
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparative isolation of maltol glycoside from Dianthus superbus and its anti-inflammatory activity in vitro†
Chen Yuan a,
Jun Dang‡
b,
Yu Hana,
Chuang Liub,
Song Yuc,
Yue Lva,
Yunbin Cuid,
Zhenhua Wanga and
Gang Li‡*a
a,
Jun Dang‡
b,
Yu Hana,
Chuang Liub,
Song Yuc,
Yue Lva,
Yunbin Cuid,
Zhenhua Wanga and
Gang Li‡*a
aCenter for Mitochondria and Healthy Aging, College of Life Sciences, Yantai University, Yantai 264005, China. E-mail: ligang@ytu.edu.cn; Fax: +86-535-6902638; Tel: +86-535-6902638
bQinghai Provincial Key Laboratory of Tibetan Medicine Research, Key Laboratory of Tibetan Medicine Research, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810001, Qinghai, China
cCollege of Pharmacy, Qinghai University, Xining 810016, Qinghai, China
dCollege of Life Sciences, Qinghai Normal University, Xining 810016, Qinghai, China
First published on 9th February 2022
Abstract
Dianthus superbus is a traditional Chinese medicine that is commonly utilized as a treatment for inflammation, pain, and immunological conditions. In this study, an anti-inflammatory maltol glycoside derived from Dianthus superbus was isolated for the first time via medium and high-pressure liquid chromatography, and at the same time, the in vitro anti-inflammatory activity of this maltol glycoside was preliminarily explored. Initially, crude samples of Dianthus superbus were preprocessed via MCI GEL® CHP20P and Spherical C18 medium-pressure chromatography, under the guidance of evaluation of in vitro anti-inflammatory activity. Fr44 was found to be the target fraction, and it was further isolated via two-dimensional reversed-phase/hydrophilic interaction liquid chromatography, yielding > 95% pure and was identified as tunicoside B. MTT assay, nitric oxide and nitric oxide synthase were used to evaluate the effects of tunicoside B on murine macrophage Raw264.7 by nitric oxide synthase assay kit, molecular docking, and western blotting. The results showed that tunicoside B did not affect the viability of cells and exhibited significant anti-inflammatory activity. As far as we know, this is the first report of tunicoside B from Dianthus superbus and the first study on the anti-inflammatory activity of tunicoside B. More importantly, the approach established in this study is expected to provide a theoretical basis for the separation and pharmacological activity study of maltol glycosides from other natural products.
1 Introduction
Traditional Chinese medicines (TCMs) exhibit a diverse array of pharmacological activities and have recently been found to represent promising drug precursors with unique chemical structures, many of which might be used as natural anti-inflammatory reagents.1,2 Inflammation is an immune-mediated response to trauma induced by physical, chemical, or pathogenic stimuli.3 Unrestrained or inappropriate inflammation, however, can contribute to the incidence of a range of diseases such as hypertension, obesity, and diabetes.4–7 Corticosteroids and non-steroidal anti-inflammatory drugs are often used to treat inflammatory conditions.8 Many recent studies have assessed the anti-inflammatory activity of different TCMs. Such activity is attributable to the multi-component nature of these medicines and to their ability to mitigate inflammation by simultaneously targeting several signaling pathways.9 The use of TCMs with anti-inflammatory activity may thus have value as a means of preventing or treating inflammatory diseases.Dianthus superbus (D. superbus) is one of the important TCMs that has been used in China for over 1000 years as a treatment for inflammation, pain, and immunological disorders.10 D. superbus is a small herb in the Caryophyllaceae family that can be found in areas of Northeastern China including Shandong Province.11 Prior chemical analyses of D. superbus had revealed it to be primarily composed of bioactive compounds, including cyclic peptides, flavones, and triterpene saponins.12–14 Nonetheless, recent research proved that the majority of studies of D. superbus focused on the discovery of new compounds,14–16 such as new quinolone alkaloids, cyclopeptides, triterpenoid saponins, etc., with just a few studies endeavored to highlight its bioactive phytochemical composition. In addition, the separation of D. superbus was mainly based on traditional separation technology such as open column (silica gel, conventional gel, and polyamide) chromatography-based separation techniques. These approaches, however, are time-consuming, complicated, not reproducible, and do not allow for online detection.15–18 There is thus an unmet need for the development of an accurate, rapid, and convenient approach for extracting compounds from D. superbus in a manner that is guided by the bioactivity of these compounds.
Preparative high-performance liquid chromatography (prep-HPLC) is an approach that can be utilized to effectively extract specific compounds from natural biological samples, achieving excellent efficiency while simultaneously enabling automated control, online detection, and reproducible separation.19–21 When conducting a prep-HPLC separation, the selection of an appropriate stationary phase is critical to ensure that compounds can be effectively isolated from complex samples.22–24 One-dimensional separation approaches are limited in their ability to isolate single compounds owing to considerations pertaining to chromatographic resolution and column efficiency. As such, two-dimensional HPLC (2D HPLC) has emerged as a more practical approach to separating individual compounds from complex natural products, enabling marked improvements in both separation selectivity and peak capacity.25–27 Owing to its reproducibility, universality, and excellent capacity, most HPLC separation approaches rely on reversed-phase liquid chromatography (RPLC) approach, which is very classical.28,29 However, due to similar separation principles, RPLC/RPLC exhibits low orthogonality. Some researchers have employed hydrophilic interaction chromatography (HILIC) for polar compound purification from complex samples or as a means of providing complementary selectivity to an RPLC approach.30,31 In this context, the coupling of RPLC and HILIC approaches represent an effective means of efficiently isolating bioactive compounds from various natural samples.32–34
In this study, an effective method was utilized to isolate an anti-inflammatory maltol glycoside from D. superbus by a combination of medium- and high-pressure liquid chromatography, which targeted in vitro anti-inflammatory activity. A novel method based on an online middle-pressure chromatographic tower containing stationary phase MCI GEL® CHP20P and Spherical C18 was used to separate the samples of D. superbus, with compounds of interest being selected under the guidance of in vitro analyses of anti-inflammatory activity, respectively. Through a high-pressure 2D RPLC/HILIC approach, the target fraction (Fr44) was subsequently isolated with high efficiency, ultimately leading to the isolation of a single maltol glycoside (Fr4411), which was identified as tunicoside B. The in vitro anti-inflammatory efficacy of tunicoside B was evaluated based on the lipopolysaccharide (LPS)-stimulated RAW 264.7 cellular model. The cytotoxicity was measured with an MTT assay, the level of NO production was determined with an NO assay kit, the binding site was predicted with molecular docking, the nitric oxide synthase (iNOS) activity was determined with iNOS assay kit, and the expression of iNOS was analyzed with western blot analysis. This bioactivity-guided method provides a new strategy for the isolation of anti-inflammatory compounds from TCMs and other natural products.
2 Experimental
2.1 Apparatus and chemicals
Preparative liquid chromatography (Hanbon Science & Technology Co., China) was used to conduct medium-pressure sample pretreatment. This system consisted of an LC workstation, two prep-HPLC pumps (NP7000), a 5 mL manual injector, and a UV-Vis detector (NU3000). The Essentia LC-16 (Shimadzu Instruments Co., China) system was composed of an LC workstation, two binary gradient pumps, a UV/Vis detector, and a fraction collector. A Shimadzu UV2401PC instrument (Shimadzu, Japan) was utilized for UV spectra acquisition. A Bruker Tensor 27 FTIR spectrometer (Bruker, Germany) was used to record IR spectra. A JASCO P-1020 digital polarimeter (Jasco, Japan) was used to obtain optical rotations, and an Agilent 6500 Q-TOF mass spectrometer (Agilent Instruments Co., USA)/Waters QDa ESI mass spectrometer (Waters Instruments Co., USA)/Q was used. The 1H and 13C NMR spectra were acquired with a Bruker Avance 600 MHz using a solvent consisting of MeOH-d4 (Bruker Instruments Co., Germany). In addition, this study made use of a microplate reader (Molecular Devices, CA, USA), 5200 Multi Luminescent Image Analyzer (Tanon Science & Technology Co., Ltd. Shanghai, China), and FACS AriaTM Flow Cytometer (Becton Dickinson, San Jose, CA, USA).MCI GEL® CHP20P (120 μm) was obtained from Mitsubishi Chemical Corporation (Japan), while Spherical C18 (50 μm) was obtained from SiliCycle (Canada). A 7-X10 preparative column (20 × 250 mm, 7 μm) and an analytical column (4.6 × 250 mm, 7 μm) were purchased from Acchrom Tech (Beijing, China). A SunFire® C18 (4.6 × 250 mm, 5 μm) analytical column was purchased from the Waters Corporation (USA). A Click XION analytical column (4.6 × 250 mm, 5 μm) and preparative columns (20 × 250 mm, 5 μm) were obtained from ACCHROM Corporation (Beijing, China).
