
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acid-mediated decarboxylative C–H coupling between arenes and O-allyl carbamates†
Camilla
Loro
 ab,
Julie
Oble
b,
Francesca
Foschi
a,
Marta
Papis
a,
Egle M.
Beccalli
c,
Sabrina
Giofrè
c,
Giovanni
Poli
*b and
Gianluigi
Broggini
*a
ab,
Julie
Oble
b,
Francesca
Foschi
a,
Marta
Papis
a,
Egle M.
Beccalli
c,
Sabrina
Giofrè
c,
Giovanni
Poli
*b and
Gianluigi
Broggini
*a
aDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell'Insubria, via Valleggio 9, 22100 Como, Italy. E-mail: gianluigi.broggini@uninsubria.it
bSorbonne Université, Faculté des Sciences et Ingénierie, CNRS, Institut Parisien de Chimie Moléculaire, IPCM, 4 place Jussieu, 75005 Paris, France. E-mail: giovanni.poli@sorbonne-universite.fr
cDISFARM, Sezione di Chimica Generale e Organica “A. Marchesini”, Università degli Studi di Milano, via Venezian 21, 20133 Milano, Italy
First published on 8th February 2022
Abstract
Treatment of O-allyl N-tosyl carbamates with aromatic compounds in the presence of Cu(OTf)2 or TMSOTf as promoters affords N-substituted 1-arylpropan-2-amines, 1,2-diarylpropanes, 1,1-diarylpropanes, or indanes, depending on the nature of the promoter and of the aryl substrates. A full mechanistic rational allowing appreciation of the outcome of these novel C–H based cascades is proposed. An initial acid promoted decarboxylative/deamidative Friedel–Crafts allylation takes place. After protonation of the allylated arene, evolution of the resulting cation may follow different paths depending on the nature of the arene partner and of the allyl moiety in the carbamate.
Introduction
In the last century, carbocation-mediated or -catalysed reactions have revolutionised the history of organic chemistry, giving rise to a set of fundamental reactions such as C–C bond formations, eliminations, and rearrangements.1 In particular, the Friedel–Crafts (FC) alkylation is one of the most powerful organic reactions that allows the selective C–H functionalization of aromatics.2 Although the FC alkylation has long been studied in many variations, its implementation in cascade reactivities that allow the multiple functionalizations of unsaturated substrates represents an extraordinary way to discover still unexplored reactivity patterns.3In the frame of our long-term study dedicated to carbamates as precursors for metal-catalysed cyclisations,4 we investigated in particular the behaviour of O-allyl N-tosyl carbamates. These compounds have been shown to undergo exo-5 or endo-trig6 cyclisations involving further functionalisation of the carbon double bond. Moreover, allyl carbamates can also allow decarboxylative O → N allylic rearrangement affording selectively anti-Markovnikov hydroamination products.7 In this work, we show that O-allyl carbamates behave as C3 1,2-, 1,1-, and 1,3-dication equivalents, allowing the generation of 1-arylpropan-2-amines, 1,2- and 1,1-diarylpropanes, as well as indane structures, in the presence of Cu(OTf)2 or TMSOTf as acid-promoters (Scheme 1).
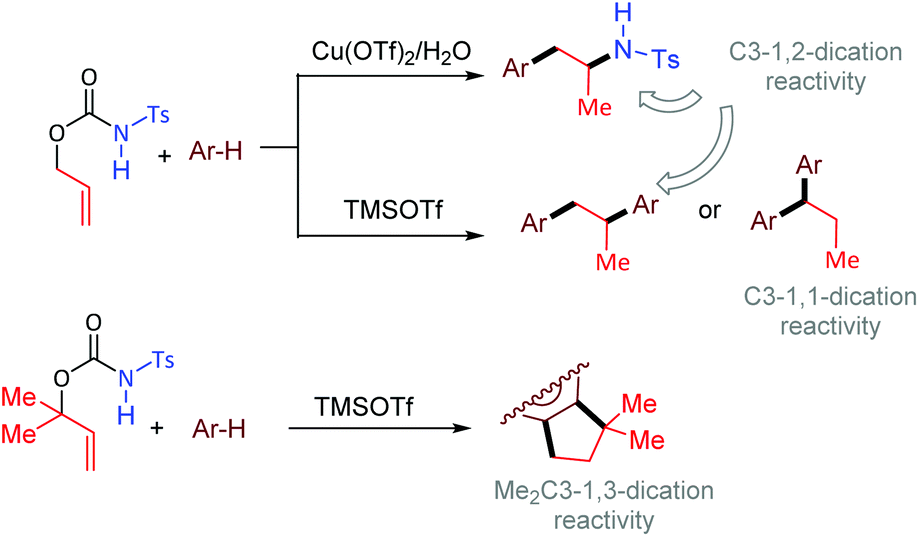 | ||
| Scheme 1 Different reactivities in the acid promoted coupling between arenes and O-allyl N-tosyl carbamates observed in the present study (bold bonds refer to forming bonds). | ||
Results and discussion
We started our study reacting carbamate 1a in mesitylene, as aromatic reaction partner and solvent, in the presence of copper(II) triflate as the promoter (Table 1, entry 1). After 6 hours at 100 °C, the reaction provided a mixture of the arylated N-tosylamide 2 (36% isolated), together with a big amount of TsNH2 (4) arising from the degradation of 1a. Unchanged starting material was found working at lower temperature, and using a catalytic amount of Cu(OTf)2 (entries 2 and 3). Although using an excess of copper salt at 130 °C led to an increased yield of 2, the formation of 4 could not be avoided (entry 4). Carrying out the reaction in chlorobenzene in the presence of mesitylene (5.0 equiv.) and H2O (96/4 v/v) allowed to obtain 2 and 4 in 77% and 22% yields, respectively (entry 5). Although this protocol does not represent an improvement in terms of yields, it shows that it is possible to work in the presence of a solvent other than the aromatic reaction partner itself.|
|
||||
|---|---|---|---|---|
| Entry | Promoter (equiv.) | Temp. (°C) | Time (h) | Product(s) (% yield)b |
| a Reaction conditions: 1a (1.0 equiv.), mesitylene (0.25 M), oil bath as heat source. b Isolated yields. c Chlorobenzene/H2O as solvent (96/4 v/v) (0.4 M) with mesitylene (5.0 equiv.). d MW irradiation at 300 W. e Chlorobenzene as the solvent (0.25 M) with mesitylene (5.0 equiv.). f DMF as the solvent (0.25 M) with mesitylene (5.0 equiv.). g DCE (0.25 M) with mesitylene (5.0 equiv.). | ||||
| 1 | Cu(OTf)2 (1.0) | 100 | 6.0 | 2 (36) + 4 (43) |
| 2 | Cu(OTf)2 (1.0) | 50 | 24 | S.M. |
| 3 | Cu(OTf)2 (0.1) | 100 | 24 | S.M. |
