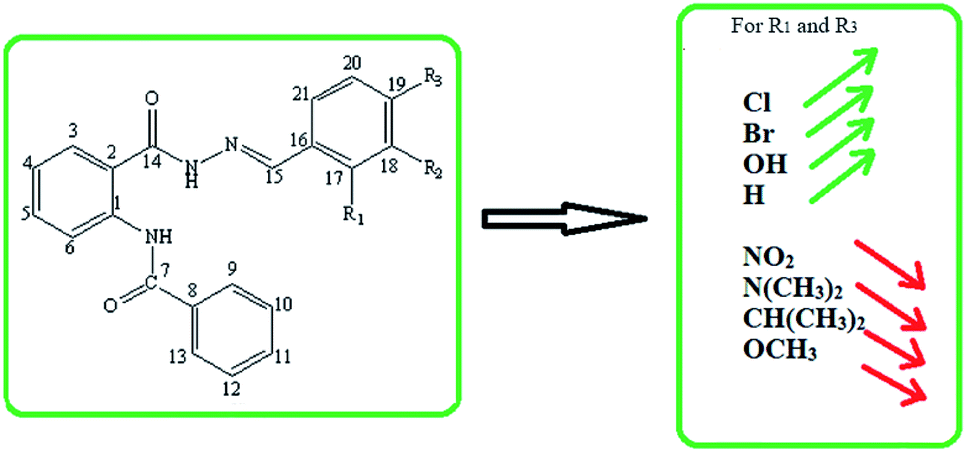
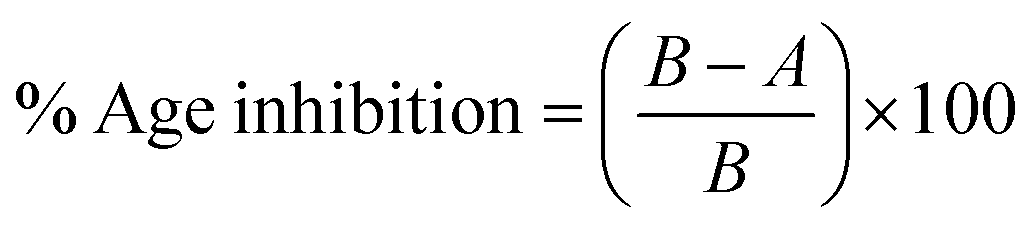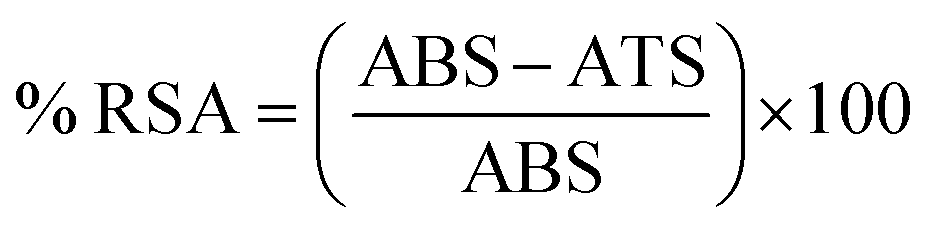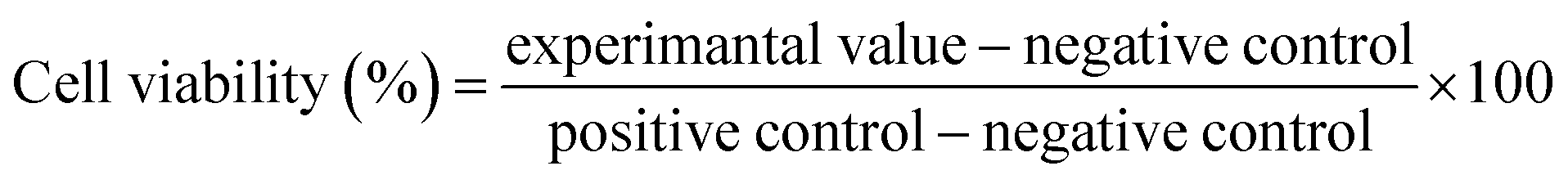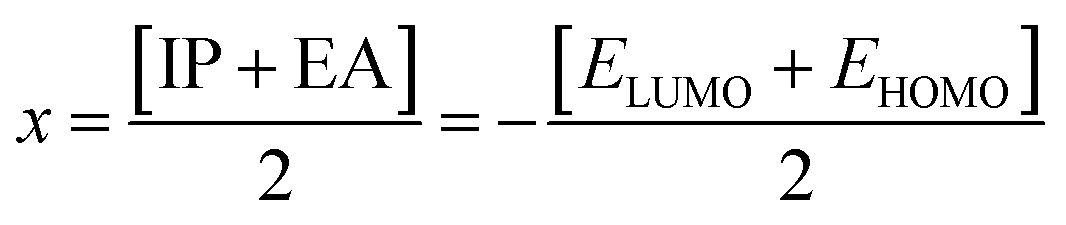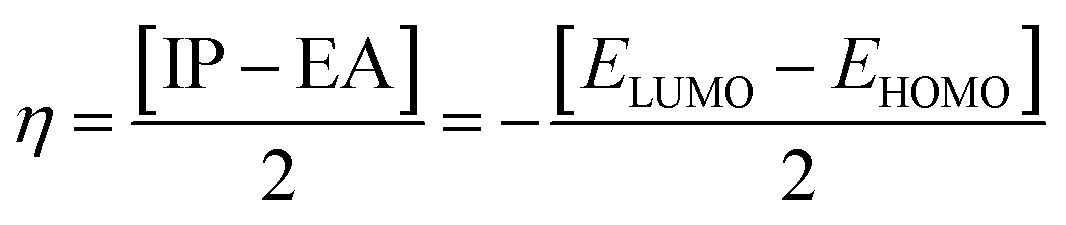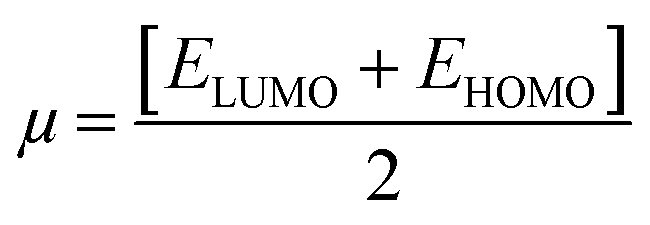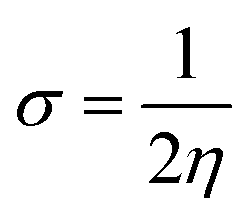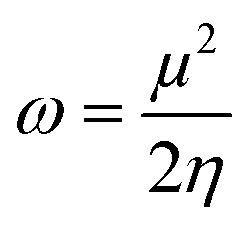DOI:
10.1039/D1RA07221H
(Paper)
RSC Adv., 2022,
12, 154-167
Design, synthesis, crystal structure, in vitro cytotoxicity evaluation, density functional theory calculations and docking studies of 2-(benzamido) benzohydrazide derivatives as potent AChE and BChE inhibitors†
Received
28th September 2021
, Accepted 21st November 2021
First published on 24th December 2021
Abstract
A series of hydrazone derivatives of 2-(benzamido) benzohydrazide was designed, synthesized, and characterized utilizing FTIR, NMR and UV spectroscopic techniques along with mass spectrometry. Compound 10 was also characterized through X-ray crystallography. These synthesized compounds were assessed for their potential as anti-Alzheimer's agents by checking their AChE and BChE inhibition properties by in vitro analysis. The synthesized derivatives were also evaluated for their antioxidant potential along with cytotoxicity studies. The results clearly indicated that dual inhibition of both the enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) was achieved by most of the compounds (03–13), showing varying IC50values. Remarkably, compound 06 (IC50 = 0.09 ± 0.05 for AChE and 0.14 ± 0.05 for BChE) and compound 13 (IC50 = 0.11 ± 0.03 for AChE and 0.10 ± 0.06 for BChE) from the series showed IC50 values comparable to the standard donepezil (IC50 = 0.10 ± 0.02 for AChE and 0.14 ± 0.03 for BChE). Moreover, the derivative 11 also exhibited selective inhibition against BChE with IC50 = 0.12 ± 0.09. Meanwhile, compounds 04 and 10 exhibited good anti-oxidant activities, showing % scavenging of 95.06% and 82.55%, respectively. Cytotoxicity studies showed that the synthesized compounds showed cell viability greater than 80%; thus, these compounds can be safely used as drugs. DFT and molecular docking studies also supported the experimental findings.
1 Introduction
Among protracted neurological diseases, Alzheimer's disease (AD) is the most important, which leads to neuropsychiatric symptoms, insufficient daily life activities and dementia.1 Almost 80% of dementia cases are due to AD and it is a major cause of death worldwide.2–4 Elderly people over 65 years of age are victims of this disease. The distribution of this disease is heterogeneous across the globe, being less dominant in Sub-Saharan African regions and more common in North America and Western Europe.5 This neurodegenerative brain disorder depends on many factors and the main cause of this disease is the increased aggregation of β-amyloid peptides (Aβ) outside the neuron cells in the brain,6 which leads to dysfunction of the cholinergic system,7,8 oxidative stress,9 inflammation10 and loss of cognitive function.11 The most appropriate target for AD drug design is the cholinergic system12,13 due to its major contribution towards regulation and memory processes.14,15 Cognitive function is lost in AD patients due to very low cholinergic neurotransmission in various parts of the human brain, especially in the cortical region, due to disturbance in the levels of acetylcholine (ACh) due to its hydrolysis by cholinesterases (ChEs). Ach, being a neurotransmitter, has its role in the inflection of memory function, both under neurodegenerative and normal conditions.16 Regulation of the levels of ACh concentration and, in turn, cholinergic signaling is regulated by hydrolytic enzymes, butyrylcholinesterase (BChE) and acetylcholinesterase (AChE). Both these enzymes belong to the ChE family of enzymes and are extremely efficient in the hydrolysis of ACh. They can hydrolyze about 10000 molecules of ACh per second.17 One of the suggested therapies for the treatment of AD patients is by inhibiting the butyrylcholinesterase (BChE) and acetylcholinesterase (AChE) enzymes by developing inhibitors of these ChE enzymes, improving the level of acetylcholine (ACh) in the bodies of AD patients and leading to improved neurotransmitter action.12 This therapy also results in the improvement of both memory and mental symptoms. Many AD patients are treated by improving the cholinergic neurotransmission level in the body using AChE inhibitor drugs such as rivastigmine, galantamine and donepezil. However, in addition to very expensive therapies using these drugs, the reoccurrence of symptoms as well as very serious side effects are observed using these drugs.
Under this situation, it is very crucial to continue working for the development of highly efficient and more potent inhibitors for these cholinesterases (ChEs). Benzohydrazide derivatives are found to possess broad biological and medicinal importance, thus providing a basic nucleus for the construction of inhibitors against these hydrolyzing enzymes. The compounds bearing benzohydrazide nucleus has been continuously explored for the treatment of many diseases, including AD. They have been found to act as analgesics,18 antimicrobial agents,19 and anticancerous agents.20 In addition, the benzohydrazide derivatives also possess good antiHIV21 and antitubercular22 activities along with significant AChE and BChE inhibition activities.23,24 Recent studies have been carried out to synthesize benzohydrazide derivatives, possessing the ortho-substituted sulfonamide moiety, which displayed potential inhibitory profile against BChE and AChE enzyme.25 In this context, we developed both amide- and sulfonamide-derived benzohydrazide derivatives and found that the amide derivatives possessed good inhibitory activity against both AChE and BChE enzymes.26 Keeping in view the previous efforts that were put to find good inhibitors for AChE and BChE, more benzohydrazide derivatives with thiophene-2-carboxamide moiety attached at the ortho position were synthesized, which showed comparatively good potency against both these enzymes.27
To further elucidate the importance of amide-derived benzohydrazide derivatives, we synthesized a series of 2-(benzamido) benzohydrazide derivatives. The characterization of the synthesized compounds was done utilizing UV, FTIR, and NMR spectroscopic techniques along with mass spectrometry and X-ray crystallographic analysis. The inhibitory activity of the synthesized compounds was assessed against human AChE and BChE. Furthermore, the experimental findings were validated by carrying out DFT analysis and molecular docking studies.
2 Materials and methods
Analytical grade benzoyl chloride, methyl anthranilate, hydrazine hydrate, and various aldehydes were acquired from Sigma Aldrich (USA) and were utilized as delivered by the company. The melting points of the compounds were taken using electrothermal melting point apparatus. Bio-rad spectrophotometer was used to obtain the FTIR spectra. The NMR spectra were recorded utilizing JEOL DELTA2_NMR Spectrometer using DMSO-d6 as the solvent (1H, 300 MHz, 13C, 75 MHz). The chemical shift values for NMR were described in ppm units while the IR data was represented in ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) units. Tandem Mass Spectrometric analysis (LTQ XL Linear Ion Trap Mass Spectrophotometer, Thermo Scientific, USA) was utilized to record the mass spectra using electrospray (ESI) ionization probe.
units. Tandem Mass Spectrometric analysis (LTQ XL Linear Ion Trap Mass Spectrophotometer, Thermo Scientific, USA) was utilized to record the mass spectra using electrospray (ESI) ionization probe.
