
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free thioesterification of α,β-unsaturated aldehydes with thiols†
Małgorzata
Bołt
 ,
Kamil
Hanek
and
Patrycja
Żak
*
,
Kamil
Hanek
and
Patrycja
Żak
*
Department of Organometallic Chemistry, Faculty of Chemistry, Adam Mickiewicz University in Poznan, Uniwersytetu Poznańskiego 8, 61-614 Poznan, Poland. E-mail: pkw@amu.edu.pl
First published on 29th July 2022
Abstract
For the first time, the synthesis of thioesters starting from enals and thiols has been performed in the presence of a bulky N-heterocyclic carbene (NHC) as a catalyst. This new method has been proved to be effective with a wide substrate scope giving selective thioesters in yields above 85% under mild and metal-free conditions. This green protocol does not require elevated temperatures, or addition of oxidants or other additives. The steric bulk of the carbene was found to markedly influence the reaction chemoselectivity. Bulky NHC carbene ligands, in contrast to those with less sterically developed ligands, completely change the chemoselectivity of the reaction leading to 1,2-addition products and not sulfa-Michael addition (SMA) adducts.
1. Introduction
Organosulfur compounds are prominent both in biological systems and organic synthesis and are found in a large number of applications in medicine, pharmaceutics, materials science and industry. One of the most interesting sulfur-containing groups is thioesters, which – due to their high reactivity – can serve as useful organic reactants and building blocks in a number of processes.1,2 Recently, much attention has been paid to polymer materials comprising a thioester group in the structure, which significantly improves their thermal properties.3,4Thioesters can be synthesized by nucleophilic substitution of the carbonyl group of acyl chlorides, carboxylic acids or acid anhydrides with thiols or disulfides.5 Besides this, a bunch of synthetic methods, exploiting aldehydes as a carbonyl group source, have been developed6 (Scheme 1a). These reactions are carried out in the presence of transition metal catalysts based on iron or copper and very often they are characterized by mediocre effectiveness, difficult reaction conditions and indispensable use of oxidants.7–9 Alternatively, thioesters can be obtained in the presence of transition metal complexes as catalysts in thiocarbonylation of alkenes and alkynes10,11 (Scheme 1b), although in most cases these processes require harsh conditions, like high temperatures and/or elevated pressures.
 | ||
| Scheme 1 C–S bond formation leading to thioesters (a, b, d, and e) and thioaldehydes (c). | ||
Thioesters can also be successfully obtained in an organocatalytic manner. Among a large number of organocatalysts, N-heterocyclic carbene (NHC) ligand precursors constitute an important class of catalysts for this transformation.12–14 Although they usually give high product yields, these methods still suffer from a number of drawbacks, such as the need for high temperature, long reaction time and inevitability of oxidant use. These routes are also limited to aryl and alkyl aldehydes and cannot be applied for α,β-unsaturated compounds because these types of aldehydes react with thiols in the presence of NHC to give SMA adducts15–19 (Scheme 1c). According to the best of our knowledge, there is only one report on the synthesis of thioethers via β-protonation of α,β-unsaturated enals using mercaptans20 (Scheme 1d). However, this process requires a large amount of base and/or strong Brønsted acid and the use of copper co-catalysts and 4 Å molecular sieves. Moreover, this protocol has been described only for a β-alkyl or CF3 substituted cinnamaldehyde, so it is difficult to say anything about its universality.
Because of great importance of thioester derivatives in various research fields, new and efficient synthetic protocols for their construction are highly desirable. In this report, we present a new method for thioester synthesis based on the reaction between enals and thiols in the presence of a bulky NHC ligand precursor. The designed and developed method is characterized by total atom economy and sustainability; besides, it can be performed under metal-free, mild conditions with excellent yields.