Chromatographic grade methanol (CH3OH) and acetonitrile (ACN) were obtained from Yunnan Xinlanjing Chemical Industry, while analytical grade versions of these same solutions were obtained from Kelon Chemical Reagent Factory (Chengdu, Sichuan, China). Chromatographic grade water was prepared using a Moore water purification station (Chongqing, China).
RPMI 1640 medium, fetal bovine serum (FBS), phosphate buffered saline (PBS), trypsin, and penicillin were obtained from Gibco (Grand Island, NY, USA). A nitric oxide assay kit, nitric oxide synthase assay kit, BCA assay kit, and enhanced chemiluminescent (ECL) assay kit were obtained from Beyotime (Beyotime Biotechnology, Shanghai, China). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and inducible NO synthase (iNOS) antibodies were purchased from Cell Signaling Technology (Shanghai, China). Horseradish peroxidase (HRP) goat anti-rabbit antibody was purchased from Abgent (Abgent, Beijing, China). Rofecoxib was purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2 Sample preparation
D. superbus was obtained in May 2019 from the Qingdao Laoshan District (altitude 1132.7 m, N 36°10′, E 120°37′) in Shandong, China, and was validated by Professor Jiantao Lv of the College of Pharmacy, Yantai University. The sample was deposited in Yantai University.In total, 1 kg of dried collected plant material was ground and extracted twice for 4 h each time with ethanol (10 L per extraction) at room temperature, after which 20 L of the total extract was pooled and concentrated at 55 °C in a reduced pressure rotary evaporator. When the extract was concentrated to a volume of approximately 500 mL, it was combined with 200 g of amorphous silica gel and fully dried in an oven at 60 °C. After the drying was complete, 338 g of the dried mixture was loaded onto a prepared 49 × 460 mm column containing a stationary phase composed of 1.2 L MCI GEL® CHP20P, with H2O/CH3OH/CH2Cl2 being used for elution as follows: 0–20 min, 100% H2O; 20–240 min, 0–100% CH3OH; 240–300 min, 100% CH3OH; 300–420 min, 0–100% CH2Cl2. During each mobile phase, the percentage of the corresponding eluant was increased from 0–100% in a linear fashion. Absorbance was assessed at 210 nm and a constant flow rate of 40 mL min−1. Following separation, the target fraction (Fr4) was collected, combined, and concentrated, yielding a final mass of 38.7 g.
Then, Fr4 was combined with amorphous silica gel and dried in an oven at 60 °C, after which the dried mixture was loaded onto a prepared 50 × 500 mm column containing a stationary phase composed of Spherical C18. Elution was conducted with H2O/CH3OH, with the percentage of methanol increasing in a linear manner from 20–65% over a 120 min period. A constant flow rate of 40 mL min−1 was maintained, and absorbance monitoring was conducted at 210 nm. Following separation, the target fraction (Fr44) was collected, combined, and concentrated, yielding a final mass of 1.4 g.
2.3 Two-dimensional high-performance liquid chromatography separated the fraction Fr44 and purity assay of Fr4411
A 7-X10 preparative column (20 × 250 mm, 7 μm) was used for 1D separation with mobile phases composed of 0.1% (v/v) formic acid in chromatographic grade H2O (A) and CH3OH (B) with a constant 19 mL min−1 flow rate and a gradient elution setting of 0–70 min, 25% B. Chromatograms were recorded at 210 nm, with the target fraction Fr441 being collected and concentrated to a final mass of 87.2 mg.A Click XION preparative column (20 × 250 mm, 5 μm) was used for 2D separation with mobile phases composed of 0.1% v/v trifluoroacetic acid in chromatographic grade H2O (A) and ACN (B). Separation was achieved at a 19 mL min−1 flow rate, with a gradient elution step of 0–60 min, 94% B. Chromatograms were recorded at 210 nm, with the target fraction Fr4411 being collected and concentrated to a final mass of 14 mg.
Fr4411 purity was determined using a SunFire® C18 analytical column (4.6 × 250 mm, 5 μm) with mobile phases composed of 0.1% (v/v) formic acid in chromatographic grade H2O (A) and CH3OH (B). Elution was conducted at a constant 1 mL min−1 flow rate with a linear gradient of 0–40 min, 20–80% B. This chromatogram was then recorded at 210 nm.
2.4 Cell culture
Murine macrophage RAW 264.7 cell lines from the cell bank of the Institute of Biochemistry and Cell Culture of Shanghai (Shanghai, China) were cultured in RPMI-1640 containing 10% FBS at 37 °C in a humidified 5% CO2 incubator.2.5 MTT assay
Cellular viability was evaluated with a conventional MTT assay. RAW 264.7 cells were added to 96-well plates (1 × 104 per well) for 24 h, and cells were then exposed to various concentrations (0, 5, 20, 80 μg mL−1 or 0, 5, 10, 20, 40, 60, 80, 100 μM) of the samples for 24 h. After that, MTT solution (5 mg mL−1, 10 μL per well) was added to the cell plates, followed by a 4 h incubation at 37 °C while protected from light. The medium was then removed and replaced with 100 μL per well DMSO, after which the absorbance at 490 nm was assessed via microplate reader.2.6 Measurement of NO concentration
A Griess reaction was used to quantify nitrite accumulation in the culture media as a means of quantifying NO production. RAW 264.7 cells were added to 96-well plates (1 × 105 per well) and allowed to adhere, after which they were treated for 24 h with LPS (1 μg mL−1) in the presence or absence of experimental samples (20 μg mL−1 or 5, 10, 20 μM) and rofecoxib (10 μM). An NO kit was then used based on the provided instructions, mixing culture supernatants with the Griess reagent and assessing the absorbance at 540 nm. A standard curve was prepared using a range of sodium nitrite concentrations.2.7 Molecular docking
A molecular docking approach was used to assess the theoretical interactions between different molecules. For this study, AutoDock was used to assess the potential interactions between the iNOS receptor and either tunicoside B or rofecoxib (as a positive control). The iNOS crystal structure (PDB code 2ORO) was obtained from the RCSB Protein Data Bank (https://www.rcsb.org/). Water molecules and ions were removed from the receptor prior to docking, after which polar hydrogen atoms and Kollman charges were added. The AutoGrid program was used to set a grid box at X, Y, and Z coordinates of 65.627, −15.4, and 49.901. Minimization was conducted with the Lamarckian genetic algorithm and the pseudo-Solis and Wets methods using default parameters. In total, 50 peptide conformations were defined based on the dock score values, with the conformation exhibiting the minimum binding energy being selected for model development.2.8 Assay of iNOS activity
The fluorescence method was used to detect the activity of total nitric oxide synthase in living cells. RAW 264.7 macrophages (4 × 105 per mL) were added to 12-well plates and allowed to adhere. After treatment with LPS (1 μg mL−1) and tunicoside B (0, 5, 10, 20 μM) and rofecoxib (10 μM) for 24 h, the culture supernatant was removed, and the nitric oxide synthase assay kit was then used based on the provided instructions. The fluorescence was measured (excitation at 495 nm and emission at 515 nm) using a FACS AriaTM Flow Cytometer (Becton Dickinson, San Jose, CA, USA). The data presented are the mean fluorescent signals for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells.
000 cells.
2.9 Western blot analysis of iNOS
RAW 264.7 macrophages (4 × 105 per mL) were added to 6-well plates for 24 h, followed by treatment for 10 h with LPS (1 μg mL−1) and with or without tunicoside B and rofecoxib (10 μM). Cells were then washed two times using chilled PBS, and proteins were extracted using 200 μL of lysis buffer, after which the lysates were centrifuged for 15 min at 14000 × g at 4 °C. BCA assay was used to measure protein levels in the collected supernatants, after which proteins were separated via SDS-PAGE and transferred onto PVDF membranes with a Bio-Rad electrophoresis apparatus. Membranes were blocked for 1 h at room temperature with 5% non-fat milk in TBST, followed by overnight incubation with iNOS antibody (1:1000, Cell Signaling Technology, Shanghai, China) at 4 °C. Membranes were then probed at room temperature for 2 h with Horseradish Peroxidase (HRP) goat anti-rabbit antibody, followed by protein detection with an enhanced chemiluminescent (ECL) assay kit based on provided instructions. A 5200 Multi Luminescent Image Analyzer was utilized to assess protein bands, with GAPDH serving as a loading control.