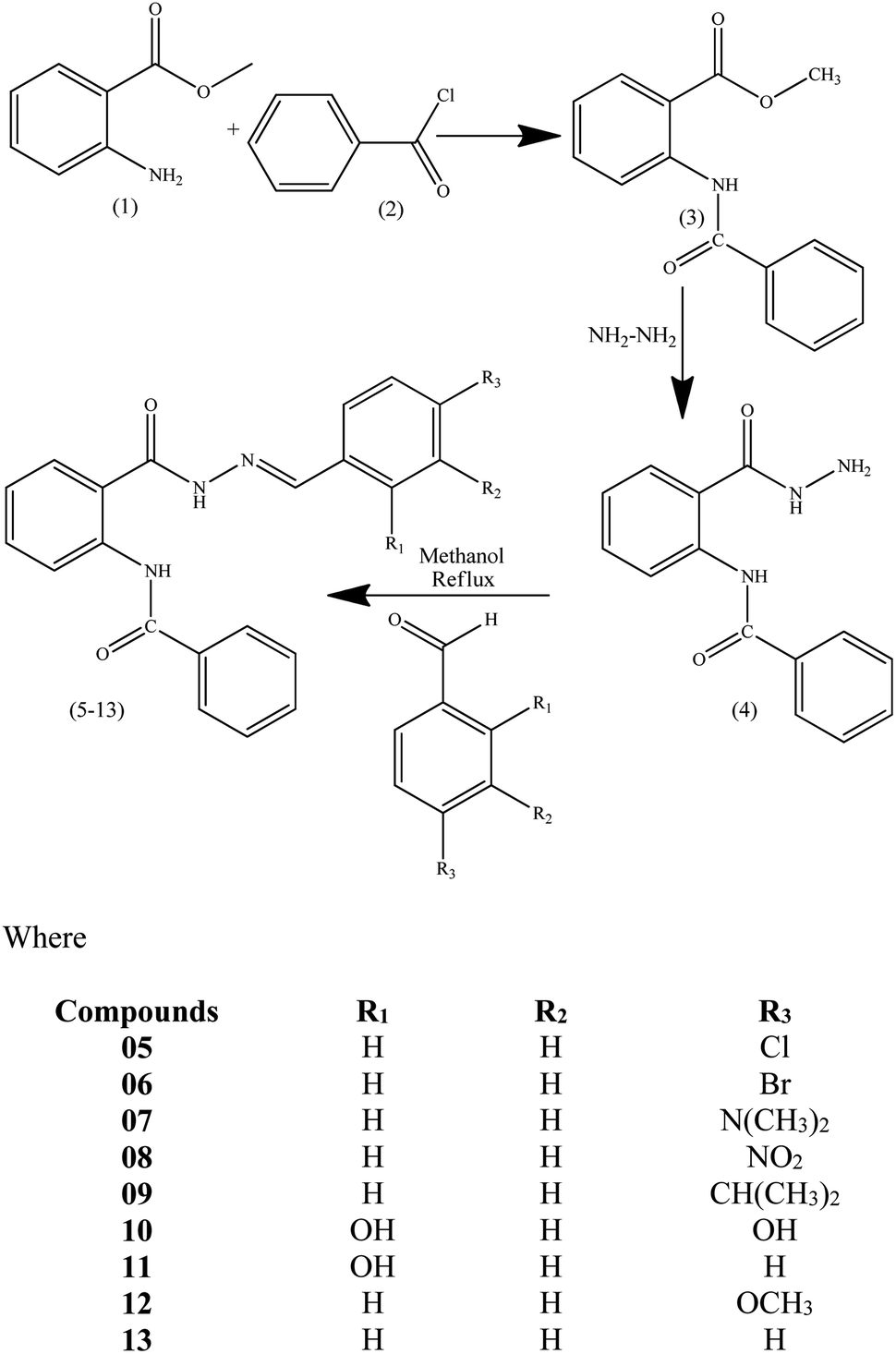
2.1. Procedure for the synthesis of hydrazone derivatives of N-(2-(hydrazinecarbonyl)phenyl)benzamide (05–13)
Methyl anthranilate (01) (1.29 mL, 10.0 mmol) was taken in methanol (5.0 mL), and benzoyl chloride (02) (1.74 mL, 15.0 mmol) in methanol (5.0 mL) was added dropwise into the methyl anthranilate solution. The reaction mixture was stirred for 2 h, followed by neutralization with sodium bicarbonate. The precipitates of methyl-2-benzamidobenzoate (03) were filtered and washed with a mixture of cold methanol and water to acquire the product as a pure white solid. A solution of methyl-2-benzamidobenzoate (03) was refluxed with excess hydrazine using ethanol as the solvent for two hours. Upon completion of the reaction, extra solvents were removed from the reaction mixture under vacuum, followed by performing ethyl acetate/water extraction of the obtained mixture to remove extra hydrazine. MgSO4 was utilized for the drying of ethyl acetate layer, followed by evaporation under reduced pressure to get the hydrazide derivative of compound 03, N-(2-(hydrazinecarbonyl)phenyl)benzamide (04). This compound 04 was further refluxed with stoichiometric amounts of differently substituted benzaldehyde derivatives using methanol as the solvent for two hours to obtain the hydrazone derivatives (05–13).26 Washing of the obtained products was done using cold methanol to get clean compounds in good yields (nearly 90%) (Scheme 1).
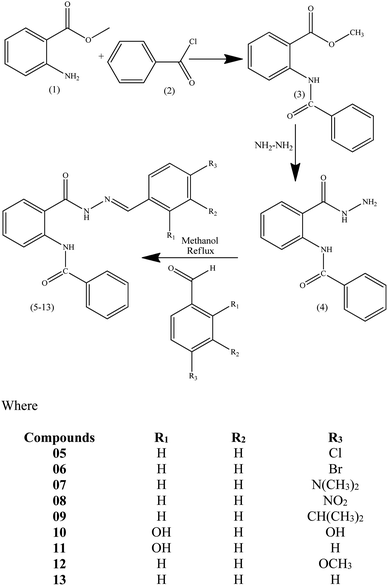 |
| | Scheme 1 Synthetic scheme of methyl-2-(benzamido) benzoate (03), 2-(benzamido) benzohydrazide (04) and Schiff base derivatives of 2-(benzamido) benzohydrazide (05–13). | |
2.2. Single crystal X-ray crystallography
The objective designed compound 10 was crystallized after the proposed synthetic steps. The plan was to realize the geometry and interactions among the molecules in the unit cell. Thus, the selected single crystal under microscope was mounted on an Agilent SuperNova (dual source) Agilent Technologies Diffractometer, equipped with microfocus Cu/Mo Kα radiation for data collection. The sample was jammed, using locally purchased glue, over glass fibers. This thin glass fiber was immersed on a copper rod, supported via a magnetic base. Data collection was accomplished using CrysAlisPro software28 at 296 K under Cu Kα radiation. The structure solution was performed using the SHELXS-97 method29 and refined by full-matrix least-squares methods on F2 using the SHELXL-97 method,29 in-built with WinGX.30 All non-hydrogen atoms were refined anisotropically by full-matrix least squares methods.29 The figures were generated through PLATON31 and ORTEP30 in built with WinGX.30
The X–H (X = C, N, and O) hydrogen atoms for aromatic and methyl hydrogen atoms were positioned geometrically and treated as the riding atoms with C–H = 0.93 Å and Uiso(H) = 1.2 Ueq(C) for aromatic carbon atoms, C–H = 0.96 Å and Uiso(H) = 1.2 Ueq(C) for methyl carbon atom. The N–H and O–H hydrogen for N1/N2/O4/O5 with N–H = 0.86 Å, O–H = 0.82 Å were positioned geometrically and treated as the riding atoms with Uiso(H) = 1.2 Ueq(N) for nitrogen atoms, and Uiso(H) = 1.5 Ueq(O) for oxygen atom. The hydrogen atom for O3 was positioned using Fourier map with O–H = 1.00 Å and Uiso(H) = 1.5 Ueq(O). The crystal data were deposited at the Cambridge Crystallographic Data Centre and the deposition number 2108057† was assigned, which is known as the CCDC number for compound 10.
2.3. Characterization
The spectral data for all the synthesized compounds is represented here.
2.3.1 (E)-N-(2-(2-(4-Chlorobenzylidene)hydrazinecarbonyl)phenyl) benzamide (05). Yield: 91%; M.F: C21H16ClN3O2; MP: 232–233 °C; FTIR (cm−1): 1601 (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, stretch), 1651 (CO, stretch), 3057 (C–H, stretch), 3327 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 7.29 (t, J = 7.6 Hz, 1H, H-4), 7.54 (d, J = 8.4 Hz, 2H, H-18, H-20), 7.58–7.67 (m, 4H, H-5, H-10, H-11, H-12), 7.79 (d, J = 8.4 Hz, 2H, H-21, H-17), 7.91 (d, J = 7.7 Hz, 1H, H-6), 7.97 (d, J = 6.6 Hz, 2H, H-9, H-13), 8.45 (1H, s, NCH), 8.57 (d, J = 8.3 Hz, 1H, H-3), 11.92 (1H, s, NH–CO), 12.21 (1H, s, NH–N). 13C NMR (75 MHz, DMSO-d6): δ = 120.95 (C-6), 121.56 (C-2), 123.63(C-4), 127.52 (2C, C-9, C-13), 129.10 (C-3), 129.39 (4C, C-10, C-12, C-18, C-20), 129.46 (2C, C-17, C-21), 132.60 (C-11), 133.15 (C-5), 133.44 (C-16), 134.83 (C-8), 135.29 (C-19), 139.77(C-1), 148.10 (C-15, CHN), 165.00 (C-14), (N–NHCO), 165.49 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 376 (20, [M]−), 239 (45), 221 (30), 153 (100), 125 (15).
CH, stretch), 1651 (CO, stretch), 3057 (C–H, stretch), 3327 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 7.29 (t, J = 7.6 Hz, 1H, H-4), 7.54 (d, J = 8.4 Hz, 2H, H-18, H-20), 7.58–7.67 (m, 4H, H-5, H-10, H-11, H-12), 7.79 (d, J = 8.4 Hz, 2H, H-21, H-17), 7.91 (d, J = 7.7 Hz, 1H, H-6), 7.97 (d, J = 6.6 Hz, 2H, H-9, H-13), 8.45 (1H, s, NCH), 8.57 (d, J = 8.3 Hz, 1H, H-3), 11.92 (1H, s, NH–CO), 12.21 (1H, s, NH–N). 13C NMR (75 MHz, DMSO-d6): δ = 120.95 (C-6), 121.56 (C-2), 123.63(C-4), 127.52 (2C, C-9, C-13), 129.10 (C-3), 129.39 (4C, C-10, C-12, C-18, C-20), 129.46 (2C, C-17, C-21), 132.60 (C-11), 133.15 (C-5), 133.44 (C-16), 134.83 (C-8), 135.29 (C-19), 139.77(C-1), 148.10 (C-15, CHN), 165.00 (C-14), (N–NHCO), 165.49 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 376 (20, [M]−), 239 (45), 221 (30), 153 (100), 125 (15).
2.3.2 (E)-N-(2-(2-(4-Bromobenzylidene)hydrazinecarbonyl)phenyl)benzamide (06). Yield: 89%; M.F: C21H16BrN3O2; MP: 243–244 °C; FTIR (cm−1): 1603 (NCH, stretch), 1650 (CO, stretch), 3055 (C–H, stretch), 3328 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 7.3 (t, J = 7.6 Hz, 1H, H-4), 7.61 (t, J = 7.4 Hz, 2H, H-10, H-12), 7.63–7.67 (m, 2H, H-5, H-11), 7.7 (dd, J = 21.9 Hz, 8.5 Hz, 4H, H-17, H-18, H-20, H-21), 7.91 (d, J = 7.0 Hz, 1H, H-6), 7.97 (d, J = 7.2 Hz, 2H, H-9, H-13), 8.44 (1H, s, NCH), 8.56 (d, J = 8.2 Hz, 1H, H-3), 11.90 (1H, s, NH–N), 12.20 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 121.01 (C-6), 121.60 (C-2), 123.66 (C-4), 124.12 (C-19), 127.53 (2C, C-9, C-13), 129.11 (C-3), 129.42 (2C, C-10, C-12), 129.61 (2C, C-17, C-21), 132.39 (2C, C-18, C-20), C-132.61 (C-11), 133.16 (C-5), 133.80 (C-16), 134.86 (C-8), 139.77 (C-1), 148.23 (C-15, CHN), 165.03 (C-14, N–NHCO), 165.50 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 420 (30, [M]−), 239 (55), 221 (30), 197 (100), 169 (15).
2.3.3 (E)-N-(2-(2-(4-(Dimethylamino)benzylidene)hydrazinecarbonyl)phenyl)benzamide (07). Yield: 88%; M.F: C23H22N4O2; MP: 220–221 °C; FTIR (cm−1): 1602 (NCH, stretch), 1648 (CO, stretch), 3057 (C–H, stretch), 3317 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 3.00 (6H, s, 2CH3, CH3–N), 6.78 (d, J = 8.9 Hz, 2H, H-18, H-20), 7.24 (t, J = 7.5 Hz, 1H, H-4), 7.48 (d, J = 7.6 Hz, 1H, H-5), 7.52 (t, J = 8.0 Hz, 1H, H-11), 7.53–7.61 (m, 5H, H-3, H-10, H-12, H-17, H-21), 7.71 (d, J = 7.1 Hz, 1H, H-6), 7.75 (m, 2H, H-9, H-13), 8.21 (1H, s, NCH), 10.94 (1H, s, NH–N), 11.60 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 40.0 (CH3N), 112.25 (2C, C-18, C-20), 121.57 (C-6), 121.72 (C-2), 123.19 (C-4), 124.78 (C-16), 129.08 (C-3), 129.19 (2C, C-9, C-13), 129.28 (2C, C-10, C-12), 129.99 (2C, C-17, C-21), 132.85 (C-11), 137.68 (C-5), 138.02 (C-8), 138.70 (C-1), 150.53 (C-15, CHN), 152.22 (C-19), 163.99 (2C, C-7, C-14, NHCO, N–NHCO). MS (ESI), 70 eV: m/z (%), 385 (20, [M]−), 239 (35), 221 (20), 162 (100), 134 (05).