2. Results and discussion
We began our research aiming to get a product from a sulfa-Michael-type addition (SMA). We knew that this transformation can occur with α,β-unsaturated esters in the presence of NHC precursors and a strong base, in toluene.14 We decided to apply similar conditions to the reaction between cinnamaldehyde (1a) and benzyl mercaptan (2a) in the presence of a bulky NHC salt – IPr*Et·HCl (A). Firstly, we carried out the reaction with an equimolar ratio of the substrates: 10 mol% of the NHC precursor and 10 mol% of KHMDS as a base. The reaction was performed in toluene at room temperature and the reaction progress was monitored by GC-MS analysis, which revealed the formation of a product with an expected mass. Surprisingly, the 1H NMR analysis did not confirm the structure of a SMA product. Instead, the obtained compound was identified as being consistent with the structure of 3a-a, which is a product of nucleophilic substitution of the carbonyl atom with hydrogenation of a double bond (Scheme 2).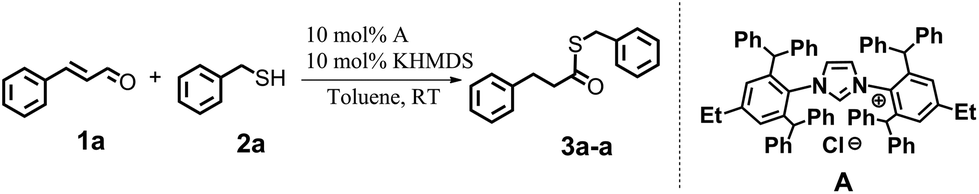 | ||
| Scheme 2 Thioesterification of cinnamaldehyde (1a) with benzyl mercaptan (2a). | ||
This unexpected result prompted us to continue further investigation. We conducted a series of optimization tests to select the type and concentration of the NHC precursor and base to be used, and a suitable solvent; to identify the right sequence for reagent introduction; and to determine the possible influence of 4 Å molecular sieves on the reaction course. All optimization tests were performed for the model reaction as shown in Scheme 1. The results are collected in Tables 1–4.
| Entry | A [mol%] | KHMDS [mol%] | Time [h] | Conv. of 1ad [%] |
|---|---|---|---|---|
Reaction conditions: toluene, RT, [1a]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[2a] = 1:1, argon.a Carbene was generated for 30 minutes before substrate addition.b 4 Å molecular sieves were added.c Reaction conducted under air.d Determined by GC-MS analysis. :[2a] = 1:1, argon.a Carbene was generated for 30 minutes before substrate addition.b 4 Å molecular sieves were added.c Reaction conducted under air.d Determined by GC-MS analysis. |
||||
| 1a | 20 | 20 | 9 | 98 |
| 2 | 10 | 10 | 12 | 72 |
| 3a | 10 | 10 | 10 | 98 |
| 4a,b | 10 | 10 | 24 | 96 |
| 5a | 5 | 5 | 24 | 45 |
| 6c | 10 | 10 | 24 | 5 |
| 7 | — | 10 | 24 | 5 |
|
|
||||
|---|---|---|---|---|
| Entry | NHC | Time [h] | Conv. of 1aa [%] | Product 3a-a:3a-a′a |
| Reaction conditions: toluene, RT, [1a]:[2a] = 1:1, [NHC·HX] = 10 mol%, [KHMDS] = 10 mol%, argon.a Determined by GC-MS analysis and confirmed by 1H NMR spectroscopy of the crude reaction mixture. |
||||
| 1 | A | 10 | 98 | 100:0 |
| 2 | B | 24 | 31 | 60:40 |
| 3 | C | 24 | 72 | 91:9 |
| 4 | D | 24 | 25 | 50:50 |
| 5 | E | 24 | 42 | 52:48 |
| 6 | F | 48 | 4 | — |
| 7 | G | 24 | 37 | 0:100 |

|
|
||||
|---|---|---|---|---|
| Entry | Base | Time [h] | Conv. of 1ac [%] | Product 3a-a:3a-a′c |
| Reaction conditions: toluene, RT, argon, [1a]:[2a] = 1:1, [A] = 10 mol%, [base] = 10 mol%.a 20 mol% of KHMDS was used.b Freshly isolated carbene IPr*Et was used as a catalyst.c Determined by GC-MS analysis and confirmed by 1H NMR spectroscopy of the crude reaction mixture. |
||||
| 1 | KHMDS | 10 | 98 | 100:0 |
| 2a | KHMDS | 10 | 97 | 100:0 |
| 3 | DABCO | 12 | 93 | 100:0 |
| 4 | K2CO3 | 24 | 4 | 100:0 |
| 5 | Cs2CO3 | 24 | 50 | 100:0 |
| 6 | tBuOK | 24 | 50 | 70:30 |
| 7 | NEt3 | 12 | 100 | 100:0 |
| 8 | — | 24 | 0 | — |
| 9b | — | 7 | 99 | 100:0 |

|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time [h] | Conv. of 1ab [%] | Product 3a-a:3a-a′b |
| Reaction conditions: RT, argon, [A] = 10 mol%, [KHMDS] = 10 mol%, [1a]:[2a] = 1:1.a Distilled water was previously degassed by three freeze–thaw cycles.b Determined by GC-MS analysis and confirmed by 1H NMR spectroscopy of the crude reaction mixture. |
||||
| 1 | Toluene | 10 | 98 | 100:0 |
| 2 | MTBE | 12 | 90 | 100:0 |
| 3 | THF | 24 | 98 | 100:0 |
| 4 | Acetone | 12 | 100 | 100:0 |
| 5 | DCE | 48 | 27 | 65:35 |
| 6 | DCM | 48 | 16 | 60:40 |
| 7 | iPrOH | 24 | 57 | 75:25 |
| 8 | H2Oa | 24 | 62 | 50:50 |
| 9 | Hexane | 24 | 50 | 90:10 |