2.10 Statistical analysis
Data were means ± SD from triplicate experiments and were compared via Student's t-tests or one-way ANOVAs using SPSS v 18.0 (SPSS, IL, USA). P < 0.05 was considered statistically significant.3 Results and discussion
3.1 Sample pretreatment under in vitro activity guidance
An ethanol extraction approach was used to process 1 kg of air-dried whole D. superbus, yielding 138 g of crude extract at a yield efficiency of approximately 14%.As chlorophyll compounds can interfere with the adsorption of the preparative column stationary phase, the removal of these chlorophylls was necessary prior to preparative isolation.21,22 In the meantime, other impurities in crude extracts can also interfere with separation, resulting in poor sample loading and column contamination. As such, further crude extract purification prior to prep-HPLC was considered necessary. To that end, the crude D. superbus extract was pretreated with two medium-pressure chromatographic columns to refine samples and enrich individual extract components. MCI GEL® CHP20P and Spherical C18 chromatographic separation were achieved by combining a medium-pressure chromatographic tower with prep-HPLC, respectively. In this study, an empty pre-column was utilized before the MCI GEL® CHP20P chromatographic tower, into which a mixture of sample and silica gel was loaded. Initially, crude sample pretreatment was performed with a large particle size (120 μm) MCI GEL® CHP20P separation medium. Four rounds of MCI GEL® CHP20P pretreatment yielded six fractions (Fr1 to Fr6) (Fig. 1A). The collected fractions Fr1 to Fr6 were concentrated and analyzed by a 7-X10 analytical column, with the analytical chromatograms corresponding to the crude extract and to Fr1 to Fr6 being shown in Fig. 1B. To guide further sample separation, in vitro anti-inflammatory analyses were next used to explore the bioactive properties of these different fractions.
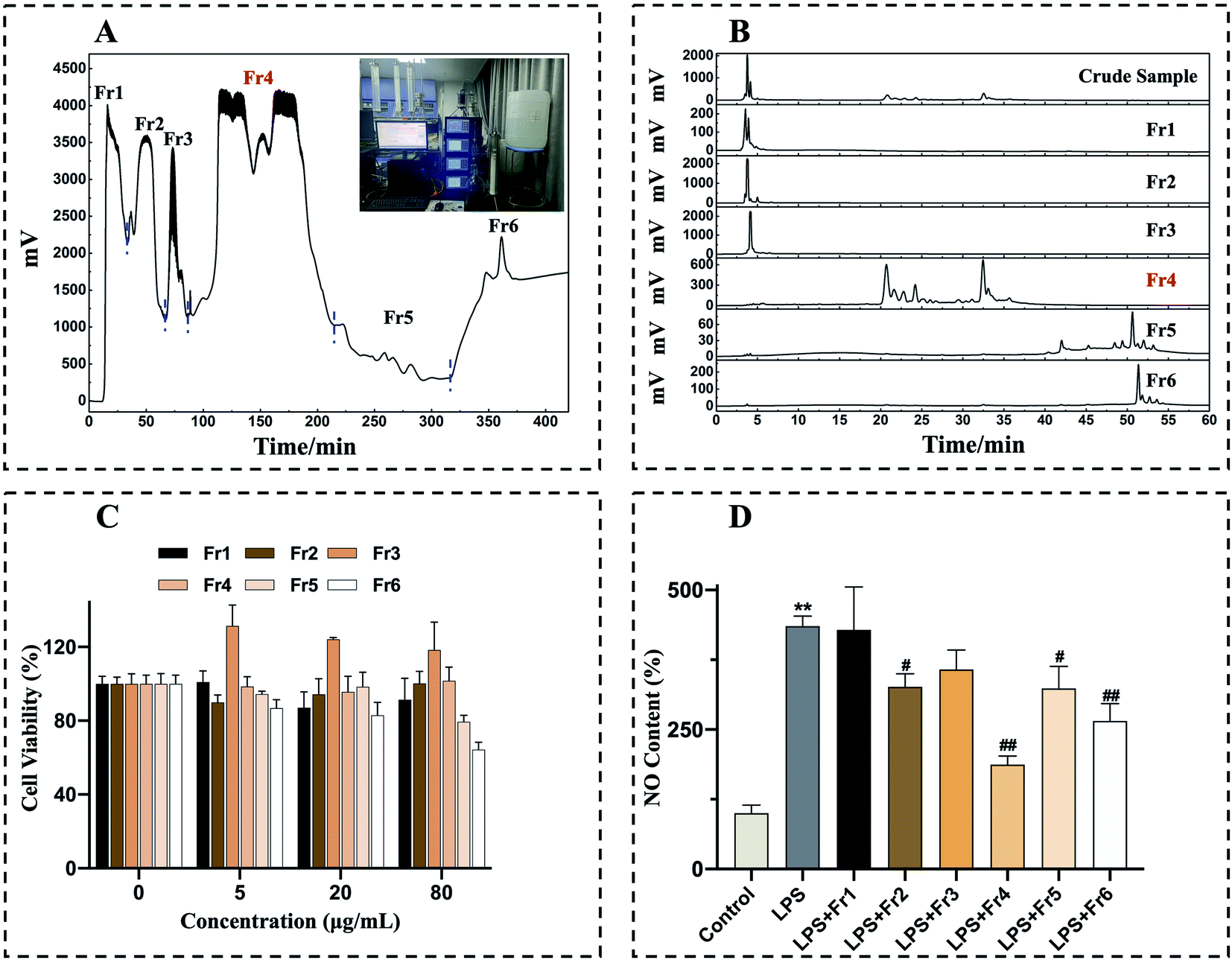 | ||
| Fig. 1 D. superbus extract pretreatment chromatograms and assessment of the effects of Fr1 to Fr6 on LPS-induced changes in RAW264.7 cell viability and NO production. Crude sample was prepared with an MCI GEL® CHP20P preparative column (49 × 460 mm, 120 μm) with the following settings: mobile phase A: H2O, B: CH3OH, C: CH2Cl2; gradient: 0–20 min, 100% A; 20–240 min, 0–100% B; 240–300 min, 100% B; 300–420 min, 0–100% C; flow rate: 40 mL min−1; chromatogram recording at 210 nm; column temperature: room temperature; sample loading: 10 g (A). Fr1 to Fr6 were analyzed via HPLC using a 7-X10 column (4.6 × 250 mm, 7 μm) with the following settings: mobile phase A: H2O, B: CH3OH; gradient: 0–50 min, 0–100% B; 50–60 min, 100% B (B). The impact of Fr1 to Fr6 (0, 5, 20, 80 μg mL−1) on RAW 264.7 cell viability was assessed via MTT assay (C). RAW 264.7 cells were treated for 24 h with Fr1 to Fr6 (20 μg mL−1) and stimulated with LPS (1 μg mL−1), after which a nitrite and nitrate assay was used to assess NO production (D). **p < 0.01 in comparison to normal cells, #p < 0.05, ##p < 0.01 in comparison to LPS-stimulated cells. Duplicate samples were assessed in a minimum of three independent experiments. | ||
Subsequently, an MTT assay was conducted to assess the cytotoxic effects of Fr1–6 by using them to treat RAW 264.7 cells for 24 h (0, 5, 20, 80 μg mL−1), revealing no significant adverse effects for any tested fraction or dose (Fig. 1C). NO can be measured as a gauge of LPS-induced inflammation in macrophages.35,36 As such, RAW 264.7 macrophages were treated with LPS (1 μg mL−1) and 20 μg mL−1 concentration of each fraction, revealing that isolate Fr4 was able to most significantly suppress NO production (Fig. 1D). The target fraction Fr4 (38.7 g) was thus selected for further isolation via Spherical C18 medium-pressure chromatography.
The fraction Fr4 exhibited optimal anti-inflammatory activity and was thus separated with a Spherical C18 medium-pressure chromatographic tower. After three consequent Spherical C18 pretreatments, six subfractions (Fr41 to Fr46) were collected (Fig. 2A). The collected subfractions Fr41 to Fr46 were concentrated and analyzed by a 7-X10 analytical column, with the analytical chromatograms for Fr4 and Fr41 to Fr46 being shown in Fig. 2B. The cytotoxicity of these subfractions was assessed by using these subfractions to treat RAW 264.7 macrophages for 24 h (0, 5, 20, 80 μg mL−1), after which an MTT assay was performed, revealing no adverse effects on cell viability (Fig. 2C). RAW 264.7 cells were then treated with 20 μg mL−1 concentration of these subfractions together with LPS (1 μg mL−1), after which NO production was examined, revealing that Fr44 (1.4 g), Fr45 (4.7 g), and Fr46 (3.5 g) all significantly suppressed the production of NO (Fig. 2D). In order to reflect the efficiency of our method, the lowest mass of Fr44 (1.4 g), which had anti-inflammatory activity, was then chosen for 2D separation.
