
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhenylalanyl tRNA synthetase (PheRS) substrate mimics: design, synthesis, molecular dynamics and antimicrobial evaluation†
Nada A. Noureldin *ab,
Jennifer Richardsc,
Hend Kothayerb,
Mohammed M. Barakab,
Sobhy M. Eladlb,
Mandy Woottonc and
Claire Simonsa
*ab,
Jennifer Richardsc,
Hend Kothayerb,
Mohammed M. Barakab,
Sobhy M. Eladlb,
Mandy Woottonc and
Claire Simonsa
aSchool of Pharmacy and Pharmaceutical Sciences, Cardiff University, Cardiff CF10 3NB, UK. E-mail: NANoureddine@pharmacy.zu.edu.eg
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Zagazig University, Zagazig P. C., 44519, Egypt
cSpecialist Antimicrobial Chemotherapy Unit, University Hospital of Wales, Heath Park, Cardiff CF14 4XW, UK
First published on 20th January 2022
Abstract
Antimicrobial resistance is a very challenging medical issue and identifying novel antimicrobial targets is one of the means to overcome this challenge. Phenylalanyl tRNA synthetase (PheRS) is a promising antimicrobial target owing to its unique structure and the possibility of selectivity in the design of inhibitors. Sixteen novel benzimidazole based compounds (5a–b), (6a–e), (7a–d), (9a–e) and three N,N-dimethyl-7-deazapurine based compounds (16a–c) were designed to mimic the natural substrate of PheRS, phenylalanyl adenylate (Phe-AMP), that was examined through flexible alignment. The compounds were successfully synthesised chemically in two schemes using 4 to 6-steps synthetic pathways, and evaluated against a panel of five microorganisms with the best activity observed against Enterococcus faecalis. To further investigate the designed compounds, a homology model of E. faecalis PheRS was generated, and PheRS-ligand complexes obtained through computational docking. The PheRS–ligand complexes were subjected to molecular dynamics simulations and computational binding affinity studies. As a conclusion, and using data from the computational studies compound 9e, containing the (2-naphthyl)-L-alanine and benzimidazole moieties, was identified as optimal with respect to occupancy of the active site and binding interactions within the phenylalanine and adenosine binding pockets.
Introduction
The continued increase in antimicrobial resistance (AMR) observed worldwide is a major challenge that needs to be addressed.1–4 Finding novel antibacterial targets, new antibiotic combination therapies and development of bacteriophages are some of the options available that may overcome AMR.5,6 Among the novel antibacterial targets that show noteworthy antibacterial activity are the aminoacyl tRNA synthetase (aaRS) enzymes,7–9 which catalyse the activation and the attachment of a specific amino acid to its cognate tRNA.10 Mupirocin (Fig. 1),11 a natural product from Pseudomonas fluorescens, was the first aaRS inhibitor available for clinical use, which inhibits isoleucine-tRNA synthetase (IleRS) and mimics the structure of the natural substrate of IleRS, isoleucyl-AMP. Mupirocin contains a hydrolysable ester bond; therefore, it is only used for topical treatment of Staphylococcus aureus and methicillin resistant S. aureus (MRSA) infections especially for surgical site infections, with MIC ≤ 4 μg mL−1 against MRSA.12 Another example of a market available aaRS inhibitor is oxaborol (AN2690) (Fig. 1), which targets the fungal lysyl-tRNA synthetase (LysRS). However, due to the lack of selectivity between human and fungal cells its use is limited to the treatment of nail fungal infections.13,14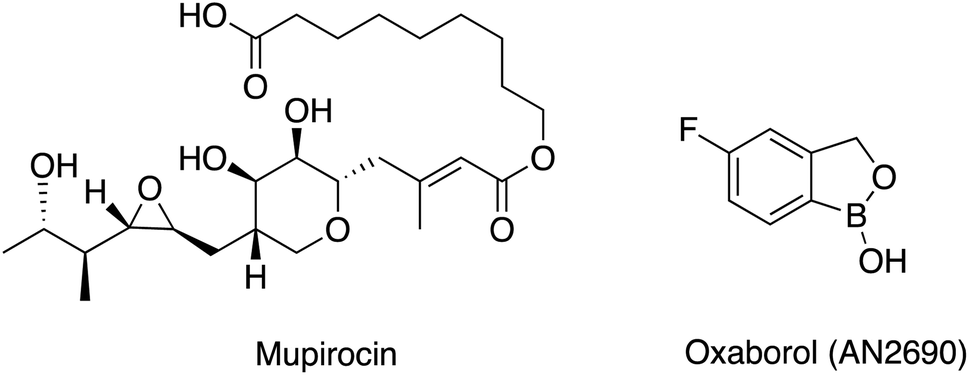 | ||
| Fig. 1 Chemical structures of mupirocin and oxaborol (AN2690). | ||
AaRS enzymes are classified into 2 main classes, class I or class II, based on the active site structure and enzyme kinetics.7 Phenylalanyl-tRNA synthetase (PheRS), a member of class II aaRS,7,8 has a heterodimeric structure (αβ)2 with two small α subunits (PheS) and two large β subunits (PheT).15 The smaller α subunit is responsible for the activation of the amino acid and the amino acylation of the cognate tRNAphe, while the larger β subunit is the site of interaction and recognition of the cognate tRNAphe and the proofreading process.15 PheRS is structurally unique among the aaRS enzymes with its tetrameric structure and, unlike other class II enzymes, it aminoacylates the 2′-OH of the terminal ribose of tRNA rather than the 3′-OH.15 Bacterial PheRS is structurally different from human PheRSs (cytoplasmic and mitochondrial) with low homology allowing for design of selective PheRS inhibitors.16 PheRS inhibitors with varying structure design have been described such as the ethanolamines,17 phenyl-thiazolyl-urea sulfonamides18 and bicyclic azetidines19 (Fig. 2). Ethanolamines displayed low nM inhibitory activity against S. aureus PheRS but did not show inhibitory activity against the whole microorganism,17 while the phenyl-thiazolylurea sulfonamides displayed potent inhibitory activity of PheRS and broad spectrum antimicrobial activity.18 The phenyl-thiazolylurea sulfonamides were found to bind in a hydrophobic auxiliary pocket adjacent to the phenylalanine binding site, however inhibitor resistant mutants were mapped to this auxiliary site indicating this pocket may not be optimal for drug design.20 Bicyclic azetidines, developed for the treatment of malaria, were found to be potent inhibitors of Plasmodium falciparum PheRS, effective in eliminating both blood-stage and liver-stage parasites.19
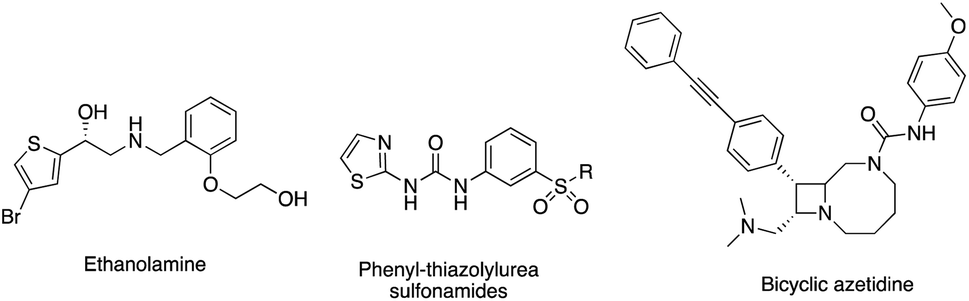 | ||
| Fig. 2 PheRS inhibitor illustrating structural diversity. | ||
This research has focused on the design of mimics of the natural substrate, phenylalanyl adenylate (Phe-AMP), as dual inhibitors of the phenylalanine and AMP binding sites. Four series were designed using flexible alignment (Fig. S1†) to map structural similarity with Phe-AMP; benzimidazole or N,N-dimethyl-7-deazapurine was included to mimic the adenosine moiety, an aryl group attached to an amide or sulfonamide linker to mimic phenylalanine or, in the case of series 3 and 4, phenylalanine derivatives were used, and a thiadiazole ring to mimic the 5-membered ribose moiety (Fig. 3).
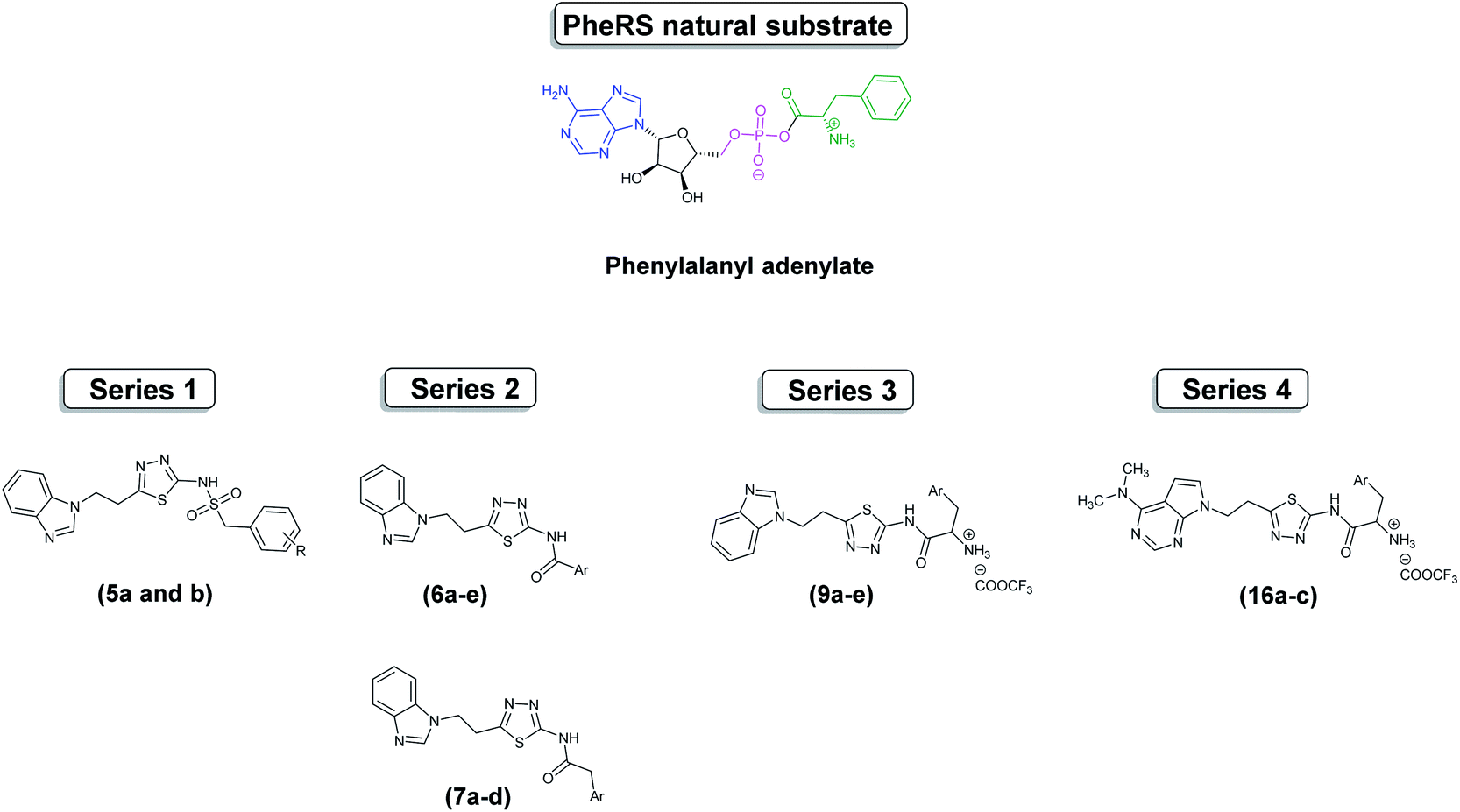 | ||
| Fig. 3 Structure of phenylalanyl adenylate (Phe-AMP) and the designed series 1–4. | ||
Recently, the acronym ESKAPEE is commonly used to describe the most life-threatening pathogens, namely Enterococcus faecalis, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp. and Escherichia coli. These were included in the WHO priority pathogen list because of their multidrug resistance behaviour and the emergence of some pan-drug resistant strains among them.21 All of the synthesised compounds were subjected to microbiological evaluation against the Gram +ive E. Faecalis and S. aureus, and the Gram −ive K. pneumoniae, P. aeruginosa and E. coli, in order to examine the activity against wide range of the most threatening organisms.
