, 4, 5585–5590 RSC.
- J. X. Jiang, A. Trewin, F. Su, C. D. Wood, H. J. Niu, J. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658–2666 CrossRef CAS.
- W. Y. Huang, Z. Q. Shen, J. Z. Cheng, L. L. Liu, K. Yang, X. R. Chen, H. R. Wen and S. Y. Liu, J. Mater. Chem. A, 2019, 7, 24222–24230 RSC.
- K. Lei, D. Wang, M. P. Kou, Z. Y. Ma, L. Wang, L. Q. Ye and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS PubMed.
- Y. O. Wang, M. K. Bayazit, S. J. Moniz, Q. Ruan, C. Lau, N. Martsinovich and J. W. Tang, Energy Environ. Sci., 2017, 10, 1643–1651 RSC.
- D. S. Yuan, W. Huang, X. R. Chen, Z. Y. Li, J. Ding, L. Wang, H. Wan, W. L. Dai and G. F. Guan, Appl. Surf. Sci., 2019, 489, 658–667 CrossRef CAS.
- X. L. Yang, Y. G. Xiang, Y. Qu, X. Ding and H. Chen, J. Catal., 2017, 345, 319–328 CrossRef CAS.
- X. H. Zhang, J. Xiao, M. Hou, Y. J. Xiang and H. Chen, Appl. Catal., B, 2018, 224, 871–876 CrossRef CAS.
- M. Ismael, Fuel, 2021, 303, 121207 CrossRef CAS.
- P. Karthik, R. Vinoth, P. Zhang, W. Y. Choi, E. Balaraman and B. Neppolian, ACS Appl. Energy Mater., 2018, 1, 1913–1923 CrossRef CAS.
- C. Cheng, C. L. Dong, J. W. Shi, L. H. Mao, Y. C. Huang, X. Kang, S. C. Zong and S. H. Shen, J. Mater. Sci. Technol., 2022, 98, 160–168 CrossRef.
- S. Li, M. F. Wu, T. Guo, L. L. Zheng, D. K. Wang, Y. Mu, Q. J. Xing and J. P. Zou, Appl. Catal., B, 2020, 272, 118989 CrossRef CAS.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- Z. W. Cui, Y. Hu, Y. K. Zhang, Q. T. Han, Y. Wang, Y. Zhou and Z. G. Zou, Polymer, 2020, 211, 123079 CrossRef CAS.
- J. Yu, X. Q. Sun, X. X. Xu, C. Zhang and X. M. He, Appl. Catal., B, 2019, 257, 117935 CrossRef CAS.
- Z. Cheng, W. Fang, T. S. Zhao, S. Q. Fang, J. H. Bi, S. J. Liang, L. Y. Li, Y. Yu and L. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 41415–41421 CrossRef CAS PubMed.
- Y. X. Diao, N. F. Xu, M. Q. Li, X. J. Zhu and Z. G. Xu, Inorg. Chem., 2020, 59, 12643–12649 CrossRef CAS PubMed.
- L. Y. Li, W. Fang, P. Zhang, J. H. Bi, Y. H. He, J. Y. Wang and W. Y. Su, J. Mater. Chem. A, 2016, 4, 12402–12406 RSC.
- S. Kuecken, A. Acharjya, L. Zhi, M. Schwarze, R. Schomäcker and A. Thomas, Chem. Commun., 2017, 53, 5854–5857 RSC.
- L. P. Guo, Y. L. Niu, H. T. Xu, Q. W. Li, S. Razzaque, Q. Huang, S. B. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- M. E. Ahmed, F. M. Ahmed, M. H. Elsayed, M. G. Mohamed, S. W. Kuo and H. H. Chou, Chem. Eng. J., 2021, 421, 129825 CrossRef.
- R. Wang, L. Gu, J. J. Zhou, X. L. Liu, F. Teng, C. H. Li, Y. H. Shen and Y. P. Yuan, Adv. Mater. Interfaces, 2015, 2, 1500037 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07916f |
| This journal is © The Royal Society of Chemistry 2022 |