DOI:
10.1039/D1RA07798H
(Paper)
RSC Adv., 2022,
12, 3313-3321
Two-step hydrothermal synthesis and conversion mechanism of zeolite X from stellerite zeolite
Received
22nd October 2021
, Accepted 17th January 2022
First published on 26th January 2022
Abstract
We investigated the conversion mechanism of stellerite zeolite to zeolite X under two-step hydrothermal conditions. To elucidate the conversion mechanism, solid products were separated from the mixtures at different crystallization times and characterized by XRD, FESEM, FT-IR, Raman, solid-state NMR, XRF, and TEM. The results indicate that in this reaction process, the Si, Al, and Na in the gel solid phases were continuously dissolved and transformed into the gel–liquid-phase. When the concentration of each component reached supersaturation in the gel–liquid-phase, Si, Al, and Na were transferred to the surface of the gel–solid-phase, and nucleation and crystallization occurred on the surface. Abundant nuclei were formed during the second hour of the crystallization. As the crystallization time increased, the nuclei rapidly grew into zeolite X crystals, and the relative crystallinity of zeolite X reached a maximum when the crystallization time reached 4 h. These phenomena indicate that the formation mechanism of zeolite X is a liquid-phase conversion mechanism.
Introduction
Zeolite X (Na2O·Al2O3·2.5SiO2·6.2H2O) is a microporous aluminosilicate material with excellent properties, such as high specific surface area, high porosity, good thermal stability, and excellent adsorption and ion exchange performance.1 As an excellent adsorbent and ion exchange agent, heavy metal ions in water are removed by zeolite X, and are widely used for environmental protection. There are many types of synthetic raw materials for the synthesis of zeolite X. The common raw materials are pure chemical-grade reagents, natural minerals, industrial waste, and agricultural waste. In particular, the use of natural minerals and industrial and agricultural wastes as raw materials for synthesis contributes to reducing production costs, reducing environmental problems, and broadening the source of raw materials. At present, many studies have been published on the synthesis of zeolite X from natural minerals such as illite,2 diatomite,3 bauxite,4 kaolin,5,6 and bentonite7 as well as agricultural and industrial waste such as bagasse8 and fly ash.9
Primarily, the influence of the synthesis conditions on the zeolite phase from natural minerals and wastes have been investigated instead of the formation mechanism, and there is limited understanding of the zeolite formation mechanism from natural minerals and wastes.10 A detailed study of the conversion mechanism of natural minerals and wastes into zeolites will not only help to find the best synthesis conditions, but also unmask the structure–activity relationships between raw materials and zeolites10 and effectively control the crystallinity of the product. By analyzing the conversion mechanism of natural minerals and wastes into zeolite, the conversion process of zeolites can be used and controlled to obtain the desired zeolite products. Stellerite zeolite, mainly formed in the fissures or almond-like pores of basaltic volcanic rocks, is a natural zeolite with the advantages of low price and wide storage. Stellerite zeolites can be converted into other zeolites because they contain a silicon source as the raw material. The conversion of stellerite zeolite to other types of zeolites is mainly achieved by hydrothermal methods (one-step and two-step) and alkali fusion–hydrothermal methods. Li et al.11,12 synthesized zeolites A, P, and Y by a fusion–hydrothermal method from stellerite zeolite. The utilization rate of raw materials is higher for the alkali fusion–hydrothermal method. However, the alkali fusion–hydrothermal method consumes more activator, such as NaOH and Na2CO3, and requires high-temperature calcination equipment, which increases the cost of zeolites synthesis. Shen et al.13–15 synthesized zeolites A, P, and X using a one-step hydrothermal method. Because quartz was an inert component in stellerite zeolite, the quartz was difficult to dissolve in the hot alkali solution. Therefore, the quartz co-existed with zeolites A, P, and X in the products. Our research group synthesized zeolite P by a one-step hydrothermal method and found that quartz often appears in the form of heterocrystals in the product.16 However, after separating quartz crystals using a two-step hydrothermal method, zeolite X and zeolite P with high purity and crystallinity can be obtained.17,18
In recent years, there have been many reports on zeolite synthesis from stellerite zeolite,13,16–18 but the conversion of highly pure and crystalline zeolite X by the two-step hydrothermal method has seldom been reported. In addition, to the best of our knowledge, no other study has discussed the mechanism of stellerite zeolite conversion into zeolite X by a two-step hydrothermal method. In this paper, the mechanism of stellerite zeolite conversion into zeolite X by a two-step hydrothermal method is discussed. It is helpful to distinguish this study from other synthesis processes that utilize pure chemical reagents, minerals, or wastes as raw materials. The results of this study are of great significance for understanding the conversion mechanism of other natural minerals to zeolites in the future.
In this work, we report the conversion of zeolite X from stellerite zeolite under two-step hydrothermal conditions. To elucidate the conversion process and species involved in nucleation and crystal growth, the crystallization products were characterized by X-ray diffraction (XRD), field-emission scanning electron microscopy (FESEM), Fourier-transform infrared spectroscopy (FT-IR), Raman spectroscopy, solid-state nuclear magnetic resonance (NMR), X-ray fluorescence (XRF), and transmission electron microscopy (TEM). Finally, a tentative model was proposed to explore the conversion mechanism of stellerite zeolite conversion into zeolite X. In this study, we report the conversion mechanism of a two-step hydrothermal method of stellerite zeolite to zeolite X for the first time to the best of our knowledge.
Experimental
Materials and reagents
Stellerite zeolite was obtained from Guilin City, Guangxi Province, China. It was ground to a size smaller than 50 mesh before use. Sodium hydroxide (96%, NaOH) and hydrochloric acid (36–38%, HCl) were obtained from Xilong Reagent Co. (Xilong, China). Sodium metaaluminate (41%, Al2O3) was purchased from Sinopharm Chemical Reagent Co., Ltd (Sinopharm Chemical, China). All chemicals were of analytical reagent grade, and deionized water was used for all experiments.
Stellerite zeolite conversion into zeolite X
Zeolite X was synthesized using a two-step hydrothermal synthesis method. Initially, 100 g of stellerite zeolite was added to the HCl solution (15%, 300 mL) at 90 °C and stirred for 2 h at a solid/liquid ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The product was filtered and washed with deionized water until a neutral pH was achieved, followed by drying at 80 °C for 12 h. Subsequently, 10 g of dry acidified stellerite zeolite was mixed with NaOH solution (50 mL, 0.075 mol L−1) under stirring at 85 °C for 15 min, when the amorphous silicon from acidified stellerite zeolite was dissolved in the solution. The solid–liquid separation of the product was carried out, and the solid product was dried at 80 °C for 12 h. A 10 g amount of stellerite zeolite was compared with the mass of the solid product, and the remainder was identified as the mass of amorphous silica. The concentration of amorphous silicon in the alkali solution was calculated according to the mass of the amorphous silicon. The amounts of NaAlO2 and NaOH solutions were determined according to the molar ratios of H2O/SiO2 = 55, Na2O/SiO2 = 1.1, and Al2O3/SiO2 = 0.33. Then, the NaOH solution and NaAlO2 solution were successively added to the alkali solution containing amorphous silicon, under vigorous stirring. After one hour of vigorous stirring, the mixture was transferred into a flask with three necks. The mixture was then aged for 48 h at room temperature. Finally, the flask with three necks was crystallized at 82 °C for 0.5–8 h, the product was filtered out, washed with deionized water and dried at 80 °C for 12 h to obtain zeolites.