| 4 | Cu(OTf)2 (4.0) | 130 | 3.0 | 2 (85) + 4 (11) |
| 5 | Cu(OTf)2 (4.0)c | 130 | 4.0 | 2 (77) + 4 (22) |
| 6 | PTSA (4.0) | 130 d | 1.5 | 2 (59) + 4 (18) |
| 7 | H2SO4 (1.0) | 130 | 4.0 | 2 (25) + 4 (14) |
| 8 | AgOTf (4.0) | 130 | 1.5 | 2 (25) + 4 (31) |
| 9 | TfOH (0.05) | 130 | 4.0 | 2 (69) + 4 (26) |
| 10 | TfOH (0.05)e | 130 | 4.0 | 2 (58) + 4 (41) |
| 11 | TfOH (0.05)f | 130 | 4.0 | degr. products |
| 12 | BF3·Et2O (1.0) | 100 d | 0.5 | 2 (21) + 3 (63) |
| 13 | BF3·Et2O (4.0) | 130d | 1.5 | 2 (15) + 3 (72) |
| 14 | TMSOTf (0.05) | 130 | 3.0 | 2 (23) + 4 (41) |
| 15 | TMSOTf (4.0) | 130 | 3.0 | 3 (89) |
| 16 | TMSOTf (4.0)g | 80 | 4.0 | 3 (79) |
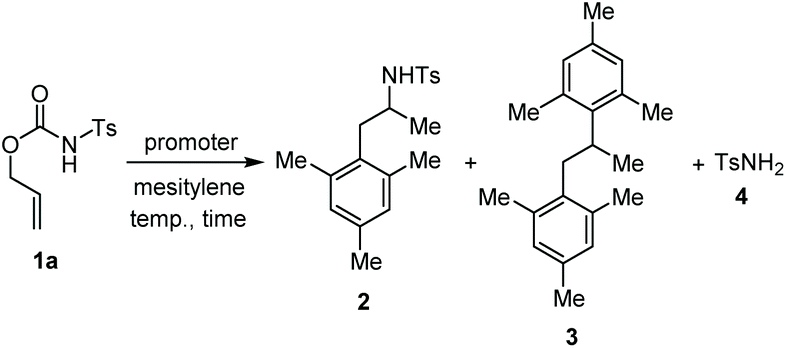
Different promoters were then tested, using either conventional heating or microwave irradiation. However, p-toluenesulfonic acid, H2SO4 and silver(I) triflate behaved analogously to Cu(OTf)2 (Table 1, entries 6–8). Assuming that the above described reaction conditions involved the in situ generation of TfOH, we also tested a catalytic amount of this acid in different solvents (entries 9–11). Working in mesitylene or in chlorobenzene with 5.0 equivalents of mesitylene, the recovery of TsNH2 was still high, whereas the use of DMF gave only intractable degradation products. The use of F3B·OEt2 disclosed a new reactivity involving the double arylation to the allyl moiety. Indeed, treatment of 1a with a stoichiometric or an excess amount of F3B·OEt2 under microwave irradiation furnished a mixture of 2 and the 1,2-diarylpropane 3 (entries 12 and 13). Switch to TMSOTf as the acid promoter was then considered. While the use of a catalytic amount of this promoter afforded a mixture of 2 and 4 (entry 14), an excess amount of it (4.0 equiv.) gladly brought about the selective formation of 3 in 89% yield (entry 15). A similar result was also obtained using DCE as solvent (entry 16).
Among the range of branched amines, N-substituted phenethylamine derivatives have been widely studied in recent years for their value in organic, bioorganic, and medicinal chemistry.8 We thus set out to test the decarboxylative arylation/hydroamination described above with other aromatic hydrocarbons (Table 2).
|
|
||
|---|---|---|
| Entry | Aryl hydrocarbon | Product(s) (% yield)b |
| a Reaction conditions: 1a (1.0 equiv.), Cu(OTf)2 (4.0 equiv.), aryl hydrocarbon (5.0 equiv.), chlorobenzene/H2O as solvent (96/4 v/v) (0.4 M), 130 °C, 4 h. b Isolated yields. Isomeric ratios calculated from the 1H-NMR of the crude reaction mixture. c Scale up: performing the reaction with 5 mmol of 1a at 130 °C, after 6 h 5 was obtained in 71% yield. | ||
| 1c | p-Xylene |
|
| 2 | o-Xylene |

|
| 3 | m-Xylene |

|
| 4 | Toluene |
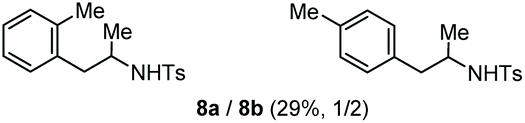
|
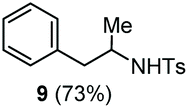
| 5 | Benzene |
|
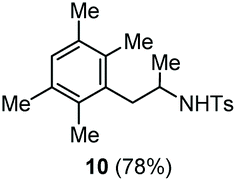
| 6 | 1,2,4,5-Tetramethylbenzene |
|
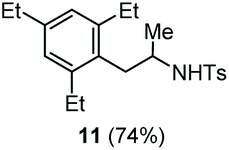
| 7 | 1,3,5-Triethylbenzene |
|
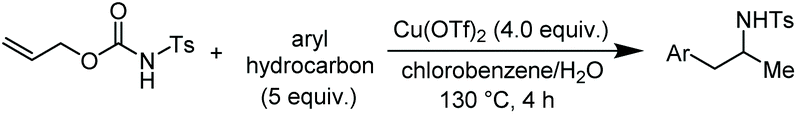
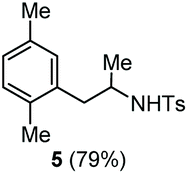
Accordingly, reacting allyl carbamate 1a in p-xylene under the conditions of Table 1, entry 5 [Cu(OTf)2 (4 equiv.), chlorobenzene9/H2O (96/4 v/v), 130 °C, 4.0 h] afforded the corresponding arylated 2-tosylaminopropane 5 as the sole product in good yield (Table 2, entry 1). The reaction carried out in o-xylene, m-xylene, or toluene gave the corresponding arylation/hydroamination products as mixtures of two regioisomers 6a/6b, 7a/7b, and 8a/8b (entries 2–4). The less electron-rich benzene also provided the expected arylation/hydroamination product 910 in acceptable yield (entry 5). Finally, the heavily alkylated arenes durene and 1,3,5-triethylbenzene gave the corresponding cascade products 10 and 11 in good yields (entries 6 and 7).
We propose for the above double coupling reactions the following mechanism, shown in the case of carbamate 1a (Scheme 2). First, we assume that triflic acid is formed in situ from Cu(OTf)2 (or TMSOTf) and water.11 Following protonation at the carbonyl oxygen atom of the carbamate function generates the activated O-allyl carbamate I, which undergoes a decarboxylative/deamidative FC alkylation by attack of arene to give intermediate III passing through an allyl carbenium ion II. Subsequent Markovnikov protonation of III generates the allyl carbenium ion IV. At this point, the nature of the additive directs the reaction path. In the presence of copper triflate, the extruded N-tosylamide can coordinate the metal, generating the amido copper-complex V, which selectively attacks the carbenium ion IV, affording the hydroamination product 2.12 Alternatively, when TMSOTf is used, intermediate IV undergoes a second FC allylation, providing the 1,2-diarylpropane 3. Finally, in the presence of excess triflic acid product 2 suffers a deamidative substitution by mesitylene via an incipient or discrete carbenium ion, to afford the diarylated product 3.