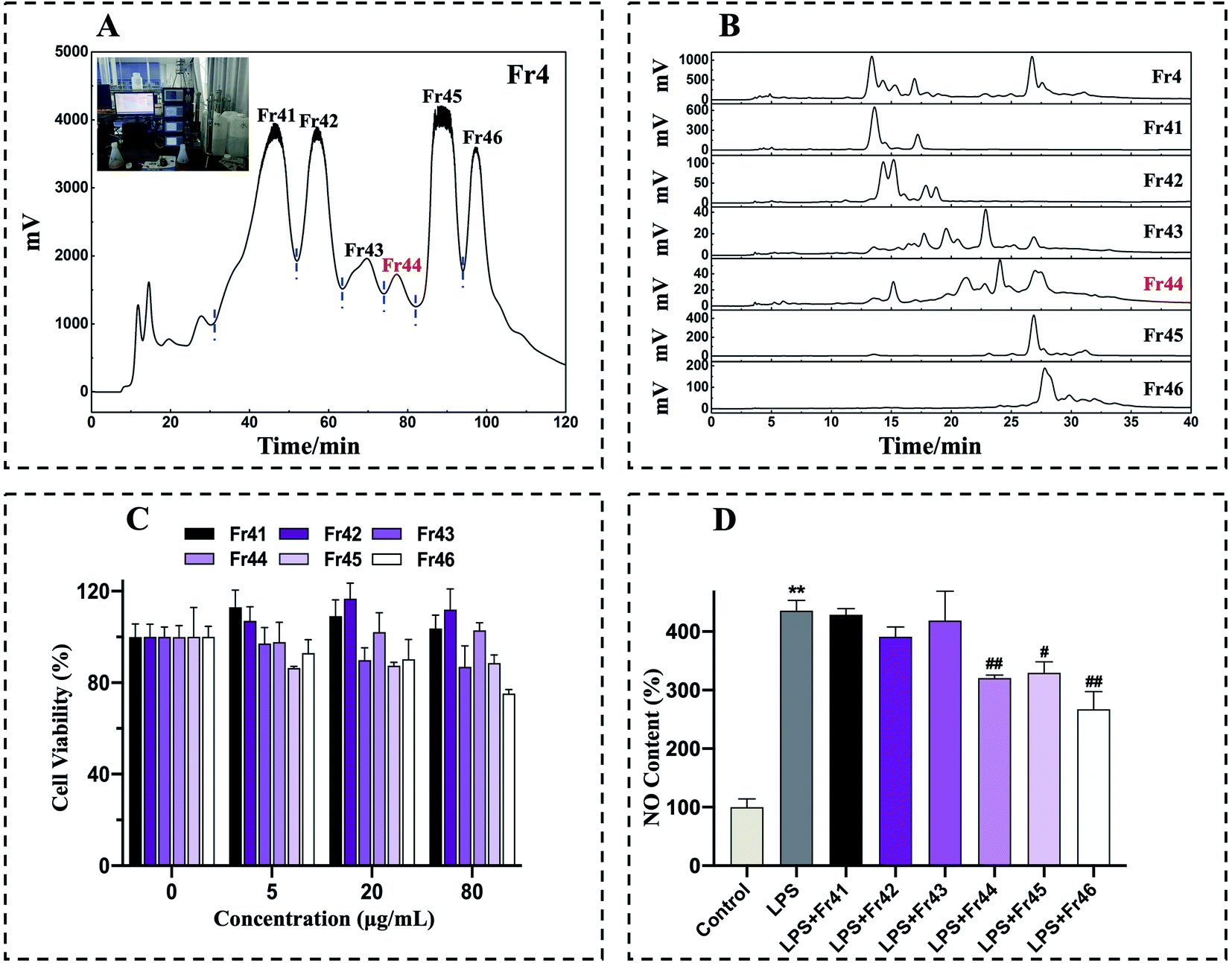 | ||
| Fig. 2 Fr4 pretreatment chromatograms and assessment of the effects of Fr41 to Fr46 on LPS-induced changes in RAW264.7 cell viability and NO production. Fr4 was prepared on a Spherical C18 preparative column (50 × 500 mm, 50 μm) with the following settings: mobile phase A: H2O, B: CH3OH, gradient: 0–120 min, 20–65% B; flow rate: 40 mL min−1; chromatogram recording at 210 nm; sample loading: 10 g; column temperature: room temperature (A). Fr41 to Fr46 were analyzed via HPLC with a 7-X10 column (4.6 × 250 mm, 7 μm) with the following settings: mobile phase A: 0.1% v/v formic acid in chromatographic grade H2O, B: CH3OH; gradient: 0–40 min, 20–80% B (B). The impact of Fr41 to Fr46 (0, 5, 20, 80 μg mL−1) on RAW 264.7 cell viability was established via MTT assay (C). RAW264.7 cells were treated for 24 h with Fr41 to Fr46 (20 μg mL−1) and LPS (1 μg mL−1), after which a nitrite and nitrate assay was used to assess NO production (D). **p < 0.01 in comparison to normal cells, #p < 0.05, ##p < 0.01 in comparison to LPS-stimulated cells. Duplicate samples were assessed in a minimum of three independent experiments. | ||
3.2 Separation of Fr44 via two-dimensional reversed-phase/hydrophilic interaction chromatography
Orthogonal purification can improve separation selectivity and peak capacity. As such, a 2D RPLC/HILIC approach was used to achieve high-purity compound separation. Initially, 1D analysis was performed with a 7-X10 analytical column (4.6 × 250 mm, 7 μm) using mobile phases consisting of 0.1% v/v formic acid in chromatographic grade H2O and CH3OH as detailed in Experimental section 2.3 (Fig. 3A1). 1D separation was conducted using a 7-X10 preparative column (20 × 250 mm, 7 μm) using the same mobile phases as above, as detailed in Experimental section 2.3 (Fig. 3A2). All chromatogram recordings were made at 210 nm. Following two repeated rounds of 1D separation, fractions were collected and dried, thereby yielding Fr441 (87.2 mg), Fr442 (0.2 g), and Fr443 (0.2 g). To ensure efficiency, the lowest mass of Fr441 (87.2 mg) was then chosen for 2D separation. However, the re-analysis of Fr44 and Fr441 on a 7-X10 analytical column revealed weak resolution for Fr441 (Fig. 3B1 and B2). As HILIC can provide complementary selectivity to RPLC, the HILIC approach was next used for 2D separation.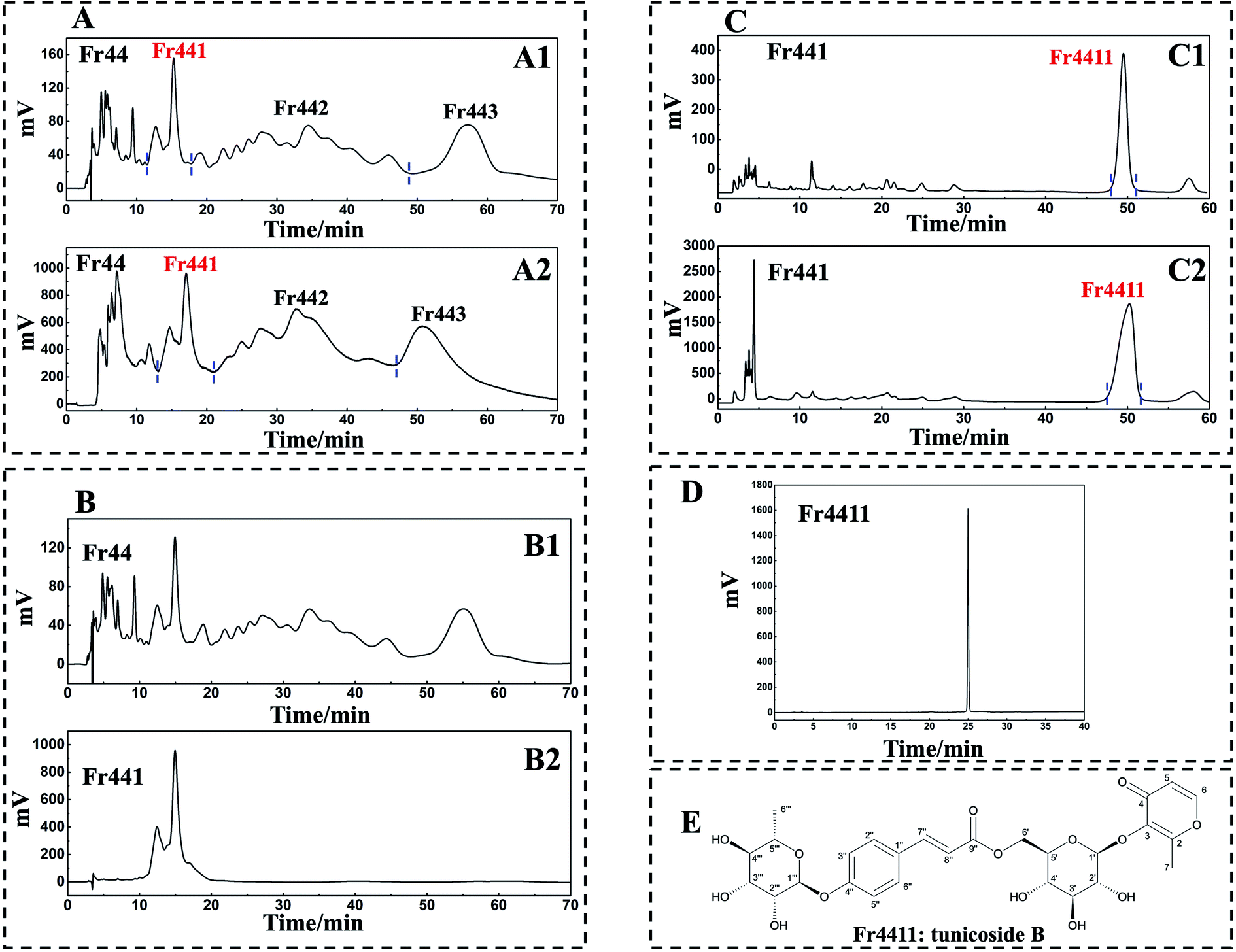 | ||
| Fig. 3 2D RPLC/HILIC mode separated Fr44. A 7-X10 analytical column (4.6 × 250 mm, 7 μm) was used to analyze Fr44 with the following settings: mobile phase A: 0.1% v/v formic acid in chromatographic grade H2O, B: CH3OH, gradient: 0–70 min, 25% B, chromatogram recording at 210 nm (A1). 1D separation of Fr44 was conducted with a 7-X10 preparative column (20 × 250 mm, 7 μm) with the following settings: mobile phase A: 0.1% v/v formic acid in chromatographic grade H2O, B: CH3OH, gradient: 0–70 min 25% B, chromatogram recording at 210 nm (A2). Fr44 and Fr441 were analyzed with a 7-X10 analytical column (4.6 × 250 mm, 7 μm) with the following settings: mobile phase A: 0.1% v/v formic acid in chromatographic grade H2O, B: CH3OH; gradient: 0–70 min, 25% B (B1 and B2). Fr441 was analyzed with a Click XION analytical column (4.6 × 250 mm, 7 μm) with the following settings: mobile phase A: 0.1% v/v trifluoroacetic acid in chromatographic grade H2O, B: ACN, gradient: 0–60 min, 94% B, chromatogram recording at 210 nm (C1). A Click XION preparative column (20 × 250 mm, 5 μm) was used for 2D separation with the following settings: mobile phase A: 0.1% v/v trifluoroacetic acid in chromatographic grade H2O, B: ACN, gradient: 0–60 min, 94% B, chromatogram recording at 210 nm (C2). Fr4411 was assessed with a SunFire® C18 column (4.6 × 250 mm, 5 μm) with the following settings: mobile phase A: 0.1% v/v formic acid in chromatographic grade H2O, B: CH3OH; gradient: 0–40 min, 20–80% B (D). Chemical structure of compound Fr4411 isolated from D. superbus (E). | ||
A Click XION analytical column (4.6 × 250 mm, 5 μm) was used to conduct 2D separation using mobile phases composed of 0.1% (v/v) trifluoroacetic acid in chromatographic grade H2O and ACN, as detailed in Experimental section 2.3 (Fig. 3C1). A Click XION preparative column (20 × 250 mm, 5 μm) was used for 2D separation using the same mobile phases as detailed in Experimental section 2.3 (Fig. 3C2). In both cases, chromatograms were recorded at 210 nm. Following the completion of such 2D RPLC/HILIC, fraction Fr4411 was obtained and concentrated to a mass of 14 mg.