:3. The product was filtered and washed with deionized water until a neutral pH was achieved, followed by drying at 80 °C for 12 h. Subsequently, 10 g of dry acidified stellerite zeolite was mixed with NaOH solution (50 mL, 0.075 mol L−1) under stirring at 85 °C for 15 min, when the amorphous silicon from acidified stellerite zeolite was dissolved in the solution. The solid–liquid separation of the product was carried out, and the solid product was dried at 80 °C for 12 h. A 10 g amount of stellerite zeolite was compared with the mass of the solid product, and the remainder was identified as the mass of amorphous silica. The concentration of amorphous silicon in the alkali solution was calculated according to the mass of the amorphous silicon. The amounts of NaAlO2 and NaOH solutions were determined according to the molar ratios of H2O/SiO2 = 55, Na2O/SiO2 = 1.1, and Al2O3/SiO2 = 0.33. Then, the NaOH solution and NaAlO2 solution were successively added to the alkali solution containing amorphous silicon, under vigorous stirring. After one hour of vigorous stirring, the mixture was transferred into a flask with three necks. The mixture was then aged for 48 h at room temperature. Finally, the flask with three necks was crystallized at 82 °C for 0.5–8 h, the product was filtered out, washed with deionized water and dried at 80 °C for 12 h to obtain zeolites.
Characterizations
The crystal structure of the material was characterized by powder XRD (X'Pert PRO, PANalytical, Almelo, Holland) under 40 kV and 40 mA conditions. The measurement was performed with an angle 2θ from 5 to 80°, with a step size of 0.02626° and a scan speed of 0.6565° s−1. X'Pert Highscore Plus software was used to process the diffraction data of the products. The data calculations of the unit cell parameters from zeolite X synthesized in this experiment were performed using UnitCell software. The solid particle size and surface morphology of the products were observed using FESEM (S-4800, Hitachi, Japan). The surface functional groups of the products were analysed FT-IR (IR Affinity, SHIMADZU, Japan) using KBr as a reference material in the range of 400–4000 cm−1, with a resolution of 0.5 cm−1. Raman spectra (RENISHAW, inVia, Britain, with the emitted green ray of wavelength = 514 nm) was used to investigate the changes in the vibration modes of the samples. Solid-state 27Al and 29Si MAS NMR experiments were performed on a Bruker 400M spectrometer with the resonance frequency of 104.3 MHz for 27Al and 79.5 MHz for 29Si. The chemical shift standards were referenced to the resonances of NaAlO2 (27Al) and tetramethylsilane (29Si). The chemical compositions of the products were determined using wavelength dispersive XRF (Zetium, PANalytical, Holland) on a Zetium apparatus. TEM (JEM-2100F, JEOL, Japan) revealed a finer morphology image of the as-synthesized zeolite X.
Results and discussion
Analysis of raw material
As shown in Fig. 1, the main mineral composition of the raw material was stellerite zeolite and quartz, indicating that the raw material was a natural mineral. The chemical composition of the raw material was characterized using energy dispersion spectroscopy (EDS) (Table 1). The SiO2 content in the stellerite zeolite was high (69.27 wt%) enough to synthesize zeolite X with the addition of enough Al source. Because calcium is a component of stellerite zeolite, it jeopardizes the synthesis of zeolite X. Therefore, calcium had to be removed with HCl solution. After the acid treatment, CaO was removed from stellerite zeolite, increasing the SiO2 to 91.63 wt% in acidified zeolite (Table 1). The XRD analysis shows that the main phases of acidified stellerite zeolite are amorphous silica and quartz.16 Quartz is difficult to dissolve in traditional hydrothermal reactions. However, the amorphous silica of stellerite can be dissolved in a hot alkali solution and then converted into zeolite X. After the acidified zeolite was dissolved by alkali, only the diffraction peaks of quartz were found, however, a broad peak of amorphous substance disappeared (Fig. 1), indicating that amorphous silica was dissolved in solution, and the amorphous silicon in solution existed in the form of sodium metasilicate. Fig. 2(a and b) shows the morphology of the raw material before and after acid treatment. The stellerite zeolite shows a lamellar structure, and the edges of stellerite zeolite are sharp. After acidification, the acidified zeolite retained its lamellar structure, but the edges of the acidified zeolite were rounded, indicating that the structure of the stellerite zeolite was destroyed.
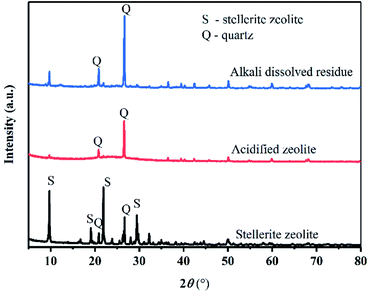 |
| | Fig. 1 XRD patterns of stellerite zeolite, acidified zeolite, and alkali dissolved residue. | |
Table 1 Chemical compositions of stellerite zeolite and acidified zeolite (wt%)
| Sample |
SiO2 |
Al2O3 |
CaO |
C |
| Stellerite zeolite |
69.27 |
16.71 |
10.23 |
3.79 |
| Acidified zeolite |
91.63 |
— |
— |
8.47 |
 |
| | Fig. 2 FESEM images of (a) stellerite zeolite and (b) acidified zeolite. | |
Synthesis of zeolite X
Fig. 3(a) demonstrates that after hydrothermal treatment at 82 °C for 0.5–1 h, only a broad diffraction peak of amorphous aluminosilicate is observed in the product. When the crystallization time was prolonged to 2 h, some weak diffraction peaks of zeolite X are observed in the product, and the broad diffraction peak of amorphous aluminosilicate disappeared gradually. These results indicate that zeolite X crystals began to grow, but that nucleation still dominated at this time. When the crystallization time was 3 h, the relative crystallinity of zeolite X increased, indicating that crystal growth was dominant at this time. The relative crystallinity of zeolite X was 100% at 4 h, indicating that the crystal structure of zeolite X was relatively complete. When the crystallization time continued to increase, the relative crystallinity of zeolite X remained unchanged. Fig. 3(b) shows the dependence of zeolite X on crystallization time. This curve indicates that crystallization of zeolite X mainly occurs at 2–4 h. Before 2 h, the nucleation induction period and after 4 h, zeolite X was fully crystallized. Previous studies have shown that the synthesis temperature of conventional zeolite X is usually 90–150 °C, and the crystallization time was 5–24 h. Shen et al. found that the optimal synthesis conditions for the synthesis of zeolite X by one-step hydrothermal method were as follows: n(H2O/SiO2) = 34–55, n(Na2O/SiO2) = 1.2–1.4, and n(Al2O3/SiO2) = 0.25–0.29, aging time 6 h, crystallization temperature 95–100 °C, and a crystallization time of 6 h.13 Our previous study found that the best synthesis conditions for zeolite X were: n(H2O/SiO2) = 33–48, n(Na2O/SiO2) = 1.1–1.2, and n(Al2O3/SiO2) = 0.33, aging time 12 h, crystallization temperature 95 °C, and a crystallization time of 7 h.18 In this study, we found that the zeolite X and be effectively synthesized at a temperature of 82 °C. We also found that when the crystallization time was 4 h, the product would achieve a higher relative crystallinity. In comparison to previous studies, stellerite zeolite can be rapidly converted into zeolite X at a relatively low temperature and in a relatively short time.
