DOI:
10.1039/D1RA07844E
(Paper)
RSC Adv., 2022,
12, 3165-3179
Utilization of transition metal fluoride-based solid support catalysts for the synthesis of sulfonamides: carbonic anhydrase inhibitory activity and in silico study†
Received
23rd October 2021
, Accepted 4th January 2022
First published on 24th January 2022
Abstract
The applications of solid support catalysts in catalyzing organic reactions are well-evident. In the present study, we explored a transition metal fluoride (FeF3) adsorbed on molecular sieves (4 Å) as a solid support catalyst for the preparation of sulfonamides 3a–3o. The solid support catalyst was characterized via X-ray diffraction and AFM analysis. The catalyst was further explored for the synthesis of indoles 6a–h, 1H-tetrazoles and 1,4-dihydropyridines. The sulfonamides prepared herein were investigated for their potential to inhibit carbonic anhydrase (hCA II, hCA IX and hCA XII). All compounds were found to be active inhibitors with IC50 values in the low micromolar range. Some compounds were even found to be highly selective inhibitors. Compound 3i only inhibited hCA II (IC50 = 2.76 ± 1.1 μM) and had <27% inhibition against hCA IX and hCA XII. Similarly, 3e (IC50 = 0.63 ± 0.14 μM) only inhibited hCA XII and showed <31% inhibition against hCA II and hCA IX. Molecular docking studies were carried out to rationalize the ligand-binding site interactions. Given the lack of selective CA inhibitors, compounds 3e and 3i can provide significant leads for the further development of highly selective CA inhibitors.
1. Introduction
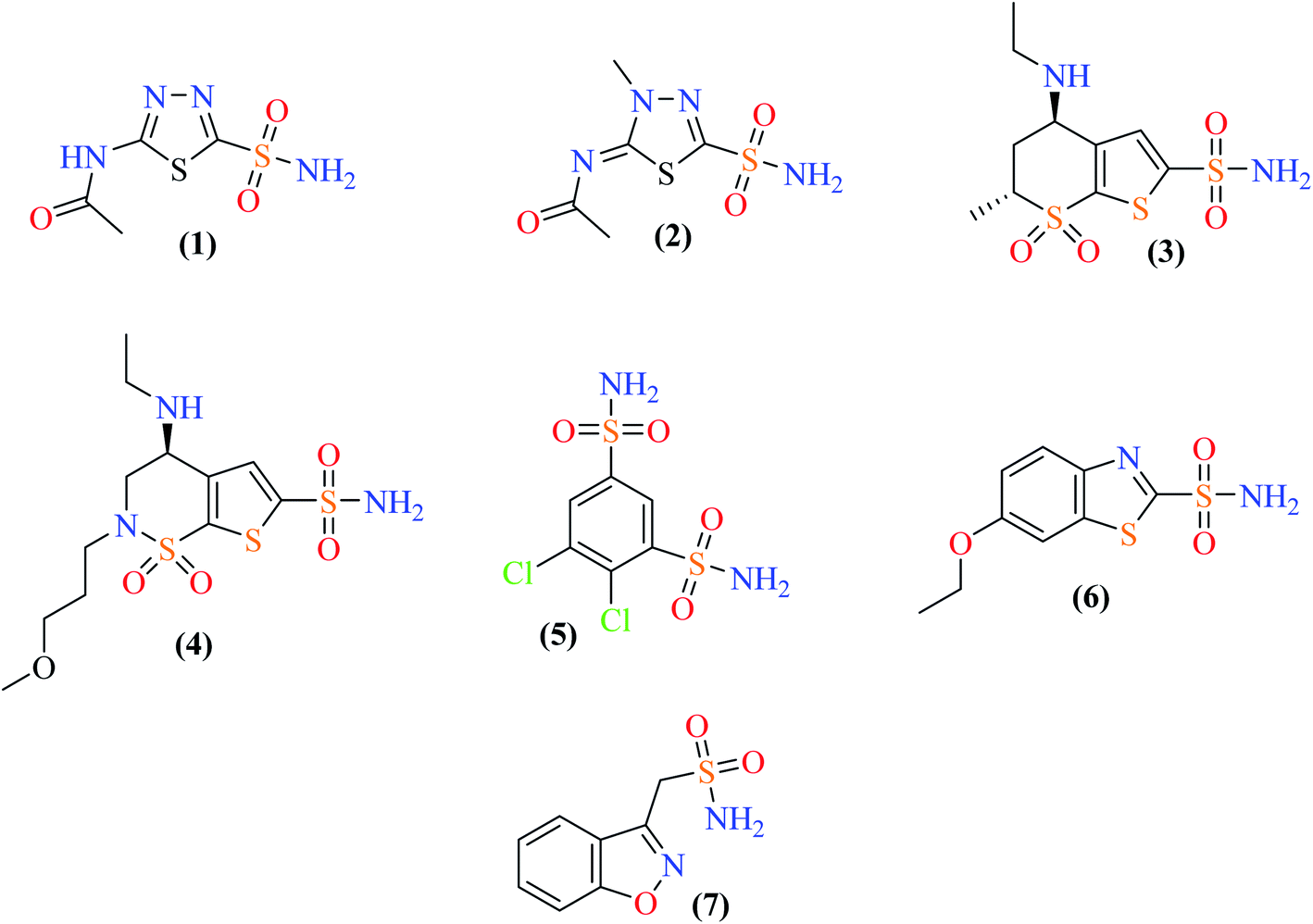
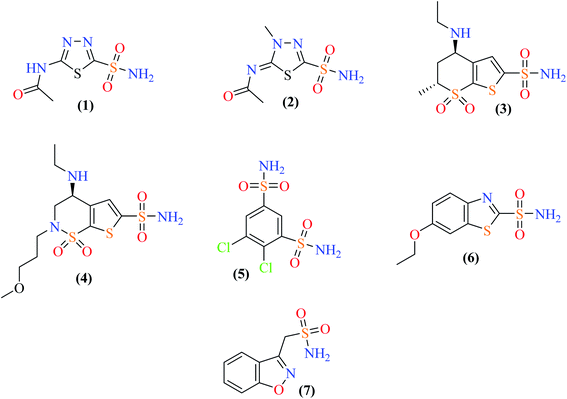
Carbonic anhydrase (CA) is a zinc-containing enzyme that is ubiquitous in nature. CA catalyzes an important physiological reaction, which is the reversible hydration of carbon dioxide to a bicarbonate ion and a proton. CA is thus crucial for maintaining homeostasis (electrolyte secretion) as well as normal physiological pH balance, lipogenesis, gluconeogenesis, and ureagenesis. In vertebrates, including humans, α-CA has several isozymes, and 16 different isozymes of CA have been found in humans. Over-expression of some CA isozymes is responsible for several disorders and diseases such as obesity, glaucoma, cancer, osteoporosis and epilepsy.1 A number of carbonic anhydrase inhibitors (CAIs) are available in the market, such as acetazolamide (1), methazolamide (2), dorzolamide (3), brinzolamide (4), dichlorfenamide (5), ethoxzolamide (6), and zonisamide (7). All these drugs are sulfonamides. Fig. 1 shows the structures of a few selected clinically used CAIs.
 |
| | Fig. 1 Structures of sulfonamide-based commercially available CAI drugs. | |
Sulfonamide is an active pharmacophore and a well-known inhibitor of the enzyme CA. It inhibits CA by direct bonding between the sulfonamide group and the Zn2+ ion of the enzyme active site. Although a number of non-sulfonamide CA inhibitors are also known, sulfonamide and its derivatives remain a popular class of compounds to explore for more potent and selective CAIs (Fig. 1).2,3
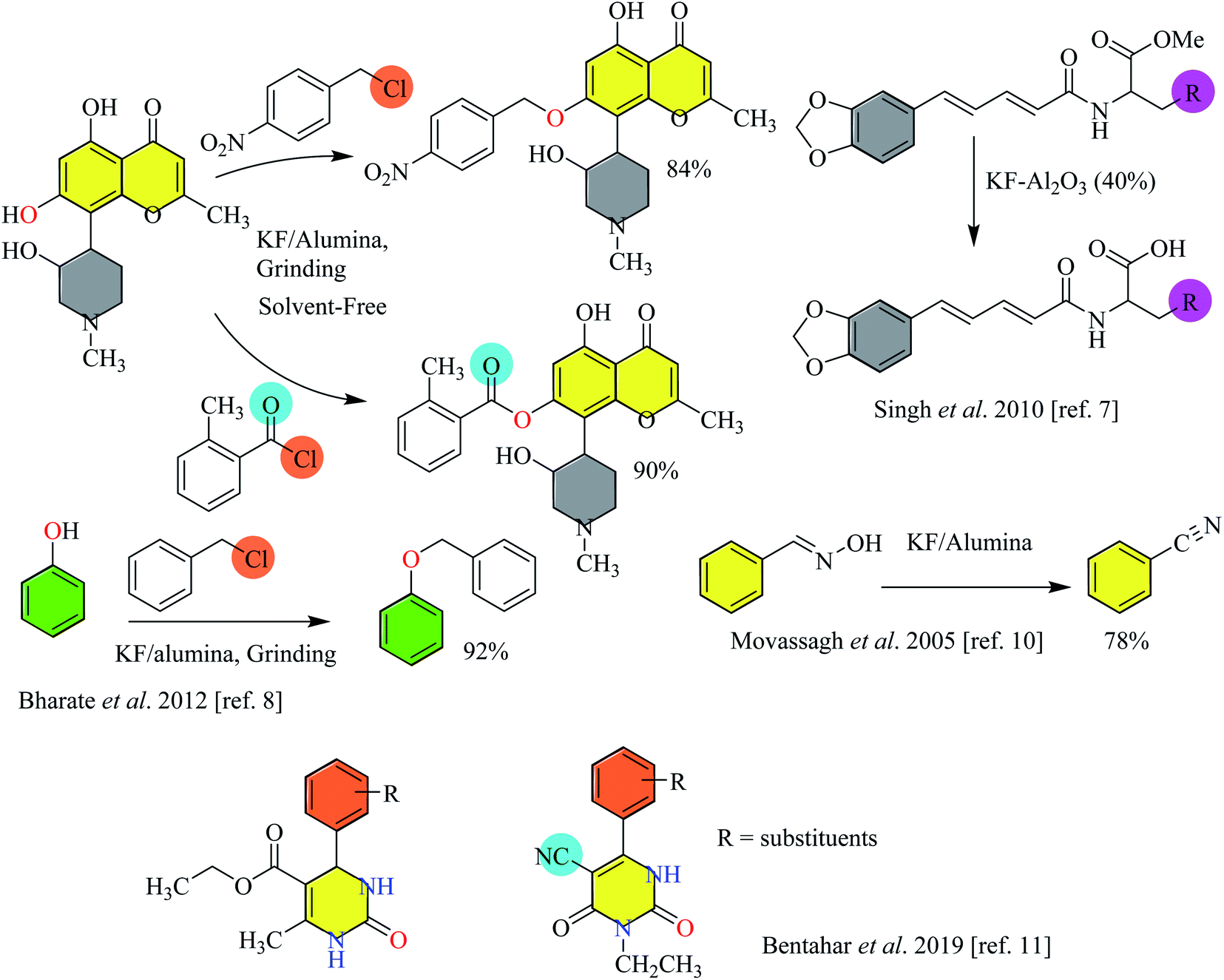
Employing fluoride ions as a base has gained much popularity in organic synthesis. In earlier applications, fluoride has been used as a mild and efficient agent to remove silyl protecting groups. The availability of a variety of fluoride salts, of both inorganic and organic origin, has tremendously increased the utility of these fluorides as catalysts in synthetic chemistry. The choice of solvent sways the basicity of fluoride salts, i.e. the use of protic solvents for alkali metal fluorides and aprotic solvents for tetraalkylammonium fluorides is generally preferred. Ionic fluorides such as potassium fluoride (KF), sodium fluoride (NaF), cesium fluoride (CsF) or tetrabutyl ammonium fluoride (TBAF) have been largely exploited for catalyzing different types of organic reactions both in advancing chemical synthesis and for the preparation of different scaffolds of medicinal interest.4,5 The application of fluoride salts such as potassium fluoride impregnated on alumina has also been considered in organic synthesis.6,7 Bharate et al. reported the selective O-benzylation/O-benzoylation, cinnamylation of substituted phenols, other aromatics, and heteroaromatics with different benzyl/heteroaryl and cinnamyl halides. Some examples with excellent yields have been shown in Scheme 1. Moreover, the methodology has also been expanded to perform N- and S-benzylation.8 Kuwano et al. carried out a one-pot synthesis of tetrahydronaphthalenes by treating the corresponding compounds with acetic anhydride and methyl acrylate in the presence of potassium fluoride.9 Further, Movassagh and Shokri reported the synthesis of nitrile from the corresponding alkyl and aryl aldoximes. For example, benzonitrile has been prepared from the corresponding aldoxime in KF/alumina via dehydration10 (Scheme 1). Recently, Bentahar et al. used KF modified clay for the synthesis of 3,4-dihydropyrimidin-2(1H)-ones via a multicomponent reaction from the corresponding starting materials.11
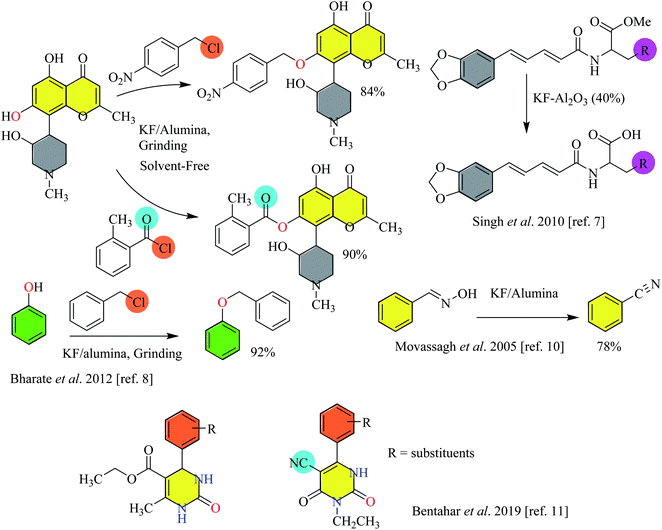 |
| | Scheme 1 Application of fluoride salts in various types of organic reactions.7–11 | |
The application of heterogeneous catalysis is well known among synthetic chemists for carrying out organic transformations, especially in the realm of green chemistry. The adsorption of fluoride salts on various solid support materials such as alumina, silica, etc., for use as heterogeneous catalysts has several advantages, such as the avoidance of tedious aqueous work-up; it also provides a reliable and quick way of executing a diverse array of synthetic reactions. In this context, the fluoride salts of alkali metals (such as KF and CsF) have been extensively studied. However, the highly reactive nature of alkali metals and their salts often encumbers their usage, therefore urging the development of mild, solid support heterogeneous catalysts. Moreover, because of the comparatively mild nature of transition metal fluorides, many undesired side reactions during the synthesis can be easily avoided, which further adds to their appeal as catalysts in organic chemistry. In the present study, we adsorbed readily available iron(III) fluoride (FeF3) on crushed molecular sieves 4 Å (Na2O·Al2O3·2SiO2·9/2H2O) to use as a catalyst for the preparation of sulfonamides. Further, to broaden the scope of the FeF3-MS 4 Å solid support catalyst, click reaction (for 1H-tetrazole formation) and multicomponent Hantzsch reaction (for the synthesis of dihydropyridines) was optimized with this catalyst.