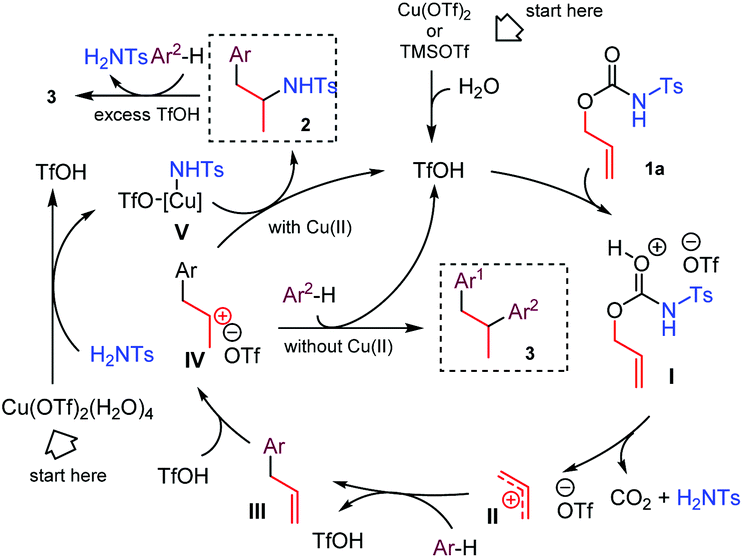 | ||
| Scheme 2 Proposed mechanisms for the acid-mediated decarboxylative C–H coupling between arenes and O-allyl carbamates. | ||
Corollary experiments using the O-allyl N-4-chlorophenyl carbamate 1b in place of 1a were carried out next (Scheme 3). Running the reaction in mesitylene/H2O (98/2 v/v) in the presence of Cu(OTf)2 as the promoter gave the diarylated product 3, and not the corresponding aniline derivative. However, carrying out the same reaction in the presence of exogenous H2NTs (2.0 equiv.) afforded a mixture of the arylated/hydroaminated derivative 2 and the diarylated derivative 3. These results corroborate the above proposed mechanism and show that tosylamine is a competent nitrogen nucleophile to trap the carbenium ion IV when Cu(OTf)2 is used as the promoter.
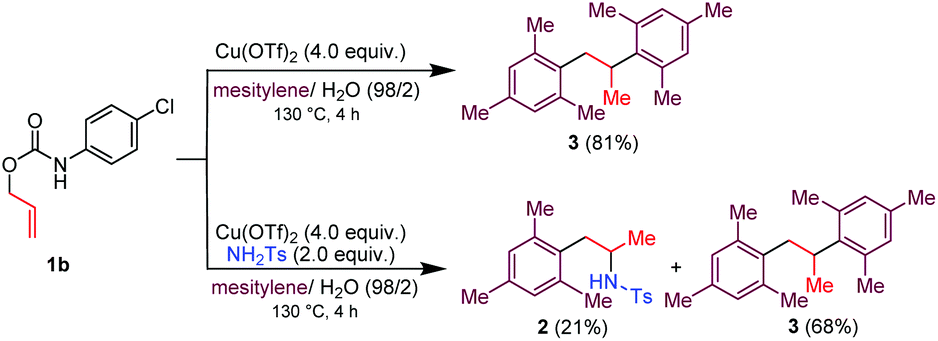 | ||
| Scheme 3 Cu(OTf)2/H2O mediated decarboxylative C–H couplings between mesitylene and O-allyl N-4-chlorophenyl carbamate. Reaction condition involves the use of 100 μL of H2O for 1 mmol of 1b. | ||
The scope of the reaction was evaluated next, keeping the promoting system Cu(OTf)2/H2O in chlorobenzene (Table 3). On the one hand, reacting O-allyl N-2-nosylcarbamate 1c with 5 equiv. of mesitylene (entry 1), and carbamate 1a with mesityl bromide (entry 2), 4-methylanisole (entry 3), 4-bromoanisole (entry 4) gave the corresponding hydroaminated products 12–15 in fair to good yields. On the other hand, strongly activated arenes such as 1,3,5-trimethoxybenzene gave the diarylated product 16 as the only product (entry 5).
|
|
|||
|---|---|---|---|
| Entry | Arene | PG | Product(s) (% yield)b |
| a Reaction conditions: N-protected O-allyl carbamate (1.0 equiv.), Cu(OTf)2 (4.0 equiv.), arene (5.0 equiv.), chlorobenzene/H2O as solvent (96/4 v/v) (0.4 M), 130 °C, 4 h. b Isolated yields. c The reaction was carried out in mesitylene as solvent (0.25 M). | |||
| 1c | Mesitylene | o-Ns |
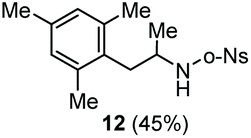
|
| 2 | Mesitylbromide | Ts |
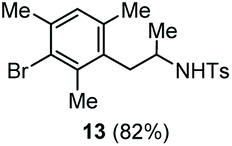
|
| 3 | 4-Methylanisole | Ts |
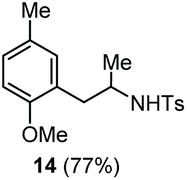
|
| 4 | 4-Bromoanisole | Ts |
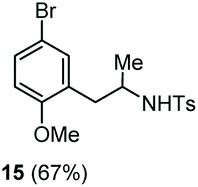
|
| 5 | 1,3,5-Trimethoxybenzene | Ts |
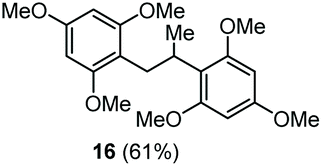
|
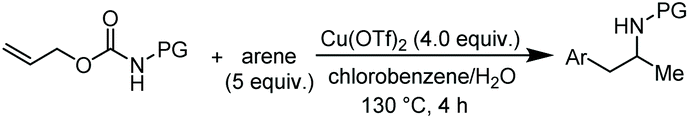
To have a better knowledge of the behaviour of this C–H cascade as a function of the adopted reaction protocol and the nature of the engaged reaction partners, other tests were undertaken. Accordingly, the C–H coupling between variously substituted N-tosyl carbamates in the presence of the Cu(OTf)2/H2O system and 5.0 equivalents of arene was next tested (Scheme 4). In the event, each isomeric carbamate 1d–f converged toward the same corresponding arylation/hydroamination product, namely 17 when reacted with mesitylene and 18 when reacted with p-xylene.