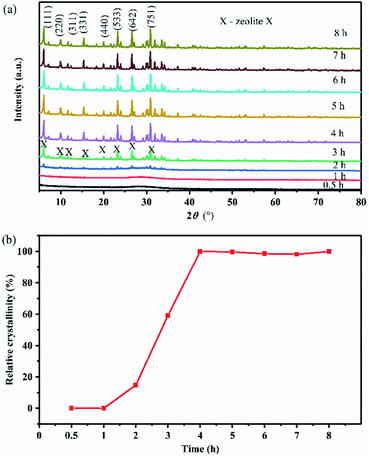 |
| | Fig. 3 (a) XRD patterns of products synthesized at various crystallization times and (b) relative crystallinity curve of the solid products. | |
Many researchers in the hydrothermal synthesis of zeolites have found that, with the extension of crystallization time, Si atoms were replaced by Al atoms, and the unit cell size of zeolites would gradually increase. When Piotr Kunecki et al.19 prepared Na–P1 zeolite from fly ash, they found that the cell size increased with increasing of crystallization time, and that the unit cell parameters of the synthesized Na–P1 zeolite conformed to standard pattern parameters. This was due to lattice parameters ripening.19 However, an entirely different phenomenon was found in this study. First, the phases of the products were characterized using XRD. Using XRD, the crystal plane index of the main diffraction peaks, the corresponding 2θ, and the crystal system of the products synthesized at various crystallization times were obtained (Table 2). Then the crystal plane index, the corresponding 2θ, and the crystal system were imported into UnitCell software, and the crystal cell parameters of different crystallization products were obtained. As shown in Table 3, the unit cell size of the synthesized zeolite X decreased at 82 °C for 2–4 h. The reason for this decrease could not be determined from our experimental results. The unit cell parameters of zeolites were correlated with framework structure, framework cation and thermal expansion. When the aluminosilicate gel was heated from room temperature to 82 °C for 4 h, the reaction system was heated and pressurized. We hypothesize that a portion of the sodium ions migrated from the β cages and hexagonal prism cages to the super cages in the crystallization stage, which led to the weakening of the filling effect of sodium ions on the crystal cell.20 Therefore, the crystal cell parameters were reduced. We plan to conduct further research to answer this question.
Table 2 Crystal plane index, corresponding 2θ, and the crystal system of products synthesized at various crystallization times
| Time (h) |
0.5 |
1 |
2 |
3 |
4 |
| (111) |
— |
— |
6.0750 |
6.0780 |
6.0957 |
| (220) |
— |
— |
9.9855 |
9.9493 |
9.9610 |
| (311) |
— |
— |
— |
11.6720 |
11.6918 |
| (331) |
— |
— |
15.3543 |
15.3798 |
15.3967 |
| (440) |
— |
— |
19.9835 |
20.0101 |
20.0242 |
| (533) |
— |
— |
23.2126 |
23.2429 |
23.2621 |
| (642) |
— |
— |
26.5670 |
26.5918 |
26.6048 |
| (751) |
— |
— |
30.8612 |
30.8770 |
30.8973 |
| Crystal system |
— |
— |
Cubic |
Cubic |
Cubic |
Table 3 Unit cell parameters (a, b, c), cell volume (V), and crystal plane spacing (d111) of the synthesized zeolite X
| |
a = b = c (Å) |
V (Å3) |
d111 (Å) |
| 0.5 h |
— |
— |
— |
| 1 h |
— |
— |
— |
| 2 h |
25.09096 |
15796.1751 |
14.53671 |
| 3 h |
25.07302 |
15762.3101 |
14.52957 |
| 4 h |
25.05453 |
15727.4709 |
14.48738 |
| Standard |
24.99000 |
15606.2600 |
14.47000 |
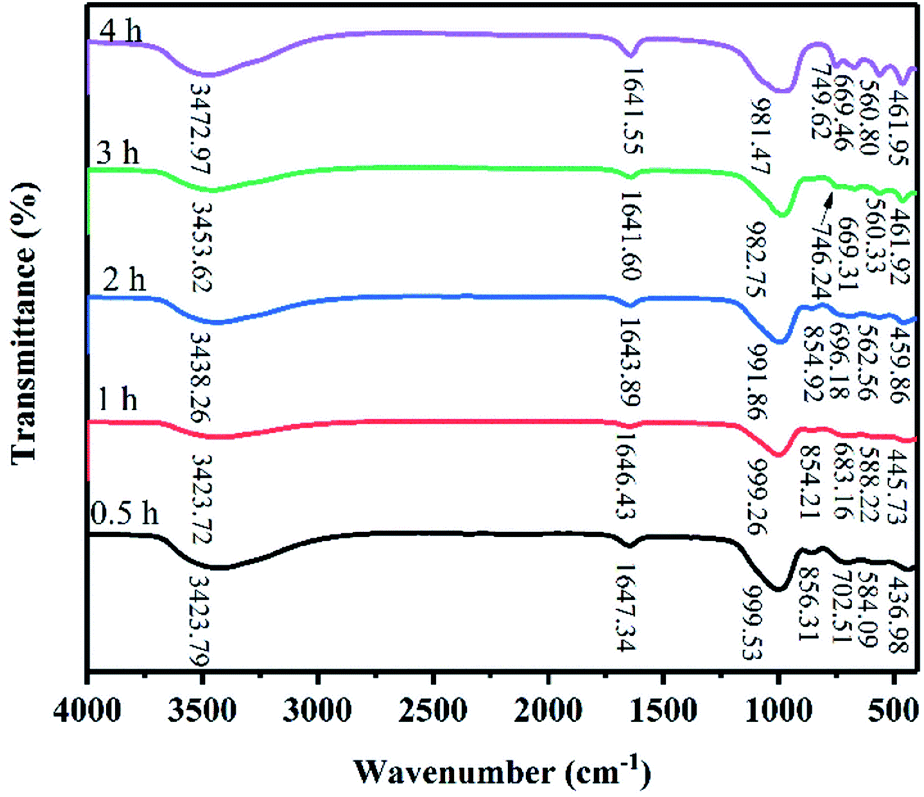
Fig. 4 shows the FT-IR spectra of the products at 82 °C for crystallization times between 0.5 h and 4 h. The absorbance bands at 1641.55–1647.34 cm−1 are attributed to the H–O–H bending vibration of adsorbed water and the broad absorbance bands at 3423.72–3472.97 cm−1 are attributed to the stretching vibration of the OH groups on the surface of zeolite X.10 The T–O (T = Si, Al) bending vibrations of the internal tetrahedron were observed in 436.98 cm−1 to 461.95 cm−1. The absorbance bands at 669.31–702.51 cm−1 are assigned to the symmetric stretching of the internal tetrahedron. The symmetric stretching vibrations of the external tetrahedron are observed at 746.24–749.62 cm−1. The bands at 854.21–856.31 cm−1 are assigned to the Si–OH bending vibration, indicating that Si and Al dissolved to form aluminosilicate gel.21,22 With increased crystallization time, the band of Si–OH bending vibration disappeared, indicating that extending the crystallization time promoted the transformation of aluminosilicate into a crystal substance. It is important to note that when the crystallization time was increased from 0.5 h to 1 h, the absorbance bands at 584.09 and 588.22 cm−1 appeared due to Si–O bending vibrations. When the crystallization time was prolonged, the bands at 584.09 and 588.22 cm−1 disappear, and the absorbance bands at approximately 560 cm−1 appeared. The absorbance bands at 560.33–562.56 cm−1 were attributed to the double six-membered rings which were the main structural subunit of zeolite X. Finally, the absorbance bands at 981.47–999.53 cm−1 are attributed to the asymmetric stretching of the internal tetrahedron. When the crystallization time was 0.5–4 h, the peak position of asymmetric stretching gradually shifted from high wave number to low wave number (999.53–981.47 cm−1). Because the peak position of asymmetric stretching gradually shifted from a high wave number to a low wave number, we suspect that the Al content in the skeleton increased. Therefore, we believe that the structure of zeolite X undergo complex changes in this system.
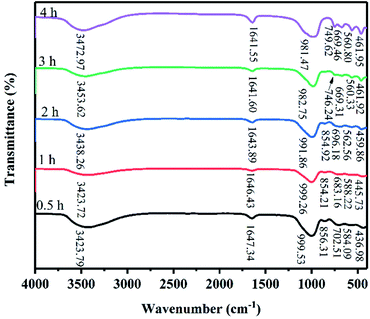 |
| | Fig. 4 FT-IR spectra of products synthesized at various crystallization times. | |
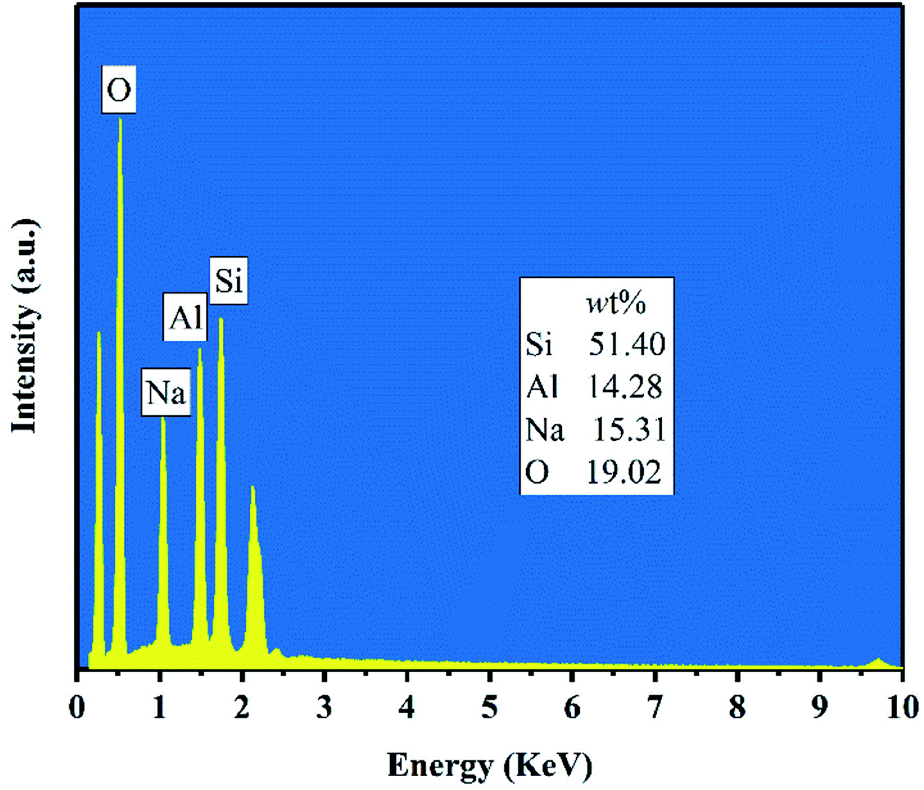
Fig. 5 shows the FESEM images of products for crystallization times between 0.5–4 h at 82 °C. Only the nanoparticles (50–100 nm) with irregular morphology were observed in the product after hydrothermal treatment for 0.5–1 h (Fig. 5(a and b)). Combined with Fig. 3(a), we can see that the nanoparticles were amorphous aluminosilicate gel. As shown in the Fig. 5(c and d), some near-spherical crystals appeared after hydrothermal treatment for 2 h, which were formed by the aggregation of many small crystals, indicating that the crystals formation of zeolite X was still in a nucleation-dominated crystal growth period. The surfaces of some of crystals were smooth and the shape was similar to a triangle. As shown in Fig. 3(a), 5(d) and Table 3, when the crystallization time reached 3 h, the diffraction peak intensity of the product at the angle of 6.08° was noticeably larger than other diffraction peak, indicating that the (111) crystal face of zeolite X was in anisotropic growth. The higher value of crystal plane spacing of the (111) crystal plane indicated that the higher the atomic density of the crystal plane, the faster the crystal growth along the (111) crystal plane. The (111) crystal face of cubic crystal system was triangle. The triangle which can be seen from the Fig. 5(d) was the (111) crystal face with rapid growth. When the crystallization time was further prolonged to 4 h, the (111) crystal faces were exposed in large area, and the eight triangular crystal faces grow into octahedral structure and finally grow into crystals with grain size of about 2 μm. At the same time, many small crystals were observed on the surface of some octahedrons (Fig. 5(e)). Because the new aggregates were formed from the Si and Al species around the octahedrons and the aggregates gradually grow into the new smaller octahedrons, the results indicate that some crystals entered the secondary growth phase. The formation of octahedron stopped when the Si and Al species were depleted or the system for synthesizing zeolite X was not under equilibrium conditions. Fig. 6 shows the EDS analysis of product after 4 h at 82 °C. The EDS analysis of zeolite X conversion from stellerite zeolite indicates the presence of Si, Al, Na, and O with their weight% of 19.02%, 15.31%, 14.28%, and 51.40%, respectively. The Si/Al was found to be 1.18 which is comparable to the stoichiometric composition of typical zeolite X.10 Nucleation of zeolites is related to liquid phase composition and gel–solid surface. Due to their nanoscale size, gel–solid particles have the characteristics of large specific surface area and high surface activity. The gel–solid-phase was dissolved in alkali solution and then a wet surface was formed with the liquid-phase, which was conducive to the nucleation of zeolite X crystals and shedding of zeolite X crystals on the surface of gel solid. When Shen et al.13 synthesized zeolites by the traditional hydrothermal method, they found that the crystal nucleus of zeolites were formed on the surface of acidified zeolite, and the finer the particle size of acidified zeolite was, the more conducive to the formation of zeolite X crystal. As can be seen from Fig. 5(c and d), zeolite X crystals were formed on the surface of the nanoscale gel–solid-phase by using the two-step hydrothermal method. The results indicate that the gel solid phase not only provides the material components needed for the hydrothermal reaction, but also provides the interface conditions for the nucleation and crystallization of zeolite X.
 |
| | Fig. 5 FESEM images of products synthesized at various crystallization times: (a) 0.5 h, (b) 1 h, (c) 2 h, (d) 3 h, (e) 4 h. | |
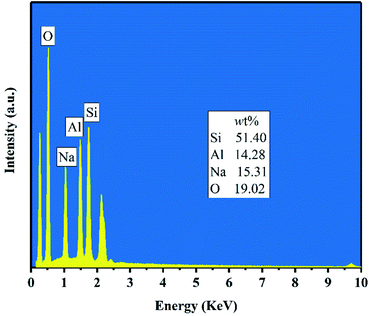 |
| | Fig. 6 EDS analysis of product after 4 h at 82 °C. | |
The Raman spectra of zeolite X in the solid-phase of crystallization are shown in Fig. 7. After 0.5 h of crystallization, the bands at 493, 567, 850, and 1009 cm−1 were observed in the Raman spectra of the solid-phase.23,24 These bands are not the characteristic Raman bands of zeolite X, indicating that the product was still amorphous aluminosilicate in the solid-phase, as confirmed by the XRD pattern (Fig. 3(a)). These Raman bands were relatively broad due to the wide distribution of the T–O–T angle of the amorphous aluminosilicate in the solid product. The band at 493 cm−1 is assigned to the bending mode of the T–O–T bond of four-membered rings.25 This result suggests that the amorphous aluminosilicate contains four-membered rings. The band at 567 cm−1 is assigned to the four-membered ring structures with branched chains.26 The band at 850 cm−1 is assigned to the symmetric stretching mode of T–O. The bands observed between 900 and 1200 cm−1 were assigned to the T–O–T asymmetric stretching mode.23 Significantly, the broad band at 1009 cm−1 in the high-frequency region indicates the presence of β cages in the product. The β cages were formed via the connection of four-membered rings and six-membered rings, which is a secondary structural unit of the zeolite X framework. However, the framework of zeolite X has not yet been formed in this stage. After 2 h of crystallization, the broad band at 493 cm−1 became sharper and more intense, indicating that more four-membered rings were formed. Additionally, the band shift from 493 to 505 cm−1 is typically assigned to four-membered rings vibration in the framework of zeolite X structure.23 Further, two new bands at 284 and 377 cm−1, which were assigned to the bending mode of a double six-membered rings appeared.23 At the same time, the bands at 1009 cm−1 split into the double bands at 987 and 1070 cm−1, which are characteristic Raman bands of zeolite X. Additionally, the band at 567 cm−1 disappear as the bands at 284 and 377 cm−1 appear,24,26 indicating that the branched four-membered rings were closely related to the formation of double six-membered rings. The double six-membered rings are formed via the connection of branched four-membered rings. The Raman bands in the higher frequency region were assigned to the symmetric stretching mode of T–O, which was very sensitive to the structure of zeolite X. According to a study by Dutta et al.,27 when Si/Al was 1.3, there were four bands in the high-frequency region, and the first three bands merged into one wide band. The bands at 987 and 1070 cm−1 were associated with different lattice oxygen atoms in the framework of zeolite X. The bands in high the region were not well resolved due to the imperfect zeolitic lattice, which was consistent with the research of Xiong et al.23 in the double six-membered rings play an important role in the synthesis of zeolite X, because the β cages are connected by double six-membered rings to form the framework of zeolite X. XRD patterns and Raman spectra indicate that the crystal nucleus of zeolite X was formed in 1–3 h, and then the crystal nucleus rapidly grew into a crystal of about 2 μm. The crystallization process of zeolite X can be described as the self-assembly process of the small fragments.26 Thus, to further understand the structures of possible small fragments formed during the crystallization of zeolite X, solid-state NMR measurements were carried out.
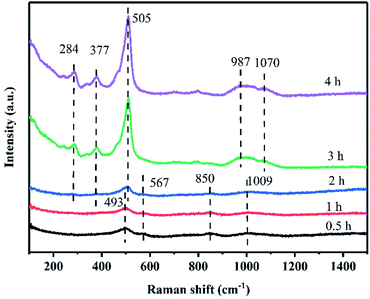 |
| | Fig. 7 Raman spectra of products synthesized at various crystallization times. | |
The 27Al MAS NMR spectra of the products crystallized at 82 °C for 0.5, 1, 2, 3, and 4 h are shown in Fig. 8(a). The spectra of the sample heated for 0.5 h exhibited a broad signal near 58.20 ppm. This peak is attributed to Al(OSi)4 or Al(OSi)3(OH) species in the amorphous aluminosilicate phase.26 Because Al(OSi)4 and Al(OSi)3(OH) species only exist in sodalite cages and 4R units, this result suggests that sodalite cages and 4R units formed.26 Heating the initial gel for 2 h or longer resulted in a sharper peak at 59.61 ppm, implying that most of the Al was tetrahedral connected to the framework Al(OSi)4.26,28 The corresponding powder XRD patterns, FT-IR spectra, and Raman spectra show the formation of crystalline zeolite X. A fully condensed Si atom connected via an oxygen bridge to n Al atoms is labeled Q4(nAl, (4-n)Si) where n = 0, 1, 2, 3, 4.10 Likewise a fully condensed Si atom connected to four Si atoms is denoted as Q4. The 29Si MAS NMR spectra of the products crystallized at 82 °C for 0.5, 1, 2, 3, and 4 h are shown in Fig. 8(b). The spectrum of the product heated for 0.5 h exhibit a broad signal centered at −85.96 ppm, it is assigned to (AlO)3SiOH, (SiO)(AlO)Si(OH), (AlO)2Si(OH)2, (SiO)2SiOH, and (SiO)2(AlO)SiOH, which was mainly due to the presence of amorphous silica.26 After crystallization for 1 h, a small shoulder at about −80.65 ppm appeared, attributable to dimer and (AlO)2Si(OH)2.26,29,30 By increasing he crystallization time to 2 h, shifts appeared at −84.85 and −89.19 ppm, corresponding to Si(OAl)4 and (SiO)1Si(OAl)3 in the sample. When the crystallization time was 3 h, the chemical shift at −80.24 ppm disappeared, attributable to the Si(OAl)4 and (SiO)1Si(OAl)3 linkage generated by the substitution of Al for Si in the tetrahedral position.26 The typical 29Si MAS NMR chemical shifts for zeolite X, centered at −84.49 and −89.45 ppm, were already quite apparent when the crystallization time reached to 3 h and became sharper with increasing crystallization time, indicating that the relative crystallinity of the product increased. The Si/Al ratio of the zeolite X framework can be calculated accurately from the relative intensities of different 29Si signals, and a Si/Al ratio of 1.14 for zeolite X was obtained from the 29Si spectrum of 4 h (Table 4). The result of the Si/Al ratio is consistent with the EDS analysis.
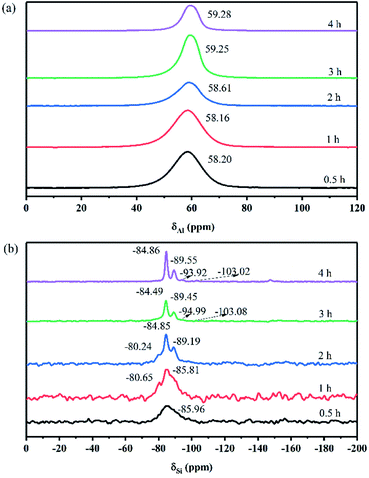 |
| | Fig. 8 27Al (a) and 29Si (b) MAS NMR spectra of the solid phase during the crystallization of zeolite X. | |
Table 4 Distribution of Al in zeolite X
| Q4(nAl, (4-n)Si) |
4 |
3 |
2 |
1 |
| δ (ppm) |
−84.62 |
−89.19 |
−93.92 |
−103.02 |
| In |
25.06 |
12.25 |
2.03 |
1.33 |
| Composition (%) |
61.61 |
30.12 |
4.99 |
3.27 |
| n(Si/Al) |
1.14 |
If the dissolution of the gel–solid-phase occurs during the crystallization process, the gel–liquid-phase directly participates in the nucleation and crystallization of zeolite X, so the conversion mechanism of zeolite X is the liquid-phase conversion mechanism. When the nucleation and crystallization of zeolite X only occurs in the gel–solid-phase, the conversion mechanism of zeolite X is the solid-phase conversion mechanism. Therefore, the conversion mechanism of zeolite X can be preliminarily determined according to the content changes of the main elements in the hydrothermal reaction process. Tables 5 and 6 present the XRF and inductively coupled plasma either optical emission spectroscopy (ICP-OES) results for the changes in Si, Al, and Na contents obtained from the solid and liquid-phase.
Table 5 Chemical compositions of the products prepared at 82 °C for various reaction times
| Crystallization time (h) |
SiO2 (wt%) |
Al2O3 (wt%) |
Na2O (wt%) |
| 0.5 |
44.287 |
35.425 |
20.202 |
| 1 |
43.484 |
35.833 |
20.589 |
| 2 |
43.364 |
35.842 |
20.701 |
| 3 |
43.104 |
35.879 |
20.92 |
| 4 |
43.189 |
35.962 |
20.751 |
Table 6 The Si, Al, and Na contents in the gel–liquid-phase at 82 °C for various reaction times
| Crystallization time (h) |
SiO2 (g) |
Al2O3 (g) |
Na2O (g) |
| 0.5 |
0.122 |
0.033 |
1.604 |
| 1 |
0.128 |
0.036 |
1.642 |
| 2 |
0.159 |
0.027 |
1.684 |
| 3 |
0.191 |
0.026 |
1.879 |
| 4 |
0.236 |
0.021 |
1.784 |
Table 5 shows that the contents of Si rapidly decreased during the crystallization, which was attributed to the dissolution of Si of gel solid phase in the alkaline solution. The contents of Al quickly increased because the Al in the solid-phase of gel is dissolved and then transported into liquid-phase. The aluminum oxide tetrahedrons are formed in the gel–liquid-phase, and further transformation from gel–liquid-phase into the nucleus and crystals of zeolite X.10 In the process of hydrothermal reaction, the contents of Na continually increased indicating that NaOH continuously participated in the reaction and also incorporated into the channel of zeolite X,10 guiding the Si–O tetrahedron and Al–O tetrahedron to connect together. A direct correlation among the contents of Si, Al, and Na in the gel–solid-phase and gel–liquid-phase was found. Table 6 shows that in the liquid phase of gel, the amounts of Si increased and the amounts of Al decreased as the crystallization time increased. And the dissolution of sodium aluminate release alumina and sodium oxide, so the content of sodium in the solution would gradually increase. According to the Tables 5 and 6, the Si, Al, and Na in the gel–solid-phase were not directly transported to zeolite X, and they are transported by the solution.
Conversion mechanism
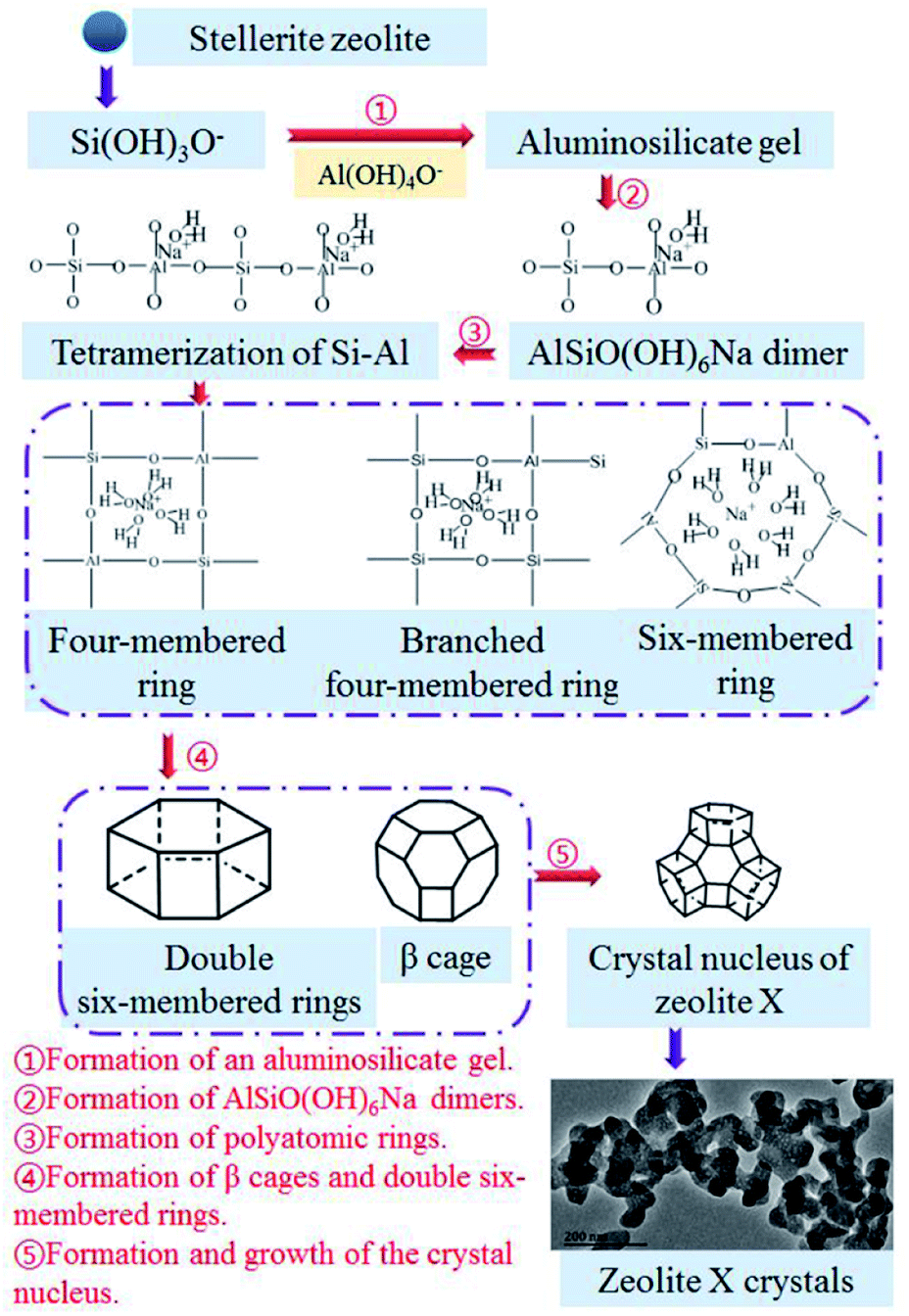
The nucleation and crystallization of zeolite X and the change in the contents of Si, Al, and Na in the gel during the hydrothermal reaction show that the Si, Al, and Na in the gel–solid-phase were transferred to zeolite X through the liquid-phase. Fig. 9 shows the conversion mechanism for zeolite X from stellerite zeolite, which can be divided into five steps.31,32 The first step was the formation of an aluminosilicate gel (Fig. 9(1)). The active silica was extracted under hydrothermal conditions at 85 °C, and stellerite zeolite started dissolving in the presence of a strong alkaline medium. The silica source was mainly composed of low modulus and low concentration of sodium silicate, and its degree of polymerization was low, which was conducive to the formation of aluminosilicate gel. The second step involved the formation of AlSiO(OH)6Na dimers (Fig. 9(2)). When some silicon atoms were replaced by Al atoms in the liquid-phase, an Al–O tetrahedron was formed, and then the AlSiO(OH)6Na dimers were formed by initial condensation from the Si–O tetrahedron and Al–O tetrahedron. Fig. 8(b) shows the presence of dimers in the sample of hydrothermal reaction for 1–2 h, and Yang et al.33 found that dimers were the key components in the formation of zeolite A by theoretical analysis, which further proves the importance of dimers. The third step involved the formation of polyatomic rings (Fig. 9(3)). The tetramerization of Si–Al is formed by AlSiO(OH)6Na dimers by condensation,33 and then the tetramerization of Si–Al further forms polyatomic rings by polymerization. Because of