Results and discussion
Chemistry
Series 1 (5a and b) and series 2 (6a–e and 7a–d) were synthesised via the route outlined in Scheme 1. Methyl 3-(1H-benzo[d]imidazol-1-yl)propanoate (2)22 was prepared by reaction of benzimidazole (1) and methyl 3-bromopropionate in the presence of K2CO3 and 18-crown-6. Excess hydrazine hydrate at room temperature in EtOH was used to convert methyl 3-(1H-benzo[d]imidazol-1-yl) propanoate (2) to 3-(1H-benzo[d]imidazol-1-yl)propane hydrazide (3). The latter was then allowed to reflux with trimethylsilyl isothiocyanate (TMSNCS) in dry isopropanol overnight, followed by evaporation of the solvent and addition of concentrated H2SO4 allowing this mixture to stir at room temperature (25 °C) for 2 hours. On neutralisation with ammonia solution, a solid of the pure cyclised product of 5-(2-(1H-benzo[d]imidazol-1-yl)ethyl)-1,3,4-thiadiazol-2-amine was obtained (4). The thiadiazol-2-amine (4) was reacted either with aryl methansulphonyl chloride to afford N-(5-(2-(1H-benzo[d]imidazol-1-yl)ethyl)-1,3,4-thiadiazol-2-yl)-1-arylmethane sulphonamide derivatives (5a and b), or with aromatic carboxylic acids to yield N-(5-(2-(1H-benzo[d]imidazol-1-yl)ethyl)-1,3,4-thiadiazol-2-yl)-2-arylamide derivatives (6a–e) and (7a–d).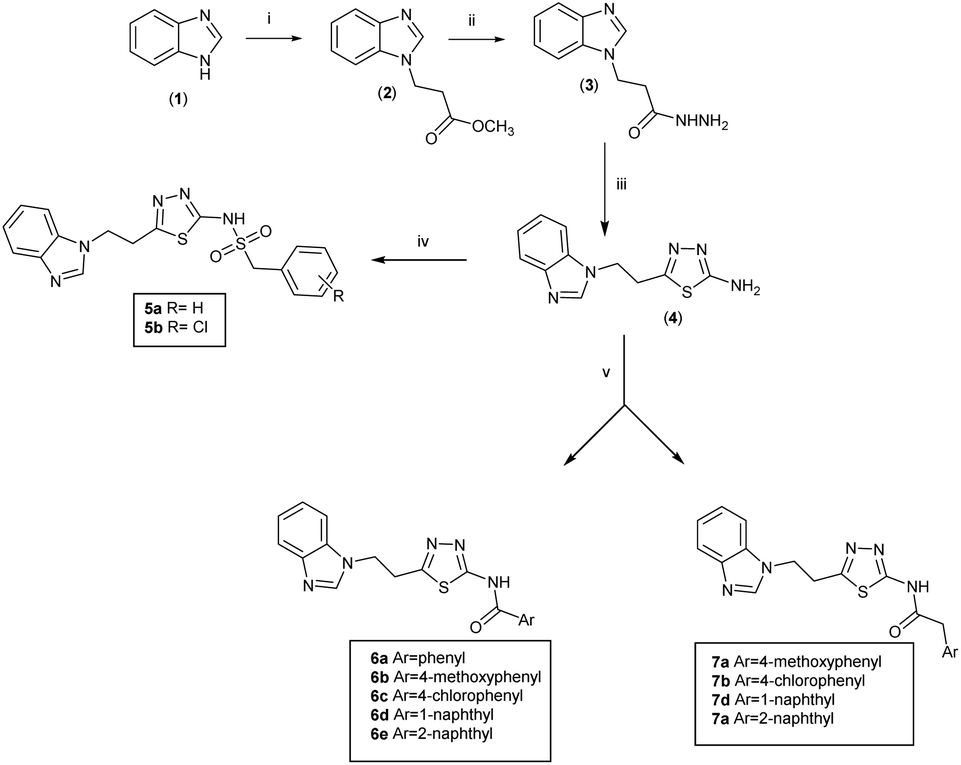 | ||
| Scheme 1 Preparation of series 1 and 2. Reagents and conditions (i) K2CO3, 18-crown-6, methyl 3-bromopropionate, DMF, 60 °C, o/n (ii) NH2NH2, EtOH, r. t. o/n (iii) TMSNCS, iPrOH, H2SO4, NH4OH, reflux o/n (iv) R-appropriate phenylmethanesulfonyl chloride, DMAP, pyridine, Ch2Cl2, r. t. o/n (v) aryl carboxylic acid, TBTU, HOBt, DMF, DIPEA, r. t. o/n. | ||
The benzimidazole derivatives of series 3 (9) were readily prepared by reaction of the thiadiazol-2-amine (4) with N-Boc protected L-amino acid to give the amide (8), which was then subject to a Boc deprotection using excess trifluoroacetic acid (TFA) to afford 1-((5-(2-(1H-benzo[d]imidazol-1-yl)ethyl)-1,3,4-thiadiazol-2-yl)amino)-1-oxo-3-phenylpropan-2-aminium salts (9a–e) (Scheme 2).
 | ||
| Scheme 2 Preparation of series 3 and 4. Reagents and conditions: (i) N-Boc-protected L-amino, TBTU, HOBt, DMF, DIPEA r. t o/n (ii) TFA, CH2Cl2, r. t. o/n (iii) DMF, KOH, 95 °C, 2 h (iv) K2CO3, 18-crown-6, methyl 3-bromopropionate, DMF, overnight, 60 °C (v) NH2NH2, EtOH, r. t. o/n (vi) TMSNCS, iPrOH, H2SO4, NH4OH, reflux o/n. | ||
The corresponding N,N-dimethyl-7-deazapurine derivatives of series 4 (16) were prepared in a 6-step pathway starting with 7-chloro-6-deazapurine (10), which was converted into 7 N,N-dimethyl-7-deazapurine (11) by reaction with DMF and 10 M aqueous KOH at 95 °C.23 The thiadiazol-2-amine (14) was prepared as described in Scheme 1 from the hydrazide (13), which was obtained on reaction of 3-(4-(dimethylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)propanoate (12) with hydrazine hydrate. The thiadiazol-2-amine (14) was coupled with the N-Boc protected L-amino acids using TBTU and HOBt to form the amides (15), which were subsequently deprotected using excess TFA to give the series 4 derivatives (16).
Antimicrobial evaluation
All the final compounds (5a–b), (6a–e), (7a–d), (9a–e) and (16a–c) were tested against a panel of Gram +ive (S. aureus, E. faecalis) and Gram −ive (Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae) microorganisms (Tables S1 and S2†), while Table 1 shows only the most promising results. All the compounds were inactive with the exception of some derivatives of series 3 (9b, 9d and 9e) and series 4 (16a and 16c), which displayed modest MIC (32–64 μg mL−1, 0.053–0.123 mM) against E. faecalis (Table 1). Compounds 9e (Ar = 2-naphthyl) and 16c (Ar = 2-naphthyl) showed optimal, although not clinically relevant, inhibitory activity (MIC 32 μg mL−1, 0.058 and 0.053 mM respectively) from this series of compounds. Surprisingly, broad spectrum activity was not obtained, although the enzyme active site is conserved among organisms (Fig S2†).| Series 3 | E. faecalis MIC (μg mL−1) | E. faecalis MIC (mM) | Series 4 | E. faecalis MIC (μg mL−1) | E. faecalis MIC (mM) |
|---|---|---|---|---|---|
| 9a | 128 | 0.253 | 16a | 64 | 0.109 |
| 9b | 64 | 0.119 | 16b | 128 | 0.227 |
| 9c | 128 | 0.239 | 16c | 32 | 0.053 |
| 9d | 64 | 0.123 | Ciprofloxacin | 0.5 | 0.0015 |
| 9e | 32 | 0.058 | |||
| Ciprofloxacin | 1 | 0.003 | |||
Computational studies
To further explore the preference for the compounds of series 3 and 4 for E. faecalis, it was necessary to develop a homology model for E. faecalis PheRS as no crystal structures are currently available. The following section describes the preparation of a homology model and generation of protein–ligand complexes for molecular dynamics and computational binding affinity studies.The protein sequence of the E. faecalis PheRS α-subunit (PheS, Uniprot code Q836J6) was downloaded from the Uniprot database24 and subject to a BLAST analysis against the resolved protein sequences from the protein data bank (PDB).25 Th. thermophilus PheS (PDB 1JJC),26 with 42% homology, was defined as an appropriate template for the homology model as it is the only solved structure with the whole Phe-AMP substrate. The model was built using Molecular Operating Environment (MOE)27 and evaluated in terms of its overall structure using Ramachandran plot, ProSA analysis and 3D structure verification and the results (Fig. S3, S4 and Table S3†) showed a good quality model.
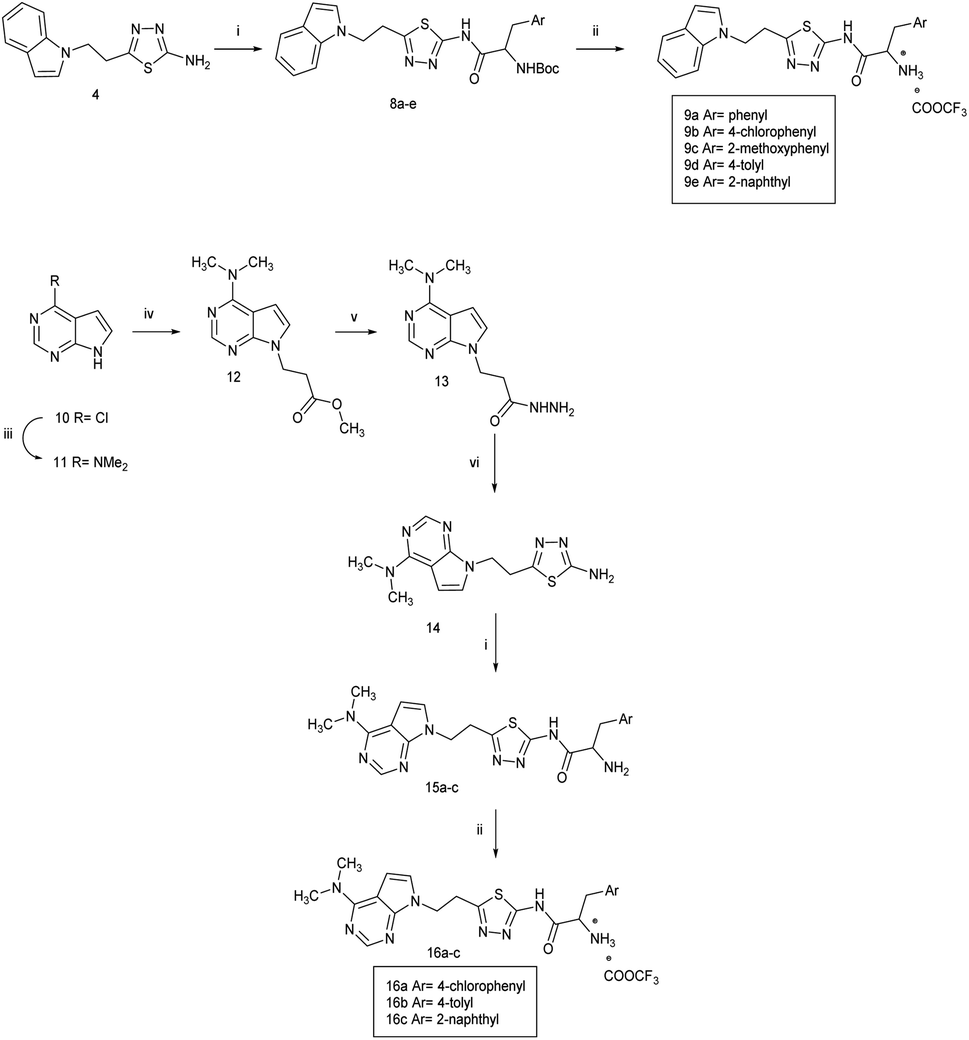
Docking of the natural substrate Phe-AMP followed by molecular dynamics simulation using the Desmond programme of Maestro,28 and comparison with previous studies16,29 enabled identification of the binding sites of the phenylalanine and AMP moieties (Fig. 4). The active site is in a deep cavity contained in a structure of antiparallel β sheets linked together with loops and helices, a conserved feature in all class II aminoacyl tRNA synthetase enzymes. The active site has pockets that specifically recognise the phenylalanine and AMP of the natural substrate Phe-AMP. The phenyl ring of phenylalanine sits within a hydrophobic pocket composed of Phe255, Pro256, Phe257, Thr258, Ala290, Ala312 and Phe313. This pocket stabilises the phenyl group and also discriminates between phenylalanine and tyrosine amino acids to prevent the false charging of tRNATyr by PheRS.26 The phenyl ring of the phenylalanine is recognised via edge-to-face hydrophobic interactions with Phe255 and Phe257. A hydrophilic environment composed of His172, Thr173, Ser174, Gln215, Gly316, Glu217 and Arg319 is suitable for anchoring of the amine group of the amino acid, the phosphate and ribose moiety. The adenine moiety of the AMP is stabilised by π–π interaction with Phe213, and by hydrogen bonding between N1 of the adenine ring and OH from the side chain of Ser210. The computational binding affinity (ΔG) for the PheS protein-PheAMP complex was −37.3960 ± 7.01 kcal mol−1.
