
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodonitrene: a direct metal-free electrophilic aminating reagent
Chunngai
Hui
ab and
Andrey P.
Antonchick
 *abc
*abc
aMax Planck Institute of Molecular Physiology, Department of Chemical Biology, Otto-Hahn-Strasse 11, 44227 Dortmund, Germany
bTechnical University Dortmund, Faculty of Chemistry and Chemical Biology, Otto-Hahn-Strasse 6, 44221 Dortmund, Germany
cNottingham Trent University, School of Science and Technology, Department of Chemistry and Forensics, Clifton Lane, NG11 8NS Nottingham, UK. E-mail: andrey.antonchick@ntu.ac.uk
First published on 14th June 2022
Abstract
The use of conventional nitrenoids and/or metal nitrenes as electrophilic aminating reagents requires a pre-activated nitrogen atom, which makes transfer of an unprotected NH-group a difficult challenge. Iodonitrene, which is generated in situ from phenyliodine(III) diacetate and an ammonia surrogate, represents a new type of reactive electrophilic aminating reagent. The novel reactivity of iodonitrene not only resulted in direct NH-group transfer to nucleophilic atoms such as sulfur and nitrogen, but also led to the development of new reactions such as diazirine synthesis via decarboxylation and contractive synthesis of cyclobutanes via nitrogen extrusion. We highlight the contemporary advances in the application of iodonitrene and discuss the current limitations and future prospects.
 Chunngai Hui | Chunngai Hui obtained his BSc degree in chemical technology from the Hong Kong Polytechnic University (China) and his MSc degree in biotechnology from the Hong Kong University of Science and Technology (China). He completed his PhD at Max Planck Institute of Molecular Physiology under the supervision of Prof. A. P. Antonchick. His research interests include the synthesis of bioactive natural products and their related biological activities. |
 Andrey P. Antonchick | Andrey P. Antonchick studied at Belarusian State University (Minsk, Belarus). He completed his PhD at the Institute of Bioorganic Chemistry of the National Academy of Sciences of Belarus and the Max Planck Institute for Chemical Ecology (Jena, Germany) under the guidance of Prof. V. A. Khripach and PD Dr B. Schneider. After a postdoctoral appointment with Prof. M. Rueping at Frankfurt University, he joined Prof. H. Waldmann at the Max Planck Institute of Molecular Physiology (Dortmund, Germany). In 2011, he was appointed as the group leader at the Max Planck Institute of Molecular Physiology and Technical University of Dortmund. Since August 2020, he has been holding the position of an Associate Professor at Nottingham Trent University (Nottingham, UK). |
1. Introduction
Direct access to nitrogen-containing functional groups is indispensable in the preparation of biologically important molecules. Amination methods such as nucleophilic substitution, reductive amination, and metal-catalyzed amination reactions (e.g. allylic substitution, hydroamination, and C–N cross-coupling reactions) are widely used for amine synthesis.1,2 Electrophilic amination requires the use of electrophilic aminating reagents, such as metal–nitrene equivalents, or oxaziridines as nitrogen sources, resulting in a net addition of an amino group to the electron-rich functionalities of the substrate.3–8 Conventionally, the dirhodium-nitrene chemistry enables intramolecular amination of the inert C(sp3)–H bond to form a C–N bond via intermolecular nitrene transfer,9 and this condition was applied to the synthesis of NH-sulfoximines from sulfoximes10 and the direct preparation of NH-aziridines and anilines from alkenes and arenes,11,12 which have been broadly applied to organic synthesis (Scheme 1A).13 On the other hand, iodonitrene14 generated in situ from the reaction between hypervalent iodine(III) reagents and ammonia or its surrogates has recently been introduced as a promising electrophilic aminating reagent (Scheme 1B). The synthesis of NH-sulfoximines (Bull, Luisi and co-workers, 2016),14 the synthesis of diazirines from unprotected amino acids (Reboul and coworkers, 2019),15 and the contractive synthesis of cyclobutanes from pyrrolidines (Antonchick and coworkers, 2021)16 revealed the novel reactivity of iodonitrene distinguishing it from the precedent metal-nitrene chemistry. The use of iodonitrene as an electrophilic aminating reagent not only provides a reactive nitrene species ready for nitrogen transfer, but also circumvents the use of metals and activated explosive reagents, such as O-mesitylenesulfonyl-hydroxylamine (MSH).17 Bull, Luisi and co-workers disclosed the evidence of iodonitrene and possible intermediates (PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH),14 for instance, iminoiodinane 1a and the unprecedented iodonitrene 1b (PhIN+) via mass spectrometric analysis and isotopic labeling using 15N18 (Scheme 1C). However, no evidence of any reactive intermediates was found throughout the NMR studies.19
NH),14 for instance, iminoiodinane 1a and the unprecedented iodonitrene 1b (PhIN+) via mass spectrometric analysis and isotopic labeling using 15N18 (Scheme 1C). However, no evidence of any reactive intermediates was found throughout the NMR studies.19
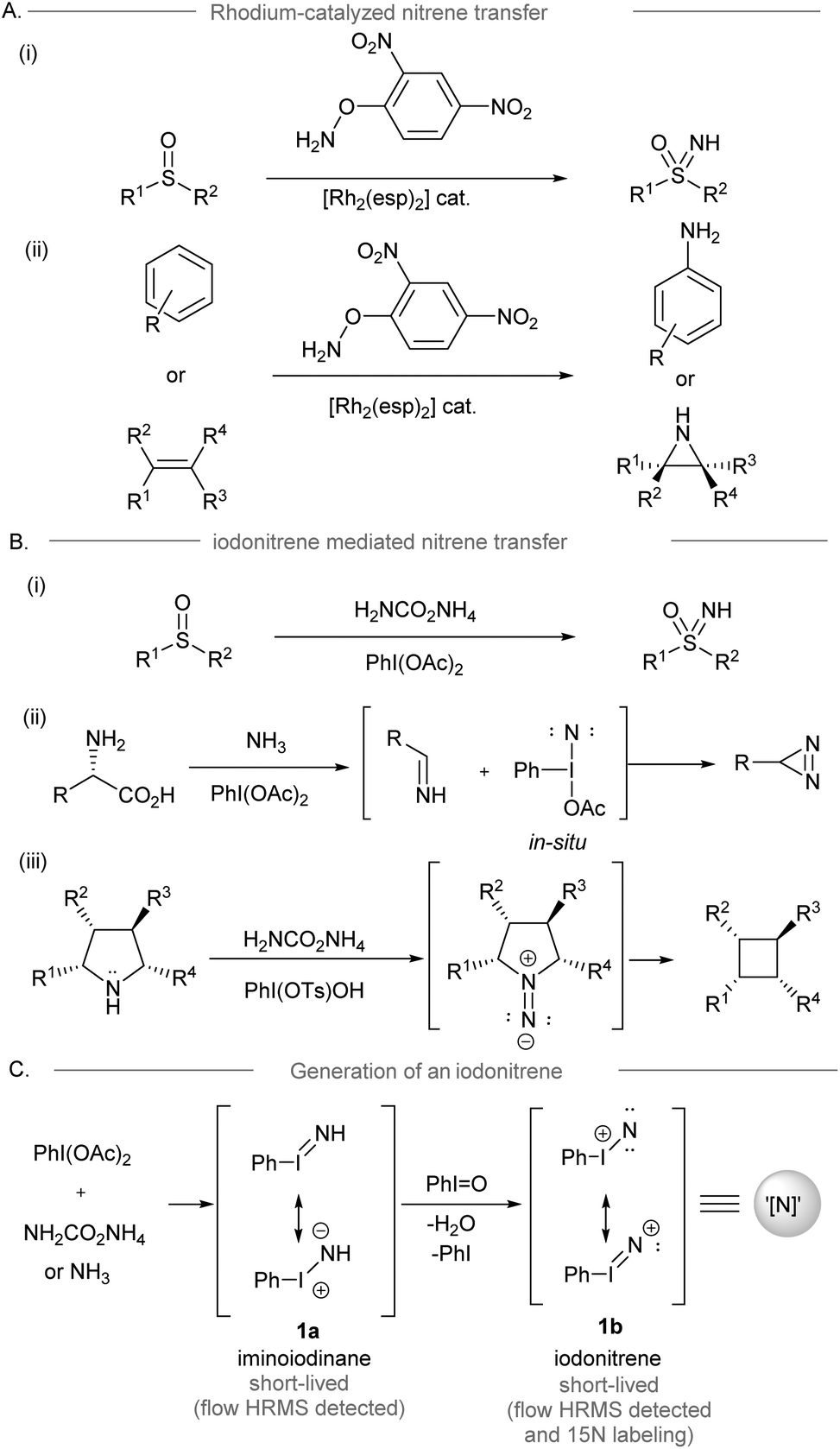 | ||
| Scheme 1 (A) Rhodium catalyzed amination reactions involving dirhodium nitrene transfer.10–12 (B) Some examples of amination reactions using iodonitrenes as electrophilic aminating reagents.9,15 (C) The proposed mechanism of iodonitrene formation.9 | ||
Our group20–27 is engaged in the novel method development using hypervalent iodine(III) chemistry.28–35 Despite a personal account and a review on NH-sulfoximines which was reported by the Bull group and the Luisi group,36–38 no review has been published, to the best of our knowledge, discussing the chemistry of iodonitrene. We are motivated to provide a concise minireview, highlighting iodonitrene and its application in organic synthesis. This minireview could be useful to synthetic scientists in method development and natural product synthesis and the pharmaceutical industry. Finally, we discuss the directions and prospects for the innovation of new reactions and potential applications in organic synthesis.
2. Electrophilic amination of sulfur-containing compounds
Sulfur-containing compounds possess significant biological profiles and appear as important elements in drug discovery.39 For instance, sulfoximines are present as essential functional groups in drug candidates such as compound AZD6738 (Scheme 2, inset) from AstraZeneca.40 Directing the transfer of the NH group from iodonitrene to sulfoxides gives NH-sulfoximines in one step. With the oxidizing power of the hypervalent iodine(III) reagent, oxidation of the sulfur atom could take place before and/or after the nitrogen transfer from iodonitrene. In this section, the direct NH-group transfer to sulfoxides and sulfonamides is discussed. Moreover, the sequential NH-group transfer accompanied by the oxidation of sulfides, thiols, and sulfonamides is elaborated.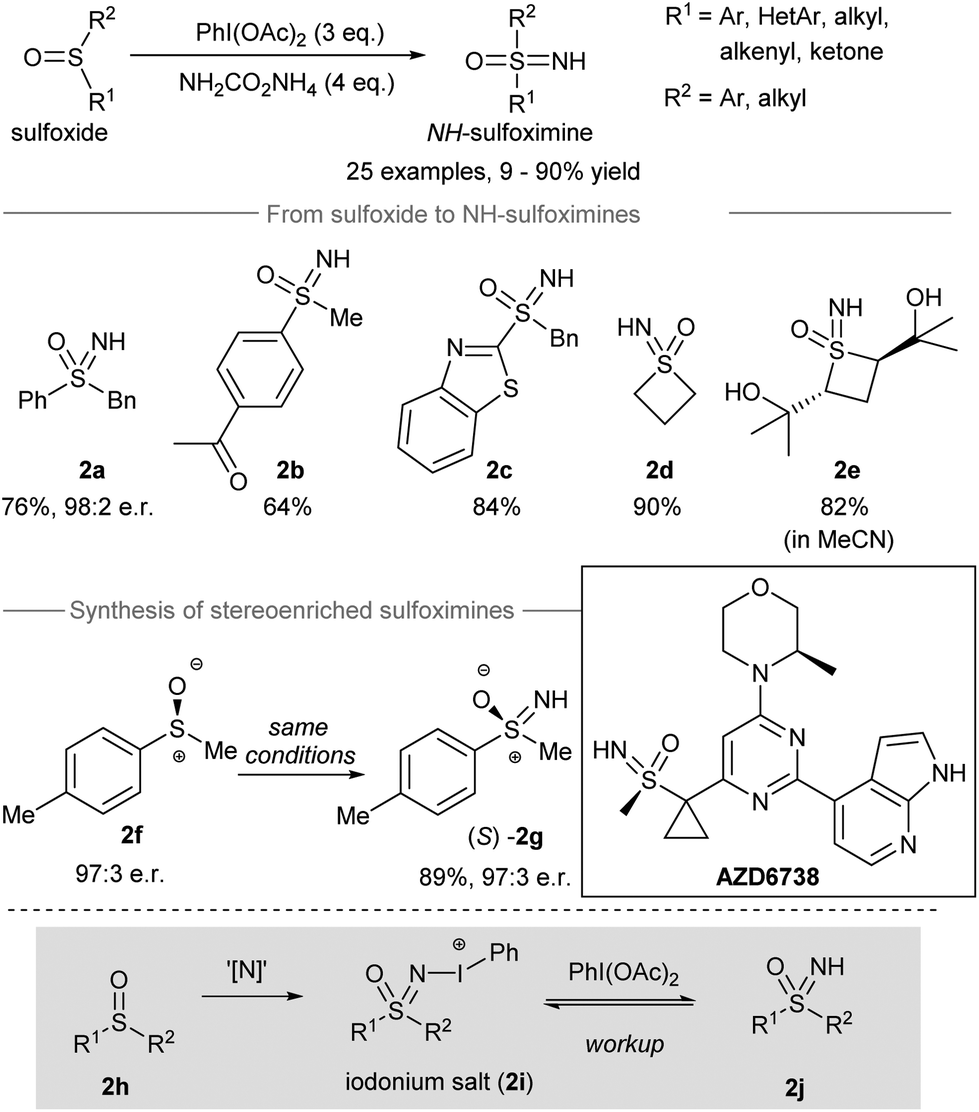 | ||
| Scheme 2 Direct NH-group transfer to sulfoxides producing NH-sulfoximines.14 | ||
2.1 Direct NH transfer to sulfoxides
In 2016, Luisi, Bull and co-workers reported the direct transfer of the NH-group from iodonitrene to sulfoxides to afford NH-sulfoximines14 (Scheme 2). Under the standard conditions, the iodonitrene generated in situ from the reaction between phenyliodine(III) diacetate (PIDA) and ammonium carbamate41 promoted a NH-group transfer to produce various NH-sulfoximines. Both aryl-substituted and alkyl-substituted sulfoxides carrying different reactive groups, such as ketones, free alcohols, and benzothiazoles, gave sulfoxides 2a–2e from decent to good yields. When methyl p-tolylsulfoxide 2f was used as the starting material, the stereospecific NH-group transfer took place under standard conditions and gave sulfoximine (S)-2g in 89% yield with 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 er. Mechanistically, the authors proposed that the nitrene transfer from iodonitrene (i.e. “[N]”) to sulfoxide 2h may generate an iodonium salt 2i. Further oxidation of 2i by free phenyliodine(III) diacetate followed by workup afforded NH-sulfoximines 2j.
:3 er. Mechanistically, the authors proposed that the nitrene transfer from iodonitrene (i.e. “[N]”) to sulfoxide 2h may generate an iodonium salt 2i. Further oxidation of 2i by free phenyliodine(III) diacetate followed by workup afforded NH-sulfoximines 2j.
2.2 Direct NH transfer to sulfinamides
Later, one-pot conversion of sulfinamides to sulfonimidamides via transfer of the electrophilic NH-group was reported by Stockman, Lücking, and co-workers (Scheme 3).42 Under the optimized conditions, sulfonimidamides (3a to 3d) were formed from sulfinamides in good to high yields. The replacement of the aryl substituents of tertiary sulfonamides by the 3-pyridinyl group (3c) or cyclohexyl group (3d) was tolerated. Importantly, the NH-group transfer to chiral sulfinamide 3e (51% ee) proceeded stereospecifically to give NH-sulfonimidamide 3f with 48% ee.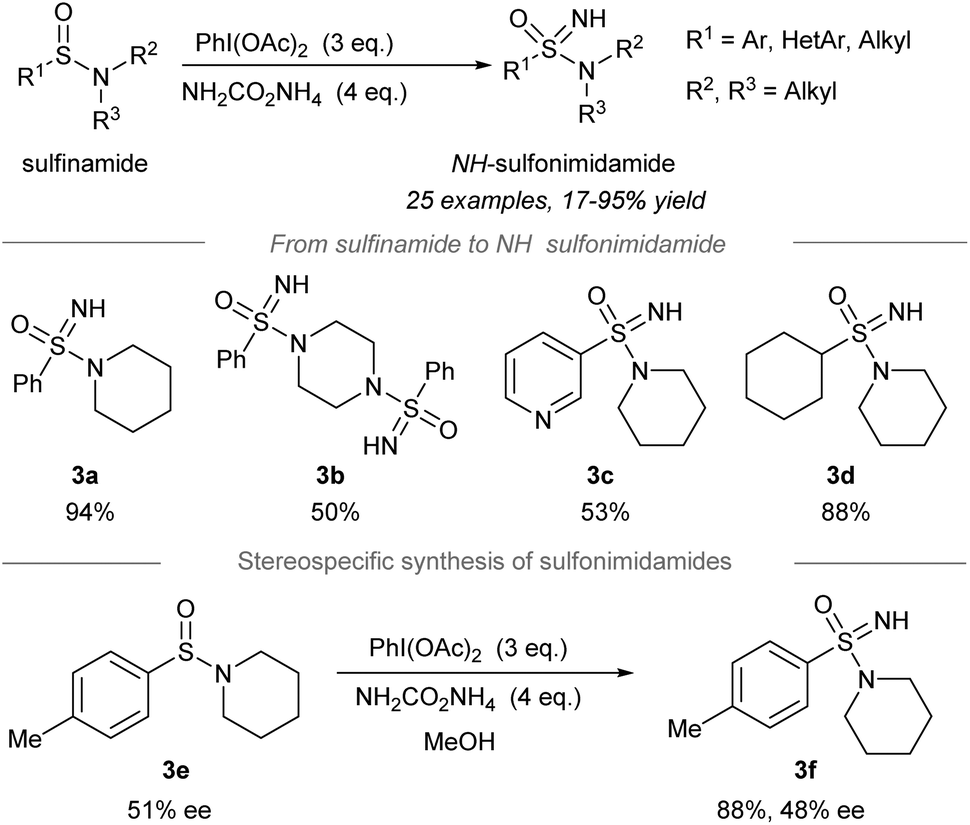 | ||
| Scheme 3 Direct NH-group transfer in the synthesis of NH-sulfonimidamides from sulfinamides.42 | ||
2.3 One-pot NH- and O-transfer to sulfides
After the pioneering synthesis of NH-sulfoximines from sulfides using iodonitrene as an electrophilic aminating reagent by Bull and Luisi in 2016,14 the synthesis of NH-sulfoximines from the corresponding sulfides via a one-pot NH- and O-transfer was realized by several research groups using iodonitrene chemistry (Scheme 4). In 2017, Bull and Luisi first reported the one-pot NH- and O-transfer to sulfides to give NH-sulfoximines (Scheme 4A).43 Aryl-, alkyl- and benzothiazole-substituted (R1) sulfides gave the corresponding NH-sulfoximines in high yield (4a–4c, 84%–94% yield). However, the low yield of vinyl substituted sulfoximine 4d is suggested to be the result of a possible polymerization of the substrate.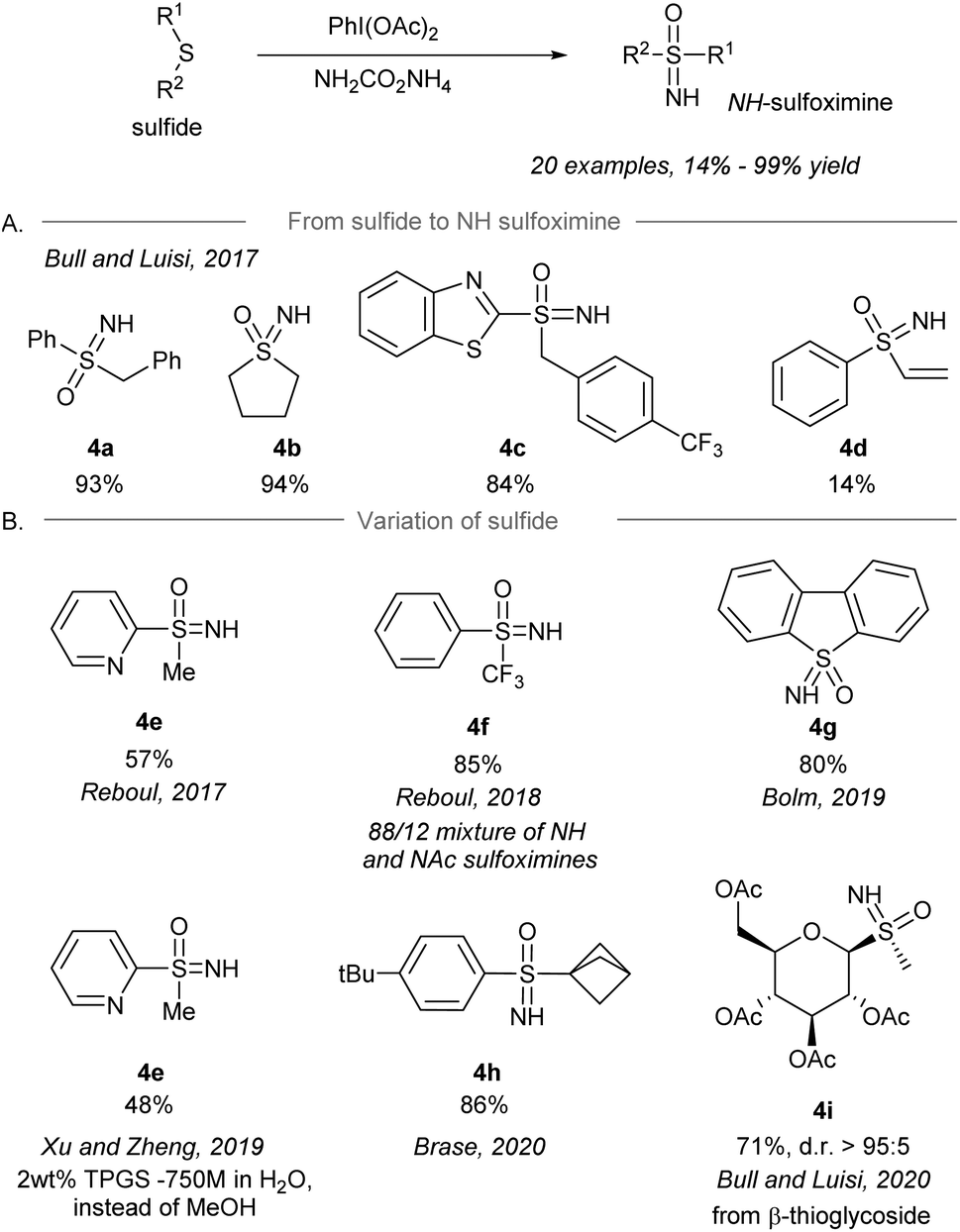 | ||
| Scheme 4 (A) Direct NH-sulfoximination of sulfides by an iodonitrene. (B) Variation of sulfides as the starting materials.43–49 | ||
Shortly after Bull and Luisi's work, other research groups reported variants of the transformation with modification of substrates and/or conditions (Scheme 4B), for instance pyridinyl sulfides 4e (Reboul's group),44,50S-perfluoroalkylated sulfides 4f (Reboul's group),51 thiophene-derived sulfides 4g (Bolm's group),48 bicyclo[1.1.1]pentyl sulfides 4h (Bräse's group),53 and β-thioglycosides 4i (Bull and Luisi's group).54 Besides, sulfoximination of sulfides could be achieved to afford 4e under aqueous micellar conditions using the surfactant TPGS-750-M as an additive.49
After the report of the direct sulfoximination of sulfides, a tandem NH-sulfoximination/C(sp2)–H amination of sulfides to give dibenzothiazines52 was developed by Chen and co-workers in 2018 (Scheme 5). Treatment of [1,1′-biaryl]-2-sulfides with PIDA and ammonium phosphate trihydrate afforded the NH- and O-transfer products NH-sulfoximines, which after intramolecular C(sp2)–H functionalization provided dibenzothiazines.55,56 The variation of the substituents R1 and R2 gave the desired dibenzothiazine products 5a and 5b in high yield. A phenyl group on R3 (5c) significantly reduced the yield of the reaction to 45% yield. Heterocyclic dibenzothiazines such as 5d and 5e were also compatible with the reaction conditions. The authors proposed that the oxidation of the newly formed NH-sulfoximine 5f with PIDA gave 5g, which cyclized to give dibenzothiazine 5h (Scheme 5).
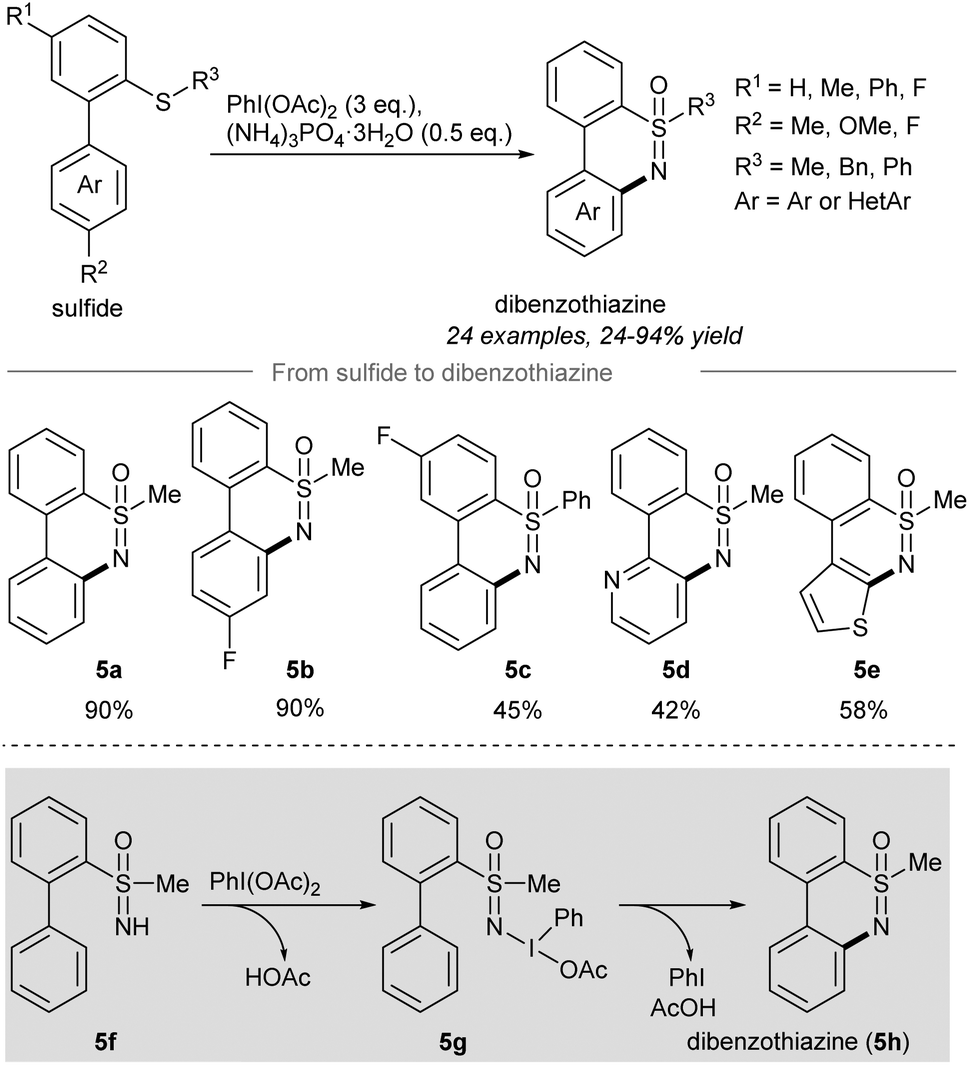 | ||
| Scheme 5 Synthesis of dibenzothiazines from sulfides through tandem NH-sulfoximination/C(sp2)–H amination.52 | ||
A mechanism of the one-pot synthesis of NH-sulfoximines from sulfides was suggested by Bull, Luisi, and co-workers (Scheme 6), based on direct nitrene NH-group transfer to sulfides and subsequent O-transfer from PIDA to afford sulfoximines43 (Scheme 6A). Later, an investigation by Reboul and co-workers44 revealed that iodonitrene could be generated when PIDA was reacted with either ammonium carbamate or ammonium carbonate (Scheme 6B). Although the sulfilimine 6a formed initially was short-lived and not detected, nucleophilic addition of methoxide or acetate to sulfilimines gave 6b and 6c, respectively, which could be detected by HRMS. The rate enhancement of the sulfoximination of S-perfluoroalkylated sulfides could be ascribed to the H-bonding between 2,2,2-trifluoroethanol (TFE) and the observed sulfanenitrile intermediate 6d (Scheme 6C). The attack of TFE on the acetate of 6d produced trifluoroethyl acetate. Acetyl group transfer from trifluoroethyl acetate to the reaction product NH-sulfoximines furnished N-Ac sulfoximines.
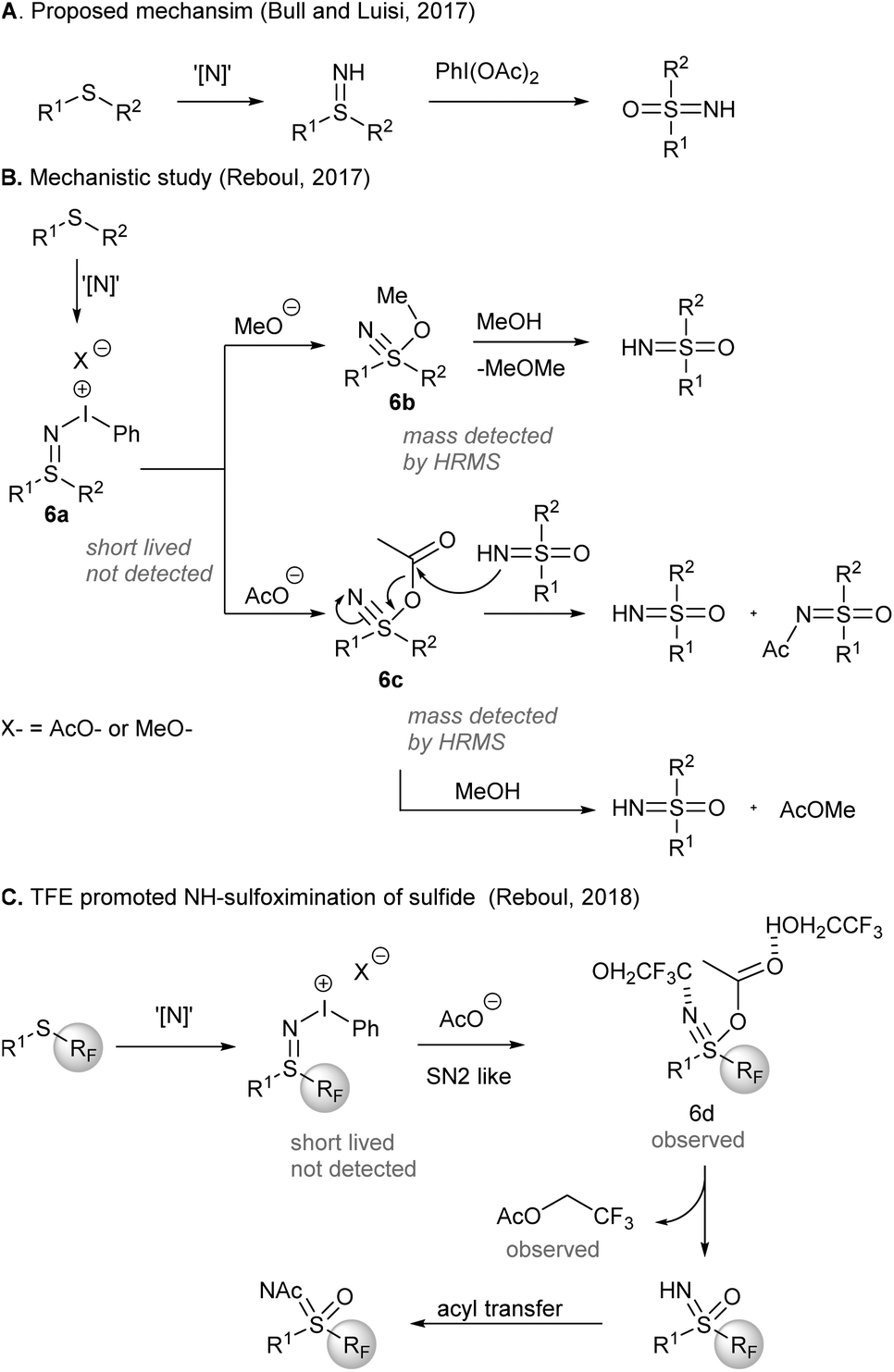 | ||
| Scheme 6 Mechanistic investigation of direct NH-sulfoximination of sulfides.42,45,46 | ||
2.4 One-pot NH- and O-transfer to thiols
In 2018, Luisi, Bull and co-workers disclosed a one-pot chemoselective NH-, O- and OR-transfer to thiols using iodonitrenes to give sulfonimidates and sulfonamides57 (Scheme 7 and 8). By reducing the amount of ammonium carbamate, the product distribution could be changed from sulfonimidates (4 equiv. of ammonium carbamate) (Scheme 7) to sulfonamides (1 equiv. of ammonium carbamate) (Scheme 8).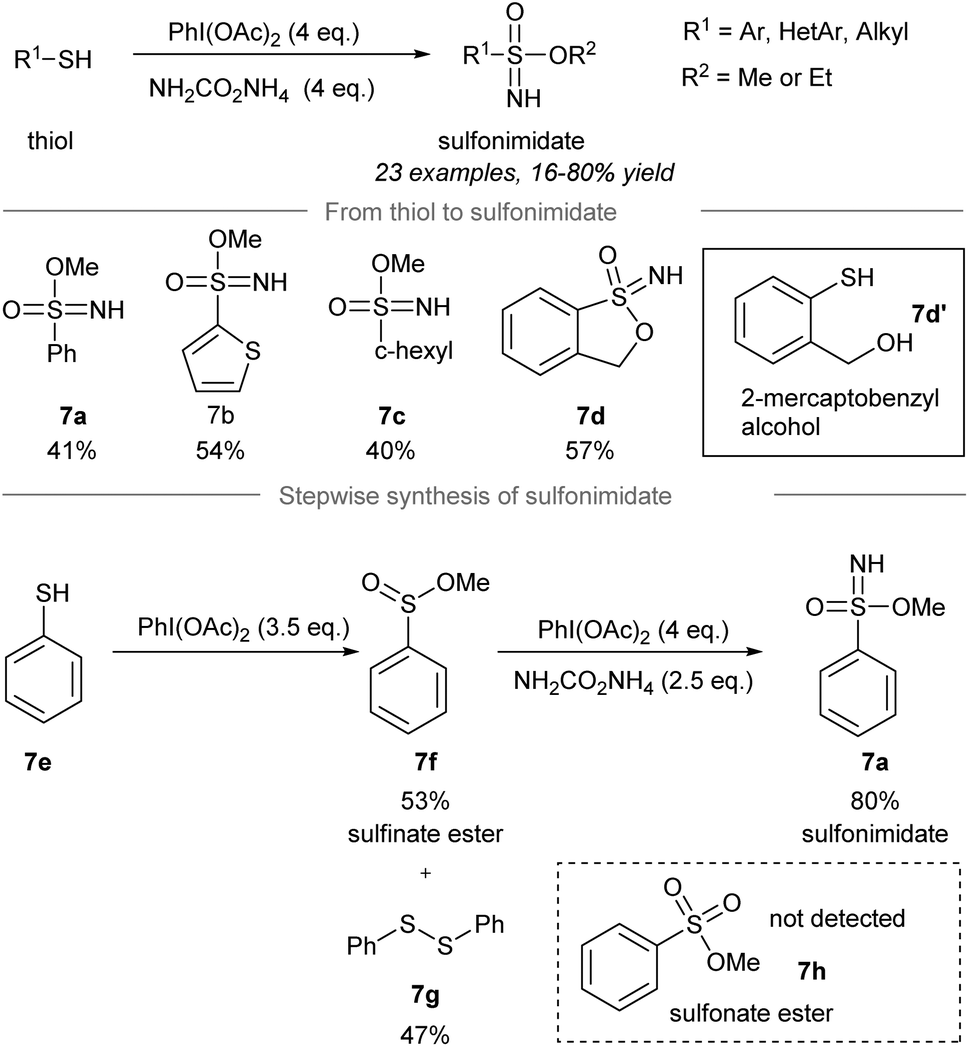 | ||
| Scheme 7 Synthesis of sulfonimidates from thiols through a one-pot NH-, O-, RO-transfer.57 | ||
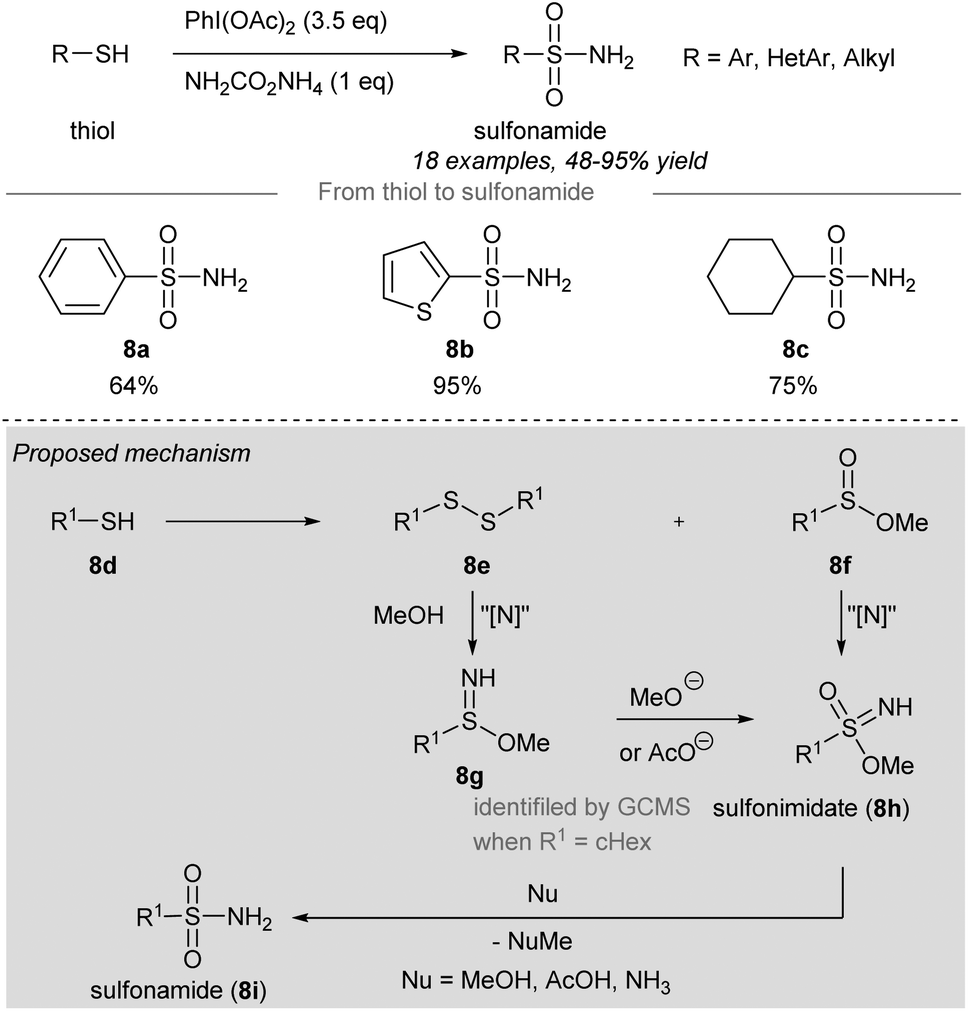 | ||
| Scheme 8 Synthesis of sulfonimidates and sulfonamides from thiols through a one-pot NH-, O-, RO-transfer.57 | ||
To prepare sulfonimidates, phenylthiol, thiophene-2-thiol and cyclohexanethanol were converted to the corresponding sulfonimidates 7a–7c in moderate yields under the standard conditions (Scheme 7). Interestingly, 2-mercaptobenzylalcohol 7d′ was transformed into cyclization product 7d in 57% yield. Treatment of phenylthiol 7e with an excess of phenyliodine(III) diacetate afforded a mixture of methyl sulfinate ester 7f and diphenyl disulfide 7g. However, sulfonate ester 7h was not observed. Exposure of methyl benzenesulfinate 7f to the in situ generated iodonitrene in acetonitrile afforded sulfonimidate 7a. Phenylthiol, thiophene-2-thiol and cyclohexanethiol afforded the corresponding sulfonamides 8a–8c in good to excellent yields (Scheme 8).
Based on the experimental evidence, the authors suggested a possible mechanism of these reactions (Scheme 8, grey box). Intermediate 8g was detected by GCMS when R was a cyclohexyl group, which was reacted with methoxide or acetate to give sulfonimidate 8h as the product. Sulfonimidate 8h could be converted to sulfonamide 8i by reaction with existing nucleophiles, including methanol, acetic acid, and ammonia.
In 2019, Bull and co-workers reported the direct one-pot NH- and O-transfer from sulfonamides to sulfonimidamides (Scheme 9A).58 Treatment of sulfonamides with 2.5 equivalents of iodosylbenzene and 2 equivalents of ammonium carbamate in the presence of 1 equivalent of AcOH as an additive gave sulfonimidamides in good yield. Phenyl-sulfenamide, 4-pyridinyl-sulfenamide, and cyclohexyl-sulfenamide performed well under the standard conditions to give the corresponding sulfonimidamides 9a–9c in decent yield. A secondary sulfenamide containing an NH moiety gave sulfonimidamide 9e in 30% isolated yield, along with the corresponding sulfinamide as the major side product.
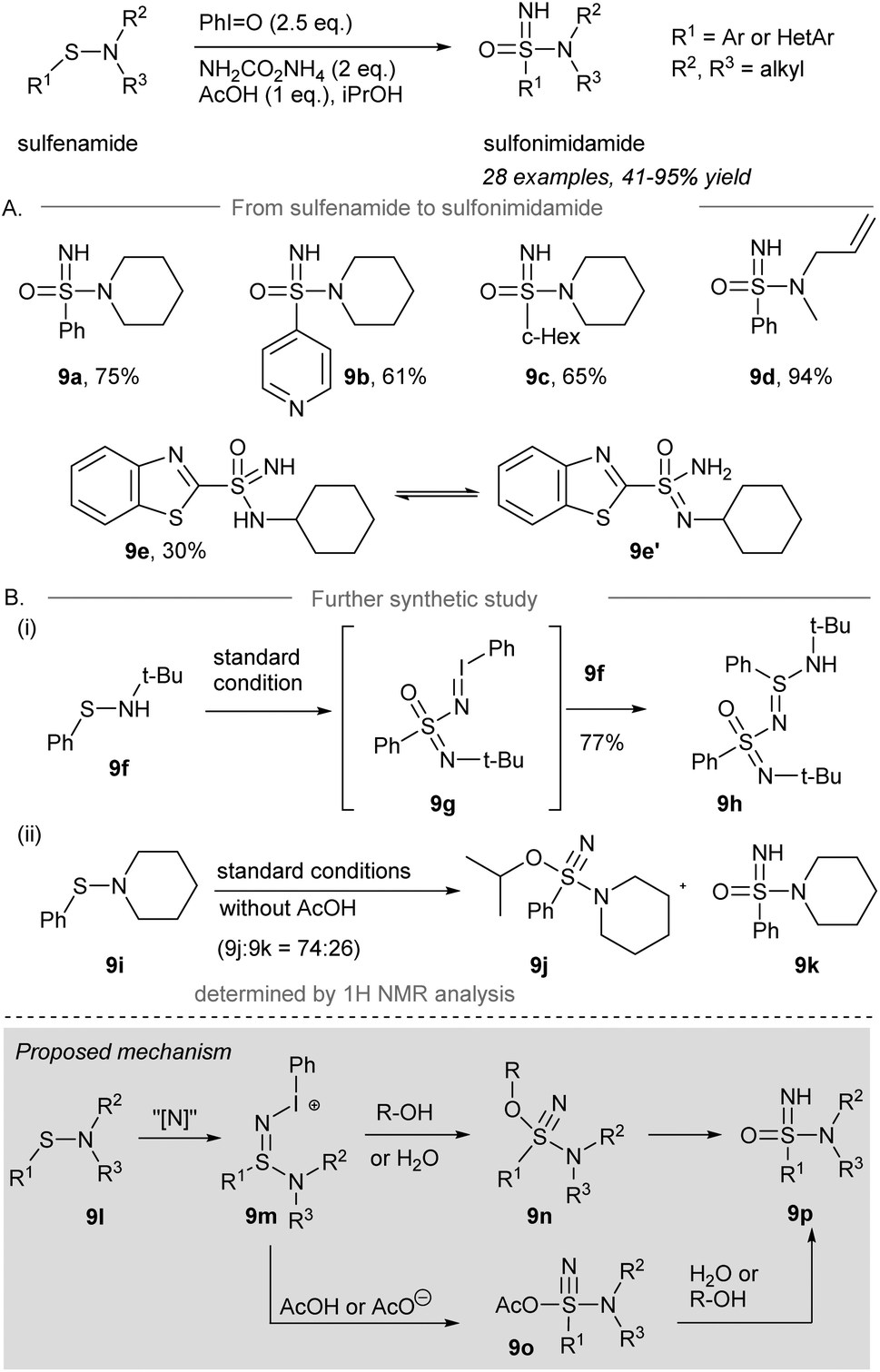 | ||
| Scheme 9 (A) Synthesis of sulfonimidamides from sulfonamides through one-pot NH- and O-transfer.58 (B) Further synthetic study of sulfonimidation. | ||
Unexpectedly, tert-butylphenylsulfenamide 9f was converted to 9h in 77% yield under the standard conditions (Scheme 9B). It is rationalized that direct one-pot NH- and O-transfer to 9f followed by activation by an excess of iodosylbenzene leads to the formation of iminoiodinane intermediate 9g. Imination of 9g by another equivalent of sulfenamide 9f produced 9h. When the reaction was performed in the absence of an acid, sulfenamide 9i was converted into λ6-sulfanenitrile 9j and sulfonimidamide 9k in a ratio of 74:26 determined by 1H NMR spectroscopy. The λ6-sulfanenitrile 9j was fully characterized by HRMS and 1H-, 13C-NMR and IR spectroscopy.
The proposed mechanism is depicted in Scheme 9 (grey box). Sulfonamide 9l is reacted with the iodonitrene to afford sulfinamidine salt 9m. Elimination of iodobenzene from 9m forming the S![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond may occur before or at the same time as an attack of a nucleophile, being either R-OH/H2O to give alkoxy-amino-λ6-sulfanenitrile 9n or AcOH to give sulfonimidamide 9o. Finally, 9n is converted to the desired sulfonimidamide 9p. Alternatively, sulfonimidamide 9o reacts with water from the solvent or the solubilization of iodosylbenzene produces sulfonimidamide 9p under the standard conditions to give the corresponding sulfonimidamides 9a–9c in decent yield, respectively.
N triple bond may occur before or at the same time as an attack of a nucleophile, being either R-OH/H2O to give alkoxy-amino-λ6-sulfanenitrile 9n or AcOH to give sulfonimidamide 9o. Finally, 9n is converted to the desired sulfonimidamide 9p. Alternatively, sulfonimidamide 9o reacts with water from the solvent or the solubilization of iodosylbenzene produces sulfonimidamide 9p under the standard conditions to give the corresponding sulfonimidamides 9a–9c in decent yield, respectively.
3. Selective electrophilic amination of tertiary amines
In 2021, Bull and Luisi disclosed the electrophilic amination of tertiary amines to give the corresponding hydrazinium salts (Scheme 10).59 Treatment of tertiary amines with 2.5 equivalents of iodosylbenzene and 2 equivalents of ammonia carbamate in the presence of p-methylbenzenesulfonic acid gave hydrazinium salts. The study of the substrate scope revealed that many reactive functional groups are well-tolerated under the standard conditions.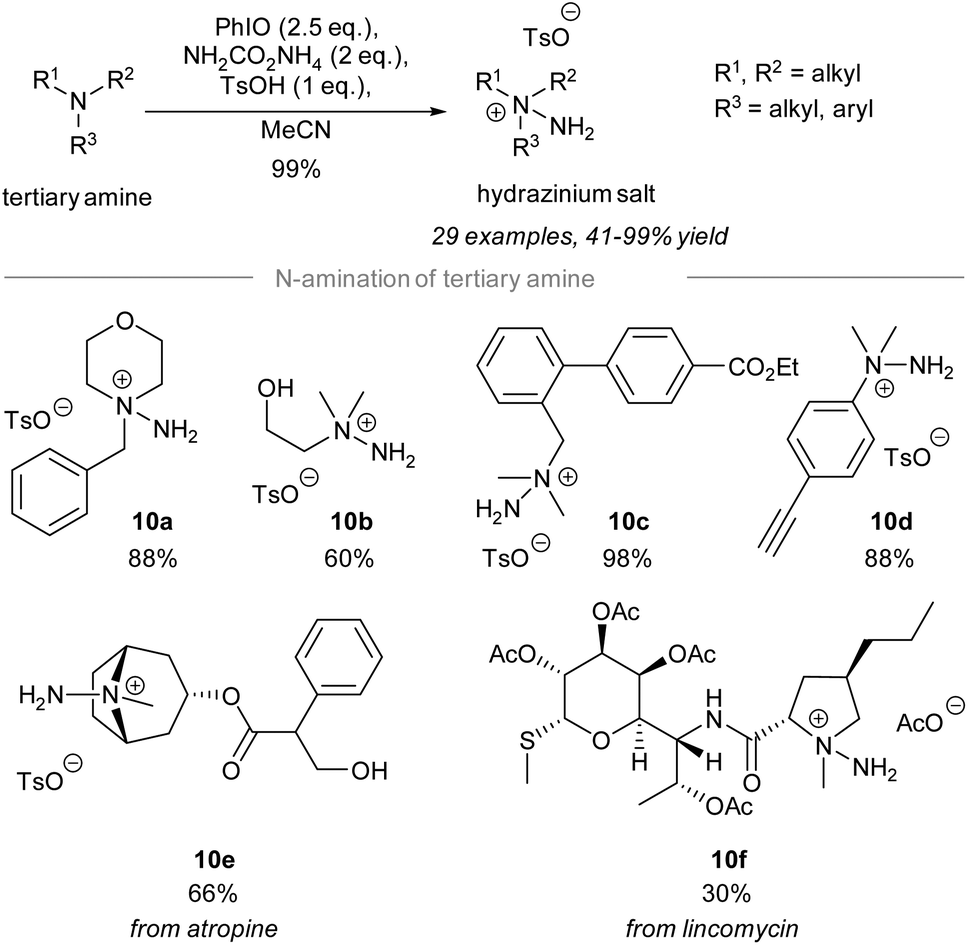 | ||
| Scheme 10 Chemoselective electrophilic amination of tertiary amines.59 | ||
For instance, the hydrazinium salts of primary alcohol 10b, ethyl ester 10c, and alkyne 10d were prepared successfully in good yield using the reported protocol. Importantly, chemoselective electrophilic amination of the tertiary amino group on atropine and a lincomycin derivative took place to give the corresponding hydrazinium salts 10e and 10f, respectively.
4. Synthesis of terminal diazirines from amino acids through tandem decarboxylation/iodonitrene transfer
In 2019, Reboul and co-workers disclosed the synthesis of terminal diazirines from amino acids through a tandem decarboxylation/iodonitrene transfer60 (Scheme 11). Treatment of amino acids with phenyliodine(III) diacetate and 7M ammonia solution produces terminal diazirines as major products accompanied by a small number of undesired nitriles that resulted from over-oxidation. Amino acids such as L-histidine, L-tyrosine, N-Boc-L-tryptophan, L-citrulline, and 4-iodo-L-phenylalanine were converted to the corresponding diazirines 11a, 11b, 11d, 11e, and 11h in high yield. However, unprotected L-tryptophan resulted in a volatile diazirine 11c in 27% yield, which was not accurate for quantification. Besides, sulfurated amino acids such as cysteine (Cys)57 and methionine (Met)50 were incompatible with the reaction conditions due to possible side reactions with PIDA. Prior protection of sulfur, for instance, in (S)-trityl-L-cysteine and (S)-Bn-L-cysteine sulfoxide gave the corresponding terminal diazirines 11f and 11g in poor to moderate yield. Noteworthily, no sulfoximination45 product was observed when the sulfoxides above were subjected to the standard conditions.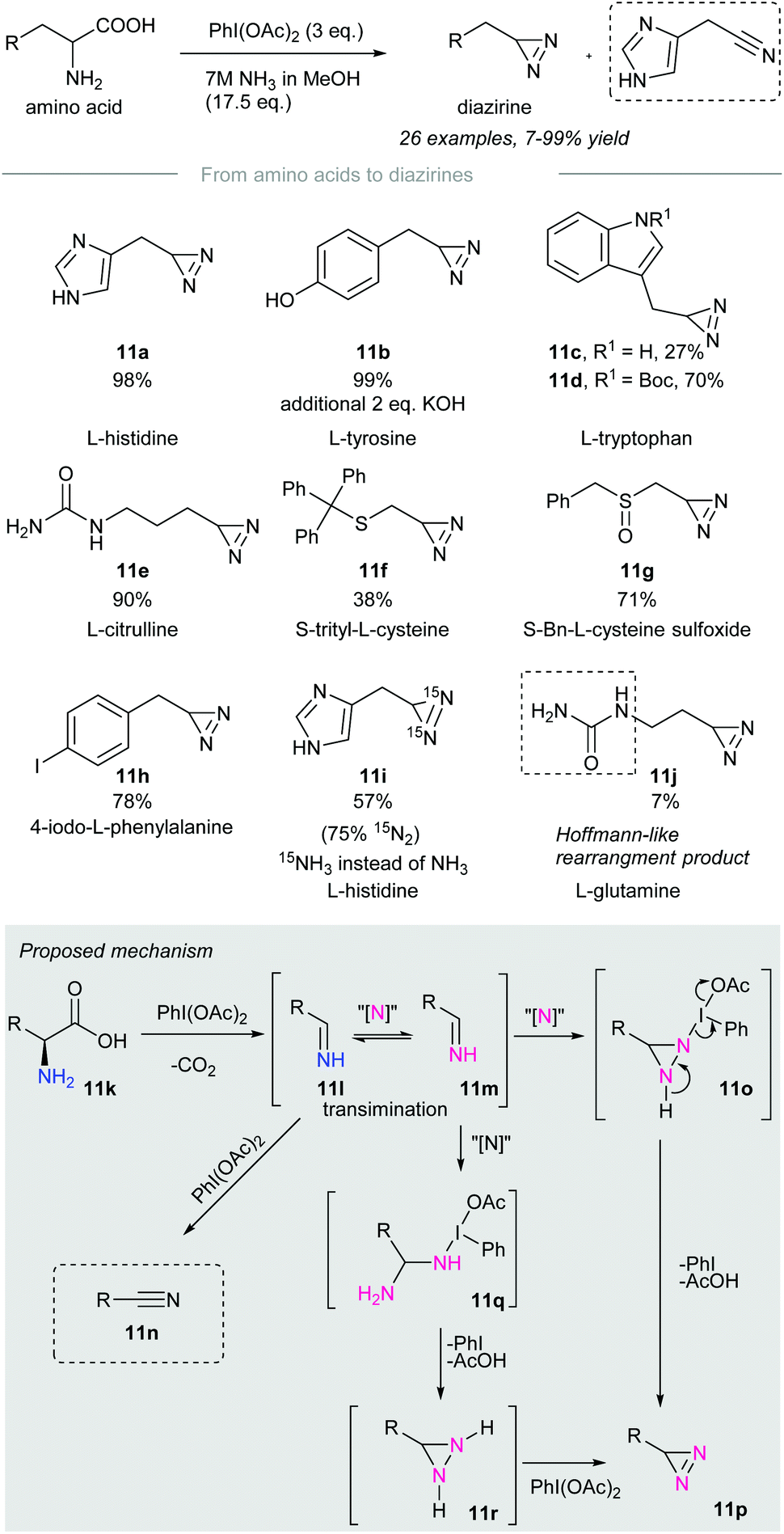 | ||
| Scheme 11 Synthesis of terminal diazirines from amino acids through a tandem decarboxylation/iodonitrene transfer.60 | ||
Treatment of L-histidine with PIDA and 15N-labeled ammonia afforded 15N2-diazirine 11i in 57% yield with 75% 15N-label incorporated. This implied that both nitrogen atoms of the newly installed diazirine group originated from the ammonia solution. Primary amides, such as L-glutamine, gave diazirines in low yield due to sublimation. In particular, a Hofmann-like rearrangement of L-glutamine took place to give urea 11j in 7% yield. This rearrangement could be alleviated by prior N-ethylation of L-glutamine.
The authors proposed a possible reaction mechanism (Scheme 11, grey box). Amino acid 11k is subjected to decarboxylation upon treatment with phenyliodine(III) diacetate to give an imine 11l.61,62 The imine 11l formed could be oxidized to nitrile 11n in the presence of an excess amount of oxidant. Transimination63 of imine 11l takes place with the iodonitrene to give 11m,14 which reacts with the second moiety of the iodonitrene via insertion to give the diaziridine intermediate 11o.64 Subsequent oxidation with the release of iodobenzene and acetic acid affords the desired diazirine 11p.65 Another possible pathway involves the nucleophilic addition of 15NH3 to give 11q,66 followed by cyclization into diaziridine 11r. Oxidation of diaziridine 11r by phenyliodine(III) diacetate afforded diazirine 11p.
Very recently, Reboul's chemistry on direct diazirine synthesis from amino acids was used to prepare a diazirine tag for chemical proteomics.67
5. Stereoselective and contractive synthesis of cyclobutanes from pyrrolidines
In 2021, stereoselective and contractive synthesis of cyclobutanes from the corresponding pyrrolidines was reported by Antonchick and co-workers16 (Scheme 12). Iodonitrene, which was generated in situ from the reaction between 2.5 equivalents of hydroxy(tosyloxy)iodobenzene (HTIB) and 8 equivalents of ammonium carbamate, acted as an electrophilic aminating reagent and converted the pyrrolidines into the corresponding cyclobutanes in a stereoselective manner. meso-Cyclobutanes carrying α-aryl and/or α-heterocyclic substituents, such as 12a and 12b, could be prepared in decent yield from the corresponding pyrrolidines under the standard conditions. The pyrrolidine possessing an α-quaternary center could be converted to the corresponding cyclobutane 12c in 48% yield. Furthermore, double ring contraction of bipyrrolidines with an additional amount of HTIB (i.e. 5 equiv.) provided polyspirocyclobutane 12d in 59% yield. The contractive synthesis of cyclobutane 12e was effected by HTIB instead of PIDA and it acted as an essential intermediate to the preparation of the cytotoxic cyclobutane natural product piperarborenine B.68–70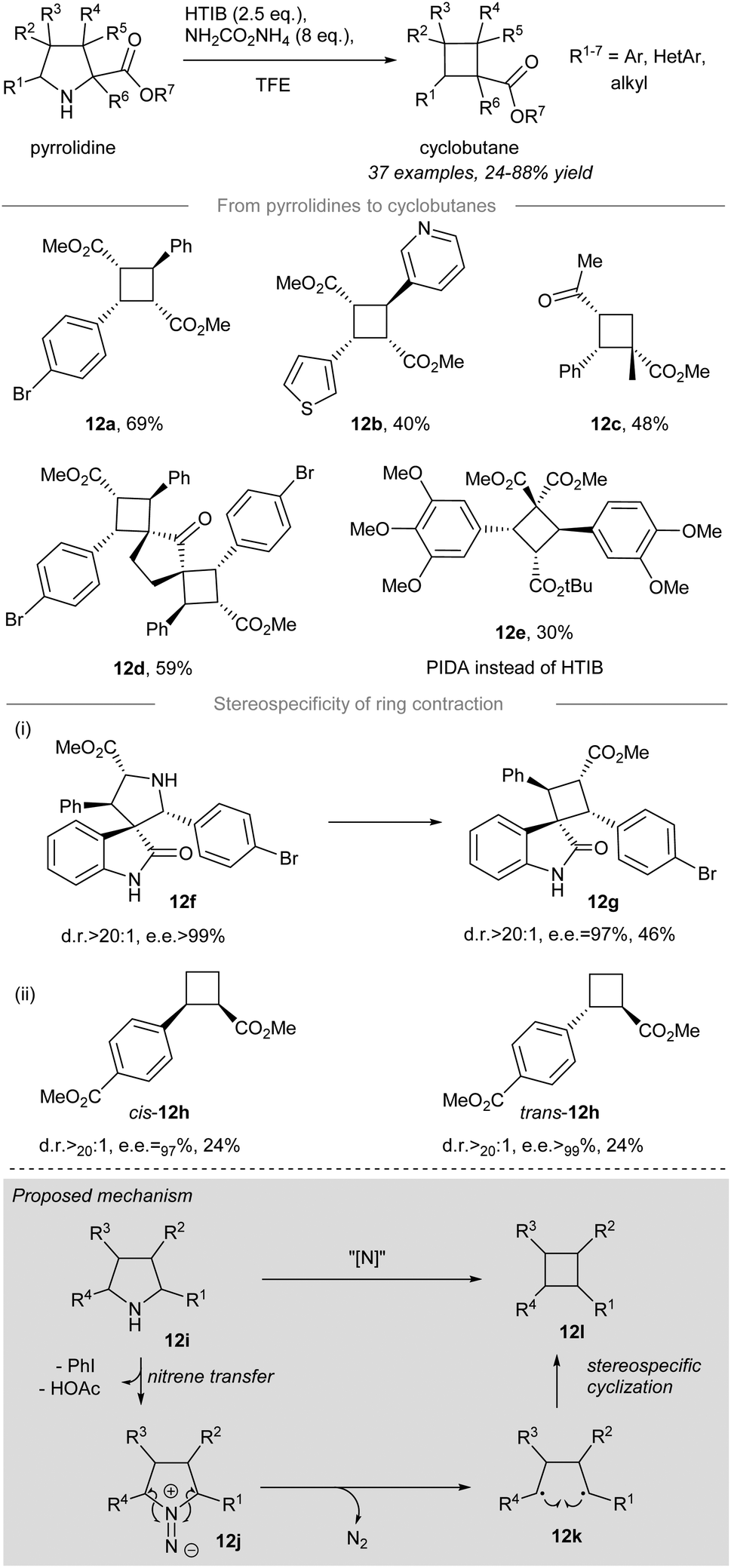 | ||
| Scheme 12 Stereospecific contraction synthesis of cyclobutanes from pyrrolidines.59 | ||
When optically-pure spirooxindole 12f was subjected to the standard conditions, spirocyclobutane 12g was formed with excellent stereocontrol (dr>20:1, ee = 97%) validating the stereospecificity of the ring contraction. More importantly, the stereospecific nature of the ring contraction was further substantiated by the formation of cyclobutanes cis-12h and trans-12h. Although both ring contractions afforded low yields (i.e. 24%), the outstanding diastereo- and enantiocontrol for cis-12h (dr > 20:1, ee > 97%) and trans-12h (dr > 20:1, ee > 99%) indicated a memory of chirality for the developed novel ring contraction allowing access to enantiopure novel cyclobutane derivatives.
The proposed reaction mechanism is depicted (Scheme 12, grey box). Treatment of pyrrolidine 12i with the in situ generated iodonitrene species leads to electrophilic amination, affording 1,1-diazene 12j as a possible intermediate. The reactive 1,1-diazene 12j proceeds further to give 1,4-biradical 12kvia dinitrogen extrusion. The intramolecular cyclization of 1,4-biradical 12k leads to C–C bond formation to give cyclobutane 12l.
6. Summary and outlook
This review provides an overview of iodonitrene chemistry and illustrates its development since 2016. Through the discovery of iodonitrene as an in situ generated reactive species from the reaction between hypervalent iodine(III) and ammonia, iodonitrene has been used extensively as an electrophilic aminating reagent in the amination of sulfides and sulfoxides. Until 2019, the unprecedented synthesis of diazirines from unprotected amino acids was achieved by Reboul making use of hypervalent iodine(III) as an oxidant for decarboxylation and an iodonitrene as a source of nitrogen. Diazirines generated by this method can be used as a tag once they are incorporated into bioactive compounds and could be used for various biological investigations. Later, our group reported the stereospecific contractive synthesis of cyclobutanes from pyrrolidines featuring iodonitrene-promoted electrophilic amination of the N-atoms of pyrrolidines followed by nitrogen extrusion. Taking into account the reaction we described above, iodonitrene not only serves as an electrophilic aminating reagent, but also shows the oxidation properties of hypervalent iodine(III). This makes iodonitrene a very interesting reagent for the development of new methods.Iodonitrene acts as a convenient and easily manageable reagent in organic synthesis. Besides its metal-free nature, the use of iodonitrene provides comparable reactivity to rhodium–carbene in the synthesis of NH-sulfoximines from sulfoxides but requires no use of a transition metal. This provides a fascinating opportunity that rhodium-catalyzed nitrene transfer reactions might be accomplished by iodonitrene chemistry, for instance, in the amination of arenes and alkenes. One major issue that needs to be addressed is the stoichiometric amounts of hypervalent iodine(III) reagent necessary to react with ammonia or its surrogate in order to generate iodonitrene. Inspired by the organocatalytic reactions developed by us and others, hypervalent iodine(III) formed from catalytic quantities of aryl iodides and m-CPBA as a stoichiometric oxidant might react with ammonia to give iodonitrenes, avoiding the use of stoichiometric amounts of hypervalent iodine(III). Furthermore, the prospect of asymmetric iodonitrene transfer might be enabled by iodonitrenes prepared from chiral hypervalent iodine(III) compounds. New method development involving the use of iodonitrene continues to be an active research area. We envision that synthetic application of the reported iodonitrene chemistry, such as the preparation of diazirine tags and the synthesis of bioactive natural products, will be flourished as practically useful chemistry applied widely in the synthetic community and the pharmaceutical industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. A. acknowledges the support of the DFG (AN 1064/4-1) and the Boehringer Ingelheim Foundation (Plus 3). C. H. acknowledges the International Max Planck Research School for Living Matter (Dortmund, Germany).References
- M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS PubMed.
- R. Dorel, C. P. Grugel and A. M. Haydl, The Buchwald–Hartwig Amination After 25 Years, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- M. Corpet and C. Gosmini, Recent Advances in Electrophilic Amination Reactions, Synthesis, 2014, 46, 2258–2271 CrossRef CAS.
- P. Starkov, T. F. Jamison and I. Marek, Electrophilic Amination: The Case of Nitrenoids, Chem. – Eur. J., 2015, 21, 5278–5300 CrossRef CAS PubMed.
- K. Muñiz, Promoting Intermolecular C–N Bond Formation under the Auspices of Iodine(III), Acc. Chem. Res., 2018, 51, 1507–1519 CrossRef PubMed.
- S. Sabir, G. Kumar and J. L. Jat, O-Substituted hydroxyl amine reagents: an overview of recent synthetic advances, Org. Biomol. Chem., 2018, 16, 3314–3327 RSC.
- R. Y. Liu and S. L. Buchwald, CuH-Catalyzed Olefin Functionalization: From Hydroamination to Carbonyl Addition, Acc. Chem. Res., 2020, 53, 1229–1243 CrossRef CAS PubMed.
- L. G. O'Neil and J. F. Bower, Electrophilic Aminating Agents in Total Synthesis, Angew. Chem., Int. Ed., 2021, 60, 25640 CrossRef PubMed.
- C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, Expanding the Scope of C−H Amination through Catalyst Design, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef CAS PubMed.
- J. Miao, N. G. J. Richards and H. Ge, Rhodium-catalyzed direct synthesis of unprotected NH-sulfoximines from sulfoxides, ChemComm, 2014, 50, 9687–9689 RSC.
- J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kürti and J. R. Falck, Direct Stereospecific Synthesis of Unprotected N-H and N-Me Aziridines from Olefins, Science, 2014, 343, 61 CrossRef CAS PubMed.
- M. P. Paudyal, A. M. Adebesin, S. R. Burt, D. H. Ess, Z. Ma, L. Kürti and J. R. Falck, Dirhodium-catalyzed C-H arene amination using hydroxylamines, Science, 2016, 353, 1144 CrossRef CAS PubMed.
- B. Darses, R. Rodrigues, L. Neuville, M. Mazurais and P. Dauban, Transition metal-catalyzed iodine(III)-mediated nitrene transfer reactions: efficient tools for challenging syntheses, ChemComm, 2017, 53, 493–508 RSC.
- M. Zenzola, R. Doran, L. Degennaro, R. Luisi and J. A. Bull, Transfer of Electrophilic NH Using Convenient Sources of Ammonia: Direct Synthesis of NH Sulfoximines from Sulfoxides, Angew. Chem., Int. Ed., 2016, 55, 7203–7207 CrossRef CAS PubMed.
- T. Glachet, H. Marzag, N. S. Rosa, J. F. P. Colell, G. Zhang, W. S. Warren, X. Franck, T. Theis and V. Reboul, Iodonitrene in Action: Direct Transformation of Amino Acids into Terminal Diazirines and 15N2-Diazirines and Their Application as Hyperpolarized Markers, J. Am. Chem. Soc., 2019, 141, 13689–13696 CrossRef CAS PubMed.
- C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, Stereoselective Synthesis of Cyclobutanes by Contraction of Pyrrolidines, J. Am. Chem. Soc., 2021, 143, 18864–18870 CrossRef CAS PubMed.
- J. Mendiola, J. A. Rincón, C. Mateos, J. F. Soriano, Ó. de Frutos, J. K. Niemeier and E. M. Davis, Preparation, Use, and Safety of O-Mesitylenesulfonylhydroxylamine, Org. Process Res. Dev., 2009, 13, 263–267 CrossRef CAS.
- C. Iacobucci, S. Reale and F. De Angelis, Elusive Reaction Intermediates in Solution Explored by ESI-MS: Reverse Periscope for Mechanistic Investigations, Angew. Chem., Int. Ed., 2016, 55, 2980–2993 CrossRef CAS PubMed.
- K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, A mechanistic analysis of the Rh-catalyzed intramolecular C–H amination reaction, Tetrahedron, 2009, 65, 3042–3051 CrossRef CAS.
- A. P. Antonchick, R. Samanta, K. Kulikov and J. Lategahn, Organocatalytic, Oxidative, Intramolecular C-H Bond Amination and Metal-free Cross-Amination of Unactivated Arenes at Ambient Temperature, Angew. Chem., Int. Ed., 2011, 50, 8605–8608 CrossRef CAS PubMed.
- A. P. Antonchick and L. Burgmann, Direct Selective Oxidative Cross-Coupling of Simple Alkanes with Heteroarenes, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS PubMed.
- K. Matcha and A. P. Antonchick, Metal-Free Cross-Dehydrogenative Coupling of Heterocycles with Aldehydes, Angew. Chem., Int. Ed., 2013, 52, 2082–2086 CrossRef CAS PubMed.
- K. Matcha, R. Narayan and A. P. Antonchick, Metal-Free Radical Azidoarylation of Alkenes: Rapid Access to Oxindoles by Cascade CN and CC Bond-Forming Reactions, Angew. Chem., Int. Ed., 2013, 52, 7985–7989 CrossRef CAS PubMed.
- S. Manna, K. Matcha and A. P. Antonchick, Metal-Free Annulation of Arenes with 2-Aminopyridine Derivatives: The Methyl Group as a Traceless Non-Chelating Directing Group, Angew. Chem., Int. Ed., 2014, 53, 9695–9695 CrossRef CAS.
- S. Manna and A. P. Antonchick, Organocatalytic Oxidative Annulation of Benzamide Derivatives with Alkynes, Angew. Chem., Int. Ed., 2014, 53, 7324–7327 CrossRef CAS PubMed.
- S. Manna, K. Matcha and A. P. Antonchick, Metal-Free Annulation of Arenes with 2-Aminopyridine Derivatives: The Methyl Group as a Traceless Non-Chelating Directing Group, Angew. Chem., Int. Ed., 2014, 53, 8163–8166 CrossRef CAS PubMed.
- L. Bering and A. P. Antonchick, Selective transition-metal-free vicinal cis-dihydroxylation of saturated hydrocarbons, Chem. Sci., 2017, 8, 452–457 RSC.
- E. M. D. Allouche, E. Grinhagena and J. Waser, Hypervalent Iodine-Mediated Late-Stage Peptide and Protein Functionalization, Angew. Chem., Int. Ed., 2022, 61, e202112287 CrossRef CAS PubMed.
- T. Wirth, Hypervalent iodine chemistry in synthesis: Scope and new directions, Angew. Chem., Int. Ed., 2005, 44, 3656–3665 CrossRef CAS PubMed.
- J. Charpentier, N. Fruh and A. Togni, Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- Y. F. Li, D. P. Hari, M. V. Vita and J. Waser, Cyclic Hypervalent Iodine Reagents for Atom-Transfer Reactions: Beyond Trifluoromethylation, Angew. Chem., Int. Ed., 2016, 55, 4436–4454 CrossRef CAS PubMed.
- A. Yoshimura and V. V. Zhdankin, Advances in Synthetic Applications of Hypervalent Iodine Compounds, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- X. Wang and A. Studer, Iodine(III) Reagents in Radical Chemistry, Acc. Chem. Res., 2017, 50, 1712–1724 CrossRef CAS PubMed.
- W. W. Chen, A. B. Cuenca and A. Shafir, The Power of Iodane-Guided C-H Coupling: A Group-Transfer Strategy in Which a Halogen Works for Its Money, Angew. Chem., Int. Ed., 2020, 59, 16294–16309 CrossRef CAS PubMed.
- R. Zhao and L. Shi, Reactions between Diazo Compounds and Hypervalent Iodine(III) Reagents, Angew. Chem., Int. Ed., 2020, 59, 12282–12292 CrossRef CAS PubMed.
- J. A. Bull, L. Degennaro and R. Luisi, Straightforward Strategies for the Preparation of NH-Sulfoximines: A Serendipitous Story, Synlett, 2017, 28, 2525–2538 CrossRef CAS.
- M. Andresini, M. Colella, L. Degennaro and R. Luisi, Hypervalent iodine(III) reagents and ammonia as useful combination for highly chemoselective N-transfer to low-valent organosulfur compounds and amines, ARKIVOC, 2021, 2021, 141–163 Search PubMed.
- M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Synthesis and Transformations of NH-Sulfoximines, Chem. – Eur. J., 2021, 27, 17293–17321 CrossRef CAS PubMed.
- C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- A. M. Weber and A. J. Ryan, ATM and ATR as therapeutic targets in cancer, Pharmacol. Ther., 2015, 149, 124–138 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Site-selective arene C-H amination via photoredox catalysis, Science, 2015, 349, 1326 CrossRef CAS PubMed.
- F. Izzo, M. Schäfer, R. Stockman and U. Lücking, A New, Practical One-Pot Synthesis of Unprotected Sulfonimidamides by Transfer of Electrophilic NH to Sulfinamides, Chem. – Eur. J., 2017, 23, 15189–15193 CrossRef CAS PubMed.
- A. Tota, M. Zenzola, S. J. Chawner, S. S. John-Campbell, C. Carlucci, G. Romanazzi, L. Degennaro, J. A. Bull and R. Luisi, Synthesis of NH-sulfoximines from sulfides by chemoselective one-pot N- and O-transfers, ChemComm, 2017, 53, 348–351 RSC.
- J.-F. Lohier, T. Glachet, H. Marzag, A.-C. Gaumont and V. Reboul, Mechanistic investigation of the NH-sulfoximination of sulfide. Evidence for λ6-sulfanenitrile intermediates, ChemComm, 2017, 53, 2064–2067 RSC.
- J. F. Lohier, T. Glachet, H. Marzag, A. C. Gaumont and V. Reboul, Mechanistic investigation of the NH-sulfoximination of sulfide. Evidence for lambda(6)-sulfanenitrile intermediates, ChemComm, 2017, 53, 2064–2067 RSC.
- A. Tota, M. Zenzola, S. J. Chawner, S. St John-Campbell, C. Carlucci, G. Romanazzi, L. Degennaro, J. A. Bull and R. Luisi, Synthesis of NH-sulfoximines from sulfides by chemoselective one-pot N- and O-transfers, ChemComm, 2017, 53, 348–351 RSC.
- S. Chaabouni, J. F. Lohier, A. L. Barthelemy, T. Glachet, E. Anselmi, G. Dagousset, P. Diter, B. Pegot, E. Magnier and V. Reboul, One-Pot Synthesis of Aryl- and Alkyl S-Perfluoroalkylated NH-Sulfoximines from Sulfides, Chem. – Eur. J., 2018, 24, 17006–17010 CrossRef CAS PubMed.
- B. Verbelen, E. Siemes, A. Ehnbom, C. Rauber, K. Rissanen, D. Woll and C. Bolm, From One-Pot NH-Sulfoximidations of Thiophene Derivatives to Dithienylethene-Type Photoswitches, Org. Lett., 2019, 21, 4293–4297 CrossRef CAS PubMed.
- G. Zhang, H. Tan, W. Chen, H. C. Shen, Y. Lu, C. Zheng and H. Xu, Synthesis of NH-Sulfoximines by Using Recyclable Hypervalent Iodine(III) Reagents under Aqueous Micellar Conditions, ChemSusChem, 2020, 13, 922 CrossRef CAS PubMed.
- H. Marzag, M. Schuler, A. Tatibouët and V. Reboul, Synthesis of Methionine-Derived Endocyclic Sulfilimines and Sulfoximines, Eur. J. Org. Chem., 2017, 896–900 CrossRef CAS.
- S. Chaabouni, J.-F. Lohier, A.-L. Barthelemy, T. Glachet, E. Anselmi, G. Dagousset, P. Diter, B. Pégot, E. Magnier and V. Reboul, One-Pot Synthesis of Aryl- and Alkyl S-Perfluoroalkylated NH-Sulfoximines from Sulfides, Chem. – Eur. J., 2018, 24, 17006–17010 CrossRef CAS PubMed.
- Y. N. Ma, C. Y. Guo, Q. Y. Zhao, J. Zhang and X. N. Chen, Synthesis of dibenzothiazines from sulfides by one-pot N,O-transfer and intramolecular C-H amination, Green Chem., 2018, 20, 2953–2958 RSC.
- R. M. Bär, L. Langer, M. Nieger and S. Bräse, Bicyclo[1.1.1]pentyl Sulfoximines: Synthesis and Functionalizations, Adv. Synth. Catal., 2020, 362, 1356–1361 CrossRef.
- A. Tota, C. Carlucci, L. Pisano, G. Cutolo, G. J. Clarkson, G. Romanazzi, L. Degennaro, J. A. Bull, P. Rollin and R. Luisi, Synthesis of glycosyl sulfoximines by a highly chemo- and stereoselective NH- and O-transfer to thioglycosides, Org. Biomol. Chem., 2020, 18, 3893–3897 RSC.
- Y. Li, Q. Ding, G. Qiu and J. Wu, Synthesis of benzosultams via an intramolecular sp2 C–H bond amination reaction of o-arylbenzenesulfonamides under metal-free conditions, Org. Biomol. Chem., 2014, 12, 149–155 RSC.
- C. Martínez, A. E. Bosnidou, S. Allmendinger and K. Muñiz, Towards Uniform Iodine Catalysis: Intramolecular C−H Amination of Arenes under Visible Light, Chem. – Eur. J., 2016, 22, 9929–9932 CrossRef PubMed.
- A. Tota, S. St John-Campbell, E. L. Briggs, G. O. Estevez, M. Afonso, L. Degennaro, R. Luisi and J. A. Bull, Highly Chemoselective NH- and O-Transfer to Thiols Using Hypervalent Iodine Reagents: Synthesis of Sulfonimidates and Sulfonamides, Org. Lett., 2018, 20, 2599–2602 CrossRef CAS PubMed.
- E. L. Briggs, A. Tota, M. Colella, L. Degennaro, R. Luisi and J. A. Bull, Synthesis of Sulfonimidamides from Sulfenamides via an Alkoxy-amino-lambda(6) -sulfanenitrile Intermediate, Angew. Chem., Int. Ed., 2019, 58, 14303–14310 CrossRef CAS PubMed.
- A. Tota, M. Colella, C. Carlucci, A. Aramini, G. Clarkson, L. Degennaro, J. A. Bull and R. Luisi, N−N Bond Formation Using an Iodonitrene as an Umpolung of Ammonia: Straightforward and Chemoselective Synthesis of Hydrazinium Salts, Adv. Synth. Catal., 2021, 363, 194–199 CrossRef CAS.
- T. Glachet, H. Marzag, N. S. Rosa, J. F. P. Colell, G. Zhang, W. S. Warren, X. Franck, T. Theis and V. Reboul, Iodonitrene in Action: Direct Transformation of Amino Acids into Terminal Diazirines and N-15(2)-Diazirines and Their Application as Hyperpolarized Markers, J. Am. Chem. Soc., 2019, 141, 13689–13696 CrossRef CAS PubMed.
- V. N. Telvekar and K. A. Sasane, Oxidative Decarboxylation of 2-Aryl Carboxylic Acids Using phenyliodine(III) diacetate for Preparation of Aryl Aldehydes, Ketones, and Nitriles, Synlett, 2010, 2778–2780 CrossRef CAS.
- R. M. Moriarty, M. Sultana and Y.-Y. Ku, Cleavage of NH2 terminal tyrosyl–peptide bonds using hypervalent iodine, J. Chem. Soc., Chem. Commun., 1985, 974–975 RSC.
- M. Ciaccia and S. Di Stefano, Mechanisms of imine exchange reactions in organic solvents, Org. Biomol. Chem., 2015, 13, 646–654 RSC.
- R. R. Mondal, S. Khamarui and D. K. Maiti, Photocatalytic Generation of Nitrenes for Rapid Diaziridination, Org. Lett., 2017, 19, 5964–5967 CrossRef CAS PubMed.
- R. D. Richardson, M. Desaize and T. Wirth, Hypervalent Iodine-Mediated Aziridination of Alkenes: Mechanistic Insights and Requirements for Catalysis, Chem. – Eur. J., 2007, 13, 6745–6754 CrossRef CAS PubMed.
- L. Lykke, K. S. Halskov, B. D. Carlsen, V. X. Chen and K. A. Jørgensen, Catalytic Asymmetric Diaziridination, J. Am. Chem. Soc., 2013, 135, 4692–4695 CrossRef CAS PubMed.
- L. P. Conway, A. M. Jadhav, R. A. Homan, W. Li, J. S. Rubiano, R. Hawkins, R. M. Lawrence and C. G. Parker, Evaluation of fully-functionalized diazirine tags for chemical proteomic applications, Chem. Sci., 2021, 12, 7839–7847 RSC.
- W. R. Gutekunst and P. S. Baran, Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C-H Arylation, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef CAS PubMed.
- R. A. Panish, S. R. Chintala and J. M. Fox, A Mixed-Ligand Chiral Rhodium(II) Catalyst Enables the Enantioselective Total Synthesis of PiperarborenineB, Angew. Chem., Int. Ed., 2016, 55, 4983–4987 CrossRef CAS PubMed.
- J. L. Hu, L. W. Feng, L. J. Wang, Z. W. Xie, Y. Tang and X. G. Li, Enantioselective Construction of Cyclobutanes: A New and Concise Approach to the Total Synthesis of (+)-Piperarborenine B, J. Am. Chem. Soc., 2016, 138, 13151–13154 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2022 |