In the current study, the iron(III) fluoride was impregnated on the crushed molecular sieves 4 Å via dissolving and evaporating ethanol as solvent. The adsorbed FeF3-MS 4 Å material was characterized using IR, X-ray diffraction, and atomic force microscopic (AFM) techniques.
Given the immense medicinal importance of sulfonamides, herein, we have designed, synthesized, and evaluated the molecules of a series of sulfonamide derivatives as carbonic anhydrase inhibitors.
2. Experimental
2.1 Catalyst preparation
The impregnation of transition metal fluoride (FeF3) on crushed molecular sieves (MS) 4 Å was carried out by a simple dissolution and evaporation technique. The iron(III) fluoride (0.1 g) was dissolved in water (3–5 mL) along with finely ground MS 4 Å (0.1 g) at room temperature. The resulting mixture was stirred well to make it homogenous. The water from the mixture was removed via a freeze dryer to get dried iron fluoride adsorbed solid support material (FeF3/crushed MS 4 Å).
2.2 Synthetic protocol for the synthesis of sulfonamides
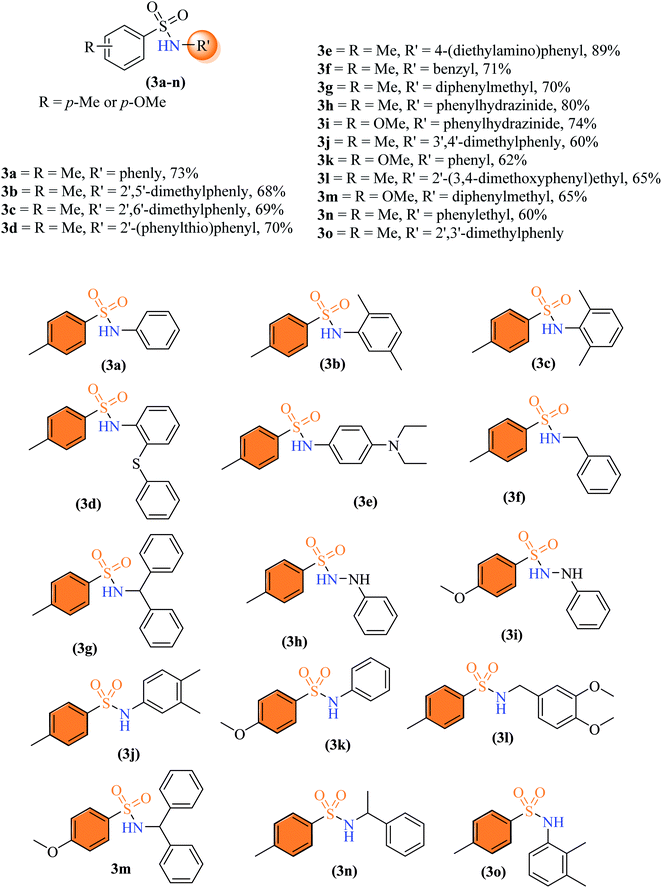
By using the optimized reaction conditions, the primary amine (1 mmol, 1.0 equiv.), p-toluenesulfonyl chloride (1.5 mmol, 1.5 equiv.), along with solid support catalyst (100 mg) was placed in a round bottom flask. Further, ethanol (3 mL) as solvent was added to homogenize the reaction mixture. The resulting reaction mixture was stirred, heated at reflux, and monitored by thin layer chromatography until the complete consumption of reactants. The reaction mixture was cooled to room temperature, and the catalyst was separated from the reaction mixture via filtration. The solvent was evaporated, and the crude product was combined with 15 mL of water before being extracted with ethyl acetate (10 mL × 3). The combined organic layers were dried with anhydrous sodium sulphate (previously heated) and filtered. The crude product was purified by column chromatography using a gradient solvent system of ethyl acetate and n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to 3:1) to get the corresponding pure products 3a–3o.
:3 to 3:1) to get the corresponding pure products 3a–3o.
2.2.1 4-Methyl-N-phenylbenzenesulfonamide (3a). Off-white solid; yield 62%, 153 mg, mp 101–103 °C, (lit. 101.5–102.2 °C).12 IR νmax, cm−1: (solid, KBr) 3239 (NH), 1658, 1598, 1483, 1338, 1158, 1089; 1H-NMR (400 MHz, DMSO-d6): δH 10.16 (1H, brs, NH), 7.63 (2H, d, ArH, J = 8.4 Hz), 7.33 (2H, d, ArH, J = 8.0 Hz), 7.20 (2H, app t, ArH, J = 7.8 Hz), 7.07 (2H, d, ArH, J = 8.0 Hz), 6.99 (1H, app t, ArH, J = 7.4 Hz), 2.31 (3H, s, Ar–CH3). EI-MS m/z (%) 247 (M+, 100), 155 (79), 91, (90).
2.2.2. N-(2,5-Dimethylphenyl)-4-methylbenzenesulfonamide (3b). Grey colour, yield 68%, 187 mg, mp 114–116 °C, IR νmax, cm−1: (solid, KBr) 3264 (NH), 1596, 1388, 1330, 1168, 1092, 947.1H-NMR (400 MHz, DMSO-d6): δH 9.37 (1H, s, NH), 7.52 (2H, d, ArH, J = 8.0 Hz), 7.33 (2H, d, ArH, J = 8.4 Hz), 6.98 (1H, d, ArH, J = 8.0 Hz), 6.88 (1H, d, ArH, J = 8.0 Hz), 6.81 (1H, s, ArH), 2.35 (3H, s, Ar–CH3), 2.15 (3H, s, Ar–CH3), 1.87 (3H, s, Ar–CH3). EI-MS m/z (%) 275 (M+, 94), 155 (7), 120 (87), 91 (16).
2.2.3 N-(2,6-Dimethylphenyl)-4-methylbenzenesulfonamide (3c). White colour, yield 69%, 190 mg, mp 136–138 °C, IR νmax, cm−1: (solid, KBr) 3270 (NH), 1596, 1376, 1158, 1091. 1H-NMR (400 MHz, DMSO-d6): δH 9.21 (1H, s, NH), 7.53 (2H, d, ArH, J = 8.0 Hz), 7.35 (2H, d, ArH, J = 8.0 Hz), 7.06–6.97 (3H, m, 3ArH), 2.37 (3H, s, Ar–CH3), 1.92 (6H, s, 2Ar–CH3). EI-MS, m/z (%) 247 (M+, 36), 120 (100), 91 (19).
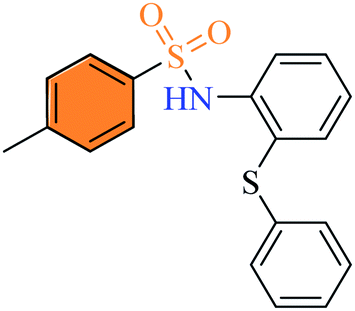
2.2.4 4-Methyl-N-(2-(phenylthio) phenyl) benzenesulfonamide (3d). White colour, yield 57%, 202 mg, mp 87–89 °C, (lit. 88 °C),13 IR νmax, cm−1: (solid, KBr) 3308 (NH), 1586, 1380, 1281, 1162, 915. 1H-NMR (400 MHz, DMSO-d6): δH 9.66 (1H, s, NH), 7.59 (2H, d, ArH, J = 8.0 Hz), 7.32–7.27 (5H, m, ArH), 7.21 (1H, td, ArH, J = 8.0, 4.0 Hz), 7.13 (2H, d, ArH, J = 8.0 Hz), 7.07–7.03 (3H, m, ArH), 2.33 (1H, s, Ar–CH3). EI-MS, m/z (%) 355.3 (95), 291.3 (4), 200.2 (100), 167.2 (36), 91.1 (17) 77.1 (19), 65.1 (10).
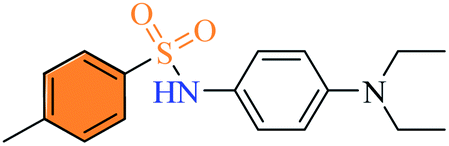
2.2.5 N-(4-(Diethylamino) phenyl)-4-methylbenzenesulfonamide (3e). Greenish blue colour, yield 89%, 0.28 g, mp 157–159 °C, IR νmax, cm−1: (solid, KBr) 3226 (NH), 1611, 1516, 1355, 1268, 1155, 1011, 924. 1H-NMR (400 MHz, DMSO-d6): δH 9.47 (1H, brs, NH), 7.54 (2H, d, ArH, J = 8.0 Hz), 7.31 (2H, d, ArH, J = 8.0 Hz), 6.80 (2H, d, ArH, J = 12.0 Hz), 6.49 (2H, d, ArH, J = 8.0 Hz), 3.24 (4H, q, –(CH2CH3)2, J = 8.0 Hz), 2.49 (3H, s, Ar–CH3), 1.00 (6H, t, –(CH2CH3)2, J = 8.0 Hz). 13C-NMR (100 MHz, DMSO-d6): δC 145.2 (C), 142.5 (C), 137.1 (C), 129.3 (2 × CH), 126.6 (2 × CH), 124.9 (C), 124.4 (2 × CH), 111.6 (2 × CH), 43.5 (2 × CH2), 20.8 (Ar–CH3), 12.2 (2 × CH3). EI-MS m/z (%) 318 (M+, 18), 163 (100), 148 (6), 91 (6).
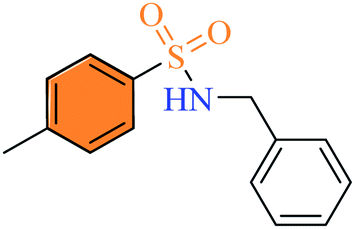
2.2.6 N-benzyl-4-methylbenzenesulfonamide (3f). Off-white to yellow colour, yield 63%, 165 mg, mp 114–116 °C (lit. 115.1–115.9 °C).12 IR νmax, cm−1 (solid, KBr) 3269 (NH), 1597, 1455, 1325, 1161, 1058, 1028, 912. 1H-NMR (400 MHz, DMSO-d6): δH 8.03 (1H, s, NH), 7.68 (2H, d, ArH, J = 8.0 Hz), 7.37 (2H, d, ArH, J = 8.0 Hz), 7.27 (3H, app t, ArH, J = 8.0 Hz), 7.22 (2H, app d, ArH, J = 8.0 Hz), 3.93 (2H, s, Ph–CH2), 2.37 (3H, s, Ar–CH3). EI-MS m/z (%) 262 (M+ 1, 14) 261 (M+, 1.9), 155 (45), 106 (100), 92 (75), 90.9992 (82).
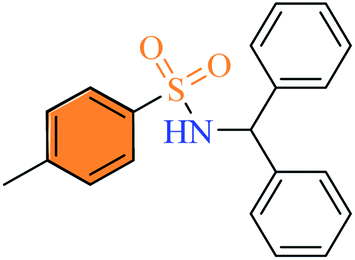
2.2.7 N-Benzhydryl-4-methylbenzenesulfonamide (3g). White color, yield 68%, 229 mg, mp 156–158 °C (lit. 156–158 °C);14 IR νmax, cm−1: (solid, KBr) 3248 (NH), 1599, 1314, 1161, ArH, J, 1027, 939. 1H-NMR (400 MHz, DMSO-d6): δH 8.69 (1H, d, NH, J = 8.0 Hz), 7.49 (2H, d, ArH, J = 8.0 Hz), 7.21–7.11 (12H, m, ArH), 5.52 (1H, d, (Ph)2–CH, J = 8.0 Hz), 2.27 (3H, s, Ar–CH3). ESI-MS m/z (M + H) 338.2.
2.2.8 4-Methyl-N′-phenylbenzenesulfonohydrazide (3h). Off-white color, yield 80%, 209 mg, mp 154–156 °C, (lit. 154–155 °C).15, IR νmax, cm−1: (solid, KBr) 3315, 3242 (NH), 1325, 1158, 1090, 1H-NMR (400 MHz, DMSO-d6): δH 9.42 (1H, brs, –NH–NH), 7.71 (2H, d, ArH, J = 8.0 Hz), 7.55 (1H, brs, –NH–NH), 7.40 (2H, d, ArH, J = 8.0 Hz), 7.08 (2H, t, ArH, J = 8.0 Hz), 6.78 (2H, d, ArH, J = 8.0 Hz), 6.68 (1H, t, ArH, J = 8.0 Hz), 2.38 (3H, s, Ar–CH3). EI-MS, m/z (%) 262 (M+, 15), 155 (2.4), 107 (100), 91 (50), 77 (76.9).
2.2.9 4-Methoxy-N′-phenylbenzenesulfonohydrazide (3i). Off-white color, yield 69%, 192 mg, mp 160–162 °C, IR νmax, cm−1: (solid, KBr) 3348, 3253 (NH), 1598, 1328, 1265, 1153, 1019, 887. 1H-NMR (400 MHz, DMSO-d6): δH 9.32 (1H, d –NH–NH, J = 0.6 Hz), 7.74 (2H, d, ArH, J = 8.0 Hz), 7.54 (1H, brs, –NH–NH), 7.11 (2H, d, ArH, J = 8.0 Hz), 7.06 (2H, t, ArH, J = 8.0 Hz), 6.76 (2H, s, ArH, J = 8.0 Hz), 6.66 (1H, t, ArH, J = 8.0 Hz), 3.82 (3H, s, CH3O). EI-MS, m/z (%) 278.1 (M+, 27), 172 (30), 155 (50), 107 (100), 92 (100), 77 (78), 64 (13).