the unbalanced charge in the Al–O tetrahedron, positive charges were needed to neutralize the charge of the reaction system, and sodium ions were important to balancing the charge of the system. Polyatomic rings such as four-membered rings and six-membered rings were further polycondensed under the bridging of sodium ions, and some of the four-membered rings with Si–Al–O oxygen tetrahedrons form branched four-membered rings. The fourth step was the formation of β cages and double six-membered rings (Fig. 9(4)). The four-membered rings and six-membered rings were connected by the effect of the oxygen bridge bond to form β cages. These processes mainly occurred during the aging stage and 1 h of the crystallization reaction. After 2–3 h of crystallization, double six-membered rings were formed from the assembly of the branched four-membered rings. The double six-membered rings played a critical role in the crystallization process because the β cages were interconnected via double six-membered rings to form the framework of zeolite X. The fifth step was the formation and growth of the crystal nucleus (Fig. 9(5)). The appearance of the double six-membered rings indicates the formation of the zeolite X framework. This suggests that zeolite X crystal nuclei were mainly formed during the 2 h of crystallization reaction. As the reaction progressed, the crystal nuclei rapidly grew into small crystals and turned into large crystals of approximately 2 μm via oriented crystalline aggregation. The TEM image in Fig. 9 shows that during the early stage of crystal growth, the small crystals of zeolite X were mainly composed of numerous nanocrystals. The TEM image further shows that the zeolite X crystal grew from small to large sizes. Interestingly, because the crystallization kinetics curve was S shaped (Fig. 3(b)) and because the phenomenon of secondary nucleation appears in the conversion process (Fig. 5(e)), zeolite X crystal nucleus formed via spontaneous nucleation and autocatalytic nucleation. The crystal growth of zeolite X was always from the surface of the gel–solid-phase to the liquid-phase. The new nuclei formed on the surface of large crystals as the concentration of Si, Al, and Na in the gel–liquid-phase became supersaturated. Crystallization did not stop until all the gel–solid-phase was consumed. Therefore, the conversion mechanism of stellerite zeolite conversion into zeolite X conformed to the liquid-phase transformation mechanism.
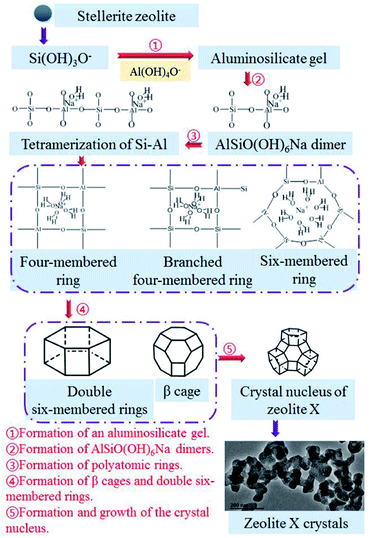 |
| | Fig. 9 Conversion mechanism for zeolite X from stellerite zeolite. | |
Conclusions
In this work, we studied the conversion mechanism of zeolite X synthesized from stellerite zeolite using a two-step hydrothermal process. The crystallization process of zeolite X was investigated by XRD, FESEM, FT-IR, Raman, solid-state NMR, XRF and TEM techniques. These analyses show that stellerite zeolite can be converted into zeolite X with high crystallinity at 82 °C using a crystallization time of 4 h. With an increase in reaction time, the crystal cell parameters (a, b, c, and volume) of the products gradually decreased. This could be a result of parts of Na ions migrating from β cages and hexagonal prism cages to super cages in the crystallization stage under the heating and pressurizing environment. The nucleation and crystallization of zeolite X were related to the gel–solid-phase surface and the concentrations of Si, Al, and Na in the gel–liquid-phase. After the gel–solid-phase was transformed into the liquid-phase, the concentrations of Si, Al, and Na in the gel–liquid-phase were supersaturated, which promoted the growth of the crystal nucleus of zeolite X on the wettable surface of the gel–solid-phase. The nucleation and crystallization of zeolite X consume Si, Al, and Na in the gel–liquid-phase and cause the gel–solid-phase to continuously dissolve until the crystal nucleus completely grows into crystals with a particle size of approximately 2 μm. A schematic was used to further explain the formation mechanism, and it was confirmed that the conversion mechanism of stellerite zeolite conversion into zeolite X conforms to the liquid-phase conversion mechanism. Therefore, the present research demonstrates the importance of understanding the formation mechanism of other natural minerals converted to zeolite.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51564008), National Natural Science Foundation of China (No. 41662005), Guangxi Innovation Research Team Project (No. 2018GXNSFGA281001), Guangxi Mid-Youth Capability Project (No. 2018KY0249), the Research Funds of Guangxi Key Laboratory of Environmental Pollution Control Theory and Technology (No. 001102216078), Foundation of Guilin University of Technology (No. GUTQDJJ2015027).