3.3 Verification of the purity and the structural identification of Fr4411
A SunFire® C18 column (4.6 × 250 mm, 5 μm) was next used to assess the purity of Fr4411, revealing it to be >95% pure (Fig. 3D). The structure of the compound present within Fr4411 was next assessed by acquiring MS, UV, IR, 1H NMR, and 13C NMR spectra and then comparing these to previously reported findings. These data are summarized below, and the full spectra are shown in the ESI,† which revealed the compound Fr4411 to be consistent with tunicoside B. The chemical structure of this compound is shown in Fig. 3E.Fr4411 (tunicoside B, yellow amorphous powder, [α]25D-168.331 (c 0.02, MeOH). UV (MeOH, λmax, nm) (logε): 210 (8.36), 297 (8.51). IR (KBr, νmax, cm−1): 3398, 2924, 1705, 1642, 1608, 1510, 1442, 1253, 1179, 1127, 1064, 1018, 984, 835, 515, 469, 406. ESI-MS m/z 603.25 [M + Na]+, calcd for C27H32O14, m/z 580.18): 1H-NMR (600 MHz, CD3OD): δ 7.84 (1H, d, J = 5.2 Hz, H-6), 7.62 (1H, d, J = 15.9 Hz, H-7′), 7.56 (2H, d, J = 8.1 Hz, H-2′′, 6′′), 7.11 (2H, d, J = 8.2 Hz, H-3′′, 5′′), 6.38 (1H, d, J = 15.8 Hz, H-8′′), 6.37 (1H, brs, H-5), 5.49 (1H, s, H-1′′′), 4.86 (1H, brs, H-1′), 4.46 (2H, t, H-6′), 4.00 (1H, s, H-2′′′), 3.83 (1H, dd, J = 9.4, 3.0 Hz, H-3′′′), 3.59 (1H, m, H-5′′′), 3.51 (1H, m, H-5′), 3.46 (1H, m, H-4′′′), 3.43 (1H, m, H-3′), 3.39 (1H, dd, J = 10.3, 5.9 Hz, H-2′), 3.36 (1H, m, H-4′), 2.38 (3H, s, H-7), 1.22 (3H, d, J = 6.1 Hz, H-6′′′); 13C-NMR (151 MHz, CD3OD): δ 177.0 (C-2), 168.5 (C-9′′), 164.6 (C-5), 159.7 (C-4′′), 157.1 (C-4), 146.1 (C-7′′), 143.2 (C-1), 131.0 (C-2′′, 6′′), 129.7 (C-1′′), 117.8 (C-3′′, 5′′), 117.3 (C-3), 116.7 (C-8′′), 104.8 (C-1′′), 99.7 (C-1′′′), 77.8 (C-3′), 76.0 (C-5′), 75.4 (C-2′), 73.7 (C-4′′′), 72.2 (C-3′′′), 71.9 (C-2′′′), 71.6 (C-4′), 70.9 (C-5′′′), 64.3 (C-6′), 18.0 (C-6′′′), 15.7 (C-6). As stated above, these data were consistent with other information in the literature pertaining to tunicoside B.37
3.4 Tunicoside B suppresses the LPS-induced production of NO by RAW 264.7 macrophages
Tunicoside B, as far as we know, is a brand-new chemical discovered in 2019 from Psammosilene tunicoides.37 In the only literature report, there is no mention of the activity of tunicoside B. Maltol has significant anti-inflammatory activity,38 and the isolation of tunicoside B from D. superbus was completed under the guidance of in vitro anti-inflammatory activity. Therefore, the anti-inflammatory activity of tunicoside B was preliminary explored in our study. The impact of isolated tunicoside B on cellular viability was further assessed by treating RAW 264.7 macrophages with various concentrations of tunicoside B for 24 h (0, 5, 10, 20, 40, 60, 80, 100 μM). No changes in macrophage survival were observed in the MTT assay following such treatment (Fig. 4A). Nitric oxide (NO) is a short-lived free radical that is both a key inflammatory mediator and an important signaling intermediate.39 Inflammatory insults of LPS in macrophages have been reported to be mediated by NO production.41 Excessive levels of the inflammatory mediator NO can induce the production of pro-inflammatory cytokines. Simultaneously, these pro-inflammatory cytokines can conversely promote the production of NO, forming a positive feedback cycle that causes an increase in inflammation.42 Thus, inhibiting the production of NO could suppress inflammation. The ability of tunicoside B to inhibit NO production was assessed by treating RAW264.7 macrophages with this compound with or without LPS (1 μg mL−1). While LPS induced substantial NO production (p < 0.01), tunicoside B treatment (5, 10, 20 μM) was sufficient to suppress this production (Fig. 4B). Therefore, we can preliminarily determine that tunicoside B can inhibit the occurrence of inflammation. The iNOS is an important cell-signaling molecule, which is involved in immune response and produces NO. The expression of NO is controlled by iNOS, which is a key upstream enzyme. It is speculated that B may show anti-inflammatory activity through iNOS related pathway. In order to further explain the anti-inflammatory mechanism of tunicoside B, the relationship between tunicoside B and iNOS will be explored.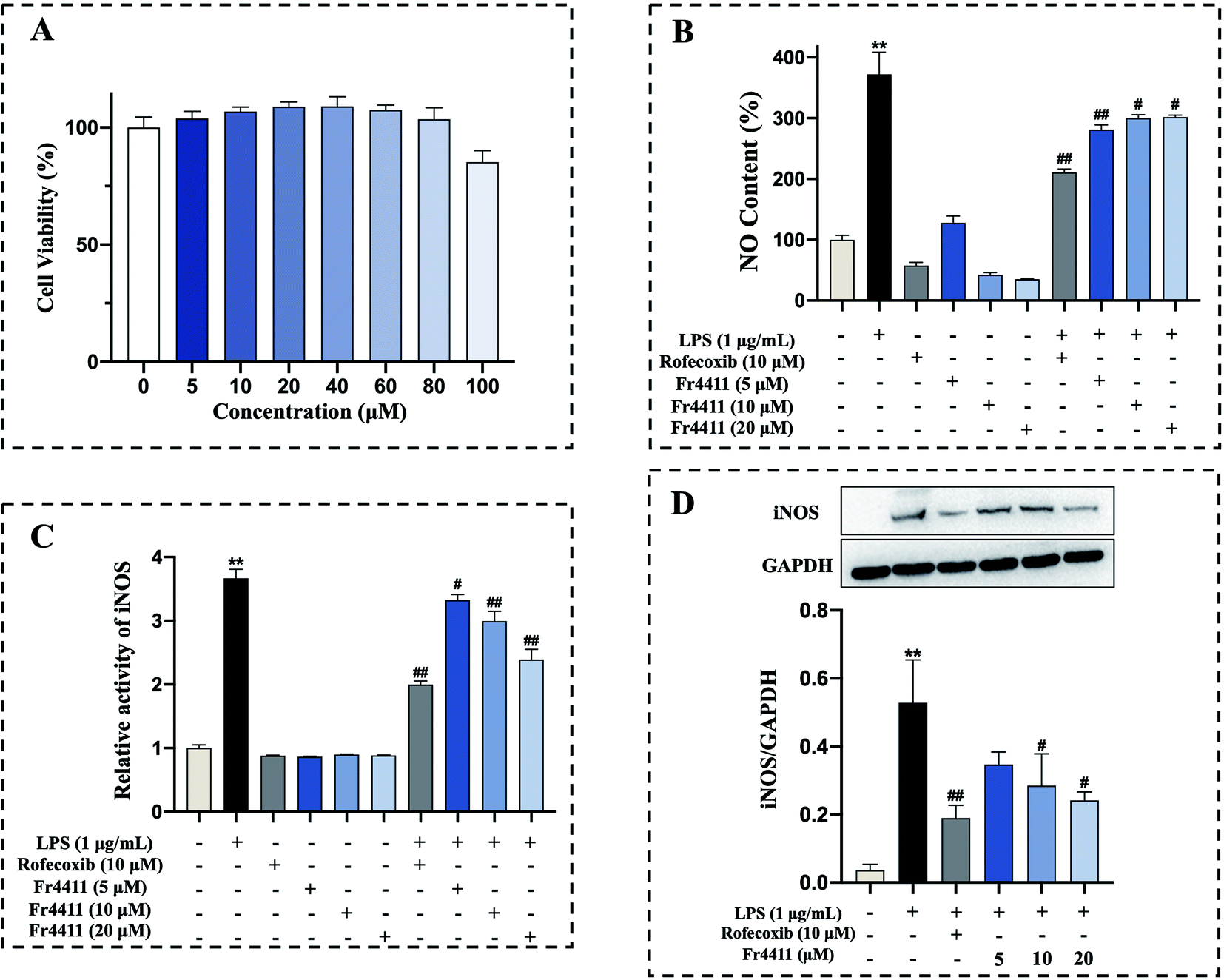 | ||