 | ||
| Fig. 4 Phe-AMP (gold) in E. faecalis PheS after MD simulation with phenylalanine positioned on the right and adenosine on the left. The key amino acid residues are indicated, and the active site is shown as a grid, with green indicating a hydrophobic region and pink a hydrophilic region. | ||
Having established the active site of Phe-AMP in E. faecalis PheS, protein–ligand complexes for series 3 and 4 were generated from docking using MOE. These protein–ligand complexes were then subject to 200 ns molecular dynamics simulations.
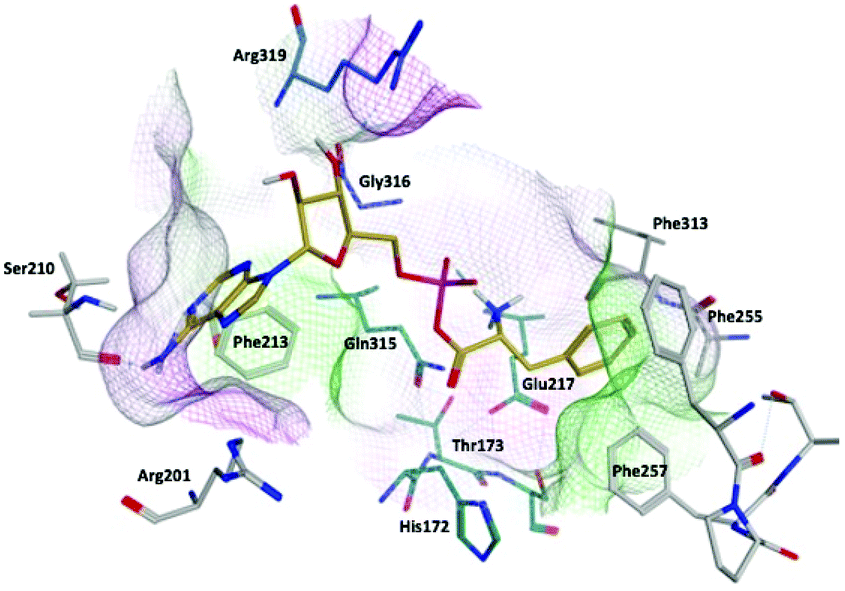
Compound 9b (Ar = 4-chlorophenyl) overlaps with the phenyl of the Phe-AMP in the phenylalanine binding site, forming π–π interaction between the 4-chlorophenyl moiety and Phe255 in the hydrophobic pocket, likewise the amine of phenylalanine interacts with Glu217 (ionic), His172 (H-bond) and via a water-mediated interaction with Ser174 (H-bond) in the hydrophilic pocket. However, a π–π interaction between the thiadiazole ring and Phe255 results in the benzimidazole being directed away from the adenosine binding site to obtain a favourable binding affinity (ΔG = −63.2862 ± 4.99 kcal mol−1) compared with the natural substrate PheAMP (Fig. 5A). The same binding profile was observed for the tolyl derivative 9d in the phenylalanine binding pocket, while the benzimidazole now extends into the entrance of the adenosine binding pocket forming a π–π interaction with His209 and the imidazole ring and a water mediated interaction (H-bond) between an imidazole N and Arg201 (Fig. 5B), however this is associated with a less favourable binding affinity (ΔG = −34.6624 ± 2.56 kcal mol−1). The bulkier naphthyl derivative 9e also forms a π–π interaction between the 2-naphthyl moiety and Phe255 in the hydrophobic pocket, however the size of the naphthyl moves the interaction with the Phe amine further along the pocket to Glu263. The benzimidazole sits in a hydrophilic pocket above the adenosine pocket and forms a water mediated interaction (H-bond) between an imidazole N and Ala207 (Fig. 5B) with a favourable binding affinity (ΔG = −61.6053 ± 2.86 kcal mol−1) (Fig. 5C).
 | ||
| Fig. 5 Alignment of (A) 9b (cyan), (B) 9d (purple) and (C) 9e (green) with Phe-AMP (gold) in E. faecalis PheS after 200 ns MD simulation, and 2D ligand interaction diagrams. | ||
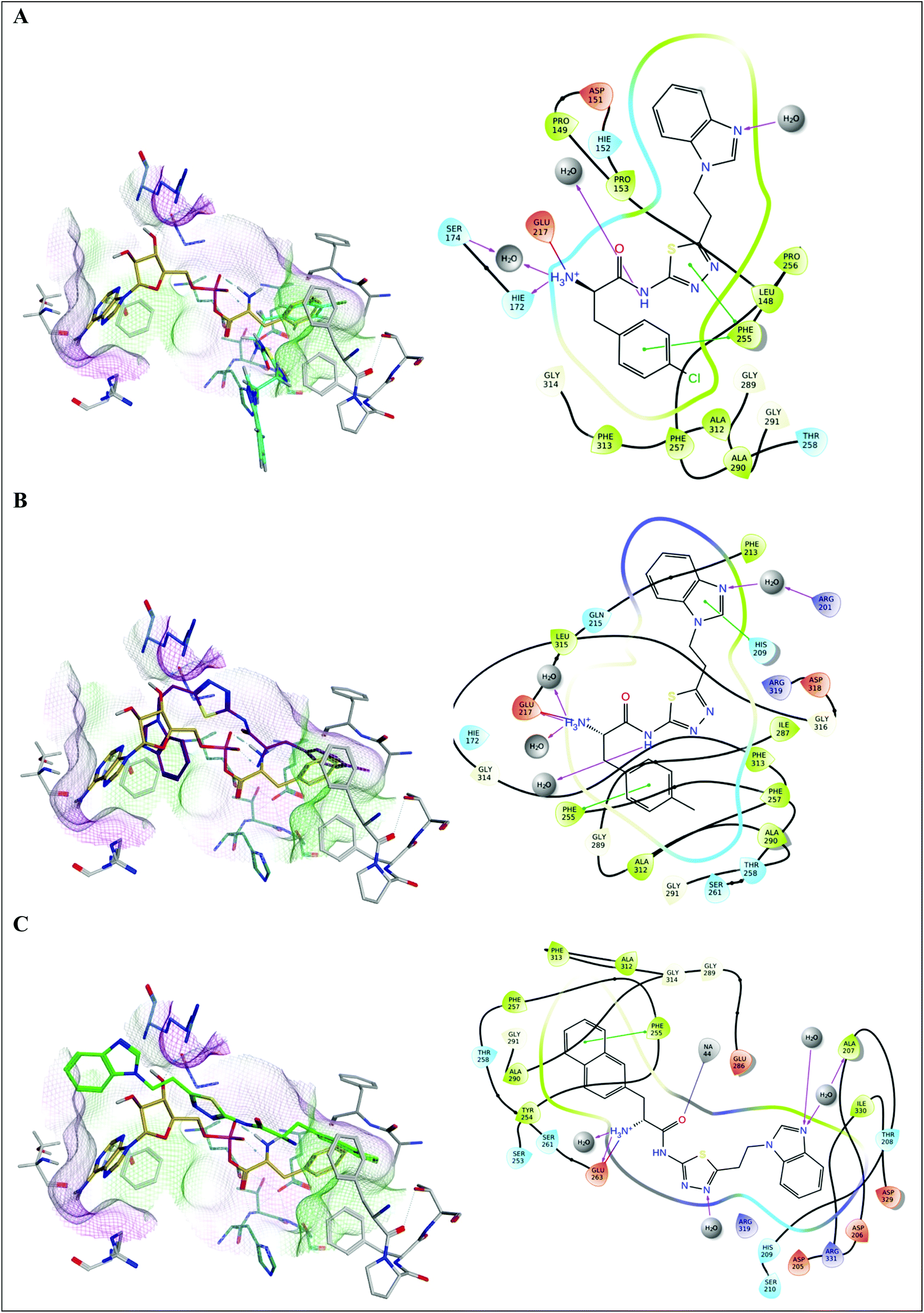
The 4-chlorophenyl derivative 16a fills the phenyl pocket with Phe255 and Phe313 in close proximity, the Phe amine forms a hydrogen bonding interaction with Glu217, and the amide chain interacts with Gln315 through the carbonyl O, and the NH forms a water mediated interaction with Glu263 (Fig. 6A). However, the N,N-dimethyl-7-deazapurine ring of 16a has moved away from the more hydrophilic pocket occupied by the adenosine of AMP, and sits in a more hydrophobic pocket composed of Met322, Val278 and Ile330 with a small loss in binding affinity observed (ΔG = −54.3843 ± 2.23 kcal mol−1). The tolyl compound 16b fills the phenyl binding pocket as observed with 16a and retains the H-bond between the Phe amine and Glu217, however the rest of the structure then angles away from the adenosine binding pocket and sits in the pocket observed for compound 9d with aryl–cation interactions between the deazapurine ring and Arg319 and water mediated binding interactions between the two nitrogen of the pyrimidine ring and Glu263 and Lys280, resulting in a favourable binding affinity (ΔG = −65.7766 ± 2.71 kcal mol−1) (Fig. 6B). For the naphthyl derivative 16c the N,N-dimethyl-7-deazapurine ring fits the adenosine binding pocket with π–π interaction between the pyrimidine ring and Phe313 and a water mediated H-bonding interaction with Arg319, but in contrast the increase in size of the 2-naphthyl moiety of 16c results in steric limitations with the naphthyl group extending outside the phenyl pocket, which moves the Phe amine away from Glu217 and closer to Glu263 that now forms the H-bonding interaction, resulting in a less favourable binding affinity (ΔG = −44.8425 ± 5.92 kcal mol−1) (Fig. 6C).
 | ||
| Fig. 6 Alignment of (A) 16a (magenta), (B) 16b (brown) and (C) 16c (cyan) with Phe-AMP (gold) in E. faecalis PheS after 200 ns MD simulation, and 2D ligand interaction diagrams. | ||
Conclusions
Nineteen thiadiazole derivatives as Phe-AMP mimics, designed using flexible alignment technique and successfully prepared using efficient 4 to 6-step synthetic pathways. Starting from either benzimidazole in compounds (5a–b), (6a–e), (7a–d) and (9a–e) or N,N-dimethyl-7-deazapurine in compounds (16a–c), following by formation of methyl propanoate analogues which then converted to the corresponding hydrazide. This hydrazide derivative was then cyclised giving thiadiazol-2-amino derivative that either was coupled with aryl methansulphonyl chloride, aromatic acids or with phenylalanine analogues to give the target compounds. Antimicrobial evaluation identified the derivatives of series 3 and 4 containing a phenylalanine or 2-naphthylalanine (9 and 16), which showed modest antimicrobial activity against E. faecalis, as compounds for further investigation through computational studies. Evaluation of the PheS protein–ligand complexes after MD simulation, provided further information with respect to binding interactions and fit within the PheS binding site, which contains the phenylalanine and adenosine/AMP binding pockets.The 4-chlorophenyl and 4-tolyl derivatives of series 3 and 4 (9b/16a and 9d/16b) preferentially bound in the phenylalanine binding site, however, they did not extend into the adenosine binding pocket. The bulkier 2-naphthyl group of derivative 9e bound in the phenylalanine binding site and the benzimidazole moiety was also able to bind in close proximity to the adenosine binding pocket. The more substituted N,N-dimethyl-7-deazapurine ring of compound 16c was the most optimally positioned in the adenosine binding pocket, however the result was a less optimal fit of the 2-naphthyl ring in the phenylalanine binding pocket. For optimal inhibitory activity both pockets should be occupied, and this is best achieved by the 2-naphthyl derivative 9e, which was also reflected in the binding affinity (ΔG = −61.6053 ± 2.86 kcal mol−1). The 2-naphthyl derivatives of series 2 (6e and 7d) with 2–3 atoms between the thiadiazole ring and the aryl ring, were shorter than series 3 and 4 and although both filled the phenylalanine binding site, they were not able to span the active site to reach the adenosine binding pocket (Fig. 7) and also lack the amino acid amine group, with overall lower binding affinities (ΔG = −54.8662 ± 7.34 and −52.8243 ± 2.48 kcal mol−1 respectively) compared with compound 9e. Therefore, from this library of compounds, 9e has been identified as a potential lead for further modification to optimise binding and antimicrobial activity.