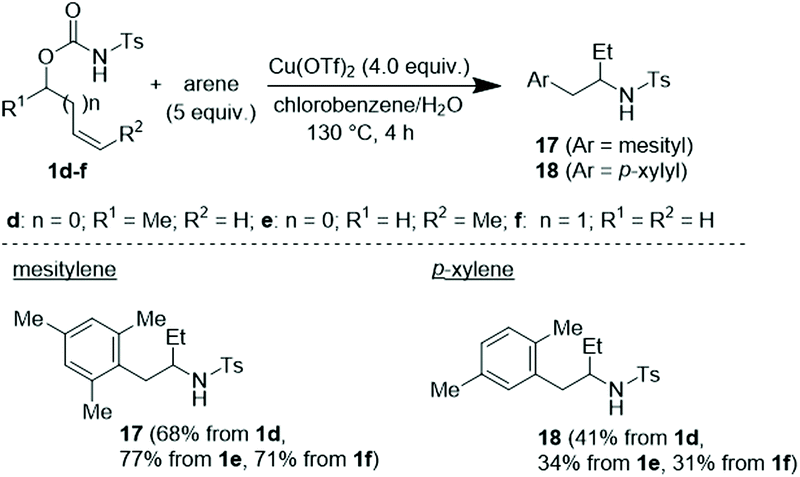 | ||
| Scheme 4 Reaction with Cu(OTf)2 with substituents on the allylic chain of the N-tosyl carbamates. | ||
Such a reactivity suggests the convergence of 1d, 1e and 1f toward the common allylic carbenium ion VI, which, after the FC reaction, undergoes a regioselective Cu-promoted hydroamination (Scheme 5).
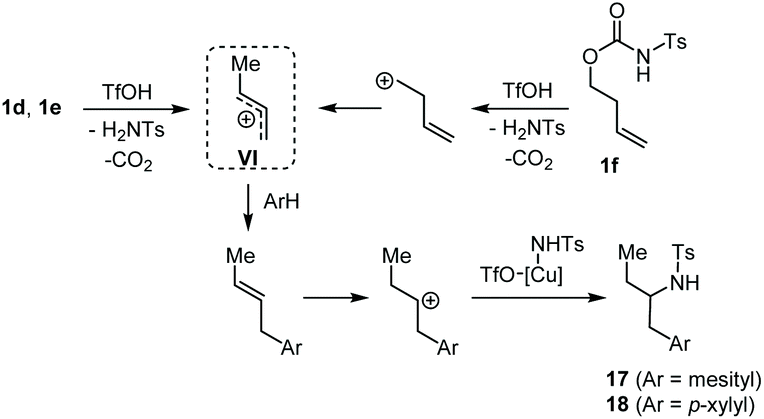 | ||
| Scheme 5 Proposed mechanism for the Cu(OTf)2 promoted transformation of 1d–f into 17 and 18. | ||
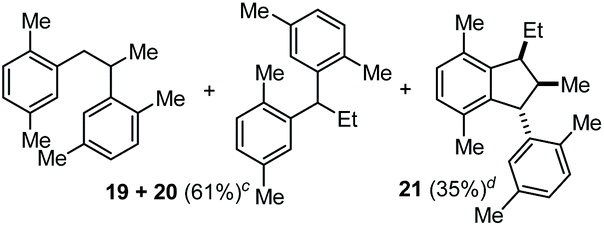
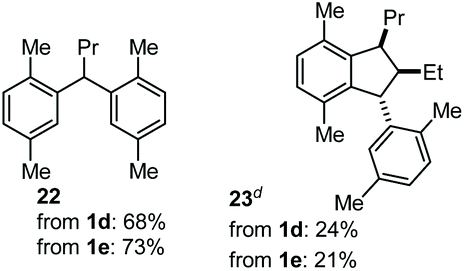
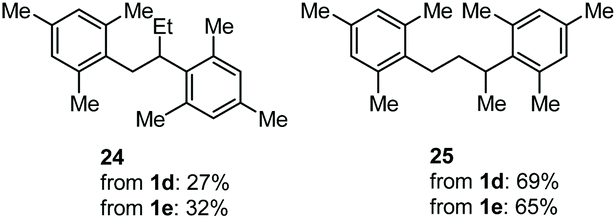
The coupling of N-tosyl carbamates 1a, and 1d–e was also tested using TMSOTf as acid promoter in DCE at 80 °C (Table 4). In this case, reaction of 1a with p-xylene gave an inseparable mixture made of the 1,2-diarylated product 19, the 1,1-diarylated product 20, and indane 21 (entry 1). Carbamates 1d and 1e converged again toward a mixture of the 1,1-diarylated product 22 and the indane 23 when reacted with p-xylene (entry 2), and toward a mixture of the 1,2-diarylated product 24 and 1,3-diarylated product 25 when reacted with mesitylene (entry 3).
|
|
|||
|---|---|---|---|
| Entryd | 1 | Arene | Product(s) (% yield)b |
| a Reaction conditions: 1a (1.0 equiv.), TMSOTf (4.0 equiv.), arene (5.0 equiv.), DCE (0.4 M), 80 °C, 4 h. b Isolated yields. Isomeric ratios calculated from the 1H-NMR of the crude reaction mixture. c 19 + 20 isomeric ratio: 1/1. d The relative configuration of indane structures 21 and 23 was determined by NOESY experiment. | |||
| 1c | 1a | p-Xylene |
|
| 2 | 1d, 1e | p-Xylene |
|
| 3 | 1d, 1e | Mesitylene |
|
The reactivity observed with the above experiments deserves further remarks. First, the formation of 19, 20, and 21 (Table 4, entry 1) can be rationalized as follows (Scheme 6, top part). After the first FC allylation of p-xylene, Markovnikov protonation and interception of the resulting carbenium ion VII by a second molecule of p-xylene generates 19. However, p-xylene – less nucleophilic than mesitylene – allows a competitive VII-to-VIII 1,2-hydride shift that takes place before the second FC reaction, generating 20. At this point, the benzylic carbocation VIII and the styrene IX could react together through a formal [3 + 2] cycloaddition giving the Wheland intermediate X which can evolve into 21.13
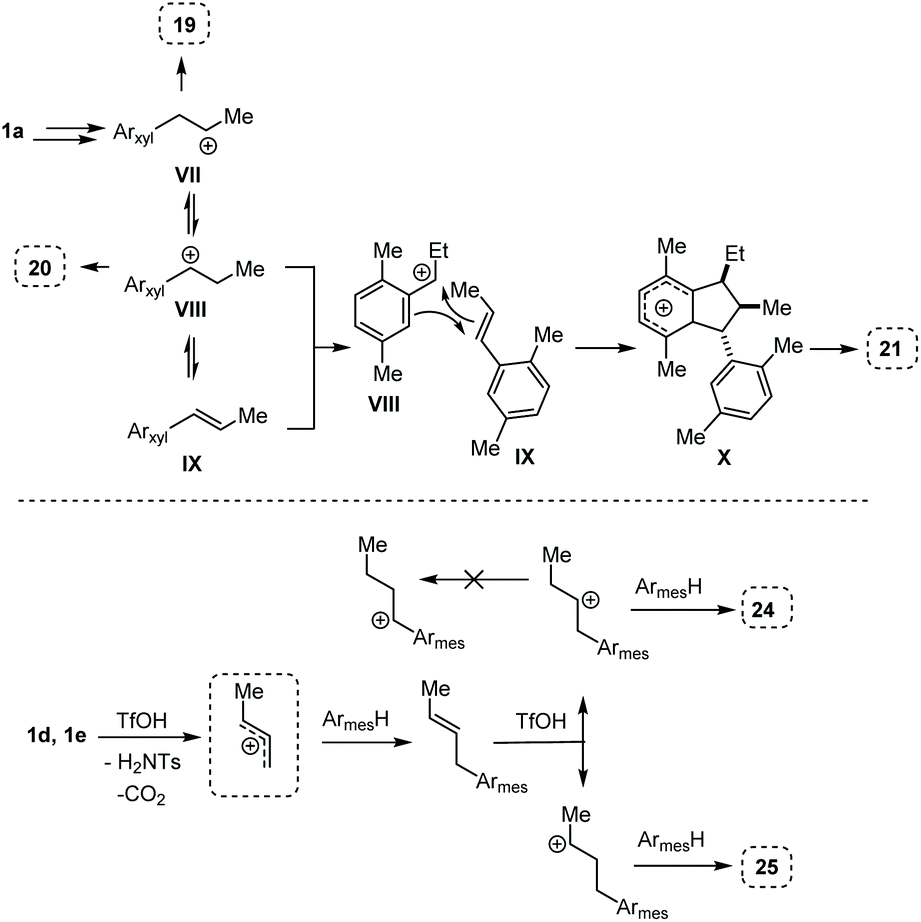 | ||
| Scheme 6 Proposed mechanisms associated to the reactions of Table 5. Top part: entry 1, from 1a to 19, 20, and 21. Bottom part: entry 3, from 1d or 1e to 24 and 25. | ||
The outcome of entry 3 in Table 4 needs further observations, too. Indeed, in this case, the protonation after the first FC reaction is non-regioselective, and the very nucleophilic mesitylene intercepts the two resulting carbenium ions to give a mixture of 24 and 25. In this case, the very reactive arene does not let the time for a homobenzylic-to-benzylic cation 1,2-hydride shift (Scheme 6, bottom part).
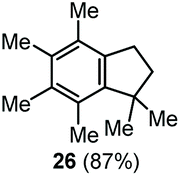
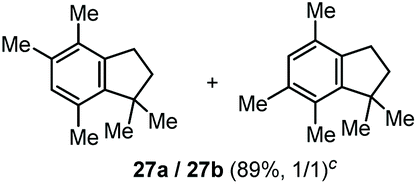
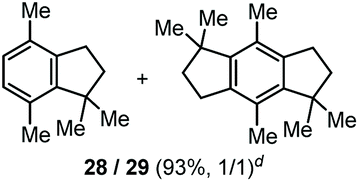
The α,α-dimethyl substituted O-allyl N-tosyl carbamate 1g was investigated next (Table 5). Thus, treatment of this carbamate with durene under the usual TMSOTf conditions gave indane 26 in 87% yield (entry 1). Analogous treatment with mesitylene afforded an inseparable mixture of the two isomeric indanes 27a and 27b in 89% yield (entry 2), while reaction with p-xylene gave an inseparable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the indane 28 and the hydrindacene 29 in 93% yield (entry 3).
:1 mixture of the indane 28 and the hydrindacene 29 in 93% yield (entry 3).
|
|
||
|---|---|---|
| Entry | Arene | Product(s) (% yield)b |
|
a Reaction conditions: 1a (1.0 equiv.), TMSOTf (4.0 equiv.), arene (5.0 equiv.), DCE (0.4 M), 80 °C, 4 h.
b Isolated yields. Isomeric ratios calculated from the 1H-NMR of the crude reaction mixture.
c 1:1 isomeric ratio.
d
28 + 29 in 1:1 ratio.
|
||
| 1 | Durene |
|
| 2c | Mesitylene |
|
| 3d | p-Xylene |
|
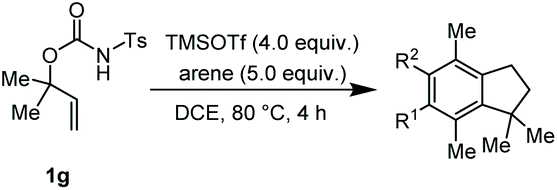
Here, in contrast to the previous cases, a one-to-one coupling between the carbamate and the arene takes place, generating indane structures. This is likely due to the fact that after the first FC allylation on the less substituted allyl terminus XI,14 the aromatic ring is favourably biased to undergo a second, intramolecular, FC reaction. It should be noted that in the cases of mesitylene and durene, 1,2-methyl migrations on the aryl moiety15 take place – before or after the first FC reaction – opening the way to the final intramolecular FC reaction (Scheme 7). However, a second carbocation species could attack the highly reactive indane 28 providing the hydrindacene 29.16
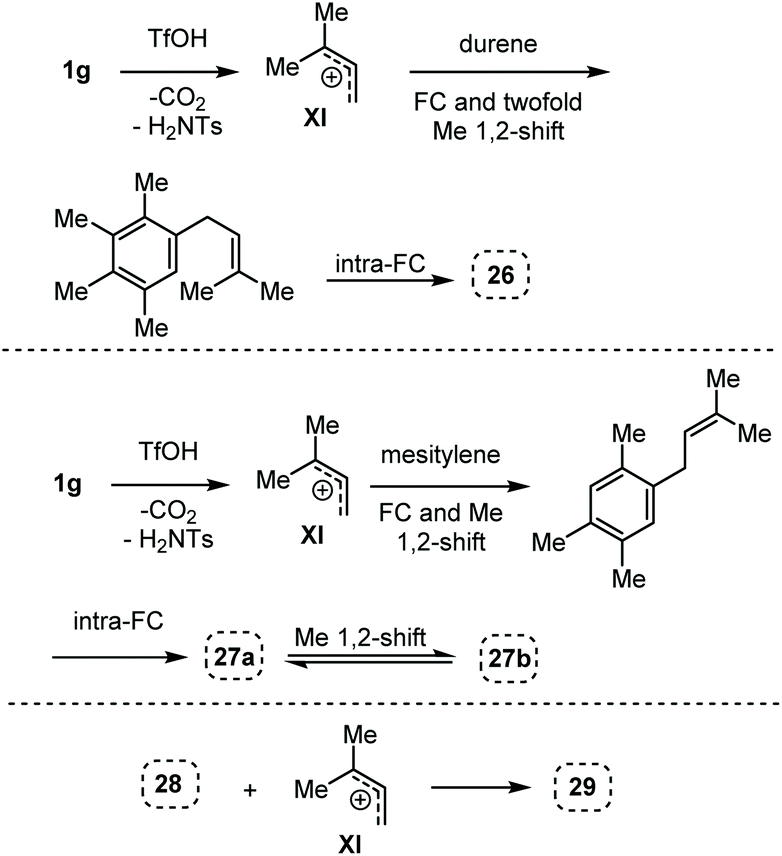 | ||
| Scheme 7 Proposed mechanisms for the generation of the indane structures 26, 27a and 27b and of hydrindacene 29 from carbamate 1g. | ||
Conclusions
In summary, this work shows for the first time that O-allyl carbamates are ideal C3 dication equivalents. Acidic conditions expected to generate in situ TfOH enable decarboxylative FC/hydromidation sequences or twofold FC alkylations, most of which proceed in synthetically useful yields. According to the nature of the arene partner and of the allyl moiety in the carbamate, the mechanisms can take different paths, all of them mechanistically justified. These results offer the chemist a rich palette of synthetic opportunities, further enriching the domains of cascade reactions in general, and FC, and hydroaminations in particular.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Università degli Studi dell'Insubria is gratefully acknowledge for financial support by CL, FF, MP and GB. GP and JO acknowledge support by Sorbonne Université and CNRS. EMB and SG thank Università degli Studi di Milano for financial support.Notes and references
- (a) G. A. Olah, in 100 Years of Carbocations and Their Significance in Chemistry, ed. G. A. Olah and G. K. Surya Prakash, John Wiley and Sons, 2004, pp. 7–41 Search PubMed; (b) R. R. Naredla and D. K. Klumpp, Contemporary carbocation chemistry: application in organic synthesis, Chem. Rev., 2013, 113, 6905–6948 CrossRef CAS PubMed; (c) S. Y. Hong, D. Kim and S. Chang, Catalytic access to carbocation intermediates via nitrenoid transfer leading to allylic lactams, Nat. Catal., 2021, 4, 79–88 CrossRef CAS; (d) G. A. Olah, in Friedel–Crafts Chemistry, Wiley, New York, 1973 Search PubMed.
