
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acetoxymethyl-BODIPY dyes: a universal platform for the fluorescent labeling of nucleophiles†
Alberto
Blázquez-Moraleja
 a,
Larissa
Maierhofer‡
a,
Enrique
Mann‡
a,
Ruth
Prieto-Montero‡
b,
Ainhoa
Oliden-Sánchez‡
b,
Lucía
Celada
c,
Virginia
Martínez-Martínez
*b,
María-Dolores
Chiara
*c and
Jose Luis
Chiara
*a
a,
Larissa
Maierhofer‡
a,
Enrique
Mann‡
a,
Ruth
Prieto-Montero‡
b,
Ainhoa
Oliden-Sánchez‡
b,
Lucía
Celada
c,
Virginia
Martínez-Martínez
*b,
María-Dolores
Chiara
*c and
Jose Luis
Chiara
*a
aInstituto de Química Orgánica General (IQOG-CSIC), Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: jl.chiara@csic.es
bDepartamento de Química Física, Universidad del País Vasco-EHU, Facultad de Ciencia y Tecnología, Apartado 644, 48080 Bilbao, Spain. E-mail: virginia.martinez@ehu.eus
cInstituto de Investigación Sanitaria del Principado de Asturias, Instituto Universitario de Oncología del Principado de Asturias (IUOPA), CIBERONC, Universidad de Oviedo, Hospital Universitario Central de Asturias, 33011 Oviedo, Spain. E-mail: mdchiara.uo@uniovi.es
First published on 22nd August 2022
Abstract
Current methods for the preparation of functional small-molecule fluorophores generally require labor-intensive, multi-step synthetic routes for all the major chromophoric groups. In spite of recent significant contributions from numerous laboratories, the paucity of rapid, straightforward and wide-scope synthetic strategies in this field is limiting the development of advanced probes for bioimaging, sensing and therapeutic applications. We describe herein a general and robust methodology for the one-step fluorescent labeling of a wide variety of molecules having C-, N-, P-, O-, S-, or halide-nucleophilic centers, using stable and readily available acetoxymethyl-BODIPYs as reagents in the presence of an acid catalyst. This modular methodology allows a very facile preparation of mono- and di-functional probes incorporating a broad assortment of biomolecules, enzyme cofactors, natural products, and other chromophores, as well as chemical functionalities for a wide range of applications including bioorthogonal conjugation, polymerization, and supramolecular chemistry, among others. The photophysical properties and preliminary applications of the new probes in live-cell imaging were also studied. The described strategy enables the high-throughput engineering of novel BODIPY dyes with diverse functionalities for basic and applied science with potential for innovative technological applications.
1. Introduction
Genetically encoded fluorescent proteins1 and small-molecule fluorescent probes2 have evolved as essential facilitating tools for the study of biology at the single cell and subcellular levels with high spatiotemporal resolution. Although small-molecule fluorophores can hardly surpass the great specificity of fluorescent proteins, their physicochemical properties and photostabilities can be readily modulated through chemical modification, further allowing the introduction of appropriate functionalities for applications in biolabeling, biotargeting, and sensing that are often beyond the reach of genetic approaches.2,3 In general, current methods for the preparation of functional small-molecule fluorophores require labor-intensive, multi-step synthetic routes for all the major chromophoric groups.2 Among these, BODIPYs have become prominent in recent years due to their high photostability, excellent photophysical properties, and high chemical versatility.4–6 The development of functionalization methods that are general, efficient, selective, and highly versatile is currently one of the main challenges in fluorophores’ chemistry in general and in BODIPYs in particular, which display a richer chemistry than other fluorophores in current use. Although different methodologies have been described for the introduction of new functionalities at every position of the BODIPY chromophore, the vast majority involve the use of multistage routes that need to be individually tailored to each final target dye, which entails greater synthetic effort, overall loss of chemical yield and increased economic costs. For these reasons, there is great interest in the development of direct functionalization methods of pre-formed BODIPYs,7,8 which ideally allow the introduction of the new functionality in a single and final reaction step.In general, most post-functionalization strategies described for BODIPYs require the presence or prior introduction of a reactive atom or group (e.g. halogen,9–19 formyl,20–24 carboxyl,25–31 thioether,32–36 alkyne,37–44 azide,45–48 or tetrazine49–53) that enables the final functionalization reaction. These reactive groups can be directly attached either to the chromophore backbone (at boron or carbon) or to peripheral positions separated by one or several bonds from the chromophoric nucleus. However, all previously described methods present limitations in terms of: (1) the variety of substituents that can be introduced under the same reaction conditions (generally, carbon and heteroatomic substituents require different conditions); (2) the type of reactive functionality that should be present in the new substituents, which affects their price, availability and structural diversity; (3) the chemical compatibility between the pre-existing functional groups on the BODIPY and on the new substituent under the reaction conditions; and (4) the yield of the post-functionalization reaction.
The objective of this work was to develop a general strategy for the post-functionalization of BODIPYs that allowed the direct and straightforward incorporation of a wide variety of molecules under similar reaction conditions. Most widely used BODIPYs have alkyl-substituted pyrrole rings, which stabilize cations, radicals and anions at the benzylic-type carbon, particularly those at C3 and C5, offering an interesting opportunity for directly incorporating new functionality at these reactive positions. Not surprisingly, this reactivity has already been exploited in the functionalization of 3/5-methyl-substituted BODIPYs (Scheme 1a) using: (a) Knoevenagel reaction with aromatic aldehydes,54–56 (b) oxidation to aldehyde with DDQ,57 (c) nitration,58 (d) electrophilic bromination followed by in situ nucleophilic substitution,59 and (e) oxidative dimerization promoted by ICl or CuI under basic conditions.60,61 Interestingly, Ulrich and Ziessel's59 two-step/one-pot protocol has been successfully used for the introduction of N-, O-, P- and S-nucleophiles. However, this approach shows some drawbacks: (i) the bromo-methylated intermediate is a very unstable compound that cannot be isolated and, consequently, the bromination and substitution steps must be carried out “in a single flask” for each transformation; (ii) reaction times are highly variable, from a few minutes to several days, depending on the nucleophile; (iii) it is limited in relation to the type of nucleophiles that can participate in the reaction. Thus, in the case of O-nucleophiles, only alcohols have been described; in the case of N-nucleophiles, only azide and secondary amines render the substitution product, while primary amines generate complex reaction mixtures; and (iv) there are no examples of introduction of C-nucleophiles, which is possibly its most important limitation.
 | ||
| Scheme 1 Post-functionalization strategies of 3-methyl-BODIPYs (substituents at other positions were removed for clarity). | ||
Our new method (Scheme 1b) was inspired by these former functionalization strategies. A key prerequisite of our approach was the incorporation of a stable reactive group that required prior activation under catalytic conditions to participate in the functionalization reaction. Thus, the reactive but non-activated BODIPY can be easily isolated and stored for later use in the substitution reaction. A functional group that fulfills these requirements is the acetoxymethyl group, widely used in the synthesis of porphyrins,62–65 but, somehow surprisingly, not yet in the chemistry of BODIPYs despite the structural similarities between both families of dyes. Acid-catalyzed activation of this group can readily generate a resonance-stabilized carbocation intermediate (A), which will considerably expand the diversity of nucleophilic groups that can participate in the SN1-type process including also different types of C-nucleophiles, in contrast to previous methods.
In this work, we describe the preparation of acetoxymethyl-BODIPYs by direct acyloxylation of the corresponding 3/5-methyl-BODIPYs and their efficient acid-catalyzed reaction with hetero- and carbon-nucleophiles to afford a wide variety of new functional BODIPYs for multiple applications. The photophysical properties of the new probes as well as some preliminary applications in live-cell microscopy will also be described.
2. Results and discussion
2.1. Synthesis of 3/5-acetoxymethyl-BODIPYs
When we started this work, there was already an example of a BODIPY dye with acetoxymethyl groups at C3 and C5 reported by Boyer et al. in 1994 in an article that is better known for first describing the regioselective oxidation of the methyl groups at C3/C5 to formyl groups with DDQ.57 The authors prepared the 3,5-di(acetoxymethyl)-BODIPY 1 by a classical method of acetoxylation using lead tetraacetate, which was originally reported for alkyl-substituted pyrroles62 and other heteroaromatic and aromatic compounds (Scheme 2a). However, attempts to hydrolyze the acetate groups under acidic or basic conditions always led to complex reaction mixtures and the compound did not awake further synthetic interest.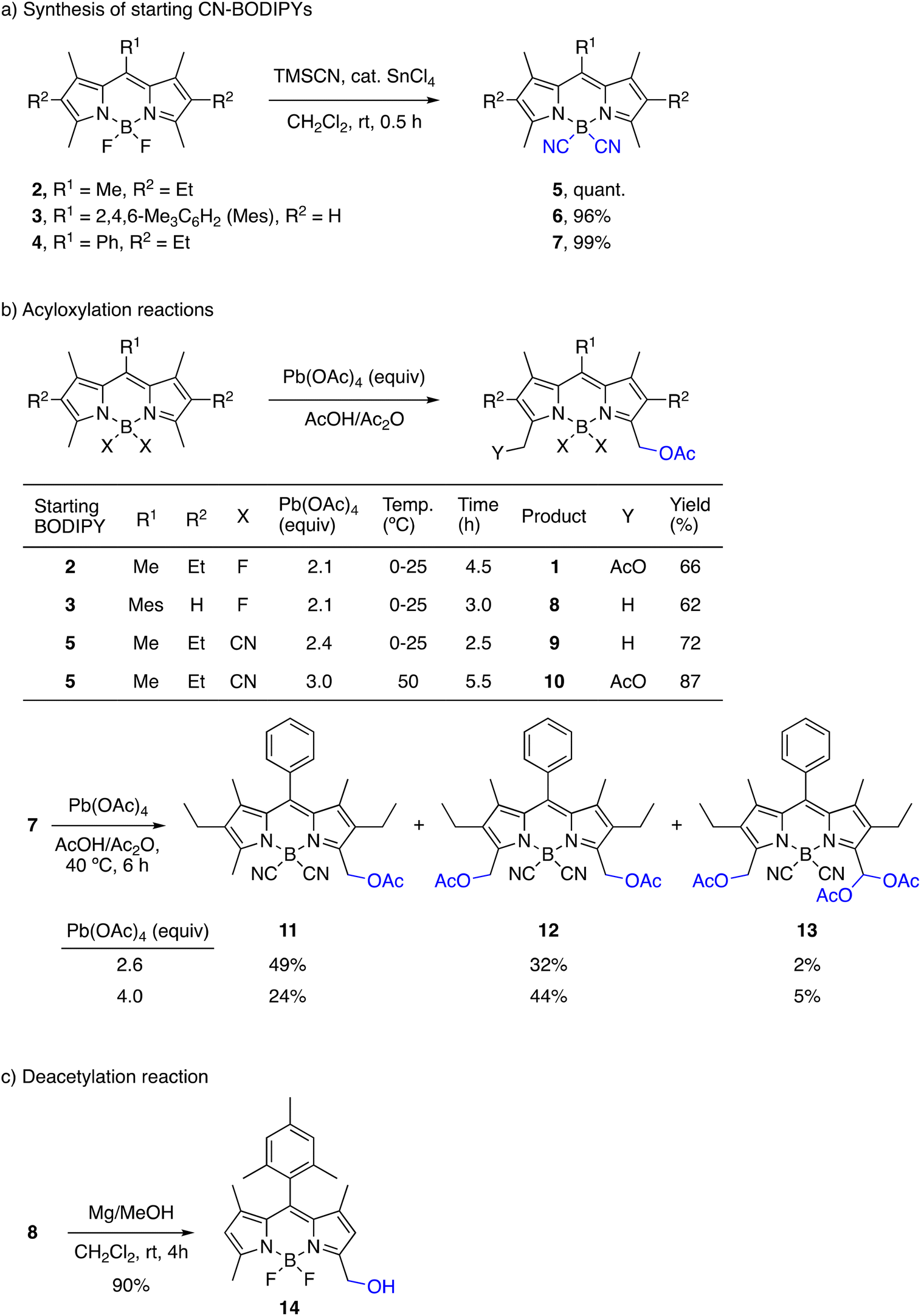 | ||
| Scheme 2 Synthesis of starting CN-BODIPYs and acetoxymethyl-BODIPYs, and deacetylation of the latter. | ||
Although F-BODIPYs show great chemical robustness under diverse reaction conditions, strong acids and bases cause decomplexation of the BF2 group.66–73 Substitution of the fluorine atoms at boron with other substituents barely affects the positions of the spectroscopic bands (absorption and emission) of the chromophore, but allows to modulate its photo- and chemo-stability. Among the substituents that have been introduced on the boron atom of BODIPYs instead of fluorine (halo-, C-, O-, and N-derivatives),7,8,68,74 the cyano group provides the greatest stability improvement in acid medium, while also enhancing the fluorescence of the chromophore.75–78 For our planned strategy, the introduction of cyano groups on the boron atom was a conceivable requirement for our acetoxymethyl-BODIPY reagent to withstand the acidic conditions of the substitution reaction. Thus, we prepared 4,4′-dicyano-BODIPYs (CN-BODIPYs) 5–7 in quantitative yield from the corresponding F-BODIPYs 2–4, respectively, following a described protocol (Scheme 2a),75,79 without any need for chromatographic purification.
We first tested the reaction of F-BODIPYs 2 and 3 with Pb(OAc)4 in acetic acid using the reported procedure,57 with minor modifications (Scheme 2b; see the ESI† for details). In the case of the peralkylated BODIPY 2, the reaction led to the regioselective formation of 3,5-bis(acetoxymethyl)-BODIPY 1, as described.57 In contrast, the reaction of BODIPY 3, with an aryl group at C8, was slower and gave exclusively the mono-acetoxy derivative 8 under the same conditions. The acyloxylation of CN-BODIPY 5 (Scheme 2b) with Pb(OAc)4 (2.4 equiv.) at room temperature exclusively yielded the mono-acetoxy product 9 in good yield, in contrast to its parent F-BODIPY 2, which generated the diacetoxy-F-BODIPY 1 under these conditions. Using a higher temperature (40–50 °C) and a higher excess of Pb(OAc)4 (3–4 equiv.) gave the diacetoxy-CN-BODIPY 10 in very good yield. Compound 10 was not stable on prolonged contact with silica gel, but it could be easily purified by recrystallization from hexane/CH2Cl2. The acyloxylation of CN-BODIPY 7 proceeded under the same thermal conditions to afford a mixture of mono- and di-acetoxy-CN-BODIPY (11 and 12, respectively), together with minor amounts of triacetoxy-CN-BODIPY 13. Interestingly, these acetoxy-CN-BODIPYs could be purified on silica gel column chromatography without decomposition.
The structure of 9 was confirmed by X-ray diffraction (Fig. 1). As previously described57 for 1, the acetyl group adopts an orthogonal orientation with respect to the median plane of the chromophore, which is the most suitable electronic prealignment for the heterolytic cleavage of the C–OAc bond after Lewis acid complexation, with generation of intermediate carbocation A (see Scheme 1). As observed for other CN-BODIPYs,76 the B–N bond lengths (1.531 and 1.538 Å) are slightly shorter than the mean value of 1.540 Å determined for its F-BODIPY precursor 2.80 The boron atom has an almost ideal tetrahedral geometry with bond angles for N–B–N, C–B–N and C–B–C of 108.4, 109.6 (mean value), and 110.0°, respectively.
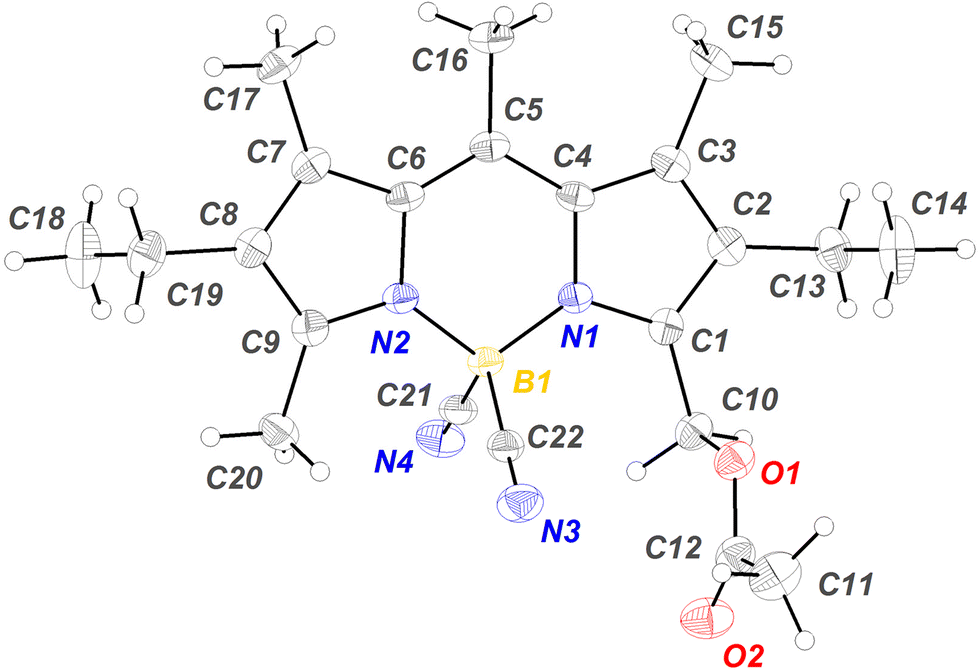 | ||
| Fig. 1 Molecular structure of compound 9 from X-ray crystal structure analysis. | ||
A plausible mechanistic proposal for this reaction is discussed in the ESI† to explain the lower reactivity observed for compounds with better electron-withdraing groups (CN- versus F-BODIPYs: 5 and 2, respectively; and 8-phenyl- versus 8-methyl-BODIPYs: 4 and 2, respectively).
With a view to subsequent chemical transformations that would require a non-acetoxy leaving group, we tried to deacetylate 8 to generate the hydroxymethyl-derivative 14, which can also be a suitable leaving group in the SN1 reaction. As observed by Boyer et al.,57 the transesterification reaction of 2 in MeOH under different basic conditions (NaOMe, K2CO3 or Et3N) always led to complex reaction mixtures. We finally found very mild deprotection conditions81 using metallic magnesium in MeOH at room temperature, which afforded 14 in very good yield (Scheme 2c). However, due to the success of our initial approach for directly activating the acetate group (see below), this alternative route was not further explored.
2.2. Reaction of 3-acetoxymethyl-BODIPYs with carbon and heteroatom nucleophiles
Preliminary studies of the substitution reaction of acetoxymethyl-F-BODIPYs 1 with isobutanol as model nucleophile in CH2Cl2 or MeCN at room temperature using either Brønsted (p-TsOH) or Lewis (Et2O·BF3, TMSOTf, Sc(OTf)3, Cu(OTf)2) acid catalysts (2.5 mol%) afforded complex reaction mixtures with generation of very polar non-fluorescent products in all cases. This result points to a probably acid-promoted decomplexation of the BF2 group generating the corresponding dipyrromethene.68–73 We next assayed the CN-BODIPY derivative 9, with an expected significantly enhanced stability under acidic conditions, as commented above. A qualitative parallel screening with a larger group of acid catalysts using the same model test reaction with 9 (see ESI† for details) showed that Sc(OTf)3 was the most efficient and general acid catalyst and was therefore selected for most of the substitution reactions described below.O-Nucleophiles. A wide variety of O-nucleophiles were incorporated at the methylene group of 3-acetoxymethyl-CN-BODIPY 9 using the general reaction conditions (Scheme 3). Primary (15–18, 20, 22, 25, 26, 28–30, 32, 33, 35), secondary (27, 31) and tertiary (23) alkanols, as well as benzylic (21, 34), allylic (24) and propargylic alcohols (19) were successfully labeled in moderate to very good yields. The reaction is compatible with the presence of ester (26), nitrile (21), carbamoyl (26), carboxamido (32, 33), imido (27–29), trialkylsilyl ether (27), isopropylidene acetal (29, 30), 1,2,4,5-tetrazino (34), disulfide (38), and chloro-/iodo-alkyl groups (20, 32, 33). Carboxylic acids were also good substrates, although a larger excess of the nucleophile (up to 5 equiv.) was generally required to obtain good product yields (36–39). In this case, the reaction is actually in thermodynamic equilibrium with the acetic acid released in the substitution reaction. This equilibrium can also explain the chemoselectivity observed in the reaction with simple hydroxy acids (25). However, in the case of more complex hydroxy acids (39) with a secondary hydroxyl group, the corresponding ester was obtained, although in moderate yield.
 | ||
| Scheme 3 Reaction with O-nucleophiles. Reaction conditions: Sc(OTf)3 (2.5–10 mol%), NuH (1.2–5.0 equiv.), 0.5–30 h (see ESI† for further details). aYield based on converted 9. bProducts formed in the same reaction. cIn MeCN. | ||
The method allowed us to fluorescently label natural products (farnesol: 24), as well as a wide variety of biomolecules, including lipids (cholesterol: 31, oleic acid: 36), carbohydrates (D-galactose: 30), nucleosides (partially protected derivatives of D-deoxyribose: 27, 28, and D-ribose: 29), amino acids (L-serine: 26), hydroxy acids with a tetraalkylammonium group (carnitine: 39), or enzyme cofactors (lipoic acid: 38). In addition, we were able to introduce functionalities for complexation with cyclodextrins (adamantyl group: 23), for polymerization (methacrylate group for radical polymerization: 37; norbornene group for ring-opening alkene metathesis-polymerization (ROMP): 22), for bioorthogonal conjugation82–84 (propargyl group for Cu(I)-catalyzed “click” cycloaddition reaction: 19; cyclooctyne group for strain-promoted alkyne–azide cycloaddition (SPAAC): 35; chloro-/iodo-acetamido group for selective labeling of cysteines: 32, 33; 1,2,4,5-tetrazine ring for inverse-electron-demand Diels–Alder cycloadditions:8534), and for selective fluorescent labeling of HaloTag fusion proteins (6-chlorohexyl chain: 20).86–88
The diversity of O-substituents included in this study allowed us to identify some limitations of the method. The first, as already mentioned, is the need to use an excess of nucleophile in the case of carboxylic acids, due to the reversibility of the substitution reaction under acidic conditions. As expected, yields decreased in the case of molecules with acid-sensitive groups, such as acetals (29, 30) or trialkylisilyl ethers (27). In the latter case, the reaction afforded the expected product 27 together with the corresponding desilylated derivative 28. Not surprisingly, polyoxygenated substrates (18, 26–30, 32, 33, 39) slowed down the reaction due to their ability to complex the metal catalyst in competition with the activation of the acetate group, which required the use of a greater amount of catalyst and/or a higher reaction temperature, including microwave irradiation in some cases (39) (see ESI† for experimental details). Other strong metal chelating groups, such as the cyclooctyne group, sequestered the scandium catalyst very effectively. The incorporation of the cyclooctyne unit 40 could only be achieved following a described strategy for transient protection of the cyclooctyne by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation with a cationic copper(I) salt, which allowed the preparation of 35 in moderate overall yield (Scheme 4).89,90
:1 complexation with a cationic copper(I) salt, which allowed the preparation of 35 in moderate overall yield (Scheme 4).89,90
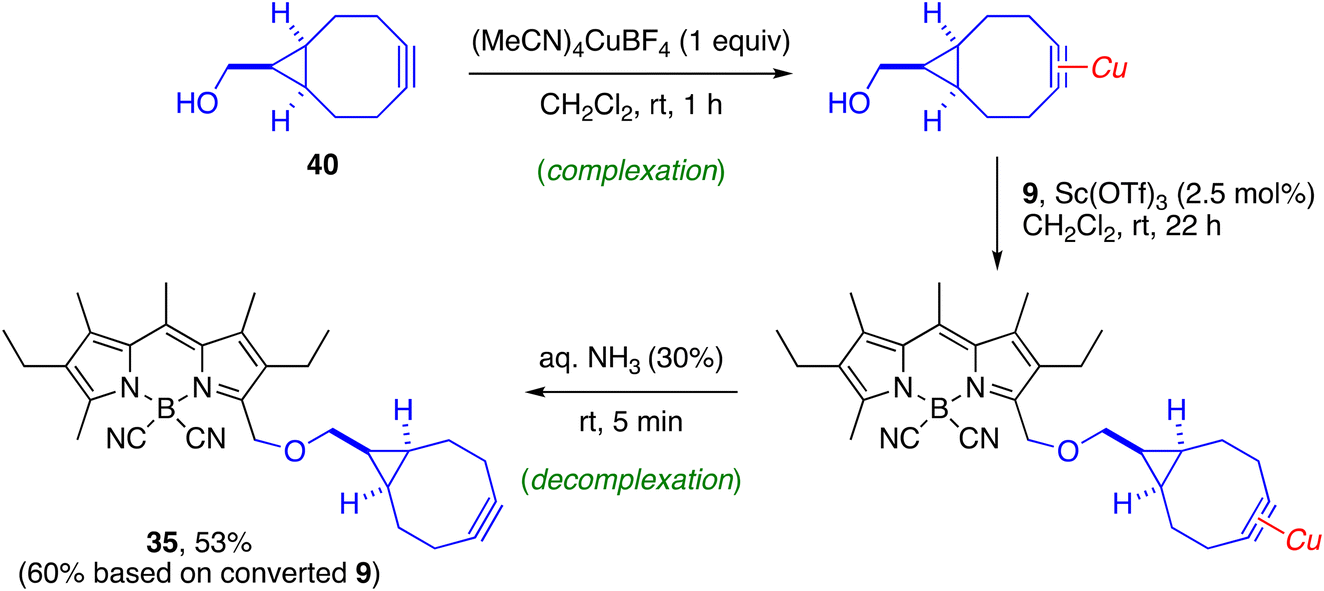 | ||
| Scheme 4 Synthesis of cyclooctyne-functionalized probe 35 from acetoxymethyl-BODIPY 9 by a transient protection strategy of the cyclooctyne. | ||
S-Nucleophiles. As expected from their higher nucleophilicity, the substitution reaction with thiols was faster and more efficient than with alcohols (Scheme 5). Aliphatic (41, 42, 45) and aromatic (43, 44) thiols were readily incorporated in high yield (70–90%), including polyfluorinated systems (42, 44) and partially protected biomolecules such as N-acetyl-L-cysteine (45). In the latter case, the observed chemoselectivity is both of kinetic origin, due to the higher nucleophilicity of the thiol group in comparison with the carboxylic acid group, and of thermodynamic origin, due to the reversibility of the reaction with the carboxylate group.
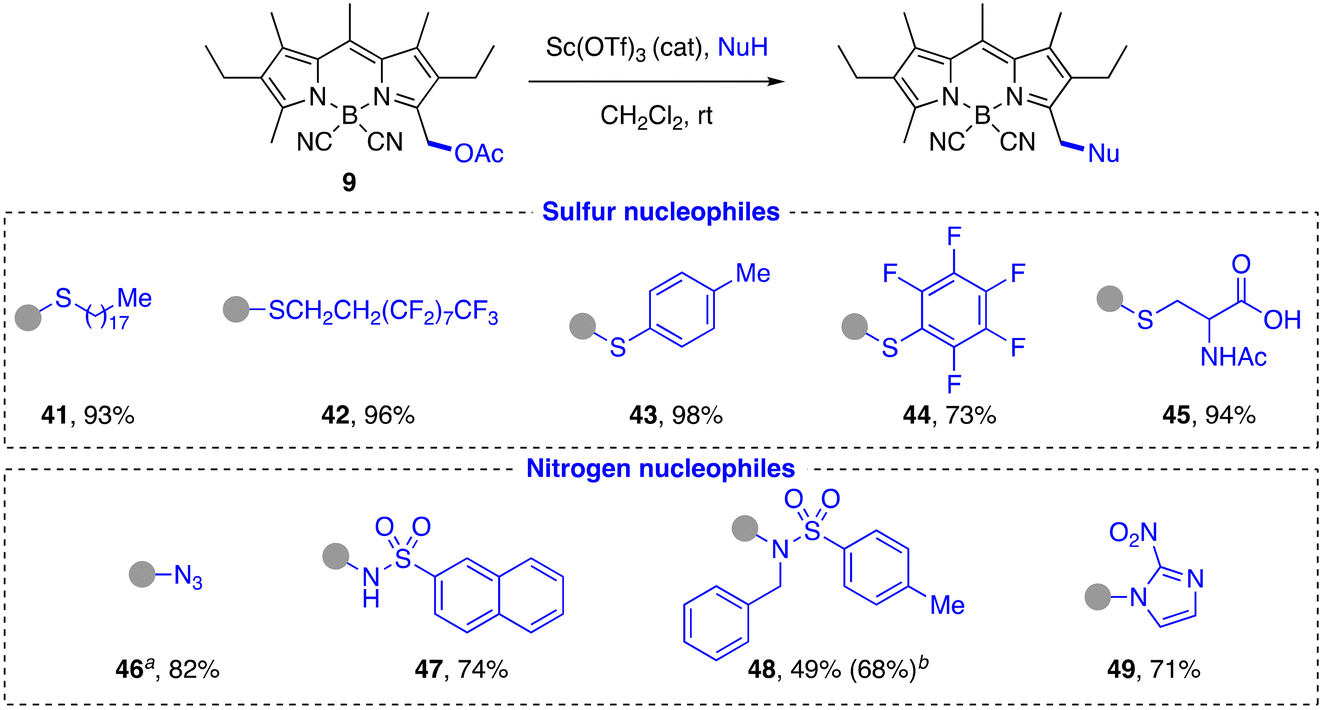 | ||
| Scheme 5 Reaction with S- and N-nucleophiles. aNuH: TMSN3. bBased on converted 9. | ||
N-Nucleophiles. As we already verified in the study of O- and S-nucleophiles, amides and carbamates are not good substrates in this substitution reaction, nor would amines be expected to react due to their incompatibility with the acid catalysis conditions. Thus, we decided to test other types of N-nucleophiles that did not present these problems, either because of their greater acidity, as in the case of sulfonamides or 2-nitroimidazole, or because of their higher nucleophilicity, as occurs with the azide anion. Thus, primary and secondary sulfonamides (47, 48, respectively), which can generate a transient nucleophilic anion, enabled the reaction in moderate to good yield, the former being more reactive due to the lower steric hindrance (Scheme 5). Trimethylsilyl azide yielded the azidomethyl-CN-BODIPY 46 in high yield and with appreciably faster kinetics than in the case of sulfonamides. Compound 46 is an interesting fluorescent reagent for bioorthogonal conjugation and sensing.46,91,92 (NH)-Heterocyles, such as 2-nitroimidazole, were efficiently alkylated under the standard conditions, affording in this case a potential nitroreductase probe (49).93
Cl- and P-Nucleophiles. The reaction of 9 with an excess of TMSCl (20 equiv.) in CH2Cl2 at room temperature quantitatively furnished the chloromethyl-derivative 50 in the absence of a metal catalyst, where the reagent acted as acid catalyst as well as nucleophile precursor (Scheme 6). Although 50 was unstable on silica gel, it could be isolated in quantitative yield and high purity by simple evaporation of the reaction mixture.59 This chlorinated compound can be directly used for SN2-type substitution transformations, as described for its bromo-analogue.59 Thus, the Arbuzov reaction with triethyl phosphite under microwave irradiation yielded the phosphonate 51 in good yield (Scheme 6).
 | ||
| Scheme 6 Reaction with halogen and phosphorous nucleophiles. | ||
C-Nucleophiles. In contrast to previously described approaches,59 the same reaction conditions that were efficient for the functionalization of heteronucleophiles proved to be equally effective for the introduction of C-nucleophiles. Thus, electron-rich aromatic (phenols: 56, 57, 58; azulene: 59) and heteroaromatic (pyrrole: 60; indole: 61) compounds efficiently yielded the BODIPY-labeled product in a Friedel–Crafts-type process (Scheme 7). The reaction with phenols was highly chemo- and regioselective, exclusively affording the 2-hydroxyphenyl-derivative in very good yields. No trace of the alternative O-alkylated derivative or the 4-hydroxyphenyl-regioisomer were detected. The method allowed us to fluorescently label partially protected L-tyrosine (57) and a polyfunctional natural product such as resveratrol (58). The reaction with the tyrosine derivative proceeded with high chemo- and regioselectivity and was fully compatible with the presence of the carbamate and the methyl ester groups. The reaction with resveratrol required the use of a greater amount of catalyst, longer reaction time and a higher temperature, but proceeded regioselectively to give the C-derivative 58 in moderate yield as major product, with the BODIPY chromophore covalently bonded to the most electron-rich aromatic ring at the most electron-rich carbon. The reaction with guaiazulene, a natural derivative of azulene, was also highly regioselective, affording exclusively the product alkylated at the electron-rich 5-membered ring. The structures of these compounds were fully confirmed by careful one- and two-dimensional 1H and 13C NMR studies. Indole and pyrrole also gave the substitution reaction with good or very good yields under the standard conditions, with the expected regioselectivity for electrophilic attack on this type of heterocycles. The good reactivity observed for pyrroles led us to also test BODIPYs with a free position on the pyrrolic rings as nucleophiles. To avoid decomposition in the presence of the acid catalyst, we used a 4,4′-dicyano-BODIPY (6) as acceptor (Scheme 2a). Despite the presence of alkyl substituents flanking the reactive CH carbon of the pyrrole rings of 6, the reaction of an equimolecular mixture of 6 and 9 under the standard conditions led to formation of the expected statistical mixture of mono- and di-substitution products 62 and 63, respectively, in close to the expected theoretical yields (50% and 25%, respectively).
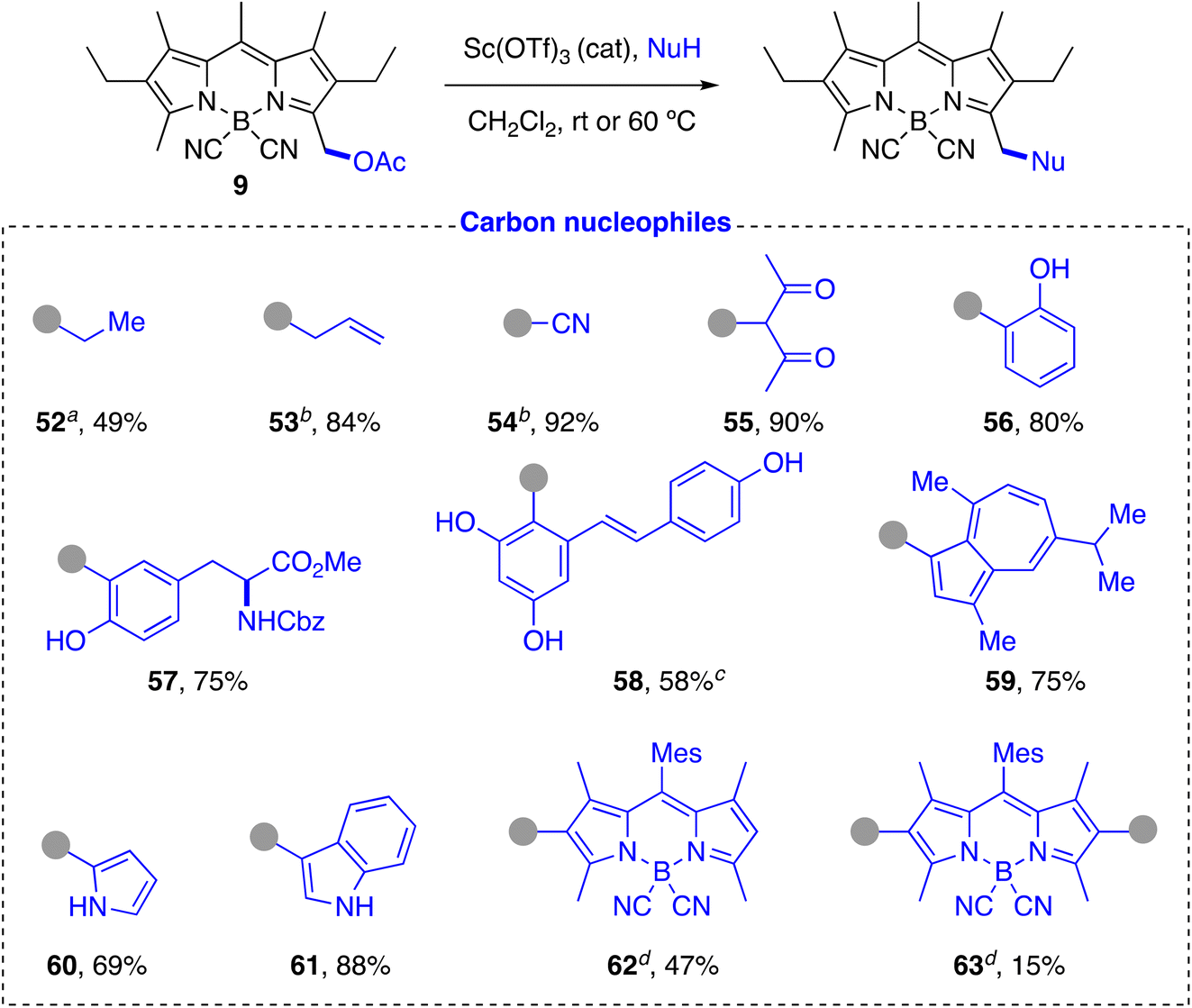 | ||
| Scheme 7 Reaction with carbon nucleophiles. aNuH: Et2Zn, using Me3SiOTf as catalyst. bNuH: Me3SiNu. cYield of the mixture of regioisomers. Major regioisomer is shown, isolated by recrystallization. dProducts formed in the same reaction (Mes: mesityl). | ||
In addition to carbocyclic and heterocyclic systems, acyclic C-nucleophiles can also be incorporated. Thus, C-trimethylsilyl derivatives, including allyltrimethylsilane and trimethylsilyl cyanide, as well as compounds with an active methylene group such as acetylacetone, reacted very efficiently to yield the expected alkylation products 53, 54, and 55, respectively, in high yield. Compound 55 presented the characteristic keto–enol equilibrium in solution, with a practically equimolecular ratio of both species in CDCl3 at room temperature, as observed by 1H NMR (see ESI†).94,95 Simple organometallic compounds such as diethylzinc were also successfully tested, although in this case it was necessary to use a stoichiometric amount of trimethylsilyl triflate as promoter to afford the alkylated product 52, which indicates the probable participation of a triflate intermediate in the substitution reaction.96
Unlike other related methods, the great versatility of the described post-functionalization strategy allowed not only to incorporate a wide diversity of heteronucleophiles in BODIPYs, but also to extend the carbon chain of the chromophore with a broad variety of cyclic and acyclic C-nucleophiles, including biomolecules (tyrosine: 57), natural products (resveratrol: 58, guaiazulene: 59), and other chromophores (62 and 63) in a direct and very efficient way using similar reaction conditions.
 | ||
| Scheme 8 Synthesis of homo- and hetero-bifunctional probes. Reaction conditions: NuH (5 equiv.), Tf2NH (15–32 mol%), MeCN, 60 °C, 1.5–7.0 h (route A); Nu1H (0.5 equiv.), Sc(OTf)3 (14 mol%), CH2Cl2, 50 °C, 24 h (route B, step 1); Nu2H: TMSN3 (1.5 equiv.), Sc(OTf)3 (10 mol%), 50 °C, 24 h (route B, step 2). aSc(OTf)3 (10 mol%) was used as catalyst. bHomo-disubstituted product with two chloroalkyl chains (69, 6% yield; see ESI† for details) and unreacted 12 (40%) were also isolated. | ||
Heterobifunctional BODIPYs can also be obtained in a two-step procedure employing two different nucleophiles, as exemplified by fluorescent probe 68 displaying a chlorohexyl chain as a selective ligand for halotagged fusion proteins,86–88,98 and an azido group for biorthogonal conjugation to a second functional molecule of interest99 (Scheme 8). The chlorohexyl substituent was first connected to a tetraethylene glycol chain, following a described method,100 in order to improve the water solubility of the probe and also to increase the distance between the chromophore and the protein to boost the efficiency of the bioconjugation reaction. A two-fold excess of diacetoxy-BODIPY 12 was used with respect to the alcohol in order to optimize the ratio of the monofunctionalization product 67. As already observed for other polyoxygenated alcohols, the reaction required the use of a larger amount of Sc(OTf)3 and a higher temperature than the standard procedure. Mono-substituted product 67 was obtained in moderate yield (42%; 70% based on total conversion of 12), along with a small amount of homodisubstituted derivative 69 (6%) and unreacted 12 (40%) (see ESI†). The azide group was introduced next under similar conditions using TMSN3 as the nucleophile to afford the heterobifunctional BODIPY 68 in a modest 25% yield.
2.3. Photophysical study of some representative compounds
With a view to their possible applications in live-cell microscopy, we carried out a primary study of the photophysical properties of some of the most representative fluorescent probes obtained. The new BODIPYs were studied in three different solvents to determine the influence of the polarity of the medium: AcOEt, as a lipophilic aprotic polar organic solvent; MeOH, as a protic polar organic solvent; and phosphate-buffered saline (PBS), as the typical aqueous medium used in live-cell microscopy experiments.Table S1† lists the photophysical properties determined in the three solvents for the most representative compounds derived from O-nucleophiles (15–39), including the starting acetoxymethyl-derivative 9 (Fig. S1†). Compound 9 shows a similar photophysical behavior as previously reported for its parent CN-BODIPY 5, without the acetoxy group.79 In aqueous medium (PBS), a considerable drop in the molar absorption coefficient and fluorescent emission accompanied by band broadening was also observed together with the appearance of a shorter lifetime (3 ns) than that typical of BODIPYs (6–7 ns). These observations can be explained by the formation of dye aggregates in this very polar medium. The same tendency was observed for other compounds in this series, with some exceptions that will be discussed below (Fig. S2†). In general, quantum yields were maintained or only slightly decreased when going from AcOEt to MeOH, but significantly drop in PBS for the compounds with the most lipophilic substituents, as observed for 9. In contrast, compounds 18, 28, 29, and 32, with more hydrophilic substituents that improve water solubility, maintained a high fluorescense in the three solvents studied. Compound 34, with a 6-phenyl-1,2,4,5-tetrazine ring, showed very poor or almost no fluorescent emission, likely assigned to a FRET process (the absorption of tetrazine overlap with the emission of BODIPY),49,101–104 but possibly also to a PET process (from BODIPY to tretazine, which is a strong electron withdrawer).105 Like other previously described tetrazine-BODIPYs, compound 34 is an excellent reagent in inverse electron demand Diels–Alder cycloadditions (IEDDA)86,106 with a wide variety of dienophiles, particularly strained alkenes and alkynes. The cycloaddition reaction, which occurs with loss of N2, deactivates the energy transfer process and restores the fluorescent emission. Thus, addition of strained cyclooctyne 40 to a solution of 34 at room temperature very rapidly (<2 min) yielded the cycloaddition product 70 with a 24-fold increase in fluorescence emission (Scheme 9). Compound 34 is an interesting fluorogenic probe for bioorthogonal conjugation in no-wash live-cell microscopy.
 | ||
| Scheme 9 Inverse electron demand Diels–Alder cycloaddition reaction of tetrazine 34 with cyclooctyne 40. | ||
In general, the S-derivatives have a higher solubility in aqueous medium and better fluorescent emission than similar O-derivatives (Table S2 and Fig. S3†). In this group, compounds with a polyfluorinated substituent (42 and 44) showed higher molar absorption coefficient and higher fluorescent emission in AcOEt than similar non-fluorinated analogs (41 and 43, respectively). The more hydrophilic N-acetyl-L-cysteine derivative 45 maintained a moderately high fluorescent efficiency in PBS medium.
The derivatives of the N-nucleophiles (46–49), showed high fluorescence quantum yields (ϕ ≥ 0.84) in both AcOEt and MeOH, with the exception of the 2-nitroimidazolyl-BODIPY 49 (Table S2†). However, these compounds were not fluorescent in PBS, unlike their O- and S-derivative analogs, possibly due to their lower solubility in water (Fig. S3†). Although it is known that nitro-derivatives are usually non-fluorescent, mainly by activation of an intersystem crossing process,107,108 compound 49 showed moderate fluorescence efficiency in AcOEt and MeOH.
The presence of chlorine (50) or a phosphonate group (51) (Table S2 and Fig. S3†) barely affected the absorption and emission bands, but significantly improved fluorescent emission in aqueous medium (PBS), probably due to the lower lipophilic nature of these derivatives.
The photophysical properties of the C-derivatives were highly dependent on the nature of the newly introduced carbon moiety (Table S3†). Thus, compounds with acyclic alkyl systems (52–55) and the phenol derivative 56 have very similar properties to their parent 9, while compounds with electron-rich aromatic systems, including guaiazulene (59) (Fig. S4†), pyrrole (60), and indole (61) derivatives, showed very low fluorescence in the three solvents studied. These results could be interpreted in terms of a photoinduced electron transfer (PET) process taking place between the newly introduced electron-rich ring and the BODIPY chromophore, which quenches fluorescence. The contribution of this PET process was theoretically modeled by DFT calculations in the case of compounds 59–61, where the guaiazulene, pyrrole and indole rings act as electron donors (reductive PET). The calculations indicate that the lowest energy electronic transition occurs from the HOMO−1 to the LUMO, both located mainly on the BODIPY chromophoric system (Fig. 2 and Fig. S5†). The thermodynamically feasible electron transfer from HOMO (mainly located on the guaiazulene and indole system) to the semi-vacant HOMO−1 after excitation prevents the excited electron from returning to its own ground state, thus causing a non-radiative decay that is responsible for the dramatic quenching of fluorescence observed for these compounds in all solvents studied (Table S3†). A similar effect on fluorescence has been described in other BODIPY derivatives with azulene109 or indole110 substituents. The multichromophoric systems 62 and 63 showed an intense absorption capacity (high molar absorption coefficient) due to the presence of two and three BODIPY units, respectively, and kept an efficient emission in AcOEt. These photophysical features are impaired in MeOH and PBS due to the inherent organophilic nature of these compounds (Table S3†).
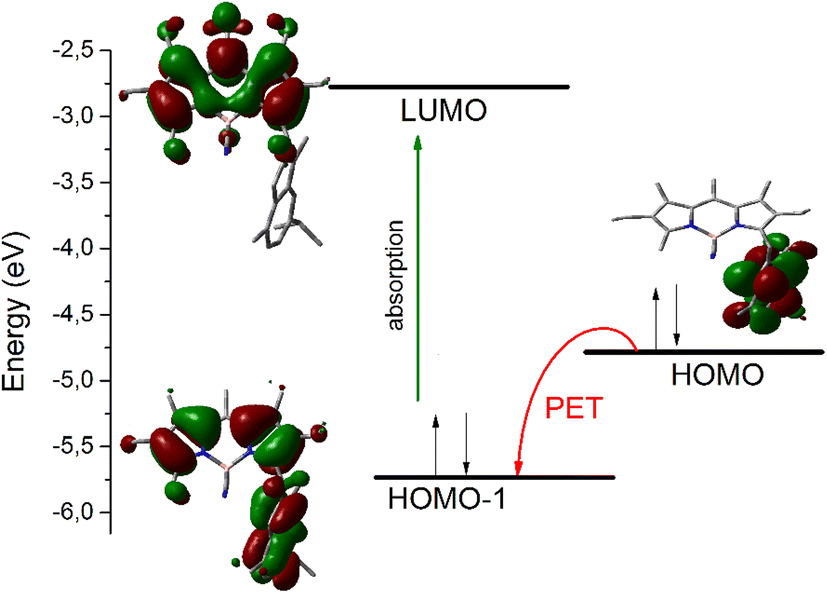 | ||
| Fig. 2 Deactivation of the fluorescence of compound 59 by photoinduced electron transfer (PET), modeled by DFT calculations (B3LYP/6-311+G*). | ||
Table S4† lists the photophysical properties of the disubstituted derivatives 64 and 66, together with their parent diacetoxy-compound 12 (Fig. S6†). As with the monosubstituted analogs, the nature of the new substituents had a negligible effect on the absorption and emission frequencies, but their lipophilic/hydrophilic character had a substantial impact on the molar absorption coefficient and fluorescent emission. Thus, a considerable drop in the magnitude of both properties was observed in PBS for the more lipophilic and less water-soluble 12 and, in particular, for 66, attributed to dye aggregation. In fact, their fluorescent decay curves became biexponential in this solvent with the appearance of a very short lifetime (<0.1 ns), than that typical for BODIPYs. In contrast, the introduction of the two hydrophilic tetraethylene glycol substituents in compound 64 endowed it with a better water solubility that aided to maintain a moderately good fluorescence quantum yield in PBS leading to a monoexponential decay curve.
2.4. Live-cell microscopy studies
We have carried out a preliminary screening of the new compounds as fluorescent probes in live-cell microscopy, using two different cancer cell lines: HeLa and SCC38 (from human cervix and larynx squamous cell carcinoma, respectively). The compounds were incubated with live cells at three different concentrations (50 nM, 100 nM and 500 nM) for 30 minutes, before washing with PBS to remove excess dye. The most suitable concentrations for visualization were 50 nM and 100 nM. In general, this screening showed that most compounds were able to cross the cell membrane and gave a specific subcellular staining without showing any cytotoxicity, with the exception of the following (the new substituent is indicated in parentheses): 9 (acetoxymethyl), 17 (hexadecanol), 18 (octaethylene glycol), 37 (methacrylic ester), 41 (octadecanethiol), 42 (heptadecafluorodecanethiol), 44 (pentafluorophenyl ring), 47 (naphthalenesulfonamide), and 55 (acetylacetone), which were found to be cytotoxic at the concentrations tested. Some compounds showed only weak cell staining due to their low water solubility and/or low permeability through the cell membrane: 36 (oleic ester), 38 (lipoic ester), 46 (azide), 51 (diethyl phosphonate), and 54 (cyano). As mentioned, some of the new probes showed specific subcellular staining in this preliminary screening (Fig. S7 and S8†). Fig. 3 shows live-cell microscopy images of compound 28 (thymidine derivative), as an illustrative example of those compounds with strong and specific subcellular staining. An interesting time-dependent staining involving the nuclear membrane and nucleoli was observed in this case, while maintaining a good cell viability over a 33 h-incubation time. This dynamic phenomenon is under investigation in our group.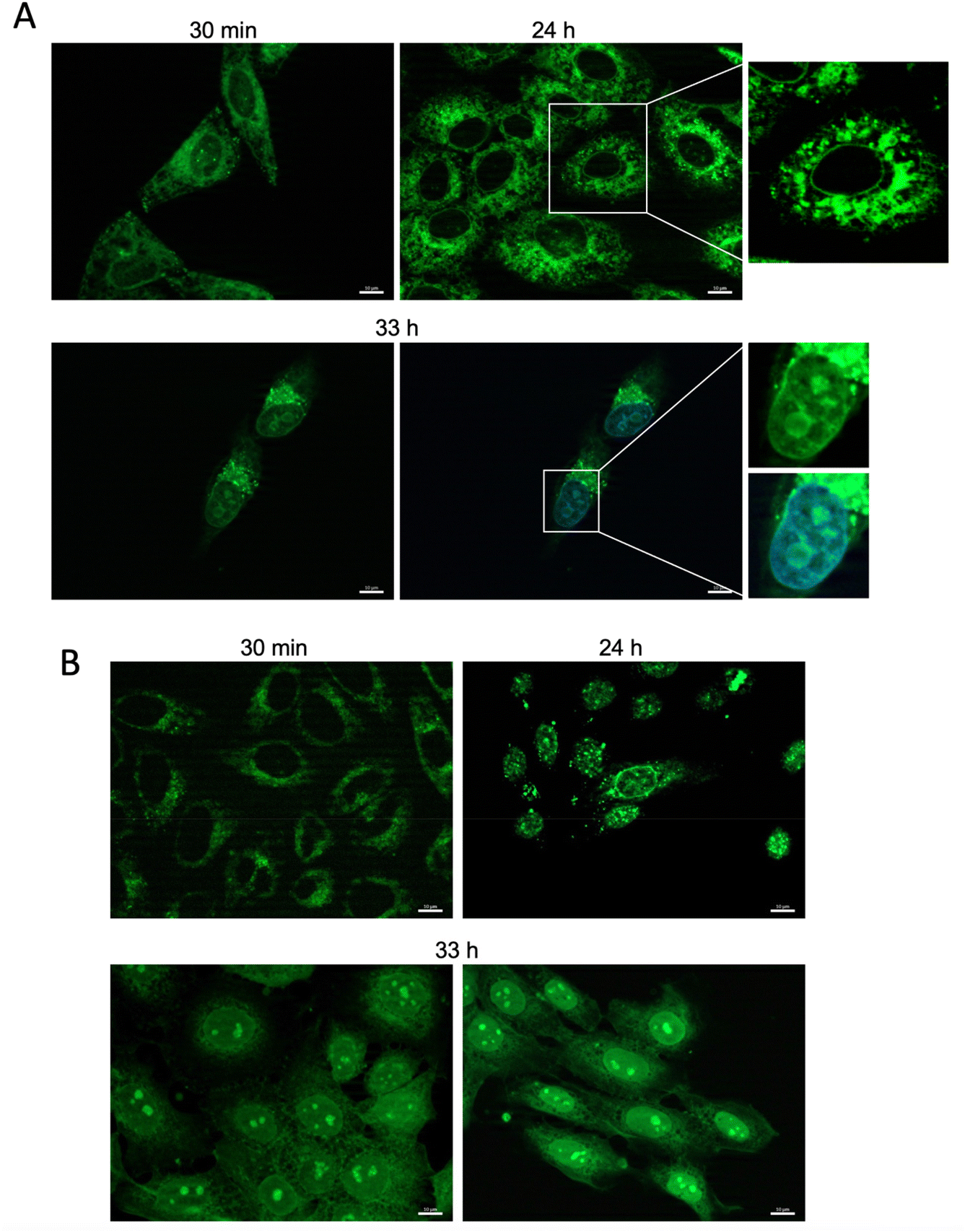 | ||
| Fig. 3 Representative images of HeLa (A) and SCC38 (B) cells stained with compound 28 (500 nM) for 30 minutes, 24 h or 33 h, as indicated. In panel A (33 h), cells were finally fixed and stained with DAPI (4′,6-diamidino-2-fenilindol), a fluorescent dye that binds to adenine and thymine enriched regions in the DNA. Insets in A show magnified images to better visualize the staining of the nuclear envelope (picture above) and the nucleoli (pictures below) in HeLa cells. Images clearly show time-dependent migration of the fluorescence from the cytoplasm (30 min) to the nuclear envelope (24 h) and the nucleoli (33 h) in the two cell lines. Scale bar: 10 μm. | ||
3. Conclusions
We have developed a general and robust methodology for the direct incorporation of a wide variety of C-, N-, P-, O-, S-, and halo-nucleophiles into functional BODIPY conjugates in a single reaction step. The new method employs stable 3-acetoxymethyl- or 3,5-diacetoxymethyl-4,4-dicyano-BODIPYs as reagents, which are readily available in two high yielding steps from the corresponding 3,5-dimethyl-4,4-difluoro-derivatives using previously described procedures. The nucleophilic substitution reaction takes place under mild acid-catalyzed conditions with remarkably broad substrate scope and functional group tolerance. Alcohols of all kinds, carboxylic acids, hydroxyacids, alkylic and aromatic thiols, primary and secondary sulfonamides, 1H-N-heterocycles, azide, chloride, and trialkylphosphites can all be easily attached to the BODIPY reagent in good yields using the same procedure. Unlike previously described approaches,59 our strategy also allows the incorporation of a broad diversity of C-nucleophiles, including electron-rich aromatic and heteroaromatic compounds, trimethylsilyl carbon nucleophiles, active methylene compounds, and organometallic reagents. Our approach provided a direct access to mono- and di-functional probes incorporating biomolecules (lipids, carbohydrates, nucleosides, amino acids, L-carnitine), enzyme cofactors (lipoic acid), and natural products (farnesol, guaiazulene), as well as chemical functionalities for a wide range of applications including bioorthogonal conjugation, polymerization, and supramolecular chemistry, among others. In addition, this strategy also proved successful in the one-step synthesis of new multichromophoric systems, including BODIPY oligomers and a BODIPY-azulene diad. We believe that this novel and simple approach will significantly expand the synthetic toolbox for the preparation of functional BODIPY-based probes and could be easily implemented by specialists and non-specialists alike to rapidly tailor and deploy advanced probes for innovative applications in bioimaging, sensing and the preparation of new photonic materials.Author contributions
J.L.C. conceived and designed the study. V.M.-M., M.-D.C. and J.L.C. supervised the research project. A.B.-M., L.M. and E.M. performed the synthesis and structural characterization of probes. R.P.-M., A.O.-S. and V.M.-M. performed the photophysical studies. L.C., M.-D.C. and A.B.-M. performed the live-cell microscopy studies. V.M.-M. and J.L.C. performed the DFT theoretical calculations. V.M.-M., M.-D.C. and J.L.C. co-wrote the paper. All authors contributed to the final interpretation of the experimental results and critically revised the manuscript. All authors have read and approved the final version of the manuscript.Data availability
Experimental procedures, X-ray diffraction data of 9, DFT computational details, photophysical characterization data, live-cell microscopy images, and copies of 1H and 13C NMR spectra of compounds (PDF) can be found in the ESI.† The datasets of 1D- and 2D-NMR spectra supporting this article are available from: https://doi.org/10.20350/digitalCSIC/14707.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from Instituto de Salud Carlos III/FEDER “A way to make Europe/Investing in your future” (project FIS 20/01754), MCIN/AEI/10.13039/501100011033 (projects PID2020-114347RB-C31 and PID2020-114347RB-C32), and Gobierno Vasco-Eusko Jaurlaritza (project IT1639-22). A. B.-M. and L. C. thank MCIN for a FPI (BES-2015-073571) and a FPU (FPU2017-01317) predoctoral contract, respectively. R. P. M. thanks MIU and NGEU for a postdoctoral contract (MARSA21/71). A. O. S. thanks UPV-EHU for a predoctoral fellowship. The authors thank the technical and human support provided by SGIker (UPV/EHU/ERDF, EU), X-rays: Molecules and Materials Unit, for the XRD structure of compound 9.References
- M. Zimmer, Green Fluorescent Protein (GFP): Applications, Structure, and Related Photophysical Behavior, Chem. Rev., 2002, 102, 759–781 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, Bright Building Blocks for Chemical Biology, ACS Chem. Biol., 2014, 9, 855–866 CrossRef CAS PubMed.
- J. B. Grimm and L. D. Lavis, Caveat fluorophore: an insiders guide to small-molecule fluorescent labels, Nat. Methods, 2022, 19, 149–158 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, The chemistry of fluorescent Bodipy dyes: versatility unsurpassed, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, BODIPY dyes and their derivatives: Syntheses and spectroscopic properties, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- J. Bañuelos, BODIPY Dye, the Most Versatile Fluorophore Ever?, Chem. Rec., 2016, 16, 335–348 CrossRef PubMed.
- R. G. Clarke and M. J. Hall, Recent developments in the synthesis of the BODIPY dyes, Adv. Heterocycl. Chem., 2019, 128, 181–261 CrossRef CAS.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Synthesis of BODIPY dyes through postfunctionalization of the boron dipyrromethene core, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- L. Jiao, W. Pang, J. Zhou, Y. Wei, X. Mu, G. Bai and E. Hao, Regioselective Stepwise Bromination of Boron Dipyrromethene (BODIPY) Dyes, J. Org. Chem., 2011, 76, 9988–9996 CrossRef CAS PubMed.
- V. Lakshmi and M. Ravikanth, Synthesis of Sterically Crowded Polyarylated Boron-Dipyrromethenes, J. Org. Chem., 2011, 76, 8466–8471 CrossRef CAS PubMed.
- X. Li, S. Huang and Y. Hu, Diversity-oriented derivatization of BODIPY based on regioselective bromination, Org. Biomol. Chem., 2012, 10, 2369–2372 RSC.
- V. Lakshmi and M. Ravikanth, Synthesis, spectral and electrochemical properties of phenylated boron-dipyrromethenes, Dyes Pigm., 2013, 96, 665–671 CrossRef CAS.
- V. Leen, P. Yuan, L. Wang, N. Boens and W. Dehaen, Synthesis of Meso-Halogenated BODIPYs and Access to Meso-Substituted Analogues, Org. Lett., 2012, 14, 6150–6153 CrossRef CAS PubMed.
- N. Zhao, S. Xuan, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, Stepwise Polychlorination of 8-Chloro-BODIPY and Regioselective Functionalization of 2,3,5,6,8-Pentachloro-BODIPY, J. Org. Chem., 2015, 80, 8377–8383 CrossRef CAS PubMed.
- N. Zhao, S. Xuan, B. Byrd, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, Synthesis and regioselective functionalization of perhalogenated BODIPYs, Org. Biomol. Chem., 2016, 14, 6184–6188 RSC.
- Z. Feng, L. Jiao, Y. Feng, C. Yu, N. Chen, Y. Wei, X. Mu and E. Hao, Regioselective and Stepwise Syntheses of Functionalized BODIPY Dyes through Palladium-Catalyzed Cross-Coupling Reactions and Direct C-H Arylations, J. Org. Chem., 2016, 81, 6281–6291 CrossRef CAS PubMed.
- F. J. Frank, P. G. Waddell, M. J. Hall and J. G. Knight, Synthesis and Reactivity of 3,5-Diiodo-BODIPYs via a Concerted, Double Aromatic Finkelstein Reaction, Org. Lett., 2021, 23, 8595–8599 CrossRef CAS PubMed.
- H. R. A. Golf, H.-U. Reissig and A. Wiehe, Nucleophilic Substitution on (Pentafluorophenyl)dipyrromethane: A New Route to Building Blocks for Functionalized BODIPYs and Tetrapyrroles, Org. Lett., 2015, 17, 982–985 CrossRef CAS PubMed.
- G. Vives, C. Giansante, R. Bofinger, G. Raffy, A. Del Guerzo, B. Kauffmann, P. Batat, G. Jonusauskas and N. D. McClenaghan, Facile functionalization of a fully fluorescent perfluorophenyl BODIPY: photostable thiol and amine conjugates, Chem. Commun., 2011, 47, 10425–10427 RSC.
- D. E. Ramírez-Ornelas, E. Alvarado-Martínez, J. Bañuelos, I. López Arbeloa, T. Arbeloa, H. M. Mora-Montes, L. A. Pérez-García and E. Peña-Cabrera, FormylBODIPYs: Privileged Building Blocks for Multicomponent Reactions. The Case of the Passerini Reaction, J. Org. Chem., 2016, 81, 2888–2898 CrossRef PubMed.
- A. Ramos-Torres, E. Avellanal-Zaballa, A. Prieto-Castaneda, F. García-Garrido, J. Bañuelos, A. R. Agarrabeitia and M. J. Ortiz, FormylBODIPYs by PCC-Promoted Selective Oxidation of α-MethylBODIPYs. Synthetic Versatility and Applications, Org. Lett., 2019, 21, 4563–4566 CrossRef CAS PubMed.
- V. Lakshmi and M. Ravikanth, Synthesis of conjugated BODIPYs via the Wittig reaction, J. Org. Chem., 2013, 78, 4993–5000 CrossRef CAS PubMed.
- L. E. Greene, R. Lincoln, K. Krumova and G. Cosa, Development of a Fluorogenic Reactivity Palette for the Study of Nucleophilic Addition Reactions Based on meso-Formyl BODIPY Dyes, ACS Omega, 2017, 2, 8618–8624 CrossRef CAS PubMed.
- S. Zhu, J. Bi, G. Vegesna, J. Zhang, F.-T. Luo, L. Valenzano and H. Liu, Functionalization of BODIPY dyes at 2,6-positions through formyl groups, RSC Adv., 2013, 3, 4793–4800 RSC.
- T. Komatsu, Y. Urano, Y. Fujikawa, T. Kobayashi, H. Kojima, T. Terai, K. Hanaoka and T. Nagano, Development of 2,6-carboxy-substituted boron dipyrromethene (BODIPY) as a novel scaffold of ratiometric fluorescent probes for live cell imaging, Chem. Commun., 2009, 7015–7017 RSC.
- D. Wang, J. Fan, X. Gao, B. Wang, S. Sun and X. Peng, Carboxyl BODIPY dyes from bicarboxylic anhydrides: One-pot preparation, spectral properties, photostability, and biolabeling, J. Org. Chem., 2009, 74, 7675–7683 CrossRef CAS PubMed.
- S. Krajcovicova, J. Stankova, P. Dzubak, M. Hajduch, M. Soural and M. Urban, A Synthetic Approach for the Rapid Preparation of BODIPY Conjugates and their use in Imaging of Cellular Drug Uptake and Distribution, Chem. – Eur. J., 2018, 24, 4957–4966 CrossRef CAS PubMed.
- T. Katoh, M. Yoshikawa, T. Yamamoto, R. Arai, N. Nii, Y. Tomata, S. Suzuki, R. Koyama, N. Negoro and T. Yogo, Parallel fluorescent probe synthesis based on the large-scale preparation of BODIPY FL propionic acid, Bioorg. Med. Chem. Lett., 2017, 27, 1145–1148 CrossRef CAS PubMed.
- H. Chen, X. He, M. Su, W. Zhai, H. Zhang and C. Li, A General Strategy Toward Highly Fluorogenic Bioprobes Emitting across the Visible Spectrum, J. Am. Chem. Soc., 2017, 139, 10157–10163 CrossRef CAS PubMed.
- Y. Jang, T.-I. Kim, H. Kim, Y. Choi and Y. Kim, Photoactivatable BODIPY Platform: Light-Triggered Anticancer Drug Release and Fluorescence Monitoring, ACS Appl. Bio Mater., 2019, 2, 2567–2572 CrossRef CAS PubMed.
- S. Jeon, T.-I. Kim, H. Jin, U. Lee, J. Bae, J. Bouffard and Y. Kim, Amine-Reactive Activated Esters of meso-CarboxyBODIPY: Fluorogenic Assays and Labeling of Amines, Amino Acids, and Proteins, J. Am. Chem. Soc., 2020, 142, 9231–9239 CrossRef CAS PubMed.
- L. Betancourt-Mendiola, I. Valois-Escamilla, T. Arbeloa, J. Bañuelos, I. López Arbeloa, J. O. Flores-Rizo, R. Hu, E. Lager, C. F. A. Gómez-Durán, J. L. Belmonte-Vázquez, M. R. Martínez-González, I. J. Arroyo, C. A. Osorio-Martínez, E. Alvarado-Martínez, A. Urías-Benavides, B. D. Gutiérrez-Ramos, B. Z. Tang and E. Peña-Cabrera, Scope and Limitations of the Liebeskind-Srogl Cross-Coupling Reactions Involving the Biellmann BODIPY, J. Org. Chem., 2015, 80, 5771–5782 CrossRef CAS PubMed.
- J. O. Flores-Rizo, I. Esnal, C. A. Osorio-Martínez, C. F. A. Gómez-Durán, J. Bañuelos, I. López Arbeloa, K. H. Pannell, A. J. Metta-Magana and E. Peña-Cabrera, 8-Alkoxy- and 8-Aryloxy-BODIPYs: Straightforward Fluorescent Tagging of Alcohols and Phenols, J. Org. Chem., 2013, 78, 5867–5877 CrossRef CAS PubMed.
- J. Bañuelos, V. Martín, C. F. A. Gómez-Durán, I. J. A. Córdoba, E. Peña-Cabrera, I. García-Moreno, A. Costela, M. E. Pérez-Ojeda, T. Arbeloa and I. L. Arbeloa, New 8-Amino-BODIPY Derivatives: Surpassing Laser Dyes at Blue-Edge Wavelengths, Chem. – Eur. J., 2011, 17, 7261–7270 CrossRef PubMed.
- I. J. Arroyo, R. Hu, G. Merino, B. Z. Tang and E. Peña-Cabrera, The Smallest and One of the Brightest. Efficient Preparation and Optical Description of the Parent Borondipyrromethene System, J. Org. Chem., 2009, 74, 5719–5722 CrossRef CAS PubMed.
- E. Peña-Cabrera, A. Aguilar-Aguilar, M. González-Domínguez, E. Lager, R. Zamudio-Vázquez, J. Godoy-Vargas and F. Villanueva-García, Simple, general, and efficient synthesis of meso-substituted borondipyrromethenes from a single platform, Org. Lett., 2007, 9, 3985–3988 CrossRef PubMed.
- C. Uriel, A. M. Gómez, E. García, M. de la Hidalga, J. Bañuelos, I. García-Moreno and J. C. López, Access to 2,6-Dipropargylated BODIPYs as “Clickable” Congeners of Pyrromethene-567 Dye: Photostability and Synthetic Versatility, Org. Lett., 2021, 23, 6801–6806 CrossRef CAS PubMed.
- P. Kele, X. Li, M. Link, K. Nagy, A. Herner, K. Lorincz, S. Beni and O. S. Wolfbeis, Clickable fluorophores for biological labeling-with or without copper, Org. Biomol. Chem., 2009, 7, 3486–3490 RSC.
- M. R. Martínez-González, A. Urías-Benavides, E. Alvarado-Martínez, J. C. López, A. M. Gómez, M. del Rio, I. García, A. Costela, J. Bañuelos, T. Arbeloa, I. López Arbeloa and E. Peña-Cabrera, Convenient Access to Carbohydrate-BODIPY Hybrids by Two Complementary Methods Involving One-Pot Assembly of “Clickable” BODIPY Dyes, Eur. J. Org. Chem., 2014, 5659–5663 CrossRef.
- T. B. Gontijo, R. P. de Freitas, F. S. Emery, L. F. Pedrosa, J. B. Vieira Neto, B. C. Cavalcanti, C. Pessoa, A. King, F. de Moliner, M. Vendrell and E. N. da Silva Jr., On the synthesis of quinone-based BODIPY hybrids: New insights on antitumor activity and mechanism of action in cancer cells, Bioorg. Med. Chem. Lett., 2017, 27, 4446–4456 CrossRef CAS PubMed.
- A. L. Nguyen, K. E. Griffin, Z. Zhou, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, Syntheses of 1,2,3-triazole-BODIPYs bearing up to three carbohydrate units, New J. Chem., 2018, 42, 8241–8246 RSC.
- M. E. Pérez-Ojeda, B. Trastoy, A. Rol, M. D. Chiara, I. García-Moreno and J. L. Chiara, Controlled Click-Assembly of Well-Defined Hetero-Bifunctional Cubic Silsesquioxanes and Their Application in Targeted Bioimaging, Chem. – Eur. J., 2013, 19, 6630–6640 CrossRef PubMed.
- M. E. Pérez-Ojeda, B. Trastoy, I. López-Arbeloa, J. Bañuelos, A. Costela, I. García-Moreno and J. L. Chiara, Click Assembly of Dye-Functionalized Octasilsesquioxanes for Highly Efficient and Photostable Photonic Systems, Chem. – Eur. J., 2011, 17, 13258–13268 CrossRef PubMed.
- B. Trastoy, M. E. Pérez-Ojeda, R. Sastre and J. L. Chiara, Octakis(3-azidopropyl)octasilsesquioxane: A Versatile Nanobuilding Block for the Efficient Preparation of Highly Functionalized Cube-Octameric Polyhedral Oligosilsesquioxane Frameworks Through Click Assembly, Chem. – Eur. J., 2010, 16, 3833–3841 CrossRef CAS PubMed.
- J. C. Er, M. K. Tang, C. G. Chia, H. Liew, M. Vendrell and Y.-T. Chang, MegaStokes BODIPY-triazoles as environmentally sensitive turn-on fluorescent dyes, Chem. Sci., 2013, 4, 2168–2176 RSC.
- J.-J. Shie, Y.-C. Liu, Y.-M. Lee, C. Lim, J.-M. Fang and C.-H. Wong, An Azido-BODIPY Probe for Glycosylation: Initiation of Strong Fluorescence upon Triazole Formation, J. Am. Chem. Soc., 2014, 136, 9953–9961 CrossRef CAS PubMed.
- H. R. A. Golf, A. M. Oltmanns, D. H. Trieu, H.-U. Reissig and A. Wiehe, Synthesis of Functionalized BODIPYs, BODIPY-Corrole, and BODIPY-Porphyrin Arrays with 1,2,3-Triazole Linkers Using the 4-Azido(tetrafluorophenyl)-BODIPY Building Block, Eur. J. Org. Chem., 2015, 4224–4237 CrossRef CAS.
- J. C. López, M. del Rio, A. Oliden, J. Bañuelos, I. López-Arbeloa, I. García-Moreno and A. M. Gómez, Solvent-Sensitive Emitting Urea-Bridged bis-BODIPYs: Ready Access by a One-Pot Tandem Staudinger/Aza-Wittig Ureation, Chem. – Eur. J., 2017, 23, 17511–17520 CrossRef PubMed.
- J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, BODIPY-Tetrazine Derivatives as Superbright Bioorthogonal Turn-on Probes, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef CAS PubMed.
- Y. Zhou, R. C. H. Wong, G. Dai and D. K. P. Ng, A bioorthogonally activatable photosensitiser for site-specific photodynamic therapy, Chem. Commun., 2020, 56, 1078–1081 RSC.
- M. Wu, X. Wu, Y. Wang, L. Gu, J. You, H. Wu and P. Feng, Alkoxy Tetrazine Substitution at a Boron Center: A Strategy for Synthesizing Highly Fluorogenic Hydrophilic Probes, ChemBioChem, 2018, 19, 530–534 CrossRef CAS PubMed.
- H. Wu, J. Yang, J. Seckute and N. K. Devaraj, In Situ Synthesis of Alkenyl Tetrazines for Highly Fluorogenic Bioorthogonal Live-Cell Imaging Probes, Angew. Chem., Int. Ed., 2014, 53, 5805–5809 CrossRef CAS PubMed.
- G. Linden and O. Vázquez, Bioorthogonal turn-on BODIPY-peptide photosensitizers for tailored photodynamic therapy, Chem. – Eur. J., 2020, 26, 10014–10023 CrossRef CAS PubMed.
- H. G. Knaus, T. Moshammer, K. Friedrich, H. C. Kang, R. P. Haugland and H. Glossman, In vivo labeling of L-type Ca2+ channels by fluorescent dihydropyridines: evidence for a functional, extracellular heparin-binding site, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3586–3590 CrossRef CAS PubMed.
- O. Buyukcakir, O. A. Bozdemir, S. Kolemen, S. Erbas and E. U. Akkaya, Tetrastyryl-Bodipy dyes: Convenient synthesis and characterization of elusive near IR fluorophores, Org. Lett., 2009, 11, 4644–4647 CrossRef CAS PubMed.
- Y.-H. Yu, A. B. Descalzo, Z. Shen, H. Rohr, Q. Liu, Y.-W. Wang, M. Spieles, Y.-Z. Li, K. Rurack and X.-Z. You, Mono- and Di(dimethylamino)styryl-substituted borondipyrromethene and borondiindomethene dyes with intense near-infrared fluorescence, Chem. – Asian J., 2006, 1, 176–187 CrossRef CAS PubMed.
- G. Sathyamoorthi, L. T. Wolford, A. M. Haag and J. H. Boyer, Selective side-chain oxidation of peralkylated pyrromethene-BF2 complexes, Heteroat. Chem., 1994, 5, 245–249 CrossRef CAS.
- L. Yang, R. S. Yalagala, S. Hutton, A. Lough and H. Yan, Reactions of BODIPY Fluorophore with Cupric Nitrate, Synlett, 2014, 25, 2661–2664 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Haefele, A General Synthetic Route to 3,5-Substituted Boron Dipyrromethenes: Applications and Properties, J. Org. Chem., 2012, 77, 4298–4311 CrossRef CAS PubMed.
- Q. Gong, K. Cheng, Q. Wu, W. Li, C. Yu, L. Jiao and E. Hao, One-Pot Access to Ethylene-Bridged BODIPY Dimers and Trimers through Single-Electron Transfer Chemistry, J. Org. Chem., 2021, 86, 15761–15767 CrossRef CAS PubMed.
- L. J. Patalag, L. P. Ho, P. G. Jones and D. B. Werz, Ethylene-Bridged Oligo-BODIPYs: Access to Intramolecular J-Aggregates and Superfluorophores, J. Am. Chem. Soc., 2017, 139, 15104–15113 CrossRef CAS PubMed.
- W. Siedel and F. Winkler, Oxidation of pyrrole derivatives with Pb tetraacetate. A new kind of porphyrin synthesis, Justus Liebigs Ann. Chem., 1943, 554, 162–201 CrossRef CAS.
- A. Boudif and M. Momenteau, A new convergent method for porphyrin synthesis based on a ‘3 + 1′ condensation, J. Chem. Soc., Perkin Trans. 1, 1996, 1235–1242 RSC.
- Q. Chen, M. T. Huggins, D. A. Lightner, W. Norona and A. F. McDonagh, Synthesis of a 10-Oxo-Bilirubin: Effects of the Oxo Group on Conformation, Transhepatic Transport, and Glucuronidation, J. Am. Chem. Soc., 1999, 121, 9253–9264 CrossRef CAS.
- M. G. H. Vincente and K. M. Smith, Porphyrins and derivatives: synthetic strategies and reactivity profiles, Curr. Org. Chem., 2000, 4, 139–174 CrossRef CAS.
- S. M. Crawford and A. Thompson, Conversion of 4,4-Difluoro-4-bora-3a,4a-diaza-s-indacenes (F-BODIPYs) to Dipyrrins with a Microwave-Promoted Deprotection Strategy, Org. Lett., 2010, 12, 1424–1427 CrossRef CAS PubMed.
- D. A. Smithen, A. E. G. Baker, M. Offman, S. M. Crawford, T. S. Cameron and A. Thompson, Use of F-BODIPYs as a Protection Strategy for Dipyrrins: Optimization of BF2 Removal, J. Org. Chem., 2012, 77, 3439–3453 CrossRef CAS PubMed.
- L. Yang, R. Simionescu, A. Lough and H. Yan, Some observations relating to the stability of the BODIPY fluorophore under acidic and basic conditions, Dyes Pigm., 2011, 91, 264–267 CrossRef CAS.
- V. Lakshmi, T. Chatterjee and M. Ravikanth, Lewis Acid Assisted Decomplexation of F-BODIPYs to Dipyrrins, Eur. J. Org. Chem., 2014, 2105–2110 CrossRef CAS.
- T. Lundrigan, T. S. Cameron and A. Thompson, Activation and deprotection of F-BODIPYs using boron trihalides, Chem. Commun., 2014, 50, 7028–7031 RSC.
- M. Yu, J. K. H. Wong, C. Tang, P. Turner, M. H. Todd and P. J. Rutledge, Efficient deprotection of F-BODIPY derivatives: removal of BF2 using Brønsted acids, Beilstein J. Org. Chem., 2015, 11, 37–41 CrossRef PubMed , 35 pp.
- J. Urieta, B. L. Maroto, F. Moreno, A. R. Agarrabeitia, M. J. Ortiz and S. de la Moya, Preparation of dipyrrins from F-BODIPYs by treatment with methanesulfonic acids, RSC Adv., 2015, 5, 68676–68680 RSC.
- C. D. Smith and A. Thompson, Facile deprotection of F-BODIPYs using methylboronic acid, RSC Adv., 2020, 10, 24273–24279 RSC.
- E. Bodio and C. Goze, Investigation of B-F substitution on BODIPY and aza-BODIPY dyes: Development of B-O and B-C BODIPYs, Dyes Pigm., 2019, 160, 700–710 CrossRef CAS.
- L. Li, B. Nguyen and K. Burgess, Functionalization of the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) core, Bioorg. Med. Chem. Lett., 2008, 18, 3112–3116 CrossRef CAS PubMed.
- A. L. Nguyen, M. Wang, P. Bobadova-Parvanova, Q. Do, Z. Zhou, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, Synthesis and properties of B-cyano-BODIPYs, J. Porphyrins Phthalocyanines, 2016, 20, 1409–1419 CrossRef CAS.
- M. Wang, M. G. H. Vicente, D. Mason and P. Bobadova-Parvanova, Stability of a Series of BODIPYs in Acidic Conditions: An Experimental and Computational Study into the Role of the Substituents at Boron, ACS Omega, 2018, 3, 5502–5510 CrossRef CAS PubMed.
- C. Uriel, C. Permingeat, J. Ventura, A. M. Gómez, J. C. López, E. Avellanal-Zaballa, J. Bañuelos and I. García-Moreno, BODIPYs as Chemically Stable Fluorescent Tags for Synthetic Glycosylation Strategies towards Fluorescently Labeled Saccharides, Chem. – Eur. J., 2020, 26, 5388–5399 CrossRef CAS PubMed.
- G. Durán-Sampedro, I. Esnal, A. R. Agarrabeitia, J. Bañuelos Prieto, L. Cerdán, I. García-Moreno, A. Costela, I. López-Arbeloa and M. J. Ortiz, First Highly Efficient and Photostable E and C Derivatives of 4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) as Dye Lasers in the Liquid Phase, Thin Films, and Solid-State Rods, Chem. – Eur. J., 2014, 20, 2646–2653 CrossRef PubMed.
- P. G. Waddell, X. Liu, T. Zhao and J. M. Cole, Rationalizing the photophysical properties of BODIPY laser dyes via aromaticity and electron-donor-based structural perturbations, Dyes Pigm., 2015, 116, 74–81 CrossRef CAS.
- C. Y. Xu, E. Lebeau and C. Walker, Selective deprotection of esters using magnesium and methanol, Tetrahedron Lett., 1994, 35, 6207–6210 CrossRef CAS.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Bioorthogonal chemistry, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioorthogonal Chemistry and Its Applications, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- H. Wu and N. K. Devaraj, Inverse Electron-Demand Diels-Alder Bioorthogonal Reactions, Top. Curr. Chem., 2016, 374, 1–22 CrossRef CAS PubMed.
- C. A. Hoelzel and X. Zhang, Visualizing and Manipulating Biological Processes by Using HaloTag and SNAP-Tag Technologies, ChemBioChem, 2020, 21, 1935–1946 CrossRef CAS PubMed.
- C. G. England, H. Luo and W. Cai, HaloTag Technology: A Versatile Platform for Biomedical Applications, Bioconjugate Chem., 2015, 26, 975–986 CrossRef CAS PubMed.
- G. V. Los and K. Wood, The HaloTag: a novel technology for cell imaging and protein analysis, Methods Mol. Biol., 2007, 356, 195–208 CAS.
- S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, Transient Protection of Strained Alkynes from Click Reaction via Complexation with Copper, J. Am. Chem. Soc., 2014, 136, 13590–13593 CrossRef CAS PubMed.
- P. Gobbo, T. Romagnoli, S. M. Barbon, J. T. Price, J. Keir, J. B. Gilroy and M. S. Workentin, Expanding the scope of strained-alkyne chemistry: a protection-deprotection strategy via the formation of a dicobalt-hexacarbonyl complex, Chem. Commun., 2015, 51, 6647–6650 RSC.
- Q. Zhao, C. Yin, J. Kang, Y. Wen and F. Huo, A viscosity sensitive azide-pyridine BODIPY-based fluorescent dye for imaging of hydrogen sulfide in living cells, Dyes Pigm., 2018, 159, 166–172 CrossRef CAS.
- C. Wang, F. Xie, N. Suthiwangcharoen, J. Sun and Q. Wang, Tuning the optical properties of BODIPY dye through Cu(I) catalyzed azide-alkyne cycloaddition (CuAAC) reaction, Sci. China: Chem., 2012, 55, 125–130 CrossRef CAS.
- X. Ao, S. A. Bright, N. C. Taylor and R. B. P. Elmes, 2-Nitroimidazole based fluorescent probes for nitroreductase; monitoring reductive stress in cellulo, Org. Biomol. Chem., 2017, 15, 6104–6108 RSC.
- B. D. Gutiérrez-Ramos, J. Bañuelos, T. Arbeloa, I. L. Arbeloa, P. E. González-Navarro, K. Wrobel, L. Cerdán, I. García-Moreno, A. Costela and E. Peña-Cabrera, Straightforward Synthetic Protocol for the Introduction of Stabilized C Nucleophiles in the BODIPY Core for Advanced Sensing and Photonic Applications, Chem. – Eur. J., 2015, 21, 1755–1764 CrossRef PubMed.
- V. Leen, M. Laine, J. M. Ngongo, P. Lipkowski, B. Verbelen, A. Kochel, W. Dehaen, M. Van der Auweraer, V. Nadtochenko and A. Filarowski, Impact of the Keto-Enol Tautomeric Equilibrium on the BODIPY Chromophore, J. Phys. Chem. A, 2018, 122, 5955–5961 CrossRef CAS PubMed.
- H. Zaimoku, T. Hatta, T. Taniguchi and H. Ishibashi, Iodine-Mediated α-Acetoxylation of 2,3-Disubstituted Indoles, Org. Lett., 2012, 14, 6088–6091 CrossRef CAS PubMed.
- W. D. G. Brittain and C. R. Coxon, Perfluoroaryl and Perfluoroheteroaryl Reagents as Emerging New Tools for Peptide Synthesis, Modification and Bioconjugation, Chem. – Eur. J., 2022, 28, 10–23 Search PubMed.
- M. Macias-Contreras and L. Zhu, The Collective Power of Genetically Encoded Protein/Peptide Tags and Bioorthogonal Chemistry in Biological Fluorescence Imaging, ChemPhotoChem, 2021, 5, 187–216 CrossRef CAS.
- H. E. Murrey, J. C. Judkins, C. W. am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, Systematic Evaluation of Bioorthogonal Reactions in Live Cells with Clickable HaloTag Ligands: Implications for Intracellular Imaging, J. Am. Chem. Soc., 2015, 137, 11461–11475 CrossRef CAS PubMed.
- C. Aonbangkhen, H. Zhang, D. Z. Wu, M. A. Lampson and D. M. Chenoweth, Reversible Control of Protein Localization in Living Cells Using a Photocaged-Photocleavable Chemical Dimerizer, J. Am. Chem. Soc., 2018, 140, 11926–11930 CrossRef CAS PubMed.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Bioorthogonal Turn-On Probes for Imaging Small Molecules inside Living Cells, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- C. Dumas-Verdes, F. Miomandre, E. Lepicier, O. Galangau, T. T. Vu, G. Clavier, R. Meallet-Renault and P. Audebert, BODIPY-Tetrazine Multichromophoric Derivatives, Eur. J. Org. Chem., 2010, 2525–2535 CrossRef CAS.
- A. M. Courtis, S. A. Santos, Y. Guan, J. A. Hendricks, B. Ghosh, D. M. Szantai-Kis, S. A. Reis, J. V. Shah and R. Mazitschek, Monoalkoxy BODIPYs - A Fluorophore Class for Bioimaging, Bioconjugate Chem., 2014, 25, 1043–1051 CrossRef CAS PubMed.
- A. Wieczorek, T. Buckup and R. Wombacher, Rigid tetrazine fluorophore conjugates with fluorogenic properties in the inverse electron demand Diels-Alder reaction, Org. Biomol. Chem., 2014, 12, 4177–4185 RSC.
- G. Beliu, A. J. Kurz, A. C. Kuhlemann, L. Behringer-Pliess, M. Meub, N. Wolf, J. Seibel, Z.-D. Shi, M. Schnermann, J. B. Grimm, L. D. Lavis, S. Doose and M. Sauer, Bioorthogonal labeling with tetrazine-dyes for super-resolution microscopy, Commun. Biol., 2019, 2, 1–13 CrossRef CAS.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Inverse electron demand Diels-Alder reactions in chemical biology, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- M.-C. Chen, D.-G. Chen and P.-T. Chou, Fluorescent Chromophores Containing the Nitro Group: Relatively Unexplored Emissive Properties, ChemPlusChem, 2021, 86, 11–27 CrossRef CAS PubMed.
- Y. M. Poronik, B. Sadowski, K. Szychta, F. H. Quina, V. I. Vullev and D. T. Gryko, Revisiting the non-fluorescence of nitroaromatics: presumption versus reality, J. Mater. Chem. C, 2022, 10, 2870–2904 RSC.
- L. Gai, J. Chen, Y. Zhao, J. Mack, H. Lu and Z. Shen, Synthesis and properties of azulene-functionalized BODIPYs, RSC Adv., 2016, 6, 32124–32129 RSC.
- E. Ozcan, H. H. Kazan and B. Cosut, Recent chemo-/biosensor and bioimaging studies based on indole-decorated BODIPYs, Luminescence, 2020, 35, 168–177 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. Supplementary tables and figures, complete experimental procedures, X-ray diffraction data of 9, computational details, live-cell microscopy images, and copies of 1H and 13C NMR spectra of all new compounds (PDF). CCDC 2177585 contains the supplementary crystallographic data for compound 9. For the corresponding 1D- and 2D-NMR datasets in the standard JCAMP-DX format and other electronic formats see DOI: https://doi.org/10.1039/d2qo01099b |
| ‡ These authors have contributed equally to this work. |
| This journal is © the Partner Organisations 2022 |