2.2.10 N-(3,4-Dimethylphenyl)-4-methylbenzenesulfonamide (3j). Off-white colour, yield 60%, 165 mg, mp 143–145 °C, IR νmax, cm−1: (solid, KBr) 3252 (NH), 1598, 1320, 1161, 1089, 952. 1H-NMR (400 MHz, DMSO-d6): δH 9.94 (1H, s, NH), 7.60 (2H, d, J = 8.0 Hz, ArH), 7.32 (2H, d, J = 8.0 Hz, ArH), 6.94 (1H, d, J = 8.0 Hz, ArH), 6.85 (1H, s, ArH), 6.78 (1H, d, J = 8.0 Hz, ArH), 2.32 (3H, s, CH3Ar), 2.08 (3H, s, CH3Ar), 2.07 (3H, s, CH3Ar). EI-MS m/z (%) 275 (M+, 94), 155 (2), 120 (100), 91 (20).
2.2.11 4-Methoxy-N-phenylbenzenesulfonamide (3k). Off-white colour, yield 62%, 163 mg, mp 106–108 °C, (lit. 107–108 °C);16 IR νmax, cm−1: (solid, KBr) 3257 (NH), 1599, 1337, 1267, 1159, 1219, 1017, 921. 1H-NMR (400 MHz, DMSO-d6): δH 10.10 (1H, s, NH), 7.67 (2H, d, J = 8.0 Hz, ArH), 7.20 (2H, t, J = 8.0 Hz, ArH), 7.07–6.97 (5H, m, ArH), 3.78 (3H, s, CH3O). EI-MS, m/z (%) 263 (M+, 88.6), 171 (100), 107 (47.7), 92 (34), 91 (2), 77 (23).
2.2.12 N-(3,4-Dimethoxyphenethyl)-4-methylbenzenesulfonamide (3l). Yellow colour, yield 65%, 218 mg, mp 135–137 °C, (lit. 136–137 °C),17 IR νmax, cm−1: (solid, KBr) 3267 (NH), 1668, 1326, 1160, 1260, 1023,1062, 936. 1H-NMR (400 MHz, DMSO-d6): δH 7.64 (2H, d, J = 8.0 Hz, ArH), 7.55 (1H, app t, NH, J = 5.6 Hz), 7.36 (2H, d, J = 8.0 Hz, ArH), 6.81 (1H, d, J = 8.0 Hz, ArH) 6.70 (1H, s, ArH), 6.63 (1H, app dd, J = 8.4, 1.2 Hz), 3.69 (6H, s, 2CH3O), 2.91 (2H, app q, J = 8.0 Hz, CH2CH2), 2.57 (2H, t, J = 8.0 Hz, CH2CH2), 2.36 (3H, s, CH3). EI-MS m/z (%) 335 (M+,59), 184 (18), 151 (100), 91 (24).
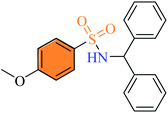
2.2.13 N-Benzhydryl-4-methoxybenzenesulfonamide (3m). Off-white colour, yield 65%, 230 mg, mp 167–169 °C, IR νmax, cm−1: (solid, KBr) 3269 (NH), 1593, 1349, 1263, 1155, 944. 1H-NMR (400 MHz, DMSO-d6): δH 8.61 (1H, d, NH, J = 12.0 Hz), 7.57 (2H, d, J = 8.0 Hz, ArH), 7.21–7.02 (10H, m, ArH),6.85 (2H, d, J = 8.0 Hz, ArH), 5.50 (1H, d, J = 12.0 Hz, CH), 3.75 (3H, s, CH3O).18 ESI-MS, M+ 1(%) 354.1.
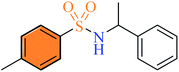
2.2.14 4-Methyl-N-(1-phenylethyl) benzenesulfonamide (3n). White color, yield 60%, 165 mg, mp 81–83 °C, (lit. 81–82 °C),12 IR νmax, cm−1: (solid, KBr) 3268 (NH), 1598, 1376, 1267, 1150, 970, 921. 1H-NMR (400 MHz, DMSO-d6): δH 7.99 (2H, d, NH) 7.61 (2H, d, J = 8.0 Hz, ArH), 7.21 (5H, m, ArH), 6.98 (2H, d, J = 8.0 Hz, ArH), 4.30 (1H, m, CH), 3.79 (3H, s, CH3), 1.18 (3H, d, J = 4.0 Hz, ArCH3). FABP-MS, M+ 1 (%), 276.2 (16).
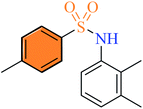
2.2.15 4-Methyl-N-(2,3-dimethylphenyl) benzenesulfonamide (3o). White color, yield 70%, mp 141–143 °C (lit. 142–143 °C),19 IR νmax, cm−1: (solid, KBr) 3252 (NH), 1598, 1499, 1391, 1320, 1246, 1161, 1089, 1008, 910. 1H-NMR (400 MHz, DMSO-d6): δH 9.41 (1H, brs, NH), 7.50 (2H, d, J = 8.0 Hz, ArH), 7.33 (2H, d, J = 8.0 Hz, ArH), 6.99 (1H, d, J = 8.0 Hz, ArH), 6.92 (1H, t, J = 8.0 Hz, ArH), 6.67 (1H, d, J = 8.0 Hz, ArH), 2.36 (3H, s, CH3), 2.15 (3H, s, CH3), 1.93 (3H, s, CH3).
3. Results and discussion
3.1 Chemistry
The solid support catalyst was prepared by adsorbing a transition metal salt (FeF3) on easily available crushed molecular sieves 4 Å via simple dissolution in water, stirring well, and evaporating the water. The catalyst was characterized with different techniques and then employed as a catalyst in performing sulfonamide preparation (Scheme 2), Fisher indole synthesis (Scheme S1†), 1H tetrazole synthesis (Scheme S2†) and multicomponent Hantzch reaction for 1,4-dihydropyridine synthesis (Scheme S3†).
 |
| | Scheme 2 Sulfonamide 3e synthesis using the solid support catalyst (FeF3/MS 4 Å). | |
3.2 Powder X-ray diffraction study
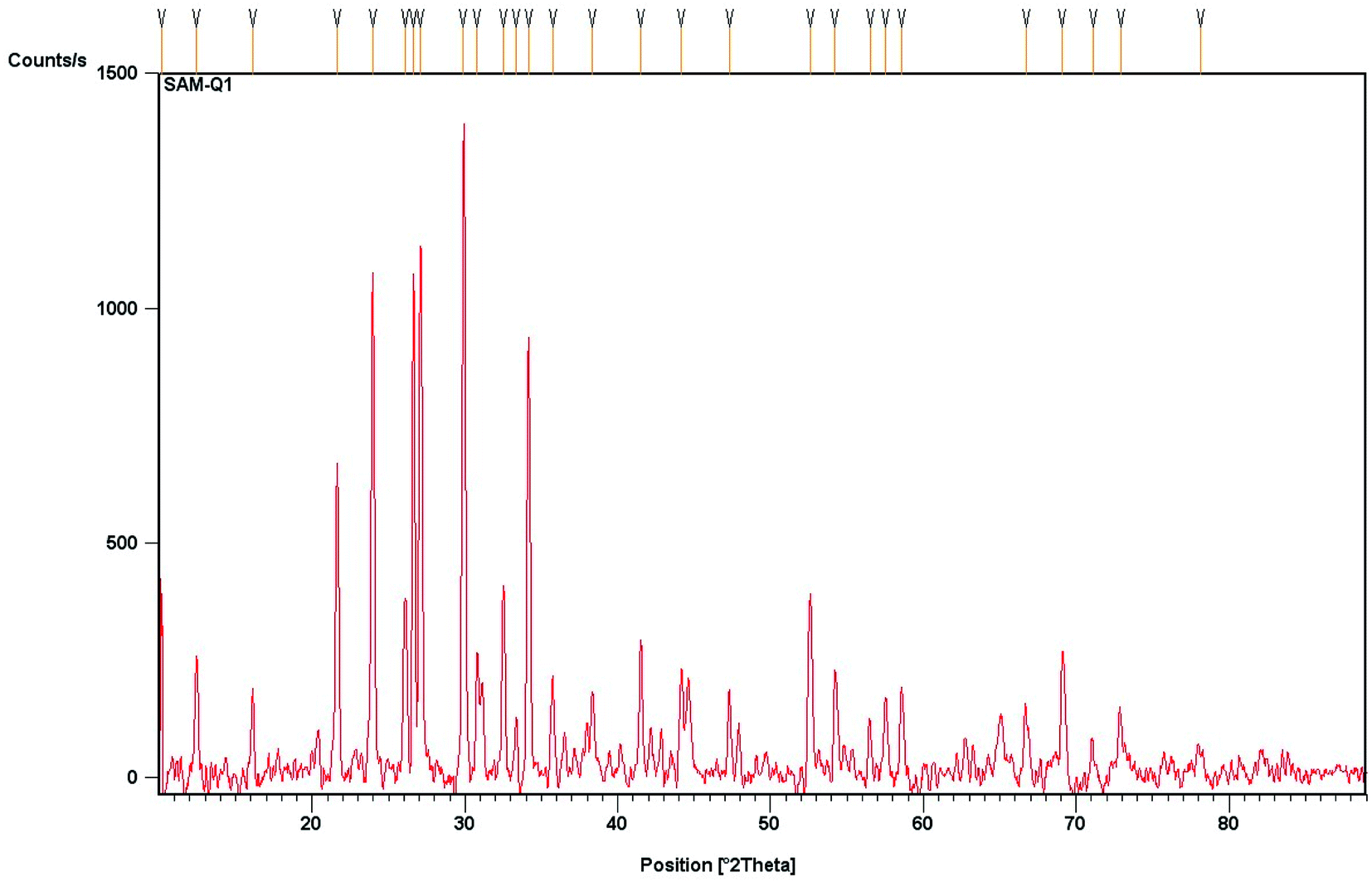
The composition studies of the solid supported material (FeF3/MS 4 Å) were carried out by using powder X-ray diffraction (XRD; Philips PW-1830) with Cu Kα radiation (λ = 1.5406 Å). The X-ray diffraction pattern revealed that the material is polycrystalline, is of composite type, and consists of FeF3 and zeolite, the typical molecular sieve constituent. The diffraction peak positions at 2θ = 10.12°, 12.44°, 16.09° are assigned to the FeF3 component. There is no peak observed for iron oxides (i.e., Fe2O3). The peaks positioned at 2θ = 21.6°, 24.0°, 26.0° and 26.65°, 27.08°, 29.85°, 30.7° are assigned typically to alumina and silica (part of zeolite). The calculated crystallite size with respect to the maximum intensity peak is 86.77 nm (Fig. 2).
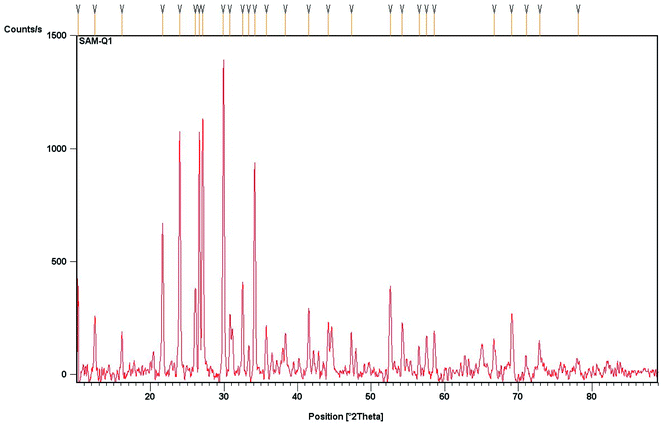 |
| | Fig. 2 Powder X-ray diffraction pattern of wt% 1/10 (FeF3/MS 4 Å). | |
3.3 AFM study of solid supported catalyst (FeF3/MS 4 Å)
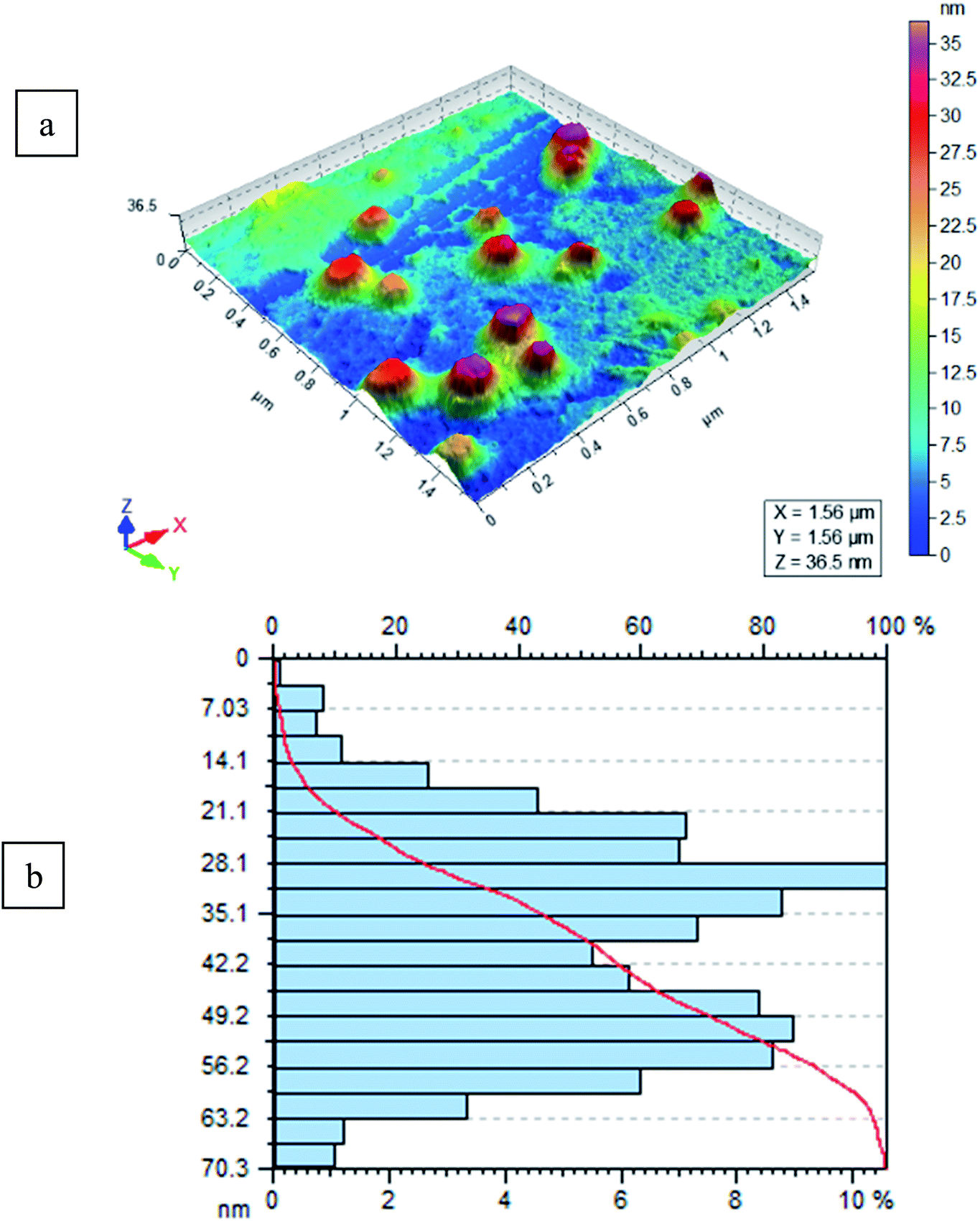
The size and morphology of FeF3/MS 4 Å were determined using AFM (AFM, Agilent 5500). A drop of FeF3 adsorbed on 4 Å molecular sieves (FeF3/MS 4 Å) in deionized water was deposited on freshly cleaved mica sheet through drop casting method, dried under inert atmosphere at room temperature, and mounted on the microscope. The slide was scanned and imaged in tapping mode by using a silicon nitride cantilever. Surface morphology, size, and size distribution of molecular sieves were studied through AFM without and with adsorption of iron(III) fluoride. Molecular sieves lacking adsorbed salt revealed spherical morphology with an average size of 80 nm, as shown in Fig. S1a,† and particle distribution from 20 to 90 nm having maximum particles in the range 40 to 50 nm (100%) is shown in Fig. S1b.† For iron adsorbed molecular sieves, the same procedure is followed. FeF3/MS 4 Å showed smooth spherical surface morphology as shown in Fig. 3a, with an average size of 100 nm. FeF3/MS 4 Å were distributed in a range of 2.5 to 70.3 nm, having maximum particles in the range of 30 nm (100%), as shown in Fig. 3b.