- (a) M. Bandini and A. Umani-Ronchi, Catalytic Asymmetric Friedel-Crafts Alkylations, Wiley-VCH, Weinheim, 2009 CrossRef; (b) T. B. Poulsen and K. A. Jørgensen, Catalytic asymmetric Friedel-Crafts alkylation reactions-copper showed the way, Chem. Rev., 2008, 108, 2903–2915 CrossRef CAS PubMed; (c) S.-L. You, Q. Cai and M. Zeng, Chiral Brønsted acid catalyzed Friedel-Crafts alkylation reactions, Chem. Soc. Rev., 2009, 38, 2190–2201 RSC; (d) M. Davoust, J. A. Kitching, M. J. Fleming and M. Lautens, Diasteroselective benzylic arylation of tetralins, Chem. – Eur. J., 2010, 16, 50–54 CrossRef CAS PubMed; (e) D. Eom, S. Park, Y. Park, T. Ryu and P. H. Lee, Synthesis of indenes via Brønsted acid catalyzed cyclization of diaryl- and alkyl aryl-1,3-dienes, Org. Lett., 2012, 14, 5392–5395 CrossRef CAS PubMed; (f) C. Zhai, D. Xing, C. Jing, J. Zhou, C. Wang, D. Wang and W. Hu, Facile synthesis of 3-aryloxindoles via Brønsted acid catalyzed Friedel-Crafts alkylation of electron-rich arenes with 3-diazooxindoles, Org. Lett., 2014, 16, 2934–2937 CrossRef CAS PubMed; (g) M. M. Heravi, V. Zadsirjan, P. Saedi and T. Momeni, Applications of Friedel-Crafts reactions in total synthesis of natural products, RSC Adv., 2018, 8, 40061–40163 RSC; (h) K. Sakaguchi, S. Kubota, W. Akagi, N. Ikeda, M. Higashino, S. Ariyoshi, T. Shinada, Y. Ohfune and T. Nishimura, Acid-catalyzed chirality-transferring intramolecular Friedel-Crafts cyclization of α-hydroxy-α-alkenylsilanes, Chem. Commun., 2019, 55, 8635–8638 RSC; (i) C.-J. Wang, Q.-Q. Yang, M.-X. Wang, Y.-H. Shang, X.-Y. Tong, Y.-H. Deng and Z. Shao, Catalytic asymmetric 1,4-type Friedel-Crafts (hetero)arylations of 1-azadienes: the highly enantioselective syntheses of chiral hetero-triarylmethanes, Org. Chem. Front., 2020, 7, 609–616 RSC; (j) H. Zheng, K. Dong, D. Wherritt, H. Arman and M. P. Doyle, Brønsted acid catalyzed Friedel-Crafts-type coupling and dedinitrogenation reactions of vinyldiazo compounds, Angew. Chem., Int. Ed., 2020, 59, 13613–13617 ( Angew. Chem. , 2020 , 132 , 13715–13719 ) CrossRef CAS PubMed.
- For a recent paper on C3 synthons, see: (a) M. Wienhold, J. J. Molloy, C. G. Daniliuc and R. Gilmour, Coumarins by direct annulation: β-borylacrylates as ambiphilic C3-synthons, Angew. Chem., Int. Ed., 2021, 60, 685–689 ( Angew. Chem. , 2021 , 133 , 695–699 ) CrossRef CAS PubMed; (b) S.-W. Xu, X.-W. Liu, X. Zuo, G. Zhou, Y. Gong, X.-L. Liu and Y. Zhou, Oxindole-chromones C3 synthons directed stereocontrolled construction of five contiguous stereocenters on spiro[tetrahydrocyclopenta[b]chromanone-oxidole]s, Adv. Synth. Catal., 2019, 361, 5328–5333 CrossRef CAS; (c) J. Chen, P. Jia and Y. Huang, Divergent domino reactions of sulfur ylides: access to functionalized six- and seven-membered nitrogen-heterocycles, Org. Lett., 2018, 20, 6715–6718 CrossRef CAS PubMed; (d) Z.-L. Jia, X.-T. An, Y.-H. Deng, H.-B. Wang, K. J. Gan, J. Zhang, X.-H. Zhao and C.-A. Fan, Palladium-catalyzed asymmetric (2 + 3) annulation of p-quinone methides with trimethylenemethanes: enantioselective synthesis of functionalized chiral spirocyclopentyl p-dieneones, Org. Lett., 2020, 22, 4171–4175 CrossRef CAS PubMed; (e) J.-X. Zou, Y. Jiang, S. Lei, G.-F. Yin, X.-L. Hu, Q.-Y. Zhao and Z. Wang, Synthesis of α-arylthioacetones using TEMPO as the C3 synthon via a reaction cascade of sequential oxidation, skeletal rearrangement and C-S bond formation, Org. Biomol. Chem., 2019, 17, 2341–2345 RSC; (f) F. Zhong, X. Han, Y. Wang and Y. Lu, Highly enantioselective [3 + 2] annulation of Morita-Baylis-Hillman adducts mediated by l-threonine-derived phosphines: synthesis of 3-spirocyclopentene-2-oxindoles having two contiguous quaternary centers, Angew. Chem., Int. Ed., 2011, 50, 7837–7841 ( Angew. Chem. , 2011 , 123 , 7983–7987 ) CrossRef CAS PubMed; (g) X. Zhou, Z. Qi, S. Yu, L. Kong, Y. Li, W. F. Tian and X. Li, Synthesis of 2-substituted quinolines via rhodium(III)-catalyzed C-H activation of imidamides and coupling with cyclopropanols, Adv. Synth. Catal., 2017, 359, 1620–1625 CrossRef CAS.
- (a) S. Giofrè, C. Loro, L. Molteni, C. Castellano, A. Contini, D. Nava, G. Broggini and E. M. Beccalli, Copper(II)-catalyzed aminohalogenation of alkynyl carbamates, Eur. J. Org. Chem., 2021, 1750–1757 CrossRef; (b) F. Foschi, C. Loro, R. Sala, J. Oble, L. Lo Presti, E. M. Beccalli, G. Poli and G. Broggini, Intramolecular aminoazidation of unactivated terminal alkenes by palladium-catalyzed reactions with hydrogen peroxide as the oxidant, Org. Lett., 2020, 22, 1402–1406 CrossRef CAS PubMed; (c) S. Giofrè, R. Sala, E. M. Beccalli, L. Lo Presti and G. Broggini, Iodoamination of alkenyl sulfonamides by potassium iodide and hydrogen peroxide in aqueous medium, Helv. Chim. Acta, 2019, 102, e1900088 CrossRef; (d) T. Borelli, S. Brenna, G. Broggini, J. Oble and G. Poli, (Diacyloxyiodo)benzenes-driven palladium-catalyzed cyclizations of unsatureted N-sulfonylamides: opportunities of path selection, Adv. Synth. Catal., 2017, 359, 623–628 CrossRef CAS; (e) F. J. S. Duarte, G. Poli and M. J. Calhorda, Mechanistic study of the direct intramolecular allylic amination reaction catalyzed by palladium(II), ACS Catal., 2016, 6, 1772–1784 CrossRef CAS; (f) J. Rajabi, M. M. Lorion, V. Linh Ly, F. Liron, J. Oble, G. Prestat and G. Poli, Dominant versus evolving aminopalladated intermediates: toward a unified mechanistic scenarion in PdII-catalyzed amination, Chem. – Eur. J., 2014, 20, 1539–1546 CrossRef CAS PubMed.
- (a) B. P. Spadafora, F. W. M. Riberio, J. E. Matsushima, E. M. Ariga, I. Omari, P. M. A. Soares, D. de Oliveira-Silva, E. Vinhato, J. S. McIndoe, T. C. Correra and A. Rodrigues, Regio- and diastereoselective Pd-catalyzed aminochlorocyclization of allylic carbamates: scope, derivatization, and mechanism, Org. Biomol. Chem., 2021, 19, 5595–5606 RSC; (b) S. T. Nguyen, Q. Zhu and R. R. Knowles, PCET-Enabled olefin hydroamidation reactions with N-alkyl amides, ACS Catal., 2019, 9, 4502–4507 CrossRef CAS PubMed; (c) H. Yang, G.-T. Fan, L. Zhou and J. Chen, Enantioselective chloro-O-cyclization of unsaturated N-tosylcarbamates, Adv. Synth. Catal., 2017, 359, 1295–1300 CrossRef CAS; (d) C.-L. Zhu, J.-S. Tian, Z.-Y. Gu, G.-W. Xing and H. Xu, Iron(II)-catalyzed asymmetric intramolecular olefin aminochlorination using chloride ion, Chem. Sci., 2015, 6, 3044–3050 RSC; (e) H. Zhu, P. Chen and G. Liu, Pd-Catalyzed intramolecular aminohydroxylation of alkenes with hydrogen peroxide as oxidant and water as nucleophile, J. Am. Chem. Soc., 2014, 136, 1766–1769 CrossRef CAS PubMed; (f) D. Huang, X. Liu, L. Li, Y. Cai, W. Liu and Y. Shi, Enantioselective bromoaminocyclization of allyl N-tosylcarbamates catalyzed by a chiral phosphine-Sc(OTf)3 c omplex, J. Am. Chem. Soc., 2013, 135, 8101–8104 CrossRef CAS PubMed; (g) S. D. R. Christie, A. D. Warrington and C. J. Lunniss, Complemetary reactions of allylic carbamates using palladium(II): formation of oxazolidinones or allylic amides from a common precursor, Synthesis, 2009, 148–154 CrossRef CAS; (h) E. J. Alexanian, C. Lee and E. J. Sorensen, Palladium-catalyzed ring-forming aminoactoxylation of alkenes, J. Am. Chem. Soc., 2005, 127, 7690–7691 CrossRef CAS PubMed.
- H. Pan, H. Huang, W. Liu, H. Tian and Y. Shi, Phosphine oxide-Sc(OTf)3 c atalyzed highly regio- and enantioselective bromoaminocyclization of (E)-cinnamyl tosylcarbamates. An approach to a class of synthetically versatile functionalized molecules, Org. Lett., 2016, 18, 896–899 CrossRef CAS PubMed.
- (a) A. Kondoh, Y. Kamata and M. Terada, Synthesis of enantioenriched γ-amino-α,β-unsaturated esters utilizing palladium catalyzed rearrangement of allylic carbamates for direct application to formal [3 + 2] cycloaddition, Org. Lett., 2017, 19, 1682–1685 CrossRef CAS PubMed; (b) J. Moritz Bauer, W. Frey and R. Peters, Dual palladium(II)/tertriary amine catalysis for asymmetric regioselective rearrangements of allylic carbamates, Chem. – Eur. J., 2016, 22, 5767–5777 CrossRef PubMed; (c) J. Moritz Bauer, W. Frey and R. Peters, Asymmetric cascade reaction to allylic sulfonamides from allylic alcohols by palladium(II)/base-catalyzed rearrangement of allylic carbamates, Angew. Chem., Int. Ed., 2014, 53, 7634–7638 ( Angew. Chem. , 2014 , 126 , 7764–7768 ) CrossRef PubMed; (d) D. Xing and D. Yang, Gold(I)-catalyzed highly regio- and stereoselective decarboxylative amination of allylic N-tosylcarbamates via base-induced aza-Claisen rearrangement in water, Org. Lett., 2010, 12, 1068–1071 CrossRef CAS PubMed; (e) O. V. Singh and H. Han, Iridium(I)-catalyzed stereospecific decarboxylative allylic amidation of chiral branched benzyl allyl imidodicarboxylates, Org. Lett., 2007, 9, 4801–4804 CrossRef CAS PubMed; (f) M. E. Synerholm, N. W. Gilman, J. W. Morgan and P. K. Hill, Thermal rearrangement of allylic phenylurethanes, J. Org. Chem., 1968, 33, 1111–1116 CrossRef CAS.
- (a) M. Chrzanowska, A. Grajewska and M. D. Rozwadowska, Asymmetric synthesis of isoquinoline alkaloids: 2004-2015, Chem. Rev., 2016, 116, 12369–12465 CrossRef CAS PubMed; (b) C. Stanciu and T. Penders, Hallucinogen persistent perception disorder induced by new psychoactive substituted phenethylamines, a review with illustrative case, Curr. Psychiatry Rev., 2016, 12, 221–223 CrossRef CAS; (c) B. V. Dean, S. J. Stellpflug, A. M. Burnett and K. M. Engebretsen, 2C or not 2C: phenethylamine designer drug review, J. Med. Toxicol., 2013, 9, 172–178 CrossRef CAS PubMed; (d) M. RodrÍguez-Mata, V. Gotor-Fernández, J. González-SabÍn, F. Rebolledo and V. Gotor, Straightforward preparation of biologically active 1-aryl and 1-heteriarylpropan-2-amines in enantioenriched form, Org. Biomol. Chem., 2011, 9, 2274–2278 RSC; (e) M. Vilches-Herrera, J. Miranda-Sepúlveda, M. Rebolledo-Fuentes, A. Fierro, S. Lühr, P. Iturriaga-Vasquez, B. K. Cassels and M. Reyes-Parada, Naphthylisopropylamine and N-benzylamphetamine derivatives as monoamine oxidase inhibitors, Bioorg. Med. Chem., 2009, 17, 2452–2460 CrossRef CAS PubMed; (f) K. W. Bentley, β-Phenylethylamines and the isoquinoline alkaloids, Nat. Prod. Rep., 2006, 23, 444–463 RSC; (g) T. Hasegawa and H. Yamamoto, Development of new chiral auxiliary derived from (S)-(-)-phenylethylamine for a synthesis of enantiopure (R,)-2-propyloctanoic acid, Synthesis, 2003, 1181–1186 CAS.