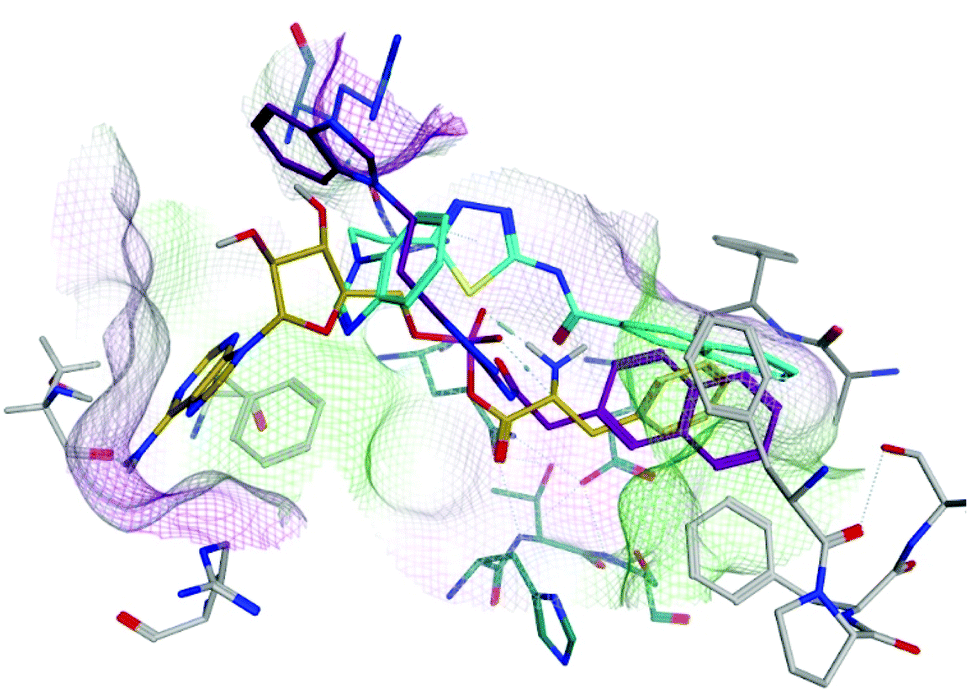 | ||
| Fig. 7 Alignment of 6e (cyan) and 7d (purple) with Phe-AMP (gold) in E. faecalis PheS after 200 ns MD simulation. | ||
Experimental
Chemistry
All reagents and solvents employed were of general purpose or analytical grade and purchased from Fluka, Acros, Alfa-Aeser chemicals and Sigma-Aldrich Chemical Company. Solvents were appropriately dried over molecular sieves (4 Å). 1H and 13C NMR spectra were recorded on a Bruker Advance DP500 spectrometer operating at 500 MHz and 125 MHz respectively. Chemical shifts are given in parts per million (ppm) relative to the internal standard tetramethylsilane (Me4Si). Coupling constants (J value) were calculated in hertz (Hz). Silica gel Fluka Kieselgel 60, particle size 35–70 μm Davisil® chromatography grade, was used for column chromatography in a glass column. Gradient column chromatography was performed with the aid of a pump. Analytical thin layer chromatography (TLC) was carried out on precoated silica plates (ALUGRAM® SIL G/UV254) with visualisation via UV light (254 nm). Melting points were determined using a Gallenkamp melting point apparatus and are uncorrected. HPLC (method A, Cardiff University) was performed on a Shimadzu LC-2030C Plus C18 Rapid Resolution 250 × 4.6 mm, 5 μm particle size using a 7–10 min gradient of water/acetonitrile 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 (method B, University of Bath) was performed on a Zorbax Eclipse Plus C18 rapid resolution 2.1 × 50 mm, 1.8 μm particle size using a 7.5 minutes gradient method 5:95 water:methanol with 0.1% formic acid as additive. Compounds 2, 3 and 12 were prepared as previously described.22,23
:60 (method B, University of Bath) was performed on a Zorbax Eclipse Plus C18 rapid resolution 2.1 × 50 mm, 1.8 μm particle size using a 7.5 minutes gradient method 5:95 water:methanol with 0.1% formic acid as additive. Compounds 2, 3 and 12 were prepared as previously described.22,23
Synthesis of 3-(4-(dimethylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)propanehydrazide (13)
To a solution of the methyl propanoate ester (12) (2.30 g, 9.26 mmol) in EtOH (80 mL) was added hydrazine monohydrate (4.49 mL, 92.6 mmol) and the reaction mixture was stirred at room temperature (25 °C) overnight. The solvent was concentrated under vacuum and the excess hydrazine monohydrate removed by co-evaporated with Et2O (2 × 60 mL) to afford the product as a pink solid, which was used without further purification in the following reaction. Yield: 1.77 g, 77%; m. p. 156–158 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.4. 1H NMR (DMSO-d6) δ: CH2CO is obscured by DMSO-d6 peak, 3.28 (s, 6H, N(CH3)2), 4.18 (bs, 2H, NHN![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 4.35 (t, J = 7.0 Hz, 2H, NCH2), 6.60 (d, J = 3.6 Hz, 1H, Ar), 7.11 (d, J = 3.6 Hz, 1H, Ar), 8.14 (s, 1H, Ar), 9.01 (bs, 1H, NNH2). 13C NMR (DMSO-d6) δ: 34.5 (
2), 4.35 (t, J = 7.0 Hz, 2H, NCH2), 6.60 (d, J = 3.6 Hz, 1H, Ar), 7.11 (d, J = 3.6 Hz, 1H, Ar), 8.14 (s, 1H, Ar), 9.01 (bs, 1H, NNH2). 13C NMR (DMSO-d6) δ: 34.5 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2CO), 39.1 (N(CH3)2), 40.8 (NCH2), 101.5 (CH), 102.9 (C), 124.2 (CH), 150.4 (C), 151.1 (CH), 157.4 (C), 169.5 (C).
H2CO), 39.1 (N(CH3)2), 40.8 (NCH2), 101.5 (CH), 102.9 (C), 124.2 (CH), 150.4 (C), 151.1 (CH), 157.4 (C), 169.5 (C).
Synthesis of 5-(2-(4-(dimethylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1,3,4-thiadiazol-2-amine (14)
To a suspension of hydrazide (13) (0.5 g, 2.01 mmol) in dry IPA (15 mL), was added (trimethylsilyl)isothiocyanate (TMSNCS) (1.14 mL, 8.04 mmol), then the mixture was heated under reflux overnight. The solvent was then concentrated under vacuum and c. H2SO4 (7 mL) was added, and the reaction stirred at room temperature (25 °C), J = 3.7 Hz, 1H, Ar), 8.13 (s, 1H, Ar). 13C NMR (CD3OD) δ: 30.1 (NCH2H2), 38.3 (N(CH3)2), 43.5 (NCH2), 101.9 (CH), 103.3 (C), 123.5 (CH), 149.6 (C), 150.2 (CH), 156.4 (C), 157.3 (C), 170.3 (C).