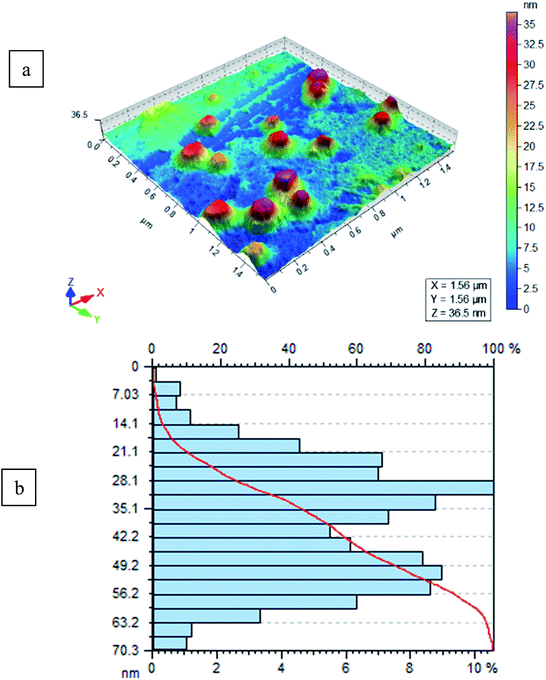 |
| | Fig. 3 (a and b) Results of FeF3-molecular sieve particle size and distribution, respectively. | |
The newly prepared solid support catalyst FeF3/MS 4 Å was employed for the synthesis of a range of sulfonamide derivatives. The synthesis of sulfonamide 3e was selected as a model reaction to get an idea about the optimal quantity of the solid support catalyst (FeF3 adsorbed on 4 Å molecular sieves) to be used for other reactions. For an optimal condition, a reaction between sulfonyl chloride 1 and the corresponding amine was carried out in the absence of the solid support catalyst; the sulfonamide 3e was obtained in a 48% yield at room temperature. While upon the addition of the catalyst (50 mg), the yield was improved to 58%, increasing the amount of catalyst (100 mg) resulted in an even improved yield (89%) in 8 h (Scheme 2, Table 1). Moreover, the optimum reaction conditions in the presence of the solid support catalyst (FeF3/MS 4 Å wt% 1/10, 100 mg) were employed to carry out other reactions, including indole, 1H-tetrazole formation, and 1,4-dihydropyridine synthesis (data in ESI†).
Table 1 Optimization of sulfonamide synthesis with different amounts of FeF3/MS 4 Å
| Entry |
FeF3/MS 4 Å (wt% 1/10) solid supported catalyst |
% Yield (3e) |
| 1 |
No catalyst |
48 |
| 2 |
50 mg |
58 |
| 3 |
100 mg |
89 |
The scope of transition metal salt (FeF3) based solid support catalyst was expanded by performing reactions between cyclohexanone and different substituted phenyl hydrazines in ethanol to get the corresponding indoles 6a–h in variable yields (Scheme S1†). Click reaction20,21 between different substituted benzonitriles and trimethylsilyl azide (TMSN3) was carried out to get the corresponding 1H-tetrazoles 8a–8c (Scheme S2†). Moreover, the transition metal based solid support catalyst (FeF3 adsorbed on 4 Å molecular sieves) was used to perform a multicomponent reaction for the synthesis of 1,4-dihydropyridines (Scheme S3†). The corresponding compounds 12a and 12b were obtained in 61% and 57% yields, respectively (Fig. 4).
 |
| | Fig. 4 Synthesis of different sulfonamides using optimal catalytic (FeF3 adsorbed on 4 Å molecular sieves) conditions. | |
3.4 Bioactivity and structure activity relationship (SAR) of sulfonamides: carbonic anhydrase inhibition activity
All compounds were found to act as active inhibitors of CAs (against at least one of three isozymes tested, that is, CA II, CA IX, and CA XII). Some compounds were even found to be selective inhibitors of CAs, as shown in Table 2. Only compounds 3b, 3e, 3i, 3k, 3l, 3m, and 3o did not inhibit all three CAs, that is, they showed selective inhibition. Whereas all other compounds indiscriminately inhibited CA II, CA IX, and CA XII.
Table 2 Carbonic anhydrase II, IX and XII inhibition activity at 100 μM
| Comp. No. |
Structures |
IC50 (μM) ± SEM/% inhibition |
| CA II |
CA IX |
CA XII |
| (3a) |
 |
1.3 ± 0.51 |
1.2 ± 0.75 |
0.56 ± 0.18 |
| (3b) |
 |
13% |
1.65 ± 1.3 |
1.03 ± 0.23 |
| (3c) |
 |
1.55 ± 0.80 |
1.06 ± 0.63 |
0.70 ± 0.20 |
| (3d) |
 |
1.02 ± 0.75 |
0.77 ± 0.59 |
1.36 ± 0.51 |
| (3e) |
 |
24% |
31% |
0.63 ± 0.14 |
| (3f) |
 |
1.07 ± 0.82 |
1.98 ± 0.61 |
0.86 ± 0.28 |
| (3g) |
 |
0.863 ± 0.54 |
0.78 ± 0.83 |
1.3 ± 0.78 |
| (3h) |
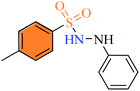 |
0.52 ± 0.44 |
0.484 ± 1.4 |
1.08 ± 0.62 |
| (3i) |
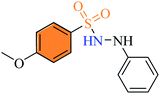 |
2.76 ± 1.1 |
19% |
27% |
| (3j) |
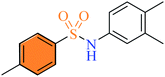 |
2.5 ± 1.5 |
0.62 ± 0.2 |
1.12 ± 1.00 |
| (3k) |
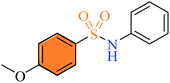 |
1.74 ± 0.54 |
21% |
1.14 ± 0.37 |
| (3l) |
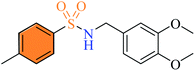 |
32% |
0.764 ± 0.54 |
1.17 ± 1.0 |
| (3m) |
 |
0.507 ± 0.4 |
16% |
1.40 ± 0.57 |
| (3n) |
 |
1.29 ± 0.6 |
2.01 ± 1.3 |
1.96 ± 0.66 |
| (3o) |
 |
1.91 ± 0.69 |
0.32 ± 0.61 |
30% |
| Reference |
Acetazolamide |
1.19 ± 0.042 |
1.08 ± 0.03 |
1.55 ± 0.037 |
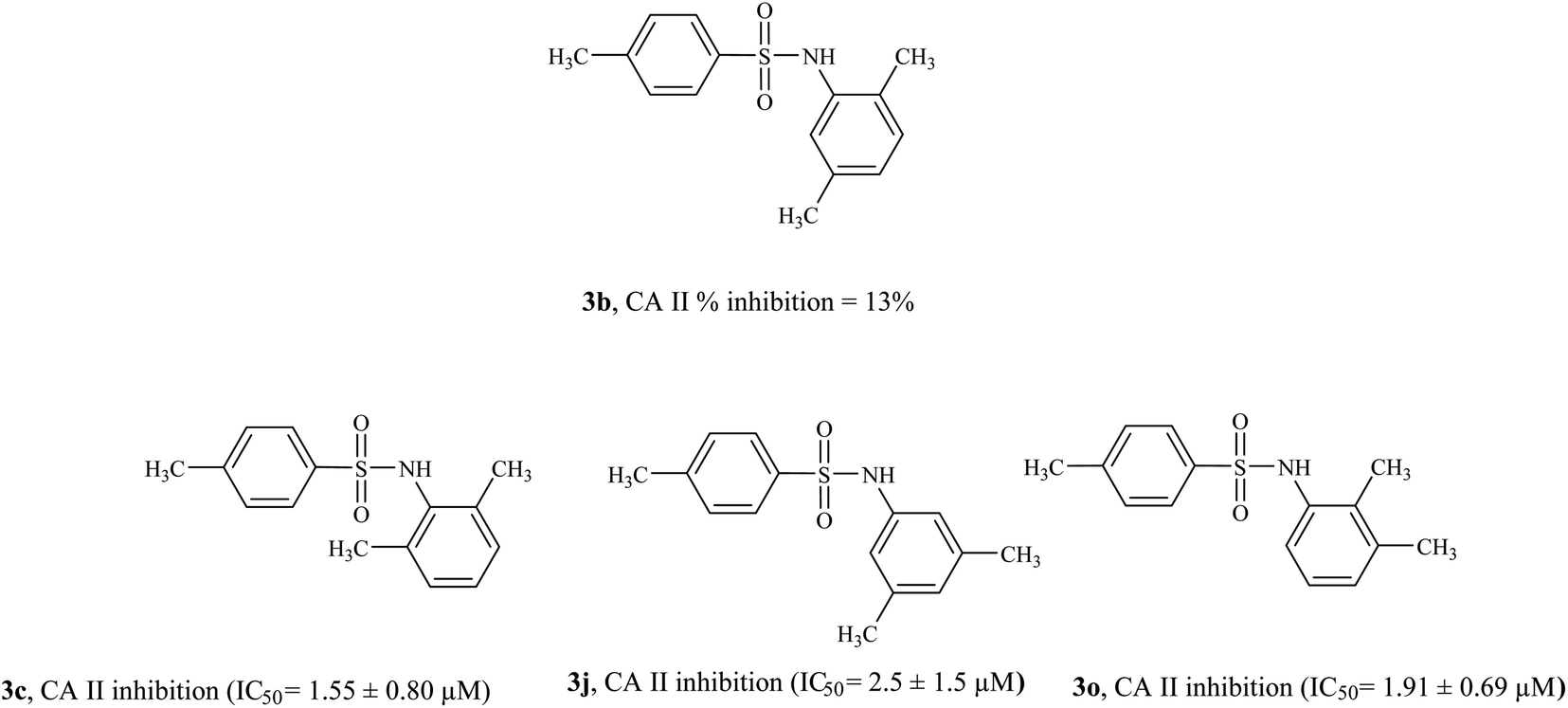
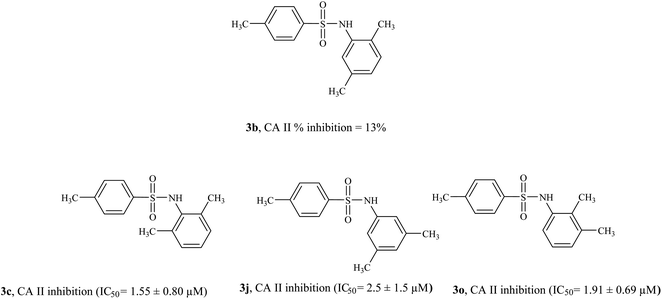
3.4.1 Structure activity relationship of CA II inhibitors. Compound 3h and 3m were found to be the most active CA II inhibitors having IC50 values 0.52 ± 0.44 μM and 0.507 ± 0.4 μM, respectively. Compound 3g also showed excellent inhibition activity (IC50 = 0.863 ± 0.54 μM). Compounds 3b, 3e, and 3l were the only compounds in the series that did not exhibit CA II inhibition activity (% inhibition < 50%). The position of the alkyl substituent on the phenyl ring seems to affect CA II inhibition activity significantly (Fig. 5). Among di-alkyl substituted sulfonamides (3b, 3c, 3j, 3o), compound 3b (having 2,5-dimethyl substituent) was the only compound not active against CA II. Interestingly, compound 3c having 2,6-dimethyl substituent on the phenyl ring was very active against CA II (IC50 = 1.55 ± 0.80 μM); similarly compounds 3j (having 3,4-dimethyl substituent, IC50 = 2.5 ± 1.5 μM) and 3o (having 2,3-dimethyl substituent, IC50 = 1.91 ± 0.69 μM) were also active CA II inhibitors. Compounds 3d, 3f, 3g, 3h, and 3m were more active than the standard inhibitor acetazolamide (IC50 = 1.19 ± 0.042 μM).