At first, the optimum concentrations of the NHC precursor and KHMDS were established, so as to ensure high conversion of the reagents (Table 1). As presented in Table 1, conducting the reaction in the presence of 10 mol% of NHC precursor A and 10 mol% of KHMDS resulted in 72% conversion of substrate 1a (Table 1, entry 2). To increase the conversion of the substrates, we decided to check if the earlier generation of free carbene would have any influence on the reaction efficiency. Indeed, when the NHC precursor was mixed only with KHMDS for 30 minutes before substrate addition, the reaction occurred with an excellent conversion within 10 hours (Table 1, entry 3). We found that the reduction of the concentration of the imidazolium salt and base to 5 mol% would decrease the substrate conversion by half (Table 1, entry 5), while an increase in their concentration to 20 mol% caused only a slight acceleration of the process (Table 1, entry 1). The presence of molecular sieves in the reaction had no effect on the overall efficiency (Table 1, entry 4). We also tested the reaction under air conditions but it produced only traces of the desired product, which confirmed that the process has to be carried out under an inert atmosphere (Table 1, entry 6). Finally, we confirmed that the presence of the NHC precursor is necessary for the reaction to proceed (Table 1, entry 7).
As the next step, the effects of using several different NHC precursors were examined to investigate the influence of various stereoelectronic properties on the reaction course. The previously optimized conditions were applied (Table 1, entry 3). The results are presented in Table 2.
As shown in Table 2, from among seven tested pre-catalysts, only one – superbulky A – was found to lead to the formation of product 3a-a with quantitative conversion of the reactants (Table 2, entry 1). The use of the other precursors directed the reaction to the Michael-addition product (3a-a′) (Table 2, entry 7) or to a mixture of adducts 3a-a and 3a-a′ (Table 2, entries 2–6). Moreover, imidazolium (B, C, E, F), benzimidazolium (D) and triazolium (G) showed mediocre or weak activity under the given reaction conditions.
Finally, we tested the effects of different bases and solvents on the reaction outcome to determine the influence of different conditions on the reaction progress (Tables 3 and 4).
From among the tested bases, KHMDS, DABCO and triethylamine provide the most effective course of the reaction, as they ensure almost quantitative conversion of substrate 1a and lead selectively to product 3a-a (Table 3, entries 1, 3 and 7). The process occurred selectively also in the presence of potassium and cesium carbonates, but then the conversion of the substrates dropped down to 4% and 50%, respectively (Table 3, entries 4 and 5). Potassium tert-butoxide also gave insufficient reactant conversion and more importantly, led to a mixture of products 3a-a and 3a-a′ at the ratio of 70 to 30 (Table 3, entry 6). Finally, we confirmed that the presence of base is necessary for the reaction to proceed (Table 3, entry 8). Moreover, to better understand the role of the base in the process we performed a test with previously isolated carbene, IPr*Et, which turned out to proceed smoothly with high conversion (Table 3, entry 9). Moreover, a twofold excess of the base was found to have no effect on the course of the process (Table 3, entry 2). Therefore, we think that the role of base in the reaction is limited only to the generation of free carbene.
The test reactions performed in different solvents confirmed that the choice of reaction medium has a significant impact on the process. Polar aprotic solvents, such as MTBE and THF, provide similar results to toluene, selectively giving product 3a-a (Table 4, entries 2 and 3). The use of acetone also allowed the reaction to run with excellent efficiency, which is particularly important due to the fact that acetone is considered to be a green-solvent, and thus the described method gains a more sustainable meaning (Table 4, entry 4). Both halogenated hydrocarbons and polar protic solvents showed rather weak performances, since their use led to low conversions and a mixture of products (Table 4, entries 5–8). On the other hand, the application of non-polar solvents, such as hexane, gave high selectivity, but the product yield remained insufficient (Table 4, entry 9).
Having the optimized conditions in hand, th range of substrates was extended to determine the versatility of the method (Scheme 3). In the first series of experiments, we probed the reactivity of a broad range of commercially available thiols (2a-j) toward cinnamaldehyde (1a). As shown in Scheme 3A, the proposed method can be successfully applied for the tested aldehyde and a series of aryl thiols with both electron-withdrawing and electron-donating substituents. Most of the products were obtained with excellent isolated yields (>85%) under very mild conditions and with 100% atom economy, which makes the reaction sustainable and environmentally friendly. When using 4-methylphenylthiol (2c), 4-bromophenylthiol (2f) and 2-naphthalenethiol (2g) a slightly increased temperature was necessary to obtain the expected products in quantitative yields. Besides its attractive advantages (room temperature in most cases, acetone as the solvent, low concentration of NHC, equimolar ratios of reagents, and high conversions), the proposed method has some limitations. Alkyl thiols containing both primary and secondary carbon connected to thiol groups, such as n-hexanethiol (2i) and cyclohexanethiol (2j), gave the product in much lower yield (3a-i and 3a-j). When using 2i, we observed 52% conversion of the substrates and the formation of an equimolar mixture of products 3a-i and 3a-i′, while 2j led only to trace amounts of the product. Also, with 3-mercaptopropionic acid (2k), the reaction occurred with a very low conversion (3a-k).
 | ||
| Scheme 3 Substrate scope (experimental conditions: (1a–l):(2a–j) = 1:1, [A] = 10 mol%, [KHMDS] = 10 mol%, acetone, RT, argon). Isolated yields and reaction times are given under the structure of each product. aReaction performed at 40 °C; byields of 3a-i, 3a-j and 3a-k were determined by GC-MS; ca mixture of 3a-i and 3a-i′ was detected in a ratio 1:1; d(1a):(2l) = 1:2. | ||
In the second series of experiments, the catalytic properties of salt A were evaluated in the thioesterification of a series of α,β-unsaturated aldehydes (1a-l) with benzyl mercaptan (2a) (Scheme 3B). Highly gratifyingly, quantitative conversions were achieved within a reasonable period of time, irrespective of the nature of the aldehyde substituent. For all the substrates tested, only 4-fluoro-cinnamaldehyde (1b) and 1-hexanal (1k) required a slightly increased temperature (40 °C). Apart from these, we did not observe a meaningful difference in the reaction course for the variety of substrates used. As presented in Scheme 3B, hex-4-en-1-al (2l), having two conjugated double bonds in its structure, was also applied in the reaction showing high activity leading to a desired product (3l-a). Under the optimized reaction conditions, only the α,β-unsaturated bond underwent the reaction, while the double bond between γ and δ carbons remained unreacted. We found that the use of a two-fold excess of thiol (1a) relative to hex-4-en-1-al (2l) and increase of the reaction temperature to 40 °C permit effective functionalization of the two unsaturated bonds, leading to a product containing two sulfur atoms (3l-a′′). This opens the possibility of synthesising organosulfur materials that may comprise different sulfur groups to meet different needs.
The successful catalytic thioesterification between equimolar amounts of α,β-unsaturated aldehyde and thiol prompted us to carry out reactions with dithiol to probe the feasibility of a procedure leading to bis-functionalized organosulfur derivatives. In this procedure, we treated 1 equiv. of 1,4-benzenedithiol (4) and 2 equiv. of the selected aldehydes (1a–g) with NHC carbene generated from salt A and the reaction mixtures were stirred at 40 °C until full conversions of the reagents were detected by GC-MS (Scheme 4).
 | ||
| Scheme 4 Thioesterification of α,β-unsaturated aldehydes (1a–f) with 1,4-benzenedithiol (4) (experimental conditions: (1a–f):(4) = 2:1, [A] = 10 mol%, [KHMDS] = 10 mol%, acetone, 40 °C, argon). Isolated yields and reaction times are given under the structure of each product. | ||
Interestingly, and highly gratifyingly, quantitative conversions were achieved within a reasonable period of time, irrespective of the nature of the aldehyde substituent. According to the best of our knowledge, the obtained derivatives (5a–f) are new compounds. It should be emphasized that all products, except 5b and 5c, precipitated in the reaction medium and their isolation required only filtration and washing with acetone.
To get some insight into the reaction mechanism, a deuterium-labeling experiment using phenylmethanethiol-d (2a′) and cinnamaldehyde (1a) was performed (Scheme 5). Phenylmethanethiol-d was obtained according to the known method described before with 67% of deuterium incorporation.
 | ||
| Scheme 5 Deuterium-labeling experiment. | ||
The experiment was conducted under standard conditions leading to full conversion of the reactants after 12 hours. GC-MS analysis revealed the formation of the expected product with the appropriate mass. 1H NMR analysis of the obtained compound showed partial disappearance of the multiplet between 2.87 and 2.91 ppm, which, based on the 1H,13C HMBC spectrum, can be assigned to alpha protons (for details see the ESI†). We did not observe any deuterium incorporation into the beta position, which confirmed the formation of the 3a′-a product.
Based on the previous research concerning ester synthesis from α,β-unsaturated aldehydes and alcohols catalyzed by NHC21–27 and on the deuterium-labelling experiment, we proposed a mechanism, which is depicted in Scheme 6.
 | ||
| Scheme 6 Proposed mechanism. | ||
The first step in the proposed mechanism is the generation of free carbene A′ through deprotonation of salt A. Then, carbene A′ reacts with the α,β-unsaturated aldehyde to give intermediate I, which then undergoes proton-transfer giving the Breslow intermediate (II).28–30 Intermediate II may remain in equilibrium with homoenolate form III.31 In the next stage, we suggest the occurrence of a direct proton transfer from the hydroxyl group to the γ carbon atom, which was earlier described in the literature.32,33 A similar process has been proposed by Chen et al. for thioester synthesis; however, in this case, the use of a proton-shuttling agent was necessary for this transfer to occur.20 We suppose that a bulky catalyst can promote intramolecular proton transfer because of the steric hindrance, and thus no proton shuttler is needed for this step. Finally, intermediate IV reacts with thiol leading to V, which after subsequent imidazole elimination generates the reaction product and regenerates the catalyst.
3. Conclusions
To sum up, we have designed a new method for thioester synthesis catalyzed by a bulky NHC carbene. The reaction has been applied for a wide range of thiols and enals giving desired products with excellent yields and chemoselectivity. However, the other less bulky NHCs tested in this reaction proved to be unsuitable for this reaction, leading to a mixture of products in poor yields. From the point of view of the utility of the presented protocol in organic synthesis, the results show many advantages. The process features high atom-economy (the lack of by-products, equimolar ratios of substrates, ease of product isolation and purification), excellent yields and a wide substrate scope. The proposed method allows one to obtain thioesters under metal-free conditions, by using green-solvents and at room temperature, which makes it sustainable and environmentally friendly. It is worth emphasizing that the methodology presented in this article provides the possibility of obtaining a new class of organosulfur compounds with potential for practical applications.4. Experimental
4.1. General methods and chemicals
All syntheses and catalytic tests were carried out under dry argon, using standard Schlenk-line and vacuum techniques. 1H NMR and 13C NMR spectra were recorded in CDCl3 on a Varian 400 operated at 402.6 and 101.2 MHz, respectively. 19F NMR spectra were recorded in CDCl3 on a Varian Mercury 300 operated at 282.6 MHz. HMBC spectra were recorded on a Bruker Avance DRX 600, operated at a frequency of 600.13 MHz (1H). GC-MS analyses were performed on a Varian Saturn 2100T equipped with a DB-1 capillary column (30 m in length and 0.25 mm in internal diameter) and an ion trap detector. Thin layer chromatography (TLC) was conducted on plates coated with a 250 μm thick silica gel layer and column chromatography was performed on silica gel 60 (70–230 mesh).NHC carbene precursors34–37 and phenylmethanethiol-d38 were prepared according to literature procedures. All the other reagents were commercially available and used as received. All the solvents, except THF, were dried over CaH2 prior to use and stored over 4 Å molecular sieves under argon. Dichloromethane was additionally passed through an alumina column and degassed by repeated freeze–pump–thaw cycles. THF was dried over sodium benzophenone ketyl and freshly distilled prior to use.
4.2. General procedure for catalytic tests
4.3. General procedure for the synthesis of thioesters
:1 v/v mixture of n-hexane and dichloromethane as eluents. Evaporation of the solvents afforded analytically pure compounds.
:1 v/v mixture of n-hexane and dichloromethane as eluents. Evaporation of the solvents afforded analytically pure compounds.
4.4. Synthesis of 3a-a on a preparative scale
A 10 mL high-pressure Schlenk vessel equipped with a magnetic stirring bar and connected to the gas and vacuum line was charged with the NHC carbene precursor A (381.3 mg, 3.90 × 10−4 mol), KHMDS (77.8 mg, 3.90 × 10−4 mol) and acetone (2 mL) under argon. The reaction mixture was stirred at RT and after 1 hour benzyl mercaptan (0.46 mL, 3.90 × 10−3 mol) and cinnamaldehyde (0.49 mL, 3.90 × 10−3 mol) were added. The reaction mixture was stirred at 40 °C for 24 h. Then, the solvent was evaporated under vacuum and the residue was purified using column chromatography (silica gel 60/petroleum ether:DCM = 5:1). Evaporation of the solvent gave the analytically pure product (yellow liquid, 0.90 g, 90%).
4.5. Deuterium-labeling experiment
A 10 mL high-pressure Schlenk vessel connected to the gas and vacuum line was charged under argon with the NHC carbene precursor (38.8 mg, 3.97 × 10−5 mol), KHMDS (7.9 mg, 3.97 × 10−5 mol) and acetone (0.5 mL). The reaction mixture was stirred at RT and after 30 minutes phenylmethanethiol-d (46.5 μL, 3.97 × 10−4 mol) and cinnamaldehyde (50 μL, 3.97 × 10−4 mol) were added. The reaction was stirred at RT in a closed vessel for 12 h. Then the solvent was evaporated under vacuum and the resulting product was isolated and purified by chromatography (silica gel 60/n-hexane:DCM = 1:5). Evaporation of the solvent gave the analytically pure product.
Author contributions
Conceptualization of paper, design of the experiments and supervision of the research, P.Ż.; preparation of NHC precursors and performance of half of the experiments, M.B.; performance of half of the experiments and isolation of all products, K.H.; analysis of data, P.Ż; writing original draft, P.Ż. and M.B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Ministry of Education and Science (project Diamond Grant No. DI2017 002647). P. Ż. acknowledges the financial support from the National Science Centre (Poland) (SONATA Project No. UMO-2016/23/D/ST5/00417). The authors are grateful to Mrs Bożena Wyrzykiewicz from the Faculty of Chemistry of Adam Mickiewicz University in Poznan for carrying out 2H NMR measurements.References
- P. Chauhan, S. Mahajan and D. Enders, Organocatalytic Carbon–Sulfur Bond-Forming Reactions, Chem. Rev., 2014, 114, 8807–8864 CrossRef CAS PubMed.
- V. Hirschbeck, P. H. Gehrtz and I. Fleischer, Metal-Catalyzed Synthesis and Use of Thioesters: Recent Developments, Chem. – Eur. J., 2018, 24, 7092–7107 CrossRef CAS PubMed.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Sulfur Chemistry in Polymer and Materials Science, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef.
- S. Aksakal, R. Aksakal and C. R. Becer, Thioester functional polymers, Polym. Chem., 2018, 9, 4507–4516 RSC.
- M. Kazemi and L. Shiri, Thioesters synthesis: recent adventures in the esterification of thiols, J. Sulfur Chem., 2015, 36, 613–623 CrossRef CAS.
- N. H. Jabarullah, K. Jermsittiparsert, P. A. Melnikov, A. Maseleno, A. Hosseinian and E. Vessally, Methods for the direct synthesis of thioesters from aldehydes: a focus review, J. Sulfur Chem., 2020, 41, 96–115 CrossRef CAS.
- C.-L. Yi, Y.-T. Huang and C.-F. Lee, Synthesis of thioesters through copper-catalyzed coupling of aldehydes with thiols in water, Green Chem., 2013, 15, 2476 RSC.
- Y.-T. Huang, S.-Y. Lu, C.-L. Yi and C.-F. Lee, Iron-Catalyzed Synthesis of Thioesters from Thiols and Aldehydes in Water, J. Org. Chem., 2014, 79, 4561–4568 CrossRef CAS PubMed.
- H.-S. Jhuang, Y.-W. Liu, D. M. Reddy, Y.-Z. Tzeng, W.-Y. Lin and C.-F. Lee, Microwave-assisted Synthesis of Thioesters from Aldehydes and Thiols in Water: Thioesters Can be Prepared in 5 Min, J. Chin. Chem. Soc., 2018, 65, 24–27 CrossRef CAS.
- F. Zhao, H.-Q. Geng and X.-F. Wu, Palladium-Catalyzed Thiocarbonylation of Alkenes toward Linear Thioesters, ACS Catal., 2021, 11, 3614–3619 CrossRef.
- X. Wang, B. Wang, X. Yin, W. Yu, Y. Liao, J. Ye, M. Wang, L. Hu and J. Liao, Palladium-Catalyzed Enantioselective Thiocarbonylation of Styrenes, Angew. Chem., Int. Ed., 2019, 58, 12264–12270 CrossRef CAS PubMed.
- T. Uno, T. Inokuma and Y. Takemoto, NHC-catalyzed thioesterification of aldehydes by external redox activation, Chem. Commun., 2012, 48, 1901 RSC.
- S. Singh and L. D. S. Yadav, The direct thioesterification of aldehydes with disulfides via NHC-catalyzed carbonyl umpolung strategy, Tetrahedron Lett., 2012, 53, 5136–5140 CrossRef CAS.
- J. Chung, U. R. Seo, S. Chun and Y. K. Chung, Poly(3,4-dimethyl-5-vinylthiazolium)/DBU-Catalyzed Thioesterification of Aldehydes with Thiols, ChemCatChem, 2016, 8, 318–321 CrossRef CAS.
- J. Chen, S. Meng, L. Wang, H. Tang and Y. Huang, Highly enantioselective sulfa-Michael addition reactions using N-heterocyclic carbene as a non-covalent organocatalyst, Chem. Sci., 2015, 6, 4184–4189 RSC.
- Y.-Z. Li, Y. Wang, G.-F. Du, H.-Y. Zhang, H.-L. Yang and L. He, N-Heterocyclic-Carbene-Catalyzed Sulfa-Michael additions, Asian J. Org. Chem., 2015, 4, 327–332 CrossRef CAS.
- P. Yuan, S. Meng, J. Chen and Y. Huang, Asymmetric Sulfa-Michael Addition of α,β-Unsaturated Esters/Amides Using a Chiral N-Heterocyclic Carbene as a Noncovalent Organocatalyst, Synlett, 2016, 27, 1068–1072 CrossRef CAS.
- Z.-S. Cong, Y.-G. Li, G.-F. Du, C.-Z. Gu, B. Dai and L. He, N-Heterocyclic carbene-catalyzed sulfa-Michael addition of enals, Chem. Commun., 2017, 53, 13129–13132 RSC.
- Z.-N. Feng, J.-Y. Luo, Y. Zhang, G.-F. Du and L. He, N-Heterocyclic carbene-catalyzed diastereoselective synthesis of sulfenylated indanes via sulfa-Michael–Michael (aldol) cascade reactions, Org. Biomol. Chem., 2019, 17, 4700–4704 RSC.
- J. Chen, P. Yuan, L. Wang and Y. Huang, Enantioselective β-Protonation of Enals via a Shuttling Strategy, J. Am. Chem. Soc., 2017, 139, 7045–7051 CrossRef CAS PubMed.
- S. S. Sohn and J. W. Bode, Catalytic Generation of Activated Carboxylates from Enals: A Product-Determining Role for the Base, Org. Lett., 2005, 7, 3873–3876 CrossRef CAS PubMed.
- A. Chan and K. A. Scheidt, Conversion of α,β-Unsaturated Aldehydes into Saturated Esters: An Umpolung Reaction Catalyzed by Nucleophilic Carbenes, Org. Lett., 2005, 7, 905–908 CrossRef CAS PubMed.
- B. E. Maki, E. V. Patterson, C. J. Cramer and K. A. Scheidt, Impact of Solvent Polarity on N-Heterocyclic Carbene-Catalyzed β-Protonations of Homoenolate Equivalents, Org. Lett., 2009, 11, 3942–3945 CrossRef CAS PubMed.
- M. H. Wang, D. Barsoum, C. B. Schwamb, D. T. Cohen, B. C. Goess, M. Riedrich, A. Chan, B. E. Maki, R. K. Mishra and K. A. Scheidt, Catalytic, Enantioselective β-Protonation through a Cooperative Activation Strategy, J. Org. Chem., 2017, 82, 4689–4702 CrossRef CAS PubMed.
- N. Wang, J. Xu and J. K. Lee, The importance of N-heterocyclic carbene basicity in organocatalysis, Org. Biomol. Chem., 2018, 16, 8230–8244 RSC.
- X. Li, J. Xu, S.-J. Li, L.-B. Qu, Z. Li, Y. R. Chi, D. Wei and Y. Lan, Prediction of NHC-catalyzed chemoselective functionalizations of carbonyl compounds: a general mechanistic map, Chem. Sci., 2020, 11, 7214–7225 RSC.
- A. Biswas, J.-M. Neudörfl, N. E. Schlörer and A. Berkessel, Acyl Donor Intermediates in N-Heterocyclic Carbene Catalysis: Acyl Azolium or Azolium Enolate?, Angew. Chem., Int. Ed., 2021, 60, 4507–4511 CrossRef CAS PubMed.
- M. Pareek, Y. Reddi and R. B. Sunoj, Tale of the Breslow intermediate, a central player in N-heterocyclic carbene organocatalysis: then and now, Chem. Sci., 2021, 12, 7973–7992 RSC.
- A. Wessels, M. Klußmann, M. Breugst, N. E. Schlörer and A. Berkessel, Formation of Breslow Intermediates from N-Heterocyclic Carbenes and Aldehydes Involves Autocatalysis by the Breslow Intermediate, and a Hemiacetal, Angew. Chem., Int. Ed., 2022, 61, e202117682 CAS.
- S.-L. Liu, X. Liu, Y. Wang and D. Wei, Unraveling the mechanism and substituent effects on the N-heterocyclic carbene-catalyzed transformation reaction of enals and imines, Mol. Catal., 2022, 519, 112122 CrossRef CAS.
- J. Mahatthananchai and J. W. Bode, The effect of the N-mesityl group in NHC-catalyzed reactions, Chem. Sci., 2012, 3, 192–197 RSC.
- Y. Reddi and R. B. Sunoj, Origin of Stereoselectivity in a Chiral N-Heterocyclic Carbene-Catalyzed Desymmetrization of Substituted Cyclohexyl 1,3-Diketones, Org. Lett., 2012, 14, 2810–2813 CrossRef CAS PubMed.
- A. Berkessel, V. R. Yatham, S. Elfert and J.-M. Neudörfl, Characterization of the Key Intermediates of Carbene-Catalyzed Umpolung by NMR Spectroscopy and X-Ray Diffraction: Breslow Intermediates, Homoenolates, and Azolium Enolates, Angew. Chem., Int. Ed., 2013, 52, 11158–11162 CrossRef CAS PubMed.
- V. Sh. Saberov, D. A. Evans, N. I. Korotkikh, A. H. Cowley, T. M. Pekhtereva, A. F. Popov and O. P. Shvaika, Exceptionally efficient catalytic hydrodechlorination of persistent organic pollutants: application of new sterically shielded palladium carbene complexes, Dalton Trans., 2014, 43, 18117–18122 RSC.
- M. Hans, J. Lorkowski, A. Demonceau and L. Delaude, Efficient synthetic protocols for the preparation of common N-heterocyclic carbene precursors, Beilstein J. Org. Chem., 2015, 11, 2318–2325 CrossRef CAS PubMed.
- R. Saravanakumar, V. Ramkumar and S. Sankararaman, Synthesis and Structure of 1,4-Diphenyl-3-methyl-1,2,3-triazol-5-ylidene Palladium Complexes and Application in Catalytic Hydroarylation of Alkynes, Organometallics, 2011, 30, 1689–1694 CrossRef CAS.
- D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, PEPPSI-Type Palladium Complexes Containing Basic 1,2,3-Triazolylidene Ligands and Their Role in Suzuki–Miyaura Catalysis, Chem. – Eur. J., 2012, 18, 6055–6062 CrossRef CAS PubMed.
- A. Guerrero-Corella, A. M. Martinez-Gualda, F. Ahmadi, E. Ming, A. Fraile and J. Alemán, Thiol–ene/oxidation tandem reaction under visible light photocatalysis: synthesis of alkyl sulfoxides, Chem. Commun., 2017, 53, 10463–10466 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qo00678b |
| This journal is © the Partner Organisations 2022 |