- Since – in contrast to mesitylene – the variously substituted aromatic reagents do not lend themselves to be used as solvents, and in view of limiting their superstochiometric use, the reactions performed in Table 3 and 4 were carried out in chlorobenzene or dichloroethane, using 5.0 equivalents of the aromatic reagent.
- C. Michon, F. Medina, F. Capet, P. Roussel and F. Agbossou-Niedercorn, Inter- and intramolecular hydroamination of unactivated alkenes catalysed by combination of copper and silver salts: the unveiling of Brønsted acid catalyst, Adv. Synth. Catal., 2010, 352, 3293–3305 CrossRef CAS.
- (a) Y.-B. Kang and L. H. Gade, The nature of the catalytically active species in olefin deoxygenation with PhI(OAc)2: metal or proton?, J. Am. Chem. Soc., 2011, 133, 3658–3667 CrossRef CAS PubMed; (b) T. T. Dang, F. Boeck and L. Hinterman, Hidden Brønsted acid catalysis: pathways of accidental or delicerate generation of triflic acid from metal triflates, J. Org. Chem., 2011, 76, 9353–9361 CrossRef CAS PubMed; (c) M. Tschan, C. M. Thomas, H. Strub and J.-F. Carpentier, Copper(II) triflate as a source of triflic acid: effective, green catalyst of hydroalkoxylation reactions, Adv. Synth. Catal., 2009, 351, 2496–2504 CrossRef CAS.
- J. G. Taylor, N. Whittall and K. K. Hii, Copper-catalyzes intermolecular hydroamination of alkenes, Org. Lett., 2006, 8, 3561–3564 CrossRef CAS PubMed.
- (a) M. Sai, Direct synthesis of indanes via iron-catalyzed dehydrative coupling/Friedel-Crafts cyclization of two different alcohols, Eur. J. Org. Chem., 2019, 1102–1106 CrossRef CAS; (b) Y. Li, L. Zhang, Z. Zhang, J. Xu, Y. Pan, C. Xu, L. Liu, Z. Li, Z. Yu, H. Li and L. Xu, Regio- and stereoselective synthesis of 1,2,3-trisubstiteted indanes from diarylmethanols and allylamides through iron(III) chloride hexahydrate, Adv. Synth. Catal., 2016, 358, 2148–2155 CrossRef CAS; (c) E. Nomura, Y. Kakimoto, T. Yamamoto, T. Noda, Y. Miyake and H. Mori, Dimerization of ferulic acid and structure determination of phenylindane derivatives, Res. Chem. Intermed., 2016, 42, 3567–3577 CrossRef CAS; (d) O. A. Ivanova, E. M. Budynina, D. A. Skortsov, M. Limoge, A. V. Bakin, A. O. Chagarovskiy, I. V. Trushkov and M. Y. Melnikov, A bioinspired route to indanes and cyclopentannulated hetarenes via, (3 + 2)-cyclodimerization of donor-acceptor cyclopropanes, Chem. Commun., 2013, 49, 11482–11484 RSC; (e) V. V. Kouznetsov and D. R. M. Arenas, First green protocols for the large-scale preparation of γ-diisoeugenol and related dihydro (1H) indenes via formal [3 + 2] cycloaddition reactions, Tetrahedron Lett., 2009, 50, 1546–1549 CrossRef CAS; (f) A. R. Katritzky, G. Zhang and L. Xie, Novel route to functionalized indans via formal [3 + 2] cycloadditions of 1-benzylbenzotriazoles with alkenes, Synth. Commun., 1997, 27, 2467–2478 CrossRef CAS; (g) S. R. Angle and D. O. Arnaiz, Formal [3 + 2] cycloaddition of benzylic cations with alkenes, J. Org. Chem., 1992, 57, 5937–5947 CrossRef CAS.
- C. L. Ricardo, X. Mo, J. A. McCubbin and D. G. Hall, A surprising substituent effect provides a superior boronic acid catalyst for mild and metal-free direct Friedel-Crafts alkylations and prenylations of neutral arenes, Chem. – Eur. J., 2015, 21, 4218–4223 CrossRef CAS PubMed.
- (a) G. K. Surya Prakash, T. Mathew, C. Panja, A. Kulkarni, G. A. Olah and M. A. Harmer, Tetraflic acid (1,1,2,2-tetrafluoroethanesulfonic acid, HC2F4SO3H) and gallium tetraflate as effective catalysts in organic synthesis, Adv. Synth. Catal., 2012, 354, 2163–2171 CrossRef; (b) G. K. Surya, Y. Zhang, L. Chen and T. Lu, A copper(II) triflate-catalyzed tandem Friedel-Crafts alkylation/cyclization process towards dihydroindenes, Adv. Synth. Catal., 2011, 353, 1055–1060 CrossRef; (c) G. A. Olah and A. Molnár, in Hydrocarbon Chemistry, John Wiley and Sons, 2003, pp. 160–214 Search PubMed; (d) M. A. Lanewala and A. P. Bolten, Isomerization of the xylenes using zeolite catalysts, J. Org. Chem., 1969, 34, 3107–3112 CrossRef CAS; (e) R. H. Allem and L. D. Yats, Kinetics of the three-compound equilibrations. II. The isomerization of xylene, J. Am. Chem. Soc., 1959, 81, 5289–5292 CrossRef; (f) H. C. Brown and H. Jungk, The isomerization of o- and p-xylenes and some related alkylbenzenes under the influence of hydrogen bromide and aluminium bromide; the relative isomerization aptitudes of alkyl groups, J. Am. Chem. Soc., 1955, 77, 5579–5584 CrossRef CAS; (g) D. A. McCaulay and A. P. Lien, Isomerization of the methylbenzenes, J. Am. Chem. Soc., 1952, 74, 6246–6250 CrossRef CAS.
- E. J. Eisenbraun, J. R. Mattox, R. C. Bansal, M. A. Wilhelm, P. W. K. Flanagan, A. B. Carel, R. E. Laramy and M. C. Hamming, Polyalkyl aromatic hydrocarbons. II. Cyclialkylation of benzenoid hydrocarbons with isoprene, J. Org. Chem., 1968, 33, 2000–2008 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2qo00114d |
| This journal is © the Partner Organisations 2022 |