 |
| | Fig. 5 Structure activity relationship of CA II inhibitors. | |
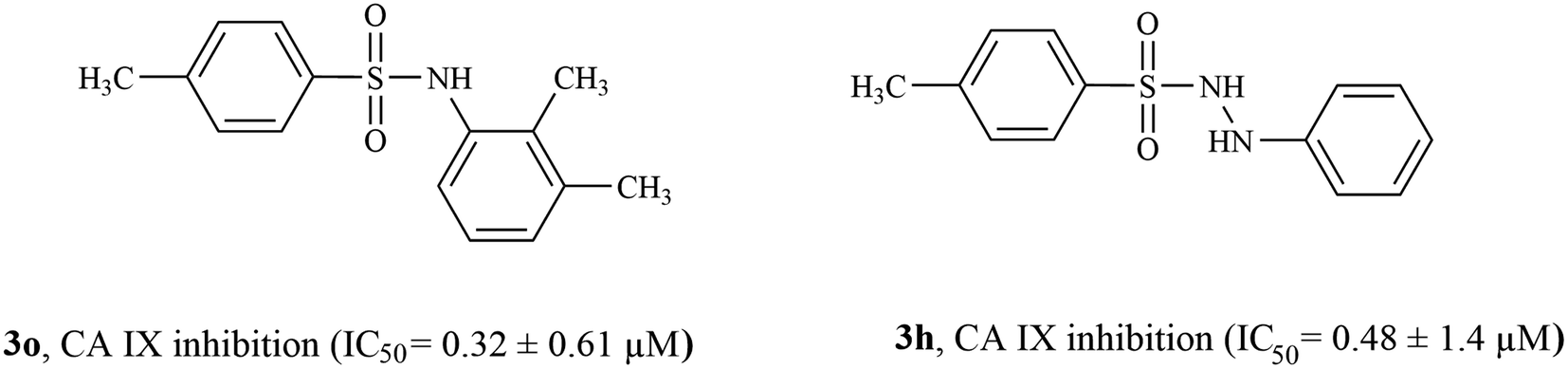
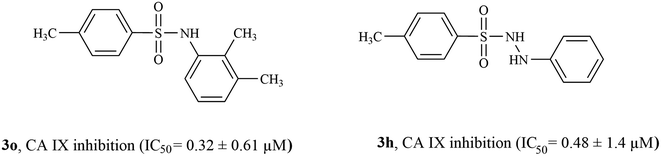
3.4.2 Structure activity relationship of CA IX inhibitors. Among inhibitors of CA IX, compounds 3o (having a 2,3-dimethyl phenyl ring) and 3h (unsubstituted phenyl ring) were the most active inhibitors exhibiting IC50 values of 0.32 ± 0.61 μM and 0.484 ± 0.14 μM, respectively (Fig. 6). Compound 3j having a 3,4-dimethyl phenyl ring also showed excellent inhibition activity having IC50 values of 0.62 ± 0.2 μM. Compounds 3d (having a diphenyl sulfide ring, IC50 = 0.77 ± 0.59 μM), 3g (having a methane diphenyl ring, IC50 = 0.78 ± 0.83 μM), and 3l (having a 3,4-dimethoxy phenyl ring, IC50 = 0.764 ± 0.54 μM) had very similar inhibition activities indicating that relatively bulky rings (such as a diphenyl ring) attached to the sulfonamide moiety are better suited for CA IX inhibition. The position of the alkyl substituent also seemed to be an important determinant for CA IX inhibition activity; while compounds 3o (having a 2,3-dimethyl phenyl ring) and 3j (having a 3,4-dimethyl phenyl ring) were highly active (IC50 = 0.32 ± 0.61 μM and 0.62 ± 0.2 μM respectively), other compounds containing a di-alkyl substituted phenyl ring were not as active in comparison to compounds 3o and 3j (such as compounds 3b containing 3,5-dimethyl phenyl ring, IC50 = 1.65 ± 1.3 μM, and 3c containing 2,6-dimethyl phenyl ring, IC50 = 1.06 ± 0.63 μM). Compounds 3e, 3i, 3k, and 3m showed <50% CA IX inhibition. Three of the inactive compounds 3i, 3k, and 3m contained a 4-methoxy phenyl sulfonamide ring, indicating that despite containing a sulfonamide moiety, the 4-methoxy phenyl sulfonamide ring is not suited for the design of CA IX inhibitors. With the only exception of compounds 3f and 3n (IC50 = 1.98 ± 0.61 μM and 2.01 ± 1.3 μM, respectively), all active CA IX inhibitors were more active than the standard inhibitor acetazolamide (IC50 = 1.08 ± 0.03 μM).
 |
| | Fig. 6 Structure activity relationship of CA IX inhibitors. | |
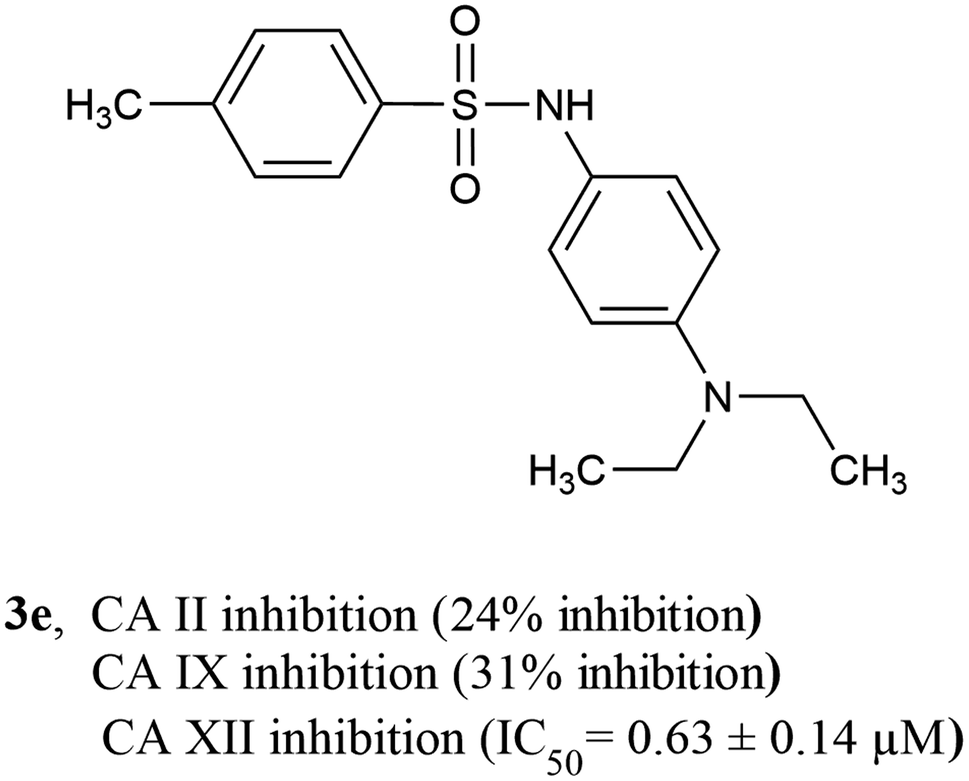
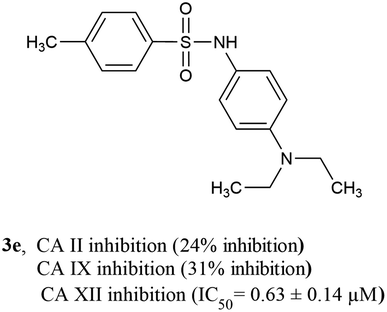
3.4.3 SAR of CA XII inhibitors. Among CA XII inhibitors, compound 3a was the most active inhibitor having an IC50 value of 0.56 ± 0.18 μM, followed by compound 3e (IC50 = 0.63 ± 0.14 μM) having an N,N-diethyl substituent on the phenyl ring. Compound 3e is exceptionally unique since it is the only compound that is a highly selective inhibitor of CA XII over both CA II and CA IX (exhibiting <50% inhibition against both CA II and CA IX, Fig. 7). Among compounds containing di-alkyl substituted phenyl ring, compound 3c having a 2,6-dimethyl phenyl ring was the most active, exhibiting an IC50 value of 0.70 ± 0.20 μM. Like before, once again, the position of the di-alkyl substituent on the phenyl ring seems to be important, while compounds 3b (having a 2,5-dimethyl phenyl ring), 3j (having a 3,4-dimethyl phenyl ring), and 3l (having a 3,4-dimethoxy phenyl ring) were all active inhibitors of CA XII (IC50 = 1.03 ± 0.23 μM, 1.12 ± 1.00 μM, and 1.17 ± 1.0 μM, respectively). Compound 3o (having a 2,3-dimethyl phenyl ring) was inactive against CA XII, exhibiting <50% inhibition. Compound 3i containing a 4-methoxy phenyl sulfonyl hydrazide moiety was inactive against CA XII (<50% inhibition), whereas other compounds containing a 4-methoxy phenyl sulfonamide moiety such as compound 3k and 3m (IC50 = 1.14 ± 0.37 μM and 1.40 ± 0.57 μM, respectively) were active CA XII inhibitors, indicating that the nature of the phenyl ring substituent also plays an important part in CA XII inhibition. All CA XII inhibitors (with the only exception of compound 3n, IC50 = 1.96 ± 0.66 μM) were more active than the standard inhibitor acetazolamide (IC50 = 1.55 ± 0.037 μM) (Table 2).
 |
| | Fig. 7 Most active and selective CA XII inhibitor. | |
3.5 Docking study
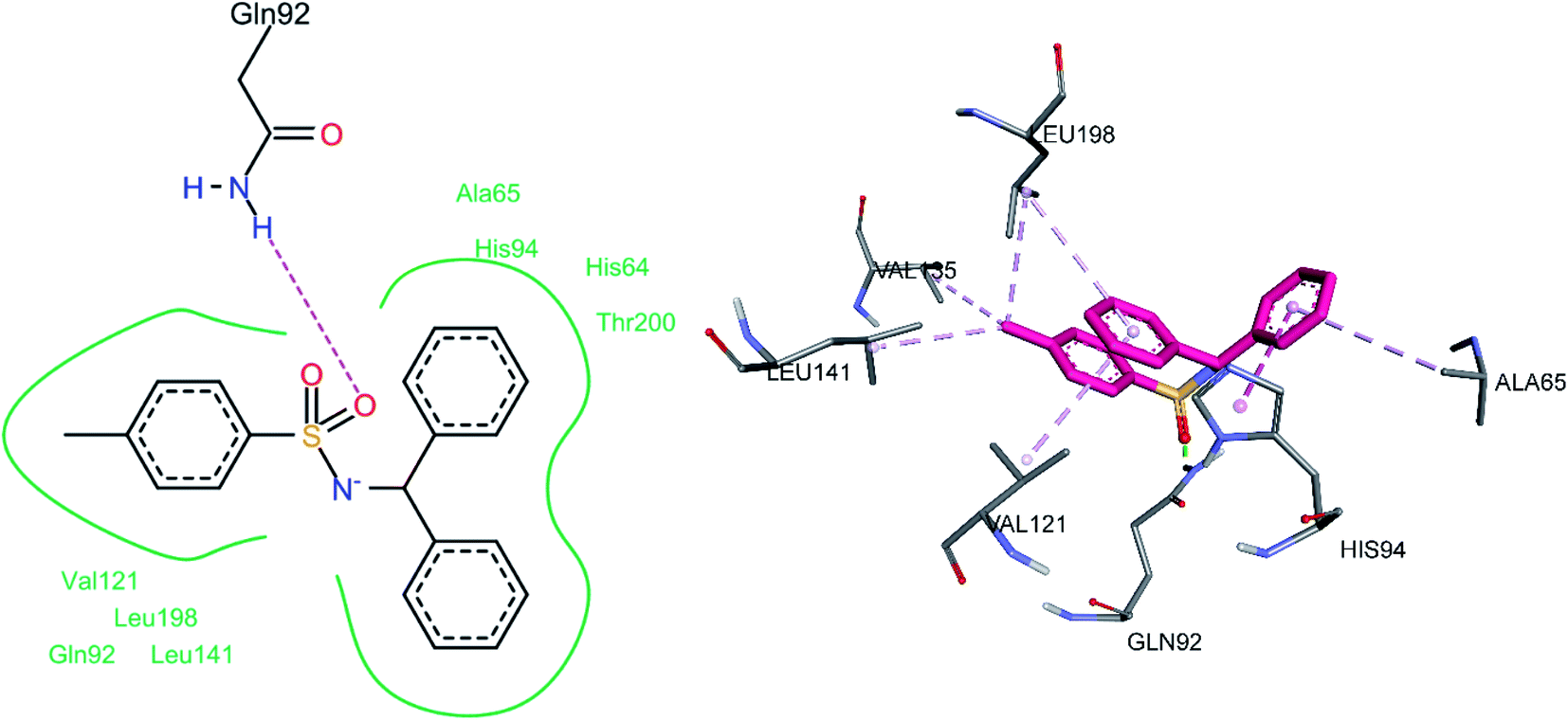
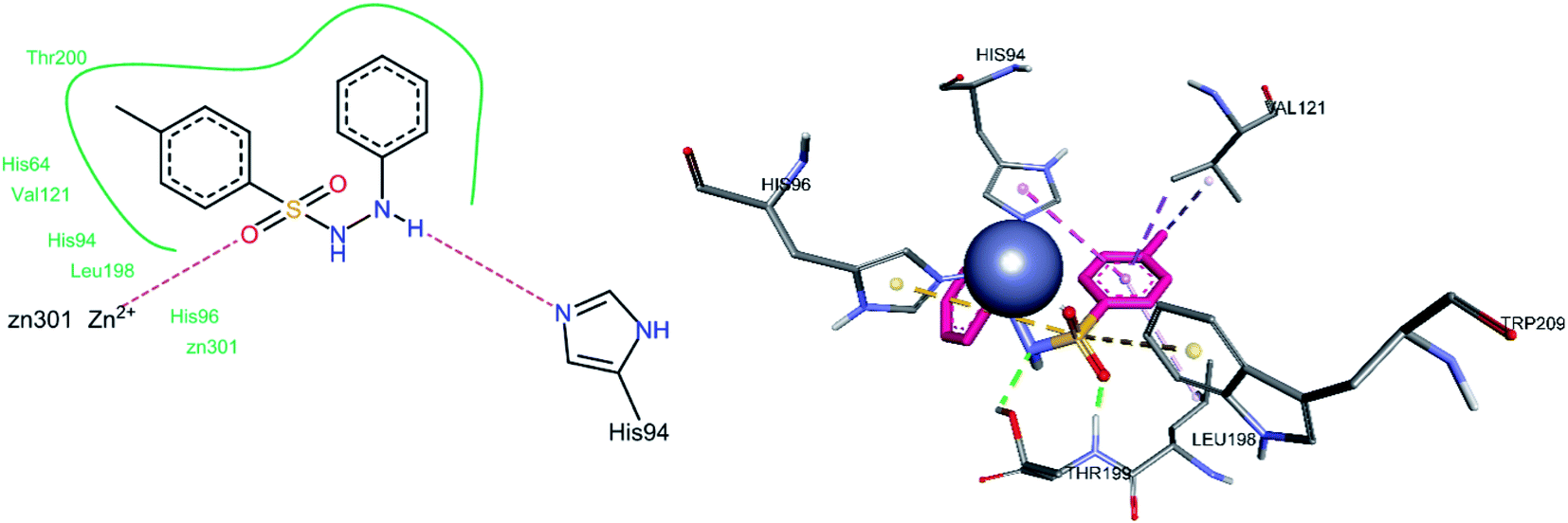
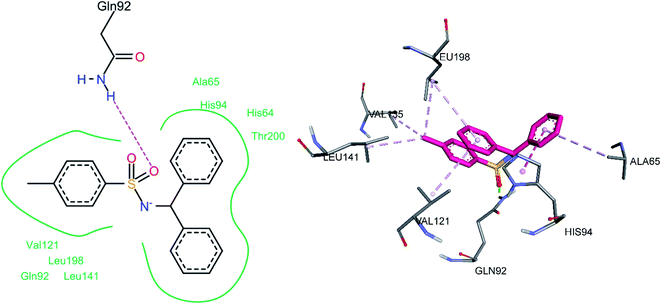
3.5.1 Docking of hCA II inhibitors. The three most active hCA II inhibitors (3g, 3h, and 3m) were selected for the docking studies. All compounds were found to bind to the active site. This is in agreement with the findings in the literature, including the single crystal structure analysis of carbonic anhydrase enzymes in complexes with sulfonamide bound inhibitors. Hence, the docking studies are in agreement with the known fact that sulfonamide moiety directly interacts with the Zn2+ ion of the enzyme's active site, thereby inhibiting its function. The common binding feature among all inhibitors was observed to be a pi–alkyl interaction with Leu198 and a hydrogen bond between the sulfonamide oxygen and Gln92. This hydrogen bond interaction is particularly interesting since a quick survey of the literature shows that the vast majority of sulfonamide containing CA inhibitors bind via sulfonamide group to the Zn2+ metal ion of the CA active site. However, our docking studies indicate the binding of sulfonamide oxygen to Gln92 instead.For compound 3g, the docking studies revealed that one of the oxygen atoms was making a hydrogen bond with Gln92 (Fig. 8). Among hydrophobic interactions, His94 made a pi–pi T-shaped interaction with one of the phenyl rings attached to the sulfonamide group, while the same phenyl ring made a pi–alkyl interaction with Ala65. The other phenyl ring attached to the sulfonamide moiety made pi–alkyl interactions with Leu198 and Val121. The methyl group made pi–alkyl interactions with amino acid residues, including Leu198, Leu141, and Val135.
 |
| | Fig. 8 Docked conformation of 3g (hCA II inhibitor). | |
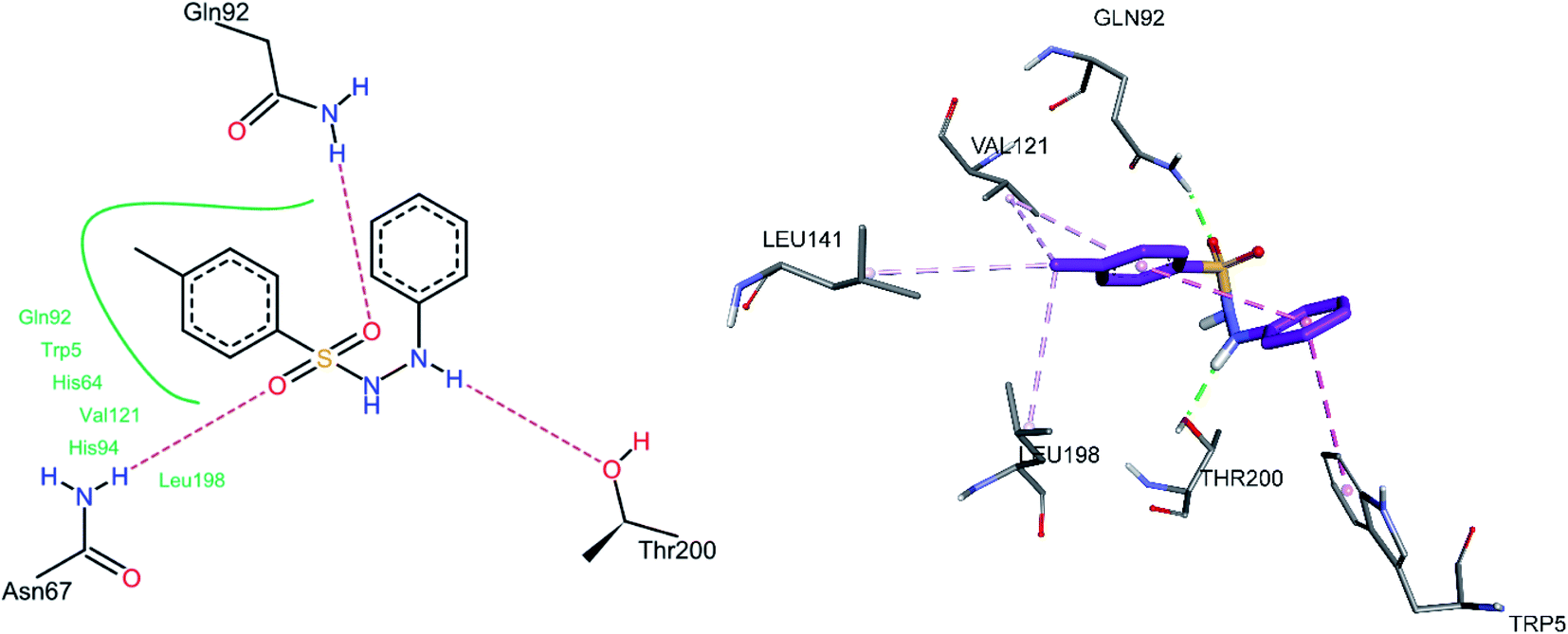
The docked conformation of 3h is given in Fig. 9. One of the sulfonamide oxygen atoms formed a hydrogen bond with Gln92, while the other oxygen with Asn67. The NH group adjacent to the phenyl group formed a hydrogen bond with Thr200, while the same phenyl group was involved in an intramolecular pi–pi stacked interaction with the 4-methyl phenyl ring and a pi–pi T-shaped interaction with Trp5. The methyl group substituted at the phenyl ring showed pi–alkyl interactions with Leu198, Val121, and Leu141, while the phenyl ring was also involved in a pi–alkyl interaction with Val121.
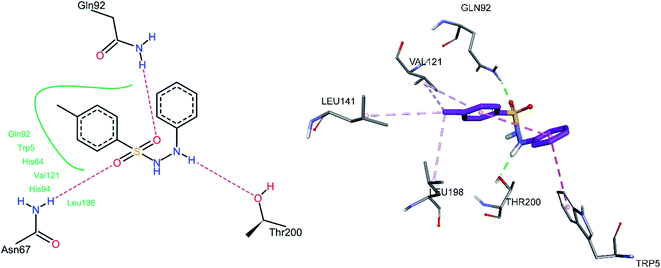 |
| | Fig. 9 Docked conformation of 3h (hCA II inhibitor). | |
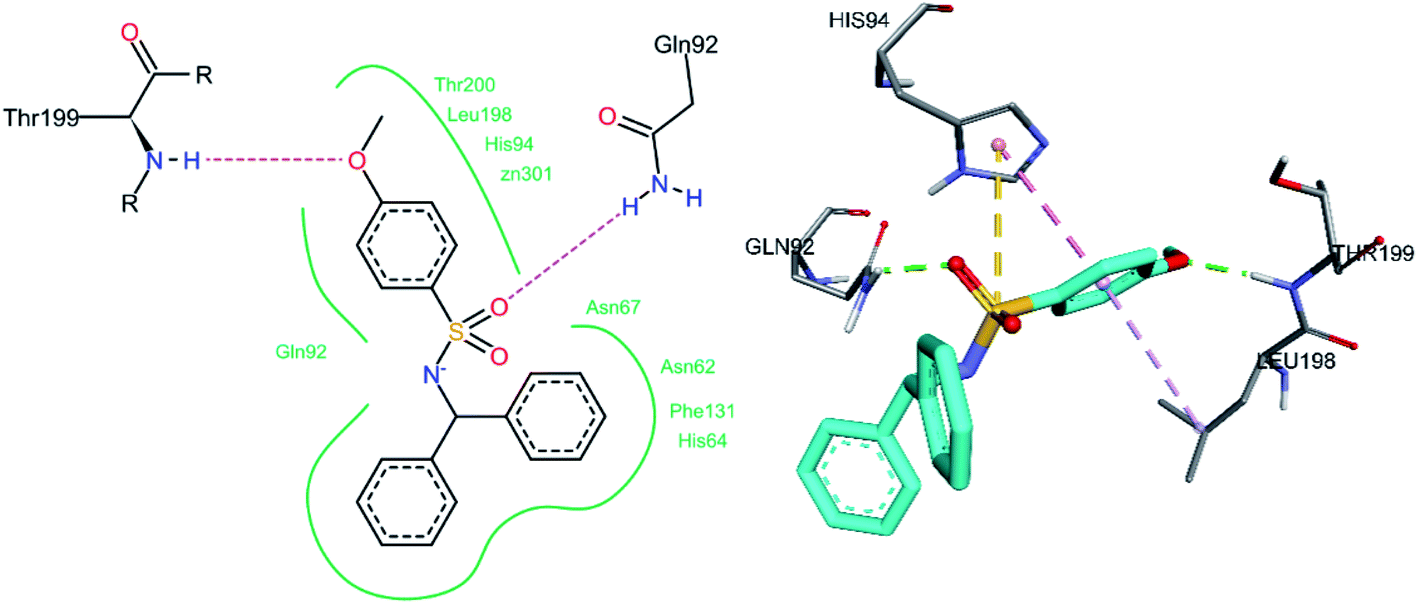
The docked conformation of hCA II inhibitor 3m is given in Fig. 10. One of the sulfonamide oxygen atoms forms a hydrogen bond with Gln92, while the oxygen atom of the methoxy group forms a hydrogen bond with Thr199. Between the sulphur atoms of the sulfonamide group and His94, a pi–sulfur interaction was observed. A number of hydrophobic interactions were also detected, which were thought to be required for effective binding. The methoxy phenyl ring exhibits a pi–pi stacked interaction with His94 and a pi–alkyl interaction with Leu198.
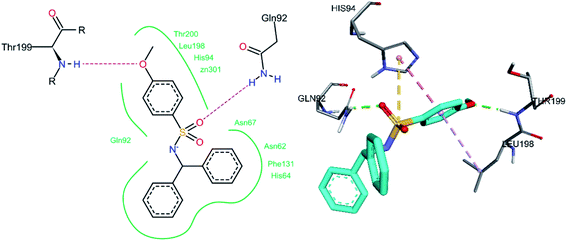 |
| | Fig. 10 Docked conformation of 3m (hCA II inhibitor). | |
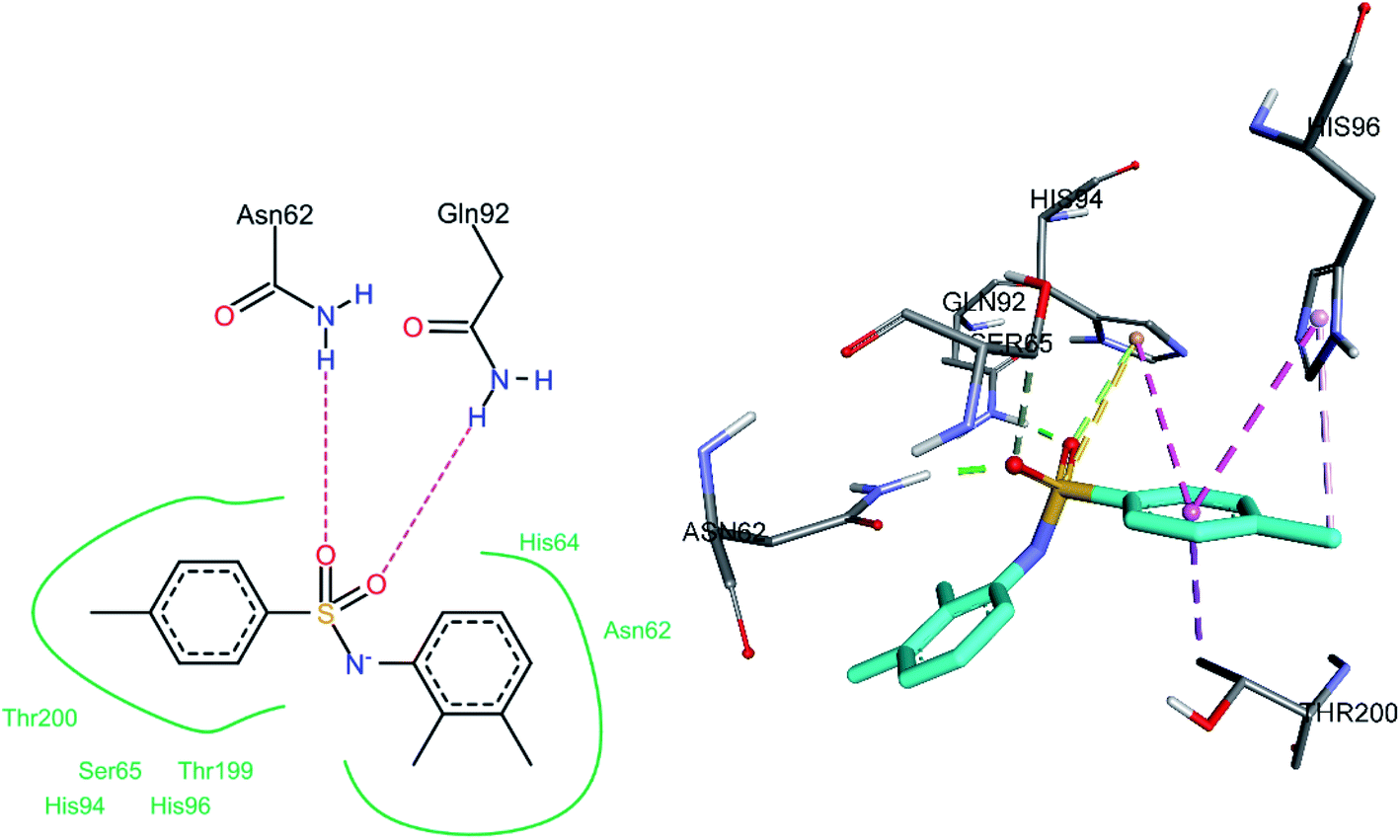
3.5.2 Docking of hCA IX inhibitors. The three most active hCA II inhibitors (3h, 3j, and 3o) were selected for the docking studies. All compounds were found to bind to the active site. The common binding feature among all inhibitors was observed to be a pi–pi stacked interaction with His94.The sulfonamide oxygen atoms of 3o formed hydrogen bonds with Asn62 and Gln92 (Fig. 11). The sulfonamide sulfur atom and His94 were involved in a pi–sulfur bond. A number of hydrophobic interactions were observed. The 4-methyl phenyl ring shows a pi–sigma interaction with Thr200 and a pi–pi stacked and a pi–pi T-shaped interaction with the amino acids His94 and His96, respectively. A pi–alkyl interaction was also observed between the methyl group (substituted on a phenyl ring) and His96.