Notes and references
- S. Sivalingam and S. Sen, Appl. Surf. Sci., 2018, 455, 903–910 CrossRef CAS.
- M. Mezni, A. Hamzaoui, N. Hamdi and E. Srasra, Appl. Clay Sci., 2011, 52, 209–218 CrossRef CAS.
- G. Y. Yao, J. J. Lei, X. Y. Zhang, Z. M. Sun and S. L. Zheng, Materials, 2018, 11, 906–919 CrossRef PubMed.
- C. Y. Wang, J. T. Zhou, Y. Wang, M. X. Yang, Y. W. Li and C. G. Meng, J. Chem. Technol. Biotechnol., 2013, 88, 1350–1357 CrossRef CAS.
- T. S. Jamil, H. H. Abdel Ghafar, H. S. Ibrahim and I. H. Abd El-Maksoud, Solid State Sci., 2011, 13, 1844–1851 CrossRef CAS.
- A. S. Kovo, Chem. Eng. Commun., 2012, 199, 786–797 CrossRef CAS.
- N. M. Musyoka, R. Missengue, M. Kusisakana and L. F. Petrik, Appl. Clay Sci., 2014, 97–98, 182–186 CrossRef CAS.
- C. W. Purnomo, C. Salim and H. Hinode, Microporous Mesoporous Mater., 2012, 162, 6–13 CrossRef CAS.
- C. Belviso, F. Cavalcante, F. Javier Huertas, A. Lettino, P. Ragone and S. Fiore, Microporous Mesoporous Mater., 2012, 162, 115–121 CrossRef CAS.
- G. Y. Yao, J. J. Lei, X. Y. Zhang, Z. M. Sun, S. L. Zheng and S. Komarneni, Mater. Res. Bull., 2018, 107, 132–138 CrossRef CAS.
- Y. Li, X. Wang, Y. C. Dong and J. W. Zhu, Trans. Nonferrous Met. Soc. China, 2002, 12, 321–325 CAS.
- Y. Li, X. Wang, X. J. Yang and S. G. Zhang, Trans. Nonferrous Met. Soc. China, 2002, 12, 1024–1030 CAS.
- S. H. Shen, S. G. Zhang, D. W. Wang and K. M. Fang, J. Cent. South Univ. Technol., 2005, 12, 315–319 CrossRef CAS.
- S. H. Shen, D. W. Wang and S. G. Zhang, J. Cent. South Univ. Technol., 2001, 32, 344–347 CAS.
- S. H. Shen, K. M. Fang, S. G. Zhang and D. W. Wang, J. Univ. Sci. Technol. Beijing, 2003, 25, 489–494 Search PubMed.
- X. J. Meng, X. W. Guo, Y. J. Zhong, Y. C. Pei, N. C. Chen and Q. L. Xie, Bull. Mater. Sci., 2019, 42, 232–239 CrossRef.
- S. S. Gao, N. C. Chen, Y. C. Pei, X. L. Wang and X. J. Meng, J. Synth. Cryst., 2018, 47, 1096–1101 CAS.
- C. Y. Zhang, N. C. Chen, X. H. Zhang and F. Zhan, J. Chin. Ceram. Soc., 2014, 42, 1332–1336 CrossRef CAS.
- P. Kunecki, R. Panek, A. Koteja and W. Franus, Microporous Mesoporous Mater., 2018, 266, 102–108 CrossRef CAS.
- L. Y. Cao, B. T. Xia, C. H. Wang, L. Zhao, P. H. Zeng and B. J. Shen, Ind. Catal., 2018, 26, 56–62 Search PubMed.
- J. L. Jiang, L. D. Feng, X. Gu, Y. H. Qian, Y. X. Gu and C. S. Duanmu, Appl. Clay Sci., 2012, 55, 108–113 CrossRef CAS.
- Z. H. Zhou, G. Jin, H. Liu, J. X. Wu and J. F. Mei, Appl. Clay Sci., 2014, 97–98, 110–114 CrossRef CAS.
- G. Xiong, Y. Yu, Z. C. Feng, Q. Xin, F. S. Xiao and C. Li, Microporous Mesoporous Mater., 2001, 42, 317–323 CrossRef CAS.
- F. T. Fan, Z. C. Feng, G. N. Li, K. J. Sun, P. L. Ying and C. Li, Chem.–Eur. J., 2008, 14, 5125–5129 CrossRef CAS PubMed.
- Y. C. Xiao, N. Sheng, Y. Y. Chu, Y. Q. Wang, Q. M. Wu, X. L. Liu, F. Deng, X. J. Meng and Z. C. Feng, Microporous Mesoporous Mater., 2017, 237, 201–209 CrossRef CAS.
- L. Zhang and Y. N. Huang, J. Porous Mater., 2015, 22, 843–850 CrossRef CAS.
- P. K. Dutta and J. Twu, J. Phys. Chem., 1991, 95, 2498–2501 CrossRef CAS.
- S. Q. Su, H. W. Ma and X. Y. Chuan, Adv. Powder Technol., 2016, 27, 139–144 CrossRef CAS.
- C. E. A. Kirschhock, R. Ravishankar, F. Verspeurt, P. J. Grobet, P. A. Jacobs and J. A. Martens, J. Phys. Chem. B, 1999, 103, 4965–4971 CrossRef CAS.
- C. T. G. Knight and S. D. Kinrade, J. Phys. Chem. B, 2002, 106, 3329–3332 CrossRef CAS.
- A. Kumar and M. K. Naskar, Int. J. Appl. Ceram. Technol., 2019, 16, 1525–1532 CrossRef CAS.
- A. Kumar and M. K. Naskar, J. Asian Ceram. Soc., 2019, 7, 355–360 CrossRef.
- C. S. Yang, J. Miguel Mora Fonz and C. R. A. Catlow, J. Phys. Chem. C, 2013, 117, 24796–24803 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yijian Zhongb,
Qinglin Xie*ac and
Nanchun Chen*d
a,
Yijian Zhongb,
Qinglin Xie*ac and
Nanchun Chen*d