2.3.4 (E)-N-(2-(2-(4-Nitrobenzylidene)hydrazinecarbonyl)phenyl)benzamide (08). Yield: 90%; M.F: C21H16N4O4; MP: 239–240 °C; FTIR (cm−1): 1586 (NCH, stretch), 1649 (CO, stretch), 3058 (C–H, stretch), 3256 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 7.31 (t, J = 7.4 Hz, 1H, H-4), 7.6 (t, J = 7.3 Hz, 2H, C-10, C-12), 7.62–7.70 (m, 2H, C-5, C-11), 7.92 (d, J = 7.7 Hz 1H, C-6), 7.97 (d, J = 7.4 Hz, 2H, C-9, C-13), 8.02 (d, J = 8.5 Hz, 2H, C-17, C-21), 8.31 (d, J = 8.5 Hz, 2H, C-18, C-20), 8.54 (d, J = 8.4 Hz, 1H, H-3), 8.56 (1H, s, NCH), 11.81 (1H, s, NH–N), 12.40 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 121.10 (C-6), 121.78 (C-2), 123.71 (C-4), 124.55 (2C, C-18, C-20), 127.56 (2C, C-17, C-21), 128.67, (2C, C-9, C-13), 129.19 (C-3), 129.39 (2C, C-10, C-12), 132.60 (C-11), 133.30 (C-5), 134.84 (C-8), 139.76 (C-1), 140.80 (C-16), 146.81 (C-15, CHN), 148.48 (C-19),165.09 (C-14, N–NHCO), 165.71 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 387 (40, [M]−), 239 (65), 221 (20), 178 (20), 164 (100).
2.3.5 (E)-N-(2-(2-(4-Isopropylbenzylidene)hydrazinecarbonyl)phenyl)benzamide (09). Yield: 92%; M.F: C24H23N3O2; MP: 229–230 °C; FTIR (cm−1): 1602 (NCH, stretch), 1657 (CO, stretch), 3030 (C–H, stretch), 3200 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 1.23 (6H, d, 2CH3–CH), 2.95 (1H, sep, CH–CH3), 7.29 (dd, J = 11.1 Hz, 4 Hz, 1H, H-4), 7.36 (d, J = 8.1 Hz, 2H, H-18, H-20), 7.62 (dd, J = 12.5 Hz, 4.8 Hz, 2H, H-5, H-11), 7.63–7.66 (m, 2H, H-10, H-12), 7.68 (d, J = 8.2 Hz, 2H, H-21, H-17), 7.92 (dd, J = 7.8 Hz, 1.1 Hz, 1H, H-9, H-13), 7.95–7.98 (m, 1H, H-6), 8.58 (d, J = 8.1 Hz, 1H, H-3), 8.44 (1H, s, NCH), 11.96 (1H, s, NH–N), 12.07 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 24.12 (2C, 2CH3C), 33.87 (1C, CH–(CH3)2), 120.96 (C-6), 121.49 (C-2), 123.62 (C-4), 127.35 (2C, C-18, C-20), 127.52 (2C, C-9, C-13), 127.88 (2C, C-17, C-21), 129.07 (C-3), 129.43 (2C, C-10, C-12) 132.18 (C-11), 132.61 (C-16), 133.06 (C-5), 134.88 (C-8), 139.79 (C-1), 149.64 (C-15, CHN), 151.53 (C-19), 165.00 (C-14, N–NHCO), 165.36 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 384 (20, [M]−), 239 (45), 221 (30), 161 (100).
2.3.6 (E)-N-(2-(2-(2,4-Dihydroxybenzylidene)hydrazinecarbonyl)phenyl)benzamide (10). Yield: 93%; M.F: C21H17N3O4; MP: 260–261 °C; FTIR (cm−1): 1601 (NCH, stretch), 1629 (CO, stretch), 3052 (C–H, stretch), 3200 (NH, stretch), 3526 (OH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 6.41–6.33 (m, 2H, H-18, H-20), 7.28 (t, J = 7.4 Hz, 1H, H-4), 7.37 (d, J = 8.4 Hz, 1H, H-17), 7.62 (m, 4H, H-5, H-10, H-11, H-12), 7.91 (d, J = 7.1 Hz, 1H, H-6), 7.93.7.98 (m, 2H, H-9, H-13), 8.55 (1H, s, NCH), 8.57 (d, J = 8.6 Hz, 1H, H-3), 10.02 (1H, p-OH), 11.30 (1H, o-OH), 11.98 (1H, s, NH–N), 12.16 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 103.08 (C-18), 108.32 (C-20), 110.97 (C-16), 120.61 (C-6), 121.50 (C-2), 123.61 (C-4), 127.50 (2C, C-9, C-13), 128.98 (C-3), 129.42 (2C, C-10, C-12), 131.56 (C-21), 132.60 (C-11), 133.07 (C-5), 134.90 (C-8), 139.79 (C-1), 150.33 (C-15), 159.95 (C-19), 161.48 (C-17), 164.80 (C-14, N–NHCO), 166.03 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 374 (30, [M]−), 356 (10), 239 (65), 221 (45), 151 (90), 123 (100).
2.3.7 (E)-N-(2-(2-(2-Hydroxybenzylidene)hydrazinecarbonyl)phenyl)benzamide (11). Yield: 89%; M.F: C21H17N3O3; MP: 236–237 °C; FTIR (cm−1): 1604 (NCH, stretch), 1648 (CO, stretch), 3053 (C–H, stretch), 3195 (HN, stretch), 3286 (OH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 6.90–6.96 (m, 2H, H-18, H-20), 7.27–7.34 (m, 2H, H-4, H-19), 7.57–7.67 (m, 5H, H-5, H-10, H-11, H-12, H-21), 7.91–7.98 (m, 3H, H-6, H-9, H-13), 8.56 (d, J = 8.4 Hz, 1H, H-3), 8.69 (1H, s, NCH), 11.12 (1H, s, 1OH), 11.89 (1H, s, NH–N), 12.32 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 116.89 (C-18), 119.22 (C-20), 119.89 (C-16), 120.75 (C-6), 121.66 (C-2), 123.67 (C-4), 127.53 (2C, C-9, C-13), 129.10 (C-3), 129.41 (2C, C-10, C-12), 129.58 (C-21), 132.17 (C-19), 132.60 (C-11), 133.20 (C-5), 134.89 (C-8), 139.78 (C-1), 149.29 (C-15), 157.90 (C-17), 165.08 (C-14, N–NHCO), 165.16 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 358 (20, [M]−), 239 (25), 221 (20), 135 (100), 107 (10).
2.3.8 (E)-N-(2-(2-(4-Methoxybenzylidene)hydrazinecarbonyl)phenyl)benzamide (12). Yield: 86%; M.F: C22H19N3O3; MP: 215–216 °C; FTIR (cm−1): 1602 (NCH, stretch), 1649 (CO, stretch), 3055 (C–H, stretch), 3309 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 3.78 (3H, s, OCH3), 7.08 (m, 2H, H-18, H-20), 7.29 (t, J = 7.6 Hz, 1H, H-4), 7.58–7.66 (m, 5H, H-5, H-10, H-12, H-17, H-21), 7.79 (dd, J = 7.7 Hz, 1H, H-11), 7.91–7.98 (m, 3H, H-6, H-9, H-13), 8.60 (d, J = 8.3 Hz, 1H, H-3), 8.73 (1H, s, NCH), 12.03 (1H, s, NH–N), 12.03 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 57.4 (CH3O), 116.10 (2C, C-18, C-20), 120.80 (C-6), 121.42 (C-2), 123.56 (C-4), 127.50 (2C, C-9, C-13), 128.97 (C-3), 129.42 (4C, C-10, C-12), 129.73 (C-16), 131.95 (2C, C-17, C-21), 132.61 (C-11), 133.08 (C-5), 134.86 (C-8), 140.34 (C-1), 148.24 (C-15, CHN), 164.97 (C-19), 165.22 (C-14, N–NHCO), 165.22 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 372 (40, [M]−), 239 (50), 221 (35), 149 (100), 121 (15).
2.3.9 (E)-N-(2-(2-Benzylidenehydrazinecarbonyl)phenyl)benzamide (13). Yield: 90%; M.F: C21H17N3O2; MP: 204–205 °C; FTIR (cm−1): 1601 (NCH, stretch), 1643 (CO, stretch), 3056 (C–H, stretch), 3212 (NH, stretch). 1H NMR (300 MHz, DMSO-d6): δ = 7.3 (t, J = 7.5 Hz, 1H, H-4), 7.46–7.52 (m, 3H, H-18, H-19, H-20), 7.61 (t, J = 7.4 Hz, 2H, H-10, H-12), 7.63–7.68 (m, 2H, H-5, H-11), 7.75–7.79 (m, 2H, H-21, H-17), 7.93 (d, J = 7.3 Hz, 1H, H-6), 7.97 (d, J = 7.3 Hz, 2H, H-9, H-13), 8.58 (d, J = 8.3 Hz, 1H, H-3), 8.48 (1H, s, NCH), 11.95 (1H, s, NH–N), 12.13 (1H, s, NH–CO). 13C NMR (75 MHz, DMSO-d6): δ = 120.97 (C-6), 121.53 (C-2), 123.63 (C-4), 127.53 (2C, C-18, C-20), 127.77 (2C, C-9, C-13), 129.09 (C-3), 129.37 (2C, C-17, C-21), 129.42 (2C, C-10, C-12) 130.87 (C-19), 132.60 (C-11), 133.11 (C-16), 134.50 (C-5), 134.87 (C-8), 139.80 (C-1), 149.56 (C-15, CHN), 165.02 (C-14, (N–NHCO), 165.46 (C-7, NHCO). MS (ESI), 70 eV: m/z (%), 342 (30, [M]−), 239 (35), 221 (20), 119 (100), 91 (10).
2.4. Biological activity
2.4.1. Enzyme inhibition essay. Ryan and Elman method32 with little variation was utilized to carry out enzyme inhibition analysis. 50 μL of every inhibitor (50 μM) (03–13) was mixed with 50 μL of enzyme (0.3 and 0.15 U mL−1 of AChE or BChE, respectively). This mixture of every sample with the enzyme was allowed to stand for 15 minutes. Then, 50 μL of the substrate, i.e., butyrylthiocholine chloride (0.2 mM) for BChE and acetylthiocholine iodide (0.71 mM) for AChE was added to the mixture. 500 μL of phosphate buffer (pH 8) was added to maintain the pH along with 50 μL of DTNB [5,5′-dithio-bis(2-nitrobenzoic acid)] (0.5 mM). The whole mixture was shaken well to mix the contents and incubated at 37 °C for 30 minutes. The hydrolysis of the substrate resulted in the production of yellow coloration. The spectrophotometric method was utilized to measure the absorbance at 412 nm for BChE and 400 nm for AChE, thus measuring the intensity of yellow coloration. The following formula was utilized to calculate the percentage enzyme inhibition for all the inhibitors eqn (1).| |
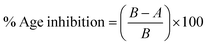 | (1) |
where A = absorption of the sample containing the test compounds, B = absorption for the samples without the test compounds.Every experiment was conducted in triplicate with average values used for calculations Donepezil was taken as the reference drug. Inhibition was also assessed by computing the IC50 values, which were calculated from the dose–effect curves by linear regression.
2.4.2. Antioxidant assay. To measure the free radical scavenging activity of antioxidants, the DPPH method is diversely being used due to its efficacy, simplicity, quick procedure, and above all, its relatively inexpensive nature. Antioxidants cause the reduction of the methanolic DPPH solution by donating hydrogen atoms or electrons to it. The antioxidant potential of the corresponding compounds was measured spectrophotometrically by measuring the decolorization of the purple color of 2,2-diphenyl-1-picrylhydrazyl free radical (DPPH) by following the Blois method.33 DPPH solution (0.004%) was prepared in methanol. 100 μL of 50 μM solution of every test compound (03–13) was mixed with 1 mL of DPPH solution. The reaction mixture was mixed carefully and incubated in the dark at 37 °C for 20 minutes. The measurement of the absorbance of the mixture was carried out spectrophotometrically at λmax = 517 nm, followed by comparison with the absorption of standard antioxidant ascorbic acid (vitamin C). The DPPH radical scavenging activity (% RSA) of the compounds was calculated according to eqn (2).| |
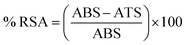 | (2) |
where ATS = absorbance of test sample, ABS = absorbance of the blank sample.
2.4.3. Cytotoxicity studies (Resazurin assay). Resazurin assay was utilized to carry out the cytotoxicity studies of the synthesized compounds. Caco-2 cells were obtained from the European Collection of Cell Cultures (ECACC, Health Protection Agency, Porton Down, Salisbury, Wiltshire, United Kingdom). Cells were seeded at a density of 2.5 × 104 cells per well in a 24-well plate for 10 days in a final volume of 500 μL of MEM with Earle's balanced salts supplemented with 2.0 mM L-glutamine, 10% FBS, and 1% penicillin-streptomycin at 37 °C in 5% CO2 environment. The culture medium was refreshed every other day.Resazurin assay was performed as an oxidation-reduction indicator to determine the in vitro cytotoxicity of the compounds (05–08). When the cells were approximately 80% confluent (80% of the surface of flask covered by the cell monolayer after 8–10 days), they were washed twice with PBS pre-warmed at 37 °C. The test solutions including the sample compounds (10 mM), positive control prepared in white MEM, and negative control (0.5% v/v Triton X-100) were added in 500 μL volumes in triplicate to the cell culture. Then, the treated cells were incubated at 37 °C in 5% CO2 environment for 3 and 24 h. Afterward, the test solutions were removed and the cells were washed twice with prewarmed phosphate buffer saline (PBS). A diluted resazurin solution (2.2 μM) in 500 μL volume was added to each well and the cells were incubated for 3 h. The supernatant (100 μL) was afterward transferred to a black 96-well plate and the fluorescence intensity was measured using a microplate reader at a wavelength of 540 nm with background subtraction at 590 nm.34 The percentage of the viable cells was calculated using the following equation eqn (3).
| |
 | (3) |
2.5. Molecular docking studies
To explore the binding interactions between the ligands and the amino acid residues from the enzyme active sites, molecular docking studies were performed. The crystal structures of both AChE and BChE enzymes were obtained from Protein Data Bank (RCSB). The molecular structures of the ligands were sketched utilizing ACD ChemSketch and 3D optimization for the sketched structures of ligands was done using 3D Pro 12.0 software packages. SYBYL mol2 file formats were used to save these 3D optimized structures. AutoDock software was used to perform the process of molecular docking. The adjustment of 100 different configurations was done and more effective and the best conformations of the compounds under analysis were visualized using Discovery Studio Visualizer v 4.0.35
2.6. Computational analysis (DFT)
Computational calculations were carried out using DFT-B3LYP/321g in Gaussian-09. The ground state geometries of compounds [04–13] were optimized using hybrid functional B3LYP and basis set 321g.
3 Results and discussion
3.1. Chemistry
2-(Benzamido) benzohydrazide derivatives (05–13) were synthesized using Scheme 1. The structures of synthetic 2-(benzamido) benzohydrazide derivatives (05–13) were interpreted using different spectroscopic techniques such as UV-Visible, NMR, and FTIR spectroscopic techniques. ESI (electrospray ionization) technique was utilized to confirm the molecular masses of the synthesized compounds.
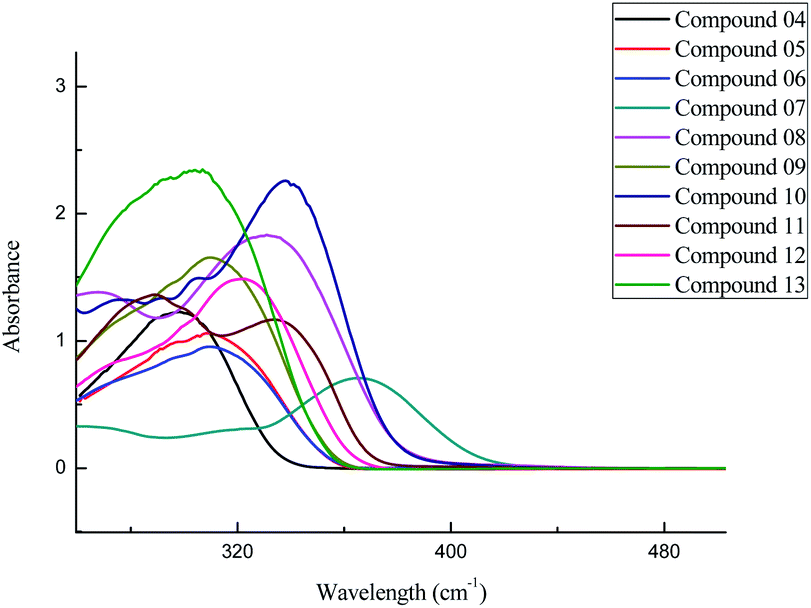
3.1.1. UV-Visible studies. The UV-Vis absorption spectra of hydrazone derivatives (05–13) of 2-(benzamido) benzohydrazide (04) were recorded using solutions of these compounds in methanol. These Schiff base derivatives showed peaks in the range of 300–365 nm, which are associated with the π → π* transitions involved due to the conjugation of the azomethine (CN) groups with the phenyl ring in these compounds (05–13). The variation in the absorption maxima (λmax) of these compounds (05–13) was associated with the attachment of different substituents in the phenyl ring attached to the azomethine group. Unsubstituted benzylidene group showed λmax at 304 nm. EWGs such as p-chloro and p-bromo showed a slight bathochromic shift (05 and 06) in comparison to EDGs such as p-N(CH3)2, o-OH, p-OH, and p-OCH3 groups (08, 10, 11, and 12). Maximum red shift was attributed to the presence of the p-NO2 group in compound 07 (Fig. 1).
 |
| | Fig. 1 Comparative absorption spectra of 2-(benzamido) benzohydrazide (04) and its derivatives (05–13). | |
3.1.2. FTIR studies. Remarkable Fourier Transform Infrared (FTIR) bands for the synthesized compounds are given in the experimental section. The appearance of the signal in the range of 1586–1604 cm−1 for the (HCN) group and the disappearance of the signal at 3316 cm−1 for the (–NH2) group in the IR spectrum clearly confirms the conversion of 2-(benzamido) benzohydrazide (04) to its Schiff bases (05–13). All these derivatives have shown significant peaks for CO stretching frequencies in the range of 1629–1657 cm−1. Furthermore, the peaks for the N–H bond appeared in the range of 3195–3328 cm−1. In addition, the peaks for the C–H stretch attached to the aromatic system appeared at 3030–3057 cm−1. The broader peaks for the –OH groups were observed for compounds (10) and (11) in the range of 3286–3526 cm−1. All the FTIR data approved the synthesis of the compounds (05–13).
3.1.3. NMR studies. Furthermore, the structures of the synthesized compounds were confirmed by 1H NMR and 13C NMR analysis. The signals detected in the range of 8.21–8.73 ppm as a singlet in all the synthesized compounds (05–13) for the (HCN) protons confirmed the formation of the imine linkage. Similarly, in the 13C spectra, the signals appearing in the range of 146.8–150.5 ppm for the imine carbon atom (HCN) in all these compounds reinforced the results. While the signals of the aromatic protons varied in the range between 6.34 ppm and 8.61 ppm in the 1H NMR spectrum, which is related to the nature of the substituents present on the aromatic ring in all the Schiff base derivatives (05–13). In compounds (07, 10, 11, and 12), the signals for aromatic protons were shifted upfield (6.34–7.08 ppm) due to the attachment of the electron donating groups (EDG), i.e., dimethylamino, hydroxyl, and methoxyl groups. These EDG groups caused a increase in the electron density at the ortho and para positions of the benzene ring in these compounds, thereby changing the position of the peaks of the corresponding protons up field in contrast to the peak of proton (7.29 ppm) in compound 13, which has the non-substituted benzylidene ring. In compound 12, the methoxyl groups attached to the aromatic ring showed proton signals at 3.78 ppm. Likewise, the signals of aromatic carbons in the 13C NMR spectra also varied in accordance with the substituents attached to the benzylidene ring in compounds (05–13) in the range of 103.0–161.4 ppm, thus approving the structures of the synthesized hydrazone derivatives. All the spectral data for 1H NMR and 13C NMR are in accordance with the structures of the synthesized compounds.
3.1.4. Mass spectroscopic studies. Mass spectral analysis was carried out utilizing the electron spray ionization (ESI) technique for all the synthesized compounds. The molecular masses of the compounds were established using mass spectroscopic analysis. The peaks related to the [M − H] ions were found for all the derivatives in the negative mode of the mass spectrum. Correspondingly, in the positive mode of mass analysis, peaks were observed for [M + H] ions for all the compounds. All the spectral data for MS (ESI) confirmed the structures of the synthesized compounds.
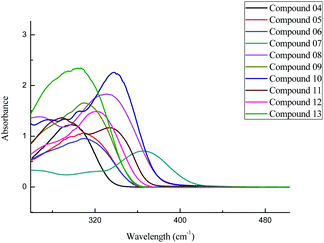
3.2. Crystal structure description
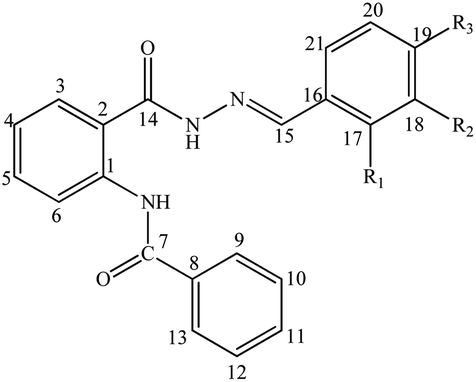
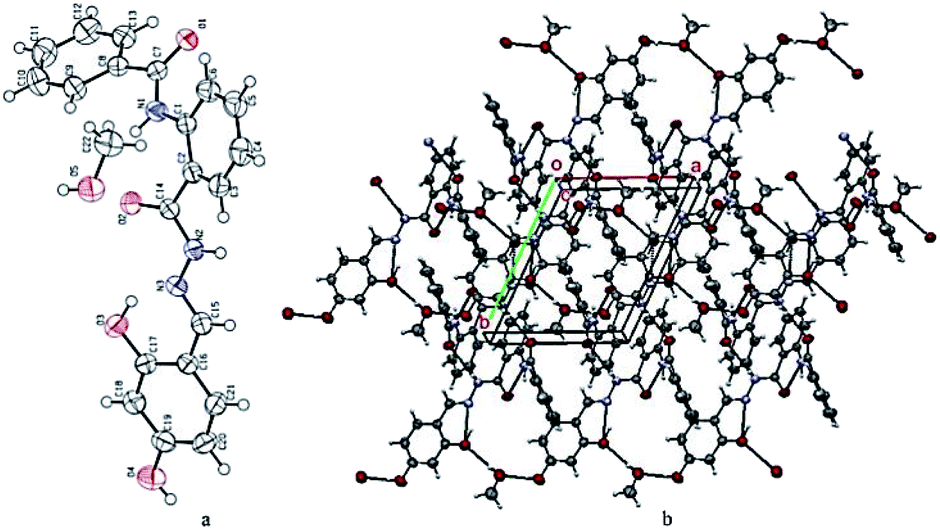
Compound 10 was crystallized along with a methanol molecule as the solvate in a triclinic crystal system (Table 1) with one independent molecule in an asymmetric unit cell Fig. 2(a). The selected bond lengths, bond angles, and torsion angles are provided in Tables S1–S3,† respectively. We have already published the crystal structure and applications of some related hydrazide compounds.36a,b The imine (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C15N3
C15N3![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ) functionality of N-[2-(2,4-dihydroxy-benzylidene-hydrazinocarbonyl)-phenyl]benzamide adopted the most stable trans geometry. The central aromatic ring (C1–C6) is occupied by the benzamide at position 1, while the C2 atom is bonded with 2,4-dihydroxy-benzylidene-hydrazinocarbonyl. The three available aromatic rings were twisted at different angles with each other, i.e., the ring (C1–C6) was oriented by 54.05(8) and 31.83(11) with respect to the (C8–C13) and (C16–C21). The dihedral angle between the rings (C8–C13) and (C16–C21) is 75.20(8). The hydrazinocarbonyl system is connected to 2,4-dihydroxyphenyl (C16–C21) and an aromatic ring (C1–C6) at its each end. The plane of the hydrazinocarbonyl system is almost planar (dihedral angle = 3.14(2)°) with 2,4-dihydroxyphenyl (C16–C21) and twisted by 32.58(9)° with respect to the aromatic ring (C1–C6). The presence of the methanol molecule as the solvent in the crystal system supports the additional hydrogen bonding interactions. Two different six-membered ring motifs were S(6) generated via the intramolecular interactions of N1–H1n⋯O2 and O3–H1O⋯N3. The N2–H2N of the hydrazine functionality is connected with the O1 of other molecule to generate a sixteen-membered ring motif R22(16),37 following the symmetry equation as 2 − X, 2 − Y, 2 − Z.
) functionality of N-[2-(2,4-dihydroxy-benzylidene-hydrazinocarbonyl)-phenyl]benzamide adopted the most stable trans geometry. The central aromatic ring (C1–C6) is occupied by the benzamide at position 1, while the C2 atom is bonded with 2,4-dihydroxy-benzylidene-hydrazinocarbonyl. The three available aromatic rings were twisted at different angles with each other, i.e., the ring (C1–C6) was oriented by 54.05(8) and 31.83(11) with respect to the (C8–C13) and (C16–C21). The dihedral angle between the rings (C8–C13) and (C16–C21) is 75.20(8). The hydrazinocarbonyl system is connected to 2,4-dihydroxyphenyl (C16–C21) and an aromatic ring (C1–C6) at its each end. The plane of the hydrazinocarbonyl system is almost planar (dihedral angle = 3.14(2)°) with 2,4-dihydroxyphenyl (C16–C21) and twisted by 32.58(9)° with respect to the aromatic ring (C1–C6). The presence of the methanol molecule as the solvent in the crystal system supports the additional hydrogen bonding interactions. Two different six-membered ring motifs were S(6) generated via the intramolecular interactions of N1–H1n⋯O2 and O3–H1O⋯N3. The N2–H2N of the hydrazine functionality is connected with the O1 of other molecule to generate a sixteen-membered ring motif R22(16),37 following the symmetry equation as 2 − X, 2 − Y, 2 − Z.
Table 1 Crystal data and structure refinement for compound 10
| CCDC number |
2108057 |
| Empirical formula |
C22H21N3O5Ho |
| Formula weight |
407.42 |
| Temperature/K |
296(2) |
| Crystal system |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å |
8.6661(9) |
| b/Å |
11.0426(8) |
| c/Å |
12.2572(12) |
| α/° |
108.672(7) |
| β/° |
102.099(8) |
| γ/° |
108.033(8) |
| Volume/Å3 |
992.66(17) |
| Z |
2 |
| ρcalc/g cm−3 |
1.363 |
| μ/mm−1 |
0.098 |
| F(000) |
428.0 |
| Crystal size/mm3 |
0.41 × 0.22 × 0.19 |
| Radiation |
MoKα (λ = 0.7107) |
| 2Θ range for data collection/° |
6.426 to 58.372 |
| Index ranges |
−10 ≤ h ≤ 11, −14 ≤ k ≤ 14, −16 ≤ l ≤ 16 |
| Reflections collected |
7704 |
| Independent reflections |
4628 [Rint = 0.0252, Rsigma = 0.0516] |
| Data/restraints/parameters |
4628/0/286 |
| Goodness-of-fit on F2 |
1.036 |
| Final R indexes [I ≥ 2σ(I)] |
R1 = 0.0560, wR2 = 0.1203 |
| Final R indexes [all data] |
R1 = 0.1014, wR2 = 0.1462 |
| Largest diff. peak/hole/e Å−3 |
0.20/−0.20 |
 |
| | Fig. 2 (a) ORTEP diagram of compound 10; thermal ellipsoids were drawn at 50% probability level. (b) A view showing the formation of the two-dimensional network along the ab-plane. | |
Table 2 Hydrogen bonds for compound 10
| D |
H |
A |
d(D–H)/Å |
d(H–A)/Å |
d(D–A)/Å |
D–H–A/° |
| 2 − X, 2 − Y, 2 − Z. 2 − X, 1 − Y, 1 − Z. 1 − X, 1 − Y, 1 − Z. |
| C6 |
H6 |
O1 |
0.93 |
2.40 |
2.923(3) |
115.7 |
| N1 |
H1N |
O2 |
0.86 |
2.04 |
2.702(2) |
133.6 |
| N2 |
H2N |
O1a |
0.86 |
2.10 |
2.882(2) |
151.8 |
| O4 |
H2O |
O5b |
0.82 |
1.90 |
2.712(3) |
173.8 |
| O5 |
H3O |
O3c |
0.82 |
2.04 |
2.852(3) |
169.9 |
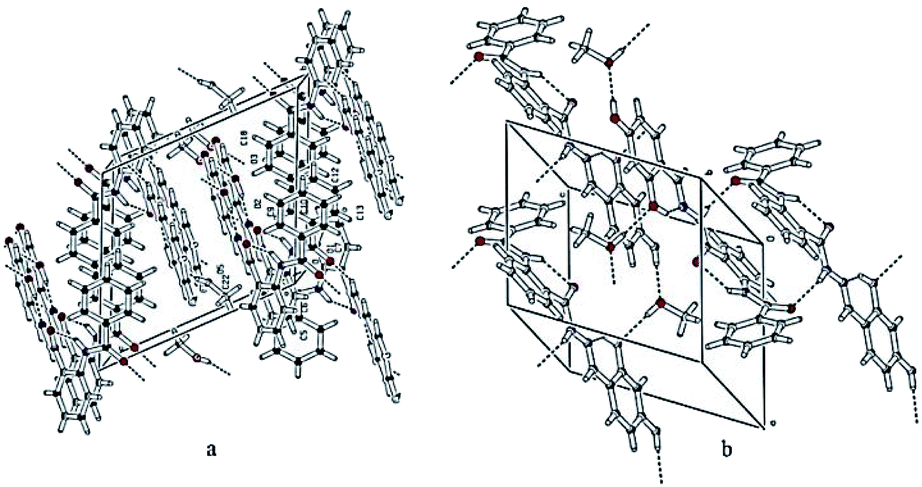
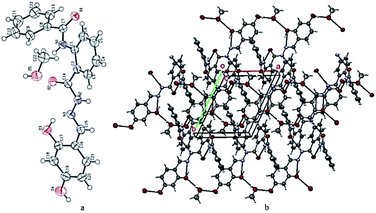
The hydroxyl group of methanol is taking part both as the donor and the acceptor for hydrogen bonding interactions. On the one hand, it is connected with the molecule via O5–H3O⋯O3 and on the other hand, it binds with the interaction of O4–H2O⋯O5, following the symmetry equations 1 − X, 1 − Y, 1 − Z and 2 − X, 1 − Y, 1 − Z, respectively Table 2. All these interactions connect the molecules and produce a two-dimensional network along the ab-plane (Fig. 2(b)). Intramolecular and inter-molecular hydrogen bonding interactions in crystal structure of compound 10 have been presented in (Fig. 3).
3.3. Antioxidant activity
The antioxidant potential of the synthesized derivatives was determined by following a well-known and commonly used DPPH assay. In recent times, the most employed form of the method, which was developed by Brand-Williams et al.,38 is an extension of the Blois33 method. The DPPH free radical shows strong absorption at 517 nm due to the delocalization of its free electron and possesses deep purple coloration. Antioxidants reduce this free radical to the more stable molecule 1,1-diphenyl-2-picrylhydrazine by donating an electron or a hydrogen atom. The purple color of the DPPH radical is altered to the pale yellow color of 1,1-diphenyl-2-picrylhydrazine molecule. This alteration in the color causes a decrease in the absorption at 517 nm, which is measured spectrophotometrically to estimate the scavenging activity. The results of the antioxidant activity of the 2-(benzamido) benzohydrazide derivatives (03–13) at the same concentration (1 μg μL−1) are described as the %scavenging of DPPH in Table 3. Ascorbic acid (vitamin C) was chosen as the standard. It can be clearly inferred from the table that many of the compounds show moderate antioxidant activities. Compounds 04 and 10 exhibited very good oxidation potentials due to free radical scavenging by hydrogen atom donation, which are attached to the –OH and –NH2 groups in their molecules.39
Table 3 Antioxidant potential of compounds 03–13 at 1 μg μL−1 concentration
| Compound # |
Absorbance |
Scavenging (%) |
| 3 |
1.570 |
37.17 |
| 4 |
0.123 |
95.07 |
| 5 |
1.615 |
35.37 |
| 6 |
2.102 |
15.88 |
| 7 |
1.400 |
43.97 |
| 8 |
1.994 |
20.20 |
| 9 |
1.656 |
33.73 |
| 10 |
0.436 |
82.55 |
| 11 |
2.003 |
67.54 |
| 12 |
1.747 |
30.09 |
| 13 |
1.744 |
30.21 |
| +ve control (AA) |
0.090 |
96.39 |
| DPPH |
2.499 |
|
 |
| | Fig. 3 Two different views showing the intra- and inter-molecular hydrogen bonding interactions in the crystal structure of compound 10. | |
3.4. Enzyme inhibition studies
To examine the importance of the benzamido moiety introduced ortho to the benzohydrazide derivatives (03–13), the inhibitory effect of these compounds was evaluated against human AChE and BChE by measuring their inhibitory potency IC50, i.e., the concentration of the inhibitor required to decrease the enzyme activity by half. The summary of the inhibition results of the compounds (03–13) against the two enzymes AChE and BChE are presented in Table 4, taking Donepezil as the reference.
Table 4 Inhibition of compounds 03–13 against AChE and BChE in terms of IC50 values
| Compounds |
AChE IC50 values ±SEMa (μM) |
BChE IC50 values ±SEMa (μM) |
| IC50 values (mean ± standard error of mean). NA: inactive. |
| 3 |
0.25 ± 0.06 |
0.21 ± 0.06 |
| 4 |
27.65 ± 0.14 |
0.17 ± 0.12 |
| 5 |
1.03 ± 0.12 |
1.24 ± 0.12 |
| 6 |
0.09 ± 0.05 |
0.14 ± 0.05 |
| 7 |
25.20 ± 0.21 |
NAb |
| 8 |
14.8 ± 0.29 |
0.19 ± 0.12 |
| 9 |
23.12 ± 0.25 |
NAb |
| 10 |
18.5 ± 0.19 |
2.15 ± 0.26 |
| 11 |
01.14 ± 0.35 |
0.12 ± 0.09 |
| 12 |
20.43 ± 0.23 |
18.34 ± 0.14 |
| 13 |
0.11 ± 0.03 |
0.10 ± 0.06 |
| Donepezil |
0.10 ± 0.02 |
0.14 ± 0.03 |
3.4.1. Structure activity relationship (SAR) studies. It is clear from the results that all the synthesized compounds showed activity against both the enzymes but they are more active against the BChE enzyme as compared to the AChE enzyme.Noteworthily, the most potent compound 06 having bromo substituent at the para position in the benzylidene ring showed dual inhibitory potential against both AChE and BChE, showing IC50 = 0.09 μM against AChE and IC50 = 0.14 μM against BChE. This is due to the presence of many important interactions with the bromo group inside the active pocket of both the enzymes, which is attributed to the presence of lone pairs of electrons on it.27 Furthermore, compound 13 showed good inhibition against both the enzymes AChE and BChE with IC50 = 0.11 μM (AChE) and IC50 = 0.10 μM (BChE). This is because of the ability of this compound to form additional hydrophobic π–π stacked interactions using its unimpeded pi-electron cloud in addition to other type of associations inside the active pockets of both the enzymes. In addition, compound 11 having hydroxyl group substituted at the ortho and para positions have shown selective inhibitory potential against BChE as compared to AChE with IC50 = 0.12 μM (BChE). We noted that all the compounds except compounds 07, 09, and 12 showed more inhibitory activity against BChE as compared to AChE.
Overall, it was found that the 2-benzamido derivatives of benzohydrazide have shown inhibitory activity against both the enzymes AChE and BChE, possessing more potency against BChE as compared to AChE. However, the inhibitory activity is mainly controlled by all the structural features present in a compound. However, the variation of the substituents on the key structural motif was actually accountable for the variation in the inhibition potential. Since the common skeleton of 2-(benzamido) benzohydrazide is present in the structures of all the synthesized compounds; therefore, the activity is mainly controlled by the type of the substituent attached to the benzylidene ring. The abovementioned findings suggest that these multi-functional hydrazone derivatives of anthranilic acid (05–13) can be used as the lead compounds for the further development toward the design and synthesis of new inhibitors against AChE and BChE enzymes. The outcome of the SAR studies are precisely presented in Fig. 4.
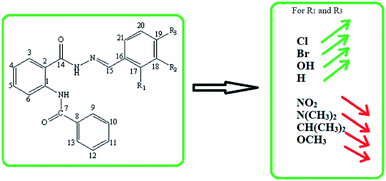 |
| | Fig. 4 Structure–activity relationship for cholinesterase enzyme activity of 2-(benzamido) benzohydrazide derivatives; downward red arrows indicate a decrease in activity; upward green arrows indicate an increase in activity. | |
3.5. Cytotoxicity
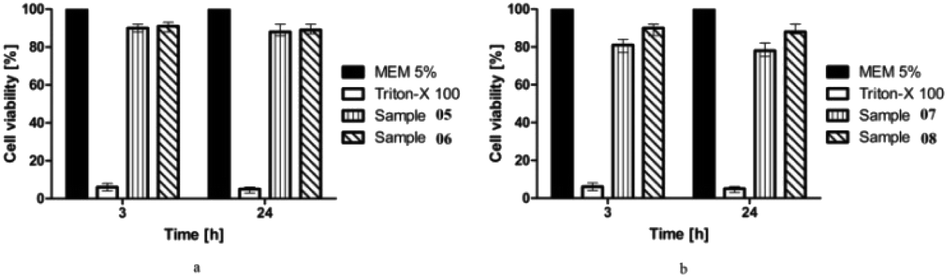
The cell viability of the synthesized compounds (05–08) on the Caco-2 cells was assessed by the resazurin assay. The principle of resazurin assay is that the viable cells reduce resazurin into resorufin. The color of the dye is converted from its blue (non-fluorescent) color to the pink (high-fluorescent) color. As healthy cells reduce resazurin more effectively than the dead cells, the water-soluble and nontoxic dye serves are the general indicator for cellular metabolic activity.40 The cells were treated with the test compounds at concentrations of 10 mM. The results showed that compounds 05, 06, and 08 showed cell viability of almost 90%, while compound 07 also depicted greater than 80% cell viability for the concentration tested after 3 and 24 h of incubation (Fig. 5). It is therefore concluded that the tested compounds 05–08 did not show significant toxicity (p < 0.05). The newly synthesized compounds can be considered as relatively safe for in vivo applications.
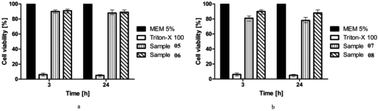 |
| | Fig. 5 Graphs showing %cell viability after 3 and 24 h for compounds 05–08 (10 mM). | |
3.6. Molecular docking studies
Computational docking studies were carried out using AutoDock to presume the most favorable binding interactions of the compounds (05–13) inside the active pocket of AChE (PDB ID: 4BDT) and BChE (PDB ID: 4BDS). The minimum binding energies of the compounds (05–13) obtained as a result of docking analysis are presented in Table 5. All the compounds exhibited binding energy values in a narrow range due to the establishment of almost similar type of binding interactions inside the active pocket of the enzymes AChE and BChE. To determine the interactions of the inhibitor with the amino acids of the active pocket of protein, the visualization of the selected poses of the most active compounds 06 and 13 was carried out.
Table 5 Binding energies of selected conformations against human AChE and BChE
| Compounds |
h AChE lowest binding energy ΔG in kJ mol−1 |
h BChE lowest binding energy ΔG in kJ mol−1 |
| 05 |
−10.34 |
−9.80 |
| 06 |
−10.63 |
−9.89 |
| 07 |
−9.55 |
−9.34 |
| 08 |
−9.35 |
−9.05 |
| 09 |
−10.27 |
−10.06 |
| 10 |
−10.29 |
−8.93 |
| 11 |
−9.47 |
−9.29 |
| 12 |
−10.15 |
−9.37 |
| 13 |
−11.51 |
−10.51 |
| 14 |
−10 (HUW) |
−6.83 (THA) |
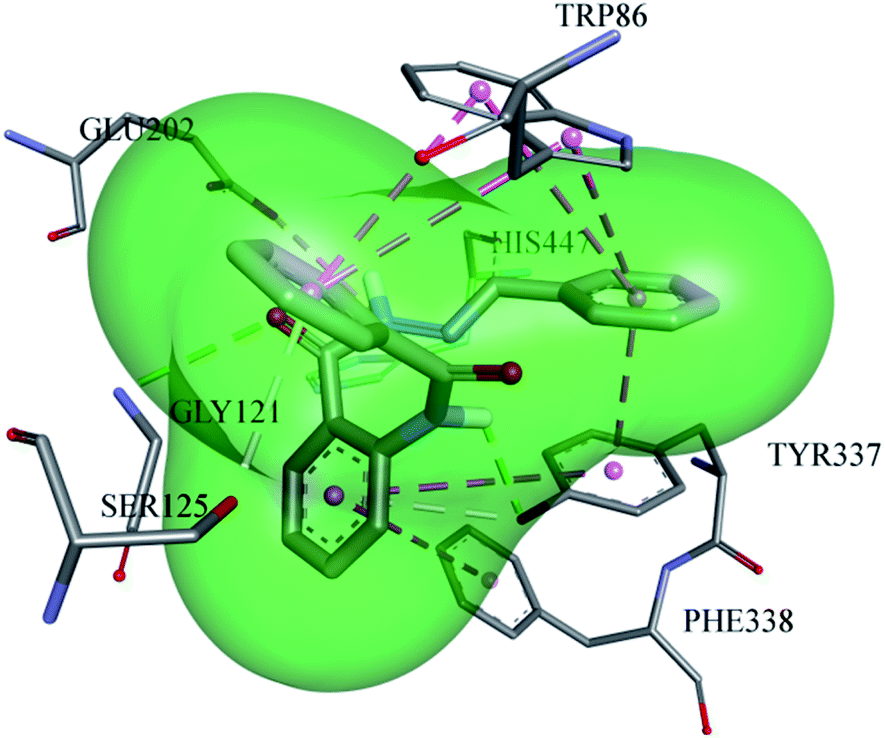
Different substitutions attached to different inhibitors caused a variation in the type of the interactions between the ligand and the proteins. A very potent inhibitor (13) is stabilized inside the active pocket of AChE by making significant associations with the amino acid residues, as depicted in Fig. 6. All the three amino acid residues (TYR-337, TRP-86, and GLU-202) of the choline binding site are involved for making important interactions with the ligand. GLU-202 made electrostatic interaction with a distance of 4.5 Å with the –NH group from the carbohydrazide part of the ligand, while the hydroxyl group of TYR-337 developed two hydrogen bond interactions, one with the amide oxygen atom (2.4 Å) and the second with the pi electrons of the aromatic system (3.5 Å). In addition, TYR-337 makes two π–π stacked interactions and TRP-86 forms 4π–π stacking with the pi electron cloud of the benzene rings, which further contribute to the stabilization of the ligand. GLY-121 from the oxyanion hole also plays a part by hydrogen bonding (3.3 Å) with the carbonyl oxygen of the carbohydrazide part of the ligand.
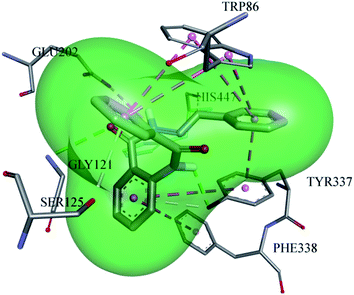 |
| | Fig. 6 Putative binding approaches of compound 13 inside the AChE enzyme. | |
In addition, HIS-447 and SER-125 both cooperate toward ligand stability inside AChE by forming hydrogen bonds with distances of 2.96 Å and 3.7 Å.
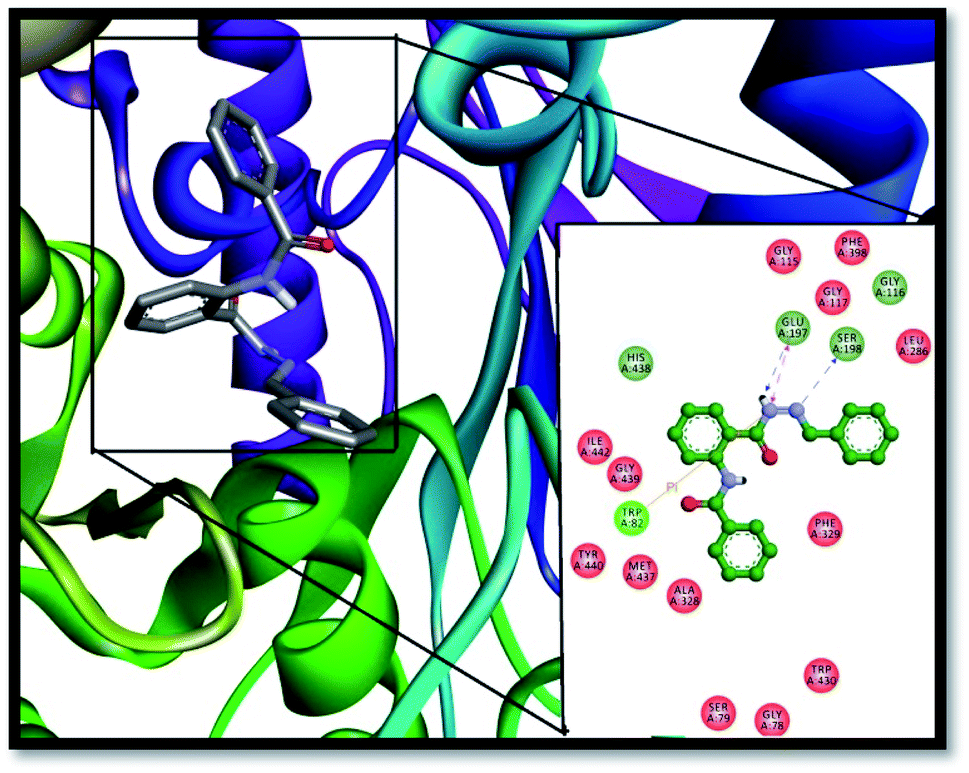
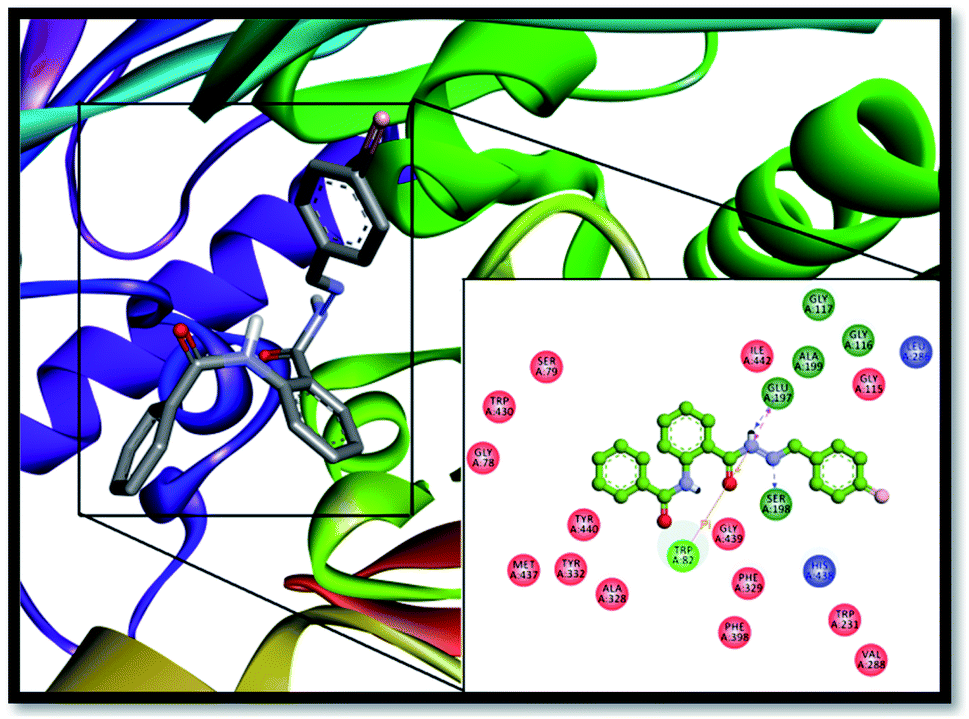
Furthermore, this inhibitor (13) resides very well inside the active pocket of BChE by establishing potent interactions with the enzyme active site residues, as depicted in Fig. 7. Hydrogen bond interactions of the ligand with the active pocket of the enzyme play an important role in its stabilization inside the active pocket such as the hydrogen atom from the NH–CO group of the hydrazone linkage is involved in the formation of two very strong H-bonds, one with oxygen atom of SER-198 from the catalytic triad of BChE with a bond distance 2.45 Å and another with the GLU-197 amino acid residue with a bond distance of 1.9 Å. In addition, SER-198 is establishes H-bond with the imine nitrogen having a bond distance of 2.7 Å. In the same way, GLY-116 and GLY-117 from the oxyanion hole of the enzyme form amide π-stacked bonding with the aromatic ring of the ligand with the bond distance of 4.3 Å. Two more hydrogen bond associations were shown by the ligand with HIS-348 and GLY-439 with bond lengths 3.4 Å and 3.3 Å, respectively. TRP-82 from the anionic site contributes to the stabilization of the ligand inside the active pocket of the enzyme by establishing two π–π stacked interactions with the π–electrons of the aromatic portion of the inhibitor (4.2 Å and 5.4 Å). Similarly, π–π T-shaped interactions were formed by PHE-329 from the anionic site (5.3 Å). ALA-328 established pi–alkyl interactions (4.2 Å) with the benzene rings of the compound 13, thus further enhancing its stabilization inside the active site of the enzyme.
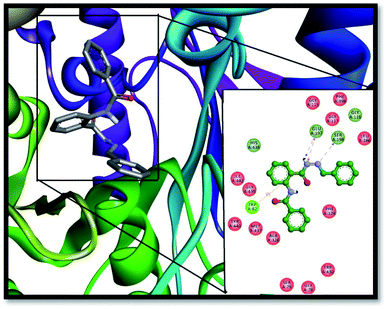 |
| | Fig. 7 Interactions of compound 13 with BChE; interactions of the compound displayed in the box are with specific amino acid residues of the enzyme. Enzyme is exemplified by 3D ribbon; the stick model is the lowest energy conformation of compound 13. | |
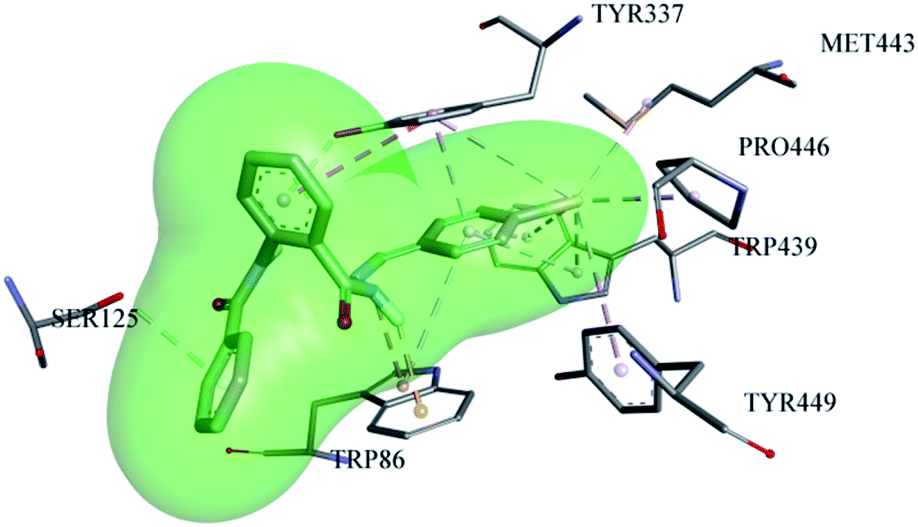
The most potent ligand (06) showed very good accommodation inside the active pocket of AChE by forming many important interactions, as shown in Fig. 8. Compound 06 shows conserved interactions with the choline binding site of the AChE enzyme, similar to that of compound 13, i.e., it makes H-bond interactions (3.32 Å) with the hydroxyl group of TYR-337 utilizing its π-electron cloud from the aromatic system. Similarly, TRP-86 also established H-bond (2.5 Å) with the hydrogen atom of the –NH group from the hydrazone part of the molecule. SER-125 also makes H-bond with the π-electrons of the benzamide part of the ligand with the bond distance of 3.98 Å. Likewise, TRP-86 contributes to the stabilization of the ligand inside the active pocket of AChE by making electrostatic π-cation interaction (3.25 Å) with the nitrogen atom of the –NH group from the hydrazone part along with one π-alkyl interaction (4.87 Å) with the aromatic portion of the benzylidene ring. Similarly, TYR-337 forms two π-alkyl interactions with bond distances of 3.5 Å and 4.9 Å along with one π–π T-shaped interaction (5.4 Å) with the π-electron cloud of the benzohydrazide ring. TRP-439 also plays its role in the stabilization of ligand 06 inside the active pocket by forming four π-alkyl interactions, two with the bromo substituent with distances of 3.5 Å and 4.1 Å, along with two π-alkyl interactions (4.6 Å and 4.8 Å) with the benzylidene ring of the ligand. The additional contribution of the bromo substituent attached at the para position of the benzylidene ring of ligand 06 is also significant because it makes two more hydrophobic alkyl interactions with MET-443 (4.2 Å) and PRO-446 (3.7 Å) along with one hydrophobic π-alkyl interaction with TYR-449 (5.2 Å).
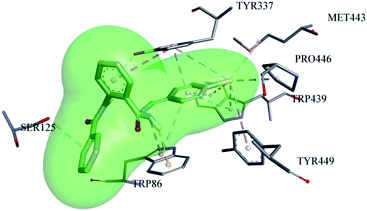 |
| | Fig. 8 Putative binding approaches of compound 06 inside the AChE enzyme. | |
Likewise, ligand 06 finds its best accommodation inside the active pocket of the BChE enzyme by making many important binding associations with the amino acid residues of the enzyme, as shown in Fig. 9. It forms conserved H-bond interactions with SER-198 from the catalytic triad (2.6 Å) and GLU-197 (2.1 Å) with the hydrogen atom from the NH–CO group of the hydrazone linkage similar to inhibitor 13. Similarly, GLY-439 established H-bond interaction with the oxygen atom of the carbonyl group of the hydrazone linkage (3.6 Å). TRP-82 from the anionic site established π–π T-shaped interaction (4.3 Å) with the phenyl ring of the benzamide part of the inhibitor. The bromine substituent at the para position of the benzylidene ring also contributes to the stabilization of the ligand by forming π–alkyl interactions with LEU-286 (4.6 Å) and VAL-288 (4.9 Å) from acyl pocket of BChE along with one more π–alkyl bonding with TRP-231 (4.7 Å) amino acid residue. ALA-328 is also making its contribution by establishing π–alkyl interaction with aromatic portion of the compound 06 with a bond distance of 4.2 Å.
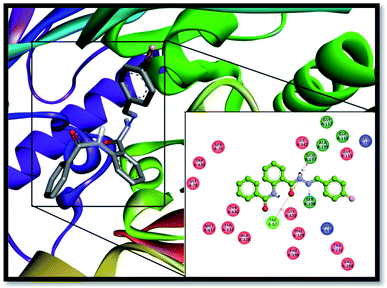 |
| | Fig. 9 Interactions of compound 06 with BChE; interactions of the compound displayed in the box are with specific amino acid residues of the enzyme. Enzyme is exemplified by the 3D ribbon; the stick model is the lowest energy conformation of the compound 06. | |
The binding modes of the known selective inhibitor (AChE), i.e., donepezil has been reported previously.41,42 The redocking of the previously known inhibitor, i.e., donepezil, was performed against the active pocket of AChE to validate the docking results and is presented in Fig. S1.† The most potent inhibitors 13 and 06 have shown hydrophobic binding interactions with TRP-86 and TYR-337 similar to the interactions shown by donepezil. The additional conserved binding interaction of compound 06 inside the AChE active pocket was with PHE-338. TYR-337 also establishes hydrogen bonding inside the AChE active pocket for compound 13, thereby further enhancing its stabilization inside AChE. Conclusively, molecular docking results have very well supported the good inhibition activities of the compounds against AChE and BChE enzymes in comparison to the standard taken, i.e., donepezil.
3.7. Density functional theory (DFT) analysis
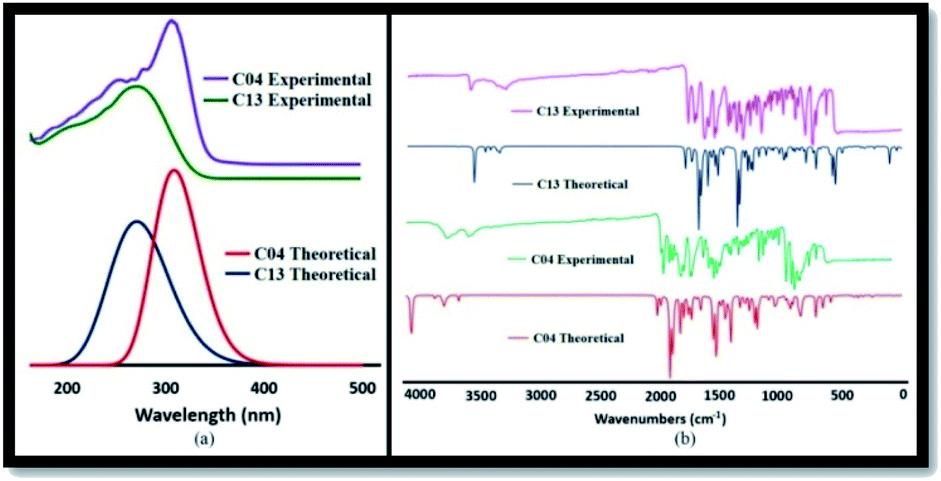
3.7.1. TD-DFT analysis for absorption spectra. The UV-Vis absorption spectra of all the derivatives (04–13) are explained in the experimental characterization. However, the theoretical spectra of the representative compounds 04 ((2-benzamido) benzohydrazide) and 13 (Schiff base derivative of 04) were calculated by the TD-DFT-B3LYP/321g method. The calculated theoretical spectra of these compounds were compared with the experimental spectra and represented in Fig. 9(a). Compound 04 has shown peaks below 300 nm, whereas 13 has shown absorption peak in the range of 300–350 nm, which represents the π → π* type transitions due to the conjugation of the azomethine (CN) groups with the phenyl ring. The red shift observed for 13 was associated with the attachment of different substituent to the azomethine group.
3.7.2. Vibrational analysis. Theoretical vibrational calculations were performed through DFT-B3LYP/321g for the representative compounds 04 ((2-benzamido) benzohydrazide) and 13 (Schiff base derivative of 04) and compared with the experimental FTIR results (Fig. 10(a)). The appearance of the (HCN) signal at 1580–1610 cm−1 in the IR spectrum unambiguously authenticates the formation of compounds, which agrees well with the theoretical spectra as well. Compound 13 shows a significant peak for CO stretching frequencies in the range of 1630–1660 cm−1. Furthermore, the peaks for the N–H bond appeared in the range of 3200–3330 cm−1. In addition, the peaks for C–H stretching attached with the aromatic system appeared at 3030–3060 cm−1. The experimental FTIR spectra agree well with the calculated theoretical spectra, as shown in Fig. 10(b).
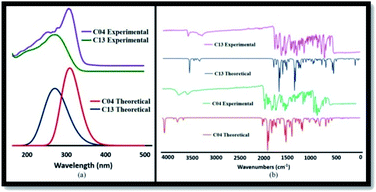 |
| | Fig. 10 (a) Experimental and TD-DFT-B3LYP/321g calculated absorption spectra of representative 04 and 13 compounds. (b) Experimental and DFT-B3LYP/321g-calculated IR spectra of the representative 04 and 13 compounds. | |
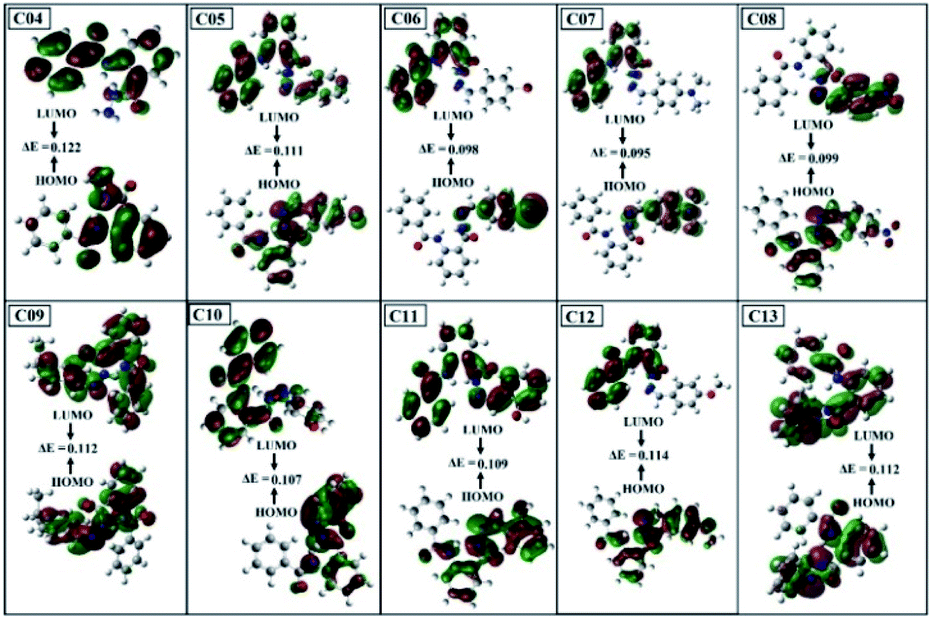
3.7.3. Frontier molecular orbitals (FMOs) analysis. The HOMO and LUMO are the electron λ donor and acceptor orbitals, and the energy gap between these orbitals indicate the stability and chemical reactivity characteristics of the compounds.43 The HOMO describes the ability to donate an electron and the LUMO indicates the capacity to accept the electron. Different functional groups in the compounds have the capability to change the positions and energy gap of HOMO–LUMO, which noticeably affects the charge–transfer transitions. This change in the energy band gap can be utilized by tuning the chemical reactivity characteristics of the compound. The B3LYP*-calculated values of the ELUMO, EHOMO, and EHOMO–ELUMO gap (ΔE) of the samples [04–13] are given in Table 6 and the participating frontier molecular orbitals are given in Fig. 11.
Table 6 DFT-B3LYP/321g calculated values of the ELUMO, EHOMO and EHOMO–ELUMO gap (ΔE) samples [C04–C13]
| Compounds |
ELUMO (Hartree) |
EHOMO (Hartree) |
ELUMO–EHOMO gap ΔE (Hartree) |
| 04 |
−0.1933 |
−0.3153 |
0.1220 |
| 05 |
−0.1931 |
−0.3039 |
0.1108 |
| 06 |
−0.1929 |
−0.2909 |
0.0980 |
| 07 |
−0.1926 |
−0.2871 |
0.0950 |
| 08 |
−0.2065 |
−0.3051 |
0.0986 |
| 09 |
−0.1985 |
−0.3101 |
0.1116 |
| 10 |
−0.1935 |
−0.3001 |
0.1066 |
| 11 |
−0.1934 |
−0.3033 |
0.1099 |
| 12 |
−0.1931 |
−0.3072 |
0.1141 |
| 13 |
−0.1993 |
−0.3106 |
0.1113 |
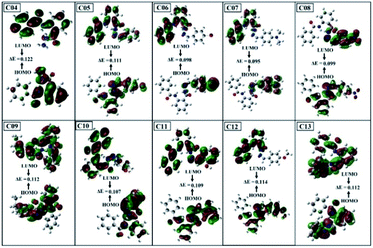 |
| | Fig. 11 DFT-B3LYP-321g-calculated frontier molecular orbitals (FMOs) of compounds 04–13. | |
3.7.4. Chemical reactivity parameters. The stability, electrical properties, and reactivity of any compound can be determined by the energy difference of the frontier molecular orbitals. The energies of HOMO and LUMO are used to gain insights into the promising global reactivity descriptors and are summarized in Table 7. Koopmans Theorem44 was used to find the electronegativity (x), chemical hardness (η), and chemical potential (μ), as given in eqn (4)–(6). IP (−EHOMO) is the ionization potential and EA (−ELUMO) is the electron affinity. The global softness (σ) can be evaluated using eqn (7).45 Parr et al.46 have given the electrophilicity index (ω), which is utilized to measure the electrophilic strength by eqn (8).47| |
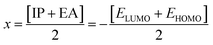 | (4) |
| |
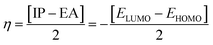 | (5) |
| |
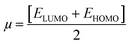 | (6) |
| |
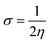 | (7) |
| |
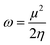 | (8) |
Table 7 The DFT-B3LYP*-calculated chemical reactivity parameters of the samples [04–13]
| Samples |
Electron affinity (EA) |
Ionization potential (IP) |
Electronegativity (x) |
Electrophilicity (ω) |
Chemical potential (μ) |
Global hardness (η) |
Global softness (σ) |
| 04 |
0.1933 |
0.3153 |
0.2543 |
0.5301 |
−0.2543 |
0.0610 |
8.1967 |
| 05 |
0.1931 |
0.3039 |
0.2485 |
0.5573 |
−0.2485 |
0.0554 |
9.0253 |
| 06 |
0.1929 |
0.2909 |
0.2419 |
0.5971 |
−0.2419 |
0.0490 |
10.204 |
| 07 |
0.1926 |
0.2871 |
0.23985 |
0.6088 |
−0.2399 |
0.0473 |
10.582 |
| 08 |
0.2065 |
0.3051 |
0.2558 |
0.6636 |
−0.2558 |
0.0493 |
10.1412 |
| 09 |
0.1985 |
0.3101 |
0.2543 |
0.5795 |
−0.2543 |
0.0558 |
8.9606 |
| 10 |
0.1935 |
0.3001 |
0.2468 |
0.5714 |
−0.2468 |
0.0533 |
9.3809 |
| 11 |
0.1934 |
0.3033 |
0.24835 |
0.5612 |
−0.2484 |
0.0549 |
9.0992 |
| 12 |
0.1931 |
0.3072 |
0.25015 |
0.5484 |
−0.2502 |
0.0571 |
8.7642 |
| 13 |
0.1993 |
0.3106 |
0.25495 |
0.5840 |
−0.2549 |
0.0557 |
8.9847 |
The chemical reactivity of a compound specifies its capability to be stabilized by attracting the charge from the environment.48 The chemical reactivity characteristics of the compounds can be elucidated by the parameters, i.e., ionization potential, electron affinity, electronegativity, electrophilicity, chemical potential, global hardness, and global softness. These parameters were calculated for all compounds (04–13) and are reported in Table 7.
The small energy gap specifies the compound to be soft and reactive while the large energy gap indicates that the molecule is hard and does not react easily.49 Compound 06 has shown greater softness characteristics (Table 7), making it the most reactive, which can be attributed to the inclusion of –Br, the functional group in its structure. The high value of global hardness compared to the softness proposed the less reactivity and high stability of the compounds, as given in Table 7. From the calculated values of chemical potential and global hardness of the synthesized compounds, the most thermally and kinetically stable compound was found to be 04. The calculated negative value of the chemical potential showed that it is comparatively easy for the compounds to gain electrons from the environment. However, the compounds found to have greater charge transfer ability and most of the compounds are more reactive. On the basis of the chemical reactivity characteristics, these compounds can be proved to be efficient antifungal and antibacterial candidates.
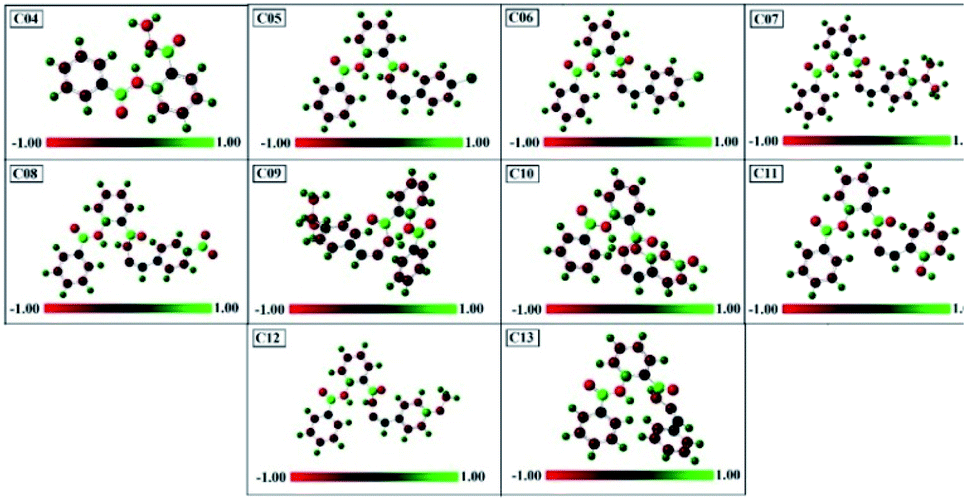
3.7.5. Mulliken atomic charges analysis. Mulliken atomic charges of compounds 04–13 were calculated using DFT/B3LYP(3-21G). The histogram of Mulliken atomic charges is represented in Fig. 12.
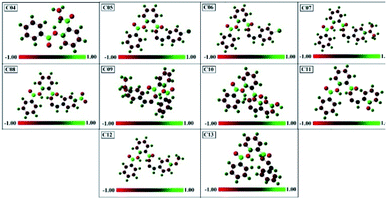 |
| | Fig. 12 DFT-B3LYP/321g-calculated Mulliken's atomic charges of compounds 04–13. | |
These charges have a very important role in quantum chemical approach and also affect the polarizability, dipole moment, and electronic structure of the molecule, and hence the reactivity of the biological targets. The measured values of the atomic charges showed the presence of highly electronegative atoms such as O, N, Cl, and Br. From this, it is clear that the positive charge is distributed on the carbon atoms, while all types of hydrogen atoms possess positive charge.
4 Conclusion
In this study, a series of 2-(benzamido) benzohydrazide derivatives was designed, synthesized, characterized, and evaluated for their inhibitory activity against the AChE and BChE enzymes. By the combination of biochemical investigation and computational analysis, we discovered two potent AChE and BChE inhibitors as compound 06 (IC50 = 0.09 μM for AChE and 0.14 μM for BChE) with the presence of the bromine substituent at the para position in the benzylidene derivative and compound 13 (IC50 = 0.11 μM for AChE and 0.10 for BChE). The BChE selective inhibition property was shown by compound 11 (IC50 = 0.12 μM for BChE) with the hydroxyl group as the substituent. It was found that 2-(benzamido) benzohydrazide derivatives show more inhibition potential against BChE as compared to AChE. The experimental results were confirmed by molecular docking simulations, which showed a conserved interaction pattern giving rise to similar interaction energy values. The cytotoxicity studies proved the safe nature of the synthesized compounds as anti-Alzheimer agents. In conclusion, by compiling the information obtained during this study, compounds 06 and 13 can be considered as effective candidates toward the design and development of drugs against Alzheimer's disease.
Author contributions
Naghmana Kausar: methodology, data curation, software, writing-original draft preparation. Shahzad Murtaza.: supervision. conceptualization, Muhammad Nadeem Arshad: crystal analysis. Rahman Shah Zaib Saleem: data curation. Abdullah M. Asiri: crystal analysis. Samia Kausar: DFT studies. Ataf Ali Altaf: DFT studies. Adina Tatheer: reviewing and editing. Ashraf Y. Elnaggar: biological evaluation, Salah M. El-Bahy: data curation, software.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by the Taif Researchers Supporting Project (TURSP-2020/32), Taif University, Taif, Saudi Arabia.
References
- A. Balkis, K. Tran, Y. Z. Lee and K. Ng, J. Agric. Sci., 2015, 7, 26–35 Search PubMed
 .
. - R. S. Wilson, E. Segawa, P. A. Boyle, S. E. Anagnos, L. P. Hizel and D. A. Bennett, Psychol. Aging, 2012, 27(4), 1008 Search PubMed .
- W. W. Barker, C. A. Luis, A. Kashuba, M. Luis, D. G. Harwood, D. Loewenstein, C. Waters, P. Jimison, E. Shepherd, S. Sevush and N. Graff-Radford, Alzheimer Dis. Assoc. Disord., 2002, 16(4), 203–212 CrossRef PubMed .
- Alzheimer’s Association, Alzheimers Dement., 2017, 13, 325–373, DOI:10.1016/j.jalz.2017.02.001 .
- C. Qiu, M. Kivipelto and E. V. Strauss, Dialogues clin. Neurosci., 2009, 11(2), 111–128 Search PubMed .
- S. Sinha and I. Lieberburg, Proc. Natl. Acad. Sci. U.S.A., 1999, 96, 11049–11053, DOI:10.1073/pnas.96.20.11049 .
- E. K. Per, Age Ageing, 1980, 9(1), 1–8 CrossRef PubMed .
- R. T. Bartus, R. Dean, B. Beer and A. S. Lippa, Science, 1982, 217(4558), 408–414 CrossRef CAS PubMed .
- M. H. Yan, X. Wang and X. Zhu, Free Radic. Biol,. Med., 2013, 62, 90–101 CrossRef CAS PubMed .
- J. M. Riphagen, I. H. Ramakers, W. M. Freeze, L. H. Pagen, B. J. Hanseeuw, M. M. Verbeek, F. R. Verhey and H. I. Jacobs, Neurobiol. Aging, 2020, 85, 96–103 CrossRef PubMed .
- A. Quincozes-Santos, L. D. Bobermin, A. C. Tramontina, K. M. Wartchow, B. Tagliari, D. O. Souza, A. T. Wyse and C. A. Gonçalves, Toxicol. in Vitro, 2014, 28(4), 544–551 CrossRef CAS PubMed .
- S. O. Bachurin, E. F. Shevtsova, G. F. Makhaeva, V. V. Grigoriev, N. P. Boltneva, N. V. Kovaleva, S. V. Lushchekina, P. N. Shevtsov, M. E. Neganova, O. M. Redkozubova and E. V. Bovina, Sci. Rep., 2017, 7(1), 1–5 CrossRef PubMed .
- E. J. Mufson, S. E. Counts, S. E. Perez and S. D. Ginsberg, Expert Rev. Neurother., 2008, 8(11), 1703–1718 CrossRef CAS PubMed .
- P. Anand and B. Singh, Arch Pharm. Res., 2013, 36(4), 375–399 CrossRef CAS PubMed .
- G. Mushtaq, N. H. Greig, J. A. Khan and M. A. Kamal, CNS Neurol. Disord.: Drug Targets, 2014, 13(8), 1432–1439 CrossRef CAS PubMed .
- M. Mesulam, A. Guillozet, P. Shaw and B. Quinn, Neurobiol. Dis., 2002, 9(1), 88–93 CrossRef CAS PubMed .
- M. Bazelyansky, E. Robey and J. F. Kirsch, Biochemistry, 1986, 25(1), 125–130 CrossRef CAS PubMed .
- A. Deep, S. Jain, P. C. Sharma, P. Phogat and M. Malhotra, Med. Chem. Res., 2012, 21(8), 1652–1659 CrossRef CAS .
- R. Manivannan and R. Shopna, Nat. Prod. Sci., 2017, 23(1), 69–74 CrossRef CAS .
- A. Katiyar, M. Hegde, S. Kumar, V. Gopalakrishnan, K. D. Bhatelia, K. Ananthaswamy, S. A. Ramareddy, E. D. Clercq, B. Choudhary, D. Schols and S. C. Raghavan, RSC Adv., 2015, 5(56), 45492–45501 RSC .
- A. Balachandran, R. Wong, P. Stoilov, S. Pan, B. Blencowe, P. Cheung, P. R. Harrigan and A. Cochrane, Retrovirology, 2017, 14(1), 1–21 CrossRef PubMed .
- V. B. Tatipamula and G. S. Vedula, J. Serbian Chem. Soc., 2019, 84(6), 555–562 CrossRef .
- M. U. Nisa, M. A. Munawar, A. Iqbal, A. Ahmed, M. Ashraf, A. G. Qurra-tul-Ann and M. A. Khan, Eur. J. Med. Chem., 2017, 138, 396–406 CrossRef CAS PubMed .
- C. Derabli, H. Boulebd, A. B. Abdelwahab, C. Boucheraine, S. Zerrouki, C. Bensouici, G. Kirsch, R. Boulcina and A. Debache, J. Mol. Struct., 2020, 1209, 127902 CrossRef CAS .
- N. Kausar, S. Muratza, M. A. Raza, H. Rafique, M. N. Arshad, A. A. Altaf, A. M. Asiri, S. S. Shafqat and S. R. Shafqat, J. Mol. Struct., 2019, 1185, 8–20 CrossRef CAS .
- N. Kausar, S. Murtaza, M. N. Arshad, R. Rashid, A. M. Asiri, N. Javid, M. H. Asim and Z. Ashraf, J. Mol. Struct., 2020, 1210, 128042 CrossRef CAS .
- N. Kausar, S. Murtaza, M. N. Arshad, R. Munir, R. S. Saleem, H. Rafique and A. Tawab, J. Mol. Struct., 2021, 1244, 130983 CrossRef CAS .
- C. P. Agilent. Agilent technologies, Yarnton, England, 2012 Search PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71(1), 3–8 CrossRef PubMed .
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45(4), 849–854 CrossRef CAS .
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65(2), 148–155 CrossRef CAS PubMed .
- G. L. Ellman, K. D. Courtney, J. V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7(2), 88–95 CrossRef CAS PubMed .
- M. S. Blois, Nature, 1958, 181(4617), 1199–1200 CrossRef CAS .
- A. Vetter, G. Perera, K. Leithner, G. Klima and A. Bernkop-Schnürch, Eur. J. Pharm. Sci., 2010, 41, 489–497 CrossRef CAS PubMed .
- K. Iftikhar, S. Murtaza, N. Kausar, A. Abbas and M. N. Tahir, Acta Pol. Pharm., 2018, 75(2), 385–396 CAS .
-
(a) M. N. Arshad, M. M. Rahman, A. M. Asiri, T. R. Sobahi and S. H. Yu, RSC Adv., 2015, 5(99), 81275–81281 RSC ;
(b) M. M. Hussain, M. M. Rahman, M. N. Arshad and A. M. Asiri, ChemistrySelect, 2017, 2(24), 7455–7464 CrossRef CAS .
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34(15), 1555–1573 CrossRef CAS .
- W. Brand-Williams, M. E. Cuvelier and C. L. Berset, LWT-Food sci. Technol., 1995, 28(1), 25–30 CrossRef CAS .
- D. Amic, D. Davidovic-Amic, D. Beslo, V. Rastija, B. Lucic and N. Trinajstic, Curr. Med. Chem., 2007, 14(7), 827–845 CrossRef CAS PubMed .
- J. O'brien, I. Wilson, T. Orton and F. Pognan, Eur. J. biochem., 2000, 267(17), 5421–5426 CrossRef PubMed .
- M. Makarian, M. Gonzalez, S. M. Salvador, S. Lorzadeh, P. K. Hudson and S. Pecic, J. Mol. Struct., 2022, 1247, 131425 CrossRef CAS .
- S. Pandey and B. K. Singh, Curr. Comput-aided Drug Des., 2020, 16(1), 54–72 CrossRef CAS PubMed .
- S. H. Sumrra, S. Kausar, M. A. Raza, M. Zubair, M. N. Zafar, M. A. Nadeem, E. U. Mughal, Z. H. Chohan, F. Mushtaq and U. Rashid, J. Mol. Struct., 2018, 1168, 202–211 CrossRef CAS .
- V. Balachandran, A. Lakshmi and A. Janaki, J. Mol. Struct., 2012, 1013, 75–85 CrossRef CAS .
- Y. R. Sharma, Elementary organic spectroscopy. S. Chand Publishing, 2007 Search PubMed .
- R. G. Parr, L. V. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121(9), 1922–1924 CrossRef CAS .
- V. Arjunan, I. Saravanan, P. Ravindran and S. Mohan, Spectrochim. Acta, Part A, 2009, 74(3), 642–649 CrossRef CAS PubMed .
- J. I. Aihara, J. Phys. Chem. A, 1999, 103(37), 7487–7495 CrossRef CAS .
- D. M. Suresh, M. Amalanathan, S. Sebastian, D. Sajan, I. H. Joe, V. B. Jothy and I. Nemec, Spectrochim. Acta, Part A, 2013, 115, 595–602 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2108057. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra07221h |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shahzad Murtaza*a,
Muhammad Nadeem Arshadbc,
Rahman Shah Zaib Saleem
*a,
Shahzad Murtaza*a,
Muhammad Nadeem Arshadbc,
Rahman Shah Zaib Saleem