| Fig. 4 Effects of tunicoside B on LPS-induced RAW 264.7 cells NO production, nitric oxide synthase activity and iNOS expression. MTT assay was used to assess the effects of a range of tunicoside B concentrations on the RAW 264.7 cell viability (A). NO production was assessed following the treatment of RAW 264.7 cells for 24 h with LPS (1 μg mL−1) and tunicoside B (5, 10, 20 μM) and rofecoxib (10 μM) (B). Nitric oxide synthase activity was assessed following the treatment of RAW 264.7 cells for 24 h with LPS (1 μg mL−1) and tunicoside B (5, 10, 20 μM) and rofecoxib (10 μM). The fluorescence excitation at 495 nm and emission at 515 nm (C). Following a 10 h treatment with LPS (1 μg mL−1) and tunicoside B (5, 10, 20 μM) and rofecoxib (10 μM), iNOS levels were assessed via western blotting, with GAPDH for normalization (D). **p < 0.01 in comparison to normal cells, #p < 0.05 in comparison to LPS-stimulated cells. Duplicate samples were assessed in a minimum of three independent experiments. | ||
3.5 Molecular docking, iNOS activity, and western blotting analyses of tunicoside B
In the context of inflammation, iNOS produces NO, which in turn regulates pro-inflammatory mediator production. In the inflamed state, excessive NO production can induce a deleterious inflammatory response by further exacerbating inflammation.40 Molecular docking analyses were used to evaluate potential interactions between iNOS and tunicoside B or the positive control drug rofecoxib. In this analysis, rofecoxib was located in a pocket surrounded by multiple amino acids (Leu203, Phe363, Trp188, Trp366, Cys194) (Fig. 5A and B), while tunicoside B was located in a pocket surrounded by multiple amino acids (Leu119, Arg193, Cys194, Phe363, Val346, Gly365, Trp366, Met368, Tyr367, Tyr485) (Fig. 5C and D). Binding energy values for rofecoxib and tunicoside B in this study were −8.83 kcal mol−1 and −6.70 kcal mol−1, respectively (Table 1). The results of this analysis suggested that the lowest predicted binding energy for tunicoside B was broadly similar to that of rofecoxib, which reflects the good activity of the docking result, and preliminarily verifies the anti-inflammatory effect of tunicoside B in iNOS from the theoretical level.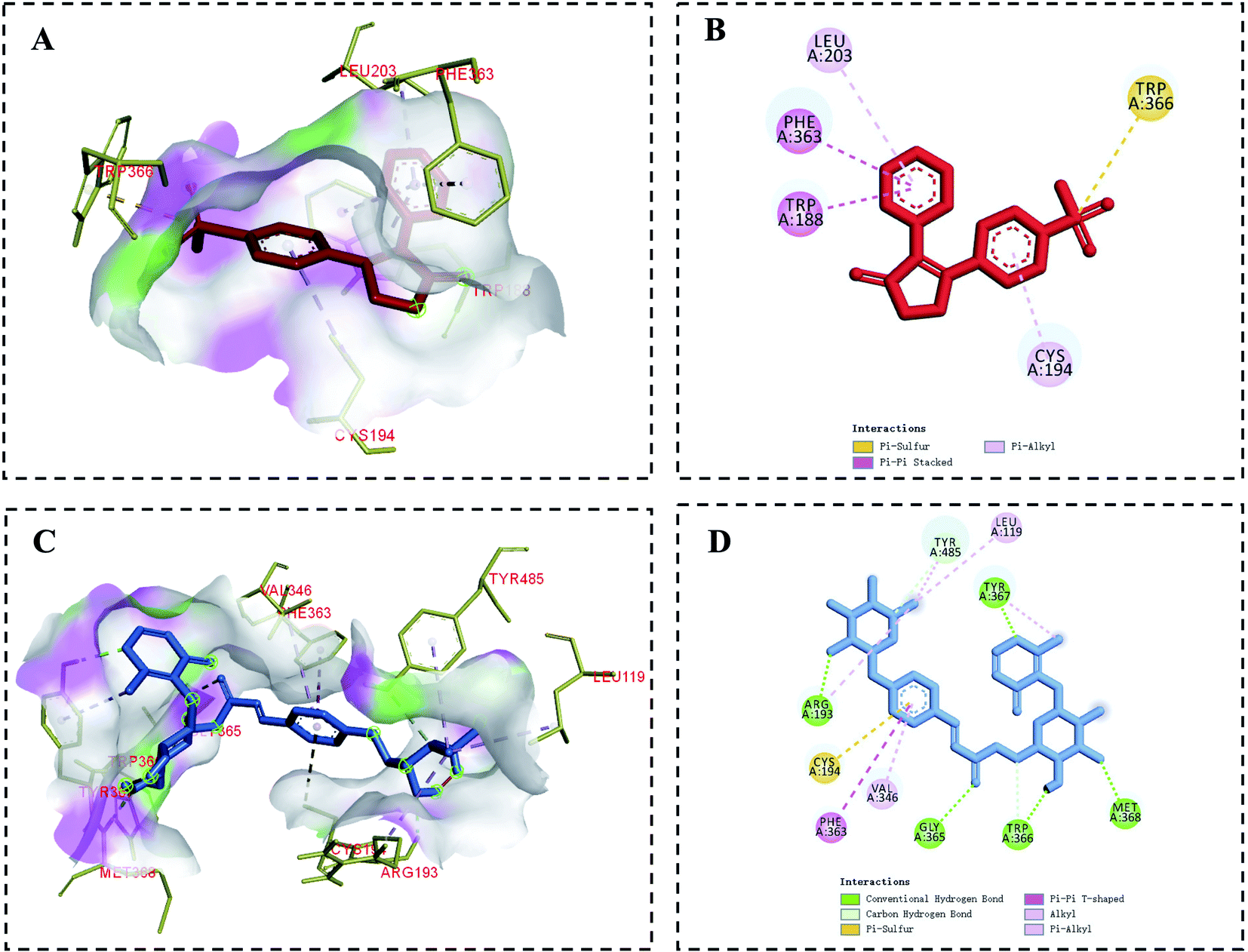 | ||
| Fig. 5 Molecular docking analysis of iNOS binding to tunicoside B and rofecoxib. Rofecoxib and tunicoside B structures are shown in blue and red, respectively. (A) and (C) Correspond to binding models, while (B) and (D) correspond to 2D-interaction diagrams for iNOS interactions with rofecoxib and tunicoside B, respectively. | ||
| Compounds | RMSD | Binding energy (kcal mol−1) |
|---|---|---|
| Tunicoside B | 82.36 | −6.70 |
| Rofecoxib | 82.60 | −8.83 |
Furthermore, in order to verify the enzymatic activity of tunicoside B on iNOS, the activity of nitric oxide synthase was detected by kits. The results showed that following LPS treatment for 24 h, the iNOS activity of the LPS group was significantly upregulated compared with that of the control group (p < 0.01), while tunicoside B treatment (5, 10, 20 μM) markedly suppressed iNOS activity in these cells in a dose-dependent manner (Fig. 4C). The results of the binding molecule docking and the nitric oxide synthase assay kit showed that tunicoside B can inhibit the activity of iNOS, laying the foundation for subsequent western blotting.
Macrophages play an important role in host defense against various infections. LPS-stimulated RAW 264.7 cells have been widely applied to carry out inflammatory model in vitro.36 RAW 264.7 macrophages treated with LPS express iNOS, and elevated iNOS expression can lead to excessive NO production. To further confirm the accuracy of our molecular docking and iNOS activity results and to better elucidate the mechanism of anti-inflammatory action for tunicoside B, western blotting was employed to assess iNOS protein levels in tunicoside B-treated RAW 264.7 cells following LPS treatment, with GAPDH as a loading control. Following LPS treatment for 10 h, iNOS was significantly upregulated compared with the control group (p < 0.01), while tunicoside B treatment (5, 10, 20 μM) markedly suppressed iNOS expression in these cells in a concentration-dependent manner (Fig. 4D).