General method for the preparation of sulfonamide derivatives (5)
To a cooled (0 °C) mixture of 5-(2-(1H-benzo[d]imidazol-1-yl)ethyl)-1,3,4-thiadiazol-2-amine (4) (0.2 g, 0.82 mmol), 4-(dimethylamino)pyridine (DMAP) (0.16 mmol) in dry pyridine (1.5 mL) and dry CH2Cl2 (3 mL) was added the appropriate phenylmethanesulfonyl chloride (1.84 mmol) in portions. The reaction was stirred at 0 °C for 10 min then at room temperature (25 °C) overnight. Then 10% aqueous NaHCO3 (150 mL) was added, and the aqueous layer extracted with of CH2Cl2 (100 mL). The combined organic layer was dried (MgSO4) and the solvent evaporated. The crude residue was purified by gradient chromatography (CH2Cl2–MeOH).:5 v/v and obtained as a brown solid. Yield: 0.030 g (9%); m. p. 214–216 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.60; HPLC (method B): 96% at tR = 3.90 min. 1H NMR (DMSO-d6) δ: 3.33 (t, J = 6.8 Hz, NCH2C2), 4.32 (s, 2H, SO2CH2), 4.56 (t, J = 6.8 Hz, 2H, NCH2), 7.20–7.32 (m, 7H, Ar), 7.63 (dd, J = 16.9 Hz, 8.0 Hz, 2H, Ar), 8.14 (s, 1H, H-imidazole). 13C NMR (DMSO-d6) δ: 30.8 (NCH2H2), 42.7 (NCH2), 59.1 (SO2CH2), 110.9 (CH), 119.9 (CH), 122.1 (CH), 122.8 (CH), 128.4 (CH), 128.6 (2× CH), 130.6 (C), 131.4 (2× CH), 134.00 (C), 143.8 (C), 144.5 (CH-imidazole), 155.3 (C), 168.9 (C).:6 v/v and obtained as a brown solid. Yield: 0.058 g (11%); m. p. 216–218 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.59; HPLC (method B): 91.5% at tR = 4.10 min. 1H NMR (DMSO-d6) δ: 3.33 (t, J = 6.9 Hz, 2H, NCH2C2), 4.36 (s, 2H, SO2CH2), 4.57 (t, J = 6.8 Hz, 2H, NCH2), 7.21 (td, J = 8.1, 1.2 Hz, 1H, Ar), 7.26 (td, J = 8.1, 1.1 Hz, 1H, Ar), 7.36 (m, 4H, Ar), 7.63 (dd, J = 12.4, 8.0 Hz, 2H, Ar), 8.16 (s, 1H, H-imidazole), 13.69 (bs, 1H, NH).13C NMR (DMSO-d6) δ: 30.9 (NCH2H2), 42.8 (NCH2), 58.2 (SO2CH2), 110.9 (CH), 119.9 (CH), 122.1 (CH), 122.9 (CH), 128.6 (2× CH), 129.9 (C), 133.2 (2× CH), 133.3 (C), 134.0 (C), 143.7 (C), 144.5 (CH-imidazole), 155.4 (C), 168.8 (C).General method for the preparation of amide derivatives (6, 7, 8 and 15)
To a solution of 1,3,4-thiadiazol-2-amine derivative (4 or 14) (1 equivalent) in dry DMF (5 mL/0.61 mmol) was added the appropriate carboxylic acid (1.1 equivalents), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) (1.6 equivalents), 1-hydroxybenzotriazole (HOBt) hydrate (1.6 equivalents) and N,N-diisopropylethylamine (DIPEA) (4 equivalents), and the reaction stirred overnight at room temperature (25 °C). The reaction was then poured into H2O (150 mL/0.61 mmol) and the product collected either by filtration or extraction with EtOAc, followed by further purification.:1 v/v, Rf 0.45; HPLC (method A): 97.3%tR = 4.03 min. 1H NMR (DMSO-d6) δ: 3.62 (t, J = 6.9 Hz, 2H, NCH2C2), 4.72 (t, J = 6.8 Hz, 2H, NCH2), 7.21 (t, J = 7.2 Hz, 1H, Ar), 7.27 (t, J = 7.6 Hz, 1H, Ar), 7.56 (t, J = 7.6 Hz, 2H, Ar), 7.66 (m, 3H, Ar), 8.08 (m, 2H, Ar), 8.19 (s, 1H, H-imidazole), 12.98 (bs, 1H, NHCO, ex).13C NMR (DMSO-d6) δ: 29.9 (NCH2H2), 43.8 (NCH2), 110.9 (CH), 119.9 (CH), 122.1 (CH), 122.9 (CH), 128.7 (2× CH), 129.1 (2× CH), 131.9 (C), 133.5 (CH), 143.8 (C), 160.0 (C), 161.15 (C), 165.6 (C).:EtOAc 50:50 v/v to afford the product as a yellow solid. Yield: 0.044 g (19%); m. p. 230–232 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.52; HPLC (method A): 96.7% tR = 4.14 min. 1H NMR (DMSO-d6) δ: 3.60 (t, J = 6.8 Hz, 2H, NCH2C2), 3.85 (s, 3H, OCH3), 4.71 (t, J = 6.8 Hz, 2H, NCH2), 7.06 (d, J = 8.8 Hz, 2H, Ar), 7.21 (t, J = 7.2 Hz, 1H, Ar), 7.27 (t, J = 7.4 Hz, 1H, Ar), 7.66 (m, 2H, Ar), 8.09 (d, J = 8.7 Hz, 2H, Ar), 8.17 (s, 1H, H-imidazole), 12.76 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 30.0 (NCH2H2), 43.7 (NCH2), 56.0 (OCH3), 110.9 (CH), 114.4 (2× CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 124.3 (C), 130.9 (2× CH), 134.1 (C), 143.9 (C), 144.6 (CH-imidazole), 147.6 (C), 160.7 (C), 163.4 (C), 165.0 (C).:5 v/v as a yellow solid. Yield: 0.108 g (46%); m. p. 254–256 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.5; HPLC (method A): 97.6% tR = 4.407.1H NMR (DMSO-d6) δ: 3.61 (t, J = 6.9 Hz, 2H, NCH2C2), 4.71 (t, J = 6.9 Hz, 2H, NCH2), 7.21 (td, J = 7.4, 1.1 Hz, 1H, Ar), 7.27 (td, J = 8.1, 1.1 Hz, 1H, Ar), 7.65 (m, 4H, Ar), 8.09 (d, J = 8.7 Hz, 2H, Ar), 8.17 (s, 1H, H-imidazole), 13.08 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 30.0 (NCH2H2), 43.7 (NCH2), 110.9 (CH), 119.7 (CH), 122.1 (CH), 122.9 (CH), 129.2 (2× CH), 130.8 (2× CH), 131.1 (C), 134.0 (C), 138.3 (C), 143.9 (C), 144.5 (CH-imidazole), 160.5 (C), 161.1 (C), 165.0 (C).:10 v/v to provide the product as a yellow solid. Yield: 0.024 g (10%); m. p. 228–230 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.68; HPLC (method A): 98.7% tR = 4.82 min. 1H NMR (DMSO-d6) δ: 3.66 (t, J = 6.9 Hz, 2H, NCH2C2), 4.76 (t, J = 6.8 Hz, 2H, NCH2), 7.24 (t, J = 7.3 Hz, 1H, Ar), 7.30 (t, J = 7.6 Hz, 1H, Ar), 7.63 (m, 3H, Ar), 7.67 (d, J = 7.9 Hz, 1H, Ar), 7.71 (d, J = 8.0 Hz, 1H, Ar), 7.90 (d, J = 6.6 Hz, 1H, Ar), 8.05 (m, 1H, Ar), 8.14 (d, J = 8.2 Hz, 1H, Ar), 8.20 (m, 1H, Ar), 8.27 (s, 1H, H-imidazole), 13.10 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 29.9 (NCH2H2), 43.9 (NCH2), 111.1 (CH), 119.8 (CH), 122.3 (CH), 123.0 (CH), 125.3 (CH), 125.3 (CH), 127.0 (CH), 127.7 (CH), 128.0 (2× CH), 129.0 (CH), 130.2 (C), 131.2 (C), 132.2 (CH-imidazole), 133.6 (C), 133.9 (C), 143.4 (C), 159.6 (C), 161.1 (C), 167.4 (C).:1 v/v, Rf 0.74; HPLC (method A): 99% tR = 5.17 min. 1H NMR (DMSO-d6) δ: 3.63 (t, J = 6.9 Hz, 2H, NCH2C2), 4.73 (t, J = 6.8 Hz, 2H, NCH2), 7.22 (td, J = 8.2, 1.10 Hz, 1H, Ar), 7.28 (td, J = 8.2, 1.2 Hz, 1H, Ar), 7.63–7.71 (m, 4H, Ar), 8.02–8.12 (m, 4H, Ar), 8.19 (s, 1H, H-imidazole), 8.79 (s, 1H, Ar), 13.12 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 30.0 (NCH2H2), 43.8 (NCH2), 110.9 (CH), 119.9 (CH), 122.1 (CH), 122.9 (CH), 125.0 (CH), 127.6 (CH), 128.2 (CH), 128.7 (CH), 129.0 (CH), 129.4 (C), 129.7 (CH), 130.0 (CH), 132.4 (C), 134.1 (C), 135.3 (C), 143.9 (C), 144.6 (CH-imidazole), 160.4 (C), 161.0 (C), 165.8 (C).:5 v/v + 1% Et3N as an off-white solid. Yield: 0.11 (46%); m. p. 222–224 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.52; HPLC (method B): 100% tR = 4.10 min. 1H NMR (DMSO-d6) δ: 3.56 (t, J = 6.9 Hz, 2H, NCH2C2), 3.70 (s, 2H, COCH2), 3.73 (s, 3H, OCH3), 4.66 (t, J = 6.8 Hz, 2H, NCH2), 6.88 (d, J = 8.8 Hz, 2H, Ar), 7.21 (m, 4H, Ar), 7.62 (d, J = 8.7 Hz, 2H, Ar), 8.13 (s, 1H, H-imidazole), 12.62 (bs, 1H, NHCO).13C NMR (DMSO-d6) δ: 29.9 (NCH2H2), 41.1 (COH2), 43.7 (NCH2), 55.5 (OCH3), 110.9 (CH), 114.3 (2× CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 126.9 (C), 130.8 (2× CH), 134.0 (C), 143.9 (C), 144.5 (CH-imidazole), 158.7 (C), 159.2 (C), 160.8 (C), 170.2 (C).:5% v/v + 1% Et3N as an off-white solid. Yield: 0.069 g (43%); m. p. 228–230 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.6; HPLC (method B): 95.9% at tR = 4.30 min. 1H NMR (DMSO-d6) δ: 3.56 (t, J = 6.9 Hz, 2H, NCH2C2), 3.80 (s, 2H, COCH2), 4.66 (t, J = 6.8 Hz, 2H, NCH2), 7.20 (td, J = 7.3, 0.7 Hz, 2H, Ar), 7.25 (td, J = 7.8, 1.3 Hz, 1H, Ar), 7.32 (d, J = 8.5 Hz, 2H, Ar), 7.38 (d, J = 8.5 Hz, 2H, Ar), 7.62 (d, J = 9.1 Hz, 2H, Ar), 8.13 (s, 1H, CH-imidazole), 12.69 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 29.9 (NCH2H2), 41.2 (COH2), 43.7 (NCH2), 110.9 (CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 128.8 (2× CH), 131.7 (2× CH), 132.1 (C), 134.0 (2× C), 143.9 (C), 144.5 (CH-imidazole), 159.2 (C), 160.8 (C), 169.6 (C).:5 v/v to provide the product as an off-white solid. Yield: 0.039 g (22%); m. p. 190–192 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.55; HPLC (method B): 96.79% tR = 4.30 min. 1H NMR (DMSO-d6) δ: 3.55 (t, J = 6.9 Hz, 2H, NCH2C2), 4.30 (s, 2H, COCH2), 4.65 (t, J = 6.8 Hz, 2H, NCH2), 7.19 (td, J = 7.2, 1.2 Hz, 1H, Ar), 7.24 (td, J = 8.3, 1.2 Hz, 1H, Ar), 7.49 (m, 2H, Ar), 7.55 (m, 2H, Ar), 7.61 (m, 2H, Ar), 7.87 (dd, J = 6.8, 2.6 Hz, 1H, Ar), 7.94 (dd, J = 7.5, 1.7 Hz, 1H, Ar), 8.04 (dd, J = 8.1, 1.0 Hz, 1H, Ar), 8.12 (s, 1H, H-imidazole), 12.84 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 30.0 (NCH2H2), COH2 peak is obscured by peak of DMSO-d6, 43.7 (NCH2), 110.9 (CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 124.5 (CH), 126.0 (CH), 126.3 (CH), 126.8 (CH), 128.1 (CH), 128.6 (CH), 129.0 (CH), 131.5 (C), 132.3 (C), 133.8 (C), 134.0 (C), 143.9 (C), 144.5 (CH-imidazole), 159.2 (C), 160.8 (C), 169.8 (C).:5 v/v to provide the product as an off-white solid. Yield: 0.038 g (23%); m. p. 230–232 °C; TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.56; HPLC (method B): 92.4% tR = 4.40 min. 1H NMR (DMSO-d6) δ: 3.55 (t, J = 6.9 Hz, 2H, NCH2C2), 3.98 (2H, s, COCH2), 4.66 (t, J = 6.8 Hz, 2H, NCH2), 7.19 (td, J = 7.2, 0.8 Hz, 1H, Ar), 7.24 (td, J = 7.8, 1.3 Hz, 1H, Ar), 7.49 (m, 3H, Ar), 7.62 (dd, J = 9.2 Hz, 1H, Ar), 7.82 (s, 1H, Ar), 7.89 (m, 3H, Ar), 8.13 (s, 1H, H-imidazole), 12.76 (s, 1H, NHCO); 13C NMR (DMSO-d6) δ: 29.9 (NCH2H2), 42.1 (COH2), 43.7 (NCH2), 110.9 (CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 126.3 (CH), 126.7 (CH), 128.0 (CH), 128.0 (CH), 128.1 (CH), 128.3 (CH), 128.4 (CH), 132.4 (C), 132.7 (C), 133.4 (C), 134.0 (C), 143.9 (C), 144.5 (CH-imidazole), 159.2 (C), 160.8 (C), 169.9 (C).:1 v/v, Rf 0.63. 1H NMR (DMSO-d6) δ: 1.31 (s, 9H, C(CH3)3), 2.80 (dd, J = 12.9, 11.5 Hz, 1H, CHC2), 2.98 (dd, J = 13.2, 2.7 Hz, 1H, CHC2), 3.58 (t, J = 6.8 Hz, 2H, NCH2C2), 4.43 (m, 1H, CCH2), 4.69 (t, J = 6.7 Hz, 2H, NCH2), 7.20–7.33 (m, 8H, Ar + CHNCO), 7.65 (m, 2H, Ar), 8.17 (s, 1H, H-imidazole), 12.68 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 28.6 (C(H3)3), 30.0 (NCH2H2), 37.2 (CHH2), 43.7 (NCH2), 56.5 (HCH2), 78.8 ((CH3)3), 110.9 (CH), 119.9 (CH), 122.1 (CH), 122.8 (CH), 126.9 (CH), 128.6 (2× CH), 129.7 (2× CH), 134.0 (C), 137.9 (C), 143.8 (C), 144.5 (CH-imidazole), 155.9 (C), 159.1 (C), 160.9 (C), 171.8 (C).:1 v/v, Rf 0.63; 1H NMR (DMSO-d6) δ: 1.31 (s, 9H, C(CH3)3), 2.79 (dd, J = 13.4, 10.9 Hz, CHC2), 2.99 (d, J = 13.5, 4.0 Hz, 1H, CHC2), 3.58 (t, J = 6.9 Hz, 2H, NCH2C2), 4.41 (m, 1H, CCH2), 4.69 (t, J = 6.9 Hz, 2H, NCH2), 7.21 (t, J = 7.4 Hz, 1H, Ar), 7.26 (t, J = 7.3 Hz, 1H, Ar), 7.35 (m, 5H, Ar + CHNCO), 7.65 (m, 2H, Ar), 8.16 (s, 1H, CH-imidazole), 12.70 (s, 1H, NHCO, ex); 13C NMR (DMSO-d6) δ: 28.6 (C(H3)3), 30.0 (NCH2H2), 36.5 (CHH2), 43.7 (NCH2), 56.4 (HCH2), 78.9 ((CH3)3), 110.9 (CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 128.5 (2× CH), 131.6 (2× CH), 134.1 (C), 137.0 (C), 143.9 (C), 144.5 (CH-imidazole), 155.9 (C), 159.1 (C), 161.0 (C), 171.6 (C).:1 v/v, Rf 0.5. 1H NMR (DMSO-d6) δ: 1.33 (s, 9H, C(CH3)3), 2.91 (m, 2H, CHC2), 3.57 (t, J = 6.9 Hz, 2H, NCH2C2), 3.68 (s, 3H, OCH3), 4.43 (m, 1H, CCH2), 4.68 (t, J = 6.8 Hz, 2H, NCH2), 6.81 (t, J = 7.4 Hz, 1H, Ar), 6.91 (d, J = 8.2 Hz, 1H, Ar), 7.01 (d, J = 7.4 Hz, 1H, Ar), 7.07 (d, J = 7.1 Hz, 1H, Ar), 7.23 (m, 3H, Ar + CHNCO), 7.64 (d, J = 8.3 Hz, 2H, Ar), 8.16 (s, 1H, H-imidazole), 12.41 (s, 1H, NHCO).13C NMR (DMSO-d6) δ: 28.6 (C(H3)3), 30.0 (NCH2H2), 32.3 (CHH2), 43.7 (NCH2), 54.8 (HCH2), 55.7 (OCH3), 78.9 ((CH3)3), 110.9 (CH), 111.0 (CH), 119.9 (CH), 120.5 (CH), 122.0 (CH), 122.8 (CH), 125.0 (C), 128.6 (CH), 130.9 (CH), 134.1 (C), 143.8 (C), 144.5 (CH-imidazole), 155.5 (C), 157.8 (C), 159.1 (C), 160.8 (C), 171.8 (C).:1 v/v, Rf 0.48. 1H NMR (DMSO-d6) δ: 1.31 (s, 9H, C(CH3)3), 2.25 (s, 3H, CH3), 2.76 (dd, J = 13.8, 10.9 Hz, 1H, CHC2), 2.93 (dd, J = 13.5, 4.3 Hz, 1H, CHC2), 3.58 (t, J = 6.9 Hz, 2H, NCH2C2), 4.39 (m, 1H, CCH2), 4.69 (t, J = 6.9 Hz, 2H, NCH2), 7.07 (m, 2H, Ar), 7.22 (m, 5H, Ar + CHNCO), 7.64 (m, 2H, Ar), 8.16 (s, 1H, H-imidazole), 12.67 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 21.13 (CH3), 28.58 (C(H3)3), 29.98 (NCH2H2), 36.84 (CHH2), 43.68 (NCH2), 56.66 (HCH2), 78.8 ((CH3)3), 110.9 (CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 129.2 (2× CH), 129.60(CH), 134.1(C), 134.8 (C), 135.9 (C), 143.9 (C), 144.5 (CH-imidazole), 155.9 (C), 159.1 (C), 160.9 (C), 171.9 (C).:1 v/v, Rf 0.53. 1H NMR (DMSO-d6) δ: 1.27 (s, 9H, C(CH3)3), 2.98 (dd, J = 13.3, 9.3 Hz, 1H, CHC2), 3.18 (dd, J = 13.4, 4.2 Hz, 1H, CHC2), 3.59 (t, J = 6.9 Hz, 2H, NCH2C2), 4.56 (m, 1H, CCH2), 4.69 (t, J = 6.9 Hz, 2H, NCH2), 7.21 (t, J = 7.5 Hz, 1H, Ar), 7.26 (t, J = 7.5 Hz, 1H, Ar), 7.38 (d, J = 8.0 Hz, 1H, CHNCO), 7.48 (m, 3H, Ar), 7.64 (m, 2H, Ar), 7.83 (m, 4H, Ar), 8.17 (s, 1H, H-imidazole), 12.74 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 28.5 (C(H3)3), 30.0 (NCH2H2), 37.4 (CHH2), 43.7 (NCH2), 56.5 (HCH2), 78.9 ((CH3)3), 110.9 (CH), 119.9 (CH), 122.0 (CH), 122.8 (CH), 126.0 (CH), 126.5 (CH), 127.8 (CH), 127.97 (CH), 128.01 (CH), 128.1 (CH), 128.2 (CH), 132.4 (C), 133.4 (C), 134.1 (C), 135.6 (C), 143.9 (C), 144.5 (CH-imidazole), 155.9 (C), 159.1 (C), 160.9 (C), 171.8 (C).:1 v/v, Rf 0.69. 1H NMR (CD3OD) δ: 1.39 (s, 9H, C(C3)3), 2.91 (dd, J = 13.8 Hz, 9.0 Hz, CHC2), 3.10 (m, 1H, CHC2), 3.37 (s, 6H, N(C3)2), 3.58 (t, J = 6.6 Hz, 2H, NCH2C2), 4.52 (t, J = 6.6 Hz, 1H, CCH2), 4.61 (t, J = 6.6 Hz, 2H, NCH2), 6.67 (d, J = 3.7 Hz, 1H, Ar), 7.03 (d, J = 3.6 Hz, 1H, Ar), 7.21 (d, J = 8.5 Hz, 2H, Ar), 7.26 (d, J = 8.5 Hz, 2H, Ar), 8.13 (s, 1H, Ar).13C NMR (CD3OD) δ: 27.2 (C(H3)3), 29.7 (NCH2H2), 36.9 (CHH2), 38.3 (N(H3)2), 43.6 (NCH2), 55.8 (HCH2), 79.5 ((CH3)3), 102.0 (CH), 103.3 (C), 123.4 (CH), 128.1 (CH), 130.6 (CH), 132.4 (C), 135.3 (C), 149.6 (C), 150.3 (CH), 157.4 (C), 159.2 (C), 162.1 (C), 170.9 (C).:10 v/v) to afford the product as a green oil, which was used without any further characterisation in the next reaction. Yield: 0.06 g (29%); TLC: CH2Cl2–MeOH 9:1 v/v, Rf 0.71.:1 v/v, Rf 0.77. 1H NMR (DMSO-d6) δ: 1.27 (s, 9H, C(CH3)3), 2.98 (m, 1H, CHC2), 3.17 (m, 1H, CHC2), 3.29 (s, 6H, N(CH3)2), 3.53 (t, J = 7 Hz, 2H, NCH2C2), 4.54 (t, J = 7 Hz, 3H, NCH2+CCH2), 6.62 (d, J = 3.6 Hz, 1H, Ar), 7.19 (d, J = 3.6 Hz, 1H, Ar), 7.37 (d, J = 7.9 Hz, 1H, Ar), 7.48 (m, 3H, Ar), 7.84 (m, 4H, Ar), 8.14 (s, 1H, Ar), 12.71 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 28.5 (C(H3)3), 30.2 (NCH![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) H2), 37.4 (CHH2), 39.1 (N(CH3)2), 43.7 (NCH2), 56.5 (HCH2), 78.9 ((CH3)3), 101.8 (CH), 103.0 (C), 124.2 (CH), 126.0 (CH), 126.5 (CH), 127.8 (CH), 128.0 (CH), 128.0 (CH), 128.1 (CH), 128.2 (CH), 132.4 (C), 133.4 (C), 135.6 (C), 150.7 (C), 151.2 (CH), 155.9 (C), 157.4 (C), 159.0 (C), 161.8 (C), 171.8 (C).
H2), 37.4 (CHH2), 39.1 (N(CH3)2), 43.7 (NCH2), 56.5 (HCH2), 78.9 ((CH3)3), 101.8 (CH), 103.0 (C), 124.2 (CH), 126.0 (CH), 126.5 (CH), 127.8 (CH), 128.0 (CH), 128.0 (CH), 128.1 (CH), 128.2 (CH), 132.4 (C), 133.4 (C), 135.6 (C), 150.7 (C), 151.2 (CH), 155.9 (C), 157.4 (C), 159.0 (C), 161.8 (C), 171.8 (C).General method for preparation of aminium salts (9) and (16)
To a flask containing the N-Boc protected compound (8 or 15) (0.53 mmol) was added CH2Cl2–TFA (75:25 v/v, 20 mL/0.53 mmol) and the reaction stirred at room temperature (25 °C) overnight. The solvent was evaporated and the remaining TFA was removed by co-evaporation with EtOH (3 × 100 mL/0.53 mmol). Et2O (75 mL/0.53 mmol) was added to the residue, which was stirred for 2 h, then the formed solid collected by filtration.
:1 v/v, Rf 0.61. HPLC (method A): 98.4% tR = 3.42 min. 1H NMR (DMSO-d6) δ: 3.08 (dd, J = 13.8, 7.7 Hz, 1H, CHC2), 3.17 (dd, J = 13.8, 6.4 Hz, 1H, CHC2), 3.68 (t, J = 6.8 Hz, 2H, NCH22), 4.31 (t, J = 6.6 Hz, 1H, CCH2), 4.83 (t, J = 6.7 Hz, 2H, NCH2), 7.20 (d, J = 6.8 Hz, 1H, Ar), 7.30 (m, 3H, Ar), 7.44 (m, 2H, Ar), 7.77 (d, J = 8.1 Hz, 1H, Ar), 7.85 (d, J = 7.2 Hz, 1H, Ar), 8.49 (bs, 3H, NH3+), 8.90 (s, 1H, H-imidazole), 13.04 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 29.5 (NCH2H2), 37.2 (CHH2), 44.6 (NCH2), 54.4 (HCH2), 112.4 (CH), 117.6 (CH), 124.6 (CH), 124.8 (CH), 127.9 (CH), 129.1 (2× CH), 129.9 (2× CH), 132.7 (C), 134.7 (2× C), 143.5 (CH-imidazole), 158.7 (C), 161.3 (C), 168.0 (C).:1 v/v, Rf 0.64; HPLC (method A): 95.2% tR = 3.78 min. 1H NMR (DMSO-d6) δ: 3.06 (dd, J = 14.0, 8.0 Hz, 1H, CHC2), 3.19 (dd, J = 14.0, 5.9 Hz, 1H, CHC2), CH2 peak is obscured by DMSO-d6 peak, 4.30 (t, J = 6.8 Hz, 1H, CCH2), 4.84 (t, J = 6.9 Hz, 2H, NCH2), 7.23 (d, J = 8.4 Hz, 2H, Ar), 7.42 (m, 5H, Ar), 7.77 (dd, J = 6.9, 1.2 Hz, 1H, Ar), 7.85 (dd, J = 7.3, 0.9 Hz, 1H, Ar), 8.48 (bs, 3H, NH3+), 8.88 (s, 1H, H-imidazole), 13.11 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 29.5 (NCH2H2), 36.4 (CHH2), 44.6 (NCH2), 54.3 (HCH2), 112.4 (CH), 117.7 (CH), 124.6 (CH), 124.8 (CH), 129.1 (2× CH), 131.8 (2× CH), 132.6 (2× C), 133.8 (2× C), 143.5 (CH-imidazole), 158.7 (C), 161.3 (C), 167.9 (C).:1 v/v, Rf 0.61; HPLC (method A): 98.8% at tR = 3.45 min. 1H NMR (DMSO-d6) δ: 3.05 (dd, J = 13.5, 7.8 Hz, 1H, CHC2), 3.17 (dd, J = 13.5, 5.4 Hz, 1H, CHC2), 3.53 (s, 3H, OCH3), 3.67 (t, J = 6.8 Hz, 2H, NCH2C2), 4.23 (t, J = 6.4 Hz, 1H, CCH2), 4.81 (t, J = 6.7 Hz, 2H, NCH2), 6.85 (td, J = 7.5, 0.7 Hz, 1H, Ar), 6.90 (d, J = 8.1 Hz, 1H, Ar), 7.26 (td, J = 7.4, 1.5 Hz, 1H, Ar), 7.26 (dt, J = 8.3, 1.7 Hz, 1H, Ar), 7.40 (quintet, 2H, Ar), 7.76 (d, J = 7.6 Hz, 1H, Ar), 7.81 (d, J = 7.5 Hz, 1H, Ar), 8.47 (bs, 3H, NH3+), 8.75 (s, 1H, H-imidazole), 12.90 (s, 1H, NHCO). 13C NMR (DMSO-d6) δ: 29.5 (NCH2H2), 37.0 (CHH2), 44.4 (NCH2), 53.2 (HCH2), 55.6 (OCH3), 111.1 (CH), 112.2 (CH), 118.1 (CH), 120.8 (CH), 122.8 (2× C), 124.2 (CH), 124.4 (CH), 129.6 (CH), 131.6 (CH), 132.9 (C), 143.6 (CH-imidazole), 157.9 (C), 158.7 (C), 161.2 (C), 168.1 (C).:1 v/v, Rf 0.51; HPLC (method B): 94.8% tR = 3.80 min; 1H NMR (DMSO-d6) δ: 2.51 (s, 3H, CH3), 3.04 (dd, J = 13.8, 7.6, 1H, 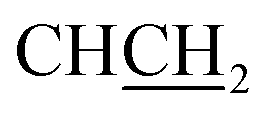 ), 3.13 (dd, J = 13.8, 6.2 Hz, 1H,
), 3.13 (dd, J = 13.8, 6.2 Hz, 1H, 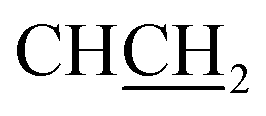 ), 3.66 (t, J = 6.7 Hz, 2H,
), 3.66 (t, J = 6.7 Hz, 2H, 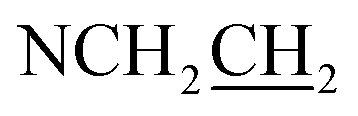 ), 4.28 (t, J = 6.7 Hz, 1H,
), 4.28 (t, J = 6.7 Hz, 1H, 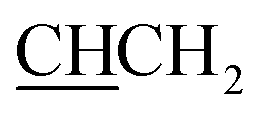 ), 4.81 (t, J = 6.4 Hz, 2H, NCH2), 7.12 (m, 4H, Ar), 7.40 (quintet, 2H, Ar), 7.75 (d, J = 7.6 Hz, 1H, Ar), 7.81 (d, J = 7.6 Hz, 1H, Ar), 8.47 (bs, 3H, NH3+), 8.75 (s, 1H, H-imidazole), 13.08 (s, 1H, NHCO).13C NMR (DMSO-d6) δ: 21.1 (CH3), 29.6 (NCH2CH2), 36.7 (CHCH2), 44.4 (NCH2), 54.4 (CHCH2), 112.2 (CH), 118.0 (CH), 124.2 (CH), 124.5 (CH), 129.7 (2× CH), 129.7 (2× CH), 131.6 (C), 132.9 (C), 137.0 (C), 138.3 (C), 143.6 (CH-imidazole), 158.7 (C), 161.3 (C), 168.1 (C).:1 v/v, Rf 0.58; HPLC (method B): 97.96% tR = 3.90 min. 1H NMR (DMSO-d6) δ: 3.24 (dd, J = 14.0 Hz, 8.0 Hz, 1H, CHC2), 3.38 (m, 1H, CHC2), 3.67 (t, J = 6.9 Hz, 2H, NCH2C2), 4.41 (t, J = 6.9 Hz, 1H, CCH2), 4.81 (t, J = 7.3 Hz, 2H, NCH2), 7.39 (m, 3H, Ar), 7.52 (m, 2H, Ar), 7.76 (m, 2H, Ar), 7.87 (m, 4H, Ar), 8.49 (bs, 3H, NH3+), 8.76 (s, 1H, H-imidazole), 13.15 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 29.5 (NCH2H2), 37.3 (CHH2), 44.7 (NCH2), 54.4 (HCH2), 112.5 (CH), 117.5 (CH), 124.8 (CH), 124.9 (CH), 126.5 (CH), 126.8 (CH), 127.7 (CH), 128.0 (2× CH), 128.7 (CH), 128.9 (CH), 132.4 (C), 132.6 (C), 132.9 (C), 133.4 (C), 143.4 (CH-imidazole), 158.8 (C), 161.2 (C), 168.1 (C), 170.9 (C).:1 v/v, Rf 0.69; HPLC (method B): 97.05% tR = 3.80 min 1H NMR (CD3OD) δ: 3. 18 (dd, J = 14.1, 8.0 Hz, 1H, CHC2), CHC2 is obscured by CD3OD peak, 3.52 (s, 6H, N(CH3)2), 3.65 (t, J = 6.7 Hz, 2H, NCH2C2), 4.38 (t, J = 7.9 Hz, 1H, CCH2), 4.76 (t, J = 6.8 Hz, 2H, NCH2), 6.97 (d, J = 3.7 Hz, 1H, Ar), 7.25 (d, J = 8.4 Hz, 2H, Ar), 7.37 (m, 3H, Ar), 8.22 (s, 1H, Ar). 13C NMR (CD3OD) δ: 29.6 (NCH2H2), 36.2 (CHH2), 39.8 (N(CH3)2), 44.0 (NCH2), 54.4 (HCH2), 102.3 (C), 104.2 (CH), 126.4 (CH), 128.9 (2× CH), 130.7 (2× CH), 132.4 (C), 133.6 (C), 142.8 (CH), 147.3 (C), 161.9 (C).:1 v/v, Rf 0.66; HPLC (method B): 94.03% tR = 3.70 min 1H NMR (CD3OD) δ: 2.20 (s, 3H, CH3), 3.01 (dd, J = 14.1, 8.1, 1H, CHC2), 3.16 (dd, J = 14.0, 6.4 Hz, 1H, CHC2), 3.41 (s, 6H, N(CH3)2), 3.53 (t, J = 6.7 Hz, 2H, NCH2C2), 4.23 (t, J = 6.5 Hz, 1H, CCH2), 4.64 (t, J = 6.7 Hz, 2H, NCH2), 6.86 (d, J = 3.7 Hz, 1H, Ar), 7.03 (m, 4H, Ar), 7.27 (d, J = 3.70 Hz, 1H, Ar), 8.10 (s, 1H, Ar). 13C NMR (CD3OD) δ: 19.7 (CH3), 29.6 (NCH2H2), 36.6 (CHH2), 39.9 (N(CH3)2), 44.0 (NCH2), 54.7 (HCH2), 102.3 (C), 104.3 (CH), 126.6 (CH), 128.9 (2× CH), 129.4 (2× CH), 130.3 (C), 137.6 (C), 142.5 (CH), 147.2 (C), 151.3 (C), 159.1 (C), 161.9 (C), 167.6 (C).:1 v/v, Rf 0.66; HPLC (method B): 91.73% tR = 3.90 min 1H NMR (DMSO-d6) δ: 3.24 (dd, J = 14.0, 8.0 Hz, 1H, CHC2), 3.32 CHC2 peak is obscured by N(CH3)2 peak, 3.36 (s, 6H, N(CH3)2), 3.59 (t, J = 7.0 Hz, 2H, NCH2C2), 4.29 (t, J = 6.7 Hz, 1H, CCH2), 4.61 (t, J = 6.9 Hz, 2H, NCH2), 6.81 (d, J = 3.3 Hz, 1H, Ar), 7.38 (m, 2H, Ar), 7.52 (m, 2H, Ar), 7.76 (s, 1H, Ar), 7.87 (m, 3H, Ar), 8.25 (s, 1H, Ar), 8.50 (bs, 3H, NH3+), 13.09 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 30.2 (NCH2H2), 37.3 (CHH2), N(CH3)2 peak is obscured by DMSO-d6 peak, 43.9 (NCH2), 54.4 (HCH2), 102.5 (C), 103.4 (CH), 125.8 (CH), 126.5 (CH), 126.8 (CH), 127.7 (CH), 128.0 (CH), 128.7 (CH), 128.9 (CH), 132.4 (C), 132.8 (C), 132.9 (CH), 133.4 (C), 148.6 (C), 161.7 (C), 168.1 (C), 171.0 (C).
), 4.81 (t, J = 6.4 Hz, 2H, NCH2), 7.12 (m, 4H, Ar), 7.40 (quintet, 2H, Ar), 7.75 (d, J = 7.6 Hz, 1H, Ar), 7.81 (d, J = 7.6 Hz, 1H, Ar), 8.47 (bs, 3H, NH3+), 8.75 (s, 1H, H-imidazole), 13.08 (s, 1H, NHCO).13C NMR (DMSO-d6) δ: 21.1 (CH3), 29.6 (NCH2CH2), 36.7 (CHCH2), 44.4 (NCH2), 54.4 (CHCH2), 112.2 (CH), 118.0 (CH), 124.2 (CH), 124.5 (CH), 129.7 (2× CH), 129.7 (2× CH), 131.6 (C), 132.9 (C), 137.0 (C), 138.3 (C), 143.6 (CH-imidazole), 158.7 (C), 161.3 (C), 168.1 (C).:1 v/v, Rf 0.58; HPLC (method B): 97.96% tR = 3.90 min. 1H NMR (DMSO-d6) δ: 3.24 (dd, J = 14.0 Hz, 8.0 Hz, 1H, CHC2), 3.38 (m, 1H, CHC2), 3.67 (t, J = 6.9 Hz, 2H, NCH2C2), 4.41 (t, J = 6.9 Hz, 1H, CCH2), 4.81 (t, J = 7.3 Hz, 2H, NCH2), 7.39 (m, 3H, Ar), 7.52 (m, 2H, Ar), 7.76 (m, 2H, Ar), 7.87 (m, 4H, Ar), 8.49 (bs, 3H, NH3+), 8.76 (s, 1H, H-imidazole), 13.15 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 29.5 (NCH2H2), 37.3 (CHH2), 44.7 (NCH2), 54.4 (HCH2), 112.5 (CH), 117.5 (CH), 124.8 (CH), 124.9 (CH), 126.5 (CH), 126.8 (CH), 127.7 (CH), 128.0 (2× CH), 128.7 (CH), 128.9 (CH), 132.4 (C), 132.6 (C), 132.9 (C), 133.4 (C), 143.4 (CH-imidazole), 158.8 (C), 161.2 (C), 168.1 (C), 170.9 (C).:1 v/v, Rf 0.69; HPLC (method B): 97.05% tR = 3.80 min 1H NMR (CD3OD) δ: 3. 18 (dd, J = 14.1, 8.0 Hz, 1H, CHC2), CHC2 is obscured by CD3OD peak, 3.52 (s, 6H, N(CH3)2), 3.65 (t, J = 6.7 Hz, 2H, NCH2C2), 4.38 (t, J = 7.9 Hz, 1H, CCH2), 4.76 (t, J = 6.8 Hz, 2H, NCH2), 6.97 (d, J = 3.7 Hz, 1H, Ar), 7.25 (d, J = 8.4 Hz, 2H, Ar), 7.37 (m, 3H, Ar), 8.22 (s, 1H, Ar). 13C NMR (CD3OD) δ: 29.6 (NCH2H2), 36.2 (CHH2), 39.8 (N(CH3)2), 44.0 (NCH2), 54.4 (HCH2), 102.3 (C), 104.2 (CH), 126.4 (CH), 128.9 (2× CH), 130.7 (2× CH), 132.4 (C), 133.6 (C), 142.8 (CH), 147.3 (C), 161.9 (C).:1 v/v, Rf 0.66; HPLC (method B): 94.03% tR = 3.70 min 1H NMR (CD3OD) δ: 2.20 (s, 3H, CH3), 3.01 (dd, J = 14.1, 8.1, 1H, CHC2), 3.16 (dd, J = 14.0, 6.4 Hz, 1H, CHC2), 3.41 (s, 6H, N(CH3)2), 3.53 (t, J = 6.7 Hz, 2H, NCH2C2), 4.23 (t, J = 6.5 Hz, 1H, CCH2), 4.64 (t, J = 6.7 Hz, 2H, NCH2), 6.86 (d, J = 3.7 Hz, 1H, Ar), 7.03 (m, 4H, Ar), 7.27 (d, J = 3.70 Hz, 1H, Ar), 8.10 (s, 1H, Ar). 13C NMR (CD3OD) δ: 19.7 (CH3), 29.6 (NCH2H2), 36.6 (CHH2), 39.9 (N(CH3)2), 44.0 (NCH2), 54.7 (HCH2), 102.3 (C), 104.3 (CH), 126.6 (CH), 128.9 (2× CH), 129.4 (2× CH), 130.3 (C), 137.6 (C), 142.5 (CH), 147.2 (C), 151.3 (C), 159.1 (C), 161.9 (C), 167.6 (C).:1 v/v, Rf 0.66; HPLC (method B): 91.73% tR = 3.90 min 1H NMR (DMSO-d6) δ: 3.24 (dd, J = 14.0, 8.0 Hz, 1H, CHC2), 3.32 CHC2 peak is obscured by N(CH3)2 peak, 3.36 (s, 6H, N(CH3)2), 3.59 (t, J = 7.0 Hz, 2H, NCH2C2), 4.29 (t, J = 6.7 Hz, 1H, CCH2), 4.61 (t, J = 6.9 Hz, 2H, NCH2), 6.81 (d, J = 3.3 Hz, 1H, Ar), 7.38 (m, 2H, Ar), 7.52 (m, 2H, Ar), 7.76 (s, 1H, Ar), 7.87 (m, 3H, Ar), 8.25 (s, 1H, Ar), 8.50 (bs, 3H, NH3+), 13.09 (bs, 1H, NHCO). 13C NMR (DMSO-d6) δ: 30.2 (NCH2H2), 37.3 (CHH2), N(CH3)2 peak is obscured by DMSO-d6 peak, 43.9 (NCH2), 54.4 (HCH2), 102.5 (C), 103.4 (CH), 125.8 (CH), 126.5 (CH), 126.8 (CH), 127.7 (CH), 128.0 (CH), 128.7 (CH), 128.9 (CH), 132.4 (C), 132.8 (C), 132.9 (CH), 133.4 (C), 148.6 (C), 161.7 (C), 168.1 (C), 171.0 (C).Microbiological evaluation
Compounds (5a–b), (6a–e), (7a–d), (9a–e) and (16a–c) were evaluated for antimicrobial activity against Staphylococcus aureus (ATCC 29213), Enterococcus faecalis (ATCC 29212), Pseudomonas aeruginosa (ATCC 29853), Escherichia coli (ATCC 25922) and Klebsiella pneumoniae (ATCC 700603) using ciprofloxacin as a reference drug. Minimum inhibitory concentrations (MICs) were determined using dilution procedures, which follow the international standard ISO/FDIS 20776-1:2006 (334).30 Briefly, compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 2560 μg mL−1, and then diluted further in Mueller–Hinton Broth to achieve a range of log2 concentrations ranging from 0.008 μg mL−1 to 128 μg mL−1. To each well of the microdilution tray containing 50 μL of diluted inhibitor in broth, a volume of 50 μL of bacterial suspension was added. Before reading results, the microdilution trays were incubated at 34 °C to 37 °C in ambient air for (18 ± 2) h. The amount of growth in each well was compared with that in the positive growth control, and the recorded MIC is the lowest concentration of the agent that completely inhibits visible growth.
Computational studies
Molecular docking and molecular dynamics simulations were performed as previously described.31,32 Briefly docking studies, using the E. faecalis PheS homology model to generate PDB files of the PheS–ligand complexes, were performed using MOE27 until a RMSD gradient of 0.01 kcal mol−1 Å−1 with the MMFF94 forcefield (ligands) and partial charges were automatically calculated. Docking was performed using the Alpha Triangle placement to determine the poses, refinement of the results was done using the MMFF94 forcefield, and rescoring of the refined results using the London ΔG scoring function was applied. The output database dock file was created with different poses for each ligand and arranged according to the final score function (S), which is the score of the last stage that was not set to zero.Molecular dynamics simulations were run on the PheS–ligand complexes with the PDB files first optimised with protein preparation wizard in Maestro by assigning bond orders, adding hydrogen, and correcting incorrect bond types. A default quick relaxation protocol was used to minimise the MD systems with the Desmond programme. The orthorhombic water box allowed for a 10 Å buffer region between protein atoms and box sides. Overlapping water molecules were deleted, and the systems were neutralised with Na+ ions and salt concentration 0.15 M. Force-field parameters for the complexes were assigned using the OPLS_2005 forcefield, that is, a 400 ns (Phe-AMP) or 200 ns (series 1–4) molecular dynamic run in the NPT ensemble (T = 300 K) at a constant pressure of 1 bar. Energy and trajectory atomic coordinate data were recorded at each 1.2 ns. Prime/MMGBAS, available in Schrödinger prime suite, was used to calculate the binding free energy of the ligands with TMPS2.
ΔG (bind) = E_complex (minimised) − (E_ligand (minimised) + E_receptor (minimised)). Mean ΔG (bind) values were calculated from each frame of the final 10 ns of the MD simulation (i.e. the equilibrated complex). The average generated ΔG was from each energy minimised frame using the equation shown above.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank the Egyptian Ministry of Higher Education-Missions Sector and the British council, Egypt, for funding this research at Cardiff University through a scholarship to NAN. Molecular dynamics simulations were undertaken using the supercomputing facilities at Cardiff University operated by Advanced Research Computing at Cardiff (ARCCA) on behalf of the Cardiff Supercomputing Facility and the HPC Wales and Supercomputing Wales (SCW) projects. We acknowledge support of the latter, which is part-funded by the European Regional Development Fund (ERDF) via the Welsh Government.References
- L. Morrison and T. R. Zembower, Gastroenterol. Clin. North Am., 2020, 30, 619–635 Search PubMed.
- R. R. Watkins and R. A. Bonomo, Infect. Dis. Clin., 2020, 34, xiii–xiv CrossRef PubMed.
- A. Cassini, L. D. Högberg, D. Plachouras, A. Quattrocchi, A. Hoxha, G. S. Simonsen, M. Colomb-Cotinat, M. E. Kretzschmar, B. Devleesschauwer and M. Cecchini, Lancet Infect. Dis., 2019, 19, 56–66 CrossRef PubMed.
- M. N. Ragheb, M. K. Thomason, C. Hsu, P. Nugent, J. Gage, A. N. Samadpour, A. Kariisa, C. N. Merrikh, S. I. Miller and D. R. Sherman, Mol. Cell, 2019, 73, 157–165 CrossRef CAS PubMed.
- T. M. Belete, Hum. Microbiome J., 2019, 11, 100052 CrossRef.
- A. R. M. Coates, Y. Hu, J. Holt and P. Yeh, Expert Rev. Anti Infect. Ther., 2020, 18, 5–15 CrossRef CAS PubMed.
- V. Rajendran, P. Kalita, H. Shukla, A. Kumar and T. Tripathi, Int. J. Biol. Macromol., 2018, 111, 400–414 CrossRef CAS PubMed.
- G. H. Vondenhoff and A. Van Aerschot, Eur. J. Med. Chem., 2011, 46, 5227–5236 CrossRef CAS PubMed.
- S.-H. Kim, S. Bae and M. Song, Biomolecules, 2020, 10, 1625 CrossRef CAS PubMed.
- M. A. R. Gomez and M. Ibba, RNA, 2020, 26, 910–936 CrossRef CAS PubMed.
- M. Parenti, S. Hatfield and J. Leyden, Clin. Pharm., 1987, 6, 761–770 CAS.
- J. G. Hurdle, A. J. O'Neill and I. Chopra, Antimicrob. Agents Chemother., 2005, 49, 4821–4833 CrossRef CAS.
- S. J. Baker, Y.-K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447–4450 CrossRef CAS PubMed.
- N. H. Kwon, P. L. Fox and S. Kim, Nat. Rev. Drug Discovery, 2019, 18, 629–650 CrossRef CAS PubMed.
- S. Chakraborty and R. Banerjee, Res. Rep. Biochem., 2016, 6, 25–38 Search PubMed.
- S. S. Elbaramawi, S. M. Ibrahim, E.-S. M. Lashine, M. E. El-Sadek, E. Mantzourani and C. Simons, J. Mol. Graph. Model., 2017, 73, 36–47 CrossRef CAS PubMed.
- D. Beyer, H.-P. Kroll, R. Endermann, G. Schiffer, S. Siegel, M. Bauser, J. Pohlmann, M. Brands, K. Ziegelbauer and D. Haebich, Antimicrob. Agents Chemother., 2004, 48, 525–532 CrossRef CAS PubMed.
- R. L. Jarvest, S. G. Erskine, A. K. Forrest, A. P. Fosberry, M. J. Hibbs, J. J. Jones, P. J. O'Hanlon, R. J. Sheppard and A. Worby, Bioorg. Med. Chem. Lett., 2005, 15, 2305–2309 CrossRef CAS PubMed.
- N. Kato, E. Comer, T. Sakata-Kato, A. Sharma, M. Sharma, M. Maetani, J. Bastien, N. M. Brancucci, J. A. Bittker and V. Corey, Nature, 2016, 538, 344–349 CrossRef CAS PubMed.
- A. Abibi, A. D. Ferguson, P. R. Fleming, N. Gao, L. I. Hajec, J. Hu, V. A. Laganas, D. C. McKinney, S. M. McLeod and D. B. Prince, J. Biol. Chem., 2014, 289, 21651–21662 CrossRef PubMed.
- Z. Yu, J. Tang, T. Khare and V. Kumar, Fitoterapia, 2020, 140, 104433 CrossRef CAS PubMed.
- A. G. Eissa, J. A. Blaxland, R. O. Williams, K. A. Metwally, S. M. El-Adl, E.-S. M. Lashine, L. W. Baillie and C. Simons, J. Enzyme Inhib. Med. Chem., 2016, 31, 1694–1697 CrossRef CAS PubMed.
- G. H. Hitchings, K. W. Ledig and R. A. West, US Pat., US3037980A, 1962 Search PubMed.
- T. U. Consortium, Nucleic Acids Res., 2018, 47, D506–D515 CrossRef PubMed Accessible from https://www.uniprot.org/.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- R. Fishman, V. Ankilova, N. Moor and M. Safro, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 1534–1544 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), Chemical Computing Group ULC, 2019, https://www.chemcomp.com/Products.htm Search PubMed.
- Schrödinger Release 2020-1; Desmond Molecular Dynamics System, D.E. Shaw Research, New York, NY, Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2020. https://www.schrodinger.com/products/desmond Search PubMed.
- R. Fishman, V. Ankilova, N. Moor and M. Safro, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 1534–1544 CrossRef CAS PubMed.
- Clinical laboratory testing and in vitro diagnostic test systems, susceptibility testing of infectious agents and evaluation of performance of antimicrobial susceptibility devices-Part 1: Reference methods for testing the in vitro activity of antimicrobial agents against bacteria involved in infectious diseases, ISO/FDIS 20776-1:2006. https://www.iso.org/standard/41630.html.
- F. A. Binjubair, J. E. Parker, A. G. Warrilow, K. Puri, P. J. Braidley, E. Tatar, S. L. Kelly, D. E. Kelly and C. Simons, ChemMedChem, 2020, 15, 1294 CrossRef CAS PubMed.
- S. M. Kishk, R. M. Kishk, A. S. Yassen, M. S. Nafie, N. A. Nemr, G. ElMasry, S. Al-Rejaie and C. Simons, Molecules, 2020, 25, 5007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06439h |
| This journal is © The Royal Society of Chemistry 2022 |