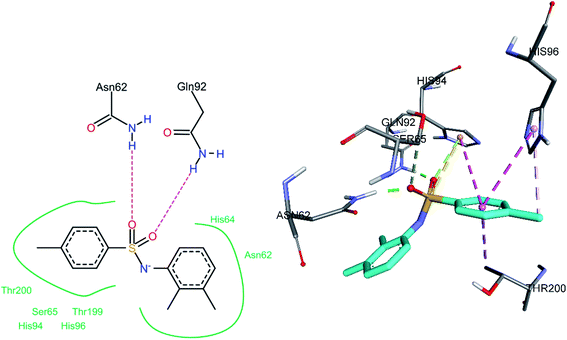 |
| | Fig. 11 Docked conformation of 3o (hCA IX inhibitor). | |
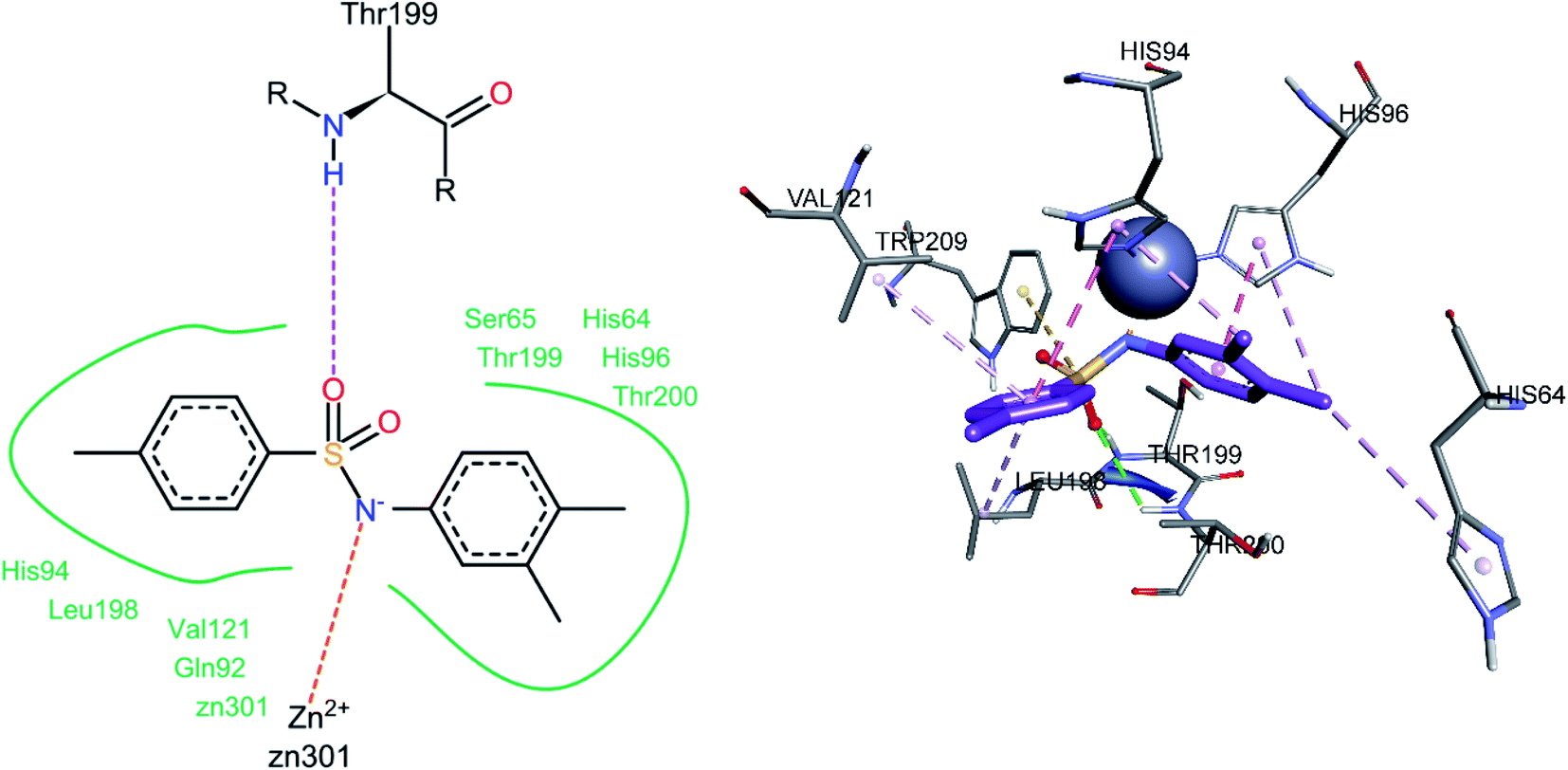
For compound 3j, the docked conformation revealed that one of the sulfonamide oxygen atoms formed a hydrogen bond with Thr199 and Thr200 (Fig. 12). A pi–sulfur bond was observed between the sulfur atom of the sulfonamide group and Trp209, while the sulfonamide nitrogen was in direct contact with the Zn2+ ion of the active site. The 4-methyl phenyl ring was involved in pi–alkyl interactions with Leu198 and Val121. The amino acid His94 showed a pi–pi stacked interaction with the 4-methyl phenyl ring and a pi–alkyl interaction with one of the methyl groups of the 3,4-dimethyl phenyl ring, while the other methyl group of the same ring was also involved in a pi–alkyl interaction with His64. The 3,4-dimethyl phenyl ring also showed a pi–pi T-shaped interaction with His96.
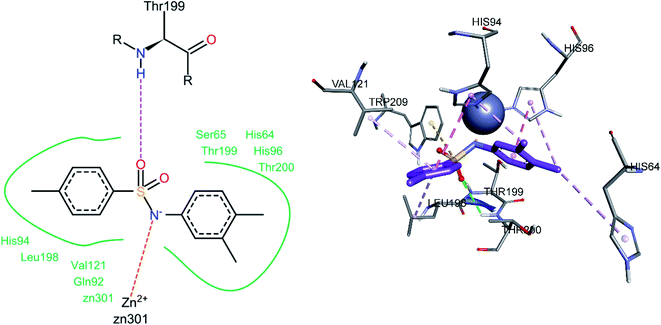 |
| | Fig. 12 Docked conformation of 3j (hCA IX inhibitor). | |
For compound 3h, one of the sulfonamide oxygen atoms formed a hydrogen bond with Thr199, while the other sulfonamide oxygen was bonded to the Zn2+ ion of the active site (Fig. 13). A hydrogen bond between Thrr199 and the sulfonamide nitrogen was observed. The sulfur atom of the sulfonamide group showed a pi–sulfur interaction with Trp209 and His96. The 4-methyl substituted phenyl ring showed a pi–sigma interaction with Val121, whereas the phenyl ring (containing 4-methyl substituent) exhibited pi–alkyl contact with Leu198 and a pi–pi stacked interaction with His94.
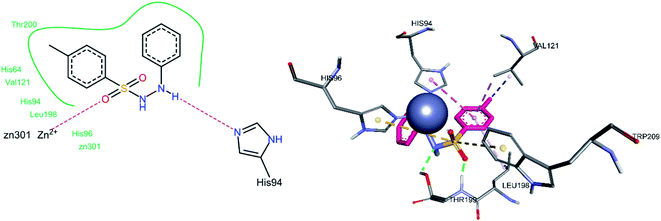 |
| | Fig. 13 Docked conformation of 3h (hCA IX inhibitor). | |
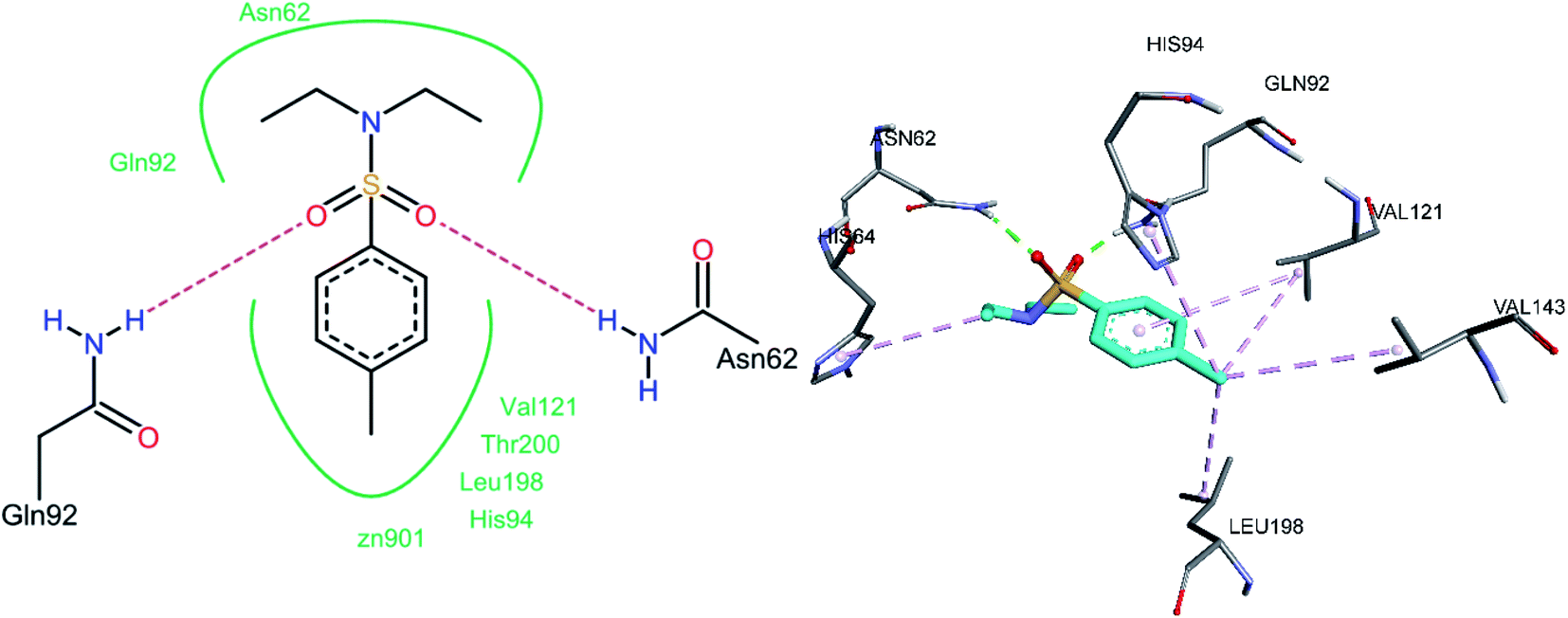
3.5.3 Docking of hCA XII inhibitors. Both oxygen atoms of the sulfonamide group of compound 3e formed hydrogen bonds with Gln92 and Asn62 (Fig. 14). Non-bonded hydrophobic pi–alkyl interactions were abundantly observed. One of the N-ethyl groups shows a pi–alkyl interaction with His64. The methyl group that was substituted on the phenyl ring was involved in pi–alkyl interactions with Leu198, Val121, Val143, and His94, whereas the phenyl ring was also observed to be involved in a pi–alkyl interaction with Val121.
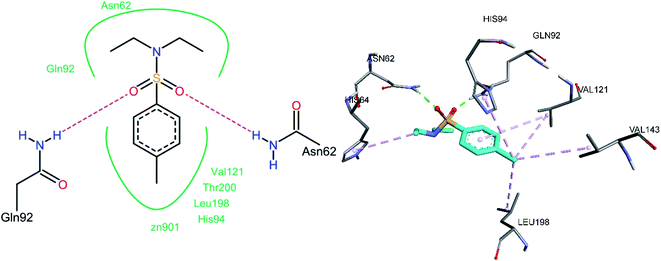 |
| | Fig. 14 Docked conformation of 3e (hCA XII inhibitor). | |
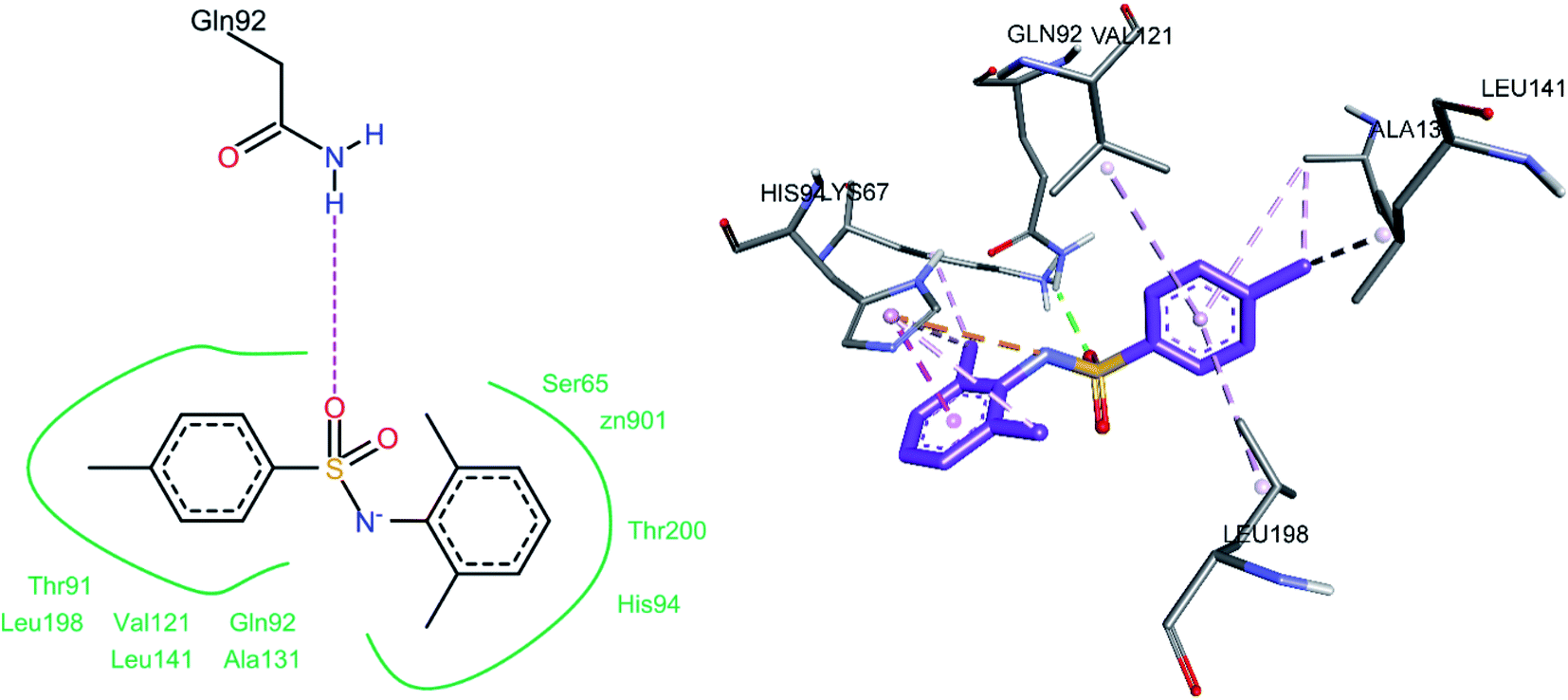
The docked conformation of compound 3c is shown in Fig. 15. One of the sulfonamide oxygen atoms formed a hydrogen bond with Gln92. A pi–anion (electrostatic) interaction was observed between the nitrogen atom of the sulfonamide group and His94. The 2,6-dimethyl phenyl ring was involved in a pi–pi stacked interaction with His94, while one of its methyl substituents showed pi–alkyl interactions with His94 and Lys67; the other methyl group (on the same ring) also showed a pi–alkyl interaction with His94. The 4-methyl substituent on the 4-methyl phenyl ring exhibited pi–alkyl interactions with Ala131 and Leu141, while the phenyl ring (on which this methyl group is substituted) showed pi–alkyl interactions with Ala131, Val121, and Leu198.
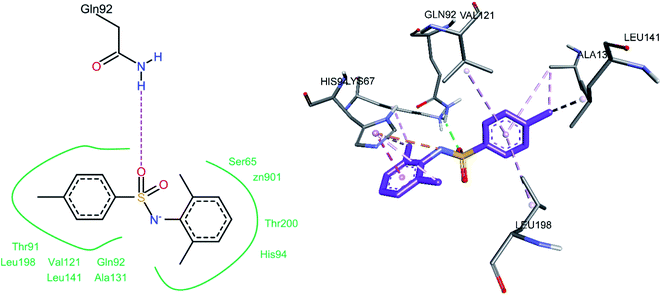 |
| | Fig. 15 Docked conformation of 3c (hCA XII inhibitor). | |
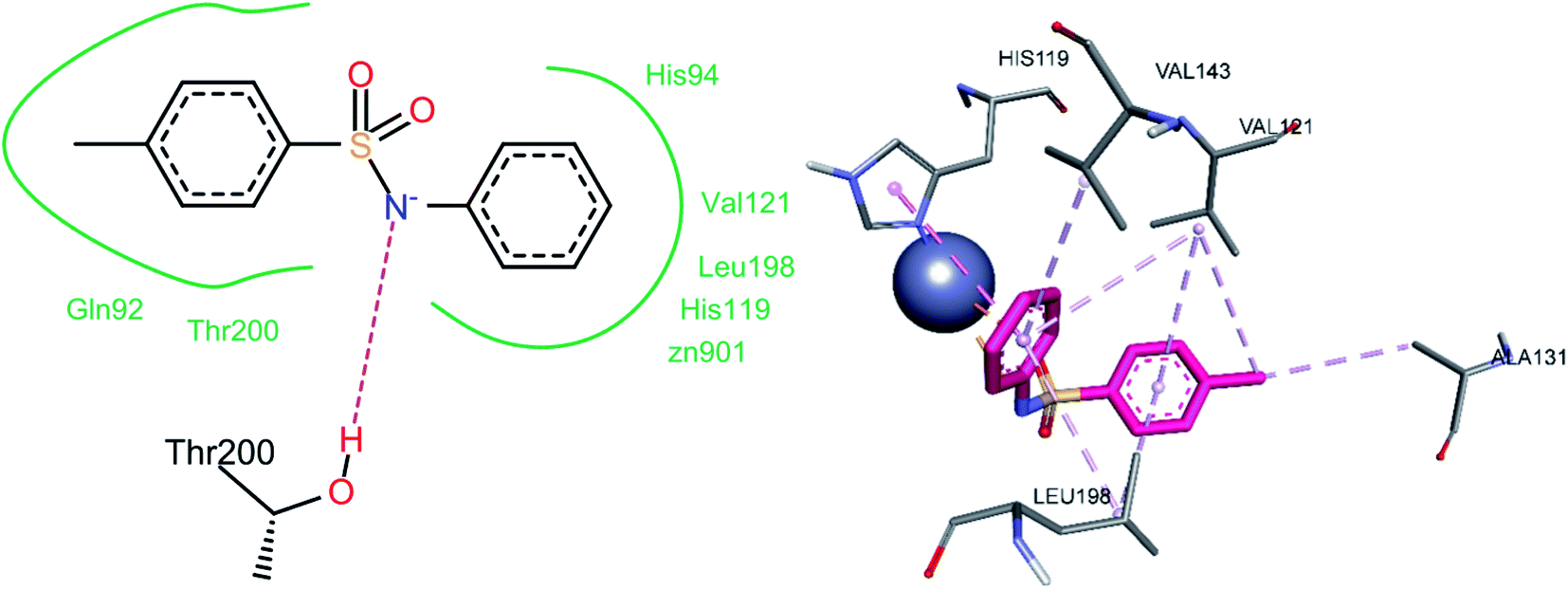
The nitrogen atom of the sulfonamide group of compound 3a showed a hydrogen bond contact with Thr200 (Fig. 16). A pi–anion interaction was observed between the phenyl ring attached to the sulfonamide moiety and the Zn2+ ion of the enzyme's active site. A pi–pi stacked interaction was observed between the same phenyl ring and His119. Two alkyl interactions were observed between the methyl group and Val121 and Ala131. The 4-methyl phenyl ring exhibited pi–alkyl interactions with Val121 and Leu198. Pi–alkyl interactions were also observed between amino acids Leu198, Val143, and Val121 and the other phenyl ring (Table 3).
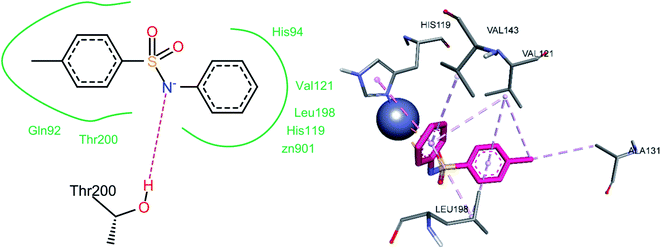 |
| | Fig. 16 Docked conformation of 3a (hCA XII inhibitor). | |
Table 3 Non-bonded interactions of selected docked hCA II, hCA IX and hCA XII inhibitors with their respective receptor
| Ligand |
Binding free energy (kJ mol−1) |
Hydrophobic interactions |
Hydrogen bond interactions |
Hydrogen bond distance (Å) |
Other interactions and distance (Å) |
| hCA II inhibitors |
| 3g |
−22 |
His94 (pi–pi T-shaped), Val135 (alkyl), Leu141 (alkyl), Leu198 (alkyl and pi–alkyl), Val121 (pi–alkyl), Ala65 (pi–alkyl) |
Gln92 (H-donor) |
1.69 |
— |
| 3h |
−24 |
Trp5 (pi–pi T-shaped), Val121 (alkyl and pi–alkyl), Leu141 (alkyl), Leu198 (alkyl), |
Thr200 (H-donor), Gln92 (H-donor) |
2.0, 2.0 |
— |
| 3m |
−25 |
His94 (pi–pi stacked), Leu198 (pi–alkyl) |
Thr199 (H-donor), Gln92 (H-donor) |
2.1, 2.0 |
His94 (pi–sulfur) |
|
| hCA IX inhibitors |
| 3h |
−26 |
Val121 (pi–sigma), His94 (pi–pi stacked), Val121 (alkyl), Leu198 (pi–alkyl) |
Thr199 (H-donor and H-acceptor) |
1.2 and 2.3 |
Zn-ligand (2.0), His96 (pi–sulfur), Trp209 (pi–sulfur) |
| 3j |
−25 |
His94 (pi–pi stacked, pi–alkyl), His96 (pi–pi T-shaped, pi–alkyl), Val121 (pi–alkyl), Leu198 (pi–alkyl), His64 (pi–alkyl), |
Thr199 (H-donor), Thr200 (H-donor) |
1.4, 2.8 |
Zn-ligand (2.0), Trp209 (pi–sulfur) |
| 3o |
−28 |
Thr200 (pi–sigma), His94 (pi–pi stacked), His96 (pi–pi T-shaped, pi–alkyl) |
Asn62 (H-donor), Gln92 (H-donor) |
1.9, 1.9 |
His94 (pi–sulfur), |
|
| hCA XII inhibitors |
| 3a |
−26 |
His119 (pi–pi stacked), Val121 (alkyl and pi–alkyl), Ala131 (alkyl), Leu198 (pi–alkyl), Val143 (pi–alkyl) |
THr200 (H-donor) |
2.0 |
Zn-ligand (electrostatic, pi–cation) |
| 3c |
−25 |
His94 (pi–pi stacked and pi–alkyl), Ala131 (alkyl and pi–alkyl), Leu141 (alkyl), Lys67 (alkyl), Val121 (pi–alkyl), Leu198 (pi–alkyl) |
Gln92 (H-donor) |
2.17 |
His94 (electrostatic, pi–anion) |
| 3e |
−25 |
Val121 (pi–alkyl and alkyl), Val143 (alkyl), Leu198 (alkyl), His64 (pi–alkyl), His94 (pi–alkyl), |
Asn62 (H-donor), Gln92 (H-donor) |
2.15, 1.89 |
— |
4. Conclusion
In conclusion, a solid support catalyst has been prepared by using transition metal salt (FeF3) adsorbed solid support and explored its catalytic efficiency in the preparation of sulfonamides (3a–3o). All compounds were prepared in good to excellent yields. The catalyst was further explored for the synthesis of indoles 6a–h, 1H-tetrazoles, and 1,4-dihydropyridines. The sulfonamides prepared herein were investigated for their potential to inhibit carbonic anhydrases (hCA II, hCA IX, and hCA XII). All compounds were found to be active inhibitors with IC50 values in the lower micro molar range. Some compounds were even found to be highly selective inhibitors. The sulfonamide 3i showed selective inhibition towards hCA II with an IC50 value of 2.76 ± 1.1 μM and 3e inhibited only hCA XII with an IC50 value of 0.63 ± 0.14 μM. Further, the molecular docking study was performed to study the putative binding interaction of sulfonamide with target enzymes. These compounds can provide significant leads for the further development of highly selective CA inhibitors.
Conflicts of interest
The authors have declared no conflict of interest.
Acknowledgements
We are thankful to HEJ, Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi and Higher Education Commission, Pakistan, for providing partial financial support. MAR is thankful to the Office of Research Innovation and Commercialization (ORIC), Forman Christian College (A Chartered University), Lahore, for providing Internal Research and Innovation Fund (IRIF).
References
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- C. T. Supuran, A. Scozzafava and A. Casini, Med. Res. Rev., 2003, 23, 146–189 CrossRef CAS PubMed.
- A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, Expert Opin. Ther. Pat., 2006, 16, 1627–1664 CrossRef CAS.
- J. H. Clark, Chem. Rev., 1980, 80, 429–452 CrossRef CAS.
- A. Hameed, R. D. Alharthy, J. Iqbal and P. Langer, Tetrahedron, 2016, 22, 2763–2812 CrossRef.
- B. Basu, P. Das and S. Das, Curr. Org. Chem., 2008, 12, 141–158 CrossRef CAS.
- I. P. Singh, S. K. Jain, A. Kaur, S. Singh, R. Kumar, P. Garg, S. S. Sharma and S. K. Arora, Eur. J. Med. Chem., 2010, 45, 3439–3445 CrossRef CAS PubMed.
- S. K. Jain, S. Meena, B. Singh, J. B. Bharate, P. Joshi, V. P. Singh, R. A. Vishwakarma and S. B. Bharate, RSC Adv., 2012, 2, 8929–8933 RSC.
- R. Kuwano and T. Shige, Chem. Lett., 2005, 34, 728–729 CrossRef CAS.
- B. Movassagh and S. Shokri, Synth. Commun., 2005, 35, 887–890 CrossRef CAS.
- S. Bentahar, M. A. Taleb, A. Sabour, A. Dbik, M. El Khomri, N. El Messaoudi, A. Lacherai and R. Mamouni, Russ. J. Org. Chem., 2019, 55, 1423–1431 CrossRef CAS.
- M. Nardi, N. H. Cano, P. Costanzo, M. Oliverio, G. Sindona and A. Procopio, RSC Adv., 2015, 5, 18751–18760 RSC.
- N. G. Kundu and B. Nandi, Tetrahedron, 2001, 57, 5885–5895 CrossRef CAS.
- B. Liu, D. Li and H. Shang, Chem. Cent. J., 2014, 8, 43 CrossRef PubMed.
- J. Chen, Y. Zhang, W. Hao, R. Zhang and F. Yi, Tetrahedron, 2013, 69, 613–617 CrossRef CAS.
- C. You, F. Yao, T. Yan and M. Cai, RSC Adv., 2016, 6, 43605–43612 RSC.
- R. Pingaew, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Med. Chem. Res., 2013, 22, 267–277 CrossRef CAS.
- Q. Xu, H. Xie, E.-L. Zhang, X. Ma, J. Chen, X.-C. Yu and H. Li, Green Chem., 2016, 18, 3940–3944 RSC.
- M. A. Abbasi, A. Sheeza, S. Z. Siddiqui, K. M. Khan, R. Malik and I. Ahmad, J. Chem. Soc. Pak., 2015, 37, 541–548 CAS.
- D. Amantini, R. Beleggia, F. Fringuelli, F. Pizzo and L. Vaccaro, J. Org. Chem., 2004, 69, 2896–2898 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, Wiley, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The NMR spectra of the synthesized compounds. See DOI: 10.1039/d1ra07844e |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Tayyaba Allamgir Sindhud,
Shafia Iftikhare,
Muhammad Raza Shaha,
Abdul Hameed
d,
Tayyaba Allamgir Sindhud,
Shafia Iftikhare,
Muhammad Raza Shaha,
Abdul Hameed