NO is a kind of gas signal molecule and active nitrogen free radical in organisms. The concentration of NO is related to the activity of various signal pathways and inflammatory responses. Excessive NO can damage the healthy cells of the body and cause oxidative stress in the tissues. iNOS is not expressed in resting cells, but when cells are stimulated by extracellular factors such as inflammatory cytokines, pathogenic microorganisms, and endotoxin LPS, iNOS is activated and catalyzes the synthesis of large amounts of NO, causing tissue damage and accelerating inflammation, producing a series of pathological effects. NO expression is controlled by iNOS, which is a key upstream enzyme. The current experimental results showed that tunicoside B can inhibit the production of NO and reduce the activity of iNOS and its protein expression. Altogether, these data indicated that tunicoside B isolated from D. superbus exhibits remarkable in vitro anti-inflammatory activity for the first time, which laid a foundation for the future investigation of the anti-inflammatory activity of tunicoside B.
4 Conclusion
In this study, under the guidance of in vitro anti-inflammatory activity, medium-pressure liquid chromatography and 2D RPLC/HILIC were used to quickly and effectively separate maltol glycoside from D. superbus, which was finally identified as tunicoside B. Tunicoside B was isolated from D. superbus for the first time and the isolation method was conducted under the guidance of anti-inflammatory activity. It is necessary to further explore the anti-inflammatory activity of tunicoside B. To test the cytotoxicity of tunicoside B, the viability of RAW 264.7 macrophages was assessed by MTT assay and found that tunicoside B had no effect on cell viability. Since tunicoside B was isolated under the guidance of anti-inflammatory activity with NO as indicator, the production of NO was used again to preliminarily explore the anti-inflammatory activity of tunicoside B, which indicated that tunicoside B can inhibit the production of NO. Since the expression of NO is controlled by iNOS, molecular docking is used to explore the relationship between tunicoside B and iNOS at the theoretical level. In order to verify the result of molecular docking, at the corresponding enzyme level, the effect of tunicoside B on the activity of iNOS was explored, and it was found that tunicoside B can inhibit the activity of iNOS. At the same time, consistent results were obtained through the study on protein level, tunicoside B can inhibit the protein expression of iNOS and exhibit anti-inflammatory activity. All of these results preliminarily indicated that tunicoside B had remarkable anti-inflammatory activity. This study is the first to our knowledge to have reported the isolation of tunicoside B from D. superbus. Similarly, it is the first preliminary study on the anti-inflammatory activity of tunicoside B. In our future study, more in-depth studies will be conducted to determine the specific anti-inflammatory mechanism of tunicoside B. While more experiments will be necessary to characterize the mechanisms underlying such anti-inflammatory activity in detail, these findings nonetheless provide a robust basis for future efforts to isolate anti-inflammatory compounds from TCMs and natural products. Overall, these results suggest that this approach offers value as a means of identifying and separating anti-inflammatory maltol glycoside from D. superbus and can be similarly employed to prepare anti-inflammatory compounds from other plant sources. Moreover, this study on anti-inflammatory activity provides a direction for studies on the activity of maltol in the future.Data availability
Data available on request from the authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The research of the present study was supported by the Shandong Provincial Science Foundation (ZR2019MH094, ZR2020MH380), Yantai Science and Technology Innovation Development Project (2021XDHZ078), Graduate Innovation Foundation of Yantai University (YDYB2118), Innovation Platform for the Development and Construction of Special Project of Qinghai Province (2021-ZJ-T05), Qinghai Province Applied Basic Research Project (2020-ZJ-905), International Cooperation Project of Qinghai Province (2019-HZ-805), Joint Research Project of Three-River Headwaters National Park, and Chinese Academy of Sciences and the People's Government of Qinghai Province (LHZX-2020-09).References
- Y. J. Tu, L. N. Li, Z. T. Wang and L. Yang, J. Pharm. Biomed. Anal., 2021, 206, 114353, DOI:10.1016/j.jpba.2021.114353.
- S. Tasneem, B. Liu, B. Li, M. I. Choudhary and W. Wang, Pharmacol. Res., 2019, 139, 126–140, DOI:10.1016/j.phrs.2018.11.001.
- W. J. Chen, X. Yu, X. R. Yuan, B. J. Chen, N. Cai, S. Zeng, Y. S. Sun and H. W. Li, Front. Pharmacol., 2021, 12, 727956, DOI:10.3389/fphar.2021.727956.
- Y. N. Lin, M. Jiang, W. J. Chen, T. J. Zhao and Y. F. Wei, Biomed. Pharmacother., 2019, 118, 109249, DOI:10.1016/j.biopha.2019.109249.
- S. Sharif, D. G. Y. Van, M. J. Cramer, L. J. Kapelle, G. J. de Borst, F. L. J. Visseren and J. Westerink, Cardiovasc. Diabetol., 2021, 20, 220, DOI:10.1186/s12933-021-01409-0.
- Y. J. Hu, L. Chi, W. M. Kuebler and N. M. Goldenberg, Cells, 2020, 9, 2338, DOI:10.3390/CELLS9112338.
- D. Artemniak-Wojtowicz, A. Kucharska and B. Pyrzak, Cent. Eur. J. Immunol., 2020, 45, 461–468, DOI:10.5114/ceji.2020.103418.
- J. W. Yang, B. Mao, R. J. Tao, L. C. Fan, H. W. Lu, B. X. Ge and J. F. Xu, J. Cell. Mol. Med., 2020, 24, 12716–12725, DOI:10.1111/JCMM.15849.
- G. T. Huo, Y. Y. Huo, J. Li, W. Chen and D. X. Jiang, Prog. Biochem. Biophys., 2020, 47, 659–674, DOI:10.16476/j.pibb.2020.0133.
- D. H. Kim, G. S. Park, A. S. Nile, Y. D. Kwon, G. Enkhtaivan and S. H. Nile, Food Chem. Toxicol., 2019, 125, 313–321, DOI:10.1016/j.fct.2019.01.013.
- S. H. Nile, D. H. Kim, A. Nile, G. S. Park, E. Gansukh and G. Kai, Food Chem. Toxicol., 2020, 135, 110985, DOI:10.1016/j.fct.2019.110985.
- Y. Yang, W. Zhang, Z. F. Xie, J. C. Lei and J. Q. Yu, Chem. Nat. Compd., 2017, 53, 740–741, DOI:10.1007/s10600-017-2105-1.
- R. Aliyazicioglu, S. Demir, M. Badem, S. O. Sener, N. Korkmaz, E. A. Demir, U. Ozgen, S. A. Karaoglu and Y. Aliyazicioglu, Rec. Nat. Prod., 2017, 11, 270–284 CAS.
- B. R. Yun, H. Yang, J. Weon, J. Lee, M. Eom and C. Ma, Pharmacogn. Mag., 2016, 12, 264–269, DOI:10.4103/0973-1296.182159.
- J. Sun, J. H. Yu, J. L. Song, C. S. Jiang, Y. Tao and H. Zhang, Tetrahedron Lett., 2018, 60, 161–163, DOI:10.1016/j.tetlet.2018.12.003.
- Y. N. Ren, X. B. Xu, Q. L. Zhang, Y. Z. Lu, X. M. Li, L. Zhang and J. K. Tian, Arch. Pharmacal Res., 2017, 40, 159–167, DOI:10.1007/s12272-014-0537-8.
- J. J. Yoon, J. H. Park, H. J. Kim, H. G. Jin, H. Y. Kim, Y. M. Ahn, Y. C. Kim, H. S. Lee, Y. J. Lee and D. G. Kang, Nutrients, 2019, 11, 553, DOI:10.3390/nu11030553.
- U. Yusupova, D. Usmanov, A. Azamatov, N. Ramazonov and J. Rejepov, Nat. Prod. Res., 2020, 1–5, DOI:10.1080/14786419.2020.1862834.
- X. D. Hou, Z. T. Cheng and J. Wang, Anal. Methods, 2020, 12, 3382–3389, 10.1039/D0AY00860E.
- M. Z. Liu, X. J. Li, Q. Liu, S. Q. Xie, M. Chen, L. M. Wang, Y. H. Feng and X. Q. Chen, J. Food Compos. Anal., 2020, 85, 103336, DOI:10.1016/j.jfca.2019.103336.
- M. M. Zhang, S. P. Cheng, Y. Liang, Y. Mu, H. J. Yan, Q. Liu, Y. L. Geng, X. Wang and H. Q. Zhao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1100, 140–147, DOI:10.1016/j.jchromb.2018.09.030.
- H. Chi, X. Qi, X. H. Wang, Y. Wang, X. H. Han, J. Wang and H. W. Wang, Anal. Methods, 2021, 13, 1939–1944, 10.1039/d1ay00186h.
- Q. Wang and S. Shen, Talanta, 2019, 19, 649–657, DOI:10.1016/j.talanta.2018.09.112.
- Q. Wang, W. J. Chen, Q. L. Wang, Y. D. Tao, R. T. Yu, G. Q. Pan and J. Dang, J. Sep. Sci., 2020, 43, 2521–2528, DOI:10.1002/JSSC.201901164.
- G. Q. Pang, J. W. Shen, Y. H. Ma, Y. F. He, Y. Bao, R. R. Li, S. S. Wang, Q. Wang, P. C. Lin and J. Dang, J. Sep. Sci., 2019, 42, 3182–3190, DOI:10.1002/jssc.201900252.
- Y. L. Cui, N. Shen, X. Yuan, J. Dang, Y. Shao, L. J. Mei, Y. D. Tao, Q. L. Wang and Z. G. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1046, 81–86, DOI:10.1016/j.jchromb.2017.01.022.
- W. D. Wang, Y. D. Tao, L. J. Jiao, M. X. Fan, Y. Shao, Q. L. Wang, L. J. Mei and J. Dang, J. Sep. Sci., 2017, 40, 3593–3601, DOI:10.1002/jssc.201700449.
- Q. Wu, X. D. Hou, H. T. Lv, H. Li, L. Zhao and H. D. Qiu, J. Chromatogr. A, 2021, 1656, 462548, DOI:10.1016/J.CHROMA.2021.462548.
- Q. Fu, H. H. Zhang, Z. S. Dai, D. S. Jiang, M. Sun, Y. X. Ke, Y. Jin and X. M. Liang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2021, 1173, 122673, DOI:10.1016/J.JCHROMB.2021.122673.
- A. Sentkowska and K. Pyrzynska, Molecules, 2021, 26, 5073, DOI:10.3390/MOLECULES26165073.
- W. Y. Yu, H. L. Jin, A. J. Shen, L. Deng, J. L. Shi, X. Y. Xue, Y. D. Guo, Y. F. Liu and X. M. Liang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1040, 47–52, DOI:10.1016/j.jchromb.2016.11.031.
- L. J. Jiao, Y. D. Tao, W. D. Wang, Y. Shao, L. J. Mei, Q. L. Wang and J. Dang, J. Sep. Sci., 2017, 40, 3808–3816, DOI:10.1002/jssc.201700675.
- J. Dang, Y. Shao, J. Q. Zhao, L. J. Mei, Y. D. Tao, Q. L. Wang and L. Zhang, J. Sep. Sci., 2016, 39, 3327–3338, DOI:10.1002/jssc.201600401.
- Z. Wang, T. T. Xie, X. Yan, S. Xue, J. W. Chen, Z. Wu and Y. K. Qiu, Chromatographia, 2019, 82, 543–552, DOI:10.1007/s10337-018-3652-8.
- M. Rezaei, H. Ghafouri, M. R. Aghamaali and M. Shourian, Iran. J. Pharm. Res., 2019, 18, 1371–1379, DOI:10.22037/ijpr.2019.1100730.
- S. S. Wang, G. Z. Cai, L. Zhao, L. M. Lai and J. Y. Gong, Int. J. Agric. Biol., 2019, 22, 313–318, DOI:10.17957/IJAB/15.1065.
- X. P. Qi, J. M. Tian, Y. H. Shen and W. D. Zhang, Chin. Tradit. Herb. Drugs, 2019, 50, 2513–2517 Search PubMed.
- M. I. G. El-Din, F. S. Youssef, R. S. Said, M. L. Ashour, O. A. Eldahshan and A. N. B. Singab, Inflammopharmacology, 2020, 29, 317–332, DOI:10.1007/s10787-020-00749-9.
- H. M. Lv, Q. M. Liu, Z. M. Wen, H. H. Feng, X. M. Deng and X. X. Ci, Redox Biol., 2017, 12, 311–324, DOI:10.1016/j.redox.2017.03.001.
- J. Y. Chen, S. Y. Lei, T. T. Li, J. Li, A. J. Zuo, D. Xu, C. X. Song and Y. Guo, Biochem. Biophys. Res. Commun., 2020, 523, 98–104, DOI:10.1016/j.bbrc.2019.12.008.
- D. Malayil, N. C. House, D. Puthenparambil, J. T. Job and A. Narayanankutty, Drug Chem. Toxicol., 2020, 1–7, DOI:10.1080/01480545.2020.1858854.
- L. L. Zhang, J. Chen, H. J. Liao, C. H. Li and M. S. Chen, J. Funct. Foods, 2020, 75, 104217, DOI:10.1016/j.jff.2020.104217.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07273k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |