
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingular value decomposition analysis of the electron density changes occurring upon electrostatic polarization of water
Hajime Torii *ab
*ab
aDepartment of Applied Chemistry and Biochemical Engineering, Faculty of Engineering, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu 432-8561, Japan. E-mail: torii.hajime@shizuoka.ac.jp; Fax: +81-53-478-1624; Tel: +81-53-478-1624
bDepartment of Optoelectronics and Nanostructure Science, Graduate School of Science and Technology, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu 432-8561, Japan
First published on 19th January 2022
Abstract
In-depth elucidation of how molecules are electrically polarized would be one key factor for understanding the properties of those molecules under various thermodynamic and/or spatial conditions. Here this problem is tackled for the case of hydrogen-bonded water by conducting singular value decomposition of the electron density changes that occur upon electrostatic polarization. It is shown that all those electron density changes are approximately described as linear combinations of ten orthonormal basis “vectors”. One main component is the interatomic charge transfer through each OH bond, while some others are characterized as the atomic dipolar polarizations, meaning that both of these components are important for the electrostatic polarization of water. The interaction parameters that reasonably well reproduce the induced dipole moments are derived, which indicate the extent of mixing of the two components in electrostatic polarization.
1. Introduction
Molecules in condensed phases are electrically polarized more or less by intermolecular interactions according to the polarity and hydrogen-bonding conditions of the surrounding environment. Such polarization often profoundly affects the system's properties. In the case of water, it is well-known that the dielectric constant changes significantly with temperature and pressure in the thermodynamic region near the critical point,1 resulting in continuously varying solubility of a wide range of solutes2 and thus extensive utility as a medium for various chemical reactions.3 It has been recognized that, to reproduce this situation computationally in molecular dynamics (MD) simulations, it is required to treat the molecules' electric polarization explicitly;4–7 fixed-charge models that include the effects of polarization in an average way are not sufficient for this purpose. Similar situations arise for water molecules on material interfaces or confined in small spaces.8,9There are many ways that have been proposed up to now to explicitly treat electric polarization of water. They are roughly categorized as (1) atomic dipolar polarization (ADP) model,10–20 (2) Drude oscillator (or shell) model,21–24 and (3) fluctuating charge (FC) model.25–28 These are different in the representation of molecular electric polarization; the polarization is completely or essentially confined within each atom with the atomic partial charges being invariant in the ADP and Drude oscillator models, while it is represented only by the interatomic charge transfer (CT) within a molecule without any atomic polarization incorporated in the FC model. In other words, the electronic responses supposed in these models are totally different. It is thus expected that these models have different effects on short- and medium-ranged intermolecular interactions, even though they may all similarly be effective in reproducing the molecular induced dipole. Recently a few combined models that take into account both the atomic dipolar polarizabilities and interatomic CT have been proposed,29–32 but the relative importance of these two factors, which is essential for correct modeling, is not sufficiently well clarified. Although it somewhat depends on the way of defining the boundaries of atoms in a molecule, analysis in this relation will be of great help in deepening our understanding on the polarization-induced behavior of electrons based on our chemical intuition. With respect to the interaction energies, inclusion of the polarization effects with many-body potential functions19,33,34 is also a powerful way, but explicit analysis of the behavior of electrons (as charged particles) is needed for better understanding on observable electric properties such as a dipole moment and infrared intensities.
In the present study, this problem is tackled by singular value decomposition (SVD) analysis35 of the electron density changes occurring upon electrostatic polarization. The electron density changes are digitally expressed as arrays of the values at sufficiently fine three-dimensional grid points, and those obtained in a number of different electrostatic situations may be analyzed statistically. Aside from a few applications to classical or quantum dynamics simulations and to spectral analysis for the purpose of decomposing a complex high-dimensional object into some low-dimensional components,36–41 SVD has been extensively used for extracting important factors in image processing,42–44 so that it is considered to be a suitable method of analysis in the present context when each array of electron density changes (calculated for each specified configuration) is regarded as an image. However, unlike usual image processing, each array of electron density changes has three (x, y, and z) spatial dimensions, so that the preferable number of grid points to represent them tends to be large. The present study shows how SVD is practically effective in analyzing the polarization-induced electron density changes.
2. Computational procedure
2.1. Calculations of electron density changes upon electrostatic polarization
Calculations were done mainly for the hydrogen-bonded pairs of water molecules in the (water)90 clusters (this cluster size is not a magic number in a scientific sense but is a rough upper limit of the size that can be treated with the available computational resources). Firstly, 13 different initial structures of the (water)90 clusters were extracted45 from the snapshots of MD simulations carried out for the liquid system of 1024 molecules using the TIP4P46 potential model at the temperature of mp +25 K (257 K),47 and then the structures were optimized at the B3LYP/6-31+G(2df,p) level of density functional theory (DFT). Hydrogen-bonded pairs of molecules were taken with the criterion of r(O⋯H) ≤ 2.5 Å and θ(O⋯H–O) ≥ 90°. There were 2035 pairs of molecules in total taken in this way. Then, after transformation of coordinates using the molecular axes of each molecule in the pairs, and symmetry operation so that the hydrogen-bond counterpart is located in the specified xy quadrant (x > 0 and y > 0 for the hydrogen-bond donating molecules, x < 0 and y < 0 for the hydrogen-bond accepting molecules), with the molecular C2 axis being taken along the z direction, 500 mutually different (to the greatest extent) configurations each of hydrogen-bond donor and acceptor were extracted and were used in the subsequent analyses. The spatial distributions of such configurations are shown in Fig. 1.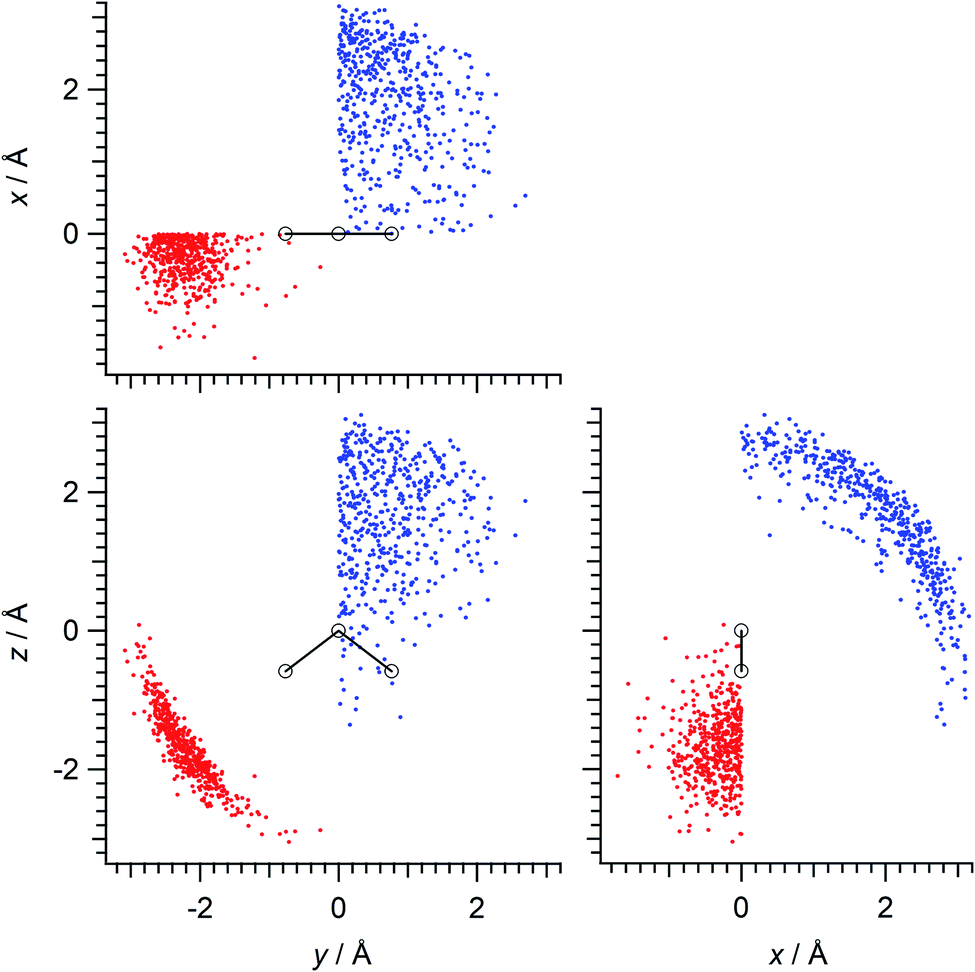 | ||
| Fig. 1 Spatial distributions of the hydrogen-bond donating (blue) and accepting (red) molecules (represented by their oxygen atoms) that are included in the SVD analysis as the source of electrostatic polarization of the water molecules (shown superimposed at the center) in the (water)90 clusters. | ||
The electron densities ρ(el)(r) of electrostatically polarized water molecules were calculated by replacing the hydrogen-bond donor or acceptor molecule with a set of atomic partial charges. Here, a fifth of the CHelpG48 charges (−0.1462764e for O and 0.0731382e for H) was employed to limit the systems within a weak perturbation regime. Then, the electron density without placing the atomic partial charges was subtracted to derive the changes upon electrostatic polarization. In calculating the electron densities, an augmented basis set of aug-cc-pVDZ49 was used for better representation of the polarization of hydrogen atoms. The evaluation points r of ρ(el)(r) were taken in a rectangular box of 10.0 × 11.6 × 10.6 Å3, so that (similarly to our previous studies of electron density analysis45,50–56) each boundary of the box is at least 5 Å from any atom in the molecule, with the interval of 0.02 Å. As a result, the number of the evaluation points r of each ρ(el)(r) is 154![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564011 (=501 × 581 × 531).
564011 (=501 × 581 × 531).
In addition, a supplementary analysis was carried out for a water molecule under dipolar electrostatic perturbation from the H atom side of the OH bond. Locating this H atom at the origin and taking the OH bond along the y axis (the O atom on the +y side), electric charges of −0.15e and +0.15e (which were made similar to those adopted above) were placed at r = ξ and ξ + 1.0 Å, respectively, of (0, −rcosθ, rsinθ) or (rsinθ, −rcosθ, 0), with ξ = 2.0, 2.4 or 2.8 Å and θ = 0° or ±30° (as a result, there are 15 configurations in total). In this case, the evaluation points r of ρ(el)(r) were taken in a rectangular box of 10.0 × 15.6 × 15.5 Å3 with the interval of 0.02 Å.
The DFT calculations described above were carried out by using the Gaussian 09 program,57 and the analyses before and after those DFT calculations were done with our own original programs.
2.2. Singular value decomposition analysis
Each of the polarization-induced electron density changes calculated in this way for the water molecules in the (water)90 clusters is regarded as a 154564011-dimensional vector (denoted as bj), and there are 1000 such vectors (j = 1, 2, …, 1000). The matrix B consisting of all these vectors (of the size of 154564011 × 1000) was subject to the SVD analysis described below.
In the SVD analysis,35 B is decomposed as
| B = UΛtV | (1) |
564011 and n = 1000), Λ is an m × n rectangular diagonal matrix [whose (non-negative) diagonal elements are denoted as λk (k = 1, 2, …, n)], and the upper left superscript t stands for transposing of the matrix. Since we have| tBB = (VtΛtU)(UΛtV) = V(tΛΛ)tV | (2) |
| (tBB)vk = λk2vk | (3) |
| BV = (UΛtV)V = UΛ | (4) |
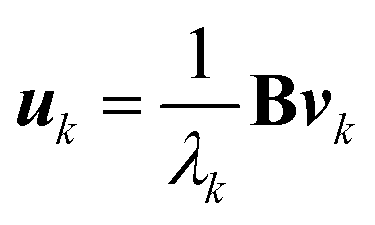 | (5) |
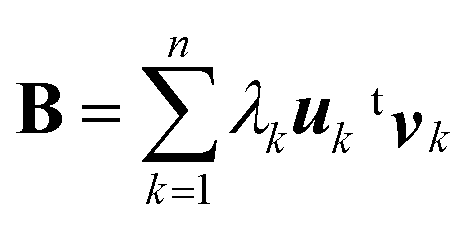 | (6) |
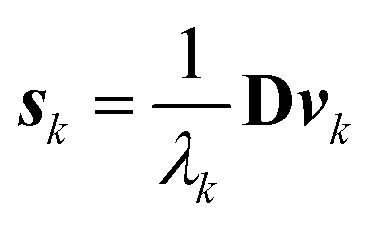 | (7) |
The polarization-induced electron density changes calculated in the supplementary analysis were treated in a similar way. In this case, SVD was conducted for fifteen 303634056-dimensional vectors bj (j = 1, 2, …, 15).
3. Results and discussion
3.1. SVD analysis of electron density changes
The eigenvalues corresponding to the first 50 eigenvectors obtained from the SVD analysis are shown in Fig. 2. It is seen that the eigenvalue decreases rather rapidly, by a factor of 100 in the first 6 eigenvectors, by another factor of 100 in the next 11 eigenvectors, and so on. This result suggests that it is possible to extract a set of a few important electron density changes that occur upon electrostatic polarization. The one- and two-dimensional plots of the electron density changes for the first and second eigenvectors are shown in Fig. 3. The electron density change corresponding to the first eigenvector shown in Fig. 3a mainly consists of the interatomic CT through the OH bond on the left-hand side, as clearly recognized from the large negative feature throughout the molecule of the one-dimensional running integral (light blue curve) along the y axis [the molecular in-plane axis taken perpendicular to the z (C2 molecular) axis] shown on the lower-left side. Inspecting the yz two-dimensional plot, it may also be recognized that the electron density change in the spatial region of the oxygen atom is not of a simple spherical character but rather of a quadrupolar nature. A similar quadrupolar nature of electron density modulation is also seen for the carbon atom of the C–Br bond involved in halogen bonding upon electrostatic polarization.55 In contrast, the electron density change corresponding to the second eigenvector shown in Fig. 3b mainly consists of the out-of-plane dipolar polarization of the oxygen atom, with a small anti-phase inner core polarization, but the interatomic CT through the OH bond on the right-hand side is also mixed to a certain extent.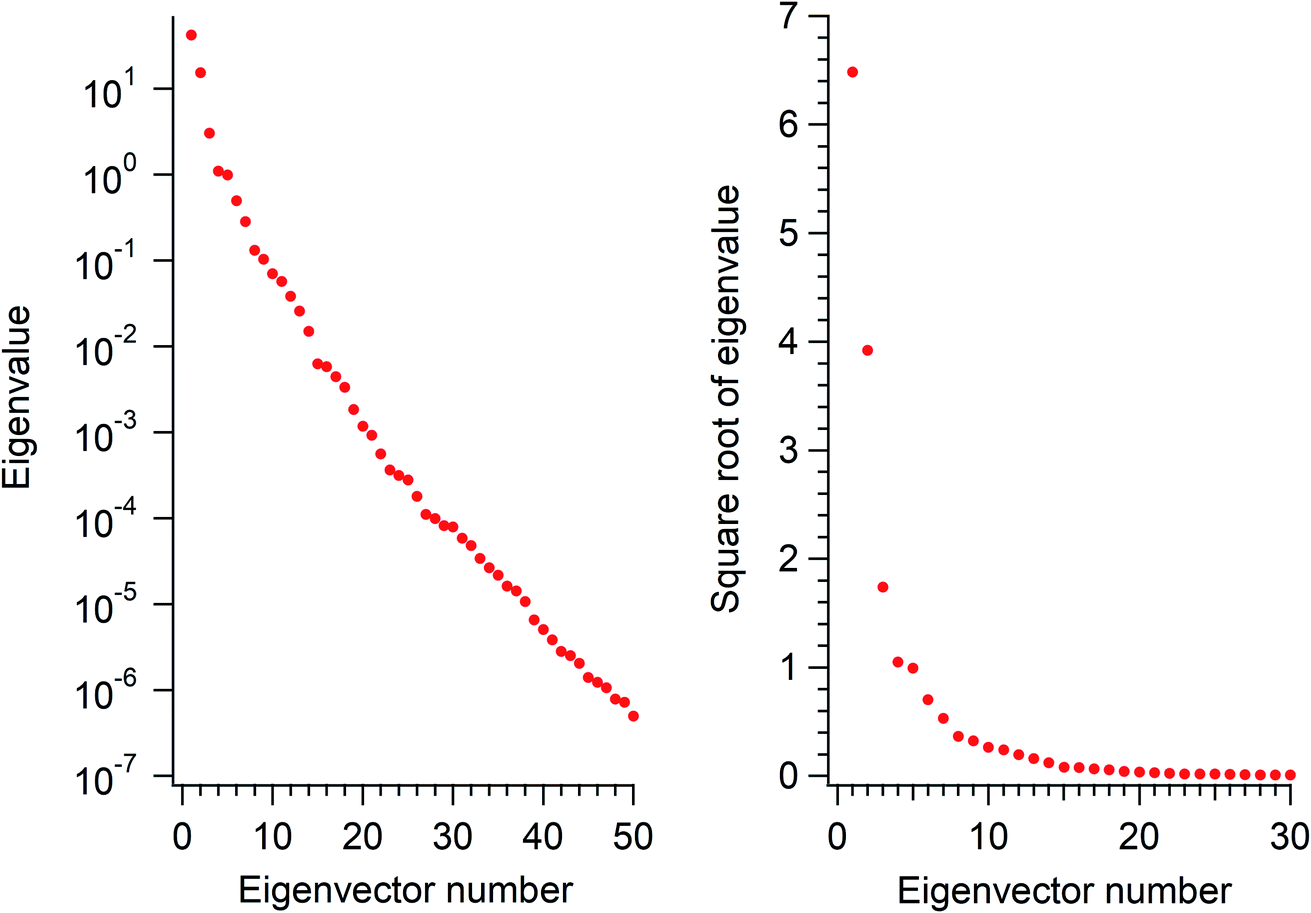 | ||
| Fig. 2 Eigenvalues (logarithmic scale, left panel) and their square roots (linear scale, right panel) obtained from the SVD analysis of the polarization-induced electron density changes of the water molecules in the (water)90 clusters. | ||
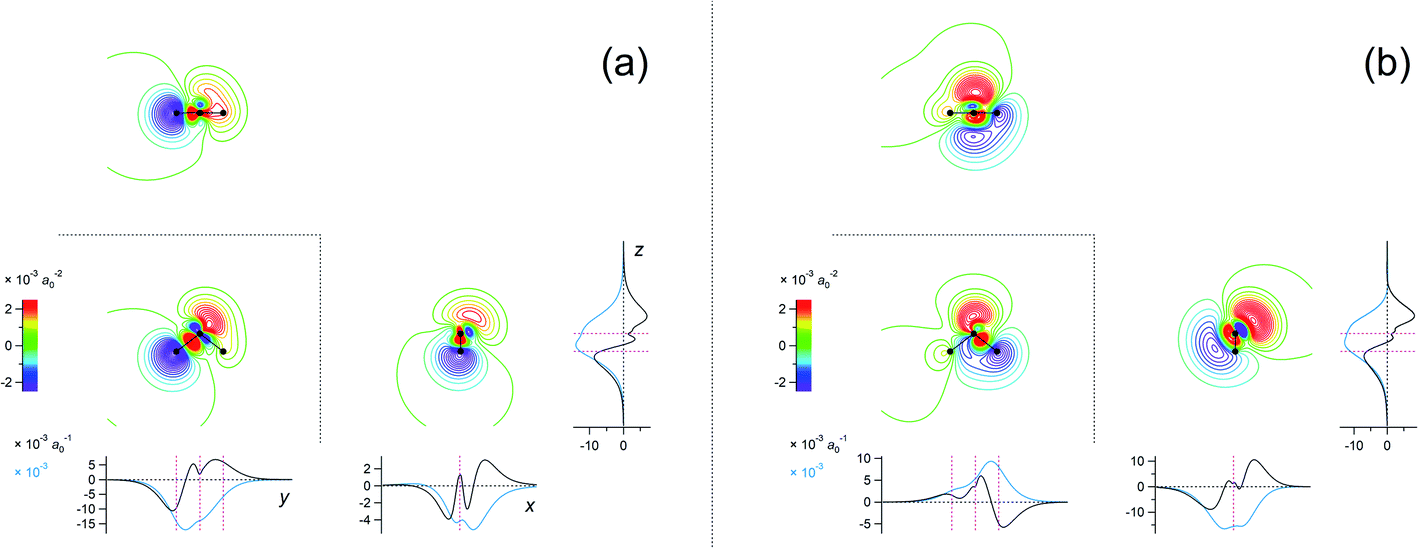 | ||
Fig. 3 Two-dimensional (xy, xz, and yz) contour plots of ∫dzρ(el)(r), ∫dyρ(el)(r), and ∫dxρ(el)(r), one-dimensional (x, y, and z) plots of 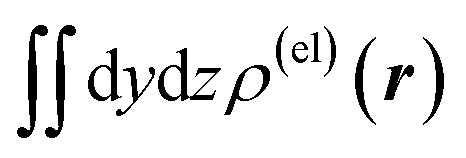 , , 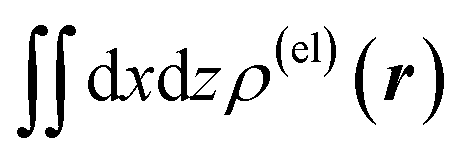 , and , and 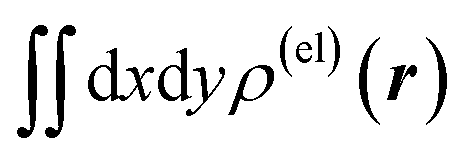 (black curves), and their running integrals along the respective axis (light blue curves) of the electron density changes corresponding to the (a) first and (b) second eigenvectors obtained from the SVD analysis. The contours in the two-dimensional plot are drawn with the interval of 0.36 × 10−3 a0−2 in the range from −9 × 10−3 a0−2 to 9 × 10−3 a0−2, with the color code shown on the left-hand side. The atomic positions of the central water molecule are indicated with black filled circles in the two-dimensional plots and with pink dotted vertical lines in the one-dimensional plots. The coordinate system is shown in Fig. 1. (black curves), and their running integrals along the respective axis (light blue curves) of the electron density changes corresponding to the (a) first and (b) second eigenvectors obtained from the SVD analysis. The contours in the two-dimensional plot are drawn with the interval of 0.36 × 10−3 a0−2 in the range from −9 × 10−3 a0−2 to 9 × 10−3 a0−2, with the color code shown on the left-hand side. The atomic positions of the central water molecule are indicated with black filled circles in the two-dimensional plots and with pink dotted vertical lines in the one-dimensional plots. The coordinate system is shown in Fig. 1. | ||
In fact, considering the structural symmetry of the water molecule, if there is an interatomic CT component through the OH bond on the left-hand side, there should also be the counterpart component through the OH bond on the right-hand side. Asymmetric distribution of the hydrogen-bond donors and acceptors adopted in the present study as shown in Fig. 1 is most suitable for obtaining localized vectors of electron density changes, but they should be partially symmetrized, handled additionally with symmetry operation, and made mutually orthogonal to obtain electron density changes that may be regarded as truly elementary components, which will be hereafter called “basis vectors”.
For example, the first basis vector (called basis vector 1a) was derived with the following procedure: (1) symmetrize the primary component obtained from the SVD analysis (shown in Fig. 3a) with respect to the yz plane; (2) conduct a symmetry operation on it with respect to the xz plane to obtain another vector; (3) calculate the normalized mutually orthogonal symmetry-related linear combinations of these two. The electron density changes thus obtained (the interatomic CT component for the OH bond on the left-hand side) is shown in Fig. 4a. The symmetry-related counterpart (not shown) is called basis vector 1b, which is the mirror image with respect to the xz plane.
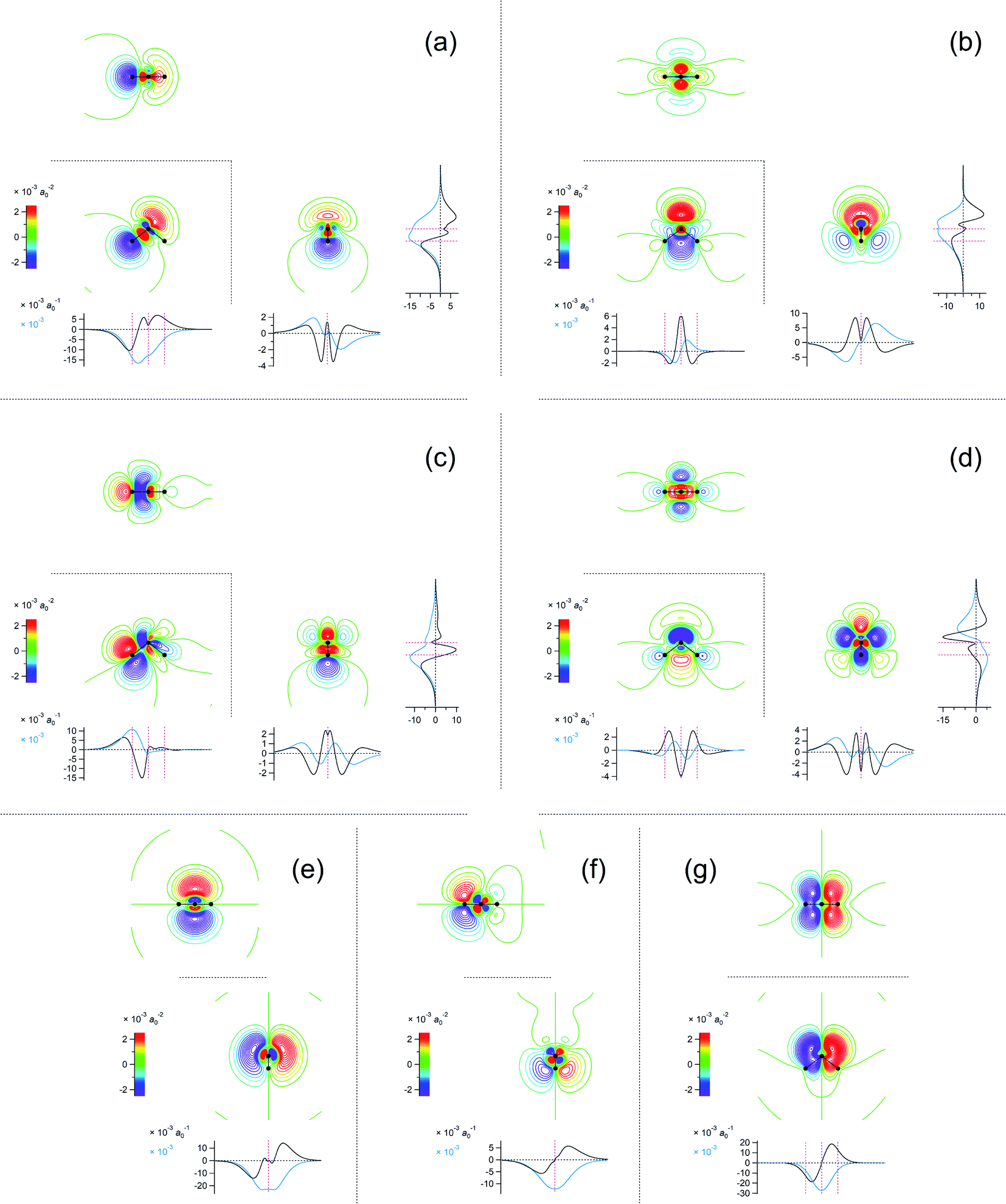 | ||
Fig. 4 Two-dimensional (xy, xz, and yz) contour plots of ∫dzρ(el)(r), ∫dyρ(el)(r), and ∫dxρ(el)(r), one-dimensional (x, y, and z) plots of 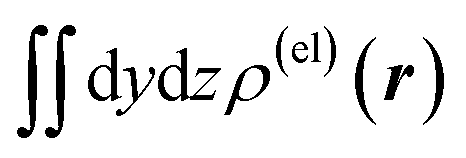 , , 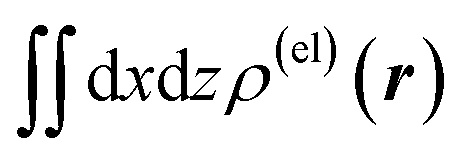 , and , and 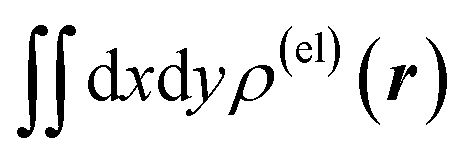 (black curves), and their running integrals along the respective axis (light blue curves) of the basis vectors of polarization-induced electron density changes of the water molecules in the (water)90 clusters: (a) vector 1a, (b) vector 3, (c) vector 6a, (d) vector 7, (e) vector 2, (f) vector 4a, and (g) vector 5 (some of the plots are omitted in the last three panels because of the symmetry). The contours in the two-dimensional plots are drawn with the interval of 0.36 × 10−3 a0−2 in the range from −14.4 × 10−3 a0−2 to 14.4 × 10−3 a0−2, with the color code shown on the left-hand side. The atomic positions of the central water molecule are indicated with black filled circles in the two-dimensional plots and with pink dotted vertical lines in the one-dimensional plots. The coordinate system is shown in Fig. 1. (black curves), and their running integrals along the respective axis (light blue curves) of the basis vectors of polarization-induced electron density changes of the water molecules in the (water)90 clusters: (a) vector 1a, (b) vector 3, (c) vector 6a, (d) vector 7, (e) vector 2, (f) vector 4a, and (g) vector 5 (some of the plots are omitted in the last three panels because of the symmetry). The contours in the two-dimensional plots are drawn with the interval of 0.36 × 10−3 a0−2 in the range from −14.4 × 10−3 a0−2 to 14.4 × 10−3 a0−2, with the color code shown on the left-hand side. The atomic positions of the central water molecule are indicated with black filled circles in the two-dimensional plots and with pink dotted vertical lines in the one-dimensional plots. The coordinate system is shown in Fig. 1. | ||
After extracting these basis vectors, projections of vectors uk onto the basis vectors were calculated and were subtracted from uk to represent the “remaining” components of the polarization-induced electron density changes (the resulting vectors, which are orthogonal to the basis vectors, are denoted as 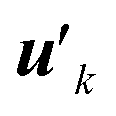 ), and the matrix of correlation among those vectors (denoted as tB′B′) was reconstructed as
), and the matrix of correlation among those vectors (denoted as tB′B′) was reconstructed as
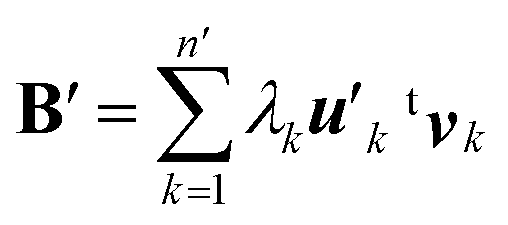 | (8) |
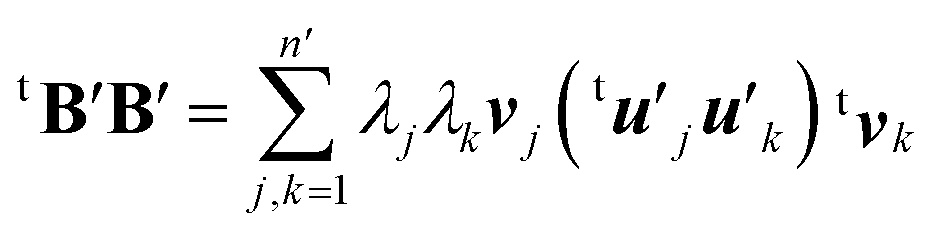 | (9) |
Here the summation was truncated to n′ = 32 (numbered from larger eigenvalues) as an approximation. In eqn (9), the scalar products 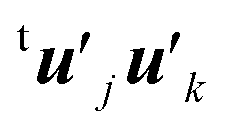 were calculated first for computational efficiency. Then, this tB′B′ was diagonalized to derive a new set of eigenvalues and eigenvectors. The vector of the largest eigenvalue this time was mainly related to the polarization of the O atom along the x axis, so that only symmetrization and normalization were conducted. The resultant basis vector (called basis vector 2) represents the truly out-of-plane (along the x axis) dipolar polarization component of the oxygen atom as shown in Fig. 4e.
were calculated first for computational efficiency. Then, this tB′B′ was diagonalized to derive a new set of eigenvalues and eigenvectors. The vector of the largest eigenvalue this time was mainly related to the polarization of the O atom along the x axis, so that only symmetrization and normalization were conducted. The resultant basis vector (called basis vector 2) represents the truly out-of-plane (along the x axis) dipolar polarization component of the oxygen atom as shown in Fig. 4e.
The remaining basis vectors (3, 4a, 5, 6a, and 7) shown in Fig. 4 and the symmetry-related ones (4b, and 6b) were obtained by repeating these procedures, which are summarized as a diagram in Chart 1. Owing to the asymmetric distribution of the hydrogen-bond donors and acceptors included in the present analysis as shown in Fig. 1, these localized and partially symmetrized basis vectors have successfully been obtained. The main characters of these may be described as rather skewed dipolar polarizations of the oxygen atom along the z and y axes (vectors 3 and 5), in-plane and out-of-plane vertical (to the bond) dipolar polarizations of one of the hydrogen atoms (vectors 6a and 4a) as well as their symmetry-related counterparts (vectors 6b and 4b), and a higher-order component with a relatively small induced dipole (vector 7). Judging from the eigenvalues of the SVD analysis, the set of these ten basis vectors covers nearly 99% of the electron density changes occurring upon electrostatic polarizations between hydrogen-bonded water molecules. In other words, the electron density changes occurring upon electrostatic polarization of water are nearly ten-dimensional, and the ten basis vectors described above (1a, 1b, 2, 3, 4a, 4b, 5, 6a, 6b, and 7) constitute an orthonormal basis of the ten-dimensional polarization “space”.
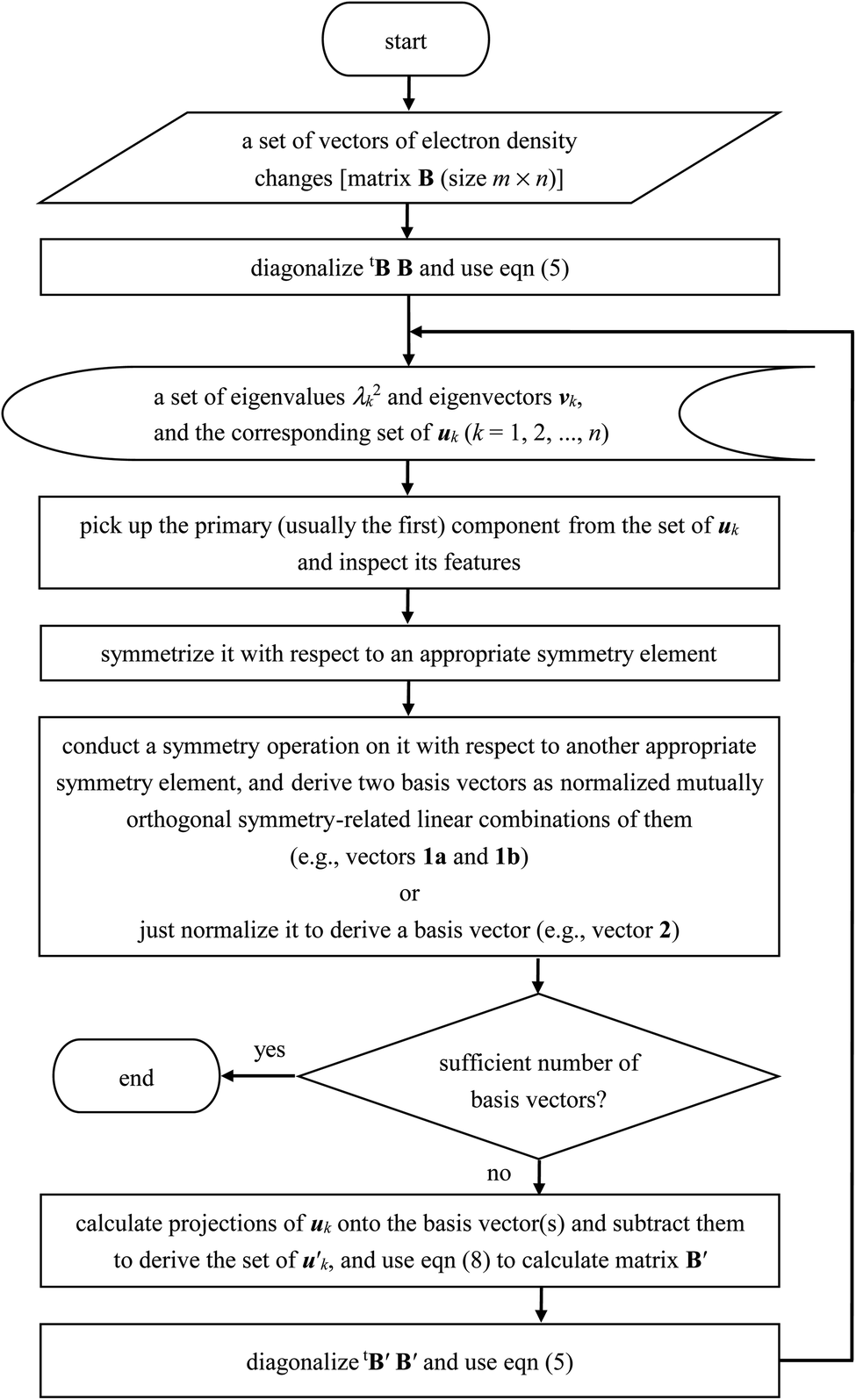 | ||
| Chart 1 Diagram of the procedures of the SVD analysis of electron density changes adopted in the present study. | ||
3.2. Electrostatic properties and induced dipole moments
The electrostatic properties and the induced dipole moments related to these basis vectors are summarized in Table 1. Then, how can we set up and derive the electrostatic interaction parameters that are usable in MD simulations? In the present case, the out-of-plane components are rather easy to handle; since there are three basis vectors (2, 4a, and 4b), with the main characters being described as the atomic dipolar polarizations, it would be most reasonable to place dipolar polarizabilities (αxx) on the three atomic sites and derive their values by linear algebra. With regard to the in-plane components, it is needed to identify the electrostatic properties related to the interatomic CT components.| Atoma/molecule | Propertyb | Electron density basis vector | ||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 2 | 3 | 4a | 5 | 6a | 7 | ||
| a H1 and H2 are located on the −y and +y side. The coordinate system is shown in Fig. 1.b In units of 10−2 Ehe−1 for the electrostatic potential (the difference from the value at the oxygen atom, ΔφOH), 10−2 Ehe−1a0−1 for the electric field (Ex, Ey, Ez), and 10−2 ea0 for the dipole moment (μx, μy, μz). | ||||||||
| Electrostatic properties | ||||||||
| O | Ex | 0 | −1.357 | 0 | −0.242 | 0 | 0 | 0 |
| Ey | −0.394 | 0 | 0 | 0 | −1.297 | 0.269 | 0 | |
| Ez | −0.508 | 0 | −1.152 | 0 | 0 | −0.116 | 0.028 | |
| H1 | ΔφOH | −1.812 | 0 | −0.445 | 0 | −0.234 | 0.502 | 0.037 |
| Ex | 0 | −0.553 | 0 | −2.275 | 0 | 0 | 0 | |
| Ey | −1.268 | 0 | 0.679 | 0 | 0.302 | 2.159 | −0.978 | |
| Ez | −0.966 | 0 | 0.085 | 0 | 0.783 | −2.676 | 0.063 | |
| H2 | ΔφOH | −0.037 | 0 | −0.445 | 0 | 0.234 | −0.115 | 0.037 |
| Ex | 0 | −0.553 | 0 | 0.121 | 0 | 0 | 0 | |
| Ey | −0.127 | 0 | −0.679 | 0 | 0.302 | 0.119 | 0.978 | |
| Ez | −0.273 | 0 | 0.085 | 0 | −0.783 | 0.134 | 0.063 | |
|
||||||||
| Induced dipole moment | ||||||||
| Molecule | μx | 0 | −8.201 | 0 | −4.312 | 0 | 0 | 0 |
| μy | −5.111 | 0 | 0 | 0 | −6.134 | 1.931 | 0 | |
| μz | −4.950 | 0 | −5.074 | 0 | 0 | −3.576 | −0.158 | |
For this purpose, a separate supplementary set of calculations was conducted for the situations of dipolar electrostatic perturbations mimicking the effect of a hydrogen-bond acceptor as described in the last part of Section 2.1. The result is shown in Fig. 5. It is clearly seen that, integrating along the two directions perpendicular to the OH bond on the left-hand side (to derive the one-dimensional plot along the y axis in this figure), the electron density changes obtained for the electrostatic perturbations with different angular settings are nearly invariant after scaling with the electrostatic potential difference between the H and O atoms. As discussed above in relation to the results shown in Fig. 3a and 4a, the component shown in Fig. 5 is characterized as the interatomic CT through the OH bond. Thus, one may conclude that it is the electrostatic potential difference between the H and O atoms rather than the electric field on the H atom along the OH bond that is most effective for the interatomic CT components, in a way similar to the through-bond polarization discussed for halogen-bonding systems55 and for hydrogen fluoride,56 and in accord with the idea of the charge response kernel.58,59
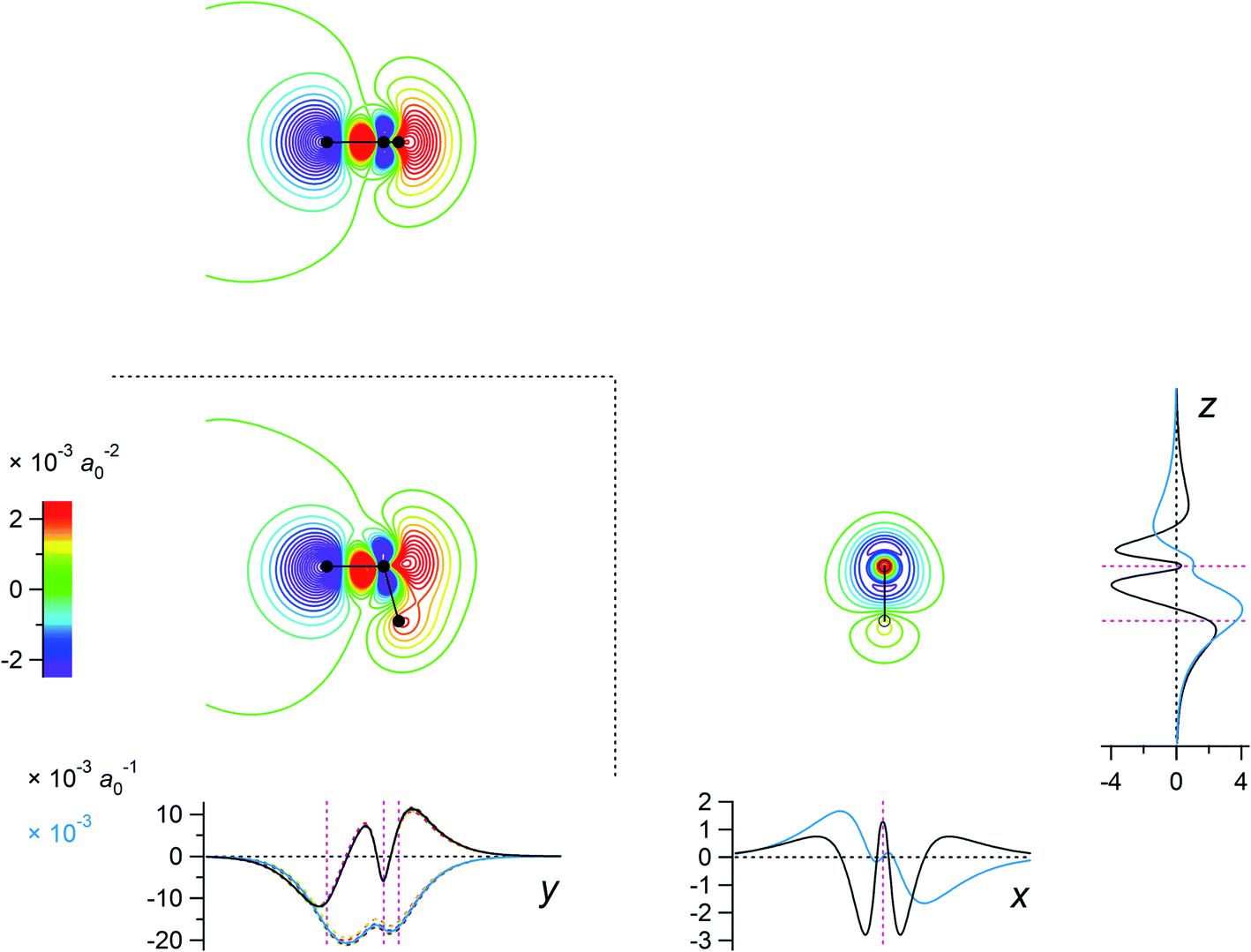 | ||
Fig. 5 Two-dimensional (xy, xz, and yz) contour plots of ∫dzρ(el)(r), ∫dyρ(el)(r), and ∫dxρ(el)(r), one-dimensional (x, y, and z) plots of 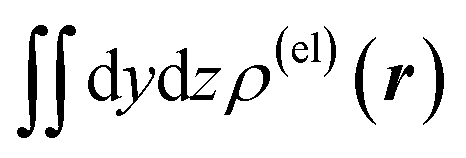 , , 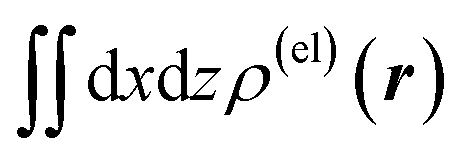 , and , and 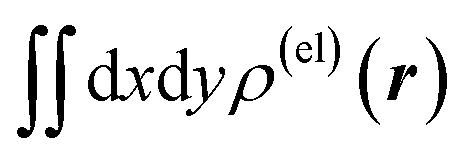 (black curves), and their running integrals along the respective axis (light blue curves) of the primary component obtained from the supplementary SVD analysis conducted for the electron density changes induced on a water molecule by the dipolar electrostatic perturbation mimicking the effect of a hydrogen-bond acceptor [i.e., from the left-hand (−y) side in the figure, as described in the last part of Section 2.1]. In the one-dimensional plot on the lower-left side (along the y axis), the plots of some important elements [with ξ = 2.0, 2.4 and 2.8 Å and θ = 0° (red, purple, and dark red dotted lines), and with ξ = 2.0 Å and θ = ±30° (yellow, green, and blue dotted lines)] included in the SVD analysis are overlapped with scaling based on the electrostatic potential difference between the primarily perturbed H and O atoms [located along the y (horizontal) axis]. The contours in the two-dimensional plot are drawn with the interval of 0.36 × 10−3 a0−2 in the range from −9 × 10−3 a0−2 to 9 × 10−3 a0−2, with the color code shown on the left-hand side. The atomic positions of the central water molecule are indicated with black filled circles in the two-dimensional plots and with pink dotted vertical lines in the one-dimensional plots. (black curves), and their running integrals along the respective axis (light blue curves) of the primary component obtained from the supplementary SVD analysis conducted for the electron density changes induced on a water molecule by the dipolar electrostatic perturbation mimicking the effect of a hydrogen-bond acceptor [i.e., from the left-hand (−y) side in the figure, as described in the last part of Section 2.1]. In the one-dimensional plot on the lower-left side (along the y axis), the plots of some important elements [with ξ = 2.0, 2.4 and 2.8 Å and θ = 0° (red, purple, and dark red dotted lines), and with ξ = 2.0 Å and θ = ±30° (yellow, green, and blue dotted lines)] included in the SVD analysis are overlapped with scaling based on the electrostatic potential difference between the primarily perturbed H and O atoms [located along the y (horizontal) axis]. The contours in the two-dimensional plot are drawn with the interval of 0.36 × 10−3 a0−2 in the range from −9 × 10−3 a0−2 to 9 × 10−3 a0−2, with the color code shown on the left-hand side. The atomic positions of the central water molecule are indicated with black filled circles in the two-dimensional plots and with pink dotted vertical lines in the one-dimensional plots. | ||
As a result, in the present case, four interaction parameters [dipolar polarizabilities αyy and αzz on O, α⊥ (vertical to the bond) on H, and the charge response parameter κOH] are selected, and their values are obtained by least-squares fitting. In this least-squares fitting, the square root of the eigenvalue related to each basis vector in the respective cycle of the repeated SVD analyses was used as the relative weight. The values of the parameters thus obtained are shown in Table 2. By using these values, the induced dipole moments corresponding to the basis vectors are reasonably well reproduced as shown in Table 3, although there is a noticeable deviation with regard to the angle of the induced dipole corresponding to basis vector 6a (by 36.3°).
The values of the parameters shown in Table 2 indicate the extent of mixing of the atomic dipolar polarization component and the interatomic CT component in the molecular electric polarization of water. For example, when the uniform electric field of 0.04 Ehe−1a0−1 is operating along the out-of-plane (x) axis, the induced dipole moment arises totally from the atomic dipolar polarizations, which amounts to 0.31 ea0 in total. In contrast, when the uniform electric field of 0.04 Ehe−1a0−1 is operating along one of the OH bond, the interatomic CT along this bond (related to κOH) gives rise to 0.19 ea0 of induced dipole moment, while the atomic dipolar polarizations of the oxygen (related to αyy and αzz) and the other hydrogen atom (α⊥) gives rise to 0.16 ea0 in almost the same direction, with a small almost perpendicular induced dipole of 0.05 ea0 due to the interatomic CT related to this other H atom. When the electric field is non-uniform, the extent of mixing of these components would vary. Since the values of the parameters shown in Table 2 are derived for the hydrogen-bonding pairs of molecules, it is expected that they are more pertinent to those non-uniform situations.
3.3. Discussion toward application to larger systems
To apply the present method to larger systems, some technical issues need to be considered. First, the number of the evaluation points r of each ρ(el)(r) increases with the system size, but not proportionally. This is because the evaluation points are taken in a rectangular box, each boundary of which is set as being at least 5 Å away from any atom in the system. For example, if we suppose a system with the frame of the atomic centers fitting in a cube of 10 × 10 × 10 Å3, the rectangular box of the evaluation points will be of the size of 20 × 20 × 20 Å3. If we retain 0.02 Å for the interval, the number of the evaluation points will be m = 109 (more exactly, 10013), which is only about 6.5 times of that for the case of water examined in the present study. Widening of the interval is not considered to be desirable. For example, in the 2D plot on the yz plane of vector 1a shown in Fig. 4a, the plotted value changes significantly within the range of 0.1 Å around the center of the oxygen atom. The computational cost is considered to scale just linearly to the number of the evaluation points. The size of matrix tBB defined in eqn (2), which is subject to diagonalization in the SVD analysis, is determined by the number of vectors bj (n = 1000 in the present case), not by their dimension. In this sense, the computational time needed for matrix diagonalization depends nonlinearly to the number of vectors bj, but diagonalization of a matrix of the size of 1000 × 1000, for example, is not a problem with the current computational power. More importantly, the size of matrix B itself, which is equal to the dimension multiplied by the number of vectors bj, affects the disk space needed to store it. In the case of the present study (m = 154564011 and n = 1000, double precision floating point numbers), it amounted to 1.24 × 1012 bytes. The number of vectors bj depends on the extent of distortion, etc., of the hydrogen-bond configurations taken into account in the analysis. If we narrow the range of them, the weight of the main components derived from the analysis will increase.
Secondly, it is expected that the eigenvalues decay more slowly (as compared to the situation shown in Fig. 2) for larger systems and, hence, the number of the basis vectors required to describe the system will increase rather proportionally to the system size. It means that the procedures described in the present study (summarized in Chart 1) will be needed to be repeated more times. To clarify the situations in this relation, however, further studies on some other typical functional groups will be needed.
4. Concluding remarks
In summary, it is shown by conducting SVD analysis on the electron density changes that both the interatomic CT through the OH bonds and the dipolar polarizations of the atoms are important for the molecular electrostatic polarization of hydrogen-bonded water. Taking only one of these factors is not sufficient to correctly represent it. The seven basis vectors (shown in Fig. 4) and the symmetry-related three basis vectors that are extracted in this SVD analysis constitute an orthonormal basis of the polarization “space” that covers nearly 99% of all the electron density changes. The parameters of the interatomic CT and the atomic dipolar polarizations derived from the present analysis (shown in Table 2) reasonably well reproduce the molecular induced dipole moments as shown in Table 3. As a result, these parameters indicate the extent of mixing and the relative importance of the two factors of molecular electrostatic polarization.In modeling electric polarizations of molecules, it has sometimes been suggested that the use of point dipoles is too simple, especially for describing the interactions between closely-located molecules, so that the use of distributed charges is more appropriate.17,18 In this sense, the electron density basis vectors derived in the present study demonstrate the spatial characteristics of the charge distributions related to the electrostatic polarizations, and thus are considered to form a basis for further developing sophisticated polarizable models of water.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by JSPS KAKENHI Grant Number JP20H05215. The calculations were carried out partly by using the computers at the Research Center for Computational Science, Okazaki, Japan.References
- D. P. Fernández, Y. Mulev, A. R. H. Goodwin and J. M. H. Levelt Sengers, A database for the static dielectric constant of water and steam, J. Phys. Chem. Ref. Data, 1995, 24, 33–69 CrossRef.
- J. S. Brown, J. P. Hallett, D. Bush and C. A. Eckert, Liquid–liquid equilibria for binary mixtures of water + acetophenone, + 1-octanol, + anisole, and + toluene from 370 K to 550 K, J. Chem. Eng. Data, 2000, 45, 846–850 CrossRef CAS.
- P. E. Savage, Organic chemical reactions in supercritical water, Chem. Rev., 1999, 99, 603–621 CrossRef CAS PubMed.
- N. Yoshii, H. Yoshie, S. Miura and S. Okazaki, A molecular dynamics study of sub- and supercritical water using a polarizable potential model, J. Chem. Phys., 1998, 109, 4873–4884 CrossRef CAS.
- P. Jedlovszky and J. Richardi, Comparison of different water models from ambient to supercritical conditions: A Monte Carlo simulation and molecular Ornstein–Zernike study, J. Chem. Phys., 1999, 110, 8019–8031 CrossRef CAS.
- P. E. M. Lopes, B. Roux and A. D. MacKerell Jr, Molecular modeling and dynamics studies with explicit inclusion of electronic polarizability: Theory and applications, Theor. Chem. Acc., 2009, 124, 11–28 Search PubMed.
- J. F. Ouyang and R. P. A. Bettens, Modelling water: A lifetime enigma, CHIMIA International Journal for Chemistry, 2015, 69, 104–111 Search PubMed.
- I. V. Leontyev and A. A. Stuchebrukhov, Polarizable mean-field model of water for biological simulations with AMBER and CHARMM force fields, J. Chem. Theory Comput., 2012, 8, 3207–3216 CrossRef CAS PubMed.
- T. A. Ho and A. Striolo, Polarizability effects in molecular dynamics simulations of the graphene–water interface, J. Chem. Phys., 2013, 138, 054117 CrossRef PubMed.
- P. Ahlström, A. Wallqvist, S. Engström and B. Jönsson, A molecular dynamics study of polarizable water, Mol. Phys., 1989, 68, 563–581 CrossRef.
- J. Caldwell, L. X. Dang and P. A. Kollman, Implementation of nonadditive intermolecular potentials by use of molecular dynamics: Development of a water–water potential and water–ion cluster interactions, J. Am. Chem. Soc., 1990, 112, 9144–9147 CrossRef CAS.
- D. N. Bernardo, Y. Ding, K. Krogh-Jespersen and R. M. Levy, An anisotropic polarizable water model: Incorporation of all-atom polarizabilities into molecular mechanics force fields, J. Phys. Chem., 1994, 98, 4180–4187 CrossRef CAS.
- L. X. Dang and T.-M. Chang, Molecular dynamics study of water clusters, liquid, and liquid–vapor interface of water with many-body potentials, J. Chem. Phys., 1997, 106, 8149–8159 CrossRef CAS.
- L.-P. Wang, T. Head-Gordon, J. W. Ponder, P. Ren, J. D. Chodera, P. K. Eastman, T. J. Martinez and V. S. Pande, Systematic improvement of a classical molecular model of water, J. Phys. Chem. B, 2013, 117, 9956–9972 CrossRef CAS PubMed.
- P. Tröster, K. Lorenzen, M. Schwörer and P. Tavan, Polarizable water models from mixed computational and empirical optimization, J. Phys. Chem. B, 2013, 117, 9486–9500 CrossRef PubMed.
- D. Sidler, M. Meuwly and P. Hamm, An efficient water force field calibrated against intermolecular THz and Raman spectra, J. Chem. Phys., 2018, 148, 244504 CrossRef PubMed.
- P. Paricaud, M. Předota, A. A. Chialvo and P. T. Cummings, From dimer to condensed phases at extreme conditions: Accurate predictions of the properties of water by a Gaussian charge polarizable model, J. Chem. Phys., 2005, 122, 244511 CrossRef PubMed.
- D. Elking, T. Darden and R. J. Woods, Gaussian induced dipole polarization model, J. Comput. Chem., 2007, 28, 1261–1274 CrossRef CAS PubMed.
- V. Babin, C. Leforestier and F. Paesani, Development of a “first principles” water potential with flexible monomers: Dimer potential energy surface, VRT spectrum, and second virial coefficient, J. Chem. Theory Comput., 2013, 9, 5395–5403 CrossRef CAS PubMed.
- C. Liu, J.-P. Piquemal and P. Ren, AMOEBA+ classical potential for modeling molecular interactions, J. Chem. Theory Comput., 2019, 15, 4122–4139 CrossRef CAS PubMed.
- P. J. van Maaren and D. van der Spoel, Molecular dynamics simulations of water with novel shell-model potentials, J. Phys. Chem. B, 2001, 105, 2618–2626 CrossRef CAS.
- H. Yu, T. Hansson and W. F. van Gunsteren, Development of a simple, self-consistent polarizable model for liquid water, J. Chem. Phys., 2003, 118, 221–234 CrossRef CAS.
- G. Lamoureux, A. D. MacKerell Jr and B. Roux, A simple polarizable model of water based on classical Drude oscillators, J. Chem. Phys., 2003, 119, 5185–5197 CrossRef CAS.
- J. A. Lemkul, J. Huang, B. Roux and A. D. MacKerell Jr, An empirical polarizable force field based on the classical Drude oscillator model: Development history and recent applications, Chem. Rev., 2016, 116, 4983–5013 CrossRef CAS PubMed.
- M. Sprik, Computer simulation of the dynamics of induced polarization fluctuations in water, J. Phys. Chem., 1991, 95, 2283–2291 CrossRef CAS.
- A. K. Rappe and W. A. Goddard III, Charge equilibration for molecular dynamics simulations, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS.
- S. W. Rick, S. J. Stuart and B. J. Berne, Dynamical fluctuating charge force fields: Application to liquid water, J. Chem. Phys., 1994, 101, 6141–6156 CrossRef CAS.
- B. A. Bauer and S. Patel, Recent applications and developments of charge equilibration force fields for modeling dynamical charges in classical molecular dynamics simulations, Theor. Chem. Acc., 2012, 131, 1153 Search PubMed.
- H. A. Stern, F. Rittner, B. J. Berne and R. A. Friesner, Combined fluctuating charge and polarizable dipole models: Application to a five-site water potential function, J. Chem. Phys., 2001, 115, 2237–2251 CrossRef CAS.
- G. S. Fanourgakis and S. S. Xantheas, Development of transferable interaction potentials for water. V. Extension of the flexible, polarizable, Thole-type model potential (TTM3-F, v. 3.0) to describe the vibrational spectra of water clusters and liquid water, J. Chem. Phys., 2008, 128, 074506 CrossRef PubMed.
- T. Hasegawa and Y. Tanimura, A polarizable water model for intramolecular and intermolecular vibrational spectroscopies, J. Phys. Chem. B, 2011, 115, 5545–5553 CrossRef CAS PubMed.
- S. Naserifar and W. A. Goddard III, The quantum mechanics-based polarizable force field for water simulations, J. Chem. Phys., 2018, 149, 174502 CrossRef PubMed.
- E. Lambros and F. Paesani, How good are polarizable and flexible models for water: Insights from a many-body perspective, J. Chem. Phys., 2020, 153, 060901 CrossRef CAS.
- C. J. Tainter, L. Shi and J. L. Skinner, Reparametrized E3B (explicit three-body) water model using the TIP4P/2005 model as a reference, J. Chem. Theory Comput., 2015, 11, 2268–2277 CrossRef CAS PubMed.
- W. H. Press, B. P. Flannery, S. A. Teukolsky and W. T. Vetterling, Numerical Recipes in Fortran 77, The Art of Scientific Computing, Cambridge University Press, Cambridge, 2nd edn, 1992 Search PubMed.
- K. Hinsen and G. R. Kneller, Projection methods for the analysis of complex motions in macromolecules, Mol. Simul., 2000, 23, 275–292 CrossRef CAS.
- B. B. Harland and P.-N. Roy, An initial value representation semiclassical approach for the study of molecular systems with geometric constraints, J. Chem. Phys., 2003, 118, 4791–4806 CrossRef CAS.
- M. Ceotto, G. Di Liberto and R. Conte, Semiclassical “Divide-and-Conquer” method for spectroscopic calculations of high dimensional molecular systems, Phys. Rev. Lett., 2017, 119, 010401 CrossRef PubMed.
- F. Gabas, G. Di Liberto and M. Ceotto, Vibrational investigation of nucleobases by means of divide and conquer semiclassical dynamics, J. Chem. Phys., 2019, 150, 224107 CrossRef PubMed.
- T. Yuzawa and H. Hamaguchi, Investigation of the photoisomerization of all-trans-retinal by singular-value-decomposition analysis of nanosecond time-resolved infrared spectra, J. Mol. Struct., 1995, 352/353, 489–495 CrossRef CAS.
- S. Yamaguchi and H. Hamaguchi, Femtosecond time-resolved absorption spectroscopy of all-trans-retinal in hexane, J. Mol. Struct., 1996, 379, 87–92 CrossRef CAS.
- Y. Li, M. Wei, F. Zhang and J. Zhao, Comparison of two SVD-based color image compression schemes, PLoS One, 2017, 12, e0172746 CrossRef PubMed.
- A. M. Rufai, G. Anbarjafari and H. Demirel, Lossy image compression using singular value decomposition and wavelet difference reduction, Digital Signal Processing, 2014, 24, 117–123 CrossRef.
- A. Anand and A. K. Singh, An improved DWT-SVD domain watermarking for medical information security, Computer Communications, 2020, 152, 72–80 CrossRef.
- H. Torii and R. Ukawa, Role of Intermolecular charge fluxes in the hydrogen-bond-induced frequency shifts of the OH stretching mode of water, J. Phys. Chem. B, 2021, 125, 1468–1475 CrossRef CAS PubMed.
- W. L. Jorgensen and J. D. Madura, Temperature and size dependence for Monte Carlo simulations of TIP4P water, Mol. Phys., 1985, 56, 1381–1392 CrossRef CAS.
- H. Torii, Cooperative contributions of the intermolecular charge fluxes and intramolecular polarizations in the far-infrared spectral intensities of liquid water, J. Chem. Theory Comput., 2014, 10, 1219–1227 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wiberg, Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- H. Torii, Intermolecular charge flux as the origin of infrared intensity enhancement upon halogen-bond formation of the peptide group, J. Chem. Phys., 2010, 133, 034504 CrossRef PubMed.
- H. Torii, Intermolecular electron density modulations in water and their effects on the far-infrared spectral profiles at 6 THz, J. Phys. Chem. B, 2011, 115, 6636–6643 CrossRef CAS PubMed.
- H. Torii, Mechanism of the secondary structure dependence of the infrared intensity of the amide II mode of peptide chains, J. Phys. Chem. Lett., 2012, 3, 112–116 CrossRef CAS.
- H. Torii, Extended nature of the molecular dipole of hydrogen-bonded water, J. Phys. Chem. A, 2013, 117, 2044–2051 CrossRef CAS PubMed.
- H. Torii, Intermolecular charge fluxes and far-infrared spectral intensities of liquid formamide, Phys. Chem. Chem. Phys., 2018, 20, 3029–3039 RSC.
- K. Saito, R. Izumi and H. Torii, Dissecting the electric quadrupolar and polarization effects operating in halogen bonding through electron density analysis with a focus on bromine, J. Chem. Phys., 2020, 153, 174302 CrossRef CAS PubMed.
- K. Saito and H. Torii, Hidden halogen-bonding ability of fluorine manifesting in the hydrogen-bond configurations of hydrogen fluoride, J. Phys. Chem. B, 2021, 125, 11742–11750 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. Morita and S. Kato, Ab initio molecular orbital theory on intramolecular charge polarization: Effect of hydrogen abstraction on the charge sensitivity of aromatic and nonaromatic species, J. Am. Chem. Soc., 1997, 119, 4021–4032 CrossRef CAS.
- T. Ishiyama and A. Morita, Analysis of anisotropic local field in sum frequency generation spectroscopy with the charge response kernel water model, J. Chem. Phys., 2009, 131, 244714 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |