
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResolution of aryl-H-phosphinates applied in the synthesis of P-stereogenic compounds including a Brønsted acid NMR solvating agent†
Bence
Varga
 *a,
Daniella
Vincze
a,
Hajnalka
Pető
a,
Levente
Buna
a,
János
Pauló
a,
Tamás
Holczbauer
b,
Béla
Mátravölgyi
a,
László
Hegedűs
a,
Elemér
Fogassy
a,
György
Keglevich
a and
Péter
Bagi
*a
*a,
Daniella
Vincze
a,
Hajnalka
Pető
a,
Levente
Buna
a,
János
Pauló
a,
Tamás
Holczbauer
b,
Béla
Mátravölgyi
a,
László
Hegedűs
a,
Elemér
Fogassy
a,
György
Keglevich
a and
Péter
Bagi
*a
aDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, Műegyetem rkp. 3., H-1111 Budapest, Hungary. E-mail: bencevarga@edu.bme.hu; bagi.peter@vbk.bme.hu
bCenter for Structural Science, Chemical Crystallography Research Laboratory and Institute for Organic Chemistry, Research Centre for Natural Sciences, Magyar tudósok körútja 2, H-1519 Budapest, Hungary
First published on 5th April 2022
Abstract
A library of racemic H-phosphinates incorporating a variety of alkoxy groups or substituted aryl groups was prepared. Starting with resolving agent screening, an efficient enantioseparation method was developed and optimized using (R,R)-(1-naphthyl)-spiro-TADDOL as the resolving agent. This method gave 12 out of 16 H-phosphinates with an ee of above 91%, and 9 of these derivatives were practically enantiopure (ee > 98%). Moreover, this resolution could be used on a gram-scale. The structures of the diastereomeric intermediates and the secondary interactions between them were investigated by single-crystal XRD, and the most important factors of enantiomeric recognition were established. (R)-1-Adamantyl phenyl-H-phosphinate was stereospecifically transformed into a few secondary and tertiary phosphine oxides. Moreover, two P-stereogenic Brønsted acids were prepared, and their applicability as chiral NMR solvating agents was assessed.
Introduction
P-Stereogenic compounds represent an important class within organophosphorus chemistry. Originally, they were used as ligands of transition metal catalysts,1 but in the past few decades their use in organocatalysis2 and medicinal chemistry emerged significantly.3 The attention that P-chirogenic remdesivir just gained in COVID-19 treatment is a recent example of the latter fact.4 The most widely used strategies are the direct enantioseparation or asymmetric synthesis of target chiral organophosphorus compounds.1b,5 Additionally, stereoselective transformations of intermediates bearing a stable P-stereogenic center can also be used, and such a modular approach may give relatively diverse compounds from a given set of intermediates.6 As the stereochemical outcome of the reactions involving a P-stereogenic center is well documented and reviewed,7 the preparation of P-chirogenic intermediates is of particular importance. From this perspective secondary phosphine oxides, phosphinates, and H-phosphinates (or their corresponding P-borane derivatives) can be regarded as ideal intermediates due to their bench and configurational stability. In this compound class, the chemistry of chiral secondary phosphine oxides received considerable attention in recent years,8 whereas the number of methods affording optically active H-phosphinates remained limited. However, H-phosphinates have higher synthetic potential, as they contain P–H and P–OR functional groups, both of which can be functionalized in a stereospecific manner by a variety of methods.9Historically, the first P-stereogenic H-phosphinates were made available in optically active form via the formation and separation of covalent diastereomers using (−)-menthol as an affordable chiral auxiliary. The corresponding alkyl-H-phosphinates were the first few derivatives that could be prepared as a pure diastereomer.10 In the past decade, several improved procedures were published on the preparation and separation of menthyl aryl-H-phosphinates.9b,11 Han et al. optimized the synthesis and crystallization of this compound class,9b,11a,b and they could solve the preparation issues caused by poor crystallinity of the racemate, which previously hindered the efficient preparation of pure diastereomers.12 The key steps of these synthetic procedures were the formation and hydrolysis of the corresponding phosphonochloridate [(−)-MentO]YPCl intermediate. Montchamp et al. prepared menthyl hydroxymethyl-H-phosphinate from hypophosphorous acid, paraformaldehyde and (−)-menthol, the procedure of which avoided the use of the corresponding P–Cl intermediates. [(−)-MentO](HOCH2)P(O)H could be transformed into a variety of H-phosphinates by P–C coupling and Corey–Kim oxidation (Fig. 1A).13
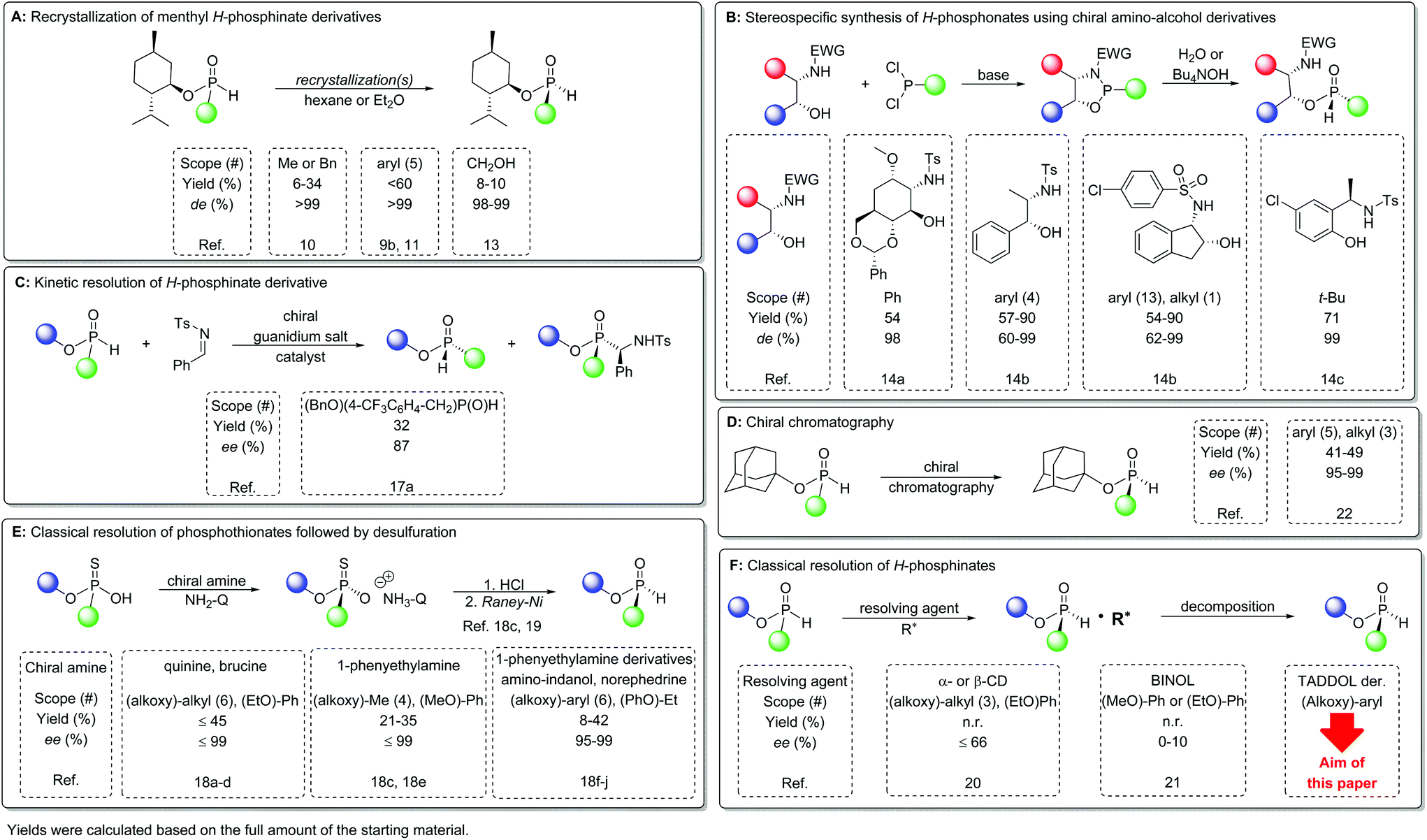 | ||
| Fig. 1 Selected literature methods for the preparation of optically active P-stereogenic H-phosphinates. | ||
P-Stereogenic oxazaphospholidines, stereoselectively prepared from readily available amino-alcohols, became key intermediates for the preparation of various P-stereogenic compounds. The P–N bond can be selectively cleaved under hydrolytic conditions to afford H-phosphinate diastereomers, and the diastereomeric purity of these intermediates could be increased by recrystallization (Fig. 1B).14
Preparation of enantiomerically enriched P*(O)H compounds using kinetic resolution became a popular method in recent years. Generally, these procedures were optimized to give enantiopure tertiary phosphine oxides,15 but in a few instances, the remaining >P(O)H starting materials were also obtained with high enantiomeric excess.16 Secondary phosphine oxides were the main starting materials in these reactions, but a few procedures are also known for H-phosphinates.17 In the latter instances, only one optically active H-phosphinate was isolated for mechanistic investigation, which leaves the scope of those procedures rather low (Fig. 1C).17a
Phosphothionates might be regarded as acidic precursors of H-phosphinates, which can be resolved using commercially available chiral bases.18 Despite the fact that the majority of the resolution procedures focus on the enantioseparation of the corresponding phosphothionates, a few studies demonstrated that desulfuration can be performed with RANEY®-nickel with the retention of the P-configuration to give optically active H-phosphinates (Fig. 1E).18c,19 This route towards enantiomerically enriched >P(O)H compounds is rather tedious, as it includes two additional steps besides the classical resolution, which increases the risk of partial racemization during the final desulfuration step.
There are only a few attempts towards the direct enantioseparation of P-stereogenic H-phosphinates. In 1970, Benschop described for the first time the partial resolution of H-phosphinates via inclusion complexation with cyclodextrins, but only low optical purities were obtained and the yields were not reported.20 Toda tried to use BINOL for the enantioseparation of methyl or ethyl phenyl-H-phosphinate, but that attempt was rather unsuccessful, despite the fact that the same method gave good results for a few phosphinates and phosphine oxides (Fig. 1F).21 Leclaire, Giordano and co-workers used semi-preparative chiral HPLC to separate the enantiomers of various 1-adamantyl H-phosphinates (Fig. 1D). They also demonstrated that the adamantyl esters are less prone to racemization during the substitution reaction at the P-stereogenic center, which can be attributed to the increased steric hindrance of the adamantyloxy moiety.22
This summary demonstrates that the literature lacks efficient separation methods to prepare P-stereogenic H-phosphinate enantiomers. Thus, the current paper aims at the development of such enantioseparation for this compound class (1). The synthesis of H-phosphinates incorporating a wide variety of sterically demanding alkoxy moieties, as well as various aryl groups, was planned (1). We intended to find the most suitable resolving agent class and crystallization conditions for H-phosphinates (1) in order to develop a robust resolution method for the target racemates. We wished to elucidate the structures of the diastereomeric associates by single crystal X-ray crystallography to study the intermolecular interactions and to determine the absolute P-configurations. In order to demonstrate the synthetic utility of H-phosphinates (1), stereoselective preparation of secondary and tertiary phosphine oxides (3a–c) as well as a few P-stereogenic Brønsted acids (3e and 3f) was also planned, and the latter derivatives were tested as chiral NMR solvating agents (Fig. 2).
 | ||
| Fig. 2 Aims of this research project. | ||
Results and discussion
In our study, sterically congested alcohols were used to prepare this H-phosphinate library (1), as previous research suggested that H-phosphinates prepared from aliphatic alcohols with low carbon chains are prone to racemization once they are resolved into their enantiomers.18c,22 Two different methods were used for the synthesis of racemic H-phosphinates (1). Phenyl-H-phosphinate esters (1a–j) were prepared from phenyl-H-phosphinic acid (4) and from the corresponding alcohol using EDC hydrochloride as the coupling agent. This synthetic strategy avoids the handling of organometallic and air-sensitive compounds, and it was used a few times for the synthesis of P-esters.23 Recently, Montchamp et al. surveyed the different coupling agents for the synthesis of adamantyl H-phosphinate esters and found that T3P is an optimal choice.24 In our study, 2 equiv. of EDC-HCl (without DMAP), and 1 equiv. of alcohol in abs. DCM gave the corresponding H-phosphinate esters (1a–f and 1h–j) generally in yields of 80–97% after an extractive workup and purification by column chromatography. The only exception was 1-methyl-cyclohexyl phenyl H-phosphinate (1g) (yield = 43%) (Scheme 1). | ||
| Scheme 1 Preparation of racemic H-phosphinates (1). | ||
For the synthesis of adamantyl aryl-H-phosphinates (1k–p), (1-AdO)PCl2 (5) was prepared in the reaction of phosphorus trichloride and 1-adamantanol.22 This compound (5) was then reacted with 1 equiv. of the corresponding Grignard reagent to give H-phosphinates (1k–p) in yields of 50–74% after work-up and column chromatography (Scheme 1).
In order to find the most suitable resolution conditions, 1-adamantyl phenyl-H-phosphinate (1a) was chosen as the model racemate from this H-phosphinate library (1), due to its high synthetic potential.22 First, a resolving agent study was conducted which included several resolving agents, successfully used for the enantioseparation of various P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O containing racemic compounds.1b,25 Using this resolving agent pool, TADDOL and spiro-TADDOL [(R,R)-2a and (R,R)-2b] showed the most promising preliminary results (see the ESI† for details). Thus, a series of TADDOL derivatives [(R,R)-2a–g] with various dioxalane scaffolds,26 and different substituted aryl or naphthyl groups were tested for the enantioseparation of 1a. Based on the results of the resolving agent study and our previous experience with this resolving agent class, a precipitation mediated resolution method was chosen using a mixture of toluene and hexane as the solvent. Only (R,R)-spiro-TADDOL [(R,R)-2b] and (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] gave crystalline diastereomeric complexes that could be purified further by additional recrystallization(s) to prepare (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a] with an ee of above 95%. Using these two resolving agents, (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] gave a significantly better yield (72% vs. 38%) (see the ESI† for details). With the most suitable resolving agent [(R,R)-2e] in hand, solvent screening was performed and the solvents were selected based on our previous studies.27 The results indicated that aromatic solvents in combination with hexane were the more suitable ones, and the best results were obtained in a mixture of toluene and hexane. It was also found that performing these resolutions according to the equivalent method is more beneficial than using a half equivalent of the resolving agent [(R,R)-2e]. It was also investigated whether the further purification of 1-adamantyl phenyl-H-phosphinate (1a) enantiomeric mixtures is feasible without any chiral additives utilizing only the SDE phenomenon (self-disproportion of enantiomers).28 The results indicated that 1-adamantyl phenyl-H-phosphinate (1a) is a racemate-forming compound having a eutectic composition of ca. 30% (see the ESI† for details). This dataset also suggested that efficient further purification of samples having an initial ee of above 70% is less practical, as the ee increases marginally and a significant amount of 1a is lost during the recrystallizations. In contrast, the recrystallization of the corresponding diastereomer [(R)-1a·(R,R)-2e] from a mixture of toluene and hexane was a more efficient purification.
O containing racemic compounds.1b,25 Using this resolving agent pool, TADDOL and spiro-TADDOL [(R,R)-2a and (R,R)-2b] showed the most promising preliminary results (see the ESI† for details). Thus, a series of TADDOL derivatives [(R,R)-2a–g] with various dioxalane scaffolds,26 and different substituted aryl or naphthyl groups were tested for the enantioseparation of 1a. Based on the results of the resolving agent study and our previous experience with this resolving agent class, a precipitation mediated resolution method was chosen using a mixture of toluene and hexane as the solvent. Only (R,R)-spiro-TADDOL [(R,R)-2b] and (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] gave crystalline diastereomeric complexes that could be purified further by additional recrystallization(s) to prepare (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a] with an ee of above 95%. Using these two resolving agents, (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] gave a significantly better yield (72% vs. 38%) (see the ESI† for details). With the most suitable resolving agent [(R,R)-2e] in hand, solvent screening was performed and the solvents were selected based on our previous studies.27 The results indicated that aromatic solvents in combination with hexane were the more suitable ones, and the best results were obtained in a mixture of toluene and hexane. It was also found that performing these resolutions according to the equivalent method is more beneficial than using a half equivalent of the resolving agent [(R,R)-2e]. It was also investigated whether the further purification of 1-adamantyl phenyl-H-phosphinate (1a) enantiomeric mixtures is feasible without any chiral additives utilizing only the SDE phenomenon (self-disproportion of enantiomers).28 The results indicated that 1-adamantyl phenyl-H-phosphinate (1a) is a racemate-forming compound having a eutectic composition of ca. 30% (see the ESI† for details). This dataset also suggested that efficient further purification of samples having an initial ee of above 70% is less practical, as the ee increases marginally and a significant amount of 1a is lost during the recrystallizations. In contrast, the recrystallization of the corresponding diastereomer [(R)-1a·(R,R)-2e] from a mixture of toluene and hexane was a more efficient purification.
As a result of this set of experiments, a resolution method using one equivalent of (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] in a mixture of toluene and hexane was developed, which afforded enantiopure (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a] in a yield of 74%, and only one recrystallization of the corresponding diastereomer was enough to achieve an ee of 99% (Scheme 2). The H-phosphinate (R)-1a could be recovered from the diastereomer by column chromatography.
 | ||
| Scheme 2 Stages of optimization for the enantioseparation of 1-adamantyl phenyl-H-phosphinate (1a) with TADDOL-derivatives [(R,R)-2]. | ||
Under optimal conditions, this resolution method showed an excellent scope among phenyl-H-phosphinates (1) (Scheme 3). The 1- and 2-adamantyl, cyclopentyl-, cyclohexyl-, 1-methylcyclohexyl-, cycloheptyl-, t-butyl- and benzyl-esters [(R)-1a–d, (R)-1f, (S)-1h and (R)-1j] could be prepared in practically enantiopure form (ee = 96–99%). (RP)-Menthyl phenyl-H-phosphinate [(RP)-1e] could also be prepared with a diastereomeric excess of 96%, demonstrating that our method can be an alternative preparation of this benchmark H-phosphinate diastereomer [(RP)-1e].
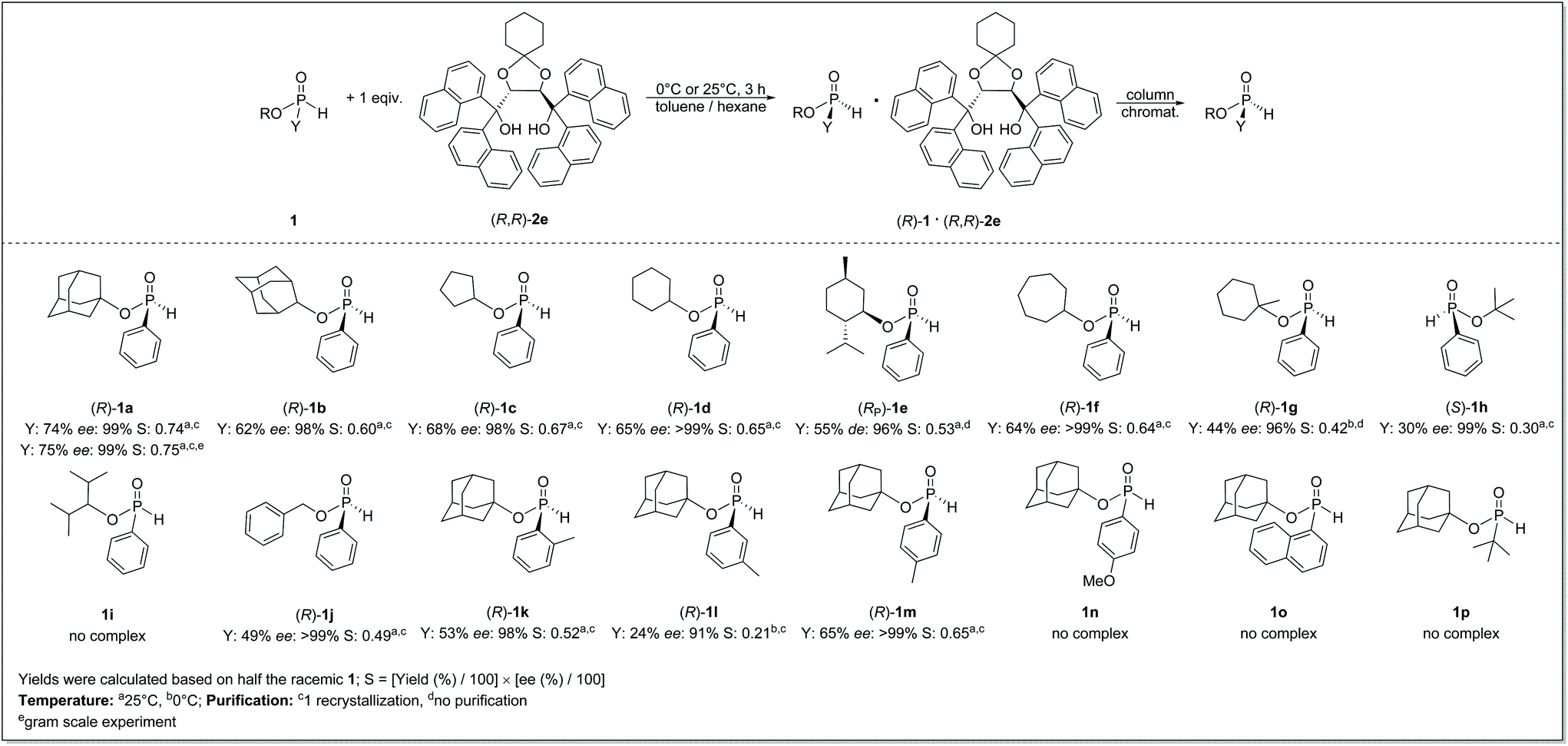 | ||
| Scheme 3 Summary of the enantioseparation of H-phosphinates (1) with (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] under optimized conditions. | ||
Using this resolution method, the crystallization issues of the corresponding diastereomeric mixtures (1e) may be avoided. For most derivatives (1), one purification was enough to increase the enantiomeric purity, and the yields were in the range of 55–74%; the three exceptions were 1-methylcyclohexyl, t-butyl and benzyl phenyl-H-phosphinates [(R)-1g, (S)-1h and (R)-1j] (yield: 30–49%). In the selected P-ester scope (1a–j), only 2,4-dimethylpentan-3-yl phenyl-H-phosphinate (1i) could not be separated into its enantiomers, as no crystalline diastereomers were formed.
The scope of this resolution method seemed more limited among 1-adamantyl H-phosphinates containing different aryl groups or a t-butyl group (1k–p). This resolution method afforded ortho- and para-tolyl-derivatives [(R)-1k and (R)-1m] in enantiopure form (ee > 98%) in yields of 53% and 65%, respectively. Moreover, 1-adamantyl (3-methylphenyl)-H-phosphinate [(R)-1l] could also be prepared with good ee but in a low yield (ee: 91%, yield: 24%). On the other hand, the enantioseparation of 4-methoxyphenyl-, 1-naphthyl- and t-butyl derivatives (1n–p) was not successful, as no crystallization occurred in the presence of a resolving agent [(R,R)-2e]. For 1-adamantyl t-butyl-H-phosphinate (1p), the underlying cause might be the lack of π–π interactions between the H-phosphinate (1p) and resolving agent [(R,R)-2e] molecules (vide infra). This resolution method could be conveniently scaled up to a gram-scale level, and enantiopure (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a] could be prepared from 4 grams of racemic 1a in the same yield as in the case of small-scale experiments (Scheme 3).
Single crystal X-ray analysis verified that the ratio of 1 to (R,R)-2e is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and it is independent of the structure of the H-phosphinate (1). Moreover, the resolving agent generally preferred the (R) enantiomer of the corresponding H-phosphinate [(R)-1a–g and (R)-1k–m]. One may assume a similar mode of binding and secondary interactions between the resolving agent [(R,R)-2e] and the corresponding H-phosphinate [(R)-1a–g and (R)-1k–m], which was also confirmed by the X-ray analysis of diastereomeric intermediates (vide infra).
:1, and it is independent of the structure of the H-phosphinate (1). Moreover, the resolving agent generally preferred the (R) enantiomer of the corresponding H-phosphinate [(R)-1a–g and (R)-1k–m]. One may assume a similar mode of binding and secondary interactions between the resolving agent [(R,R)-2e] and the corresponding H-phosphinate [(R)-1a–g and (R)-1k–m], which was also confirmed by the X-ray analysis of diastereomeric intermediates (vide infra).
It is noteworthy that the corresponding diastereomers [1·(R,R)-2e] could be stored at room temperature in air for years without any decomposition of the corresponding H-phosphinate (1) or the deterioration of its enantiomeric excess. Additionally, 1-adamantyl, 2-adamantyl, cyclohexyl- and benzyl phenyl-H-phosphinates [(R)-1a, (R)-1b, (R)-1d or (R)-1j] were selected to test the configurational stability of the corresponding enantiomers (ee: 99%) by stirring the given toluene solutions either at 25 °C or at 110 °C under an inert atmosphere. The results showed that the ee value of the corresponding H-phosphinates [(R)-1a, (R)-1b, (R)-1d or (R)-1j] did not change at 25 °C in 3–8 days, whereas the enantiomeric purity of (R)-1-adamantyl and (R)-2-adamantyl phenyl-H-phosphinate [(R)-1a and (R)-1b] decreased only to 96% or 93%, respectively, after 3 days of reflux, which is another indication of the excellent configurational stability of the adamantyl H-phosphinates [(R)-1a and (R)-1b] as was originally proposed by Leclaire, Giordano and co-workers (see the ESI† for additional details).22
Besides the scalability, we intended to demonstrate the robustness of the resolution method, and to develop a complete preparation and enantioseparation procedure for 1a in which the purification steps are simplified. The racemic 1-adamantyl phenyl-H-phosphinate (1a) was synthesized from 1.0 g of phenyl-H-phosphinic acid (4) using EDC-HCl. This crude product (with a purity of ca. 85%) was transferred to the enantioseparation step without any additional purification. Our optical resolution of 1a was proved to be robust enough to give enantiopure (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a]. Similarly to the small-scale experiments, one crystallization and an additional recrystallization of the corresponding diastereomer [(R)-1a·(R,R)-2e] were also sufficient using crude H-phosphinate (1a), but the yield was somewhat lower than the one obtained with the purified starting material (1a) (74% vs. 63%), which might be attributed to the impurities originating from EDC-coupling. The chromatographic isolation of optically active H-phosphinates (1) from the corresponding diastereomers was tedious on a larger scale due to the significant (molecular) weight difference of 1 and the resolving agent [(R,R)-2e]. It was observed during the optimization of the resolution that (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] forms a stable complex with ethanol (see the ESI† for additional details of decomposition). Utilizing this phenomenon, the (R)-1a·(R,R)-2e diastereomeric complex was treated with hot ethanol, and the majority of the resolving agent [(R,R)-2e] was recovered as its crystalline ethanol complex leaving only a minor amount of [(R,R)-2e] besides the (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a]. Another portion of the resolving agent [(R,R)-2e] also could be recycled from the mother liquor of the resolution using a similar ethanolic decomposition. From the two residual mixtures containing (R,R)-2e and (R)/(S)-1a, the enantiopure (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a] (ee: 99%, Y: 61%) and the corresponding (S) enantiomer [(S)-1a] with an ee of 60% were isolated by column chromatography. The purity of the enantiomeric mixture containing (S)-1a in excess was increased by a simple recrystallization from ethyl acetate utilizing the SDE phenomenon, and in this manner (S)-1-adamantyl phenyl-H-phosphinate [(S)-1a] could be obtained with an ee of 91% and in a yield of 41% without using the antipode of the resolving agent [(S,S)-2e] (Scheme 4).
 | ||
| Scheme 4 Gram scale preparation of (R)- and (S)-1-adamantyl phenyl-H-phosphinate [(R)- and (S)-1a] using a simplified preparation and enantioseparation procedure. | ||
It is worth mentioning that the final column chromatographic separation of the (R)-1a and (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] mixtures was performed by a simplified DCM to DCM–MeOH gradient elution. Our previous study also demonstrated that organic solvent nanofiltration may be an alternative to column chromatography during the isolation of optically active products and the recovery of the resolving agent.27a
X-Ray quality crystals could be prepared from the diastereomeric intermediates of all successful resolutions, and the two exceptions were (R)-1g·(R,R)-2e and (R)-1l·(R,R)-2e diastereomers. These measurements allowed the unambiguous assignation of absolute P-configurations and the study of non-bonding interactions responsible for enantiomeric recognition within this wide scope of H-phosphinates (1a–h and 1j–m). All of the 10 diastereomeric complexes crystallized in the orthorhombic crystal system space group of P212121. The unit cell parameters are isostructural, except for the (RP)-1e·(R,R)-2e and (S)-1h·(R,R)-2e structures where the b and c unit cell parameters are smaller. The contents of the asymmetric unit (Z′) are the (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] host molecule with a known absolute configuration and the corresponding optically active H-phosphinate guest molecules [(R)-1a–f, (S)-1h, (R)-1j, (R)-1k and (R)-1m]. Moreover, a toluene solvent molecule is also incorporated into the asymmetric unit in all but two instances [(RP)-1e·(R,R)-2e and (S)-1h·(R,R)-2e], which is in accordance with the experimental observation that aromatic solvents, especially toluene, gave the best enantioseparation results.
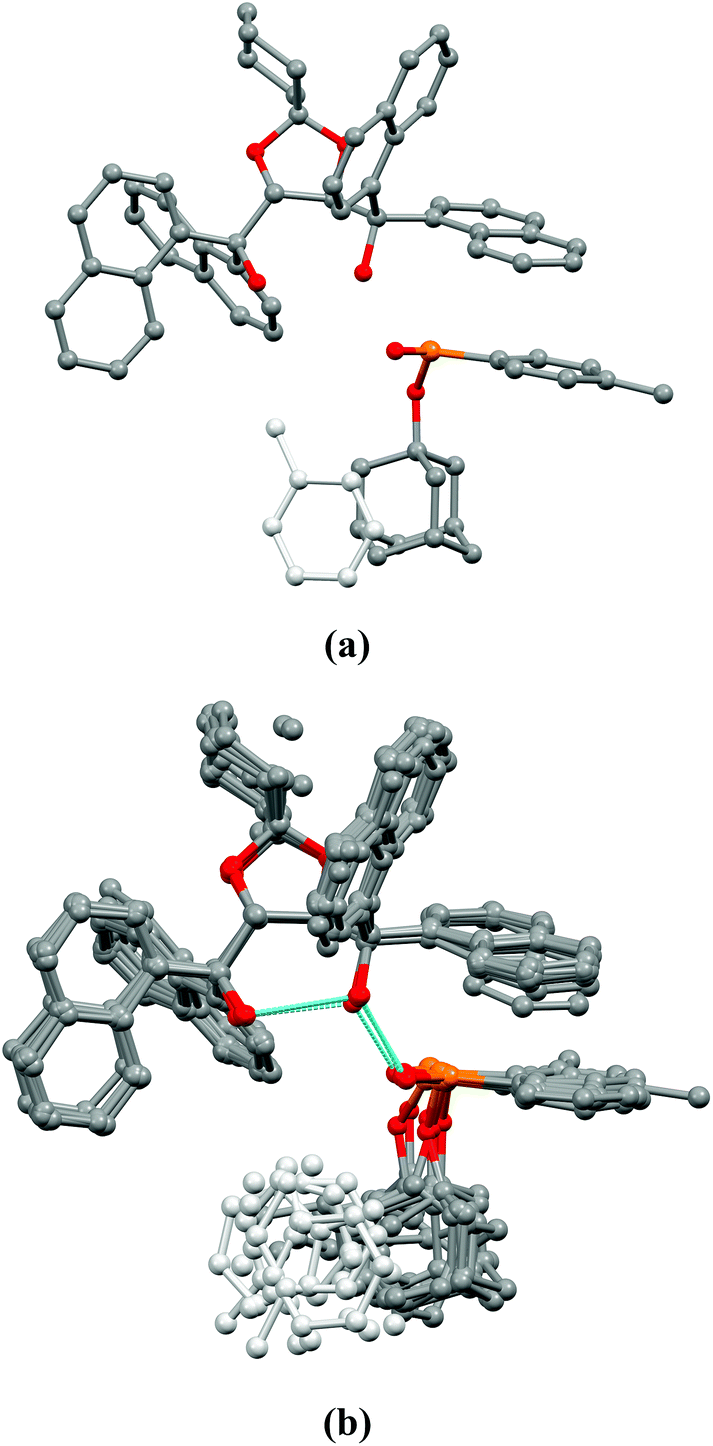
It is worth mentioning that the disordered regions of toluene and alkoxy-groups can be observed in most crystal structures. The diastereomeric complex (R)-1m·(R,R)-2e was selected to illustrate the non-bonding interactions responsible for enantiomeric recognition, and these interactions were similar regardless of the structure of the H-phosphinate (1) (vide infra). In this crystal structure, only half of the toluene solvent was refinable in the solvent cavity due to the above-mentioned disorder phenomenon. No other cavity is observed in the crystal lattices, which is indicated by a KPI packing index of 68.8, and a density of 1.270 g cm−3. (R,R)-(1-Naphthyl)-spiro-TADDOL [(R,R)-2e] and 1-adamantyl (4-methylphenyl)-H-phosphinate [(R)-1m] are associated with many weak secondary intermolecular connections, and DFT calculations were used to reveal the main contacts within the neighboring molecules (calculated using CrystalExplorer with B3LYP/6-31G(d,p); see the ESI† for details). The main H-phosphinate [(R)-1m] and (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] dimer molecules have the strongest interaction based on Coulomb interactions with a hydrogen bond system between the ((1-naphthyl)-spiro-TADDOL)O1–H1⋯((1-naphthyl)-spiro-TADDOL)O2–H2⋯O5(P(1-AdO)(4-Me-C6H4)) atoms (distances are 2.66(1) Å and 2.65(1) Å, respectively). Moreover, a parallel π⋯π interaction is also present between the 1-naphthyl ring of (R,R)-2e and the 4-methylphenyl moiety of the H-phosphinate [(R)-1m] and the corresponding π⋯π centroid distance is 3.89(1) Å (Fig. 3a). Two other (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] molecules connect to the same H-phosphinate [(R)-1m] with strong dispersion intermolecular interactions: T-shape π⋯π, PH⋯π and CH⋯π interactions (Fig. 3b). The bulky alkoxy group fills the cavities, and toluene also fits into the remaining space, which stabilizes the crystal structure. There are a few weaker interactions that can be associated with the contacts of the adamantyloxy group of the H-phosphinates [(R)-1m], the space-filling solvent and the resolving agent [(R,R)-2e].
 | ||
| Fig. 3 (a) The main interactions within the (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] and the 1-adamantyl (4-methylphenyl)-H-phosphinate [(R)-1m]. (b) DFT calculated weaker interactions of (R)-1m with the neighboring (R,R)-2e molecules. | ||
This overall filled chiral environment might be the underlying reason why the (R) enantiomer of the corresponding H-phosphinates [(R)-1a–f, (R)-1j, (R)-1k and (R)-1m] could be prepared in most of the instances. The above-mentioned interaction pattern also suggests that different aromatic substituents of the H-phosphinate (1) may hinder the tight π⋯π interactions between the host and guest molecules [1 and (R,R)-2e], which consequently leads to a destabilized crystal structure. This may explain the lower scope of this resolution method towards (substituted) aryl- and t-butyl-substituted adamantyl H-phosphinates (1n–p).
These non-bonding interactions were also characteristic of the crystal structure of the diastereomeric complexes incorporating other H-phosphinates [(R)-1a–f, (R)-1j and (R)-1k]. Fig. 4 shows the a sole (R)-1m·(R,R)-2e dimer (Fig. 4a) and its overlay with the other diastereomers incorporating the corresponding (R)-H-phosphinate enantiomers [(R)-1a–f, (R)-1j and (R)-1k] (Fig. 4b).
 | ||
| Fig. 4 (a) Asymmetric unit of the diastereomer incorporating 1-adamantyl (4-methylphenyl)-H-phosphinate [(R)-1m] and (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e]. (b) Overlaid structures of the diastereomeric complexes [(R)-1·(R,R)-2e] except the structure of [(S)-1h·(R,R)-2e]. The toluene atoms are shown in front, white-coloured. Hydrogens are omitted for clarity. | ||
The overlaid structures indicate similar positions in the crystal lattices, and thus similar spatial arrangements and main secondary intermolecular connections between the host [(R,R)-2e] and guest molecules (1), which serves as an explanation for why the range of H-phosphinates (1) could be resolved effectively by this enantioseparation method despite the structural variety of the alkoxy moiety.
In order to demonstrate the synthetic utility of (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a], a few stereoselective transformations were performed to obtain a variety of valuable P-stereogenic products (3). (R)-(1-AdO)PhP(O)H [(R)-1a] was first reacted with (2-methoxyphenyl)lithium to give (R)-(2-methoxyphenyl)-phenylphosphine oxide [(R)-3a], which indicated that the H-phosphinate (R)-1a can also be substituted with aromatic moieties in excellent yield (89%) and without significant erosion of enantiomeric purity (ee: 95%) (Scheme 5A). It is noteworthy that (2-methoxyphenyl)lithium was prepared by the direct ortholithiation of anisole using n-BuLi and TMEDA. A halogen lithium exchange by means of n-BuLi was not feasible for the preparation of the corresponding aryl-lithium,29 as the bromobutane by-product would have caused a side reaction via the formation of the corresponding tertiary phosphine oxides (vide infra).
 | ||
| Scheme 5 Stereospecific synthesis of various P-stereogenic compounds (3a–f) from (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a]. | ||
In a separate attempt, the anionic intermediate formed in the reaction of (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a] and (2-methoxyphenyl)lithium was quenched with iodomethane to give (R)-PAMP-oxide [(R)-3b] (Scheme 5B). The initial enantiomeric excess of (R)-3b (ee: 93%) was increased to an ee of 97% by the recrystallization of the corresponding enantiomeric mixture. Then, the phenyl ring was selectively saturated by heterogeneous catalytic hydrogenation to prepare (R)-CAMP-oxide [(R)-3c] (yield: 80%, ee: 96%) as another well-known P-stereogenic compound (Scheme 5C).
The (R)-(1-AdO)PhP(O)H [(R)-1a] was converted under Atherton–Todd conditions to the corresponding P-chloride which immediately reacted with NH4OH to give (R)-1-adamantyl phenylphosphonamidate [(R)-3d] (Scheme 5D). It was assumed that the sequence of these two transformations involves an inversion of the P-configuration.30 The corresponding sulfonamide [(R)-3e] was also prepared in the reaction of (R)-3d and 2,4,6-triisopropylbenzenesulfonyl chloride (Scheme 5E).
Moreover, the H-phosphinate (R)-1a was also reacted with elemental sulfur in the presence of triethylamine to prepare the corresponding thiophosphonic acid [(S)-3f] with the retention of the configuration (Scheme 5F).11b
As the last step, we sought the applications of the compounds prepared in this study. (S)-(1-Adamantyl)-phenylthiophosphonic acid [(S)-3f] seemed like a promising NMR chiral solvating agent (CSA), as it is a structural analogue of (R)-t-butyl-phenylthiophosphinic acid,31 and a few chiral phosphonothioic acids are known to behave as CSAs, as well.18d,32 The optimization of the NMR parameters was performed with 1-(1-naphthyl)ethylamine (6) as the racemate, and using 1–1.5 equiv. of (S)-3f in CDCl3 with a final concentration of 25 mM gave the best peak separation. The 31P NMR signals of the (S)-3f·6 intermediates coalesced into one peak making enantiomeric purity determination impossible. On the other hand, 1H NMR could be used to differentiate between the given signals. (S)-(1-Adamantyl)-phenylthiophosphonic acid [(S)-3f] as a CSA showed a good scope among chiral amino-alcohols (7–10) and secondary phosphine oxides (11–13) differentiating the >CH(OH), >CH(NH2) or P–H protons, respectively (Fig. 5). Among the chiral amines, 1-(1-naphthyl)ethylamine (6) was the only derivative that could be analyzed.
 | ||
| Fig. 5 Utilization of (S)-(1-adamantyl)-phenylthiophosphonic acid [(S)-3f] as a NMR chiral solvating agent. | ||
Conclusions
In this paper, the enantioseparation of P-stereogenic H-phosphinates (1) was demonstrated. Altogether 16 examples of H-phosphinates (1) with structurally different alkoxy groups or substituted aryl groups were prepared in racemic form by two methods. A resolution method was then developed, and the crystallization conditions were investigated. Under optimized conditions, (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] was found to be the most suitable resolving agent affording 12 out of 16 examples with an ee above 91%, and 9 of these derivatives were obtained in practically enantiopure form (ee > 98%). The enantioseparation method developed showed a good scope for the preparation of phenyl-H-phosphinates incorporating various alkoxy-groups (1a–j). The scalability of the resolution method was demonstrated. It was also found that crude H-phosphinates (1) can also be used as starting racemates, which does not influence the overall efficiency of the resolution method significantly. Moreover, the decomposition of the diastereomeric intermediates can also be simplified by precipitating most of the (R,R)-(1-naphthyl)-spiro-TADDOL [(R,R)-2e] as an ethanol complex. The SDE phenomenon in the case of 1-adamantyl phenyl-H-phosphinate (1a) and the thermal stability of a few selected H-phosphinates were also assessed (1a, 1b, 1d and 1j). Single crystal XRD allowed us to determine the absolute P-configurations of most of the H-phosphinates, and the most important non-bonding interactions responsible for the enantiomeric recognition. The crystal structures showed similarities, which explained well the experimental findings regarding the H-phosphinate scope (1) and the role of the aromatic solvents.A few stereoselective transformations of (R)-1-adamantyl phenyl-H-phosphinate [(R)-1a] were also elaborated. Using 2-methoxyphenyl-lithium, the nucleophilic substitution of the adamantyloxy group was facilitated to prepare (2-methoxyphenyl)-phenylphosphine oxide [(R)-3a] and PAMP-oxide [(R)-3b]. The latter derivative was also transformed into CAMP-oxide [(R)-3c]. (R)-N-(1-Adamantyl-phenylphosphonyl)-2,4,6-triisopropylbenzenesulfonamide [(R)-3e] and (S)-(1-adamantyl)-phenylthiophosphonic acid [(S)-3f] were also prepared in a stereoselective manner from H-phosphinate [(R)-1a]. (S)-(1-Adamantyl)-phenylthiophosphonic acid [(S)-3f] was applicable as an NMR solvating agent for chiral amino-alcohols (7–10) and secondary phosphine oxides (11–13).
Author contributions
Conceptualization: B. V. and P. B.; investigation: B. V., D. V., H. P., L. B., J. P., T. H., B. M., L. H., and P. B.; formal analysis: B. V., T. H., B. M., and P. B.; data curation: B. V. and P. B.; visualization: B. V. and T. H.; supervision: P. B.; resources: P. B.; validation: P. B.; funding acquisition: B. V. and P. B.; writing—original draft preparation: B. V. and T. H.; and writing—review and editing: E. F., G. K., and P. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. V. acknowledges the financial support from the New National Excellence Program of the Ministry of Human Capacities (ÚNKP-21-3-II-BME-299). T.H. is grateful for the support from the National Research, Development and Innovation Office-NKFIH (Grant No. OTKA PD 128504).Notes and references
- (a) A. Börner, Phosphorus Ligands in Asymmetric Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) A. Grabulosa, P-Stereogenic Ligands in Enantioselective Catalysis, The Royal Society of Chemistry, Cambridge, 2010 Search PubMed; (c) Phosphorus(III)Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. Van Leeuwen, John Wiley & Sons, New York, 2012 Search PubMed; (d) T. Imamoto, Searching for Practically Useful P-Chirogenic Phosphine Ligands, Chem. Rec., 2016, 16, 2655–2669 CrossRef CAS PubMed.
- (a) A. Golandaj, A. Ahmad and D. Ramjugernath, Phosphonium Salts in Asymmetric Catalysis: A Journey in a Decade's Extensive Research Work, Adv. Synth. Catal., 2017, 359, 3676–3706 CrossRef CAS; (b) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine Organocatalysis, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed; (c) H. Ni, W.-L. Chan and Y. Lu, Phosphine-Catalyzed Asymmetric Organic Reactions, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed; (d) T. Ayad, A. Gernet, J.-L. Pirat and D. Virieux, Enantioselective Reactions Catalyzed by Phosphine Oxides, Tetrahedron, 2019, 75, 4385–4418 CrossRef CAS.
- (a) U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed; (b) J. B. Rodriguez and C. Gallo-Rodriguez, The Role of the Phosphorus Atom in Drug Design, ChemMedChem, 2019, 14, 190–216 CAS.
- (a) R. T. Eastman, J. S. Roth, K. R. Brimacombe, A. Simeonov, M. Shen, S. Patnaik and M. D. Hall, Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19, ACS Cent. Sci., 2020, 6, 672–683 CrossRef CAS PubMed; (b) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Evolution of the Synthesis of Remdesivir. Classical Approaches and Most Recent Advances, ACS Omega, 2021, 6, 19356–19363 CrossRef CAS PubMed.
- (a) M. Dutartre, J. Bayardon and S. Jugé, Applications and Stereoselective Syntheses of P-Chirogenic Phosphorus Compounds, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC; (b) S. Lemouzy, L. Giordano, D. Hérault and G. Buono, Introducing Chirality at Phosphorus Atoms: An Update on the Recent Synthetic Strategies for the Preparation of Optically Pure P-Stereogenic Molecules, Eur. J. Org. Chem., 2020, 3351–3366 CrossRef CAS; (c) X. Ye, L. Peng, X. Bao, C.-H. Tan and H. Wang, Recent Developments in Highly Efficient Construction of P-Stereogenic Centers, Green Synth. Catal., 2021, 2, 6–18 CrossRef.
- D. Xu, N. Rivas-Bascón, N. M. Padial, K. W. Knouse, B. Zheng, J. C. Vantourout, M. A. Schmidt, M. D. Eastgate and P. S. Baran, Enantiodivergent Formation of C-P Bonds: Synthesis of P-Chiral Phosphines and Methylphosphonate Oligonucleotides, J. Am. Chem. Soc., 2020, 142, 5785–5792 CrossRef CAS PubMed.
- (a) O. I. Kolodiazhnyi, Asymmetric Synthesis in Organophosphorus Chemistry, Wiley-VCH Verlag, Weinheim, Germany, 2016 Search PubMed; (b) O. I. Kolodiazhnyi and A. Kolodiazhna, Nucleophilic Substitution at Phosphorus: Stereochemistry and Mechanisms, Tetrahedron: Asymmetry, 2017, 28, 1651–1674 CrossRef CAS.
- (a) A. Gallen, A. Riera, X. Verdaguer and A. Grabulosa, Coordination Chemistry and Catalysis with Secondary Phosphine Oxides, Catal. Sci. Technol., 2019, 9, 5504–5561 RSC; (b) D. S. Glueck, Asymmetric Synthesis of P-Stereogenic Secondary Phosphine -Oxides (SPOs), Synthesis, 2022, 54, 271–280 CrossRef CAS; (c) F. A. Kortmann, M.-C. Chang, E. Otten, E. P. A. Couzijn, M. Lutz and A. J. Minnaard, Consecutive dynamic resolutions of phosphine oxides, Chem. Sci., 2014, 5, 1322–1327 RSC.
- (a) J. L. Montchamp, Phosphinate Chemistry in the 21st Century: A Viable Alternative to the Use of Phosphorus Trichloride in Organophosphorus Synthesis, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed; (b) T. Chen and L.-B. Han, Optically Active H-Phosphinates and Their Stereospecific Transformations into Optically Active P-Stereogenic Organophosphoryl Compounds, Synlett, 2015, 26, 1153–1163 CrossRef CAS.
- (a) H. P. Benschop, D. H. J. M. Platenburg, F. H. Meppelder and H. L. Boter, Stereospecific Reactions of Optically Pure Menthyl Methylphosphinate, J. Chem. Soc. D, 1970, 1, 33b RSC; (b) R. Bodalski and J. Koszuk, Synthesis and Absolute Configuration of Diastereomeric Menthyl Benzylphosphinates, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 44, 99–102 CrossRef CAS.
- (a) L.-B. Han, C.-Q. Zhao, S. Onozawa, M. Goto and M. Tanaka, Retention of Configuration on the Oxidative Addition of P–H Bond to Platinum (0) Complexes: The First Straightforward Synthesis of Enantiomerically Pure P-Chiral Alkenylphosphinates via Palladium-Catalyzed Stereospecific Hydrophosphinylation of Alkynes, J. Am. Chem. Soc., 2002, 124, 3842–3843 CrossRef CAS PubMed; (b) Q. Xu, C.-Q. Zhao and L.-B. Han, Stereospecific Nucleophilic Substitution of Optically Pure H-Phosphinates: A General Way for the Preparation of Chiral P-Stereogenic Phosphine Oxides, J. Am. Chem. Soc., 2008, 130, 12648–12655 CrossRef CAS PubMed; (c) D. Gatineau, L. Giordano and G. Buono, Bulky, Optically Active P-Stereogenic Phosphine–Boranes from Pure H-Menthylphosphinates, J. Am. Chem. Soc., 2011, 133, 10728–10731 CrossRef CAS PubMed.
- (a) T. L. Emmick and R. L. Letsinger, Unsymmetrical Secondary Phosphine Oxides. Synthetic, Isotopic Exchange, and Stereochemical Studies, J. Am. Chem. Soc., 1968, 90, 3459–3465 CrossRef CAS; (b) W. B. Farnham, R. K. Murray and K. Mislow, Stereospecific Alkylation of Menthyl Phenylphosphinate, J. Am. Chem. Soc., 1970, 92, 5809–5810 CrossRef CAS.
- (a) O. Berger and J.-L. Montchamp, A General Strategy for the Synthesis of P-Stereogenic Compounds, Angew. Chem., Int. Ed., 2013, 52, 11377–11380 CrossRef CAS PubMed; (b) O. Berger and J.-L. Montchamp, General Synthesis of P-Stereogenic Compounds: The Menthyl Phosphinate Approach, Org. Biomol. Chem., 2016, 1, 7552–7562 RSC.
- (a) L. Copey, L. Jean-Gérard, B. Andrioletti and E. Framery, Synthesis of P-Stereogenic Secondary Phosphine Oxides Using α-d-Glucosamine as a Chiral Precursor, Tetrahedron Lett., 2016, 57, 543–545 CrossRef CAS; (b) Z. S. Han, H. Wu, Y. Xu, Y. Zhang, B. Qu, Z. Li, D. R. Caldwell, K. R. Fandrick, L. Zhang, F. Roschangar, J. J. Song and C. H. Senanayake, General and Stereoselective Method for the Synthesis of Sterically Congested and Structurally Diverse P -Stereogenic Secondary Phosphine Oxides, Org. Lett., 2017, 19, 1796–1799 CrossRef CAS PubMed; (c) S.-G. Li, M. Yuan, F. Topic, Z. S. Han, C. H. Senanayake and Y. S. Tsantrizos, Asymmetric Library Synthesis of P-Chiral t-Butyl-Substituted Secondary and Tertiary Phosphine Oxides, J. Org. Chem., 2019, 84, 7291–7302 CrossRef CAS PubMed.
- (a) R. Beaud, R. J. Phipps and M. J. Gaunt, Enantioselective Cu-Catalyzed Arylation of Secondary Phosphine Oxides with Diaryliodonium Salts toward the Synthesis of P-Chiral Phosphines, J. Am. Chem. Soc., 2016, 138, 13183–13186 CrossRef CAS PubMed; (b) Q. Dai, W. Li, Z. Li and J. Zhang, P-Chiral Phosphines Enabled by Palladium/Xiao-Phos-Catalyzed Asymmetric P–C Cross-Coupling of Secondary Phosphine Oxides and Aryl Bromides, J. Am. Chem. Soc., 2019, 141, 20556–20564 CrossRef CAS PubMed; (c) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, Ni-Catalyzed Asymmetric Allylation of Secondary Phosphine Oxides, J. Am. Chem. Soc., 2019, 141, 16584–16589 CrossRef CAS PubMed; (d) Q. Dai, L. Liu, Y. Qian, W. Li and J. Zhang, Construction of P–Chiral Alkenylphosphine Oxides through Highly Chemo–, Regio–, and Enantioselective Hydrophosphinylation of Alkynes, Angew. Chem., Int. Ed., 2020, 59, 20645–20650 CrossRef CAS PubMed; (e) Z.-H. Wu, A.-Q. Cheng, M. Yuan, Y.-X. Zhao, H.-L. Yang, L.-H. Wei, H.-Y. Wang, T. Wang, Z. Zhang and W.-L. Duan, Cobalt–Catalysed Asymmetric Addition and Alkylation of Secondary Phosphine Oxides for the Synthesis of P –Stereogenic Compounds, Angew. Chem., Int. Ed., 2021, 60, 27241–27246 CrossRef CAS PubMed.
- (a) H. Qiu, Q. Dai, J. He, W. Li and J. Zhang, Access to P -Chiral Sec - and Tert -Phosphine Oxides Enabled by Le-Phos-Catalyzed Asymmetric Kinetic Resolution, Chem. Sci., 2020, 11, 9983–9988 RSC; (b) Q. Dai, L. Liu and J. Zhang, Palladium/Xiao–Phos–Catalyzed Kinetic Resolution of Sec –Phosphine Oxides by P –Benzylation, Angew. Chem., Int. Ed., 2021, 60, 27247–27252 CrossRef CAS PubMed.
- (a) X. Fu, W.-T. Loh, Y. Zhang, T. Chen, T. Ma, H. Liu, J. Wang and C.-H. Tan, Chiral Guanidinium Salt Catalyzed Enantioselective Phospha-Mannich Reactions, Angew. Chem., Int. Ed., 2009, 48, 7387–7390 CrossRef CAS PubMed; (b) Z. Yang, X. Gu, L.-B. Han and J. (Joelle) Wang, Palladium-Catalyzed Asymmetric Hydrophosphorylation of Alkynes: Facile Access to P -Stereogenic Phosphinates, Chem. Sci., 2020, 11, 7451–7455 RSC; (c) Q. Zhang, X.-T. Liu, Y. Wu and Q.-W. Zhang, Ni-Catalyzed Enantioselective Allylic Alkylation of H –Phosphinates, Org. Lett., 2021, 23, 8683–8687 CrossRef CAS PubMed.
- (a) H. S. Aaron, T. M. Shryne and J. I. Miller, The Stereochemistry of Asymmetric Phosphorus Compounds. I. The Resolution of O-Ethyl Ethylphosphonothioic Acid, J. Am. Chem. Soc., 1958, 80, 107–110 CrossRef CAS; (b) H. S. Aaron, J. Braun, T. M. Shryne, H. F. Frack, G. E. Smith, R. T. Uyeda and J. I. Miller, The Stereochemistry of Asymmetric Phosphorus Compounds. III. The Resolution of a Series of O-Alkyl Alkylphosphonothioic Acids 1, J. Am. Chem. Soc., 1960, 82, 596–598 CrossRef CAS; (c) K. E. DeBruin, C. I. W. Tang, D. M. Johnson and R. L. Wilde, Kinetic Facial Selectivity in Nucleophilic Displacements at Tetracoordinate Phosphorus: Kinetics and Stereochemistry in the Reaction of Sodium Ethoxide with O,S-Dimethyl Phenylphosphonothioate, J. Am. Chem. Soc., 1989, 111, 5871–5879 CrossRef CAS; (d) K. Matsumoto, J. Sawayama, S. Hirao, N. Nishiwaki, R. Sugimoto and K. Saigo, Enantiopure O -Ethyl Phenylphosphonothioic Acid: A Solvating Agent for the Determination of Enantiomeric Excesses, Chirality, 2014, 26, 614–619 CrossRef CAS PubMed; (e) H. L. Boter and D. H. J. M. Platenburg, Organophosphorus Compounds. Part V: The Resolution of O -Alkyl Hydrogen Methylphosphonothioates with (+)- and (−)-α-Phenylethylamine, Recl. Trav. Chim. Pays-Bas, 1967, 86, 399–404 CrossRef CAS; (f) Y. Kobayashi, F. Morisawa and K. Saigo, A New Hydrogen-Bonding Motif for Chiral Recognition in the Diastereomeric Salts of Racemic 1-Phenylethylamine Derivatives with Enantiopure O -Ethyl Phenylphosphonothioic Acid, Org. Lett., 2004, 6, 4227–4230 CrossRef CAS PubMed; (g) Y. Kobayashi, F. Morisawa and K. Saigo, Enantiopure O -Substituted Phenylphosphonothioic Acids: Chiral Recognition Ability during Salt Crystallization and Chiral Recognition Mechanism, J. Org. Chem., 2006, 71, 606–615 CrossRef CAS PubMed; (h) Y. Kobayashi, J. Maeda, F. Morisawa and K. Saigo, Synthesis and Chiral Recognition Ability of O-Phenyl Ethylphosphonothioic Acid with a Conformationally Flexible Phenoxy Group for CH/π Interaction, Tetrahedron: Asymmetry, 2006, 17, 967–974 CrossRef CAS; (i) Y. Kobayashi, J. Maeda and K. Saigo, Synthesis and Chiral Recognition Ability of O-Ethyl (2-Naphthyl)Phosphonothioic Acid, Tetrahedron: Asymmetry, 2006, 17, 1617–1621 CrossRef CAS; (j) Y. Kobayashi, J. Maeda, T. Ando and K. Saigo, Halogen-Bonding Interaction Stabilizing Cluster-Type Diastereomeric Salt Crystals, Cryst. Growth Des., 2010, 10, 685–690 CrossRef CAS.
- (a) L. P. Reiff and H. S. Aaron, Stereospecific Synthesis and Reactions of Optically Active Isopropyl Methylphosphinate, J. Am. Chem. Soc., 1970, 92, 5275–5276 CrossRef CAS; (b) L. J. Szafraniec, L. L. Szafraniec and H. S. Aaron, Reaction of (R)-(+)-Isopropyl Methylphosphinate with Methyl Triflate. Stereospecific Synthesis of (R)-(+)-Isopropyl Methyl Methylphosphonite, J. Org. Chem., 1982, 47, 1936–1939 CrossRef CAS; (c) B. Krawiecka and E. Wojna-Tadeusiak, Reaction of Thiolo and Selenolo Esters of Phosphorus Acids with Halogens. Part 3. Interaction of O, O,S-Trialkyl Phosphorothioates and O,S-Dialkyl t-Butylphosphonothioates with Sulphuryl Chloride and Halogens, J. Chem. Soc., Perkin Trans. 1, 1991, 229–237 RSC; (d) A. Ferry, X. Guinchard, P. Retailleau and D. Crich, Synthesis, Characterization, and Coupling Reactions of Six-Membered Cyclic P-Chiral Ammonium Phosphonite–Boranes; Reactive H -Phosphinate Equivalents for the Stereoselective Synthesis of Glycomimetics, J. Am. Chem. Soc., 2012, 134, 12289–12301 CrossRef CAS PubMed.
- H. P. Benschop and G. R. Van den Berg, Stereospecific Inclusion in Cycloamyloses: Partial Resolution of Isopropyl Methylphosphinate and Related Compounds, J. Chem. Soc. D, 1970, 1431–1432 RSC.
- F. Toda, K. Mori, S. Zafra and I. Goldberg, Optical Resolution of Phosphinates and Phosphine Oxides by Complex Formation with Optically Active 2,2′-Dihydroxy-1,1′-Binaphthyl and Crystallographic Study of Two Diastereomeric Complexes with (CH3)(C6H5)(OCH3)PO, J. Org. Chem., 1988, 53, 308–312 CrossRef CAS.
- D. Gatineau, D. H. Nguyen, D. Hérault, N. Vanthuyne, J. Leclaire, L. Giordano and G. Buono, H-Adamantylphosphinates as Universal Precursors of P-Stereogenic Compounds, J. Org. Chem., 2015, 80, 4132–4141 CrossRef CAS PubMed.
- (a) G. Morales, W. Li and P. F. Jackson, Process for Preparing Enantiomer-Enriched Hydroxyphosphinyl Derivatives, WO1999033848A1, 1998 Search PubMed; (b) N. S. Sampson and P. A. Bartlett, Synthesis of Phosphonic Acid Derivatives by Oxidative Activation of Phosphinate Esters, J. Org. Chem., 1988, 53, 4500–4503 CrossRef CAS; (c) S. Chen and J. K. Coward, Investigations on New Strategies for the Facile Synthesis of Polyfunctionalized Phosphinates: Phosphinopeptide Analogues of Glutathionylspermidine, J. Org. Chem., 1998, 63, 502–509 CrossRef CAS PubMed; (d) I. Wilkening, G. Del Signore and C. P. R. Hackenberger, Synthesis of Phosphonamidate Peptides by Staudinger Reactions of Silylated Phosphinic Acids and Esters, Chem. Commun., 2011, 47, 349–351 RSC.
- K. R. Winters, C. Ricke and J. Montchamp, Synthesis of Adamantyl H –Phosphinate Esters, Eur. J. Org. Chem., 2022, 2022, 44–49 CrossRef.
- P. Bagi and R. Herbay, Resolution of Phosphine Oxides, in Organophosphorus Chemistry - Novel Developments, ed. G. Keglevich, De Gruyter, Berlin, 2018, pp. 66–90 Search PubMed.
- D. Seebach, A. K. Beck and A. Heckel, TADDOLs, Their Derivatives, and TADDOL Analogues: Versatile Chiral Auxiliaries, Angew. Chem., Int. Ed., 2001, 40, 92–138 CrossRef CAS PubMed.
- (a) B. Varga, R. Herbay, G. Székely, T. Holczbauer, J. Madarász, B. Mátravölgyi, E. Fogassy, G. Keglevich and P. Bagi, Scalable Enantiomeric Separation of Dialkyl-Arylphosphine Oxides Based on Host-Guest Complexation with TADDOL-Derivatives, and Their Recovery, Eur. J. Org. Chem., 2020, 1840–1852 CrossRef CAS; (b) B. Varga, P. Szemesi, P. Nagy, R. Herbay, T. Holczbauer, E. Fogassy, G. Keglevich and P. Bagi, Enantioseparation of P-Stereogenic Secondary Phosphine Oxides and Their Stereospecific Transformation to Various Tertiary Phosphine Oxides and a Thiophosphinate, J. Org. Chem., 2021, 86, 14493–14507 CrossRef CAS PubMed.
- J. Han, O. Kitagawa, A. Wzorek, K. D. Klika and V. A. Soloshonok, The Self-Disproportionation of Enantiomers (SDE): A Menace or an Opportunity?, Chem. Sci., 2018, 9, 1718–1739 RSC.
- A. Leyris, J. Bigeault, D. Nuel, L. Giordano and G. Buono, Enantioselective Synthesis of Secondary Phosphine Oxides from (RP)-(-)-Menthyl Hydrogenophenylphosphinate, Tetrahedron Lett., 2007, 48, 5247–5250 CrossRef CAS.
- B. Xiong, Y. Zhou, C. Zhao, M. Goto, S. F. Yin and L. B. Han, Systematic Study for the Stereochemistry of the Atherton-Todd Reaction, Tetrahedron, 2013, 69, 9373–9380 CrossRef CAS.
- J. Drabowicz, P. Pokora-Sobczak, D. Krasowska and Z. Czarnocki, Optically Active t-Butylphenylphosphinothioic Acid: Synthesis, Selected Structural Studies and Applications as a Chiral Solvating Agent, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 977–991 CrossRef CAS.
- K. Kuwabara, Y. Maekawa, M. Minoura and T. Murai, Hydrolysis of Phosphonothioates with a Binaphthyl Group: P -Stereogenic O -Binaphthyl Phosphonothioic Acids and Their Use as Optically Active Ligands and Chiral Discriminating Agents, Org. Lett., 2018, 20, 1375–1379 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General methods and synthetic procedures; additional experimental data; X-ray data; 31P, 19F, 1H and 13C NMR spectra; and HPLC traces. CCDC 2143598–2143607. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo00241h |
| This journal is © the Partner Organisations 2022 |